
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel star-shaped molecules based on a 1,3,5-triazine core linked to different heterocyclic systems as novel hybrid molecules†
Hadeer M. Diab,
Mostafa E. Salem,
Ismail A. Abdelhamid * and
Ahmed H. M. Elwahy*
* and
Ahmed H. M. Elwahy*
Department of Chemistry, Faculty of Science, Cairo University, Giza 12613, Egypt. E-mail: ismail_shafy@yahoo.com; aelwahy@hotmail.com
First published on 14th December 2020
Abstract
The synthesis of novel star-shaped compounds based on an s-triazine core and linked to hexahydroacridinediones, pyrimido[4,5-b]quinolones, 1H-isoquinolino[2,1-a]quinolines, tetrahydro-4H-chromenes, dihydropyrano[2,3-c]pyrazoles, thiazole, or benzothiazole as new hybrid molecules through Michael and Hantzsch reactions is reported. For this purpose, 2,4,6-tris(4-formylphenoxy)benzaldehyde was used as a versatile precursor.
Introduction
Star-shaped molecules have attracted considerable attention in recent decades because of their large application as promising compounds for use in optoelectronics and electrochromic devices1–3 as well as in organic solar cells (OSCs).4–6 They are also considered as building blocks for the creation of mesophases with interesting mesomorphic and photophysical properties.7–9 Applications of star-shaped molecules as building units for dendrimers10 as well as in supramolecular host-guest chemistry have also been reported.11 In this regard, we recently reviewed the synthesis and application of star-shaped molecules.12In particular, star-shaped molecules that contain 1,3,5-triazine as a central core have been found to play an important role as powerful chelating agents13 and many of their derivatives have been employed in combinatorial and supramolecular chemistry.14 The planarity of 1,3,5-triazine moiety and its symmetric nature plays a key role in self-organizing ability which enhances their use in the development of organic light-emitting diodes,15,16 liquid crystalline materials,6,17,18 dendrimers,19,20 and nonlinear optical materials.21–23 In addition, some s-triazine derivatives were reported as corrosion inhibitors for mild steel in 1 M HCl solution.24 Moreover, some s-triazine derivatives have recently found extensive use as reagents in the conversion of functional groups.25
Moreover, s-triazine derivatives are an interesting class of compounds due to their diverse pharmacological activities as antibacterial,26–28 antifungal,29 antiviral,30,31 antimalarials,32,33 antiprotozoals,34 anti-asthmatic activity,35 estrogen receptor modulators,36 cyclindependent kinase inhibitors.37,38 In addition, the use of s-triazine derivatives as anticancer agents has also been extensively reported. In this regard, 1,3,5-triazine scaffold is present in some anticancer drugs, such as altretamine, trimelamol and irsogladine (Fig. 1).39–42
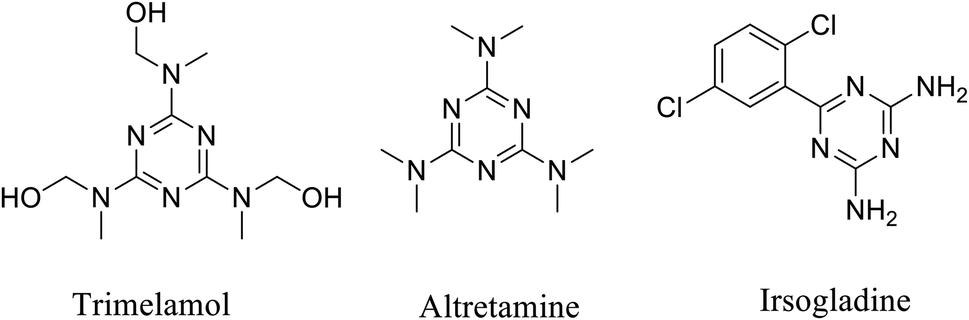 | ||
| Fig. 1 Some anticancer drugs incorporating 1,3,5-triazine scaffold. | ||
Furthermore, the development of hybrid molecules through the combination of different pharmacophores in one molecule may improve their biological efficacy and overcoming drug resistance.43–45 In this aspect, heterocyclic hybrid skeleton comprising 1,3,5-triazine and different heterocyclic systems were found to exhibit modified therapeutic activities.33,46–49 Synthetic chemistry is a highly creative discipline due to its ability to create new methodologies to contribute to the discovery of new drugs, and to enable the synthesis of important molecules with novel properties and functions in reasonable yields with a direct impact on the welfare of the world. Motivated by these findings and in conjunction with our ongoing research work on poly(heterocycles) as well as the new concept in drug design,50–67 we report herein on the synthesis of novel hybrid molecules containing 1,3,5-triazine linked to different heterocyclic systems.
Results and discussion
In general, the synthesis of star-shaped compounds bearing the s-triazine core was achieved starting from 2,4,6-trichloro-1,3,5-triazine due to the simple displacement of chlorine atoms by different nucleophiles. These reactions open access to many useful molecules of important applications in medicinal chemistry as well as in material science.12 We utilized this strategy to synthesize 4,4′,4′′-((1,3,5-triazine-2,4,6-triyl)tris(oxy))tribenzaldehyde (3), by a modified procedure to some reported methods,68–70 as a precursor for a variety of star-shaped compounds based on s-triazine. Thus, the reaction of three equivalents of potassium 4-formylphenolate (obtained upon treatment of p-hydroxybenzaldehyde 2 with KOH in ethanol) with one equivalent of 2,4,6-trichloro-1,3,5-triazine 1 in dimethylformamide at 0 °C afforded 3 in 82% yield (Scheme 1).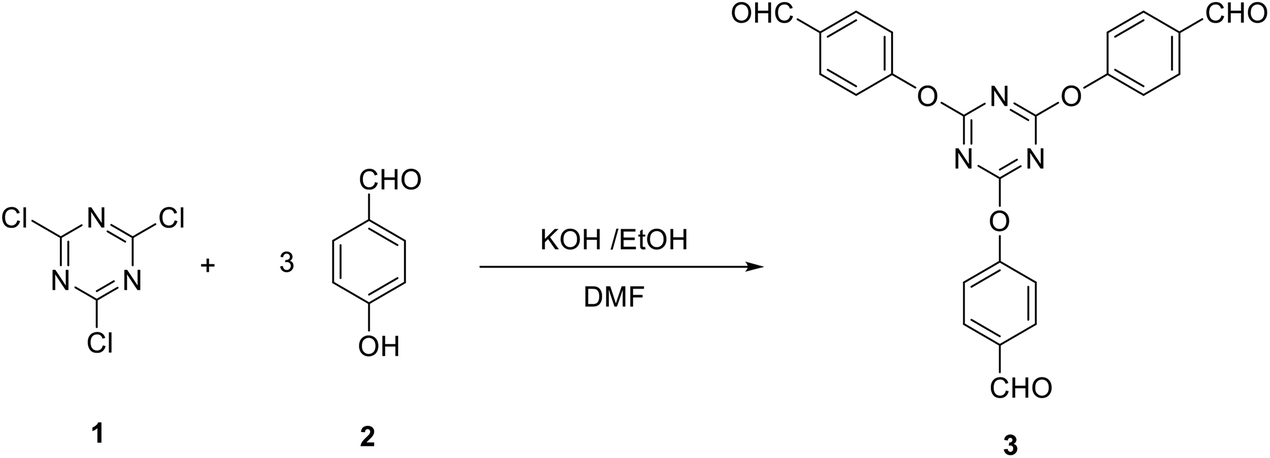 | ||
| Scheme 1 Synthesis of tris-aldehyde 3. | ||
Firstly, we investigate the reactivity of tris(aldehyde) 3 towards the synthesis of tris(2,6-dimethyl-1,4-dihydropyridine-3,5-dicarbonitrile) 5 through reaction with 6 equivalents of 3-aminobut-2-enenitrile 4 in acetic acid under reflux. Compound 5 was successfully obtained in 88% yield (Scheme 2).
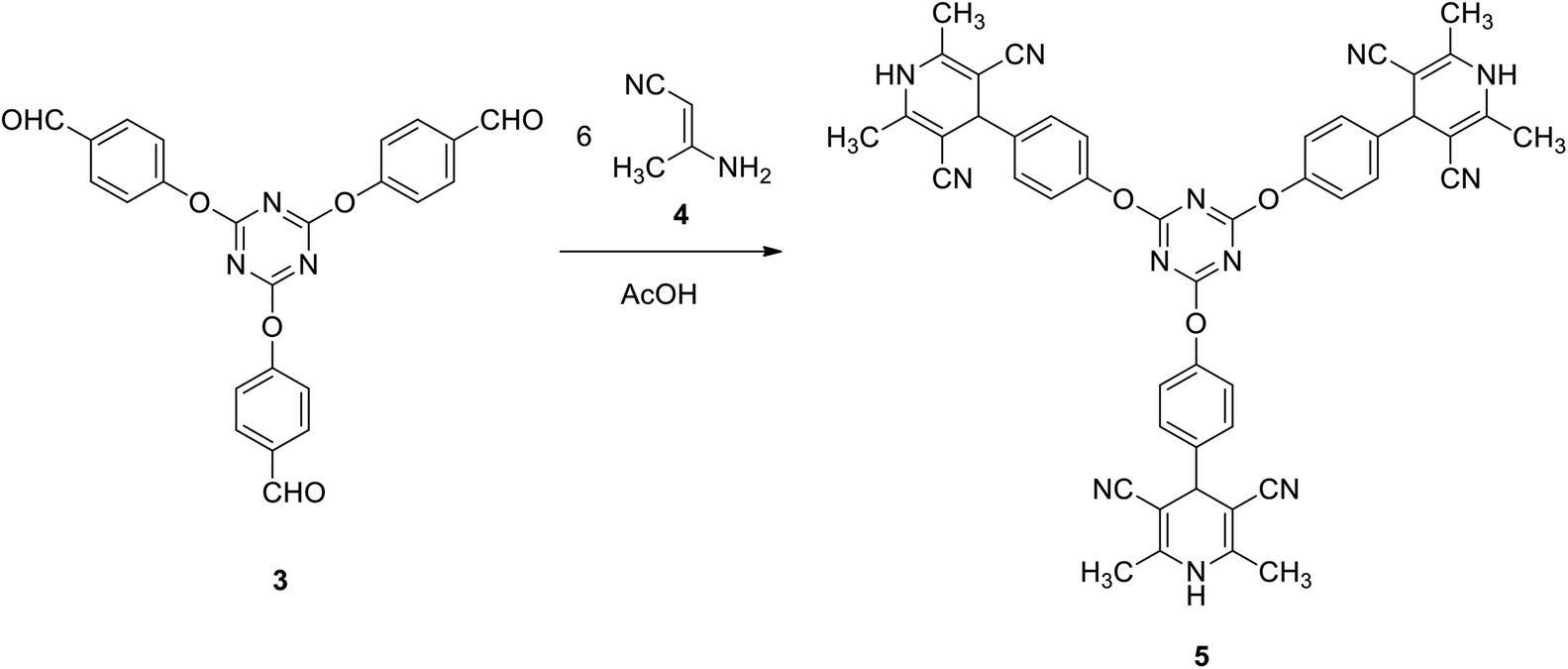 | ||
| Scheme 2 Synthesis of tris(2,6-dimethyl-1,4-dihydropyridine-3,5-dicarbonitrile) 5. | ||
The structure of compound 5 was confirmed spectroscopically, as the 1H NMR spectrum revealed a characteristic singlet integrated by 18H at δ 2.06 for the six methyl groups. It also showed a singlet signal at δ 4.47 for the pyridine-H. In addition, it exhibited a singlet signal characteristic for the NH group at δ 9.58. Furthermore, the 13C NMR spectrum of 5 was found to be in agreement with the proposed structure, it showed the methyl signal at δ 18.3 and the pyridine-C at δ 40.7.
Moreover, tris(hexahydroacridine-1,8-diones) 7 in which the acridinedione moiety is connected to 1,3,5-triazine core via ether linkage can also be obtained in good yield via multi-component reaction of dimedone 6 with tris(aldehyde) 3 and ammonium acetate in acetic acid at reflux (Scheme 3).
