
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of the physicochemical features of TiO2 nanoparticles on the formation of a protein corona and impact on cytotoxicity†
Ozge Kose,
Marion Stalet,
Lara Leclerc and
Valérie Forest *
*
Mines Saint-Etienne, Univ Lyon, Univ Jean Monnet, INSERM, U1059 Sainbiose, Centre CIS, F-42023 Saint-Etienne Cedex 2, France. E-mail: vforest@emse.fr
First published on 10th December 2020
Abstract
Due to their unique properties TiO2 nanoparticles are widely used. The adverse effects they may elicit are usually studied in relation to their physicochemical features. However, a factor is often neglected: the influence of the protein corona formed around nanoparticles upon contact with biological media. Indeed, although it is acknowledged that it can strongly influence nanoparticle toxicity, it is not systematically considered. The aim of this study was to characterize the formation of the protein corona of TiO2 nanoparticles as a function of the main nanoparticle properties and investigate potential relationship with the cytotoxicity nanoparticles induce in vitro in human lung cells. To that purpose, five TiO2 nanoparticles differing in size, shape, agglomeration state and surface charge were incubated in cell culture media (DMEM or RPMI supplemented with 10% fetal bovine serum) and the amount and profile of adsorbed proteins on each type of nanoparticle were compared to their toxicological profile. While nanoparticle size and surface charge were found to be determinant factors for protein corona formation, no clear impact of the shape and agglomeration state was observed. Furthermore, no clear relationship was evidenced between the protein corona of the nanoparticles and the adverse effect they elicited.
1. Introduction
Titanium dioxide (TiO2) nanoparticles are among the most produced and industrially used nanoparticles.1 Because their small size confers unique properties, they are used in a wide range of application fields. They are for instance characterized by their brightness/whiteness, high refractive index, opacifying strength, resistance to discoloration, self-cleaning and antifogging property, ultraviolet block capacity, catalytic properties and antimicrobial activity.2–5 Many applications can take advantage of these properties, both in the industrial sector and in daily-life consumer products. Thus, TiO2 nanoparticles can be used as pigment, catalyst, in paints, papers, inks, plastics, rubber industry, solar cells, construction material, self-cleaning roof tiles and windows, anti-fogging car mirrors, textile, as food additive (E171), in personal care products (toothpaste, cosmetics, sunscreens), in environmental or biomedical applications (photocatalytic degradation of pollutants, air and water purification, biosensing, drug delivery) and even in pharmaceuticals.1,2,4–6Initially considered as biologically inert, titanium dioxide was classified as “possibly carcinogen to humans” (Group 2B) by the International Agency for Research on Cancer (IARC).2 However, it remains a controversial issue as there is not only one type of TiO2 nanoparticles but several depending on their physicochemical features and conclusions can hardly be generalized. Indeed, the biological impact of nanoparticles is directly influenced by their physicochemical characteristics.7,8 We previously assessed the cytotoxicity induced by TiO2 nanoparticles on human lung cell lines in relation to their physicochemical features.9 We observed a higher toxicity with bigger sized, less agglomerated, rod-shaped, and positively charged TiO2 nanoparticles, confirming that nanoparticle toxicity is directly correlated with their physicochemical features.
However, to fully understand this relationship, another factor has to be considered: the formation of a protein corona. Indeed, upon contact with biological media, the biomolecules they contain, and especially proteins, rapidly adsorb on the nanoparticle surface forming a crown, the so-called corona. This formation is directly influenced both by the biological environment (medium composition and factors such as temperature, pH, duration of incubation, etc.) and by the nanoparticle physicochemical features (especially, size, shape, surface charge and hydrophobicity).10–21 The protein corona represents the new interface between nanoparticles and biological systems and will thus have a strong impact on their interactions and the subsequent cell response. The pristine nanoparticle being “screened” by the proteins adsorbed on its surface, interactions with biological systems will be made through this protein layer and not with bare materials.22 It will consequently determine the physiological behavior of nanoparticles, triggering vast biological outcomes.13,23,24
It is commonly acknowledged that the protein corona generally mitigates the cytotoxicity induced by nanoparticles23–28 as a consequence of a lower cell uptake.29–37 However, some studies have shown on the contrary that the presence of a protein corona can enhance the nanoparticle cytotoxicity.38
In any way, a better characterization of the nanoparticle corona is of paramount importance to better understand the biological effects of nanoparticles. The aim of this study was to characterize the formation of the protein corona of TiO2 nanoparticles as a function of the main nanoparticle properties and investigate potential relationship with the cytotoxicity nanoparticles induce in vitro in human lung cell lines. To that purpose, 5 TiO2 nanoparticles differing in size, shape, agglomeration state and surface charge were incubated in cell culture media and the amount and profile of adsorbed proteins on each type of nanoparticle were compared to their toxicological profile.
