
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of new thienylpicolinamidine derivatives and possible mechanisms of antiproliferative activity†
Mohamed A. Ismail *a,
Mohamed H. Abdel-Rhmana,
Ghada A. Abdelwahaba,
Wafaa S. Hamamaa,
Heba M. El-Shafeaia and
Wael M. El-Sayed*b
*a,
Mohamed H. Abdel-Rhmana,
Ghada A. Abdelwahaba,
Wafaa S. Hamamaa,
Heba M. El-Shafeaia and
Wael M. El-Sayed*b
aDepartment of Chemistry, Faculty of Science, Mansoura University, Mansoura 35516, Egypt. E-mail: mismail@mans.edu.eg
bDepartment of Zoology, Faculty of Science, University of Ain Shams, Abbassia 11566, Cairo, Egypt. E-mail: wael_farag@sci.asu.edu.eg
First published on 11th November 2020
Abstract
Three thienylpicolinamidine derivatives 4a–c were prepared from their corresponding picolinonitriles 3a–c on treatment with lithium trimethylsilylamide, LiN(TMS)2, followed by a de-protection step using ethanol/HCl (gas). DFT calculations were used to optimize the geometric structure of the newly synthesized picolinamidines. The comparison of DFT calculated spectral data with the experimental data (1H-NMR and 13C-NMR) showed a good agreement. The in vitro antiproliferative activity of the cationic compounds 4a–c was determined against 60 cancer cell lines representing nine types of cancer. The tested picolinamidines were highly active with compounds 4a and 4b eliciting mainly cytotoxic activity with GI values ranging from −7.17 to −86.03. Leukemia (SR and K-562), colon (SW-620 and HT29), and non-small cell lung cancer (NCI-H460) cell lines were the most responsive to the investigated picolinamidines. In particular, 4-methoxyphenyl derivative 4a showed a profound growth deterring power with GI50 of 0.34 μM against SR, 0.43 μM against SW-620, and 0.52 μM against NCI-H460. The three tested picolinamidines elicited potent GI50 values against all tested cell lines at low micromolar to sub-micromolar level. The new picolinamidines were selective and did not affect normal human fibroblasts. The selectivity index ranged from 13–21 μM. The novel picolinamidines downregulated the expression of key genes in the cell cycle, cdk1 and topoII, but did not affect p53 or txnrd1. Compounds 4b and 4c caused a significant reduction in the concentrations of TopoII and MAPK proteins but were devoid of any effect on the activity of caspase 3. Taken together, these promising anticancer candidates are effective at very low concentrations and safe to normal cells, and most probably work through arresting the cell cycle, and therefore, they deserve further investigations.
1. Introduction
Cancer is a fast growing global threat. According to the WHO health observatory report in 2018, about 9.6 million people have died from cancer worldwide. This number is projected to double by 2030. With about 26 million new cancer cases worldwide expected by 2030, cancer may precede cardiovascular diseases in affecting the humans. Although progress has been achieved regarding the 5-year survival, the increase of incidence is still ongoing at a rate exceeding this progress.1 At present, surgical treatment, combined with radiotherapy and chemotherapy are the main treatments for colorectal cancer.2,3 Over the recent years, technical advances have led to better response to cancer therapies and survival rates, but still substantial number of patients cannot be cured, develop metastasis, and/or suffer from severe early and late side effects. Therefore, the discovery of new anticancer agents is of great importance.Pyridine derivatives possess a diverse chemotherapeutic profile as anticancer, antimicrobial, or antiprotozoal agents. The pyridine ring mostly contributes an effective role in the pharmacokinetic properties of these biologically active compounds.4–8 Furanylnicotinamidine derivatives have been reported for their potent antiproliferative activity.4 Symmetrical furanylnicotinamidine compound DB829 has successfully passed the preclinical studies as antitrypanosomal agent.5–7 Cationic arylchalcophenes have been reported for their wide range of biological activities.9–13 Cationic arylthiophene derivatives have been reported for their anticancer activity.14,15 Recently, a bithiophene-fluorobenzamidine compound showed a very promising antitumor activity against colorectal cancer in rats reducing the incidence and multiplicity of polyps without causing hepato- or nephro-toxicity.16
Based on the biological importance of the reported pyridines and cationic arylthiophenes, three picolinamidine derivatives including methoxy and chloro-substituted phenyl moiety were synthesized and evaluated for in vitro antiproliferative activity against 60 cancer cell lines at the National Cancer Institute (NCI). The most responsive two types were leukemia and colon cancer cell lines (six cell lines for leukemia & seven for colon cancer). The selectivity of these compounds was assessed using WI-38 normal human fibroblasts. To understand the molecular pathways through which these compounds could exert their anticancer activity, the effect of these compounds on the expression of some of the key genes and proteins in the cell cycle and apoptosis was studied in HepG2 cells. These genes and proteins include p53, cyclin-dependent kinase 1 (cdk1), caspase 3, topoisomerase II (topo II), thioredoxin reductase 1 (txnrd1), and p38 mitogen-activated protein kinase (MAPK). Also, the quantum chemical calculation at DFT/B3LYP level was studied to investigate the structural, electronic, and spectral properties of the new picolinamidine derivatives.
2. Experimental
2.1. Chemistry
Melting points were measured in degree centigrade on Gallenkamp apparatus and are uncorrected. The infrared spectra (KBr) were recorded on Thermo Scientific Nicolet iS10 FT-IR spectrometer. Reaction monitoring was made using thin layer chromatography on silica gel pre-coated aluminum sheets (60 F254), and visualized under ultraviolet light. 1H-NMR and 13C-NMR spectra were measured in dimethyl sulfoxide (DMSO-d6) as a solvent and self-internal reference (2.49 parts per million (ppm) for proton and 39.50 ppm for carbon using 500 MHz JEOL's spectrometer). A GC-MS (Shimadzu Qp-2010 Plus) spectrometer was used for recording mass spectra. The elemental analyses were measured using PerkinElmer 2400 CHN analyzer and were within ±0.4 of the theoretical values.2.1.1.1. 5-[5-(4-Methoxyphenyl)thiophen-2-yl]picolinonitrile (3a). Compound 3a was obtained in 66% yield as a golden-yellow solid, mp 153–154 °C. Rf = 0.40, petroleum ether (60–80 °C)–EtOAc (7
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). IR (KBr) ν′/cm−1; 3080, 2957 (CH, stretch), 2228 (CN, stretch), 1602, 1573, 1507 (C
:3). IR (KBr) ν′/cm−1; 3080, 2957 (CH, stretch), 2228 (CN, stretch), 1602, 1573, 1507 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, CC, stretch) cm−1. 1H-NMR (DMSO-d6); δ/ppm = 3.80 (s, 3H, OCH3), 7.02 (d, J = 8.5 Hz, 2H), 7.56 (d, J = 4.0 Hz, 1H of thiophene ring), 7.66 (d, J = 8.5 Hz, 2H), 7.86 (d, J = 4.0 Hz, 1H of thiophene ring), 8.05 (d, J = 8.5 Hz, 1H of pyridyl ring), 8.26 (dd, J = 8.5, 2.0 Hz, 1H of pyridyl ring), 9.09 (d, J = 2.0 Hz, 1H of pyridyl ring). 13C-NMR; δ 55.32, 114.70 (2C), 117.70, 124.36, 125.57, 126.98 (2C), 128.96, 129.33, 130.04, 132.76, 133.08, 135.56, 146.36, 147.19, 159.57. MS (EI) m/z (rel. int.); 292 (M+, 100), 277 (M+–CH3, 63). Anal. calcd for: C17H12N2OS (292.35): C, 69.84; H, 4.14; N, 9.58 found: C, 69.73; H, 4.20; N, 9.37%.
N, CC, stretch) cm−1. 1H-NMR (DMSO-d6); δ/ppm = 3.80 (s, 3H, OCH3), 7.02 (d, J = 8.5 Hz, 2H), 7.56 (d, J = 4.0 Hz, 1H of thiophene ring), 7.66 (d, J = 8.5 Hz, 2H), 7.86 (d, J = 4.0 Hz, 1H of thiophene ring), 8.05 (d, J = 8.5 Hz, 1H of pyridyl ring), 8.26 (dd, J = 8.5, 2.0 Hz, 1H of pyridyl ring), 9.09 (d, J = 2.0 Hz, 1H of pyridyl ring). 13C-NMR; δ 55.32, 114.70 (2C), 117.70, 124.36, 125.57, 126.98 (2C), 128.96, 129.33, 130.04, 132.76, 133.08, 135.56, 146.36, 147.19, 159.57. MS (EI) m/z (rel. int.); 292 (M+, 100), 277 (M+–CH3, 63). Anal. calcd for: C17H12N2OS (292.35): C, 69.84; H, 4.14; N, 9.58 found: C, 69.73; H, 4.20; N, 9.37%.
