
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen adsorption mechanism of MOF-74 metal–organic frameworks: an insight from first principles calculations
Trang Nguyen-Thuy *ab,
Phong Le-Hoanga,
Nam Hoang Vugh,
Thong Nguyen-Minh Lecde,
Tan Le Hoang Doanb,
Jer-Lai Kuoc,
Toan T. Nguyen*a,
Thang Bach Phanbg and
Duc Nguyen-Manhf
*ab,
Phong Le-Hoanga,
Nam Hoang Vugh,
Thong Nguyen-Minh Lecde,
Tan Le Hoang Doanb,
Jer-Lai Kuoc,
Toan T. Nguyen*a,
Thang Bach Phanbg and
Duc Nguyen-Manhf
aKey Laboratory for Multiscale Simulation of Complex Systems, University of Science, Vietnam National University – Hanoi, Hanoi, Vietnam. E-mail: nguyenthuytrang@hus.edu.vn
bCenter for Innovative Materials and Architectures, Vietnam National University Ho Chi Minh City, Ho Chi Minh City, Vietnam
cInstitute of Atomic and Molecular Sciences, Academia Sinica, Daan District, Taipei City 10617, Taiwan
dMolecular Science and Technology Program, Taiwan International Graduate Program, Academia Sinica, Nankang District, Taipei City 11529, Taiwan
eDepartment of Physics, National Central University, Zhongli District, Taoyuan City 32001, Taiwan
fCCFE, United Kingdom Atomic Energy Authority, Abingdon, OX14 3DB, UK
gVietnam National University, HoChiMinh City, Vietnam
hFaculty of Materials Science and Technology, University of Science, Ho Chi Minh City, Vietnam
First published on 10th December 2020
Abstract
The microscopic mechanism of the H2 adsorption of two Mg-MOF-74 isoreticular frameworks, one with a benzenedicarboxylate (BDC) linker and the other with a dihydroxyfumarate (DHF) linker, were studied on the basis of density functional theory (DFT) method. Possible adsorption sites on the internal surface of the two MOFs were detected using ab initio molecular dynamics (AIMD) annealing simulations. The simulations were able to reproduce all adsorption sites which have been experimentally observed for the BDC-based M-MOF-74 frameworks with M = Ni and Zn. In descending order of binding strengths, they are the adsorption sites primarily induced by the open metal sites P1, the oxygen atoms of the oxido groups P2 and the aromatic rings P3. The H2–framework binding strengths were properly evaluated by taking into account the vibrational zero-point energy (ZPE) contribution. An additional type of adsorption sites induced by the oxygen atoms of the carboxyl groups P4 is predicted for the Mg-MOF-74 framework. Two types of adsorption sites primarily induced by the open metal sites P1 and oxygen atoms of the carboxyl groups P2 were predicted for the DHF-based Mg-MOF-74 framework. Detailed analysis of the electron density showed that the electrostatic interaction of the H2 molecule with the charge distribution of the local framework environment within a radius of ∼3.5 Å is a key factor to define adsorption positions and binding strength. The absence of the P4 sites in the BDC-based Zn-MOF-74 framework is caused by the lower charge density at the oxygen atoms induced by less electro-positive metal. The substitution of the nonaromatic DHF linker for the aromatic BDC linker reduces the binding strength at the metal induced adsorption sites by 1.45 kJ mol−1 due to the absence of the aromatic ring.
1. Introduction
In recent decades, metal–organic frameworks (MOFs) have emerged as an advanced generation of nanoporous materials.1–3 They have crystalline structures that are composed of metal clusters linked to each other by organic molecules to form coordination networks. The large variation in chemical compositions and conformations of both metal-cluster nodes and organic linkers allows a great flexibility in designing and adjusting MOFs to achieve desired internal surface area, void volume as well as other physicochemical properties.2 Consequently, designing MOFs for a wide range of applications such as catalysis,4 gas storage,5 gas sensors,6 and biomedicine7 etc. is a very active research area.Numerous research studies have been devoted to designing MOFs for solid-state hydrogen storage applications.8,9 One of the big challenges is the weak interaction between non-polar covalent hydrogen molecules and MOFs which typically can maintain high hydrogen uptake only at extreme conditions of low temperature and high pressure.8 Using Grand Canonical Monte Carlo (GCMC) simulations of hydrogen adsorption in the IRMOF series, Frost and Snurr suggested that a reasonable H2 uptake at ambient condition could be obtained with an isosteric heat of adsorption of 15 kJ mol−1 (0.155 eV).10 Particular interests have been paid for MOFs with open metal sites which can attract H2 molecules via stronger electrostatic interactions. For example, the low loading heat of adsorption in Co-based SNU-15 MOF with open Co(II) sites is up to 15 kJ mol−1.11 However, its hydrogen uptake at T = 77 K and P = 1 bar is very low 0.74 wt% due to the low metal site density 0.0012 sites per Å3.
Considerable attention has been paid for MOF-74 series since they have higher open metal site density 0.0045 sites per Å3 which corresponds to hydrogen uptake 2.5 wt% at T = 77 K and P = 1 bar.12–15 The experimental values of low loading heat of H2 adsorption of these materials are between 8 and 13 kJ mol−1.13 GCMC simulations using classical force field demonstrated that the primary adsorption sites which are responsible for such loading heat of adsorption are induced by the electrostatic interaction between the positively charged open metal sites and the induced dipole moment of H2 molecules.12 This interaction was demonstrated to be strongly correlated with the ionic charge and the polarizability of the metal. The role of the linker was also revealed with the combination of experimental measurements and theoretical calculations. Kapelewski et al. were able to increase the low loading heat of adsorption by 1.5 kJ mol−1 with a substitution of the original linker 2,5-dioxido-1,4-benzenedicarboxylate (BDC) using an isomer 4,6-dioxido-1,3-benzenedicarboxylate (mBDC).16 Their electronic structure calculations based on the density functional theory (DFT) suggest that the extra contribution of mBDC linker enhances the metal site–H2 interaction. Moreover, the neutron powder diffraction (NPD) experiments evidence that the change from para (BDC linker) to meta (mBDC linker) configurations of the carboxylic acid functionalities and hydroxyl substituents of the linker lead to the change in secondary adsorption sites.16,17 The hydrogen uptake of mBDC-Co/Ni-MOF-74 frameworks at P = 100 bar, T = 25 °C is up to 13 g L−1 and 1.2 wt%, slightly higher than their BDC-based counterparts. These frameworks were believed to be among the best of hydrogen physisorptive adsorbents.17 However, they are still much lower than the targets of the US Department of Energy for practical fuel cell which require a volumetric uptake at least 50 g L−1 in the temperature range −40 ÷ 60 °C at maximum delivery pressure 12 bar.18 Based on classical GCMC simulations, Witman and coworkers suggested that substituting the aromatic linker with a shorter aliphatic linker, 2,3-dihydroxyfumarate (DHF) can strongly enhance the volumetric uptake owing to the higher open-metal site density.19
In order to improve MOFs for practical hydrogen storage applications, it is crucial to understand the underlying mechanism of the H2 adsorption. Various adsorption sites on the internal surface of MOF-74 frameworks have been reported by experimental works.16,20,21 However, previous theoretical calculations primarily focused on the role of the open metal sites.12,13 In this work, basing on ab initio molecular dynamics (AIMD) simulations, all possible adsorption sites including both metal-induced and linker-induced sites were predicted for the original BDC-based Mg-MOF-74 framework and its isoreticular DHF-based framework. Detail electronic structure analysis demonstrates that changing the electro-positivity of the metal affects the stability of linker-induced adsorption sites whereas the linker substitution gives significant influences on the binding strength of metal-induced adsorption sites.