2. Materials and methods
Our approach is summarized in Fig. 1.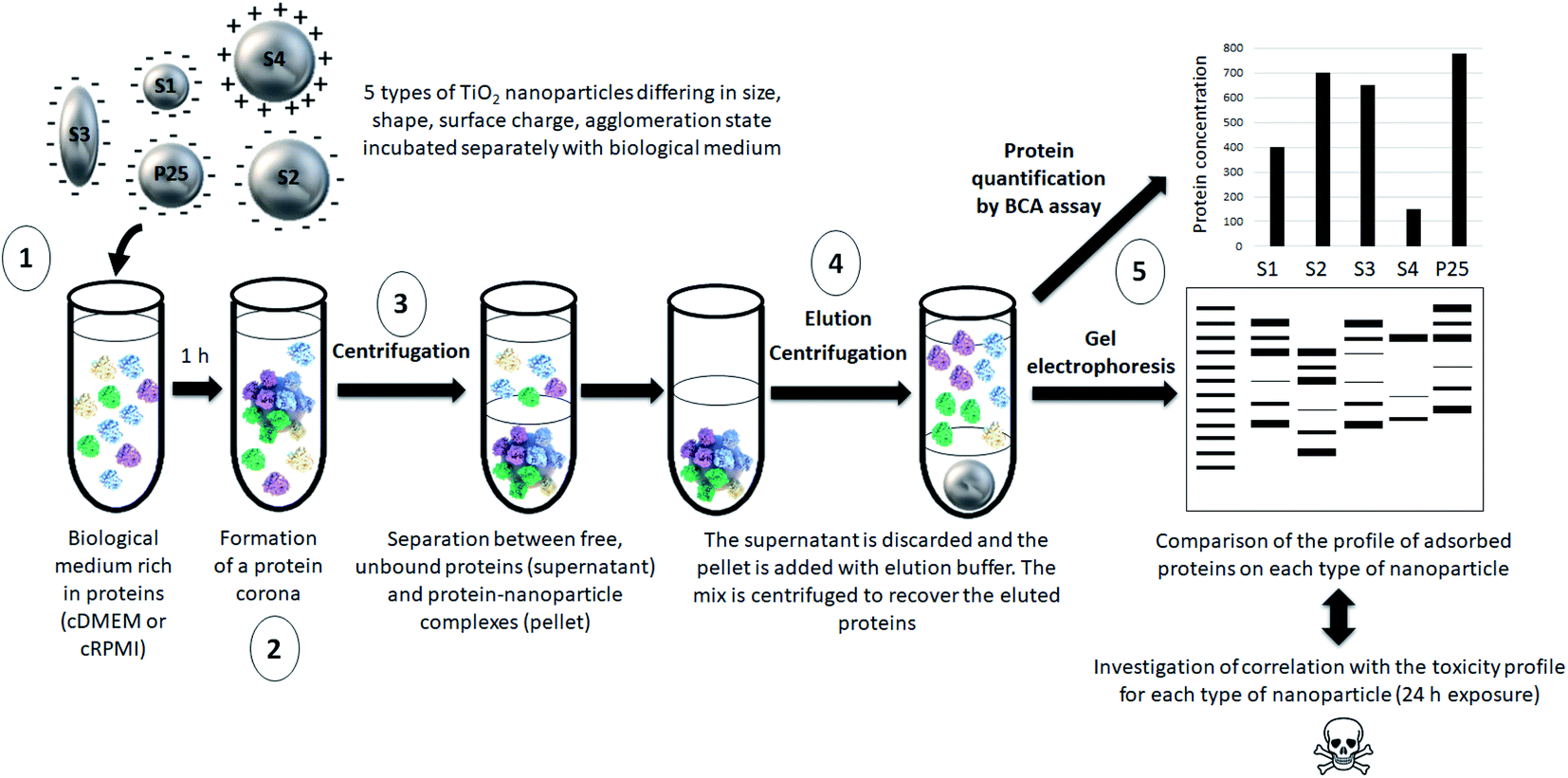 | ||
| Fig. 1 Schematic representation of the main steps of our approach. (1) 5 types of nanoparticles characterized by different physicochemical features were incubated in a biological fluid (cell culture medium) for 1 h. (2) Proteins from this biological environment adsorbed at the nanoparticle surface forming the so-called protein corona. (3) The nanoparticle–protein complexes were separated from unbound proteins by centrifugation. (4) Proteins were then eluted from nanoparticles and (5) quantified by a protein assay and analyzed by gel electrophoresis. The profile of adsorbed proteins was discussed with regard to the physicochemical features of the nanoparticles and potential correlations with their previously assessed toxicity profile were investigated. | ||
The protocol used for the semi-quantitative analysis of the protein corona composition of TiO2 nanoparticles was inspired from Docter et al.39 but was adapted to our nanoparticles and conditions.
2.1. Nanoparticles
Five types of TiO2 nanoparticles were used in this study. In addition to commercial P25 nanoparticles (Evonik P25 CAS: 1317-70-0, Sigma-Aldrich, France) used as a reference, four types of TiO2 nanoparticles were synthesized with different and well-controlled physicochemical properties. They were referred to as S1 to S4 and differed in size, shape, agglomeration state and surface functionalization/charge. They were synthesized using Chen et al. method.40 Basically, titanium(IV) butoxide (CAS: 5593-70-4 reagent grade 97%, Sigma-Aldrich, Saint-Quentin-Fallavier, France) was mixed with triethanolamine (CAS: 102-71-6, Analytical reagent 97%, VWR International, Fontenay-sous-Bois, France) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio. The mixture was put in Teflon lined sealed autoclave and then heated at 150 °C during 24 h. The pH values of the synthesis medium were adjusted using HCl or NH4OH to tune particle size and morphology. Finally, the solutions were washed by three centrifugations using de-ionized water and the resulting products were dried in an oven at 40 °C. Surface functionalization of S2 nanoparticles was generated by aminopropyltriethoxysilane (APTES) using Zhao et al. method.41 Briefly, 0.25 g of S2 nanopowder was dispersed in 25 mL de-ionized water by ultrasonication for 10 min. Then, the silane coupling agents APTES were added in the dispersion (molar ratio of 1:1). The mixture was sonicated until a clear solution was obtained and then refluxed at 80 °C for 4 h. After that, dispersed particles were separated from solvent by centrifugation (10 min at 1200g) followed by washing with water at least 2 times. The final functionalized samples were then prepared in de-ionized water and labeled as S4.