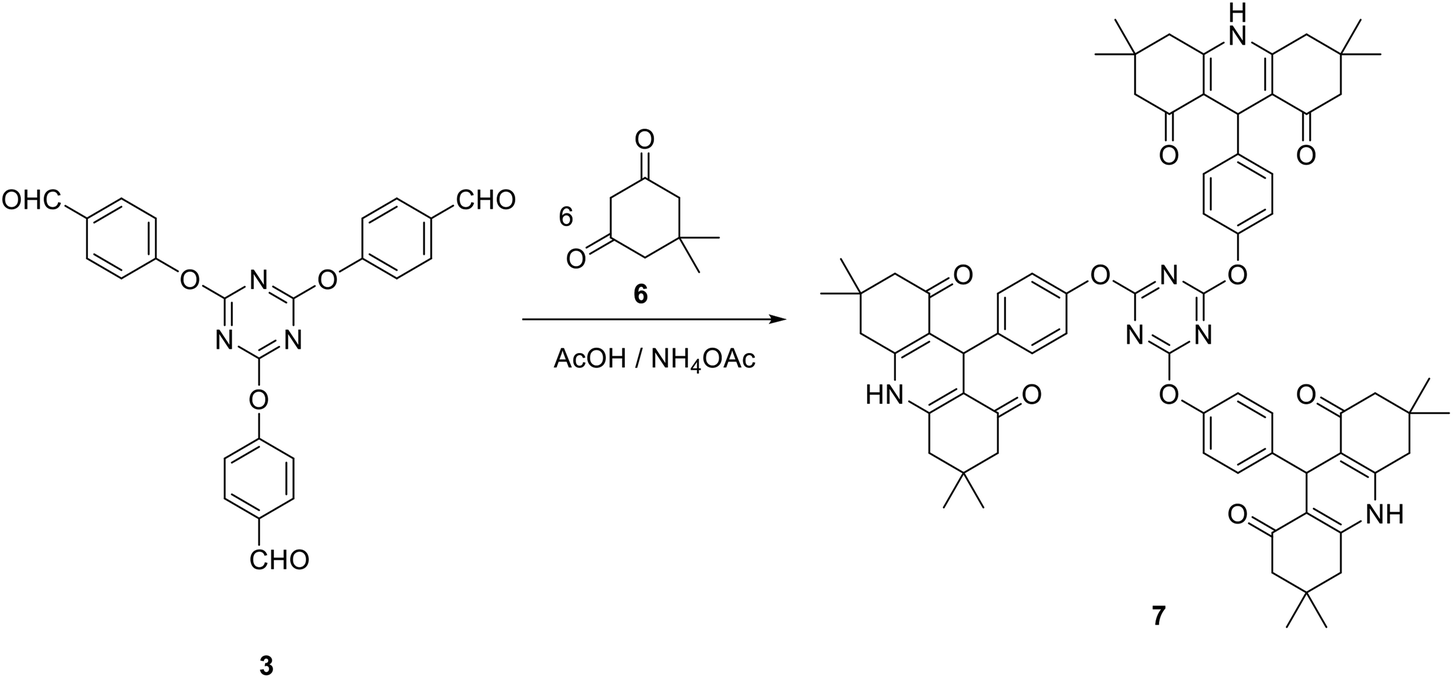 | ||
| Scheme 3 Synthesis of tris(hexahydroacridine-1,8-diones) 7. | ||
The IR spectra of compound 7 suggested the presence of NH group at 3277 cm−1. In addition, the carbonyl group appeared at 1712 cm−1. The 1H NMR spectrum of 7 suggested the existence of two singlets integrated by 36 protons at δ 0.87 and δ 1.01 allocated to twelve CH3 groups. Moreover, the singlet signal at δ 4.70 is corresponding to H9. The NH group appeared as a broad singlet signal at δ 9.20. All other signals appeared at their expected positions. Moreover, the 13C NMR spectrum of 7 was found to be in accordance with the proposed structure, it showed the C9 at δ 32.1 and the carbonyl group at δ 194.8. All other carbon signals appeared at their expected positions.
Besides, the three-component Hantzsch-like reaction of tris (aldehyde) 3 with three equivalents of each of 6-aminouracil 8 and 5,5-dimethyl-1,3-cyclohexanedione 6 in acetic acid at reflux in the presence of p-TSA as a catalyst yielded the corresponding tris(pyrimido[4,5-b]quinolines) 9 in 72% yield. In this case, the uncyclized adduct 10 has not been obtained (Scheme 4).
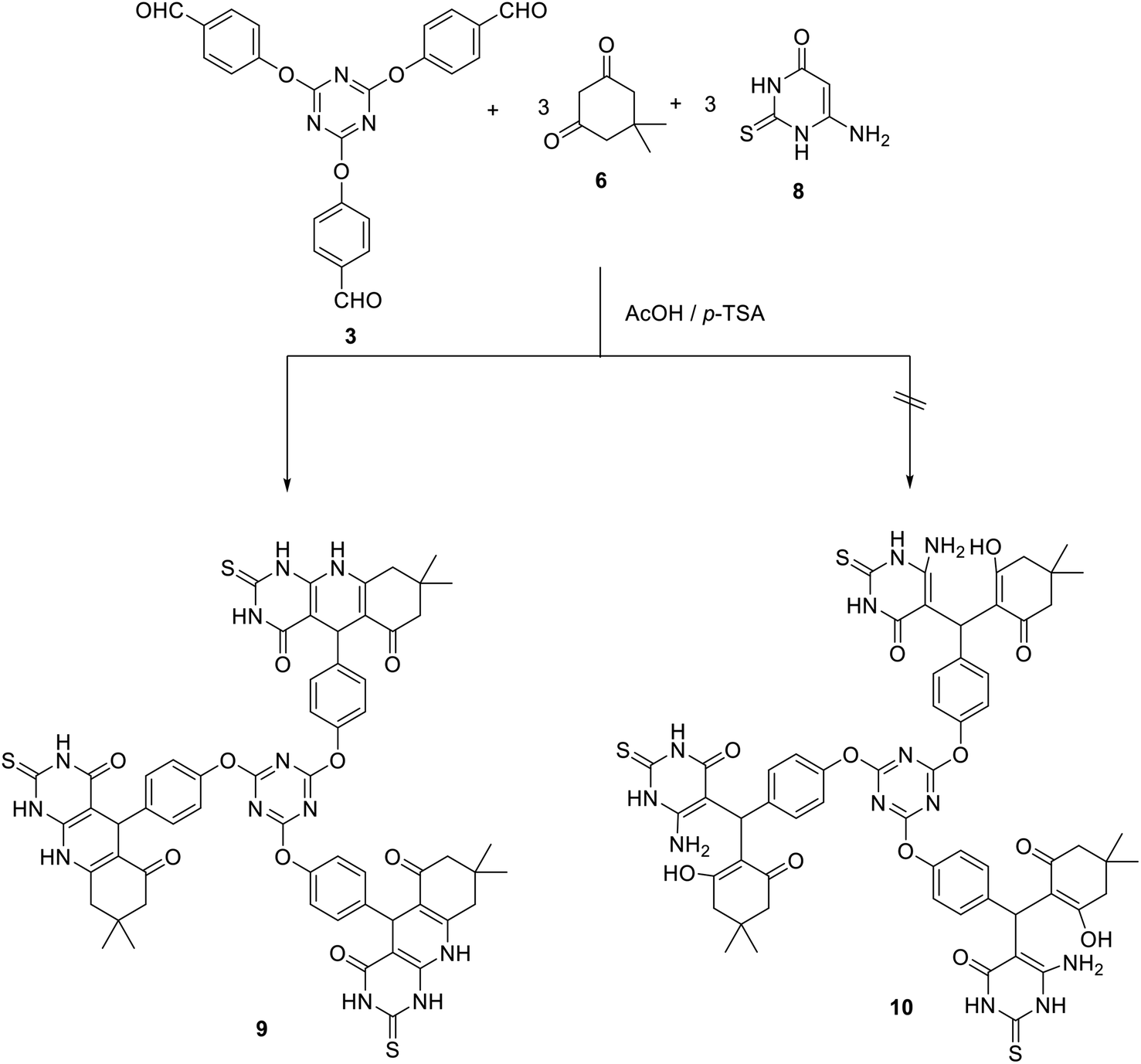 | ||
| Scheme 4 Synthesis of tris(pyrimido[4,5-b]quinolines) 9. | ||
The IR spectrum of compound 9 suggested the presence of NH groups at 3200 cm−1. In addition, the carbonyl groups appeared around 1668 cm−1. The 1H NMR spectrum of 9 suggested the existence of a singlet signal at δ 4.81 corresponding to H5. The NH group appeared as broad signals at δ 8.59, 11.89, and 12.01. All other signals appeared at their expected positions. Moreover, the 13C NMR spectrum of 9 showed the C5 at δ 40.2 and the carbonyl group at δ 173 and 194.4.
The synthesis of tris(1H-isoquinolino[2,1-a]quinoline-12-carbonitrile) 12 was also studied by the reaction of tris-aldehyde 3 with 2-(6,7-dimethoxy-3,4-dihydroisoquinolin-1-yl)acetonitrile 1171 and dimedone 6 in acetic acid at reflux. The cyclocondensation reaction can also lead to the formation of tris(5H-isoquinolino[3,2-a]isoquinoline-13-carbonitrile) 13 (Scheme 5).
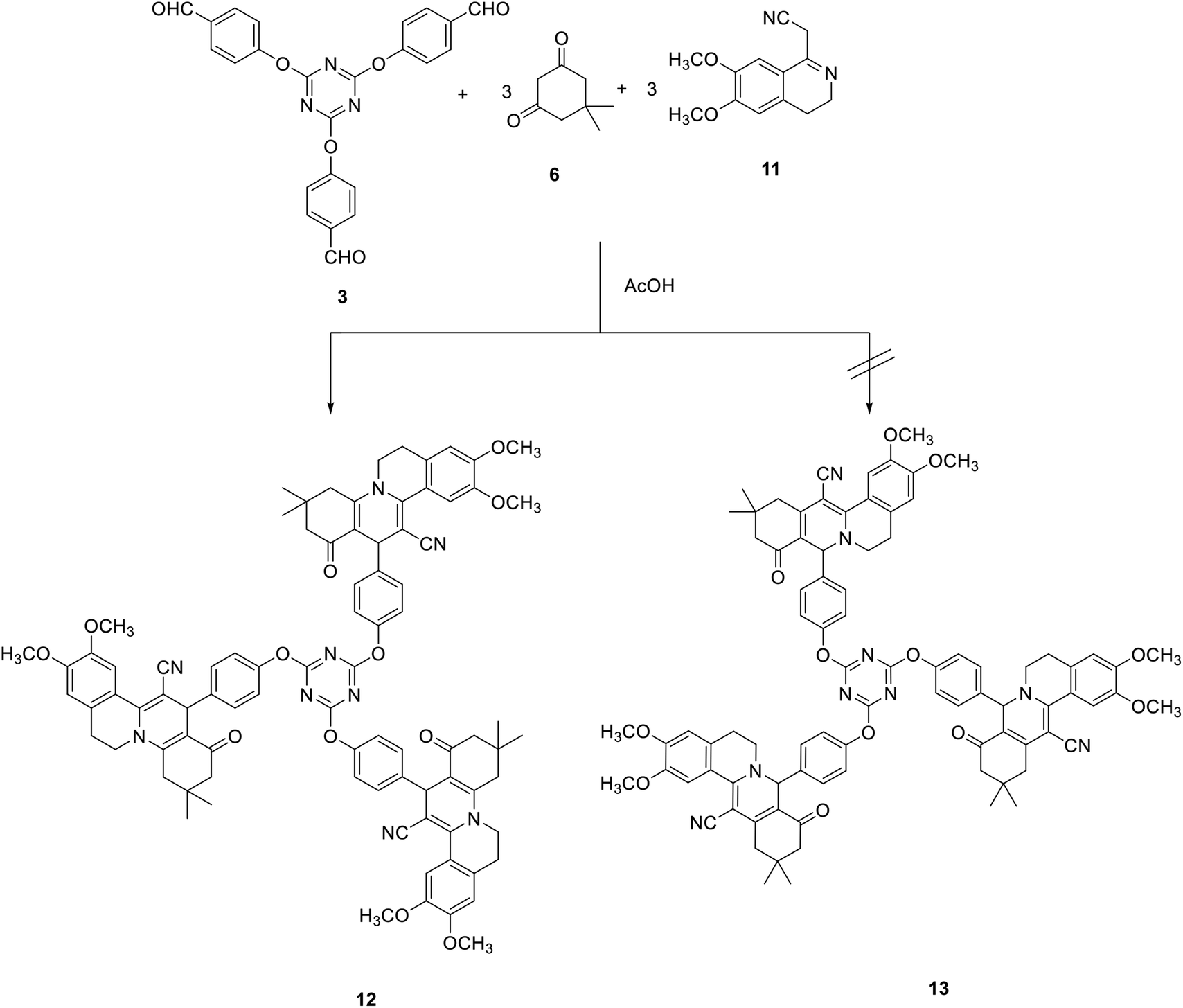 | ||
| Scheme 5 Synthesis of tris(1H-isoquinolino[2,1-a]quinoline-12-carbonitrile) 12. | ||
The regioselectivity was approved on the basis of recent literature supporting the formation of 9,10-dimethoxy-13-(4-methoxyphenyl)-3,3-dimethyl-1-oxo-2,3,4,6,7,13-hexahydro-1H-isoquinolino[2,1-a]quinoline-12-carbonitrile 14 (Fig. 2) using X-ray crystallography and 2D-HMBC spectroscopy via the Hantzsch-like reaction of 4-methoxybenzaldehyde with 2-(6,7-dimethoxy-3,4-dihydroisoquinolin-1-yl)acetonitrile and dimedone in the presence of acetic acid.72
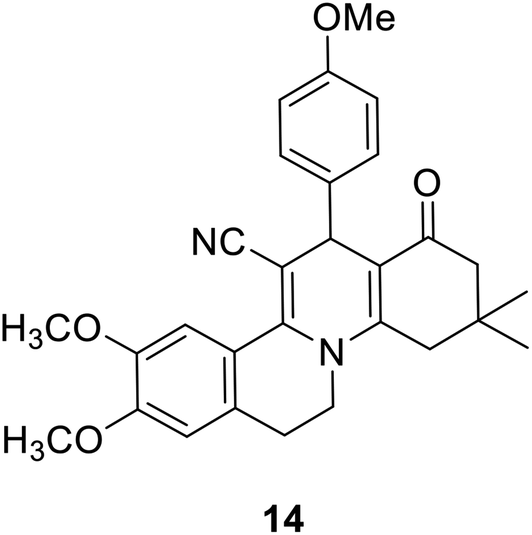 | ||
| Fig. 2 Structure of hexahydro-1H-isoquinolino[2,1-a]quinoline-12-carbonitrile 14. | ||
The IR spectra of compound 12 indicated the presence of the cyano group at 2188 cm−1 and the carbonyl group as a broad band at 1629 cm−1. The 1H NMR spectra of 12 revealed a characteristic singlet signal at δ 4.71 for H-13. In addition, the 13C NMR spectrum of compound 12 featured the pyridine-C13 at 37.7 ppm.