2. Computational details
The crystal structures of MOF-74 family are based on rhombohedral lattice (Fig. 1a and b). Both BDC- and DHF-based frameworks belong to R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group. Their crystal structures were fully optimized on the base of static DFT calculations. There are 3 equivalent 1-dimensional (1D) void channels along c direction within a unit cell of each framework. Thus, adsorption sites in one channel are assumed to be the same as those in the others so that we only need to probe one channel.
space group. Their crystal structures were fully optimized on the base of static DFT calculations. There are 3 equivalent 1-dimensional (1D) void channels along c direction within a unit cell of each framework. Thus, adsorption sites in one channel are assumed to be the same as those in the others so that we only need to probe one channel.
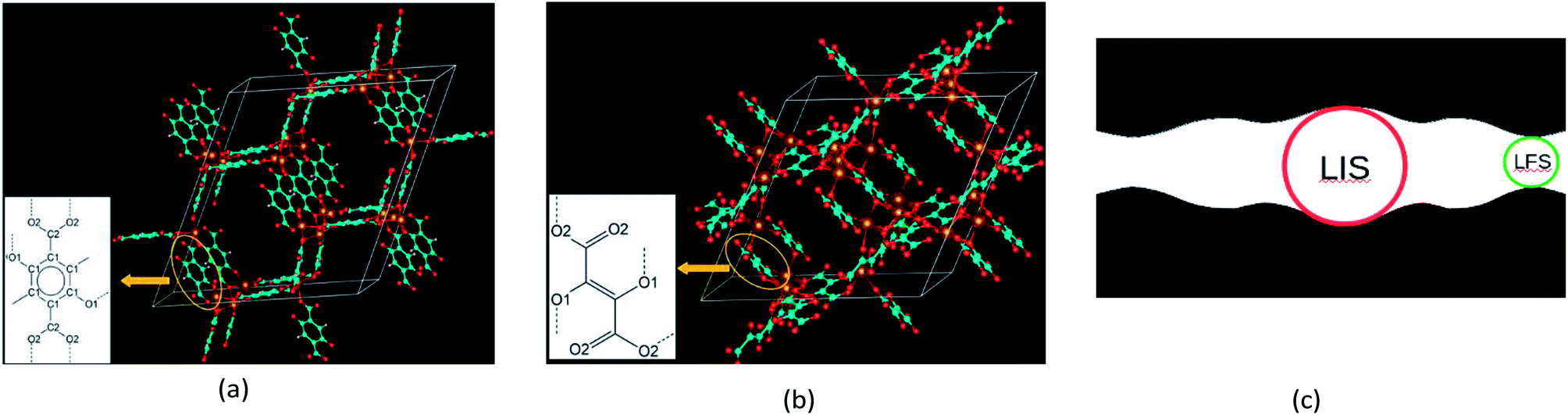 | ||
Fig. 1 (a) and (b) represent the crystal structures of the BDC-Mg-MOF-74 and DHF-Mg-MOF-74 frameworks, respectively. Red, brown, cyan and white spheres represent oxygen, magnesium, carbon and hydrogen atoms, respectively. The insets are schematic representations of corresponding linkers, i.e. (–O)2C6H2–1,4-(COO–)2 linker derived from 2,5-dioxido-1,4-benzenedicarboxylic acid (BDC) in (a) and (–COO)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–2,3-(O–)2 linker derived from 2,3-dihydroxyfumaric acid (DHF) in (b). Dash lines in the insets denote dangling bonds which can be attached to metal atoms. The carbon atoms of the aromatic ring are labeled C1 and the carbon atoms of the carboxyl groups are labeled C2. The oxygen atoms of the oxido groups are labeled O1 and the oxygen atoms of the carboxyl groups are labeled O2. (c) A schematic representation of the largest included sphere (LIS) and the largest free sphere (LFS). The red circle, green circle and black shaded area denote the LIS, LFS and the space filled by the framework atoms, respectively. C–2,3-(O–)2 linker derived from 2,3-dihydroxyfumaric acid (DHF) in (b). Dash lines in the insets denote dangling bonds which can be attached to metal atoms. The carbon atoms of the aromatic ring are labeled C1 and the carbon atoms of the carboxyl groups are labeled C2. The oxygen atoms of the oxido groups are labeled O1 and the oxygen atoms of the carboxyl groups are labeled O2. (c) A schematic representation of the largest included sphere (LIS) and the largest free sphere (LFS). The red circle, green circle and black shaded area denote the LIS, LFS and the space filled by the framework atoms, respectively. | ||
Molecular dynamics simulated annealing method has been adopted to locate minima of the potential energy surface of adsorption systems.22–24 In this work, n hydrogen molecules were randomly put into one void channel with n = 12 for the DHF-based framework and 25 for the BDC-based framework. These systems are so-call framework–nH2 systems. The total number of atoms in a unit cell is 132 in the case of the DHF-based framework–12H2 system and 212 in the case of the BDC-based framework–25H2 system. All framework–nH2 systems were equilibrated at 300 K for 10 ps by AIMD simulations in NVT ensembles and then annealed from 300 K to 0 K for 10 ps. The H2 molecules were expected to reach various adsorption sites on the potential energy surfaces induced by the frameworks at the end of the annealing processes. In order to evaluate the influence of initial conditions on the annealing processes, three initial configurations sampled from 300 K NVT ensembles were considered.