:2 molar ratio. The mixture was put in Teflon lined sealed autoclave and then heated at 150 °C during 24 h. The pH values of the synthesis medium were adjusted using HCl or NH4OH to tune particle size and morphology. Finally, the solutions were washed by three centrifugations using de-ionized water and the resulting products were dried in an oven at 40 °C. Surface functionalization of S2 nanoparticles was generated by aminopropyltriethoxysilane (APTES) using Zhao et al. method.41 Briefly, 0.25 g of S2 nanopowder was dispersed in 25 mL de-ionized water by ultrasonication for 10 min. Then, the silane coupling agents APTES were added in the dispersion (molar ratio of 1:1). The mixture was sonicated until a clear solution was obtained and then refluxed at 80 °C for 4 h. After that, dispersed particles were separated from solvent by centrifugation (10 min at 1200g) followed by washing with water at least 2 times. The final functionalized samples were then prepared in de-ionized water and labeled as S4.
Stock suspensions of all nanoparticles (1600 μg mL−1) were prepared in de-ionized water (MilliQ systems, Millipore, Bedford, MA, USA) and sonicated with Branson Sonifier S-450 for 10 min at 89% amplitude. Nanoparticles were then extensively characterized.9 The morphology and size distribution of the nanoparticles were analyzed by transmission electron microscopy (TEM) using a FEI TECNAI 20FST operating at 200 kV and scanning electron microscopy (SEM) at 2–3 kV on a Zeiss Sigma 300 microscope using a secondary electron detector. Fig. 2 illustrates the morphology of the nanoparticles. After each TEM image of each sample were chosen, the size distribution and the mean diameter were measured by ImageJ software.
 | ||
| Fig. 2 Transmission electron microscope images of S1 (A), S2 (B), and S3 (C) TiO2 nanoparticles. | ||
The hydrodynamic size and the agglomeration status of the TiO2 nanoparticles (120 μg mL−1) in de-ionized water and in culture media were determined by using dynamic light scattering (DLS, Zetasizer Nano ZS Malvern Instruments, Worcestershire, UK) measurements. Surface charge of the nanoparticles was monitored using electrophoretic light scattering (ELS, Zetasizer Nano ZS Malvern Instruments, Worcestershire, UK). Specific surface areas (SSA) were measured by linearizing the physisorption isotherm of N2 at 77 K with the classical method of Brunauer, Emmett and Teller (BET) (Volumetric Adsorption ASAP 2020, Micrometrics, USA).42
Tables 1 and 2 summarize the main physicochemical features of the nanoparticles. DLS graphs can be found in ESI.†
| Primary size (nm) | SSA (m2 g−1) | Particle shape | Crystal structure | Surface coating | |
|---|---|---|---|---|---|
| a Minimum and maximum Feret diameters.b APTES coating was carried out on S2 nanoparticles to obtain S4 nanoparticles. | |||||
| S1 | 15 | 146 | Spherical | Anatase | No |
| S2 | 30 | 61 | Spherical | Anatase | No |
| S3 | 20–250a | 41 | Rod | Anatase | No |
| S4 | 30 | 61 | Spherical | Anatase | APTESb |
| P25 | 21 | 55 | Spherical | Anatase: Rutile (90:10) |
No |
| DI H2O | cDMEM | cRPMI | ||||
|---|---|---|---|---|---|---|
| Average hydrodynamic sizea (nm) (PDI) | Zeta potential (mV) pH 7.5 | Average hydrodynamic sizea (nm) (PDI) | Zeta potential (mV) pH 7.5 | Average hydrodynamic sizea (nm) (PDI) | Zeta potential (mV) pH 7.5 | |
| a Dynamic light scattering (DLS) measurements are the mean of at least 3 runs each containing 20 sub-measurements. | ||||||
| S1 | 211.4 ± 2.3 (0.15) | −15.8 ± 4.0 | 226 ± 9.1 (0.28) | −33.8 ± 1.8 | 241.4 ± 3.0 (0.22) | −12.6 ± 1.6 |
| S2 | 969.3 ± 39.5 (0.27) | −13.8 ± 4.4 | 1094 ± 46.4 (0.36) | −31.0 ± 0.3 | 1138 ± 35.9 (0.328) | −11.8 ± 0.6 |
| S3 | 1409 ± 89.6 (0.11) | −13.2 ± 4.2 | 1275 ± 66.6 (0.18) | −32.0 ± 3.0 | 1267 ± 111.5 (0.42) | −10.9 ± 0.1 |
| S4 | 1204 ± 59.9 (0.16) | +12.3 ± 0.5 | 1398 ± 54.9 (0.48) | −36.6 ± 4.5 | 1515 ± 16.01 (0.312) | −11.4 ± 0.6 |
| P25 | 256.4 ± 136.6 (0.27) | −15.2 ± 5.3 | 325 ± 4.1 (0.26) | −28.1 ± 0.6 | 434.6 ± 21.1 (0.33) | −11.9 ± 0.5 |
2.2. Biological media
Two types of cell culture media were used: Dulbecco's Modified Eagle Medium (DMEM) high glucose (4.5 g L−1) with stable glutamine and sodium pyruvate (cat. no. L0103-500, VWR International, France) and Roswell Park Memorial Institute (RPMI 1640) with L-glutamine (cat. no. L0500-500, Gibco, Life Technologies, France). Both media were supplemented with 10% (v/v) fetal bovine serum (FBS, cat. no. S1810-500, Biowest, France, for the reproducibility of the results, we ensured that all FBS aliquots came from the same batch) and 1% penicillin-streptomycin (VWR International, France). Once supplemented, media were called complete and referred to as cDMEM and cRPMI respectively.2.3. Nanoparticles/biological media contact
To ensure comparability between the results the ratio of total particle–surface area to the biological medium volume has to be kept constant for all nanoparticles. After preliminary experiments, we established that the optimal ratio was 0.1 m2 mL−1 in a final volume of 4 mL. Based on the nanoparticle surface specific area (SSA), we calculated for each nanoparticle type the volume of stock solution to dilute in culture medium as reported in Table 3. TiO2 nanoparticles were incubated either in cRPMI or cDMEM for 1 h at 37 °C.| NP type | SSA (m2 g−1) | NP stock solution concentration (μg mL−1) | NP stock volume (μL) | Cell culture medium volume (μL) | Total volume (μL) |
|---|---|---|---|---|---|
| S1 | 146.5 | 16000 |
171 | 3829 | 4000 |
| S2 | 61.02 | 16000 |
410 | 3590 | 4000 |
| S3 | 40.78 | 16000 |
613 | 3387 | 4000 |
| S4 | 61.02 | 16000 |
410 | 3590 | 4000 |
| P25 | 55.00 | 16000 |
455 | 3545 | 4000 |
2.4. Removal of unbound proteins
Samples were centrifuged 1 h at 14100g (Mini-centrifuge MiniSpin Plus, VWR International, Fontenay-sous-Bois, France) at room temperature to separate the nanoparticle–protein complexes from the cell culture medium. The supernatant was discarded and the pellets were washed with 1 mL PBS to remove the proteins that did not bound tightly to the nanoparticles, the tubes were sonicated (Branson Sonifier S-450, amplitude 89%, pulse 2 s and intervals 2 s). Two additional washes consisting of a 20 min centrifugation at 14100g, room temperature were carried out. The supernatant was finally discarded and the pellet containing the nanoparticle–protein complexes was used for the next step.