Our study was expanded to include the use of Michael addition reactions as an effective method for the synthesis of novel tris(2-amino-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile) 16. Thus, compound 16 was successfully obtained by a multicomponent reaction of one equivalent of tris-aldehydes 3 with three equivalents of both malononitrile 15 and dimedone 6 in EtOH/piperidine (Scheme 6).
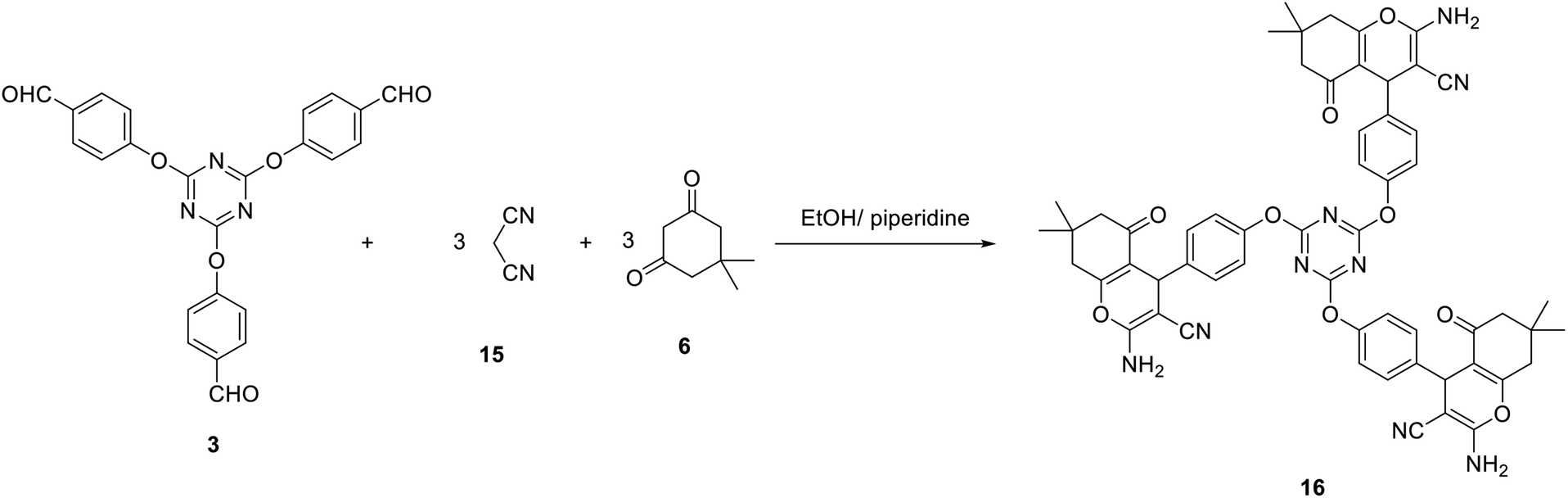 | ||
| Scheme 6 Synthesis of tris(5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile) 16. | ||
The constitution of compound 16 was spectroscopically determined based on the basis of elemental analysis and spectral data. The IR spectra of compound 16 indicated the presence of amino group at 3365 and 3313 cm−1. In addition, it revealed the cyano group at 2190 cm−1. The carbonyl group appeared as a broad band at 1684 cm−1. The 1H-NMR spectrum of 16 suggested the existence of two singlets integrated by 18 protons at δ 0.94 and δ 1.04 assigned to six CH3. In addition, it revealed the pyran-H4 as a singlet signal at δ 4.22 ppm.
Likewise, we have also successfully demonstrated the synthesis of tris(6-amino-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile) 19 and tris(benzene-4,1-diyl)tris(2-amino-5-oxo-4,5-dihydropyrano[3,2-c]chromene-3-carbonitrile) 20 in 73 and 82% yields by a three-component reaction of one mole of tris-aldehyde 3 with three moles of both of malononitrile 15 and pyrazolone 17 (in ethanol in the presence of catalytic amount piperidine) or 4-hydroxycoumarin 18 (in the presence of acetic acid/sodium acetate) (Scheme 7).
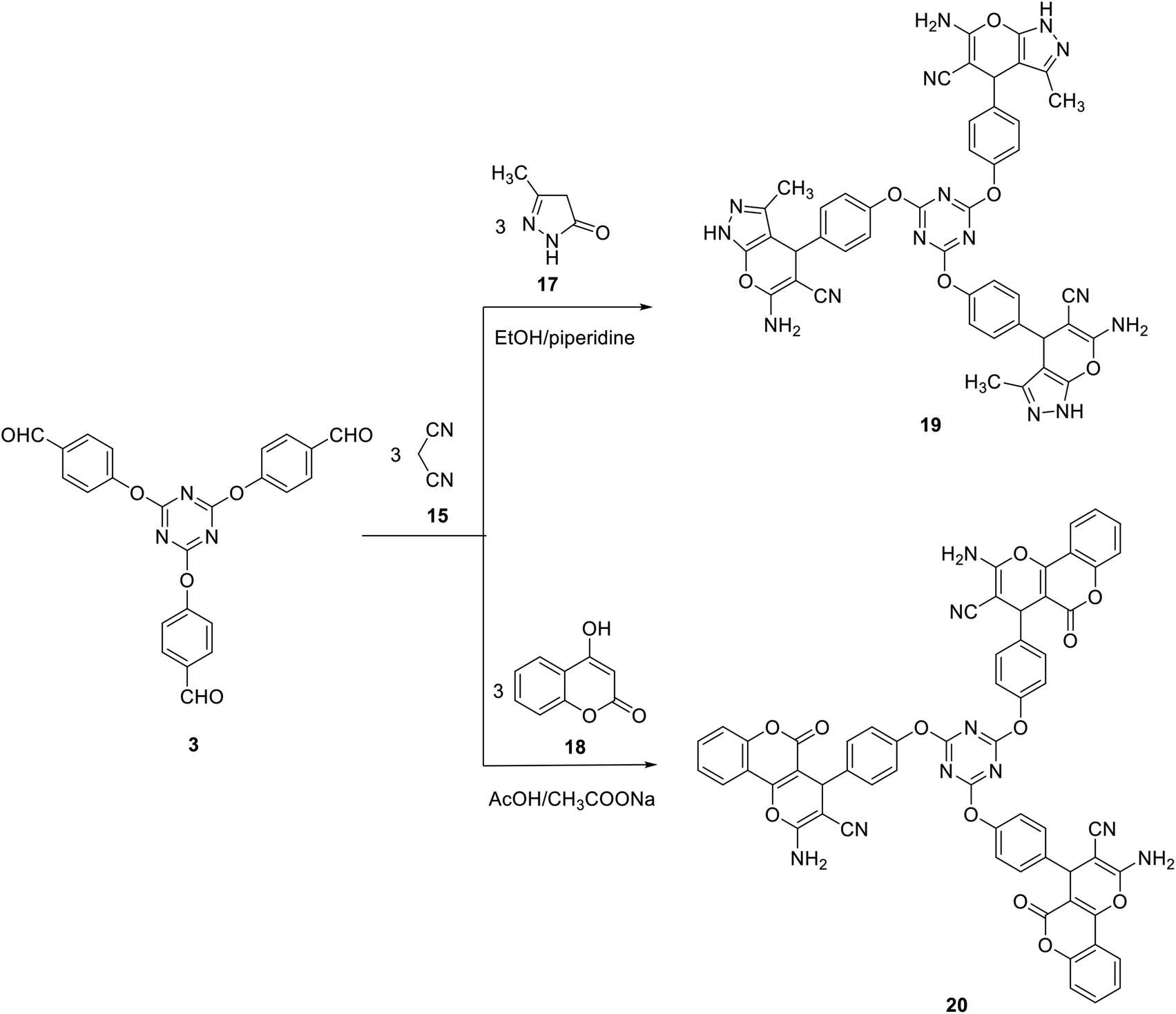 | ||
| Scheme 7 Synthesis of tris(dihydropyrano[2,3-c]pyrazole-5-carbonitrile) 19 and tris(dihydropyrano[3,2-c]chromene-3-carbonitrile) 20. | ||
The structure of compound 19 was confirmed by their elemental analysis and spectral results. Thus, IR spectrum of compound 19 indicated the presence of amino group at ν 3295 and 3168 cm−1 and a cyano group at ν 2187 cm−1. The 1H NMR spectrum indicated the presence of the pyran-H4 and the pyrazole methyl protons as two singlet signals at δ 4.64 and δ 1.78, respectively.
The infrared spectra of compound 20 indicated the presence of amino group at ν 3255 and 3184 cm−1. In addition, it revealed the cyano band at ν 2197 cm−1. The carbonyl group appeared as a broad band at ν 1715 cm−1. The 1H NMR spectrum of 20 indicated the presence of the pyran-H4 as a singlet signal at δ 4.49.
The utility of tris-aldehyde 3 as a building block for novel tris(hexahydro-1H-xanthene-1,8(2H)-dione) 22 has also been attempted. Unfortunately, the reaction of one equivalent of 3 with six equivalents of dimedone (6) in the presence of 15 mol% of p-TSA in a mixture of ethanol/H2O (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) or DCE as solvents did not lead to the formation of 22 and instead the interesting tris(tetraketone) 21 was obtained in 83% yield (Scheme 8).
:1) or DCE as solvents did not lead to the formation of 22 and instead the interesting tris(tetraketone) 21 was obtained in 83% yield (Scheme 8).