The frameworks with a single H2 molecule at various adsorption sites, which are so-called single molecule adsorption models, were further optimized from the annealed structures with static DFT calculations. The electronic adsorption energies were estimated via the following formula:
| ΔE0i = Efr+gasi − Egas − Efr | (1) |
| ΔEi = (Efr+gasi + ZPEfr+gasi) − (Egas + ZPEgas) − (Efr + ZPEfr) | (2) |
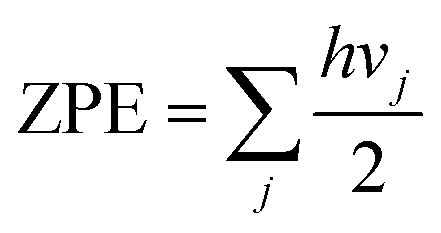 | (3) |
The sum is taken over all vibrational modes of the system, h is the Planck's constant and vj is the frequency of the jth vibrational mode. The adsorption energies calculated using the single molecule adsorption model correspond to the zero loading adsorption energies.
First principles calculations were introduced within the framework of DFT implemented within the Vienna ab initio simulation package (VASP).26,27 Exchange–correlation potentials were expressed within the generalized gradient approximation (GGA) using Perdew–Burke–Ernzerhof (PBE) functional.28 The dispersion correction was added to original Kohn–Sham DFT equations in terms of atom-pairwise sum-over C8R−8 potentials with cut-off distances and dispersion coefficients explicitly computed by S. Grimme et al. from first principles.29 This method is commonly called DFT-D3. The valence electrons' wave functions, i.e. the wave functions of Mg 3s, C and O 2s, 2p and H 1s electrons were represented in plane wave basis set with cut-off energy of Ecut-off = 550 eV. Highly oscillating core parts were simulated with projector augmented wave (PAW) method.30,31 Since both BDC-based and DHF-based Mg-MOF-74 frameworks have large unit cells of sizes ∼26.08 × 26.08 × 6.87 Å3 and ∼18.76 × 18.76 × 6.87 Å3 respectively,19 total energies were calculated on a Γ-center 1 × 1 × 3 Monkhorst–Pack mesh in reciprocal space. Both cut-off energy and k-mesh were chosen according to the convergence of the electronic adsorption energy of the strongest site within an error <1 meV per atom in comparison to the case using Ecut-off = 1000 eV and k-mesh of 4 × 4 × 12.
Self-consistent-field (SCF) convergence criterion for electronic structure optimization was 10−6 eV. For crystal structural optimization, all atomic positions were relaxed until all atomic forces smaller than 10−3 eV Å−1. The vibrational frequencies required for calculating ZPE corrections were obtained from normal mode analysis within harmonic approximation. The elements of the Hessian matrix were calculated from the DFT forces using finite difference method with the atomic step size 0.015 Å. The frequencies were then calculated as the square roots of the eigenvalues of the Hessian matrix. For reducing computational costs, only the vibrations of hydrogen molecules were considered. Benchmarks on two adsorption sites of two different types in DHF-based framework showed that the ZPE contributions of the framework to the adsorption energies are less than 2 meV.
AIMD simulations were carried out within the framework of Born–Oppenheimer approximation.32 The temperature was controlled using Nosé–Hoover thermostat while the optimized volume of the unit cell of the frameworks were kept constant.33 During the AIMD simulations, all displacements of the frameworks and gas molecules were allowed. The time step is 0.2 fs – 1/40 of the period of H–H stretching mode.
3. Results and discussion
3.1. Crystal structure and porous geometry
The porosity characteristics including accessible surface area per gram (ASA), accessible volume per gram (AV), largest included sphere (LIS) radius (RI) and largest free sphere (LFS) radius (RF) were examined using Voronoi-mesh method with Zeo++ package.34 A schematic representation of the LIS and the LFS is shown in Fig. 1c. In Table 1, calculated structure parameters and porosity characteristics of the two isoreticular Mg-MOF-74 frameworks are summarized and compared with those derived from powder neutron diffraction (PND) data35 and previous PBE calculations using the same basis set and pseudopotential method.23 According to this table, the dispersion correction is important to reproduce experimental lattice constants of the BDC-based framework. It should be noted that the NPD data show a contraction of the lattice constant a and extension of the lattice constant c when the temperature increases from 20 K to ∼470 K.35| BDC-based Mg-MOF-74 framework | DHF-based Mg-MOF-74 framework | |||
|---|---|---|---|---|
| PBE-D3/plane wave + PAW (this work) | PBE/plane wave + PAW23 | PND35 | PBE-D3/plane wave + PAW (this work) | |
| a = b (Å) | 26.06 | 26.22 | 25.92 (T = 20 K), 25.88 (T = 298 K) | 18.80 |
| c (Å) | 6.86 | 6.96 | 6.86 (T = 20 K), 6.88 (T = 298 K) | 6.84 |
| ASA (m2 g−1) | 1763 | — | — | 1042 |
| AV (cm3 g−1) | 0.340 | — | — | 0.088 |
| RI (Å) | 11.7 | — | — | 7.6 |
| RF (Å) | 10.9 | — | — | 6.2 |
The resulted unit cell volume change is a tiny oscillation ∼0.09% around the average value ∼3393 Å3. The lattice constant changes are also very small within this temperature range, i.e. <0.3%. Hence, the optimized lattice unit cell can be used for the NVT AIMD simulation within the temperature range from 300 K down to 0 K.
There is no experimental data for the porosity characteristics except from the ASA which can be compared with Langmuir surface area calculated from the experimental N2 adsorption isotherms. However, only experimental N2 adsorption isotherms for the BDC-based Mn, Fe, Co and Ni-MOF-74 frameworks were reported with the deduced Langmuir surface areas 1797, 1535, 1432 and 1574 m2 g−1 respectively.16 The theoretical ASA value for the BDC-based Mg-MOF-74 framework 1763 m2 g−1 is well within the experimental range. The shorter linker of the DHF-based framework lead to smaller lattice constant a (b) and narrower void channel with smaller LIS and LFS radii but does not significantly change the lattice constant c. The narrower void channel, in turn, lead to the smaller ASA and AV. The ASA of the BDC-based framework is 1.69 times as large as that one of the DHF-based framework. Whereas the AV is 3.86 times larger accordingly.