2.5. Recovery of the proteins adsorbed at the surface of the nanoparticles
Proteins were eluted from the nanoparticle–protein complexes by adding 300 μL of Laemmli buffer containing 62.5 mM Tris, 5% (vol/vol) glycerol, 2% (wt/vol) SDS. After a 5 min incubation at 95 °C, the samples were centrifuged for 15 min at 14100g at room temperature. The supernatant containing the eluted corona proteins was transferred into an Eppendorf Protein Lobind tube and used for protein quantification and gel electrophoresis as described below.
2.6. Determination of protein concentration
The protein concentration was assessed in the supernatant using a BCA protein assay kit (Pierce, Thermo Fisher Scientific, France) according to the manufacturer's instructions. Briefly, in a microplate 25 μL of solution were transferred per well and added with 200 μL of reagents from the kit. The plate was shaken for 30 s, covered and incubated at 37 °C for 30 min. After cooling to room temperature, the absorbance at 562 nm was read with a microplate spectrophotometer (Multiskan, Thermo Fisher Scientific, France). Calculation of protein concentrations was based on the use of a standard curve established with BSA (protein standard included in the kit). Results are means of three independent experiments each performed in duplicate.2.7. Determination of the profile of proteins adsorbed at the surface of the nanoparticles by gel electrophoresis
30 μL of sample was added with 10 μL of 4× Bolt LDS sample buffer Novex (Invitrogen, Thermo Fisher Scientific, France). The mix was heated at 70 °C for 10 min. Samples were loaded in precast polyacrylamide gels (Invitrogen Bolt Bis-Tris Plus 4–12%, 10 wells, Thermo Fisher Scientific, France). A well was filled with 10 μL protein markers (SeeBlue Plus2 pre-stained protein standard Novex, Thermo Fisher Scientific, France). 1× running buffer was prepared by mixing 50 mL of 20× Bolt MES SDS Novex (Invitrogen, Thermo Fisher Scientific, France) with 950 mL of deionized water. The gel was run at room temperature, at 200 V constant for 22 min. It was then directly transferred into the Instant Blue staining solution (cat. no. ISB1L, Expedeon) for 1 h at room temperature with gentle shaking. A picture of the gel was taken. Three independent experiments were performed.2.8. Nanoparticle cytotoxicity assessment
The cytotoxicity of the TiO2 nanoparticles was previously assessed9 on a human lung coculture system consisting of A549 epithelial cells and THP-1 differentiated macrophages cultivated in cDMEM. After a 24 h cell exposure to 15, 30, 60 or 120 μg mL−1 of nanoparticles, cytotoxicity was assessed in terms of cell viability (Trypan blue exclusion assay), pro-inflammatory response (production of the IL-8 and TNF-α cytokines assessed by commercial ELISA kits) and oxidative stress (production of reactive oxygen species, ROS detected by the cell-permeable fluorogenic probe 2′,7′-dichlorodihydrofluorescin diacetate, DCFH-DA).2.9. Statistical analysis
For protein quantification, unless otherwise stated, results are expressed as mean of 3 independent experiments, each performed in duplicate ± standard deviation (SD). Statistical analyses were performed using GraphPad Prism® (version 8.0, GraphPad Software, San Diego, CA, USA). Statistical analyses were conducted using a one-way Anova analysis followed by a Tukey's multiple comparison test. Differences were considered statistically significant when P < 0.05. The amount of proteins found in the corona of S1 and S3 was compared to that of the P25 reference. The amount of proteins found in the corona of S2 was compared to that of S4, its APTES-functionalized counterpart. Finally, the protein amount was compared between protein coronas formed in cDMEM and those formed in cRPMI for each sample.3. Results
3.1. Protein concentration
Fig. 3 reports the mean concentrations of proteins eluted from the corona of the different nanoparticles incubated either in cDMEM or cRPMI. | ||
| Fig. 3 Concentrations of proteins eluted from the protein corona of S1, S2, S3, S4 and P25 nanoparticles that were incubated either in cDMEM or cRPMI. Controls are cDMEM or cRPMI alone. Results are means of 3 independent experiments (except for S3 where we did not have enough nanoparticle sample to perform the last repetition so results are means of 2 independent experiments for this sample), standard deviation is also indicated. Statistically different from P25 (*) P < 0.05, (****) P < 0.0001. Statistical difference between S2 and S4 (§§§§) P < 0.0001. Statistical difference between cDMEM and cRPMI (#) P < 0.05. | ||
For both media, proteins were less abundantly found in the corona from S4 nanoparticles. S2, S3 and P25 nanoparticles exhibited similar amounts of proteins in their corona. A slightly less important protein concentration was observed in the S1 corona.