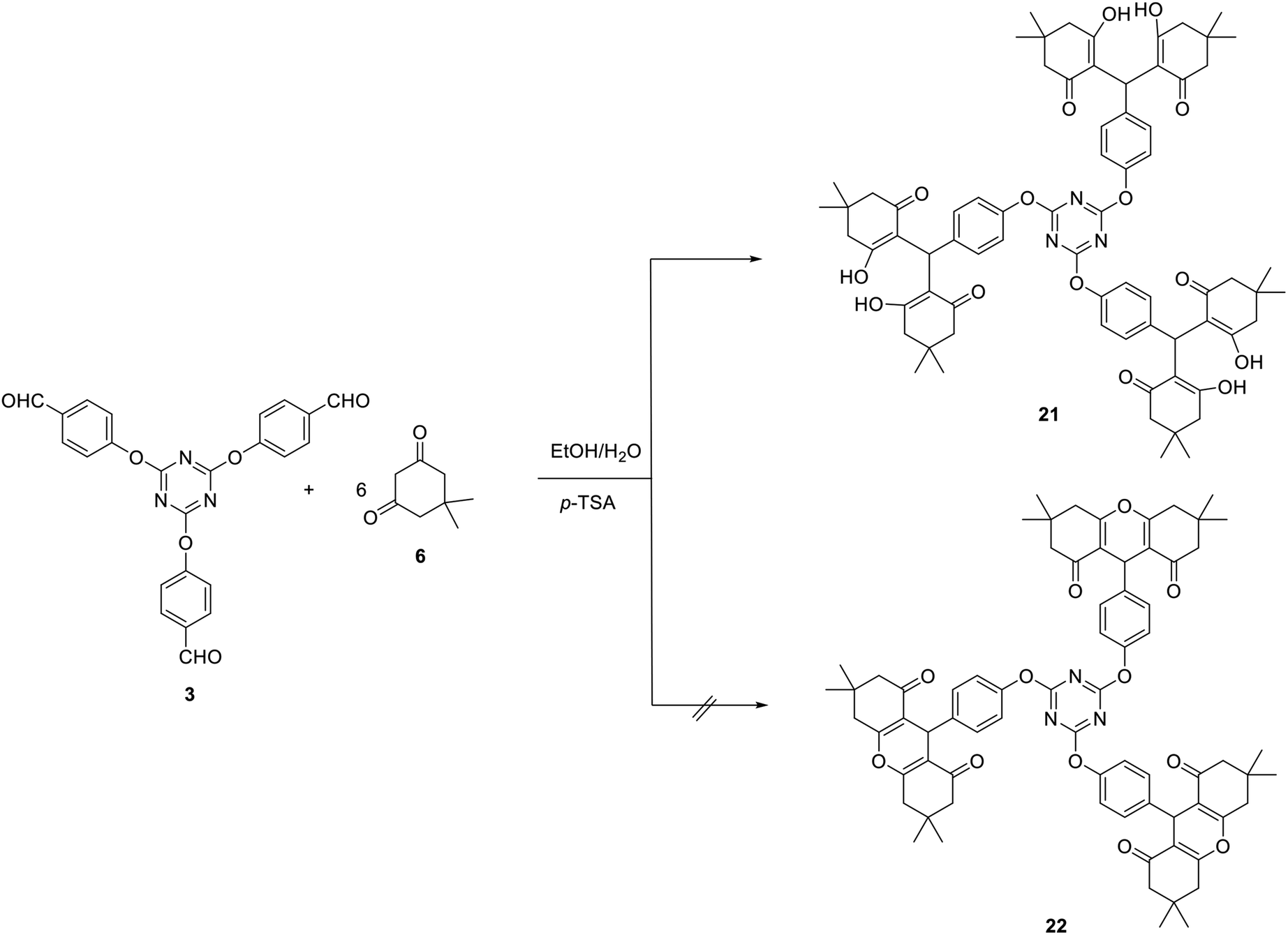 | ||
| Scheme 8 Synthesis of tris(tetraketone) 21. | ||
Tetraketone derivatives are considered not only as an important class of biologically active compounds but also as significant precursors for the synthesis of various fused heterocyclic.73
The structure of compound 21 was spectroscopically verified. In the IR spectrum, the carbonyl and the hydroxyl stretching frequencies were noticed at 1650 and 3345 cm−1, respectively. The 1H NMR spectrum of 21 displayed a broad signal at δ 9.20 characteristic for the OH protons in addition to the methine-H at δ 4.42.
Subjecting the tris(aldehyde) 3 to the cyclocondensation reaction with 2-aminothiophenol 23 in ethanol at reflux in the presence of NaHSO3 afforded the tris(benzothiazole) 24 in 61% yield (Scheme 9). Moreover, the three-component cyclocondensation reaction of tris-aldehyde 3 with 9,10-phenanthrenequinone 25 and ammonium acetate afforded the corresponding tris(1H-phenanthro[9,10-d]imidazole) 26 as a new building block for blue light-emitting materials (Scheme 9).74
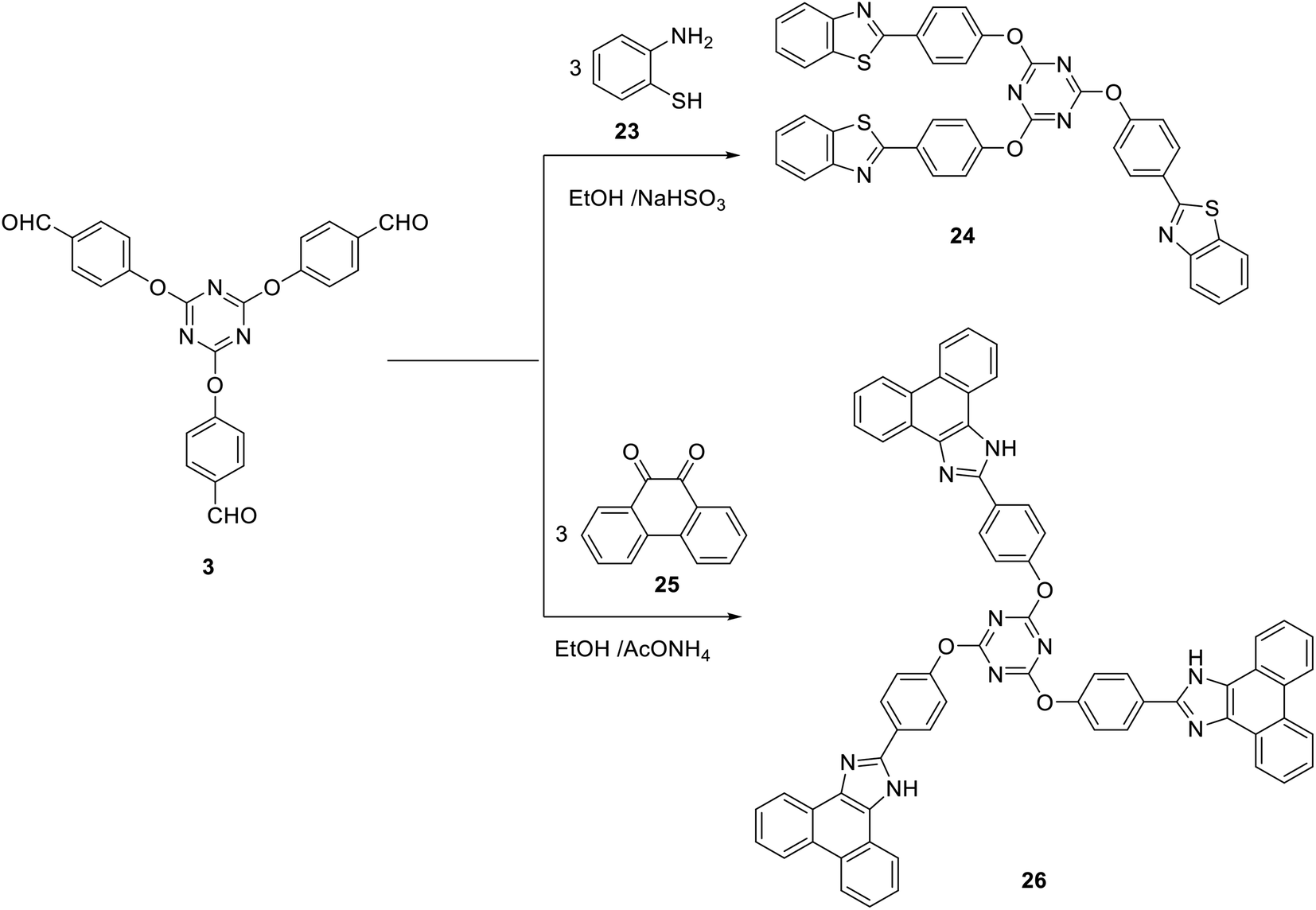 | ||
| Scheme 9 Synthesis of tris(benzothiazole) 24 and tris(1H-phenanthro[9,10-d] imidazole) 25. | ||
Compound 24 was confirmed by the absence of characteristic absorption bands or signals for CHO, NH2, or SH in its IR or 1H NMR spectra. The structure of the tris(imidazole) 26 was defined on the basis of spectral data. Thus, its IR spectra indicated the absence of a peak characteristic for a carbonyl group. In addition, it revealed the absorption of the NH group at 3305 cm−1. The 1H NMR spectrum of 26 indicated the presence of a singlet signal integrated by three protons at δ 13.47 assigned to the NH protons.
The synthetic utility of tris-aldehyde 3 as building blocks for novel tris(thiazoles) through Hantzsch thiazole synthesis was also investigated. Thus, the tris(aldehyde thiosemicarbazone) 28 was first synthesized in 90% yield, by acid-catalyzed condensation of thiosemicarbazide 27 with tris(aldehyde) 3.75 Reaction of 28 with the corresponding N-(4-chlorophenyl)-2-oxopropanehydrazonoyl chloride 29 in refluxing ethanol/DMF in the presence of TEA as a catalyst gave the corresponding tris(arydiazenyl)thiazole 30 in 85% yield (Scheme 10).
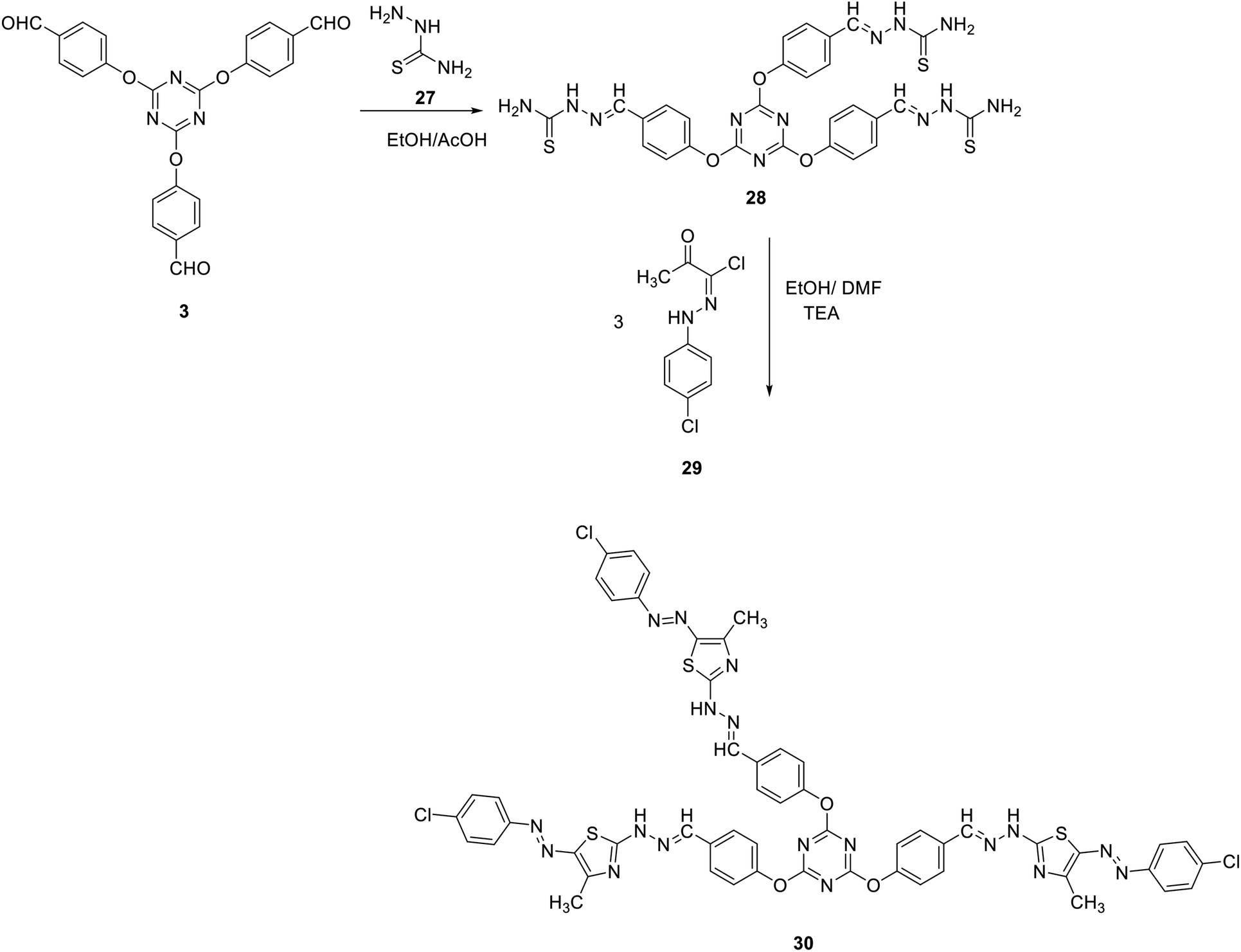 | ||
| Scheme 10 Synthesis of tris(arydiazenyl)thiazole 30. | ||
The structure of compound 30 has been verified by spectral as well as elemental analyses. The IR spectra of 30 as a representative example showed an absorption band at 3424 cm−1 due to the NH group. Moreover, its 1H NMR spectrum showed a D2O-exchangeable signal at δ 10.53 due to NH protons together with sharp singlet signals at δ 2.56 and δ 8.61 attributed to the 4-CH3 of thiazole group and the methine protons (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), respectively. All other protons appeared at the predicted chemical shifts and integral values.
CH), respectively. All other protons appeared at the predicted chemical shifts and integral values.
Furthermore, the reaction of 28 with three equivalents of each of 2-bromo-1-phenylethanone 31 and 2-bromo-1-(4-chlorophenyl)ethanone 32 in ethanol at reflux in the presence of few drops of TEA afforded 2,4,6-tris(4-(-(2-(4-arylthiazol-2-yl)hydrazono)methyl)phenoxy)-1,3,5-triazines 33 and 34 in 79 and 81% yields, respectively (Scheme 11).