3.2. Annealing processes
Fig. 2 shows the annealed structures of framework–nH2 systems starting from three different initial configurations sampling within 300 K NVT ensembles. A general agreement between these annealing simulations is that there are five types of H2 adsorption sites in the BDC-based framework and three types of adsorption sites in the DHF-based framework. In the Fig. 2, adsorption positions of the same type are represented by the same color of hydrogen atoms. Before going to analyse in detail of the adsorption positions, we first discuss the occupation temperature range within which H2 molecules gradually occupy adsorption sites of each type. Table 2 is a summary of the occupation temperature ranges of various types with three different initial configurations. According to this, different initial configurations lead to a slight fluctuation ∼5 ÷ 15 K of the occupation temperature range of each type. This fluctuation can be considered as a result of the thermal fluctuation of the system during the annealing process. The initial configuration does not affect the occupation order between various types. | ||
| Fig. 2 Annealed structures of the BDC-based Mg-MOF-74–25H2 system (on the left) and DHF-based Mg-MOF-74–12H2 systems (on the right) starting from three different initial configurations sampling within the 300 K NVT ensemble. Red, brown, cyan and white spheres represent oxygen, magnesium, carbon and hydrogen atoms of the frameworks. Hydrogen atoms of the molecules adsorbed at P1, P2, P3, P4 and P5 sites of the BDC-based framework are represented by blue, yellow, green, pink and white spheres respectively. Hydrogen atoms of the molecules adsorbed at P1, P2 and P3 sites of the DHF-based framework are represented by blue, pink and white spheres respectively. | ||
| P1 | P2 | P3 | P4 | P5 | |
|---|---|---|---|---|---|
| BDC based | 300–210 K | 205–55 K | 70–30 K | 45–20 K | 25–15 K |
| 300–215 K | 220–55 K | 85–30 K | 55–20 K | 30–20 K | |
| 300–175 K | 170–70 K | 80–45 K | 55–10 K | 40–15 K | |
| DHF based | 300–170 K | 260–50 K | 50 K | ||
| 300–210 K | 155–65 K | — | |||
| 300–210 K | 150–20 K | 20 K |
For the BDC-based MOF–25H2 system, during the simulated annealing processes, the blue sites in Fig. 2, start to be occupied first, and then yellow sites, green sites, pink sites and white sites which are called P1, P2, P3, P4 and P5 sites respectively. Due to the symmetry of honeycomb-like lattice, there are 6 sites of each type, and hence totally 30 adsorption sites, in one void channel of a R unit cell. The P1, P2 and P3 sites have been experimentally found for the BDC-based Zn- and Ni-MOF-74 frameworks via NPD data.20,21 Interestingly, the NPD experiments showed that these sites can be fully occupied in the BDC-based Zn-MOF-74 framework at T down to 30 K (ref. 20) which is the same as the lowest temperature for the P3 sites to be fully occupied in our simulated annealing processes. The P5 sites were coincidentally observed via the NPD experiments for the BDC-based Zn-MOF-74 framework at T = 15 K. It should be noted that these sites were not experimentally observed for the Ni-based framework even at T = 4 K because, in this case, the NPD data were collected at low loadings (0.5 and 1.5H2/Ni).21 The P4 sites were not experimentally observed for the BDC-based Zn-MOF-74 framework at loadings up to 4.2H2/Zn. The absence of P4 sites in this framework will be discussed with the electronic structure analysis later. It should be noted that the number of H2 molecules in the simulations, 25 molecules, is smaller than the total number of all adsorption sites, 30 sites. The first three types of adsorption sites have higher probability to be occupied so that they are fully occupied at the end of the annealing processes. The P4 and P5 sites are not fully occupied so the final temperatures of the corresponding occupation temperature ranges are the temperatures at which H2 molecules populate P4 and P5 sites the last time.
For the DHF-based MOF–12H2 system, the occupation order is the blue sites first, then yellow sites and white sites in the Fig. 2, mentioned as P1, P2 and P3 sites respectively. The occupation temperature ranges of P1 and P2 sites shown in the Table 2 are similar to the P1 and P2 sites in the BDC-based framework, respectively. However, the dependence of the occupation temperature range of the P2 sites on the initial configuration is stronger, denoting by a stronger thermal fluctuation ∼100 K. There are 6 P1 and 6 P2 sites but only one P3 site, and hence 13 adsorption sites in one void channel of a unit cell while there are 12H2 molecules. The P1 sites are fully occupied first at temperature down to 170 K. In two of the simulated annealing processes, only 5 of 6 P2 sites are occupied, the last H2 molecule go to P3 site instead of populating the last P2 site. The P3 site is occupied at the same time with the final occupation of P2 sites at temperature down to 20 K.
In general, P1, P2, P3 and P4 sites of the BDC-based Mg-MOF-74 framework and P1 and P2 sites of the DHF-based framework are direct adsorption sites since they are directly attached to the frameworks. The P5 sites of the BDC-based framework and P3 sites of the DHF-based framework are indirect adsorption sites. In the following, direct adsorption sites will be discussed in detail to clarify the mechanism of the interaction between the frameworks and the H2 molecules.
3.3. Adsorption positions and binding mechanisms
 | ||
| Fig. 3 Foreground illustrations of (a) P1, (b) P2, (c) P3 and (d) P4 adsorption positions in the BDC-Mg-MOF-74 framework. The P1 and P2 sites in the DHF-Mg-MOF-74 are illustrated in (e) and (f) respectively. Red, brown, cyan and white spheres represent oxygen, magnesium, carbon and hydrogen atoms of the framework respectively. P1, P2, P3 and P4 hydrogen molecules in the BDC-based framework are represented by blue spheres in (a), yellow spheres in (b), green spheres in (c) and pink spheres in (d) respectively. P1 and P2 hydrogen molecules in the DHF-based framework are represented by blue spheres in (e) and pink spheres in (f) respectively. Dash lines denote the connection between the adsorbed H2 molecules and the nearest framework atoms. Dot lines in (b) denote the oxygen triangle close to the P2 site of the BDC-based Mg-MOF-74 framework. | ||
A P2 site of the BDC-based Mg-MOF-74 framework shown in Fig. 3b is located above a triangle of three oxygen atoms which connect to the same metal ion, one from an oxido group called O1 and two from two carboxyl groups called O2 (see Fig. 1b). This site locates the nearest to O1 atoms with a distance 3.21 Å. The experimental values for this distance are 3.5 Å from the NPD data collected at 77 K.36 At this site, the H2 molecule is likely in head-on position with respect to the O1 atom. This is also the common position for a H2 molecule when it is attached to an isolated negative ion in order to optimize the negative charge–quadrupole interaction.37 The BDC ligand environment of the framework also gives rise to a slight tilt by an angle 6° from the ideal head-on position. A P2 site of the DHF-based framework shown in Fig. 3f is the nearest to the oxygen atom of a carboxyl group O2 at a distance 2.47 Å. The H2 molecule populate this site nearly at head-on position to the O2 atom with a significant tilting angle 22.5° induced by the DHF ligand environment.