For each nanoparticle type, a similar concentration of proteins was found in the coronas formed either in cDMEM or cRPMI (no statistically significant differences).
3.2. Profile of proteins adsorbed at the surface of the nanoparticles by gel electrophoresis
Fig. 4 illustrates the protein profile of the corona of the different nanoparticle types. The picture is representative of the three experiments performed. | ||
| Fig. 4 SDS-PAGE of S1, S2, S3, S4 and P25 nanoparticles incubated either in cDMEM (A) or in cRPMI (B). As controls, the complete biological media alone were also run (last lane). | ||
Both in cDMEM and cRPMI we clearly observed that the corona of S2, S3 and P25 had very similar protein profiles, S1 presented a slightly lower amount of proteins whereas S4 corona exhibited the smallest protein content. These results are in perfect agreement with those of protein quantification (Fig. 3). In qualitative terms, interestingly and as highlighted by the rectangles on Fig. 4, we could observe that some proteins largely bound to the nanoparticles were not necessarily the most abundant in the culture medium (control lane).
3.3. Nanoparticles toxicity profiles
Table 4 reports the cytotoxicity patterns of the different nanoparticle types as previously assessed.9| Cytotoxicity | Pro-inflammatory response | Oxidative stress | |
|---|---|---|---|
| S1 | + | + | − |
| S2 | ++ | + | − |
| S3 | ++++ | ++ | − |
| S4 | ++ | ++ | − |
| P25 | +++ | ++ | − |
We clearly observed different patterns of toxicity depending on the nanoparticle considered in relation with its physicochemical characteristics.9
4. Discussion
Fig. 5 summarizes the findings of this paper as well as previous data for discussion. | ||
| Fig. 5 Schematic summary of the protein corona and cytotoxicity profiles for each type of TiO2 nanoparticle in relation with its physicochemical features. | ||
4.1. Impact of the nanoparticle physicochemical features on the formation of the protein corona
It has been extensively reported in the literature that the nanoparticle physicochemical features play a key role in the formation of the protein corona, especially, size, shape, surface charge and hydrophobicity.10–21,43 In this study, by comparing the protein corona formation around 5 types of TiO2 nanoparticles differing in size, shape, surface charge and agglomeration state, we aimed to shed light on the respective influence of such parameters.By comparing S1 and P25 we could highlight the influence of nanoparticle size/SSA on the formation of the protein corona. We observed that smaller S1 nanoparticles (15 nm) showed a slightly less important amount of proteins in their corona compared to bigger P25 nanoparticles. This observation can be easily explained by the fact that when nanoparticle size varies, the curvature of the nanoparticle–protein interface is altered consequently affecting the protein adsorption at the nanoparticle surface. Similarly, a change in nanoparticle size/SSA induces an alteration of the deflection angle between adjacent proteins, thus when the size of the nanoparticle decreases, more steric repulsion occurs between the proteins of the corona.12 In other words, smaller nanoparticles increase the deflection angle of the proteins and have a higher curvature than bigger nanoparticles, which can strongly impact qualitatively and quantitatively the composition of the protein corona.20 It has even been shown that highly curved surfaces (very small nanoparticles) can suppress protein adsorption to the point where it no longer occurs.44 Moreover, Tenzer et al.18 demonstrated that even a 10 nm particle size difference significantly determined the protein corona of silica nanoparticles.
The influence of the nanoparticle shape on the formation of the protein corona was investigated by comparing rod-shaped S3 and spherical P25 nanoparticles. The amount and profile of proteins in the corona were similar, suggesting no impact of the shape. This finding is not in agreement with previous studies where nanorods were reported to adsorb more proteins than nanospheres of similar size, thanks to their small curvature.19,45 This discrepancy can be due to the fact that the nanoparticles used in these studies were of different chemical nature (gold and silica) and of different sizes (10 and 270 nm for nanospheres and 10–35 and 270–1100 nm for nanorods). The nanoparticle–surface area ratios used were also different (0.003 and 0.05 m2 mL−1). This argues for a complex picture where several parameters are concomitantly involved.
S2 and P25 nanoparticles exhibited similar pattern suggesting that nanoparticle agglomeration state does not play a major role in the formation of the protein corona.
On the contrary, by comparing the protein coronas of S2 and S4 (amine-functionalized form of S2) nanoparticles, we clearly observed a significant impact of the nanoparticle surface charge on the formation of a protein corona, confirming previous studies. However, it was quite surprising to observe that protein coronas were preferentially formed on negatively charged nanoparticles. Indeed, it is generally admitted that charged particles tend to adsorb more proteins than neutral nanoparticles.12,16,17,20 The logical explanation is that, negatively charged particles attract positively charged proteins and vice versa. But, proteins contained in FBS are largely negative for a pH around 8, it should thus be reasonable to expect a larger protein binding to nanoparticles which surface is positively charged. However, this simplified statement does not fit all circumstances.12,46,47 And our finding is consistent with those of Bewersdorff et al.48 who reported that serum proteins preferably bound to negatively charged nanoparticles compared to positively charged ones. Tenzer et al.18 also demonstrated that, at physiological pH (7.3), proteins in general with negative charge were preferentially bound by negatively charged SiO2 nanoparticles irrespective of their relative plasma abundance. As nanoparticle surface chemistry has been shown to deeply influence the evolution and composition of a protein corona, it could be assumed that the nature of the chemical groups used for functionalization can contribute to controversial results. In our case, we can hypothesize a smaller protein corona for positively charged S4 nanoparticle as a result of a potential steric repulsion between the serum proteins and the amine groups of the APTES.