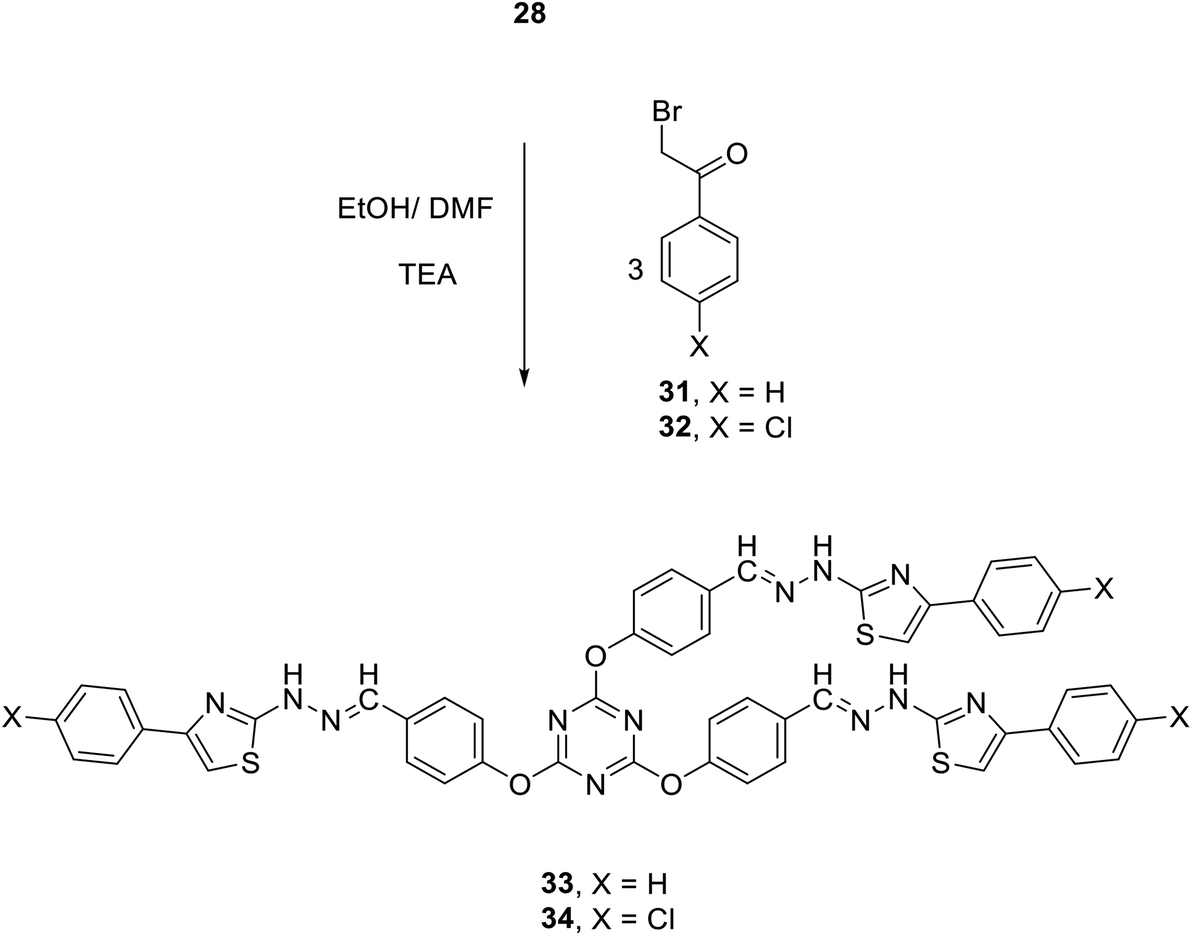 | ||
| Scheme 11 Synthesis of 2,4,6-tris(4-arylthiazol-2 ylhydrazonomethylphenoxy)-1,3,5-triazines 33 and 34. | ||
In analogy, reaction of compound 28 with 2-bromo-1-(5-methyl-1-phenyl-1H-pyrazol-4-yl)ethanone 35 in ethanol at reflux in the presence of few drops of TEA afforded 2,4,6-tris(4-(-(2-(4-(3-methyl-1-phenyl-1H-pyrazol-4-yl)thiazol-2yl) hydrazono)methyl)phenoxy)-1,3,5-triazine 36 in 66% yield (Scheme 12). Compound 35 was synthesized by the reaction of phenylhydrazine with ((dimethyl-amino) methylene)pentane-2,4-dione, obtained upon treatment of acetylacetone with dimethylformamide dimethylacetal (DMF/DMA), followed by bromination through treatment with Br2 in AcOH.52,76
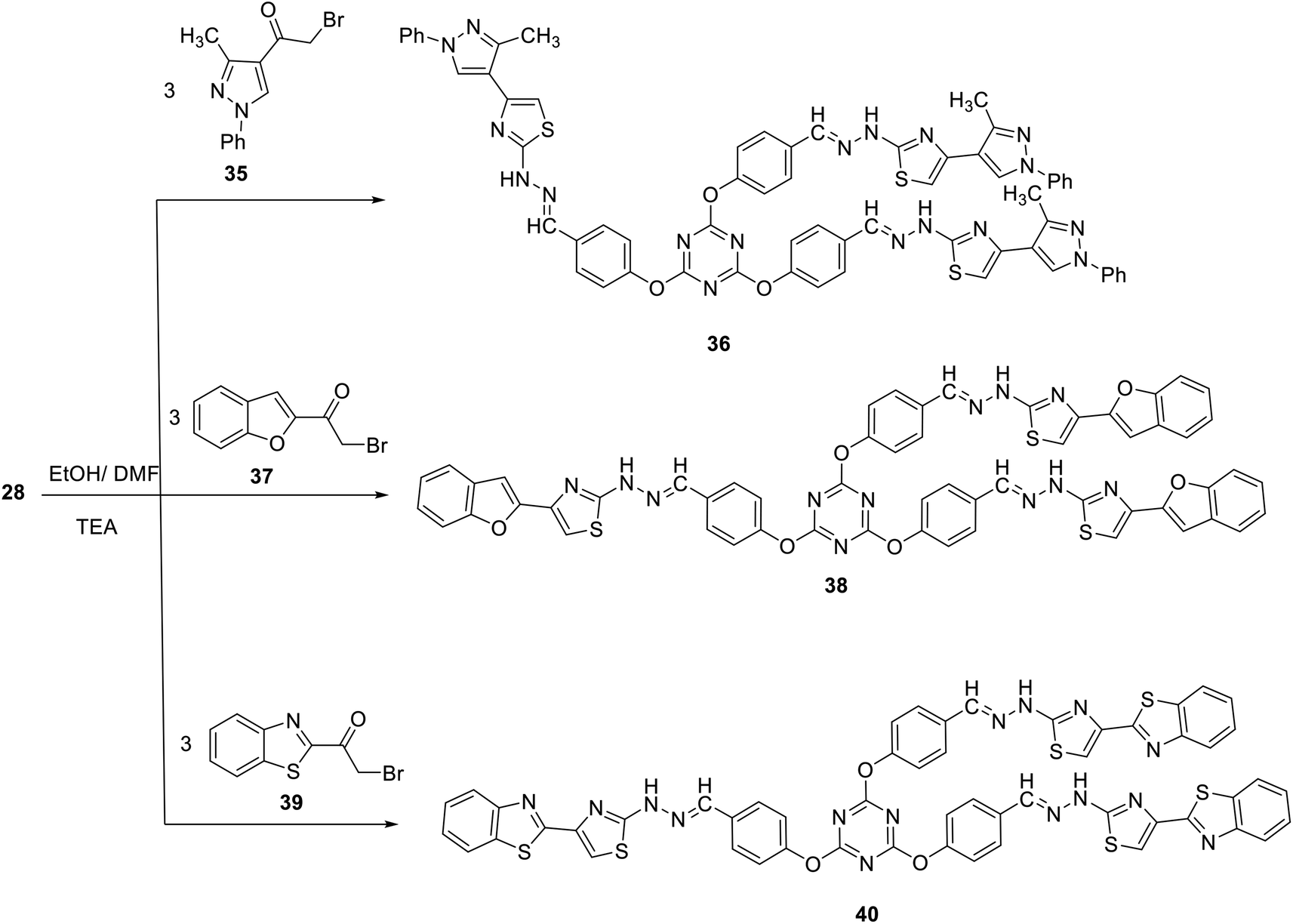 | ||
| Scheme 12 Synthesis of tris(thiazoles) 36, 38 and 40. | ||
In an attempt to construct novel tris(thiazole) linked to other heterocylic moieties aiming at achieving the concept of molecular hybridization, we studied the synthesis of novel 2,4,6-tris(4-(-(2-(4-(benzofuran-2-yl)thiazol-2-yl)hydrazono)-methyl) phenoxy)-1,3,5-triazine 38 and 2,4,6-tris(4-(-(2-(4-(benzo[d]thiazol-2-yl)thiazol-2-yl)hydrazono)methyl)phenoxy)-1,3,5-triazine 40 using a similar strategy. Thus, reaction of tris(thiosemicarbazone) 28 with each of 1-(benzofuran-2-yl)-2-bromoethanone 37 and 1-(benzo[d]thiazol-2-yl)-2-bromoethanone 39 in ethanol at reflux in the presence of TEA afforded 38 and 40 in 62 and 76% yields, respectively (Scheme 12).
The structures of the newly synthesized compounds have been confirmed by spectral data as well as elementary analyses. The IR spectrum of tris(pyrazole) 36 as a representative example of this class of compounds showed an absorption band at 3427 cm−1 because of (NH) together with the absence of absorption band characteristic for CS group.77 The symmetry of compound 36 is represented by a characteristic set of signals within its 1H NMR spectrum. It revealed the presence of a D2O-exchangeable singlet signal at δ 12.18 attributable to NH protons, a singlet signal at δ 6.84 attributed to C-5 protons of the thiazole rings, and a singlet signal at δ 8.03 because of methine protons (NCH). All other protons were observed at the predicted chemical shifts and integral values.
Conclusions
The most popular strategy in drug design as well as in the construction of important molecules is the synthesis of analogs of existing active molecules. The aim of this work was to develop new methodologies to tackle synthetic problems encountered in the synthesis of star-shaped molecules that contain 1,3,5-triazine as a central core with improved pharmacological and photophysical properties. In this respect, a simple protocol for the preparation of some star-shaped compounds based on s-triazine core linked to hexahydroacridinediones, pyrimido[4,5-b]quinolones, 1H-isoquinolino[2,1-a] quinolines, tetrahydro-4H-chromenes, dihydropyrano[2,3-c]pyrazoles, thiazole, or benzothiazole was developed. Hantzch and Michael reactions have been used as effective strategies for the synthesis of the target compounds from easily accessible precursors under mild reaction conditions. Moreover, multicomponent reactions (MCRs), which are associated with a range of advantages such as procedural efficiency, shorter reaction times, energy savings, and lower costs and time-consuming, were successfully used in this manuscript to synthesize the target compounds. The combination of two fused heterocyclic cores in a single molecular structure is supposed to take advantage of the pharmacological and physical properties of the resulting ligands. The successful synthesis of these compounds should open an access to a variety of star shaped molecules with interesting applications. Further analysis is underway to investigate the biological activities of the novel compounds. We believe also that some of the new star-shaped molecules should exhibit useful NLO properties on account of preliminary theoretical calculation of their polarizability and hyperpolarizability parameters. The theoretical as well as the experimental investigations are still underway and will be published separately because of the large quantity of data accumulated.Experimental
General
Melting points were determined in open glass capillaries with a Gallenkamp apparatus. The infrared spectra were recorded in potassium bromide disks on a Pye Unicam SP 3-300 and Shimadzu FTIR 8101 PC infrared spectrophotometer. NMR spectra were recorded with a Varian Mercury VXR-300 NMR spectrometer operating at (300 MHz and 75 MHz) or Bruker AVS NMR spectrometer at (400 MHz and 101 MHz), respectively, using TMS as an internal standard. Chemical shifts were reported as δ values in ppm. Mass spectra (EI) were obtained at 70 eV with a type Shimadzu GCMQP 1000 EX spectrometer. Analytical thin-layer chromatography was performed using pre-coated silica gel 60778 plates (Fluka), and the spots were visualized with UV light at 254 nm. Elemental analyses were performed on a Perkin-Elmer 240 micoanalyser at the Micro analytical Center of Cairo University. All chemicals were purchased from Sigma-Aldrich and used without further purification.