2.1.1.2. 5-[5-(3-Chloro-4-methoxyphenyl)thiophen-2-yl]picolinonitrile (3b). Compound 3b was obtained in 71% yield as a yellow solid, mp 195–196.5 °C. Rf = 0.41, petroleum ether (60–80 °C)–EtOAc (7
:3). IR (KBr) ν′/cm−1; 3050, 2945 (CH, stretch), 2225 (CN, stretch), 1603, 1573, 1544 (CC, stretch) cm−1. 1H-NMR (DMSO-d6); δ/ppm = 3.89 (s, 3H, OCH3), 7.23 (d, J = 8.5 Hz, 1H), 7.64–7.67 (m, 2H), 7.83 (d, J = 2.5 Hz, 1H), 7.88 (d, J = 4.0 Hz, 1H of thiophene ring), 8.06 (d, J = 8.5 Hz, 1H), 8.28 (dd, J = 8.5, 2.0 Hz, 1H), 9.10 (d, J = 2.0 Hz, 1H of pyridyl ring). MS (EI) m/z (rel. int.); 326, 328 (M+, 100, 38; chlorine isotopes), 311, 313 (M+–CH3, 82, 31; chlorine isotopes). Anal. calcd for: C17H11ClN2OS (326.80): C, 62.48; H, 3.39; N, 8.57 found: C, 62.31; H, 3.52; N, 8.35%.
2.1.1.3. 5-(5-(3,4,5-Trimethoxyphenyl)thiophen-2-yl)picolinonitrile (3c). Compound 3c was obtained in 84% yield as a yellow solid, mp 170–171 °C. Rf = 0.22, petroleum ether (60–80 °C)–EtOAc (7
:3). IR (KBr) ν′/cm−1; 3060, 2934 (CH, stretch), 2226 (CN, stretch), 1579, 1509 (CC, stretch) cm−1. 1H-NMR (DMSO-d6); δ/ppm = 3.69 (s, 3H, OCH3), 3.86 (s, 6H, 2 × OCH3), 6.98 (s, 2H), 7.67 (d, J = 4.0 Hz, 1H of thiophene ring), 7.91 (d, J = 4.0 Hz, 1H of thiophene ring), 8.06 (d, J = 8.5 Hz, 1H of pyridyl ring), 8.30 (dd, J = 8.5, 2.0 Hz, 1H of pyridyl ring), 9.11 (d, 2.0 Hz, 1H of pyridyl ring). 13C-NMR; δ 56.07 (2C), 60.15, 103.17 (2C), 117.63, 125.71, 128.62, 128.82, 129.33, 130.21, 132.88, 132.96, 136.40, 137.89, 146.36, 147.22, 153.36 (2C). MS (EI) m/z (rel. int.); 352 (M+, 100), 337 (M+–CH3, 83). Anal. calcd for: C19H16N2O3S (352.40): C, 64.76; H, 4.58; N, 7.95 found: C, 64.52; H, 4.65; N, 7.72%.
2.1.2.1. 5-[5-(4-Methoxyphenyl)thiophen-2-yl]picolinamidine hydrochloride salt (4a). Compound 4a was obtained in 82% yield as a yellowish-brown solid, mp 257–259 °C. IR (KBr) ν′/cm−1; 3430, 3383, 3274 (NH, stretch), 3071, 2963 (CH, stretch), 1679, 1605, 1577 (C
N & CC, stretch) cm−1. 1H-NMR (DMSO-d6); δ/ppm = 3.79 (s, 3H, OCH3), 7.02 (d, J = 8.5 Hz, 2H Ar of 4-methoxyphenyl-), 7.56 (d, J = 4.0 Hz, 1H of thiophene ring), 7.67 (d, J = 8.5 Hz, 2H Ar of 4-methoxyphenyl-), 7.92 (d, J = 4.0 Hz, 1H of thiophene ring), 8.37 (d, J = 8.5 Hz, 1H of pyridyl ring), 8.40 (dd, J = 8.5, 2.0 Hz, 1H of pyridyl ring), 9.11 (d, J = 2.0 Hz, 1H of pyridyl ring), 9.39 (s, 2H, NH2), 9.59 (s, 2H, +NH2). 13C-NMR; δ 55.32, 114.68 (2C), 123.77, 124.33, 125.58, 126.95 (2C), 128.96, 133.14, 133.77, 135.66, 141.60, 145.62, 146.23, 159.54, 161.43. MS (EI) m/z (rel. int.); 309 (M+, 100), 294 (M+–CH3, 15), 292 (M+–NH3, 62). Anal. calcd for: C17H15N3OS-1.0HCl (345.84): C, 59.04; H, 4.66; N, 12.15 found: C, 58.84; H, 4.70; N, 11.91%.
2.1.2.2. 5-[5-(3-Chloro-4-methoxyphenyl)thiophen-2-yl]picolinamidine hydrochloride salt (4b). Compound 4b was obtained in 76% yield as a golden-yellow solid, mp 266–267.5 °C. IR (KBr) ν′/cm−1; 3361 (NH, stretch), 3059, 2950 (CH, stretch), 1683, 1579, 1531 (C
N & CC, stretch) cm−1. 1H-NMR (DMSO-d6); δ/ppm = 3.89 (s, 3H, OCH3), 7.24 (d, J = 8.5 Hz, 1H), 7.65–7.67 (m, 2H), 7.83 (d, J = 2.0 Hz, 1H), 7.94 (d, J = 4.0 Hz, 1H of thiophene ring), 8.36 (d, J = 8.5 Hz, 1H), 8.41 (dd, J = 8.5, 2.0 Hz, 1H), 9.12 (d, J = 2.0 Hz, 1H), 9.38 (s, 2H, NH2), 9.59 (s, 2H, +NH2). 13C-NMR; δ 56.35, 113.44, 121.88, 123.73, 125.37, 125.62, 126.53, 126.68, 129.00, 133.33, 133.60, 136.45, 141.85, 144.44, 145.71, 154.58, 161.31. MS (EI) m/z (rel. int.); 343, 345 (M+, 100, 43: chlorine isotopes), 328, 330 (M+–CH3, 43, 9: chlorine isotopes), 326, 328 (M+–NH3, 72, 43: chlorine isotopes). Anal. calcd for: C17H14ClN3OS-1.0HCl (380.29): C, 53.69; H, 3.98; N, 11.05 found: C, 53.43; H, 4.10; N, 10.79%.
2.1.2.3. 5-[5-(3,4,5-Trimethoxyphenyl)thiophen-2-yl]picolinamidine hydrochloride salt (4c). Compound 4c was obtained in 75% yield as a golden-yellow solid, mp 280–281.5 °C. IR (KBr) ν′/cm−1; 3426, 3250 (NH, stretch), 3054, 2990, 2954 (CH, stretch), 1684, 1632, 1582 (C
N & CC, stretch) cm−1. 1H-NMR (DMSO-d6); δ/ppm = 3.69 (s, 3H, para OCH3), 3.86 (s, 6H, meta 2 OCH3), 6.99 (s, 2H), 7.70 (d, J = 4.0 Hz, 1H of thiophene ring), 7.97 (d, J = 4.0 Hz, 1H of thiophene ring), 8.38 (d, J = 8.5 Hz, 1H of pyridyl ring), 8.44 (dd, J = 8.5, 2.0 Hz, 1H of pyridyl ring), 9.14 (d, 2.0 Hz, 1H of pyridyl ring), 9.41 (s, 2H, NH2), 9.60 (s, 2H, +NH2). 13C-NMR; δ 56.09 (2C), 60.16, 103.17 (2C), 123.76, 125.73, 128.67, 128.82, 133.33, 133.69, 136.54, 137.89, 141.82, 145.69, 146.26, 153.37 (2C), 161.37. MS (EI) m/z (rel. int.); 369 (M+, 100), 354 (M+–CH3, 65), 352 (M+–NH3, 37), 337 (35). Anal. calcd for C19H19N3O3S-1.0HCl (405.89): C, 56.22; H, 4.97; N, 10.35 found: C, 55.98; H, 4.99; N, 10.12%.
2.2. Computational studies
Quantum chemical calculations were achieved using Gaussian 09W program18 to optimize the geometry at DFT/B3LYP level19–21 with standard 6-311++G(d,p) basis set. In DMSO, 1H and 13C-NMR chemical shifts were calculated by the gauge-invariant atomic orbital (GIAO) method22 by polarizable continuum model using the integral equation formalism variant (IEF-PCM).23,24 The NMR spectral data were assigned by GaussView 6.0 program,25 using corresponding TMS shielding calculated at the same theoretical levels as the reference. Fukui indices have been calculated using DMol3 module of Materials Studio package from Accelrys Inc.26 The B3LYP functions have been used with a double numeric plus polarization (DNP version 3.5) basis set which has comparable accuracy to 6-31G** Gaussian basis set.272.3. Biology
2.3.3.1. Effect of the new picolinamidine derivatives on p38 mitogen-activated protein kinase (MAPK) and topoisomerase II (Topo II). The concentration of MAPK and Topo II was determined in HepG2 cells after treatment with doxorubicin and the novel compounds (4a, 4b, and 4c) based on the sandwich ELISA methods using kits from Invitrogen (Carlsbad, CA, USA) and MyBioSource (San Diego, CA, USA), respectively.
2.3.3.2. Measurement of the active cleaved caspase 3 activity. The activity of the cleaved caspase-3 was determined in HepG2 cells after incubation with the GI50 concentration of compounds 4a, 4b, and 4c for 12 hours has described elsewhere33 using kit obtained from Abcam (UK).