4.2. Impact of biological media on the formation of nanoparticle corona
As it is widely acknowledged that the protein corona formation also depends upon the biological environment13,49–51 we compared the protein profiles obtained when TiO2 nanoparticles were incubated in two types of standard cell culture media, DMEM and RPMI, supplemented by 10% FBS. From the protein quantification as well as the protein profiles observed on electrophoresis gels, we could conclude that the nature of the medium did not really impact the protein concentration of the corona of any nanoparticles. This finding is not in agreement with Maiorano et al.36 who exposed various sized citrate-capped gold nanoparticles to cDMEM and cRPMI and reported that the protein corona formed in cDMEM was more abundant and stable compared to that formed in cRPMI. This discrepancy can be partly explained by the fact that systems are highly sensitive to even minor changes of some parameters. Here the chemical nature of the nanoparticle was different (TiO2 and gold) and this calls for caution in generalizing conclusions.We should also be careful in our conclusions as the zeta potential of nanoparticles varied greatly depending on the biological medium nanoparticles were incubated in (Table 2). Values were all negative when nanoparticles were in cDMEM and cRPMI, it even turned from positive (in water) to negative for S4. More important changes in zeta potential values were observed when nanoparticles were incubated in cDMEM compared to cRPMI. This could be due to a different protein composition of the two cell culture media resulting in the binding of proteins of various nature, differently charged on nanoparticles, making the zeta potential vary accordingly. Thus this observation is not inconsistent with a similar amount of proteins bound to nanoparticles irrespective of the biological medium (Fig. 3). Further investigations are needed, especially a qualitative characterization of the protein corona composition.
4.3. Correlation with the toxicity profile of nanoparticles
Among the numerous parameters that influence nanoparticle–cell interactions, the presence of a protein corona plays a key role. In this regard, Lesniak et al. showed that for identical particles and cells, under identical conditions, the nanoparticle–cell interactions and the biological outcomes could vary greatly in the presence or in the absence of a preformed corona in serum.34 Because the protein corona defines the biological identity of nanoparticles and affects their biological response, it is now accepted that the types of proteins and their abundance on the nanoparticle surface encode information that predicts nanoparticle bioactivity.24 And it can predict it more accurately than using models based only on the physicochemical properties of the nanoparticles.20 Usually, the protein corona has a protective role against cell uptake and nanoparticles exhibiting a protein corona are less taken up than their bare counterparts.30–33,35,37 The protein corona could decrease nanoparticle adhesion to the cell membrane due to the decreased surface-free energy after binding with proteins, thus decreasing particle–cell interactions and resulting in reduced uptake.31,33 But some studies have shown the contrary, i.e. the presence of the protein corona enhancing nanoparticle uptake by cells.52,53 These discrepancies can be explained by different cell internalization pathways: in the presence of a corona, non-specific uptake seems to be decreased whereas specific uptake seems to be promoted.29,33,52,54 Because of a lower cell uptake the presence of a protein corona generally reduces the cytotoxic effects of nanoparticles.14,25,27,29,32,34,36 Furthermore, the presence of a protein corona by masking the nanoparticle surface can mitigate its reactivity in interfacing cellular membranes and consequently prevent potential adverse effects.34,55 At this point it is interesting to note that the five types of nanoparticles possessed comparable values in zeta potential in the culture media (Table 2), thus, the role of surface charge is expected to be screened by the protein corona. This observation seems to be in contradiction with the general knowledge that cationic nanoparticles tend to be more toxic than anionic nanoparticles. To reconcile these apparently diverging statements we should be very careful in the terms we use. Indeed, we should systematically specify if we are talking of the nanoparticle charge in cell culture medium (i.e. in the medium where the cytotoxicity assays are carried out and thus in the presence of the protein corona) or if we are talking of the pristine nanoparticle charge (as characterized after synthesis in DI water and thus in the absence of a protein corona). This crucial distinction is not always made and can lead to misinterpretations. Finally, the presence of a protein corona can increase the safety of nanoparticles by inhibiting the generation of radical oxygen species by which several compounds exert their cytotoxic activity.56In this context, the characterization of a nanoparticle protein corona is of paramount importance to better understand the mechanisms underlying its toxicity. As shown by Fig. 5, we were not able to evidence a clear relationship between protein corona pattern and the toxicity elicited by the nanoparticles. For instance, S4 exhibited the smallest protein corona and a significant cytotoxic activity. While S2, S3 and P25 had similar protein corona profiles, the intensity of the adverse effects they induced ranged from low to very high.
However, we should be cautious with this result as the nanoparticle doses used for the protein corona characterization and the cytotoxicity assays were different. Indeed, to allow comparison between the protein coronas of different nanoparticles, we used a constant total particle–surface area to biological medium volume ratio. We first tried concentrations equivalent to those used for cytotoxicity assays but we did not get enough proteins for analysis, therefore we increased the doses to get exploitable results. Such concentrations allowed us to conclude on the impact of the nanoparticle physicochemical features on the formation of the protein corona as discussed before. Although lower nanoparticle doses were used for cytotoxicity assays, we were able to rank the nanoparticles with respect to their toxicity and in Fig. 5 we reasoned in terms of relative and not absolute values making the comparison still meaningful. Similarly, another parameter to consider is the incubation time of the nanoparticles in the cell culture media (1 h in the present study of the protein corona and 24 h in the cytotoxicity assessment). This difference may result in a slightly different protein corona composition as this latter is known to evolve over time. Indeed, the proteins the most abundant in the medium and with high mobility first adsorb on the nanoparticle surface but they are progressively replaced by proteins exhibiting a higher affinity.12,57 However, it seems to be a really fast process as Tenzer et al. demonstrated that an interaction between nanoparticle surface and plasma proteins is established as early as after 30 s of contact and that protein corona nature did not change over time.17 It is thus reasonable to assume that protein corona composition remains quite stable after 1 h of nanoparticle/culture medium contact and we can confidently assume that it is representative of protein corona that would be observed after 24 h.