Synthesis of 4,4′,4′′-((1,3,5-triazine-2,4,6-triyl)tris(oxy))tribenzaldehyde (3)
A solution of p-hydroxybenzaldehyde (3 mmol) and KOH (3 mmol) in ethanol (10 ml) was stirred for 10 min at room temperature. The solvent was then removed in vacuo and the remaining potassium salt was collected, dissolved in DMF (5 ml), and stirred for 10 min at 0 °C. A solution of the latter salt and 2,4,6-trichloro-1,3,5-triazine (1 mmol) in DMF (5 ml) was allowed to stir for 15 min at 0 °C during which time KBr was precipitated. The solvent was then removed in vacuo and the remaining material was washed with water (20 ml), collected and crystallized from H2O/EtOH (3:1) to give 3 as a colorless powder; yield: 82%; mp 170–175 °C. IR (KBr) ν 2839, 2746, 1705 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 7.46–7.49 (d, J 8.4 Hz, 6H, Ar–H), 7.95–7.98 (d, J 8.1 Hz, 6H, Ar–H), 9.98 (s, 3H, 3CHO). 13C NMR (100 MHz, DMSO-d6) δ 122.2, 131.1, 134.1, 155, 172.6, 191.8 ppm. MS (EI, 70 eV): m/z (%) 441 [M+]. Anal. calcd for C24H15N3O6: C, 65.31; H, 3.43; N, 9.52. Found: C, 65.02; H, 3.29; N, 9.34.
4,4′,4′′-(((1,3,5-Triazine-2,4,6-triyl)tris(oxy))tris(benzene-4,1-diyl))tris(2,6-dimethyl-1,4-dihydropyridine-3,5-dicarbonitrile) (5)
To a warm solution of tris-aldehyde 3 (1 mmol) in glacial acetic acid (5 ml) was added 3-aminocrotononitrile 4 (6 mmol). The resulting solution was heated at reflux for 1 h. The solid obtained was collected and crystalized from DMF EtOH to give 5 as pale yellow crystals; yield: 88%; mp > 300 °C. IR (KBr) ν 3317 (NH), 2198 (CN) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 02.06 (s, 18H, 6CH3), 4.47 (s, 3H, 3CH), 7.29–7.36 (m, 12H, Ar–H), 9.58 (s, 3H, 3NH). 13C NMR (100 MHz, DMSO-d6) δ 18.3, 40.7, 82.9, 119.8, 122.4, 129.2, 142.2, 147.5, 151.1, 173.5. MS (EI, 70 eV): m/z (%) 828 [M+]. Anal. calcd for C48H36N12O3: C, 69.55; H, 4.38; N, 20.28. Found: C, 69.27; H, 4.11; N, 20.02.9,9′,9′′-(((1,3,5-Triazine-2,4,6-triyl)tris(oxy))tris(benzene-4,1-diyl))tris(3,3,6,6-tetramethyl-3,4,6,7,9,10-hexahydroacridine-1,8(2H,5H)-dione) (7)
A mixture of tris-aldehyde 3 (1 mmol), dimedone 6 (6 mmol) and ammonium acetate (5 mmol) in glacial acetic acid (3 ml) was heated at reflux for 6 h. The obtained crude solid was collected and crystalized from DMF/EtOH to give 7 as yellow crystals; yield: 83%; mp > 300 °C. IR (KBr) ν 3277 (NH), 1712 (CO) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 0.87 (s, 18H, 6CH3), 1.01 (s, 18H, 6CH3), 2.00–2.18 (m, 12H, 6 CH2), 2.32–2.49 (m, 12H, 6CH2), 4.70 (s, 3H, 3CH), 6.51–6.53 (d, J 8.4 Hz, 6H, Ar–H), 6.91–6.94 (d, J 8.4 Hz, 6H, Ar–H), 9.20 (s, 3H, 3NH). 13C NMR (100 MHz, DMSO-d6) δ 26.9, 29.6, 32.1, 32.6, 50.8, 112.3, 114.8, 128.9, 138.4, 149.3, 155.5, 172.5, 194.8. MS (EI, 70 eV): m/z (%) 1170 [M+]. Anal. calcd for C72H78N6O9: C, 73.82; H, 6.71; N, 7.17. Found: C, 73.66; H, 6.49; N, 7.03.
5,5′,5′′-(((1,3,5-Triazine-2,4,6-triyl)tris(oxy))tris(benzene-4,1-diyl))tris(8,8 dimethyl-2-thioxo-2,3,5,8,9,10-hexahydropyrimido[4,5-b]quinoline-4,6(1H,7H)-dione) (9)
A mixture of tris-aldehyde 3 (1 mmol), dimedone 6 (3 mmol) and 6-aminothiouracil 8 (3 mmol) in glacial acetic acid (3 ml) was heated at reflux for 3 h. The solid formed was collected and crystalized from DMF to give 9 as creamy powder; yield: 72%; mp > 300 °C. IR (KBr) ν 3200 (NH), 1668 (CO) cm−1; 1H NMR (300 MHz, DMSO-d6) δ 0.92 (s, 9H, 3CH3), 1.01 (s, 9H, 3CH3), 2.06–2.22 (m, 6H, 3CH2), 2.45–2.48 (m, 6H, 3CH2), 4.81 (s, 3H, 3CH), 7.05–7.08 (d, J 8.4 Hz, 6H, Ar–H), 7.22–7.25 (d, J 8.4 Hz, 6H, Ar–H), 8.59 (br, 3H, 3NH), 11.89 (br, 3H, 3NH), 12.01 (br, 3H, 3NH). 13C NMR (75 MHz, DMSO-d6) δ 26.9, 28.6, 32.3, 32.6, 40.2, 50.1, 94, 110.7, 120.8, 128.6, 143.6, 144.2, 149.5, 160.8, 173, 194.4. MS (EI, 70 eV): m/z (%) 1182 [M+]. Anal. calcd for C60H54N12O9S3: C, 60.90; H, 4.60; N, 14.20. Found: C, 60.84; H, 4.33; N, 14.03.
13,13′,13′′-(((1,3,5-Triazine-2,4,6-triyl)tris(oxy))tris(benzene-4,1-diyl))tris(9,10-dimethoxy-3,3-dimethyl-1-oxo-2,3,4,6,7,13-hexahydro-1H-isoquinolino[2,1-a] quinoline-12-carbonitrile) (12)
A mixture of tris-aldehyde 3 (1 mmol), dimedone 6 (3 mmol) and 2-(6,7-dimethoxy-3,4-dihydroisoquinolin-1-yl)acetonitrile 11 (3 mmol) in glacial acetic acid (3 ml) was heated at reflux for 6 h. The formed crude solid was collected, washed with ethanol and crystalized from DMF to give 12 as yellow crystals; yield: 77%; mp 260–262 °C; (DMF). IR (KBr) ν 2188 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1629 (CO) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 0.99 (s, 9H, 3CH3), 1.02 (s, 9H, 3CH3), 2.18 (m, 6H, 3CH2), 2.50 (m, 6H, 3CH2), 2.83–2.91 (m, 6H, CH2), 3.51–3.57 (m, 3H, CH2), 3.75 (s, 9H, 3OCH3), 3.82 (s, 9H, 3OCH3), 3.89–3.92 (m, 3H, CH2), 4.71 (s, 3H, pyridine-H13), 7.02 (s, 3H, Ar–H), 7.14–7.31 (m, 12H, Ar–H), 7.66 (s, 3H, Ar–H). 13C NMR (100 MHz, DMSO-d6) δ 28.2, 28.7, 28.8, 32.7, 37.4, 43.6, 49.8, 56.1, 56.2, 84.9, 110.4, 110.9, 111.2, 114.3, 120.2, 122.1, 128.5, 131.3, 142.4, 147.2, 151.0, 152.3, 158.6, 172.5, 195.1. MS (EI, 70 eV): m/z (%) 1443 [M+]. Anal. calcd for C87H81N9O12: C, 72.33; H, 5.65; N, 8.73. Found: C, 72.07; H, 5.49; N, 8.62.
N), 1629 (CO) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 0.99 (s, 9H, 3CH3), 1.02 (s, 9H, 3CH3), 2.18 (m, 6H, 3CH2), 2.50 (m, 6H, 3CH2), 2.83–2.91 (m, 6H, CH2), 3.51–3.57 (m, 3H, CH2), 3.75 (s, 9H, 3OCH3), 3.82 (s, 9H, 3OCH3), 3.89–3.92 (m, 3H, CH2), 4.71 (s, 3H, pyridine-H13), 7.02 (s, 3H, Ar–H), 7.14–7.31 (m, 12H, Ar–H), 7.66 (s, 3H, Ar–H). 13C NMR (100 MHz, DMSO-d6) δ 28.2, 28.7, 28.8, 32.7, 37.4, 43.6, 49.8, 56.1, 56.2, 84.9, 110.4, 110.9, 111.2, 114.3, 120.2, 122.1, 128.5, 131.3, 142.4, 147.2, 151.0, 152.3, 158.6, 172.5, 195.1. MS (EI, 70 eV): m/z (%) 1443 [M+]. Anal. calcd for C87H81N9O12: C, 72.33; H, 5.65; N, 8.73. Found: C, 72.07; H, 5.49; N, 8.62.
4,4′,4′′-(((1,3,5-Triazine-2,4,6-triyl)tris(oxy))tris(benzene-4,1-diyl))tris(2-amino-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile) (16)
To a mixture of tris-aldehyde 3 (1 mmol), malononitrile 15 (3 mmol) and dimedone 6 (3 mmol) in absolute ethanol (4 ml), piperidine (0.2 ml) was added. The reaction mixture was heated at reflux for 3 h. The crude solid formed was collected and crystalized from DMF/EtOH to give 16 as buff powder; yield: 88%; mp 285–290 °C. IR (KBr) ν 3365, 3313 (NH2), 2190 (CN), 1684 (CO) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 0.94 (s, 9H, 3CH3), 1.04 (s, 9H, 3CH3), 2.10–2.14 (m, 6H, 3CH2), 2.24–2.51 (m, 6H, 3CH2), 4.22 (s, 3H, 3CH), 7.02 (br, 6H, 3NH2), 7.11–7.13 (d, J 8.4 Hz, 6H, Ar–H), 7.17–7.19 (d, J 8.4 Hz, 6H, Ar–H). 13C NMR (100 MHz, DMSO-d6) δ 27.2, 28.9, 32.3, 35.5, 50.4, 58.6, 113.2, 120.1, 122.0, 128.5, 142.3, 150.8, 159, 162.8, 165.9, 172.3, 196.1. MS (EI, 70 eV): m/z (%) 1005 [M+]. Anal. calcd for C57H51N9O9: C, 68.05; H, 5.11; N, 12.53. Found: C, 67.76; H, 5.03; N, 12.24.
4,4′,4′′-(((1,3,5-Triazine-2,4,6-triyl)tris(oxy))tris(benzene-4,1-diyl))tris(6-amino-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile) (19)
To a mixture of tris-aldehyde 3 (1 mmol), malononitrile 15 (3 mmol) and pyrazolone 17 (3 mmol) in absolute ethanol (4 ml), piperidine (0.2 ml) was added. The reaction mixture was heated at reflux for 3 h. The crude solid formed was collected and crystalized from EtOH to give 19 as creamy powder; yield: 73%; mp 246–250 °C; (EtOH). IR (KBr) ν 3453 (NH), 3295, 3168 (NH2), 2187 (CN) cm−1; 1H NMR (300 MHz, DMSO-d6) δ 1.78 (s, 9H, 3CH3), 4.64 (s, 3H, 3CH), 6.85 (s, 6H, 3NH2), 7.12–7.15 (d, J 8.7 Hz, 6H, Ar–H), 7.19–7.21 (d, J 8.4 Hz, 6H, Ar–H), 12.08 (s, 3H, 3NH). 13C NMR (75 MHz, DMSO-d6) δ 9.6, 35.6, 57.1, 97.6, 120.7, 121.7, 128.4, 135.6, 141.5, 150.5, 154.7, 160.8, 172. MS (EI, 70 eV): m/z (%) 879 [M+]. Anal. calcd for C45H33N15O6: C, 61.43; H, 3.78; N, 23.88. Found: C, 61.22; H, 3.59; N, 23.65.
4,4′,4′′-(((1,3,5-Triazine-2,4,6-triyl)tris(oxy))tris(benzene-4,1-diyl))tris(2-amino-5-oxo-4H,5H-pyrano[3,2-c]chromene-3-carbonitrile) (20)
A mixture of tris-aldehyde 3 (1 mmol), malononitrile 15 (3 mmol), 4-hydroxycoumarin 18 (3 mmol) and sodium acetate (3 mmol) in glacial acetic acid (3 ml) was heated at reflux for 3 h. The crude solid formed was collected and crystalized from DMF/EtOH to give 20 as grey powder; yield: 82%; mp 255–265 °C; (DMF/EtOH). IR (KBr) ν 3323, 3184 (NH2), 2197 (CN), 1715 (CO) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 4.49 (s, 3H, 3CH), 7.17–7.91 (m, 30H, Ar–H + 3NH2).13C NMR (100 MHz, DMSO-d6) δ 36.8, 58.3, 104.2, 113.5, 117.1, 119.7, 122.1, 122.9, 125.2, 129.3, 133.4, 141.5, 150.7, 152.7, 154, 158.5, 160.1, 173.5. MS (EI, 70 eV): m/z (%) 1071 [M+]. Anal. calcd for C60H33N9O12: C, 67.23; H, 3.10; N, 11.76. Found: C, 66.94; H, 2.88; N, 11.47.