A P3 site of the BDC-based Mg-MOF-74 framework shown in Fig. 3c is above an aromatic ring of a linker at a distance 3.05 Å from the ring plane. This distance is in good agreement with the NPD value 3.08 Å for Co-based framework. It was shown that when a hydrogen molecule interacts with a molecule of a benzene dicarboxylic acid derivative, it prefers to bind head-on above the center of the aromatic ring at a distance ∼3 Å with its axes perpendicular to the ring plane.37 However, in the BDC-based framework, the H2 molecule is significantly inclined by an angle of 25° with the normal of the ring plane denoting significant influence of metal cluster nodes on this linker-induced adsorption site. A P4 site of the BDC-based Mg-MOF-74 framework shown in Fig. 3d is the nearest to the oxygen atoms of a carboxyl group O2 at a distance 3.01 Å. Similar to the P2 site, the H2 molecule is inclined by an angle of 6° from the ideal head-on position to the O2 atom because of the BDC ligand environment.
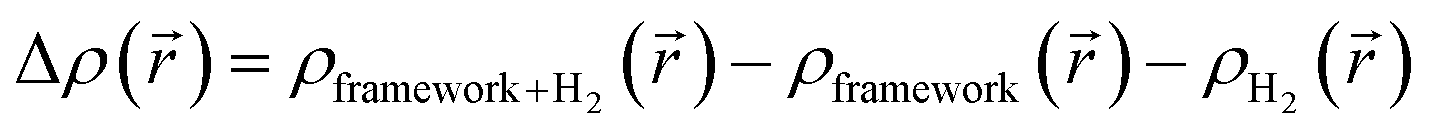 | (4) |
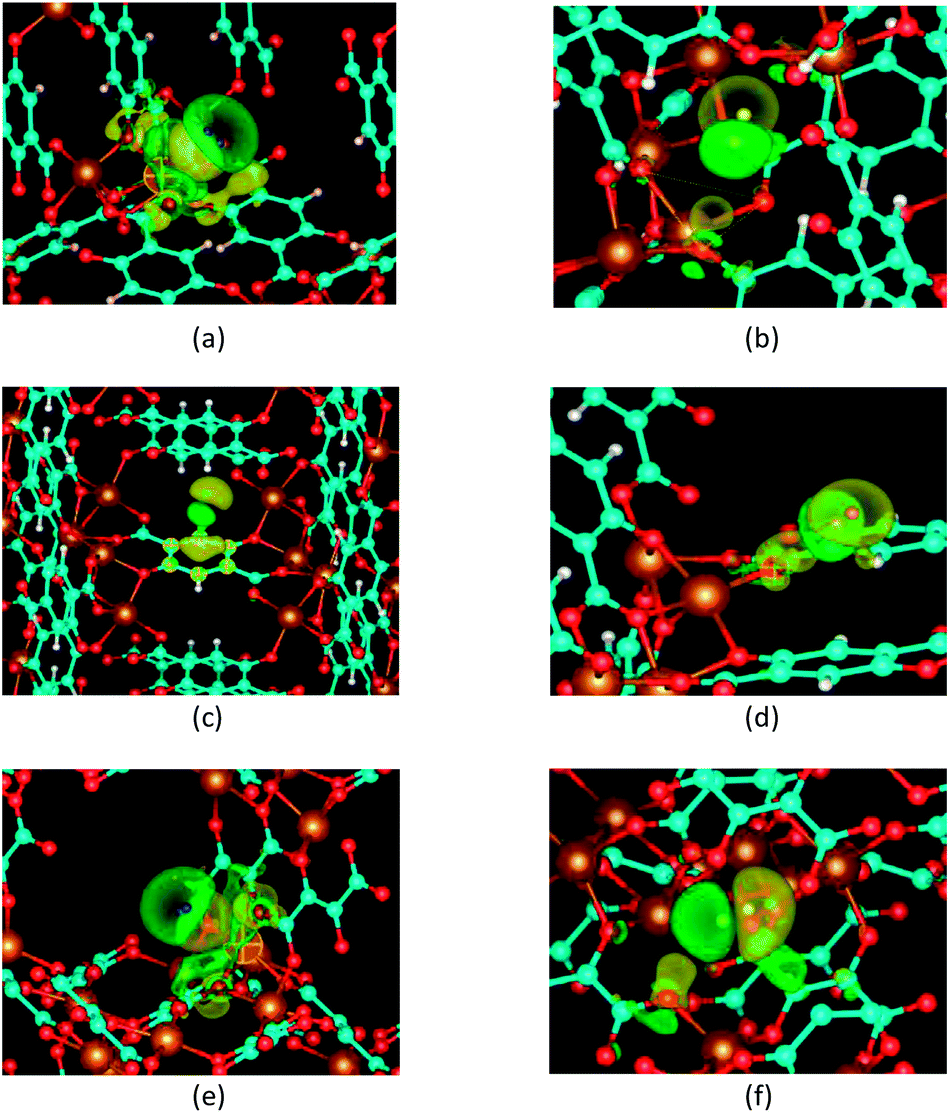 | ||
| Fig. 4 Isosurfaces of the electron density deformations upon hydrogen adsorption onto (a) P1, (b) P2, (c) P3 and (d) P4 sites in the BDC-based Mg-MOF-74 framework, (e) P1 and (f) P2 sites in the DHF-based Mg-MOF-74 framework. The isovalues is 0.0001|e| Å−3. Yellow and green denote electron donated and loss surfaces respectively. Red, brown, cyan and white spheres represent oxygen, magnesium, carbon and hydrogen atoms of the frameworks respectively. P1, P2, P3 and P4 hydrogen molecules in the BDC-based framework are represented by blue, yellow, green and pink spheres respectively. P1 and P2 hydrogen molecules in the DHF-based framework are represented by blue and pink spheres respectively. | ||
For a P1 site in both frameworks, the electron density is increased in an extended area on the Mg side of the H2 molecule and decreased on the other side. This electron density deformation clearly evidences that the positive charge of the Mg2+ cation polarizes the hydrogen molecule perpendicularly to its axis. Thus, the interaction of the Mg2+ charge with the hydrogen induce-dipole is the dominating force to hold H2 molecules at a P1 site in both frameworks. Additionally, there are electron loss in the area between the oxygen and magnesium atoms, but the electron enhancements in the p-shaped area around the oxygen atoms and on the aromatic ring carbon atoms called C1 (see Fig. 1a). These atoms are located within a radius 3.5 Å around the P1 site. This indicates remarkable contribution of the surrounding ligand environment within the radius 3.5 Å to the hydrogen attraction of the P1 site.