Furthermore, several assumptions can be made to explain the absence of clear correlation between the nanoparticle protein corona and toxicity. It is mainly related to our incomplete characterization of the protein corona. Indeed, because of technical challenges, we could only study the hard corona, composed of tightly bound proteins. The soft corona, consisting of less tightly bound proteins cannot be preserved during the analysis process. However, the soft corona represents the most external layer of proteins, which is thus likely to be in contact with biological systems. The unexplored role of soft protein corona may be a determinant factor for a better understanding of nanoparticle–cell interactions. One limitation of the present study was to consider the protein corona mainly through a quantitative aspect. In addition, we have to keep in mind that it is a preliminary study aiming to draw general patterns, to gain a first impression about the amount and composition of the corona proteins and that further investigations, including more sensitive techniques such as mass spectrometry analyses are required to refine our conclusions. Indeed, many studies evaluating the protein corona of nanoparticles by 1D or 2D gel electrophoresis are followed by mass spectrometry.58,59 In particular, liquid chromatography-mass spectrometry (LC-MS) could be employed to determine protein corona composition qualitatively and quantitatively.
Indeed, to draw firm conclusion further investigations on the nature of the proteins should be conducted. It cannot be excluded that some proteins present at a minor level could be responsible for major biological consequences and vice versa.29 Finally, it should be kept in mind that protein corona formation is very sensitive to many parameters both from the nanoparticle and the biological environment and minor changes in experimental conditions can have a deep impact on the protein corona formation/composition.21,30 It is an interplay of different forces in a competing environment.12
5. Conclusions
Protein corona is a major issue for the study of the nanoparticle fate and consequences especially in vivo but in vitro studies are very useful to better understand what happens at a cellular level. In our model and experimental conditions the parameters influencing the protein corona formation are mainly nanoparticle size and surface charge, no clear impact of the shape and agglomeration state was observed. Although protein corona nature may undoubtedly influence nanoparticle cytotoxicity, no clear relationship was evidenced between the protein corona of 5 types of TiO2 nanoparticles and the adverse effects they elicited in human lung cells. More comprehensive studies (considering qualitative/quantitative aspects, hard/soft corona) are needed to better understand the relationship between nanoparticle protein corona and toxicity.Conflicts of interest
There are no conflicts to declare.Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.References
- E. Baranowska-Wójcik, D. Szwajgier, P. Oleszczuk and A. Winiarska-Mieczan, Biol. Trace Elem. Res., 2020, 193, 118–129 CrossRef.
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, International Agency for Research on Cancer and World Health Organization, Carbon black, titanium dioxide, and talc, International Agency for Research on Cancer, Distributed by WHO Press, Lyon, France, Geneva, 2010 Search PubMed.
- M. Skocaj, M. Filipic, J. Petkovic and S. Novak, Radiol. Oncol., 2011, 45, 227–247 CAS.
- M. S. Waghmode, A. B. Gunjal, J. A. Mulla, N. N. Patil and N. N. Nawani, SN Appl. Sci., 2019, 1, 310 CrossRef.
- A. Weir, P. Westerhoff, L. Fabricius and N. von Goetz, Environ. Sci. Technol., 2012, 46, 2242–2250 CrossRef CAS.
- X. Chen and A. Selloni, Chem. Rev., 2014, 114, 9281–9282 CrossRef CAS.
- A. Albanese, P. S. Tang and W. C. W. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS.
- A. Kurtz-Chalot, J. P. Klein, J. Pourchez, D. Boudard, V. Bin, G. B. Alcantara, M. Martini, M. Cottier and V. Forest, J. Nanopart. Res., 2014, 16, 1–15 CrossRef CAS.
- O. Kose, M. Tomatis, L. Leclerc, N.-B. Belblidia, J.-F. Hochepied, F. Turci, J. Pourchez and V. Forest, Chem. Res. Toxicol., 2020, 33, 2324–2337 Search PubMed.
- E. Casals and V. F. Puntes, Nanomed, 2012, 7, 1917–1930 CrossRef CAS.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS.
- D. Chen, S. Ganesh, W. Wang and M. Amiji, Nanomed, 2017, 12, 2113–2135 CrossRef CAS.
- V. Forest, in Nanoparticle–Protein Corona, 2019, pp. 31–60 Search PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. B. Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS.
- S. Tenzer, D. Docter, S. Rosfa, A. Wlodarski, J. Kuharev, A. Rekik, S. K. Knauer, C. Bantz, T. Nawroth, C. Bier, J. Sirirattanapan, W. Mann, L. Treuel, R. Zellner, M. Maskos, H. Schild and R. H. Stauber, ACS Nano, 2011, 5, 7155–7167 CrossRef CAS.
- R. M. Visalakshan, L. E. G. García, M. R. Benzigar, A. Ghazaryan, J. Simon, A. Mierczynska-Vasilev, T. D. Michl, A. Vinu, V. Mailänder, S. Morsbach, K. Landfester and K. Vasilev, Small, 2020, 2000285 CrossRef.
- C. D. Walkey, J. B. Olsen, F. Song, R. Liu, H. Guo, D. W. H. Olsen, Y. Cohen, A. Emili and W. C. W. Chan, ACS Nano, 2014, 8, 2439–2455 CrossRef CAS.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- G. Caracciolo, O. C. Farokhzad and M. Mahmoudi, Trends Biotechnol., 2017, 35, 257–264 CrossRef CAS.
- G. Caracciolo, S. Palchetti, V. Colapicchioni, L. Digiacomo, D. Pozzi, A. L. Capriotti, G. La Barbera and A. Laganà, Langmuir, 2015, 31, 10764–10773 CrossRef CAS.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS.
- C. Ge, J. Du, L. Zhao, L. Wang, Y. Liu, D. Li, Y. Yang, R. Zhou, Y. Zhao, Z. Chai and C. Chen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16968–16973 CrossRef CAS.
- P. Jain, R. S. Pawar, R. S. Pandey, J. Madan, S. Pawar, P. K. Lakshmi and M. S. Sudheesh, Biotechnol. Adv., 2017, 35, 889–904 CrossRef CAS.
- N. P. Mortensen, G. B. Hurst, W. Wang, C. M. Foster, P. D. Nallathamby and S. T. Retterer, Nanoscale, 2013, 5, 6372–6380 RSC.
- M. Pearson, V. V. Juettner and S. Hong, Front. Chem., 2014, 2, 108 Search PubMed.
- E. Brun and C. Sicard-Roselli, Cancer Nanotechnol., 2014, 5, 7 CrossRef.
- P. Chandran, J. E. Riviere and N. A. Monteiro-Riviere, Nanotoxicology, 2017, 11, 507–519 CrossRef CAS.
- X. Cheng, X. Tian, A. Wu, J. Li, J. Tian, Y. Chong, Z. Chai, Y. Zhao, C. Chen and C. Ge, ACS Appl. Mater. Interfaces, 2015, 7, 20568–20575 CrossRef CAS.
- D. Docter, C. Bantz, D. Westmeier, H. J. Galla, Q. Wang, J. C. Kirkpatrick, P. Nielsen, M. Maskos and R. H. Stauber, Beilstein J. Nanotechnol., 2014, 5, 1380–1392 CrossRef.
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. W. Radomski, K. A. Dawson and C. Åberg, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Åberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS.
- A. Lesniak, A. Campbell, M. P. Monopoli, I. Lynch, A. Salvati and K. A. Dawson, Biomaterials, 2010, 31, 9511–9518 CrossRef CAS.
- G. Maiorano, S. Sabella, B. Sorce, V. Brunetti, M. A. Malvindi, R. Cingolani and P. P. Pompa, ACS Nano, 2010, 4, 7481–7491 CrossRef CAS.
- S. R. Saptarshi, A. Duschl and A. L. Lopata, J. Nanobiotechnol., 2013, 11, 26 CrossRef CAS.
- Z. J. Deng, M. Liang, M. Monteiro, I. Toth and R. F. Minchin, Nat. Nanotechnol., 2011, 6, 39–44 CrossRef CAS.
- D. Docter, U. Distler, W. Storck, J. Kuharev, D. Wünsch, A. Hahlbrock, S. K. Knauer, S. Tenzer and R. H. Stauber, Nat. Protoc., 2014, 9, 2030–2044 CrossRef CAS.
- D. Chen, J. Shi, J. Yan, Y. Wang, F. Yan, S. Shang and J. Xue, Chem. Res. Chin. Univ., 2008, 24, 362–366 CrossRef CAS.
- J. Zhao, M. Milanova, M. M. C. G. Warmoeskerken and V. Dutschk, Colloids Surf., A, 2012, 413, 273–279 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- J. Piella, N. G. Bastús and V. Puntes, Bioconjugate Chem., 2017, 28, 88–97 CrossRef CAS.
- I. Lynch and K. A. Dawson, Nano Today, 2008, 3, 40–47 CrossRef CAS.
- J. E. Gagner, M. D. Lopez, J. S. Dordick and R. W. Siegel, Biomaterials, 2011, 32, 7241–7252 CrossRef CAS.
- V. Forest, M. Cottier and J. Pourchez, Nano Today, 2015, 10, 677–680 CrossRef CAS.
- V. Forest and J. Pourchez, Mater. Sci. Eng., C, 2017, 70, 889–896 CrossRef CAS.
- T. Bewersdorff, J. Vonnemann, A. Kanik, R. Haag and A. Haase, Int. J. Nanomed., 2017, 12, 2001–2019 CrossRef CAS.
- C. Gräfe, A. Weidner, M. V. D. Lühe, C. Bergemann, F. H. Schacher, J. H. Clement and S. Dutz, Int. J. Biochem. Cell Biol., 2016, 75, 196–202 CrossRef.
- V. Mirshafiee, R. Kim, M. Mahmoudi and M. L. Kraft, Int. J. Biochem. Cell Biol., 2016, 75, 188–195 CrossRef CAS.
- D. Pozzi, G. Caracciolo, A. L. Capriotti, C. Cavaliere, G. La Barbera, T. J. Anchordoquy and A. Laganà, J. Proteomics, 2015, 119, 209–217 CrossRef CAS.
- Y. Qiu, Y. Liu, L. Wang, L. Xu, R. Bai, Y. Ji, X. Wu, Y. Zhao, Y. Li and C. Chen, Biomaterials, 2010, 31, 7606–7619 CrossRef CAS.
- M. M. Yallapu, N. Chauhan, S. F. Othman, V. Khalilzad-Sharghi, M. C. Ebeling, S. Khan, M. Jaggi and S. C. Chauhan, Biomaterials, 2015, 46, 1–12 CrossRef CAS.
- F. Zhao, Y. Zhao, Y. Liu, X. Chang, C. Chen and Y. Zhao, Small, 2011, 7, 1322–1337 CrossRef CAS.
- C. Corbo, R. Molinaro, A. Parodi, N. E. Toledano Furman, F. Salvatore and E. Tasciotti, Nanomed, 2016, 11, 81–100 CrossRef CAS.
- H. Yin, R. Chen, P. S. Casey, P. C. Ke, T. P. Davis and C. Chen, RSC Adv., 2015, 5, 73963–73973 RSC.
- L. Vroman and A. L. Adams, Surf. Sci., 1969, 16, 438–446 CrossRef CAS.
- M. Wang, O. J. R. Gustafsson, E. H. Pilkington, A. Kakinen, I. Javed, A. Faridi, T. P. Davis and P. C. Ke, J. Mater. Chem. B, 2018, 6, 6026–6041 RSC.
- F. S. M. Tekie, M. Hajiramezanali, P. Geramifar, M. Raoufi, R. Dinarvand, M. Soleimani and F. Atyabi, Sci. Rep., 2020, 10, 9664 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08429h |
| This journal is © The Royal Society of Chemistry 2020 |