2,2′,2′′,2′′′,2′′′′,2′′′′′-((((1,3,5-Triazine-2,4,6-triyl)tris(oxy))tris(benzene-4,1-diyl)) tris (methanetriyl))hexakis(3-hydroxy-5,5-dimethylcyclohex-2-en-1-one) (21)
A mixture of tris-aldehyde 3 (1 mmol), dimedone 6 (6 mmol) and p-TSA (15 mol%) in ethanol/H2O (15 ml, 2:1) was heated at reflux for 6 h. The solid formed was collected and crystalized from EtOH to give 21 as colorless crystals; yield: 83%; mp 245–248 °C; (EtOH). IR (KBr) ν 3345 (OH), 1650 (CO) cm−1; 1H NMR (300 MHz, DMSO-d6) δ 0.90 (s, 18H, 6CH3), 1.02 (s, 18H, 6CH3), 2.04–2.27 (m, 12H, 6 CH2), 2.48–2.52 (m, 12H, 6CH2), 3.33 (br, 3H, 3OH), 4.42 (s, 3H, methine-H), 6.56–6.59 (d, J 8.7 Hz, 6H, Ar–H), 6.92–6.95 (d, J 8.7 Hz, 6H, Ar–H), 9.20 (br, 3H, 3OH). 13C NMR (75 MHz, DMSO-d6) δ 26.4, 28.7, 30.1, 31.8, 40.1, 50.1, 114.6, 114.8, 128.9, 134.8, 155.6, 162.5, 196.1. MS (EI, 70 eV): m/z (%)1227 [M+]. Anal. calcd for C72H81N3O15: C, 70.40; H, 6.65; N, 3.42. Found: C, 70.17; H, 6.38; N, 3.19.
2,4,6-Tris(4-(benzo[d]thiazol-2-yl)phenoxy)-1,3,5-triazine (24)
To a mixture of 2-aminothiophenol (3 mmol) 23, tris-aldehyde 3 (1 mmol) in absolute ethanol (10 ml), sodium hydrogen sulfite (3 mmol) was added. The reaction mixture was heated at reflux for 4 h. The obtained solid was collected and crystalized from acetic acid to give 24 as pale yellow powder; yield: 61%; mp 200–202 °C. IR (KBr) ν 1568 (CN) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 7.42–8.13 (m, 24H, Ar–H).13C NMR (100 MHz, DMSO-d6) δ 121.5, 122.7, 123.1, 123.3, 126, 127.1, 129, 131.2, 135, 153.9, 166.6, 173.4. MS (EI, 70 eV): m/z (%) 756 [M+]. Anal. calcd for C42H24N6O3S3: C, 66.65; H, 3.20; N, 11.10. Found: C, 66.33; H, 3.03; N, 10.84.
2,4,6-Tris(4-(1H-phenanthro[9,10-d]imidazol-2-yl)phenoxy)-1,3,5-triazine (26)
To a solution of tris-aldehyde 3 (1 mmol) in absolute ethanol (10 ml), 9,10-phenanthrenequinone 25 (3 mmol) and ammonium acetate (5 mmol) were added. The reaction mixture was heated at reflux for 3 h. The formed crude solid was collected and crystalized from AcOH to give 26 as orange powder; yield: 81%; mp > 300 °C; (AcOH). IR (KBr) ν 3305 (NH), 1590 (CN) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 7.48–8.88 (m, 36H, Ar–H), 13.47 (s, 3H, 3NH).13C NMR (100 MHz, DMSO-d6) δ 122.3, 122.9, 124.2, 124.9, 125.6, 125.8, 127.4, 127.9, 128.3, 129.6, 129.8, 131.7, 135.7, 135.9, 137.5, 149.0, 153.1, 169.9, 172.6, 179.4. MS (EI, 70 eV): m/z (%)1005 [M+]. Anal. calcd for C66H39N9O3: C, 78.79; H, 3.91; N, 12.53. Found: C, 78.46; H, 3.69; N, 12.32.
2,4,6-Tris(4-((2-(5-((4-chlorophenyl)diazenyl)-4-methylthiazol-2-yl) hydrazineylidene)methyl)phenoxy)-1,3,5-triazine (30)
To a solution of tris(aldehyde thiosemicarbazone) 28 (1 mmol) in ethanol/DMF (20 ml, 3:1) containing TEA (0.1 ml, 1 mmol), N-(4-chlorophenyl)-2-oxopropanehydrazonoyl chloride 29 was added. The reaction mixture was heated at reflux for 6 h. The formed solid was collected and crystalized from DMF to give 30 as crimson red crystals; yield: 85%; decompose: 218–224 °C; (DMF). IR (KBr) ν 3424 (NH) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 2.56 (s, 9H, 3CH3), 7.28–7.35 (m, 18H, Ar–H), 7.84–7.86 (m, 6H, Ar–H), 8.62 (s, 3H, 3CH), 10.53 (s, 3H, 3NH). MS (EI, 70 eV): m/z (%) 1190 [M+]. Anal. calcd for C54H41C13N18O3S3: C, 54.39; H, 3.47; N, 21.14. Found: C, 54.17; H, 3.23; N, 21.02.
General method for the synthesis of tris(thiazoles) 33, 34, 36, 38 and 40
To a solution of tris(aldehyde thiosemicarbazone) 28 (1 mmol) in ethanol/DMF (20 ml, 3:1) containing TEA (0.1 ml, 1 mmol), the appropriate 2-bromoethanones 31, 32, 35, 37 and 39 (3 mmol) were added. The reaction mixture was heated at reflux for 3 h. The formed crude solid was collected and crystalized from the proper solvents to give 33, 34, 36, 38 and 40.
2,4,6-Tris(4-((2-(4-phenylthiazol-2-yl)hydrazineylidene)methyl)phenoxy)-1,3,5-triazine (33)
Pale yellow powder; yield: 79%; mp 250–255 °C; (DMF). IR (KBr) ν 3434 (NH), 1564 (CN) cm−1; 1H NMR (300 MHz, DMSO-d6) δ 7.20 (s, 3H, thiazole-H), 7.33–7.80 (m, 27H, Ar–H), 8.03 (s, 3H, CH), 12.21 (br, 3H, 3NH).13C NMR (75 MHz, DMSO-d6) δ 103.7, 121.8, 125.4, 127.2, 127.5, 128.5, 132.3, 134.6, 140.1, 151.8, 168.3, 172.9. MS (EI, 70 eV): m/z (%) 960 [M+]. Anal. calcd for C51H36N12O3S3: C, 63.73; H, 3.78; N, 17.49. Found: C, 63.49; H, 3.47; N, 17.23.
2,4,6-Tris(4-((2-(4-(4-chlorophenyl)thiazol-2-yl)hydrazineylidene)methyl) phenoxy)-1,3,5-triazine (34)
Pale yellow powder; yield: 81%; mp 265–267 °C; (DMF). IR (KBr) ν 3432 (NH), 1566 (CN) cm−1; 1H NMR (300 MHz, DMSO-d6) δ 7.25 (s, 3H, thiazole-H), 7.32–7.35 (d, J 8.7 Hz, 6H, Ar–H), 7.42–7.39 (d, J 8.7 Hz, 6H, Ar–H), 7.68–7.71 (d, J 8.7 Hz, 6H, Ar–H), 7.77–7.80 (d, J 8.7 Hz, 6H, Ar–H), 8.03 (s, 3H, CH), 12.21 (br, 3H, 3NH).13C NMR (75 MHz, DMSO-d6) δ 104.4, 121.8, 127.1, 127.2, 128.5, 131.9, 132.3, 133.4, 140.2, 149.2, 151.8, 168.4, 172.9. MS (EI, 70 eV): m/z (%) 1062 [M+]. Anal. calcd for C51H33Cl3N12O3S3: C, 57.55; H, 3.13; N, 15.79. Found: C, 57.36; H, 3.01; N, 15.44.
2,4,6-Tris(4-((2-(4-(3-methyl-1-phenyl-1H-pyrazol-4-yl)thiazol-2-yl) hydrazineylidene)methyl)phenoxy)-1,3,5-triazine (36)
Pale yellow powder; yield: 66%; mp 245–250 °C; (DMF). IR (KBr) ν 3427 (NH), 1565 (CN) cm−1; 1H NMR (300 MHz, DMSO-d6) δ 2.47 (s, 9H, 3CH3), 6.84 (s, 3H, thiazole-H) 7.31–7.90 (m, 30H, Ar–H), 8.03 (s, 3H, CH), 12.18 (br, 3H, 3NH).13C NMR (75 MHz, DMSO-d6) δ 11.9, 101.8, 116.7, 121.8, 122.4, 124.9, 127.2, 127.7, 129.1, 132.4, 135.4, 138.7, 139.3, 168.1, 172.9. MS (EI, 70 eV): m/z (%) 1200 [M+]. Anal. calcd for C63H48N18O3S3: C, 62.99; H, 4.03; N, 20.99. Found: C, 62.71; H, 3.84; N, 20.68.
2,4,6-Tris(4-((2-(4-(benzofuran-2-yl)thiazol-2-yl)hydrazineylidene)methyl) phenoxy)-1,3,5-triazine (38)
Brown powder; yield: 62%; mp 270–275 °C; (DMF). IR (KBr) ν 3423 (NH), 1563 (CN) cm−1; 1H NMR (300 MHz, DMSO-d6) δ 7.03 (s, 3H, thiazole-H), 7.24–7.73 (m, 27H, Ar–H), 8.05 (s, 3H, 3CH), 12.26 (br, 3H, 3NH). MS (EI, 70 eV): m/z (%)1080 [M+]. Anal. calcd for C57H36N12O6S3: C, 63.32; H, 3.36; N, 15.55. Found: C, 63.11; H, 3.07; N, 15.23.
2,4,6-Tris(4-((2-(4-(benzo[d]thiazol-2-yl)thiazol-2-yl)hydrazineylidene) methyl)phenoxy)-1,3,5-triazine (40)
Green crystals; yield: 76%; mp 265–270 °C; (DMF). IR (KBr) ν 3432 (NH), 1559 (CN) cm−1; 1H NMR (300 MHz, DMSO-d6) δ 7.32–8.05 (m, 27H, Ar–H), 8.05 (s, 3H, 3CH), 12.47 (s, 3H, 3NH). MS (EI, 70 eV): m/z (%)1131 [M+]. Anal. calcd for C54H33N15O3S6: C, 57.28; H, 2.94; N, 18.56. Found: C, 57.03; H, 2.77; N, 18.29.
Conflicts of interest
There are no conflicts to declare.References
- Z. Luo, W. Xiong, T. Liu, W. Cheng, K. Wu, Y. Sun and C. Yang, Org. Electron., 2016, 41, 1–7 Search PubMed.
- Z. Hao, Y. Liu, Y. Huang, F. Meng, Y. Wang, H. Tan, S. Su and W. Zhu, J. Organomet. Chem., 2017, 835, 52–59 CrossRef CAS.
- A. Irfan, S. Muhammad, A. R. Chaudhry, A. G. Al-Sehemi and R. Jin, Optik, 2017, 149, 321–331 CrossRef CAS.
- K. Rundel, S. Maniam, K. Deshmukh, E. Gann and C. R. Mcneill, J. Mater. Chem. A, 2017, 5, 12266–12277 RSC.
- C. Lu, I. T. Choi, J. Kim and H. K. Kim, J. Mater. Chem. A, 2017, 5, 20263–20276 RSC.
- H. Feng, X. Geng, J. Lin, H. Guo and F. Yang, Liq. Cryst., 2018, 45, 1470–1476 CrossRef CAS.
- F. A. Olate, M. L. Parra, J. M. Vergara, J. Barberá and M. Dahrouch, Liq. Cryst., 2017, 44, 1173–1184 CrossRef CAS.
- S. K. Pathak, S. Nath, J. De, S. K. Pal and A. S. Achalkumar, New J. Chem., 2017, 41, 4680–4688 RSC.
- Y. N. Luponosov, J. Min, A. N. Solodukhin, O. V. Kozlov, M. A. Obrezkova, S. M. Peregudova, T. Ameri, S. N. Chvalun, M. S. Pshenichnikov, C. J. Brabec and S. A. Ponomarenko, Org. Electron., 2016, 32, 157–168 CrossRef CAS.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS.
- D. L. Reger, R. F. Semeniuc and M. D. Smith, Inorg. Chem. Commun., 2003, 42, 8137–8139 CrossRef CAS.
- H. M. Diab, A. M. Abdelmoniem, M. R. Shaaban, I. A. Abdelhamid and A. H. M. Elwahy, RSC Adv., 2019, 9, 16606–16682 RSC.
- P. Gamez, P. De Hoog, M. Lutz, A. L. Spek and J. Reedijk, Inorg. Chim. Acta., 2003, 351, 319–325 CrossRef CAS.
- G. Giacomelli, A. Porcheddu and L. Luca, Curr. Org. Chem., 2005, 8, 1497–1519 CrossRef.
- L. Hu, J. Li, J. Huang and J. Yin, Chin. J. Chem., 2017, 35, 93–97 CrossRef CAS.
- M. Jung, K. H. Lee, J. Y. Lee and T. Kim, Mater. Horiz., 2020, 7, 559–565 RSC.
- P. Bhagavath, R. Shetty and D. Sunil, Crit. Rev. Solid State Mater. Sci., 2019, 45, 1–32 Search PubMed.
- K. C. Majumdar, N. De, B. Roy and A. Bhaumik, Liq. Cryst., 2010, 37, 1459–1464 CrossRef CAS.
- S. Matsumura, A. R. Hlil, C. Lepiller, J. Gaudet, D. Guay, Z. Shi, S. Holdcroft and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7207–7224 CrossRef.
- K. K. Bansal, D. Kakde, U. Gupta and N. K. Jain, J. Nanosci. Nanotechnol., 2010, 10, 8395–8404 CrossRef CAS.
- B. N. Veerabhadraswamy, H. K. Dambal, D. S. S. Rao and C. V. Yelamaggad, ChemPhysChem, 2016, 17, 2225–2237 CrossRef CAS.
- A. Laventure, G. De Grandpré, A. Soldera, O. Lebel and C. Pellerin, Phys. Chem. Chem. Phys., 2016, 18, 1681–1692 RSC.
- Y. Wang, T. Li, Y. Yin, Y. Jiang, G. Wang, D. Liu and J. Hua, Optik, 2017, 142, 163–167 CrossRef CAS.
- A. El-Faham, K. A. Dahlous, Z. A. A. L. Othman, H. A. Al-Lohedan and G. A. EL-Mahdy, Molecules, 2016, 21, 1–11 Search PubMed.
- G. Blotny, Tetrahedron, 2006, 62, 9507–9522 CrossRef CAS.
- Z. E. Koc, H. Bingol, A. O. Saf, E. Torlak and A. Coskun, J. Hazard. Mater., 2010, 183, 251–255 CrossRef CAS.
- R. V. Patel, P. Kumari, D. P. Rajani and K. H. Chikhalia, Eur. J. Med. Chem., 2011, 46, 4354–4365 CrossRef CAS.
- P. Gahtori and S. K. Ghosh, J. Enzyme Inhib. Med. Chem., 2012, 27, 281–293 CrossRef CAS.
- U. P. Singh, H. R. Bhat, P. Gahtori and R. K. Singh, Silico pharmacol., 2013, 1, 1–9 CrossRef.
- V. K. Pandey, S. Tusi, Z. Tusi, M. Joshi and S. Bajpai, Acta Pharm., 2004, 54, 1–12 CAS.
- V. Lozano, L. Aguado, B. Hoorelbeke, M. Renders, M. J. Camarasa, D. Schols, J. Balzarini, A. San-Félix and M. J. Pérez-Pérez, J. Med. Chem., 2011, 54, 5335–5348 CrossRef CAS.
- A. Agarwal, K. Srivastava, S. K. Puri and P. M. S. Chauhan, Bioorg. Med. Chem. Lett, 2005, 15, 3133–3136 CrossRef CAS.
- P. Gahtori, S. K. Ghosh, P. Parida, A. Prakash, K. Gogoi, H. R. Bhat and U. P. Singh, Exp. Parasitol., 2012, 130, 292–299 CrossRef CAS.
- A. Baliani, G. J. Bueno, M. L. Stewart, V. Yardley, R. Brun, M. P. Barrett and I. H. Gilbert, J. Med. Chem., 2005, 48, 5570–5579 CrossRef CAS.
- F. Leroux, B. J. Van Keulen, J. Daliers, N. Pommery and J. P. Hénichart, Bioorg. Med. Chem., 1999, 7, 509–516 CrossRef CAS.
- B. R. Henke, T. G. Consler, N. Go, R. L. Hale, D. R. Hohman, S. A. Jones, A. T. Lu, L. B. Moore, J. T. Moore, L. A. Orband-Miller, R. G. Robinett, J. Shearin, P. K. Spearing, E. L. Stewart, P. S. Turnbull, S. L. Weaver, S. P. Williams, G. B. Wisely and M. H. Lambert, J. Med. Chem., 2002, 45, 5492–5505 CrossRef CAS.
- G. H. Kuo, A. DeAngelis, S. Emanuel, A. Wang, Y. Zhang, P. J. Connolly, X. Chen, R. H. Gruninger, C. Rugg, A. Fuentes-Pesquera, S. A. Middleton, L. Jolliffe and W. V. Murray, J. Med. Chem., 2005, 48, 4535–4546 CrossRef CAS.
- G. H. Kuo, A. DeAngelis, S. Emanuel, A. Wang, Y. Zhang, P. J. Connolly, X. Chen, R. H. Gruninger, C. Rugg, A. Fuentes-Pesquera, S. A. Middleton, L. Jolliffe and W. V. Murray, J. Med. Chem., 2005, 48, 4535–4546 CrossRef CAS.
- J. Lloyd, H. J. Finlay, W. Vacarro, T. Hyunh, A. Kover, R. Bhandaru, L. Yan, K. Atwal, M. L. Conder, T. Jenkins-West, H. Shi, C. Huang, D. Li, H. Sun and P. Levesque, Bioorg. Med. Chem. Lett., 2010, 20, 1436–1439 CrossRef CAS.
- B. Kolesinska, K. Barszcz, Z. J. Kaminski, D. Drozdowska, J. Wietrzyk and M. Switalska, J. Enzyme Inhib. Med. Chem., 2012, 27, 619–627 CrossRef CAS.
- J. K. Srivastava, G. G. Pillai, H. R. Bhat, A. Verma and U. P. Singh, Sci. Rep., 2017, 7, 1–18 CrossRef CAS.
- M. Shanmugam, K. Narayanan, K. H. Hari Prasad, D. Karthikeyan, L. Chandrasekaran, R. Atchudan and V. Chidambaranathan, New J. Chem., 2018, 42, 1698–1714 RSC.
- O. Prakash, D. K. Aneja, K. Hussain, P. Lohan, P. Ranjan, S. Arora, C. Sharma and K. R. Aneja, Eur. J. Med. Chem., 2011, 46, 5065–5073 CrossRef CAS.
- M. Henary, S. Paranjpe and E. A. Owens, Heterocycl. Commun., 2013, 19, 89–99 CAS.
- M. M. Kamel and N. Y. Megally Abdo, Eur. J. Med. Chem., 2014, 86, 75–80 CrossRef CAS.
- N. Mibu, K. Yokomizo, K. Yamada, J. Matsuyama, S. Tomonaga, I. Sakai, R. Sato, Y. Kawano, Y. Matsumoto, Y. Fujita, Y. Inoue, M. Iida, K. Hashiguchi, J. R. Zhou, M. Furutachi and K. Sumoto, Heterocycles, 2019, 98, 489–508 CrossRef CAS.
- H. R. Bhat, U. P. Singh, P. Gahtori, S. K. Ghosh, K. Gogoi, A. Prakash and R. K. Singh, New J. Chem., 2013, 37, 2654–2662 RSC.
- R. Kaur, P. Kaur, S. Sharma, G. Singh, S. Mehndiratta, P. M. S. Bedi and K. Nepali, Recent Pat. Anticancer, Drug Discovery, 2014, 10, 23–71 Search PubMed.
- A. Makowska, F. Saczewski, P. J. Bednarski, J. Saczewski and Ł. Balewski, Molecules, 2018, 23, 1–16 CrossRef.
- M. E. Salem, A. F. Darweesh, A. M. Farag and A. H. M. Elwahy, J. Heterocycl. Chem., 2017, 54, 586–595 CrossRef CAS.
- A. H. M. Elwahy, R. M. Sarhan and M. A. Badawy, Curr. Org. Synth., 2013, 10, 786–790 CrossRef CAS.
- M. E. Salem, A. F. Darweesh, A. M. Farag and A. H. M. Elwahy, Tetrahedron, 2016, 72, 712–719 CrossRef CAS.
- N. A. Abd El-Fatah, A. F. Darweesh, A. A. Mohamed, I. A. Abdelhamid and A. H. M. Elwahy, Tetrahedron, 2017, 73, 1436–1450 CrossRef CAS.
- M. F. Mohamed, A. F. Darweesh, A. H. M. Elwahy and I. A. Abdelhamid, RSC Adv., 2016, 6, 40900–40910 RSC.
- I. A. Abdelhamid, A. F. Darweesh and A. H. M. Elwahy, Tetrahedron Lett., 2015, 56, 7085–7088 CrossRef CAS.
- M. E. Salem, M. Hosny, A. F. Darweesh and A. H. M. Elwahy, Synth. Commun., 2019, 49, 2319–2329 CrossRef CAS.
- H. M. Diab, I. A. Abdelhamid and A. H. M. Elwahy, Synlett, 2018, 29, 1627–1633 CrossRef CAS.
- M. E. Salem, A. F. Darweesh, M. R. Shaaban and A. H. M. Elwahy, Arkivoc, 2019,(part v), 73–88 Search PubMed.
- M. Hosny, M. E. Salem, A. F. Darweesh and A. H. M. Elwahy, J. Heterocycl. Chem., 2018, 55, 2342–2348 CrossRef CAS.
- A. M. S. Hebishy, M. S. Abdelfattah, A. Elmorsy and A. H. M. Elwahy, Synth. Commun., 2020, 50, 980–996 CrossRef CAS.
- M. E. Salem, A. A. Ahmed, A. F. Darweesh, O. Kühn and A. H. M. Elwahy, J. Mol. Struct., 2019, 1176, 19–30 CrossRef CAS.
- M. E. Salem, A. F. Darweesh and A. H. M. Elwahy, Synth. Commun., 2020, 50, 256–270 CrossRef CAS.
- A. M. Abdelmoniem, S. A. S. Ghozlan, D. M. Abdelmoniem, A. H. M. Elwahy and I. A. Abdelhamid, J. Heterocycl. Chem., 2017, 54, 2844–2849 CrossRef CAS.
- A. M. Abdella, M. F. Mohamed, A. F. Mohamed, A. H. M. Elwahy and I. A. Abdelhamid, J. Heterocycl. Chem., 2018, 55, 498–507 CrossRef CAS.
- N. A. A. El-Fatah, A. F. Darweesh, A. A. Mohamed, I. A. Abdelhamid and A. H. M. Elwahy, Monatsh. Chem., 2017, 148, 2107–2122 CrossRef.
- S. K. Salama, M. F. Mohamed, A. F. Darweesh, A. H. M. Elwahy and I. A. Abdelhamid, Bioorg. Chem., 2017, 71, 19–29 CrossRef CAS.
- O. M. Sayed, A. E. M. Mekky, A. M. Farag and A. H. M. Elwahy, J. Sulfur Chem., 2014, 36, 124–134 CrossRef.
- F. G. Khan, M. V. Yadav and A. D. Sagar, Med. Chem. Res., 2014, 23, 2633–2638 CrossRef CAS.
- H. Duan, L. Wang, D. Qin, X. Li, S. Wang and Y. Zhang, Synth. Commun., 2011, 41, 380–384 CrossRef CAS.
- M. Bashiri, A. Jarrahpour, B. Rastegari, A. Iraji, C. Irajie, Z. Amirghofran, S. Malek-Hosseini, M. Motamedifar, M. Haddadi, K. Zomorodian, Z. Zareshahrabadi and E. Turos, Monatsh. Chem., 2020, 151, 821–835 CrossRef CAS.
- E. M. Awad, N. M. Elwan and H. M. Hassaneen, Helv. Chim. Acta, 2001, 84, 1172–1180 CrossRef CAS.
- F. M. Saleh, H. M. Hassaneen, H. Butenschön, G. Dräger and I. A. Abdelhamid, Tetrahedron Lett., 2019, 60, 151265 CrossRef CAS.
- T. Josephrajan and V. T. Ramakrishnan, Can. J. Chem., 2007, 85, 572–575 CrossRef CAS.
- M. Idris, C. Coburn, T. Fleetham, J. Milam-Guerrero, P. I. Djurovich, S. R. Forrest and M. E. Thompson, Mater. Horiz., 2019, 6, 1179–1186 RSC.
- Z. Chang, X. Jing, C. He, X. Liu and C. Duan, ACS Catal., 2018, 8, 1384–1391 CrossRef CAS.
- C. J. J. Goddard, Heterocycl. Chem., 1991, 28, 1607–1612 CrossRef CAS.
- I. Mohammed-Ziegler, A. Hamdi, R. Abidi and J. Vincens, Supramol. Chem., 2006, 18, 219–234 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09025e |
| This journal is © The Royal Society of Chemistry 2020 |