The electron density deformations upon hydrogen adsorption to P2 and P4 sites in the BDC-based framework and a P2 site in the DHF-based framework demonstrate the dominating role of the interaction between the H2 induced-dipole and oxygen ions. The electron donated and loss regions on the two heads of the hydrogen molecule indicate a polarization of the hydrogen molecule along its axis. Obvious electron density deformations can be seen around the nearest framework atoms, i.e. O1 for the P2 site, O2 for the P4 site in the BDC-based framework and O2 for the P2 site in the DHF-based framework. There are also other noticeable electron density deformation regions. For the P2 site of the BDC-based framework, they are the regions around the two other oxygen atoms O2 of the oxygen triangle and the carbon atoms of the related carboxyl group C2 at distance. For the P4 site of the BDC-based framework, they are the regions around the H atom of the aromatic ring. For the P2 site of the DHF-based framework, they are the region between two carbon atoms within the radius 3.5 Å. These deformations indicate the contributions of the surrounding ligand environment within the radius 3.5 Å. The electron deformation upon the H2 adsorption at a P3 site in the BDC-based framework shows an electron donated region above the aromatic ring and a polarization of the H2 molecule along its axis. This demonstrates the interaction between the H2 induced dipole and the electron cloud of the ring at this site.
| BDC-based Mg-MOF-74 framework | DHF-based Mg-MOF-74 framework | |||||
|---|---|---|---|---|---|---|
| P1 | P2 | P3 | P4 | P1 | P2 | |
| ΔE0 (eV) | −0.192 | −0.119 | −0.105 | −0.099 | −0.183 | −0.128 |
| ΔE (eV) | −0.121 | −0.088 | −0.066 | −0.062 | −0.106 | −0.080 |
| ZPE (eV) | 0.070 | 0.031 | 0.038 | 0.037 | 0.077 | 0.048 |
| ΔE0f (eV) | −0.171 | −0.110 | −0.090 | −0.074 | — | — |
| ΔEf (eV) | −0.100 | −0.057 | −0.042 | −0.027 | — | — |
| ZPEf (eV) | 0.071 | 0.053 | 0.048 | 0.047 | — | — |
As discussed above, the electronic origin of the H2 attraction is the electrostatic interaction between the H2 molecule and the surrounding charged fragments of the MOF-74 frameworks within the radius ∼3.5 Å. Thus, charge distribution should be quantitatively considered to explain for the binding strength at various adsorption sites. The atomic charges were calculated from the DFT-derived charge density using Bader charge analysis method which defined atomic charge as total charge within zero charge flux surface.39–42 Table 4 show the calculated Bader atomic charges of the MOF-74 frameworks.
| BDC-based Mg-MOF-74 framework | DHF-based Mg-MOF-74 framework | BDC-based Zn-MOF-74 framework | |
|---|---|---|---|
| Metal | +2|e| | +2|e| | +1.36|e| |
| O1 | −1.93 ± 0.02|e| | −1.93 ± 0.02|e| | −1.19 ± 0.02|e| |
| O2 | −1.88 ± 0.01|e| | −1.88 ± 0.01|e| | −1.16 ± 0.01|e| |
Both P1 sites in the BDC- and DHF-based framework are primarily based on the interaction of the positive Mg ion and its surrounding oxygens with the H2 molecule. As shown in Table 4, the Bader atomic charges of these ions are the same for both frameworks. However, the binding strength of a P1 site in the DHF-based framework is smaller than in the BDC-based framework by 0.015 eV – 1.45 kJ mol−1. This difference can be assigned to the contribution of the aromatic ring in the BDC linker as denoted by the electron deformation on the ring, which is absent from the nonaromatic DHF linker. Similar linker-induced binding strength variation was also experimentally observed when the configuration of the BDC linker was changed from para to meta (mBDC linker). In this case, the extra contribution of the mBDC linker lead to an enhancement of the binding strength at the metal-induced site by 1.5 kJ mol−1.16
In the BDC-based Mg-MOF-74 framework, the Bader charge of oxygen ion in the oxido group O1 is slightly more negative than that of the oxygen ion in the carboxyl group O2 by 0.05|e| as seen in Table 4. However, the binding strength of the O1 induced adsorption site P2 is significantly larger than the O2 induced adsorption site P4, 0.023 eV (2.22 kJ mol−1). This is due to the fact that, within the radius 3.5 Å, there are more framework atoms surrounding the P2 site (one O1, two O2 and one C2 atom) than the P4 site (only one O2 and one H atom). The Bader charge of the O2 ion in the DHF-based Mg-MOF-74 framework is the same as that of the O2 atom and less negative than that of the O1 atom in the BDC-based Mg-MOF-74 framework. However, the binding strength of the O2 induced site P2 in the DHF-based framework is larger than the O2 induced site P4 and very close to the O1 induced site P2 in the BDC-based framework. The reason is that this site is also surrounded by more framework atoms (O2 and carbon atoms) than the O2 induced site P4 in the BDC-based framework.