3. Results and discussion
3.1. Chemistry
The preparation of the new thienylpicolinamidines 4a–c (Fig. 1) begins with Suzuki coupling reaction of bromothienyl derivative 117 with the appropriate phenylboronic acid 2a–c to afford thienylpicolinonitriles 3a–c. The new picolinonitriles 3a–c were converted to the target thienylpicolinamidines 4a–c by the action of LiN(TMS)2, followed by de-protection with hydrogen chloride and subsequent neutralization with NaOH. Picolinamidines hydrochloride salts were prepared by treatment of the proper free base of monoamidines with hydrogen chloride in ethanol.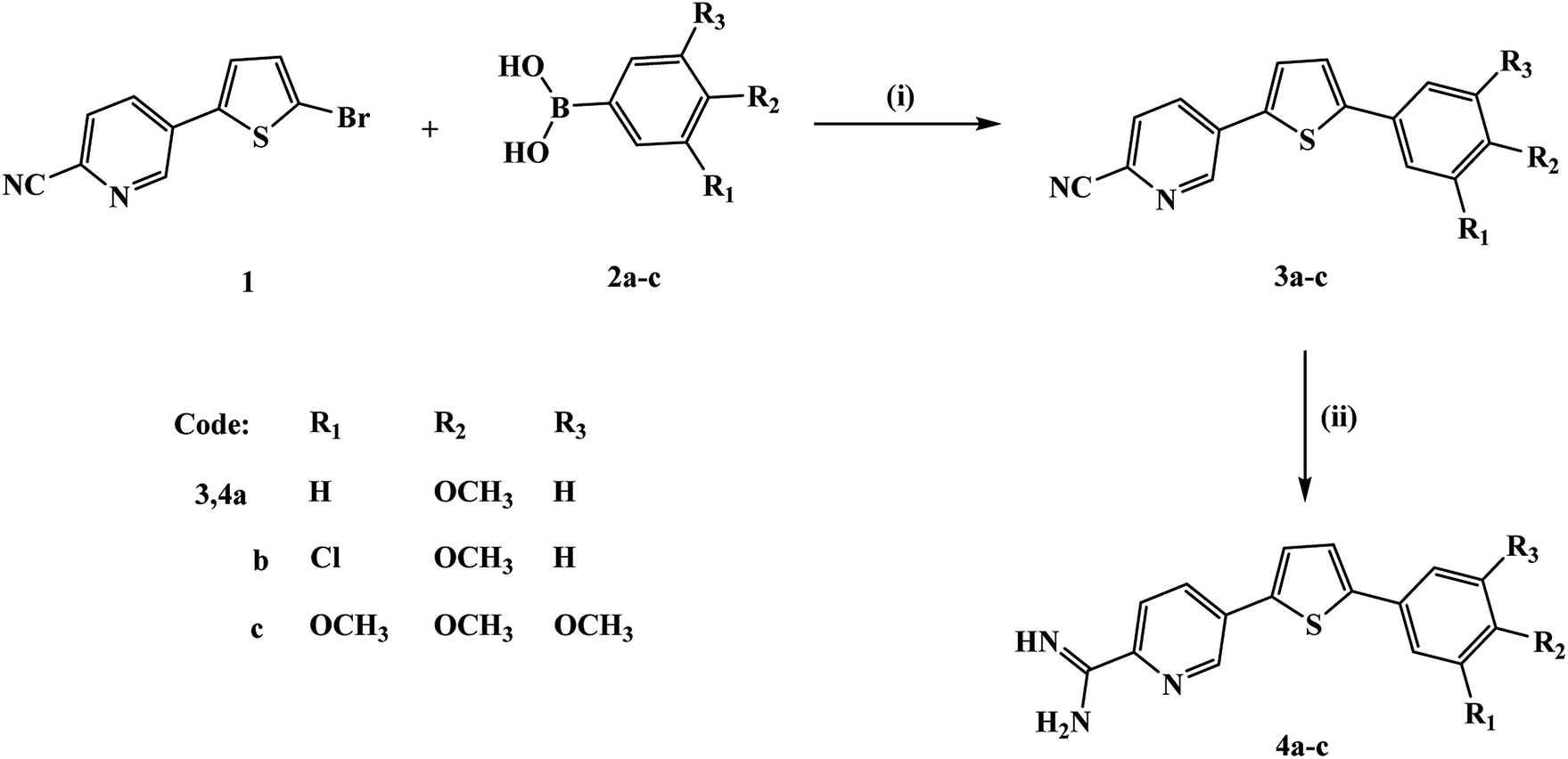 | ||
| Fig. 1 Synthesis scheme for the new thienylpicolinamidine derivatives. Reagents and conditions: (i) Pd(PPh3)4, anhydrous K2CO3, 1,4-dioxane; (ii) (a) LiN(TMS)2, (b) ethanol/HCI (gas). | ||
The structures of the new thienylpicolinonitriles 3a–c and thienylpicolinamidine derivatives 4a–c were assigned based on their spectral and elemental analyses. The IR spectra of all mononitriles 3a–c confirmed the appearance of nitrile group in each case with stretching vibrations values of 2228, 2225, and 2226 cm−1, respectively. Whereas, the IR spectra of all monoamidines indicated the disappearance of the nitrile group in each case and displayed new peaks corresponding for N–H stretching vibrations (ν′ 3430, 3383 & 3274 cm−1 for compound 4a). 1H NMR spectra of all monoamidines showed the characteristic signals corresponding to the cationic amidine group and were deuterium exchangeable. Thus, 1H NMR spectrum of monocationic compound 4a showed two singlet signals at δ 9.39 (2H) and 9.59 (2H) referring to the cationic amidine group, the presence of ABX system splitting pattern for three protons of 2,5-disubstituted pyridyl moiety, two doublet signals of thiophene ring coupling at δ 7.56 (1H), 7.92 (1H), in addition to three signals; one singlet signal at δ 3.79 (1 × OCH3), two aromatic doublets at δ 7.02 & 7.67 ppm of AA′BB′ system corresponding to 4-methoxyphenyl moiety. The carbon skeleton of compound 4a was confirmed by the presence of 15 carbon-lines in its 13C NMR spectrum with two characteristic carbon signals at δ 55.32 (for methoxy carbon), and 161.43 (for amidinic carbon). Moreover, picolinamidine derivative 4a mass spectrum gave a m/z peak at 309 referring to its molecular ion peak (M+), and m/z peak at 292 corresponding for a fragment produced from the loss a molecule of ammonia. The structure determination of monocationic compound 4c was confirmed by its 1H NMR spectrum which displayed two singlets at δ 9.40 (2H, NH2) & 9.60 (2H, +NH2) characteristic for the amidinic group, splitting pattern of 2,5-disubstituted pyridyl moiety confirmed by the presence of ABX system (one doublet of doublet at δ 8.44, one large doublet signal at δ 8.38, and small doublet signal at δ 9.14), along with two doublet signals of thiophene ring at δ 7.70 (1H), 7.97 (1H), in addition to three singlet signals at δ 3.69 (1 × OCH3), 3.86 (2 × OCH3), 6.99 (2H) referring to 3,4,5-trimethoxyphenyl moiety. The carbon skeleton of 4c was confirmed by its 13C NMR spectrum with three characteristic carbon signals at δ 56.09 (carbon of para methoxy), δ 60.16 (carbons of two equivalent meta methoxy groups), and 161.37 (amidinic carbon). Mass fragmentation of picolinamidine derivative 4c was inconsistent with the mass fragmentation of its nicotinamidine analogue.34 Compound 4c gave a m/z peak at 369 of its molecular ion peak (M+), m/z peak at 354 corresponding for a fragment produced from the loss of a methyl group, and m/z peak at 352 corresponding for a fragment produced from the loss of a molecule of ammonia, in addition to a fragment with m/z peak at 337 due to the loss of a methyl group and a molecule of ammonia.
3.2. Computational studies
The difficulty to obtain a single crystal of the newly synthesized cationic picolinamidine derivatives promoted us to carry out quantum chemical calculation at DFT/B3LYP level to investigate their structural, electronic and spectral properties.The dihedral angle data of the cationic picolinamidines 4a–c showed that the pyridyl ring was titled on the thienyl moiety plane by 3.00–4.50°, i.e. in 3-chloro-4-methoxyphenyl derivative 4b, S(5)–C(4)–C(7)–C(13) = 4.37° (Table S3†). Moreover, the amidinic group and pyridyl ring of 4-methoxyphenyl derivative 4a exhibited ∼1.30° for C(14)–C(15)–C(18)–N(20) and C(14)–C(15)–C(18)–N(19) while in 3-chloro-4-methoxyphenyl derivative 4b and 3,4,5-trimethoxyphenyl derivative 4c, these angles were in 0.14–0.32° range. Furthermore, the data indicated that the phenyl ring is more titled on thienyl moiety than pyridyl one where the S(5)–C(1)–C(6)–C(8) is ∼20.25° for compounds 4a and 4b while it reaches ∼24.5° in compound 4c.
Both thienyl and phenyl rings bond angle data of the investigated cationic picolinamidines almost coincided with those obtained from X-ray single crystal of analogue compounds.35–38 For example, cationic compound 4c data showed that C(4)–S(5)–C(1), C(4)–C(3)–C(2) and C(9)–C(8)–C(6) angles were 92.40, 113.94 and 121.15°, where their X-ray values were 92.8, 112.6–113.4 and 119.9–120.0°,35–38 respectively. Meanwhile, the pyridyl ring exhibited a little deviation from the X-ray values ranged from 0.47° to 2.67°. For instance, in case of cationic compound 4a, the angles C(17)–N(16)–C(15) and C(17)–C(7)–C(13) were 119.34 and 115.63° while the corresponding reported values were 118.5 and 118.3–122.1°,35–38 respectively. The cationic amidine bond angle H–N+–H and H–N–H were ∼116.88 and 120.40°, respectively. Comparison with the corresponding X-ray reported values indicated a difference by ±3.81° for both angles35–38 (Table S4†).
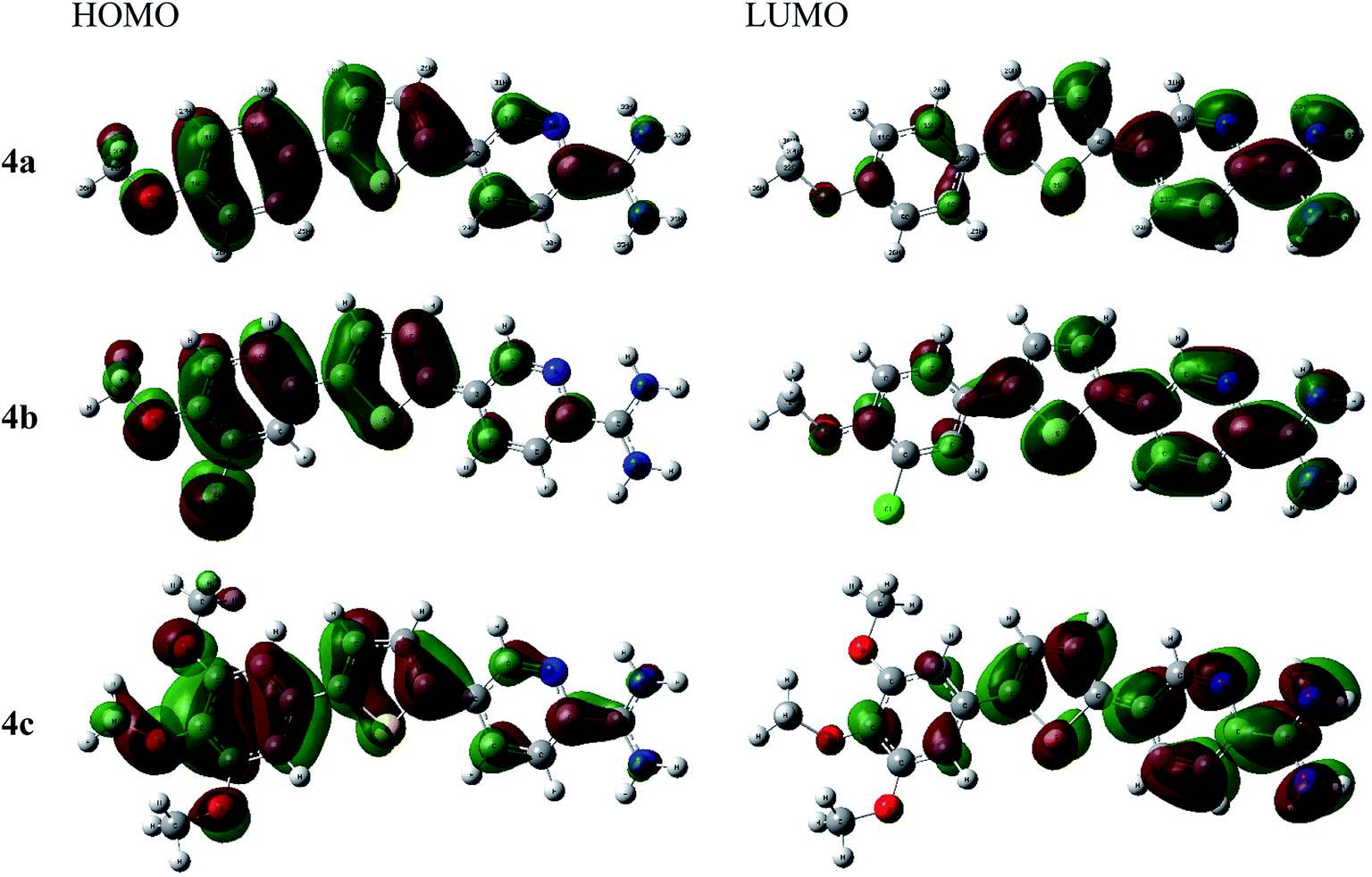 | ||
| Fig. 2 The frontier molecular orbital of cationic picolinamidines 4a–c. | ||
As shown in Fig. 2, the thienyl ring is involved in HOMO and LUMO orbitals formation in all derivatives. For example, thienylpicolinamidine derivative 4a, its HOMO spread mainly over the 4-methoxyphenyl moiety while LUMO orbital is on the pyridyl ring and cationic amidine group. This behaviour was observed in all other derivatives (4b and 4c). The methoxy group and chloro substituents participate strongly in HOMO and slightly in LUMO formation in all derivatives. Thus, it could be concluded that the methoxy or chlorophenyl moieties serve as electron donor sites while the amidinic group at pyridyl moiety acts as electron acceptor. Accordingly, the HOMO–LUMO charge transfer within the 4a–c derivatives may be expressed as electrons migration from donor site, methoxy or chlorophenyl, to acceptor site, positively charged amidinic group on pyridyl moiety.
As shown in Table 1, the trisubstituted derivative 4c exhibited the lowest EHOMO −7.94 eV while the chloro derivative 4b was the highest with −8.17 eV. Moreover, the ELUMO data showed that 4a and 4c have the same value, −5.89 eV; while 4b was a bit higher −5.91 eV. Furthermore, the energy gap data, ΔEHOMO–LUMO, indicated that the trisubstituted derivative 4c, 2.05 eV, was the lowest while the disubstituted derivative 4b, was the highest, 2.27 eV where the three investigated picolinamidines ordered as 4c < 4a < 4b.
| Compound | HOMO | LUMO | ΔEH–L | χ | η | δ | ω |
|---|---|---|---|---|---|---|---|
| 4a | −8.08 | −5.89 | 2.19 | 6.99 | 1.09 | 0.91 | 22.30 |
| 4b | −8.17 | −5.91 | 2.27 | 7.04 | 1.13 | 0.88 | 21.87 |
| 4c | −7.94 | −5.89 | 2.05 | 6.92 | 1.03 | 0.98 | 23.33 |
The EHOMO and ELUMO have been utilized for determining electronegativity (χ) to evaluate the Lewis acid character, global hardness (η) and softness (δ) as a measure of the charge transfer resistance and the capacity of a molecule to receive electrons, respectively (Table 1). In addition to electrophilicity (ω) which is a measure of energy lowering due to electron flow between HOMO and LUMO41 where:
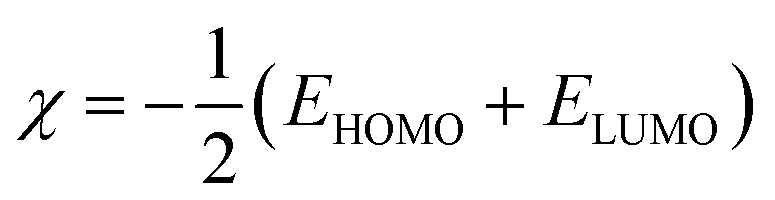
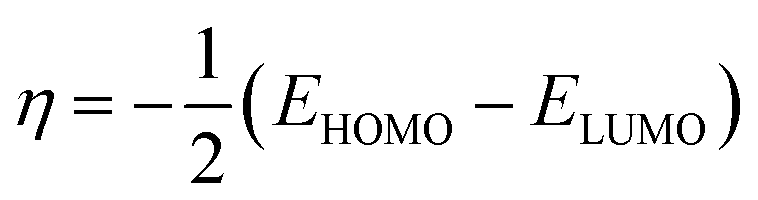
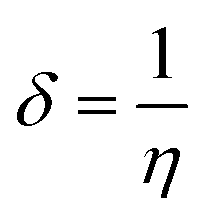
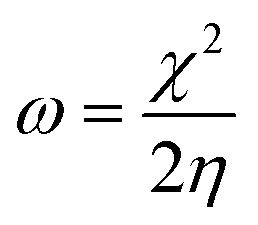
The data showed that the hardest derivative was 4b while the softest one was 4c, where according to softness, the three investigated picolinamidines were ordered as 4b > 4a > 4c. The Mulliken's charges (Table S5†) and Fukui's and relative nucleophilicity indices (Table S6†) were estimated and discussed in details in the ESI section.