It is worth noting from experiments of H2 adsorption in the BDC-based Mg-MOF-74 framework that the heat of adsorption depend on the loading rate.13 The heat of adsorption at zero loading is a bit higher than at nonzero loading rate below 1H2/metal. In this loading rate range, the P1 sites provide the main contribution to the hydrogen uptake.20,21 The slight reduction of the heat of adsorption can be assigned to the site–site interaction. In the present work, the adsorption energy of each type of adsorption sites was calculated at zero loading and full loading rate of P1 sites (1H2/metal). The absolute values of the ZPE corrected adsorption energy of a P1 site at the zero loading and 1H2/metal loading in the BDC-based Mg-MOF-74 framework are 0.121 eV and 0.100 eV, respectively. These values are in good agreement with the experimental data of zero loading and low loading enthalpies of adsorption 0.119 eV and 0.109 eV.15 At loading rate larger than 1H2/metal, the main contributions to the experimental enthalpies come from the P2 and P3 sites. The absolute values of the ZPE corrected adsorption energies of a P2 and P3 site at 1H2/metal loading are 0.057 eV and 0.042 eV respectively, which are around the experimental enthalpies of adsorption at loading rate >1H2/metal ∼0.052 eV.
Concerning the absence of P4 sites in the BDC-based Zn-MOF-74 framework suggested by the NPD experiment,20 it is worth noting that the main origin of these sites is the O2 negative charge. The more negative the O2 ion is, the stronger the H2 attraction is. On the other hand, Mg is more electro-positive than Zn so that the oxygen atoms surrounding Mg are expected to be more negatively charged than those surrounding Zn. The reducing in charge density at the O2 site is expected to reduce the binding strength at the P4 site or even destabilize this site. In this work, the charge distribution of the BDC-based Zn-MOF-74 was examined using DFT derived electron density and Bader charge analysis. The results are also shown in Table 4. It is clear that the charge of Mg ion in the Mg-MOF-74 framework is +2|e|, larger that of Zn ion in the Zn-MOF-74 framework +1.36|e|. The ionic charges of neighbouring O2 atoms are −1.9 and −1.2|e| in the Mg- and Zn-based framework respectively. The ZPE corrected adsorption energy of a P4 site in the BDC-based Zn-MOF-74 was also calculated at zero loading and 1H2/metal loading. The zero loading value is −0.042 eV, 0.02 eV – 1.93 kJ mol−1 weaker than the case of the Mg-MOF-74. At loading rate 1H2/metal, the adsorption energy of a P4 site is −0.027 eV and −0.007 eV for Mg- and Zn-MOF-74 framework, respectively. The predicted smaller values of the adsorption energy of the P4 site agree with the fact that it has been absent in the Zn-based framework. However, due to the higher adsorption energy it would be interesting to check experimentally whether the H2 molecules could be occupied at the P4 sites in Mg-based framework.
The electronic adsorption energy at the ring induced site P3 in the BDC-based MOF-74 framework is 0.105 eV (10.13 kJ mol−1), higher than those of the BDC derivatives ∼4 kJ mol−1.37 The ZPE corrected adsorption energy of this site is −0.066 eV (6.37 kJ mol−1), stronger than that of the ring site in the MOF-5 framework 5.1 kJ mol−1.22 The enhancement of the binding strength between the ring and the H2 molecule in the MOF-74 can be explained basing on the fact that adding electron-donated substituents such as –OH, –CH3, –NH2 can increase the binding strength.37 In the case of MOF-74, the enhancement of H2 attraction can come from the fact that there are two oxido substituents on the ring which directly connect to strong electron-donating metal atoms. Whereas, there is no such substituent on the BDC linker in the MOF-5.
4. Conclusions
In conclusion, the microscopic origin of H2 adsorption onto the internal surface of the MOF-74 frameworks were explored on the base of AIMD simulations and DFT electronic structure calculations. The guest molecules show significant electrostatic interactions with the local framework environment within a radius 3.5 Å. Four types of direct adsorption sites on the internal surface of the BDC-based Mg-MOF-74 framework were specified. With shorter nonaromatic linker, the DHF-based MOF-74 framework provides only two types of direct adsorption sites. The primary adsorption sites P1 in both frameworks are due to the interaction of the H2 molecules with the positively charged open metal sites and their surrounding oxygen sites. In the case of the aromatic BDC linker, the contribution of the high electron density aromatic rings within the distance 3.5 Å from these sites enhances the binding strength by 1.45 kJ mol−1 in comparison to the case of nonaromatic DHF linker. The secondary adsorption sites originate from the interactions of the H2 molecule with the negatively charged oxygen sites and the high density electron clouds of the aromatic rings. The reduction of the electro-positivity of the metal can induce reductions of the negative charges of the oxygen sites which in turn destabilize the oxygen induced adsorption sites.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like acknowledge the financial supports of the World Bank and the Ministry of Science and Technology of Vietnam, grant number 13/FIRST/1.a/VNU1; and Vietnam National University Ho Chi Minh city, grant number TX-2020-50-01.References
- S. T. Meek, J. A. Greathouse and M. D. Allendorf, Metal-organic frameworks: a rapidly growing class of versatile nanoporous materials, Adv. Mater., 2011, 23, 249–267 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The chemistry and applications of metal-organic frameworks, Science, 2013, 341(6149), 1230444 CrossRef.
- S. Ahmad, A. Ansari, W. A. Siddiqi and M. K. Akram, Nanoporous Metal-Organic Framework, Mater. Res. Found., 2019, 58, 107–139 CAS.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Catalysis and photocatalysis by metal organic frameworks, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- R. B. Getman, Y.-S. Bae, C. E. Wilmer and R. Q. Snurr, Review and Analysis of Molecular Simulations of Methane, Hydrogen, and Acetylene Storage in Metal–Organic Frameworks, Chem. Rev., 2012, 112, 703–723 CrossRef CAS.
- H. Lee, T.-B. Nguyen, D.-K. Nguyen, J.-H. Kim, J.-Y. Kim, B. T. Phan and S. S. Kim, Gas Sensing Properties of Mg-Incorporated Metal–Organic Frameworks, Sensors, 2019, 19(15), 3323 CrossRef.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Ferey, R. E. Morris and C. Serre, Metal–Organic Frameworks in Biomedicine, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Hydrogen Storage in Metal–Organic Frameworks, Chem. Rev., 2012, 112, 782–835 CrossRef CAS.
- A. Ahmed, S. Seth, J. Purewal, A. G. Wong-Foy, M. Veenstra, A. J. Matzger and D. J. Siegel, Exceptional hydrogen storage achieved by screening nearly half a million MOFs, Nat. Commun., 2019, 10, 1568 CrossRef.