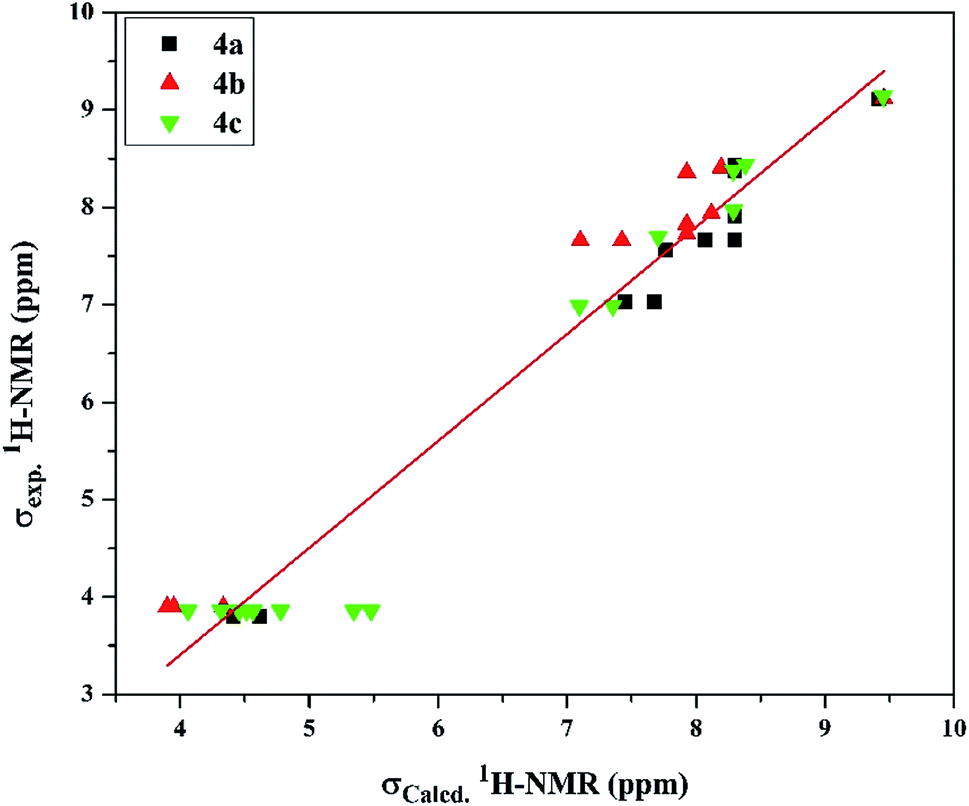 | ||
| Fig. 3 The linear regression between experimental and DFT calculated 1H NMR chemical shifts of the investigated cationic picolinamidines 4a–c. | ||
On the other hand, the 13C-NMR DFT calculated signals for the amidinic carbon atom were observed at ∼164.02 ppm while its experimental were at ∼161.37 ppm. The methoxy carbon atom in all the derivatives was estimated at ∼58.30 ppm exhibiting average difference 1.89 ppm with the experimental values (Table S8†). The DFT calculation for the pyridyl and phenyl carbons showed signals in the ranges of 126.73–148.78 and 102.64–167.13 ppm, respectively, exhibiting average differences 4.58 and 5.89 ppm, respectively, where the experimental signals observed for the former at 123.73–146.26 ppm and for the latter at 103.17–159.54 ppm. Moreover, the calculated chemical shifts of thienyl carbons, 125.94–164.77 ppm, displayed less agreement with experimental data, 125.58–141.85 ppm, with average difference 10.34 ppm (Table S8†). Examining the relationship between the experimental and calculated chemical shifts showed a linear relationship expressed as σExp. = 7.157 + 0.909 σCalcd with regression coefficient (R2) 0.934 and root mean square error (RMSE) 6.791 (Fig. 4).
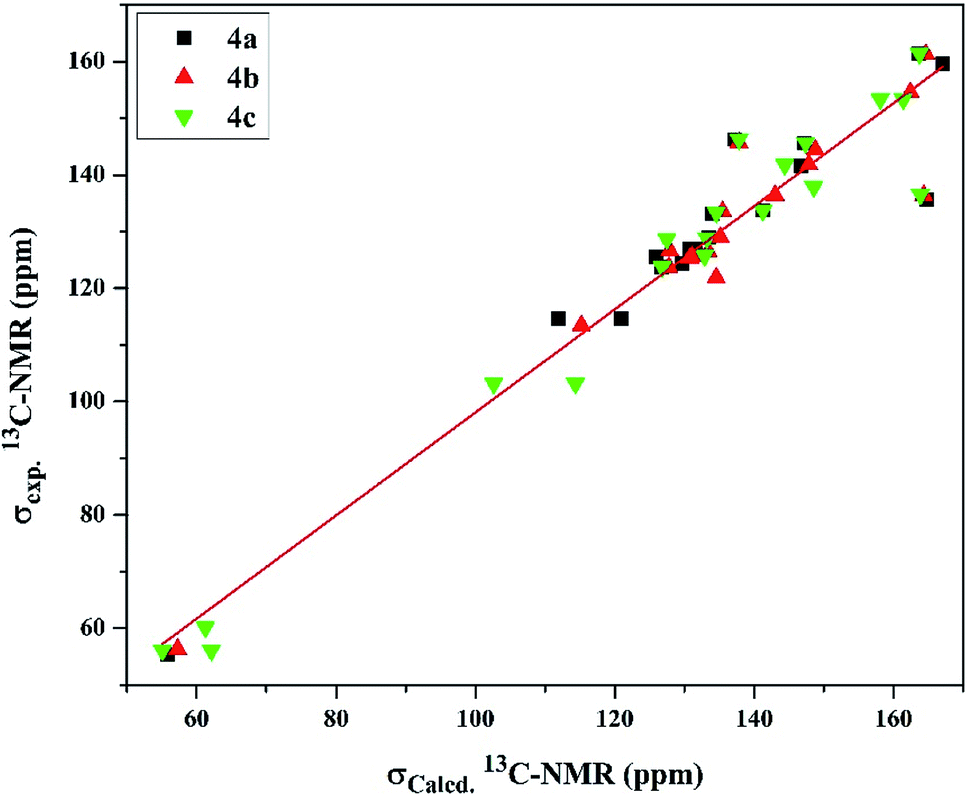 | ||
| Fig. 4 The linear regression between experimental and DFT calculated 13C NMR chemical shifts of the investigated cationic picolinamidines 4a–c. | ||
3.3. Biology
Three thienylpicolinamidine derivatives 4a–c were evaluated for their in vitro antiproliferative activity against nine types of cancer cell lines viz leukemia, prostate, non-small cell lung, melanoma, colon, ovarian, CNS, renal, and breast cancer at the NCI, USA. First, the chosen compounds were evaluated at an initial high dose of 10 μM. Mean percent growth inhibition (MPGI) of 4-methoxyphenyl derivative 4a (−28.84) and 3-chloro-4-methoxyphenyl derivative 4b (−19.95) showed cytotoxic activities. Whereas, 3,4,5-trimethoxyphenyl derivative 4c (52.58) was cytostatic in action. The individual percent growth inhibition results for the initial screening of the three tested picolinamidines 4a–c against the most responsive two types of cancer (13 cancer cell lines; six subtypes for leukemia and seven for colon cancer) are presented in Table 2. The tested picolinamidines were highly active with compounds 4a and 4b eliciting cytotoxic activity with GI% values ranging from −7.17 to −86.03, and cytostatic activity with 99.71 inhibition for compound 4a against SR leukemia cancer cell line and 97.48 inhibition for compound 4b against HT29 colon cancer cell line. Whereas, compound 4c showed mainly cytostatic activity against all tested cell lines with inhibition activity ranging from 50.93 to 99.05, except for HL-60 (TB) for which 4c was cytotoxic with GI% of −40.59.| Leukemia | 4a | 4b | 4c | Colon cancer | 4a | 4b | 4c |
|---|---|---|---|---|---|---|---|
| CCRF-CEM | −43.35 | −48.98 | 89.10 | COLO 205 | −52.23 | −56.05 | NT |
| HL-60 (TB) | −47.33 | −48.71 | −40.59 | HCC-2998 | −44.90 | −43.85 | 87.93 |
| K-562 | −26.99 | −29.78 | 94.42 | HCT-116 | −60.38 | −65.49 | 99.05 |
| MOLT-4 | −53.72 | −49.30 | 76.19 | HCT-15 | −76.97 | −77.39 | 78.61 |
| RPMI-8226 | −41.90 | −45.37 | NT | HT29 | −43.08 | 97.48 | 85.79 |
| SR | 99.71 | −7.17 | 93.69 | KM12 | −73.71 | −86.03 | 50.93 |
| SW-620 | −65.69 | −79.80 | 70.82 |
According to the NCI protocol, the compounds showing high antiproliferative activity in the initial screen, all test picolinamidines herein, are to be subjected to a five dose screen for determining their GI50 values. The individual GI50 values of the tested picolinamidines 4a–c against a panel of cancer cell lines are presented in Table 3. Among the leukemia and colon cancer cell lines, the most responsive two types tested in the five-dose screen, SR and K-562 leukemia cancer cell lines, and SW-620 and HT29 colon cancer cell lines were the most responsive cell lines. In specific, the most active compound was 4-methoxyphenyl derivative 4a that showed a profound growth deterring power with an GI50 of 0.34 μM against SR and 0.43 μM against SW-620, with an LC50 of >100 μM and 4.98 μM, respectively. Furthermore, picolinamidine 4a exhibited GI50 of 0.52 μM against NCI-H460 (non-small cell lung cancer). The 3-chloro-4-methoxyphenyl derivative 4b comes second with GI50 of 0.58 against SR and 0.90 μM against K-562 cell lines, with an LC50 of >100 μM for both tested cancer cells. 3,4,5-Trimethoxyphenyl derivative 4c exhibited GI50 ranging from 1.37 to 2.78 μM (for Leukemia and colon) and was the least active in the initial screen among the three tested picolinamidines. According to the cut off set by the NCI at 10 μM, all the three picolinamidines are considered active antiproliferative agents against the 60 cell lines (except compound 4c against ovarian SK-OV-3 cell line).
| Leukemia | 4a | 4b | 4c | Colon cancer | 4a | 4b | 4c |
|---|---|---|---|---|---|---|---|
| a Data represent GI50 in μM against the tested cell lines.b This table includes the data for the most two responsive types of cancer (for the full data of the nine types of cancer, see Table S9 ESI). | |||||||
| CCRF-CEM | 1.43 | 1.18 | 2.49 | COLO 205 | 1.89 | 1.94 | 1.60 |
| HL-60 (TB) | 1.78 | 1.66 | 1.87 | HCC-2998 | 1.82 | 1.86 | 2.00 |
| K-562 | 1.09 | 0.90 | 1.39 | HCT-116 | 1.62 | 1.82 | 1.47 |
| MOLT-4 | 1.90 | 1.72 | 2.53 | HCT-15 | 1.27 | 1.57 | 2.18 |
| RPMI-8226 | 2.13 | 2.25 | 1.67 | HT29 | 0.88 | 1.35 | 1.37 |
| SR | 0.34 | 0.58 | 2.48 | KM12 | 1.81 | 1.81 | 2.78 |
| SW-620 | 0.43 | 1.79 | 2.09 | ||||
These in vitro results reflect that while all three compounds elicit potent GI50 values against all tested cell lines at the low micromolar to sub-micromolar level, the parent compound 4a stands out for possessing LC50 at low micromolar concentration against five of the seven examined colon cancer cell lines (COLO 205, HCC-2998, HCT-15, KM12, SW-620 with LC50 values of 8.13, 6.38, 5.71, 6.03, and 4.98 μM, respectively). On the other hand, picolinamidine derivative 4c (possessing three methoxy groups on the phenyl ring) demonstrated somewhat better LC50 values in both leukemia and colon cancer (HL-60, RPMI-8226, COLO 205, HCC-2998 and HCT-116 with LC50 values of 9.44, 9.40, 6.66, 7.61, and 5.60 μM, respectively). Finally, the m-chloro substitution at the phenyl ring exemplified by analogue 4b led to relatively similar growth inhibition profile to that displayed by the parent derivative 4a, yet it led the compound to have much higher LC50 values against all the tested cell lines with the exception of KM12 and SW-620 colon cancer cell lines.
Table 4 presents the median values of GI50, TGI and LC50 for the investigated picolinamidine compounds 4a–c. Compound 4a was the most active compound giving median GI50 values at 1.62 μM. Trimethoxyphenyl derivative 4c was the least potent growth inhibitor (GI50 and TGI values of 2.39 and 6.16 μM, respectively), moreover, it was the least cytotoxic (LC50 of 19.49 μM). The HOMO and LUMO, highest occupied and the lowest unoccupied molecular orbitals, have significant role in understanding the chemical reactivity of the molecule to electronic transport and the establishment of biological activity with intramolecular charge transfer. Moreover, the decrease in the HOMO–LUMO energy gap indicates the ease of charge transfer interactions within the molecule, which may be responsible for the bioactivity of the molecules.41–43 Linking biology data with ΔEHOMO–LUMO values herein this manuscript, ΔEHOMO–LUMO indicated that the trimethoxy derivative 4c, 2.05 eV, is the lowest while the chloro-methoxy derivative 4b, is the highest, 2.27 eV where the three investigated picolinamidines were ordered as 4c < 4a < 4b. Whereas, the median GI50's were ordered as 4c < 4b < 4a, therefore, the previous model is valid for compounds 4a and 4b, but not for 4c. This finding suggests that the biological activity could not be correlated to a single factor but it is the interplay between different factors e.g. lipophilicity, interaction with nuclear factors and DNA, fitting to different key growth factors and modulators, ability to induce apoptosis, and others.
| MG-MIDa | 4a | 4b | 4c |
|---|---|---|---|
| a MG-MID = mean graph midpoint representing mean sensitivity of all examined cell lines to the test compound.b GI50 = compound concentration causing 50% growth inhibition of tested cells.c TGI = compound concentration causing 100% growth inhibition of tested cells.d LC50 = compound concentration causing 50% lethality of tested cells. | |||
| GI50b | 1.62 | 1.73 | 2.39 |
| TGIc | 3.31 | 3.54 | 6.16 |
| LC50d | 8.12 | 8.31 | 19.49 |
The ability of the investigated cationic picolinamidines 4a–c to discriminate between cancer cell and normal cells is one of the essential goals in the anticancer drug development. Therefore, selectivity index (SI) was determined for the three picolinamidines and ranged from 13.0 to 21.5 fold (Table 5). The most selective compound was 4c (SI = 21.5). These results support the safety of these picolinamidine derivatives and their selectivity against cancer cells.
| Compounds | WI38 | SIc |
|---|---|---|
| a The viability of the cells was measured after 48 h of incubation with different concentrations of the investigated compounds by MTT assay. The dose–response curves from as the mean of two parallel experiments were used to determine the IC50; three wells were assigned for every concentration. Doxorubicin (Dox) was used as a positive control. The data are presented as mean ± SEM.b Significant toxicity as compared to the untreated cells.c SI: selectivity index = WI-38 IC50/median GI50. | ||
| Dox | 2.9 ± 0.8b | NA |
| 4a | 21.1 ± 3.1b | 13.0 |
| 4b | 23.7 ± 2.5b | 13.7 |
| 4c | 51.3 ± 9.0 | 21.5 |
| Treatment | p53 | cdk1 | caspase 3 | topoII | txnrd1 |
|---|---|---|---|---|---|
| a Significant difference (p < 0.05) as compared to untreated cells. The data are expressed as mean ± SEM for two independent experiments. | |||||
| Fold induction from untreated cells | |||||
| DOX | 3.14 ± 0.81a | 0.31 ± 0.09a | 4.17 ± 0.91a | 0.47 ± 0.08a | 0.47 ± 0.10a |
| 4a | 1.19 ± 0.43 | 0.37 ± 0.04a | 1.57 ± 0.76 | 1.23 ± 0.05 | 1.32 ± 0.14 |
| 4b | 1.03 ± 0.08 | 0.51 ± 0.15a | 1.85 ± 0.11 | 0.57 ± 0.08a | 1.29 ± 0.13 |
| 4c | 1.18 ± 0.10 | 0.30 ± 0.04a | 3.37 ± 0.25a | 0.27 ± 0.02a | 1.07 ± 0.12 |
p53 (the guardian of the genome) is mutated in more than 50% of human cancer types.51 It is the main regulator of cell cycle progression and apoptosis. When DNA is damaged, p53 expression increases arresting the cell cycle at the G1/S checkpoint, induces the DNA repair mechanisms, or initiates apoptosis if the damage is severe52 but apparently, none of the tested picolinamidines acts through this gene. Apoptosis usually occurs through intrinsic or extrinsic pathways. In both pathways caspase 3 is the main execution enzyme.53
Cdk1 controls the cell cycle progression to S phase.54 The downregulation of cdk1 by test compounds 4a, 4b, and 4c suggests the cell cycle arrest. Therefore, we believe that these picolinamidines directly acted on the cdk1 gene without affecting p53, but this assumption needs further investigation. Thioredoxin reductase (Txnrd1) is the only known enzyme that reduces thioredoxin.55 The role of thioredoxin is very essential for the structure and activity of proteins, cell growth, and survival. Thioredoxin is responsible for the reduction of proteins and formation of the disulfide bridges. Therefore, this enzyme is upregulated in many types of cancer.56 Inhibition of txnrd1 leads to cell death.57 Unfortunately, none of the tested picolinamidines significantly inhibited the expression of this gene.
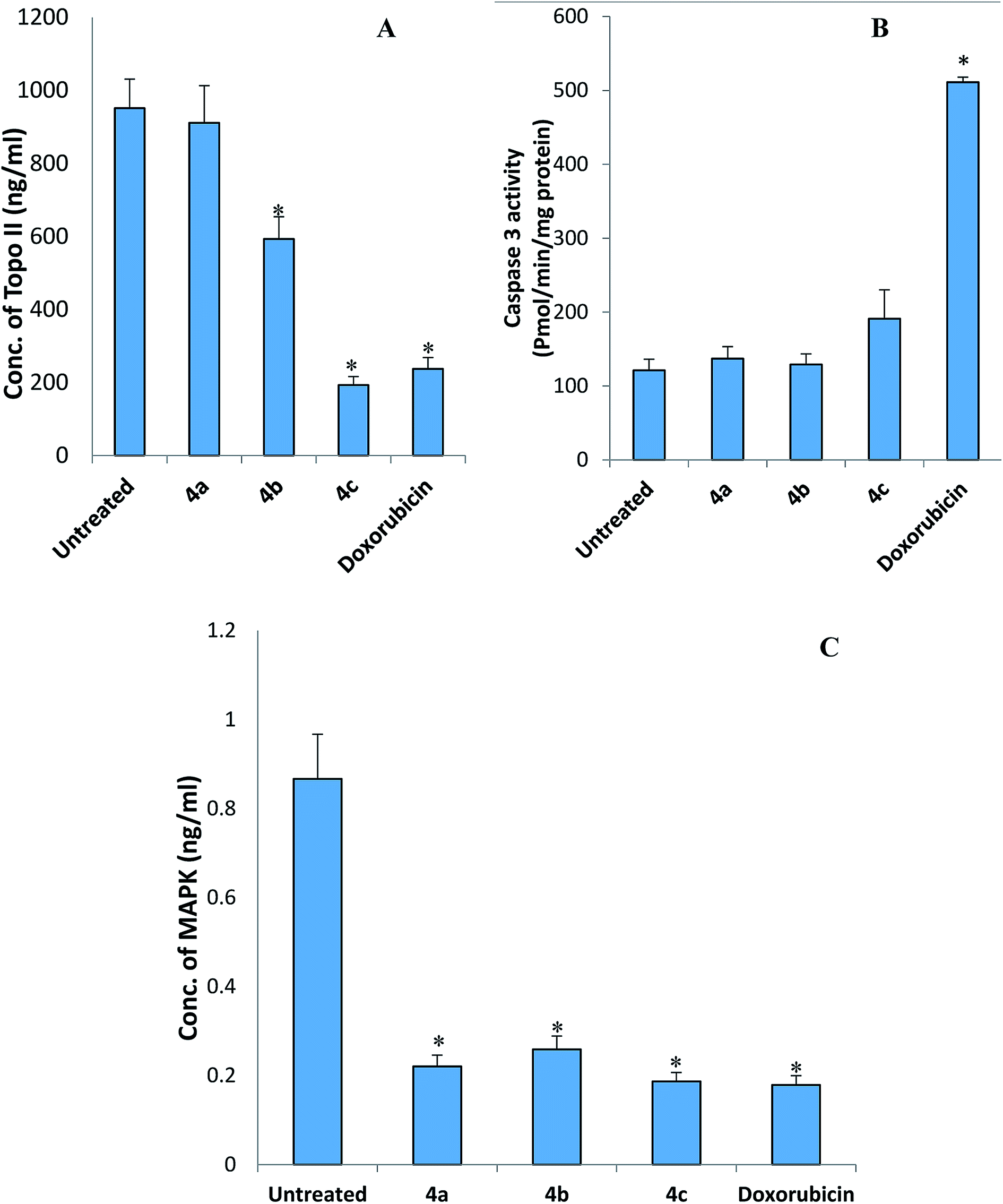 | ||
| Fig. 5 The effect of the picolinamidine derivatives 4a–c on (A) topoisomerase II (Topo II) concentration, (B) cleaved caspase 3 activity, and (C) MAPK concentration in HepG2 cells. Each compound has been assigned three wells and the experiment was performed twice. Data are expressed as mean ± SEM. *Significant change compared to untreated cells. | ||
4. Conclusions
The frontier molecular orbitals of the new thienylpicolinamidines 4a–c, the energy gap data, ΔEHOMO–LUMO, indicated that the trimethoxy derivative 4c, is the lowest while the chloro-methoxy derivative 4b, is the highest, where the three investigated picolinamidines were ordered as 4c < 4a < 4b. The comparison of DFT calculated NMR spectral data with the experimental data showed a good agreement. The in vitro antiproliferative data of compounds 4a–c revealed that the leukemia and colon cancer were the most responsive types of the nine types tested. Picolinamidines 4a and 4b exhibited mainly cytotoxic activity, whereas compound 4c displayed mainly cytostatic activity against leukemia and colon cancer cells. The most active compound was 4-methoxyphenyl derivative 4a that showed potent antiproliferative activity with GI50 values of 0.34 μM, 0.43 μM and 0.88 μM against SR, SW-620 and HT29, respectively. 3,4,5-Trimethoxyphenyl derivative 4c was less toxic than the other two derivatives. The three compounds were more selective towards the cancer cells while safer to the normal fibroblasts. The tested picolinamidines downregulated the expression of key genes in the cell cycle, cdk1 and topoII, but did not affect the expression of p53 or txnrd1. Compounds 4b and 4c caused a significant reduction in the concentrations of Topo II and MAPK proteins. Taken together, these promising anticancer candidates are effective at very low concentrations, safe to normal cells and most probably work through arresting the cell cycle and therefore, they deserve further investigations as potential targeted small anticancer molecules.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The antiproliferative activity was evaluated at the National Cancer Institute (NCI, USA). This work was supported by the Mansoura research unit, Mansoura University, Egypt [grant number MU-SCI-18-13].References
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer statistics, Ca-Cancer J. Clin., 2019, 69, 7–34 CrossRef.
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer statistics, Ca-Cancer J. Clin., 2018, 68, 7–30 CrossRef.
- L. Xu, L. Shi, S. Qiu, S. Chen, M. Lin, Y. Xiang, C. Zhao, J. Zhu, L. Shen and Z. Zuo, Design, synthesis, and evaluation of cyanopyridines as anti-colorectal cancer agents via inhibiting STAT3 pathway, Drug Des., Dev. Ther., 2019, 13, 3369–3381 CrossRef CAS.
- M. M. Youssef, R. K. Arafa and M. A. Ismail, Synthesis, antimicrobial, and antiproliferative activities of substituted phenylfuranylnicotinamidines, Drug Des., Dev. Ther., 2016, 10, 1133–1146 CAS.
- J. K. Thuita, K. K. Wolf, G. A. Murilla, A. S. Bridges, D. W. Boykin, J. N. Mutuku, Q. Liu, S. K. Jones, C. O. Gem, S. Ching, R. R. Tidwell, M. Z. Wang, M. F. Paine and R. Brun, Chemotherapy of Second Stage Human African Trypanosomiasis: Comparison between the Parenteral Diamidine DB829 and Its Oral Prodrug DB868 in Vervet Monkeys, PLoS Neglected Trop. Dis., 2015, 9, e0003409 CrossRef.
- J. K. Thuita, K. K. Wolf, G. A. Murilla, Q. Liu, J. N. Mutuku, Y. Chen, A. S. Bridges, R. E. Mdachi, M. A. Ismail, S. Ching, D. W. Boykin, J. E. Hall, R. R. Tidwell, M. F. Paine, R. Brun and M. Z. Wang, Safety, Pharmacokinetic, and Efficacy Studies of Oral DB868 in a First Stage Vervet Monkey Model of Human African Trypanosomiasis, PLoS Neglected Trop. Dis., 2013, 7, e2230 CrossRef CAS.
- T. Wenzler, D. W. Boykin, M. A. Ismail, J. E. Hall, R. R. Tidwell and R. Brun, New treatment option for second-stage African sleeping sickness: in vitro and in vivo efficacy of aza analogs of DB289, Antimicrob. Agents Chemother., 2009, 53, 4185–4192 CrossRef CAS.
- L. Hu, R. K. Arafa, M. A. Ismail, T. Wenzler, R. Brun, M. Munde, D. W. Wilson, S. Nzimiro, S. Samyesudhas, K. A. Werbovetz and D. W. Boykin, Azaterphenyl diamidines as antileishmanial agents, Bioorg. Med. Chem. Lett., 2008, 18, 247–251 CrossRef CAS.
- M. A. Ismail, W. M. El-Sayed, S. Shaaban, G. A. Abdelwahab and W. S. Hamama, A Review of Cationic Arylfurans and their Isosteres: Synthesis and Biological Importance, Curr. Org. Chem., 2019, 23, 2751–2782 CrossRef CAS.
- A. A. Farahat, M. A. Ismail, A. Kumar, T. Wenzler, R. Brun, A. Paul, W. D. Wilson and D. W. Boykin, Indole- and benzimidazole Bichalcophenes: Synthesis, DNA binding and Antiparasitic activity, Eur. J. Med. Chem., 2018, 143, 1590–1596 CrossRef CAS.
- S. A. Ohnmacht, E. Varavipour, R. Nanjunda, I. Pazitna, G. Di Vita, M. Gunaratnama, A. Kumar, M. A. Ismail, D. W. Boykin, W. D. Wilson and S. Neidle, Discovery of new G-quadruplex binding chemotypes, Chem. Commun., 2014, 50, 960–963 RSC.
- M. Munde, A. Kumar, P. Peixoto, S. Depauw, M. A. Ismail, A. A. Farahat, A. Paul, M. V. Say, M. H. David-Cordonnier, D. W. Boykin and W. D. Wilson, The Unusual Monomer Recognition of Guanine-Containing Mixed Sequence DNA by a Dithiophene Heterocyclic Diamidine, Biochemistry, 2014, 53, 1218–1227 CrossRef CAS.
- R. Nhili, P. Peixoto, S. Depauw, S. Flajollet, X. M. Dezitter, M. Munde, M. A. Ismail, A. Kumar, A. A. Farahat, C. E. Stephens, M. Duterque-Coquillaud, W. D. Wilson, D. W. Boykin and M. H. David-Cordonnier, Targeting the DNA-binding activity of the human ERG transcription factor using new heterocyclic dithiophene diamidines, Nucleic Acids Res., 2013, 41, 125–138 CrossRef CAS.
- S. Depauw, M. Lambert, S. Jambon, A. Paul, P. Peixoto, R. Nhili, L. Morongiu, M. Figeac, C. Dassi, C. Paul-Constant, B. Billoré, A. Kumar, A. A. Farahat, M. A. Ismail, E. Mineva, D. P. Sweat, C. E. Stephens, D. W. Boykin, W. D. Wilson and M. David-Cordonnier, Heterocyclic Diamidines DNA ligands as HOXA9 Transcription Factor Inhibitors: Design, Molecular Evaluation and Cellular Consequences in HOXA9-Dependant Leukemia Cell Model, J. Med. Chem., 2019, 62, 1306–1329 CrossRef CAS.
- M. A. Ismail, M. M. Youssef, R. K. Arafa, S. S. Al-Shihry and W. M. El-Sayed, Synthesis and antiproliferative activity of monocationic arylthiophene derivatives, Eur. J. Med. Chem., 2017, 126, 789–798 CrossRef CAS.
- M. Abdel-Rasol, N. M. El-Beih, S. M. Yahya, M. A. Ismail and W. M. El-Sayed, The antitumor activity of a novel fluorobenzamidine against dimethylhydrazine-induced colorectal cancer in rats, Anti-Cancer Agents Med. Chem., 2020, 20, 450–463 CrossRef CAS.
- R. R. Tidwell, D. W. Boykin, C. E. Stephens, M. A. Ismail, A. Kumar, W. D. Wilson and R. Brun and K. Werbovetz, 2,5-Diarylselenophene compounds, aza 2,5-diarylthiophene compounds, and their prodrugs as antiprotozoal agents, US Pat., 2010/0331368A1, December 30, 2010.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Gaussian 09, Revision A.1, Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- J. P. Perdew and Y. Wang, Pair-distribution function and its coupling-constant average for the spin-polarized electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 12947–12954 CrossRef.
- R. Ditchfield, Self-consistent perturbation theory of diamagnetism: I. A Gauge-Invariant LCAO method for NMR chemical shifts, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- M. Cossi and V. Barone, Time-dependent density functional theory for molecules in liquid solutions, J. Chem. Phys., 2001, 115, 4708–4717 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS.
- R. Dennington, T. Keith and J. Millam, GaussView, version 5, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- D. S. Biovia, Materials Studio, Dassault Systèmes, San Diego, 2017 Search PubMed.
- B. Delley, Ground-state enthalpies: evaluation of electronic structure approaches with emphasis on the density functional method, J. Phys. Chem. A, 2006, 110, 13632–13639 CrossRef CAS; B. Delley, An all-electron numerical method for solving the local density functional for polyatomic molecules, J. Chem. Phys., 1990, 92, 508–517 CrossRef.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, New colorimetric cytotoxicity assay for anticancer-drug screening, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS.
- T. A. McCaffrey, L. A. Agarwal and B. B. Weksler, A rapid fluorometric DNA assay for the measurement of cell density and proliferation in vitro, In Vitro Cell. Dev. Biol., 1988, 24, 247–252 CrossRef CAS.
- A. Monks, D. Scudiero, P. Skehan, R. Shoemaker, K. Paull, D. Vistica, C. Hose, J. Langley, P. Cronise, A. Vaigro-Wolff, M. Gray-Goodrich, H. Campbell, J. Mayo and M. Boyd, Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines, J. Natl. Cancer Inst., 1991, 83, 757–766 CrossRef CAS.
- S. A. El-Metwally, A. K. Khalil and W. M. El-Sayed, Design, molecular modeling and anticancer evaluation of thieno[2,3-d]pyrimidine derivatives as inhibitors of topoisomerase II, Bioorg. Chem., 2020, 94, 103492 CrossRef CAS.
- K. J. Livak and T. D. Schmittgen, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method, Methods, 2001, 25, 402–408 CrossRef CAS.
- M. H. Hekal, A. M. El-Naggar, F. S. M. Abu El-Azm and W. M. El-Sayed, Synthesis of new oxadiazol-phthalazin derivatives with anti-proliferative activity; molecular docking, pro-Apoptotic, and enzyme inhibition profile, RSC Adv., 2020, 10, 3675–3688 RSC.
- M. A. Ismail, M. H. Abdel-Rhman, G. A. Abdelwahab and W. S. Hamama, Synthesis and spectroscopic studies of methoxysubstituted phenylthienylnicotinamidines, Synth. Commun., 2020, 50, 2355–2375 CrossRef CAS.
- B. J. Ramulu, S. Koley and M. S. Singh, Metal-free Brønsted acid mediated synthesis of fully substituted thiophenes via chemo-and regioselective intramolecular cyclization of α,α′-bis (β-oxodithioesters) at room temperature, Org. Biomol. Chem., 2016, 14, 434–439 RSC.
- P. Pouzet, I. Erdelmeier, D. Ginderow, P. M. Dansette, D. Mansuy and J. P. Mornon, Thiophene-1oxides. V. Comparison of the crystal structures and thiophene ring aromaticity of 2,5-diphenylthiophene, its sulfoxide and sulfone, J. Heterocycl. Chem., 1997, 34, 1567–1574 CrossRef CAS.
- I. Nohara, A. Prescimone, D. Häussinger, C. E. Housecroft and E. C. Constable, [Cu(POP)(N^S)][PF6] and [Cu(xantphos)(N^S)][PF6] compounds with 2-(thiophen-2-yl)pyridines, RSC Adv., 2019, 9, 13646–13657 CAS.
- G. Portalone, Benzamidinium 2-methoxybenzoate, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, o1114–o1115 CAS.
- D. Sajan, L. Joseph, N. Vijayan and M. Karabacak, Natural bond orbital analysis, electronic structure, non-linear properties and vibrational spectral analysis of L-histidinium bromide monohydrate: a density functional theory, Spectrochim. Acta, Part A, 2011, 81, 85–98 CrossRef CAS.
- F. A. Bulat, E. Chamorro, P. Fuentealba and A. Toro-Labbe, Condensation of frontier molecular orbital Fukui functions, J. Phys. Chem. A, 2004, 108, 342–349 CrossRef CAS.
- S. Xavier, S. Periandy and S. Ramalingam, NBO, conformational, NLO, HOMO–LUMO, NMR and electronic spectral study on 1-phenyl-1-propanol by quantum computational methods, Spectrochim. Acta, Part A, 2015, 137, 306–320 CrossRef CAS.
- M. M. Makhlouf, A. S. Radwan and B. Ghazal, Experimental and DFT insights into molecular structure and optical properties of new chalcones as promising photosensitizers towards solar cell applications, Appl. Surf. Sci., 2018, 452, 337–351 CrossRef CAS.
- A. Bouchoucha, S. Zaater, S. Bouacida, H. Merazig and S. Djabbar, Synthesis and characterization of new complexes of nickel(II), palladium(II) and platinum(II) with derived sulfonamide ligand: structure, DFT study, antibacterial and cytotoxicity activities, J. Mol. Struct., 2018, 1161, 345–355 CrossRef CAS.
- V. Arjunan, P. Balamourougane, M. Kalaivani, A. Raj and S. Mohan, Experimental and theoretical quantum chemical investigations of 8-hydroxy-5-nitroquinoline, Spectrochim. Acta, Part A, 2012, 96, 506–516 CrossRef CAS.
- M. Karabacak, M. Cinar and M. Kurt, DFT based computational study on the molecular conformation, NMR chemical shifts and vibrational transitions for N-(2-methylphenyl)methanesulfonamide and N-(3-methylphenyl)methanesulfonamide, J. Mol. Struct., 2010, 968, 108–114 CrossRef CAS.
- M. I. Orief and M. H. Abdel-Rhman, Molecular modeling, spectroscopic and structural studies on newly synthesized ligand N-benzoyl-2-isonicotinoylhydrazine-1-carboxamide, J. Mol. Struct., 2018, 1173, 332–340 CrossRef CAS.
- Y. Sun, P. Xia, H. Zhang, B. Liu and Y. Shi, P53 is required for doxorubicin-induced apoptosis via the TGF-β signaling pathway in osteosarcoma-derived cells, Am. J. Cancer Res., 2016, 6, 114–125 CAS.
- M. A. Ismail, A. Negm, R. K. Arafa, E. Abdel-Latif and W. M. El-Sayed, Anticancer activity, dual prooxidant/antioxidant effect and apoptosis induction profile of new bichalcophene-5-carboxamidines, Eur. J. Med. Chem., 2019, 169, 76–88 CrossRef CAS.
- J. Herrero-Ruiz, M. Mora-Santos, S. Giráldez, C. Sáez, M. Á. Japón, M. Tortolero and F. Romero, βTrCP controls the lysosome-mediated degradation of CDK1, whose accumulation correlates with tumor malignancy, Oncotarget, 2014, 5, 7563–7574 CrossRef.
- J. J. Liu, Q. Liu, H. L. Wei, J. Yi, H. S. Zhao and L. P. Gao, Inhibition of thioredoxin reductase by auranofin induces apoptosis in adriamycin-resistant human K562 chronic myeloid leukemia cells, Pharmazie, 2011, 66, 440–444 CAS.
- T. Ozaki and A. Nakagawara, p53: the attractive tumor suppressor in the cancer research field, J. Biomed. Biotechnol., 2011, 3, 994–1013 CAS.
- E. Senturk and J. J. Manfredi, p53 and cell cycle effects after DNA damage, Methods Mol. Biol., 2013, 962, 49–61 CrossRef.
- M. Olsson and B. Zhivotovsky, Caspases and cancer, Cell Death Differ., 2011, 18, 1441–1449 CrossRef CAS.
- J. M. Enserink and R. D. Kolodner, An overview of Cdk1-controlled targets and processes, Cell Div., 2010, 5, 1–41 CrossRef.
- E. S. Arnér and A. Holmgren, Physiological functions of thioredoxin and thioredoxin reductase, Eur. J. Biochem., 2000, 267, 6102–6109 CrossRef.
- P. Nguyen, R. T. Awwad, D. D. Smart, D. R. Spitz and D. Gius, Thioredoxin reductase as a novel molecular target for cancer therapy, Cancer Lett., 2005, 2361, 64–74 Search PubMed.
- K. F. Tonissen and G. Di Trapani, Thioredoxin system inhibitors as mediators of apoptosis for cancer therapy, Mol. Nutr. Food Res., 2009, 53, 87–103 CrossRef CAS.
- G. Ambrosini, H. S. Cheema, S. Seelman, E. B. Sambol, S. Singer and G. K. Schwartz, Sorafenib Inhibits Growth and MAPK Signaling in Malignant Peripheral Nerve Sheath Cells, Mol. Cancer Ther., 2008, 7, 890–896 CrossRef CAS.
- J. L. Nitiss, Targeting DNA topoisomerase II in cancer chemotherapy, Nat. Rev. Cancer, 2009, 9, 327–337 CrossRef CAS.
- E. Willmore, S. de Caux and N. J. Sunter, A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia, Blood, 2004, 103, 4659–4665 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08796c |
| This journal is © The Royal Society of Chemistry 2020 |