- H. Frost and R. Q. Snurr, Design Requirements for Metal-Organic Frameworks as Hydrogen Storage Materials, J. Phys. Chem. C, 2007, 111, 18794 CrossRef CAS.
- Y. E. Cheon and M. P. Suh, Selective gas adsorption in a microporous metal–organic framework constructed of Co4II clusters, Chem. Commun., 2009, 45, 2296 RSC.
- T. Pham, K. A. Forrest, R. Banerjee, G. Orcajo, J. Eckert and B. Space, Understanding the H2 Sorption Trends in the M-MOF-74 Series (M = Mg, Ni, Co, Zn), J. Phys. Chem. C, 2015, 119, 1078–1090 CrossRef CAS.
- W. Zhou, H. Wu and T. Yildirim, Enhanced H2 Adsorption in Isostructural Metal–Organic Frameworks with Open Metal Sites: Strong Dependence of the Binding Strength on Metal Ions, J. Am. Chem. Soc., 2008, 130, 15268–15269 CrossRef CAS.
- J. L. C. Rowsell and O. M. Yaghi, Effects of Functionalization, Catenation, and Variation of the Metal Oxide and Organic Linking Units on the Low-Pressure Hydrogen Adsorption Properties of Metal–Organic Frameworks, J. Am. Chem. Soc., 2006, 128, 1304–1315 CrossRef CAS.
- P. D. Dietzel, P. A. Georgiev, J. Eckert, R. Blom, T. Strassle and T. Unruh, Interaction of hydrogen with accessible metal sites in the metal–organic frameworks M2(dhtp) (CPO-27-M; M = Ni, Co, Mg), Chem. Commun., 2010, 46, 4962–4964 RSC.
- M. T. Kapelewski, S. J. Geier, M. R. Hudson, D. Stück, J. A. Mason, J. N. Nelson, D. J. Xiao, Z. Hulvey, E. Gilmour, S. A. FitzGerald, M. H. Gordon, C. M. Brown and J. R. Long, M2(m-dobdc) (M = Mg, Mn, Fe, Co, Ni) Metal–Organic Frameworks Exhibiting Increased Charge Density and Enhanced H2 Binding at the Open Metal Sites, J. Am. Chem. Soc., 2014, 136, 12119–12129 CrossRef CAS.
- M. T. Kapelewski, T. Runčevski, J. D. Tarver, H. Z. H. Jiang, K. E. Hurst, P. A. Parilla, A. Ayala, T. Gennett, S. A. FitzGerald, C. M. Brown and J. R. Long, Record High Hydrogen Storage Capacity in the Metal–Organic Framework Ni2(m dobdc) at Near-Ambient Temperatures, Chem. Mater., 2018, 30(22), 8179–8189 CrossRef CAS.
- https://www.energy.gov/eere/fuelcells/hydrogen-storage.
- M. Witman, S. Ling, A. Gladysiak, K. C. Stylianou, B. Smit, B. Slater and M. Haranczyk, Rational Design of a Low-Cost, High-Performance Metal–Organic Framework for Hydrogen Storage and Carbon Capture, J. Phys. Chem. C, 2017, 121, 1171–1181 CrossRef CAS.
- Y. Liu, H. Kabbour, C. M. Brown, D. A. Neumann and C. C. Ahn, Increasing the density of adsorbed hydrogen with coordinatively unsaturated metal centers in metal-organic frameworks, Langmuir, 2008, 24, 4772–4777 CrossRef CAS.
- C. M. Brown, A. J. R. Cuesta, J.-H. Her, P. S. Wheatley and R. E. Morris, Structure and spectroscopy of hydrogen adsorbed in a nickel metal-organic framework, Chem. Phys., 2013, 427, 3–8 CrossRef CAS.
- K. Sillar, A. Hofmann and J. Sauer, Ab Initio Study of Hydrogen Adsorption in MOF-5, J. Am. Chem. Soc., 2009, 131, 4143–4150 CrossRef CAS.
- P. Suksaengrat, V. Amornkitbamrung, P. Srepusharawoot and R. Ahuja, Density Functional Theory Study of Hydrogen Adsorption in a Ti-Decorated Mg-Based Metal–Organic Framework-74, ChemPhysChem, 2016, 17, 879–884 CrossRef CAS.
- M. Dixit, D. T. Major and S. Pal, Hydrogen adsorption in ZIF-7: a DFT and ab initio molecular dynamics study, Chem. Phys. Lett., 2016, 651, 178–182 CrossRef CAS.
- D. A. Mc Quarrie and J. D. Simon, Molecular Thermodynamics, University Science Books, Sausalito, CA, 1999 Search PubMed.
- G. Kresse and J. Furthmuller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev., 1999, 59, 1758 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- P. E. Blochl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- D. Marx and J. Hutter, Ab initio Molecular Dynamics: Basic Theory and Advanced Methods, Cambridge UP, 2012 Search PubMed.
- D. Frenkel and B. Smit, Understanding Molecular Simulation, Academic Press, London, 1996 Search PubMed.
- T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza and M. Haranczyk, Algorithms and tools for high-throughput geometry-based analysis of crystalline porous materials, Microporous Mesoporous Mater., 2012, 149, 134–141 CrossRef CAS.
- W. L. Queen, C. M. Brown, D. K. Britt, P. Zajdel, M. R. Hudson and O. M. Yaghi, Site-Specific CO2 Adsorption and Zero Thermal Expansion in an Anisotropic Pore Network, J. Phys. Chem. C, 2011, 115, 24915–24919 CrossRef CAS.
- K. Sumida, C. M. Brown, Z. R. Herm, S. Chavan, S. Bordigad and J. R. Long, Hydrogen storage properties and neutron scattering studies of Mg2(dobdc)—a metal–organic framework with open Mg2+ adsorption sites, Chem. Commun., 2011, 47, 1157–1159 RSC.
- R. C. Lochan and M. Head-Gordon, Computational studies of molecular hydrogen binding affinities: the role of dispersion forces, electrostatics, and orbital interactions, Phys. Chem. Chem. Phys., 2006, 8, 1357–1370 RSC.
- D. E. Stogryn and A. P. Stogryn, Molecular multipole moments, Mol. Phys., 1966, 11, 371 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, A grid-based Bader analysis algorithm without lattice bias, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, An improved grid-based algorithm for Bader charge allocation, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- M. Yu and D. R. Trinkle, Accurate and efficient algorithm for Bader charge integration, J. Chem. Phys., 2011, 134, 064111 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |