
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new synthetic route for the preparation of [Os3(CO)10(μ-OH)(μ-H)] and its reaction with bis(diphenylphosphino)methane (dppm): syntheses and X-ray structures of two isomers of [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)] and [Os3(CO)7(μ3-CO)(μ3-O)(μ-dppm)]†
Md. Tuhinur R. Joya,
Nikhil C. Bhoumika,
Shishir Ghosh *a,
Michael G. Richmondb and
Shariff E. Kabir*a
*a,
Michael G. Richmondb and
Shariff E. Kabir*a
aDepartment of Chemistry, Jahangirnagar University, Savar, Dhaka 1342, Bangladesh. E-mail: sghosh_006@yahoo.com; skabir_ju@yahoo.com
bDepartment of Chemistry, University of North Texas, 1155 Union Circle, Box 305070, Denton, TX 76203, USA
First published on 17th December 2020
Abstract
The triosmium cluster [Os3(CO)10(μ-OH)(μ-H)] containing bridging hydride and hydroxyl groups at a common Os–Os edge was obtained in good yield (ca. 75%) from the hydrolysis of the labile triosmium cluster [Os3(CO)10(NCMe)2] in THF at 67 °C. [Os3(CO)10(μ-OH)(μ-H)] reacts with dppm at 68 °C to afford the isomeric clusters 1 and 2 with the general formula [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)] that differ by the disposition of bridging dppm ligand. Cluster 1 is produced exclusively from the reaction of [Os3(CO)10(μ-OH)(μ-H)] with dppm in CH2Cl2 at room temperature in the presence of added Me3NO. Heating cluster 1 at 81 °C furnishes 2 in a process that likely proceeds by the release of one arm of the dppm ligand, followed by ligand reorganization about the cluster polyhedron and ring closure of the pendent dppm ligand. The oxo-capped [Os3(CO)7(μ3-CO)(μ3-O)(μ-dppm)] (3) has been isolated starting from the thermolysis of either 1 or 2 at 139 °C. Reactions of [Os3(CO)10(μ-dppm)] with ROH (R = Me, Et) in the presence of Me3NO at 80 °C furnish [Os3(CO)8(μ-OH)(μ,η1,κ1-OCOR)(μ-dppm)] (4, R = Me; 5, R = Et). Clusters 1–5 have been characterized by a combination of analytical and spectroscopic studies, and the molecular structure of each product has been established by X-ray crystallography. The bonding in these products has been examined by electronic structure calculations, and cluster 1 is confirmed as the kinetic product of substitution, while cluster 2 represents the thermodynamically favored isomer.
1. Introduction
The hydroxy-bridged triosmium cluster [Os3(CO)10(μ-OH)(μ-H)] was first reported by Johnson and Lewis in 1968 as a minor by-product (1.6% yield) in the synthesis of [Os3(CO)12] via high-pressure carbonylation of OsO4 in methanol at 125 °C.1 The same report also described three alternative syntheses for [Os3(CO)10(μ-OH)(μ-H)] starting from the parent dodecacarbonyl. Treatment of [Os3(CO)12] with KOH in methanol, sodium amalgam reduction, followed by hydrolytic workup, and reaction with NaBH4 in THF all furnish [Os3(CO)10(μ-OH)(μ-H)] in varying yields. Complicating these initial routes to [Os3(CO)10(μ-OH)(μ-H)] was the presence of tri- and/or tetra-nuclear osmium hydride species1 that severely hampered the isolation of pure [Os3(CO)10(μ-OH)(μ-H)]. Subsequently, [Os3(CO)10(μ-OH)(μ-H)] was isolated from the hydrolysis of the reactive species [Os3(CO)10(μ-OCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)(μ-H)],2 [Os3(CO)10(μ-NCHNMe2)(μ-H)],3 and [Os3(CO)10(κ4-C6H8)]4 in 36, 60 and 20% yield, respectively. More recently, Roberto and coworkers have developed a high-yield synthetic route for the preparation of [Os3(CO)10(μ-OH)(μ-H)],5 but this method requires prior activation of [Os3(CO)12] by silica through a surface-assisted reaction where the surface silanol groups afford the anchored intermediate [Os3(CO)10(μ-H)(μ-OSi
CH2)(μ-H)],2 [Os3(CO)10(μ-NCHNMe2)(μ-H)],3 and [Os3(CO)10(κ4-C6H8)]4 in 36, 60 and 20% yield, respectively. More recently, Roberto and coworkers have developed a high-yield synthetic route for the preparation of [Os3(CO)10(μ-OH)(μ-H)],5 but this method requires prior activation of [Os3(CO)12] by silica through a surface-assisted reaction where the surface silanol groups afford the anchored intermediate [Os3(CO)10(μ-H)(μ-OSi![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) )], which is readily hydrolyzed with aqueous HF (56% yield)6 or H2O (up to 81% yield)5 to give [Os3(CO)10(μ-OH)(μ-H)].
)], which is readily hydrolyzed with aqueous HF (56% yield)6 or H2O (up to 81% yield)5 to give [Os3(CO)10(μ-OH)(μ-H)].
Although [Os3(CO)10(μ-OH)(μ-H)] has been known for more than 50 years, relatively few studies, especially in comparison to the commercially available cluster [Os3(CO)12], exist on its reactivity with various substrates.5,7–10 For example, [Os3(CO)10(μ-OH)(μ-H)] reacts with HX to afford [Os3(CO)10(μ-X)(μ-H)] (where X = Cl, Br, I, OBun, OMe, OPh, RCO2; R = H, Me, CF3), but when X is a non-coordinating anion as with HBF4, the cation [Os3(CO)10(NCMe)2(μ-H)]+ is formed when MeCN is employed as a solvent.5,7,8 The dearth of studies based on [Os3(CO)10(μ-OH)(μ-H)] may be attributed to the absence of an easy and high-yield synthetic route to this hydroxy cluster from the commercially available parent cluster [Os3(CO)12]. We have developed a new synthetic route for the preparation of this hydroxyl-substituted cluster in good yield from hydrolysis of the triosmium cluster [Os3(CO)10(NCMe)2]; the latter is a well-known precursor for many different Os3 clusters and is easily prepared by treating [Os3(CO)12] with Me3NO in MeCN at room temperature.11
Metal hydroxides have been investigated as catalysts in the electro-oxidation of ethanol, conversion of benzene to phenol, and hydrogenation of ethylene to ethane.12–14 In the case of the latter reaction, functionalization of the hydroxyl group in [Os3(CO)10(μ-OH)(μ-H)] using silica gives the grafted cluster [Os3(CO)10(μ-H)(μ-OSi)] whose use as a catalyst for the hydrogenation of ethylene to ethane has been reported to proceed via intact Os3 intermediates.14 The stability of a metal cluster is greatly enhanced during autogenous catalysis by using a bridging diphosphine ligand. Incorporation of a rigid diphosphine such as bis(diphenylphosphino)methane (dppm) and 1,2-bis(diphenylphosphino)benzene (dppbz) in the coordination sphere of metal clusters has been demonstrated to prevent cluster fragmentation.15
Our interest in [Os3(CO)10(μ-OH)(μ-H)] centers on its reactivity with diphosphines and the coordination mode adopted by the ligand as a function of the bite angle. The reaction of this particular cluster and dppm was first reported by Johnson and Lewis to yield [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)] where the ancillary diphosphine was shown to bridge the two osmium centers tethered by the hydroxyl and hydride ligands.10 The ligand distribution in the product gives rise to a cluster that possesses idealized Cs symmetry. Following this report, we published our data on the reaction of [Os3(CO)10(μ-OH)(μ-H)] with the larger backbone diphosphines dppe and dppp and confirmed the formation of the analogous phosphine-bridged clusters [Os3(CO)8(μ-OH)(μ-H)(μ-PP)].16 In connection with our ligand reactivity studies involving metal cluster compounds, we needed [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)] and synthesized it from the thermolysis of [Os3(CO)10(μ-OH)(μ-H)] with dppm. Interestingly, we isolated two products from the reaction; the major product corresponded to the known cluster [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)] reported by Johnson and Lewis,10 and the minor product was subsequently confirmed as an isomer of the major product whose dppm ligand bridged a different metallic edge. Accordingly, we have explored the reaction that yields [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)] in more detail and report our results herein. Our attempts to prepare the related alkoxy-bridged cluster [Os3(CO)8(μ-OR)(μ-H)(μ-dppm)] (where R = Me, Et) starting from [Os3(CO)10(μ-OR)(μ-H)] and [Os3(CO)10(μ-dppm)] are also described.
2. Experimental section
2.1. General remarks
All reactions were carried out under an inert atmosphere of nitrogen using standard Schlenk techniques unless otherwise noted, and reagent grade solvents were dried by the standard methods and freshly distilled before use. The reagents [Os3(CO)12] and bis(diphenylphosphino)methane (dppm) were purchased from Strem Chemical Inc. and Acros Organics, respectively, and both were used as received. The starting cluster [Os3(CO)10(NCMe)2] was prepared according to the published procedure11 and employed in the preparation of [Os3(CO)10(μ-OH)(μ-H)] and [Os3(CO)10(μ-dppm)]. Infrared spectra were recorded on a Shimadzu IR Prestige-21 spectrophotometer, while NMR spectra were recorded on a Bruker Avance III HD (400 MHz) instrument. The NMR chemical shifts are reported in δ units and are referenced to the residual protons of the deuterated solvent (1H) or external 85% H3PO4 (31P), as appropriate. Elemental analyses were performed by the Microanalytical Laboratories of the Wazed Miah Science Research Centre at Jahangirnagar University. All products were separated in the air on TLC plates coated with 0.25 mm of silica gel (HF254-type 60, E. Merck, Germany).2.2. Preparation of [Os3(CO)10(μ-OH)(μ-H)]
A few drops of water were added to a THF solution (20 mL) of [Os3(CO)10(NCMe)2] (0.18 g, 0.19 mmol). The mixture was heated to reflux for 90 min and then allowed to cool to room temperature. The solvent was removed under reduced pressure, and the residue chromatographed by TLC on silica gel. Elution with cyclohexane/CH2Cl2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) developed two bands. The contents of the first band were too small for complete characterization, while the second band afforded [Os3(CO)10(μ-OH)(μ-H)] (0.13 g, 75%) as orange crystals after recrystallization from n-hexane/CH2Cl2 at 4 °C. Data for [Os3(CO)10(μ-OH)(μ-H)]: IR (νCO, CH2Cl2): 2110 vw, 2070 s, 2060 m, 2019 vs, 2000 m, 1983 w cm−1. 1H NMR (CDCl3): δ 0.57 (s, 1H), −12.59 (s, 1H).
:1, v/v) developed two bands. The contents of the first band were too small for complete characterization, while the second band afforded [Os3(CO)10(μ-OH)(μ-H)] (0.13 g, 75%) as orange crystals after recrystallization from n-hexane/CH2Cl2 at 4 °C. Data for [Os3(CO)10(μ-OH)(μ-H)]: IR (νCO, CH2Cl2): 2110 vw, 2070 s, 2060 m, 2019 vs, 2000 m, 1983 w cm−1. 1H NMR (CDCl3): δ 0.57 (s, 1H), −12.59 (s, 1H).
2.3. Reaction of [Os3(CO)10(μ-OH)(μ-H)] with dppm
:3, v/v) developed four bands. The third band afforded the previously reported [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)] (1)10 (43 mg, 62%), while the fourth band gave [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)] (2) (10 mg, 15%) as yellow crystals after recrystallization from n-hexane/CH2Cl2 at 4 °C. The contents of the first and second bands were too small for complete characterization. Data for 1: anal. calc. for C33H24O9Os3P2: C, 33.11; H, 2.02. Found: C, 33.43; H, 2.08%. IR (νCO, CH2Cl2): 2066 s, 2000 vs, 1962 s, 1931 w cm−1. 1H NMR (CDCl3): δ 7.85 (m, 1H), 7.49 (m, 1H), 7.40–7.34 (m, 13H), 7.26 (m, 4H), 7.09 (m, 1H), 3.59 (m, 1H), 2.57 (m, 1H), 0.43 (t, J 4.0 Hz, 1H), −11.75 (t, J 12.0 Hz, 1H). 31P{1H} NMR (CDCl3): δ −11.3 (s). Data for 2: anal. calc. for C33H24O9Os3P2: C, 33.11; H, 2.02. Found: C, 33.38; H, 1.98%. IR (νCO, CH2Cl2): 2068 s, 2033 m, 2021 m, 1992 vs, 1962 m, 1919 w cm−1. 1H NMR (CDCl3): δ 7.69 (m, 2H), 7.53 (m, 4H), 7.45–7.32 (m, 10H), 7.18 (m, 4H), 6.13 (m, 1H), 4.38 (m, 1H), 0.12 (s, 1H), −11.12 (d, J 36 Hz, 1H). 31P{1H} NMR (CDCl3): δ −19.2 (d, JPP 44 Hz, 1P), −22.8 (d, JPP 44 Hz, 1P).:3, v/v) developed two bands. The first band afforded [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)] (1) (51 mg, 75%) as yellow crystals after recrystallization from n-hexane/CH2Cl2 at 4 °C while the second band (not 2) was too small for complete characterization.2.4. Thermolysis of 1 at 81 °C
A cyclohexane solution (10 mL) of 1 (25 mg, 0.021 mmol) was heated to reflux at 81 °C for 18 h. Usual workup and chromatographic separation, as described above, afforded unreacted 1 (19 mg, 72%) and isomer 2 (4 mg, 16%) in an overall mass balance of 88%.2.5. Thermolysis of 1 at 139 °C
A xylene solution (15 mL) of 1 (30 mg, 0.025 mmol) was heated at 139 °C for 30 min. Upon cooling, the crude product mixture was chromatographically separated, allowing the isolation of six bands developed on TLC plates. The fourth and sixth bands afforded [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)] (2) (4 mg, 13%) as yellow crystals and [Os3(CO)7(μ3-CO)(μ3-O)(μ-dppm)] (3) (13 mg, 43%) as pale yellow crystals, respectively, after recrystallization from n-hexane/CH2Cl2 at 4 °C. The contents of the other bands were too small for complete characterization. Data for 3: anal. calc. for C33H22O9Os3P2: C, 33.16; H, 1.86. Found: C, 33.42; H, 1.92%. IR (νCO, CH2Cl2): 2073 s, 2016 vs, 1991 s, 1956 m, 1643 w cm−1. 1H NMR (CDCl3): δ 7.70 (m, 2H), 7.55 (m, 5H), 7.41 (m, 8H), 7.22 (m, 5H), 5.20 (m, 1H), 4.32 (m, 1H). 31P{1H} NMR (CDCl3): δ 16.7 (d, JPP 58 Hz, 1P), −4.1 (d, JPP 58 Hz, 1P).2.6. Thermolysis of 2 at 139 °C
A xylene solution (15 mL) of 2 (30 mg, 0.025 mmol) was heated to reflux for 5 min. Usual workup and chromatographic separation described above afforded several bands on the TLC plates. The fourth and sixth bands afforded 1 (4 mg, 13%) as yellow crystals and [Os3(CO)7(μ3-CO)(μ3-O)(μ-dppm)] (3) (8 mg, 27%) as pale yellow crystals after recrystallization from n-hexane/CH2Cl2 at 4 °C. The contents of the other minor bands were too small for complete characterization.2.7. Preparation of [Os3(CO)8(μ-OH)(μ,η1,κ1-OCOMe)(μ-dppm)] (4)
A methanol (5 mL) solution of Me3NO (12 mg, 0.16 mmol) was added dropwise to a benzene solution (15 mL) of [Os3(CO)10(μ-dppm)] (0.10 g, 0.081 mmol). The mixture was heated to reflux for 4 h and then allowed to cool at room temperature, after which time the solvent was removed under reduced pressure and the residue chromatographed by TLC on silica gel. Elution with cyclohexane/CH2Cl2 (7:3, v/v) developed three bands. The fastest-moving major band corresponded to unreacted [Os3(CO)10(μ-dppm)] (51 mg) and the third band to [Os3(CO)8(μ-OH)(μ,η1,κ1-OCOMe)(μ-dppm)] (4) (11 mg, 11%), which was isolated as yellow crystals after recrystallization from n-hexane/CH2Cl2 at 4 °C. The contents of the second band were too small for complete characterization. Data for 4: anal. calc. for C35H26O11Os3P2: C, 33.49; H, 2.09. Found: C, 33.86; H, 2.15%. IR (νCO, CH2Cl2): 2072 s, 2010 m, 1989 vs, 1964 m, 1938 s, 1917 m cm−1. 1H NMR (CD2Cl2): δ 7.70 (m, 2H), 7.53 (m, 5H), 7.44 (m, 9H), 7.26 (m, 4H), 4.97 (m, 1H), 4.08 (m, 1H), 3.64 (s, 3H), −0.39 (s, 1H). 31P{1H} NMR (CD2Cl2): δ 18.1 (d, JPP 84 Hz, 1P), −0.9 (d, JPP 84 Hz, 1P).
2.8. Preparation of [Os3(CO)8(μ-OH)(μ,η1,κ1-OCOEt)(μ-dppm)] (5)
An ethanol (5 mL) solution of Me3NO (12 mg, 0.160 mmol) was added dropwise to a benzene solution (15 mL) of [Os3(CO)10(μ-dppm)] (0.10 g, 0.081 mmol). The mixture was then heated to reflux for 4 h. The solution was then allowed to cool at room temperature and then worked up as described above for cluster 4. The first band corresponding to unreacted [Os3(CO)10(μ-dppm)] (56 mg) and the third band was established as [Os3(CO)8(μ-OH)(μ,η1,κ1-OCOEt)(μ-dppm)] (5) (8 mg, 8%), which was isolated as yellow crystals after recrystallization from n-hexane/CH2Cl2 at 4 °C. The contents of the second band were too small for complete characterization. Data for 5: anal. calc. for C36H28O11Os3P2: C, 34.07; H, 2.22. Found: C, 34.44; H, 2.31%. IR (νCO, CH2Cl2): 2070 s, 2010 m, 1989 vs, 1964 m, 1938 s, 1917 m cm−1. 1H NMR (CDCl3): δ 7.67 (m, 2H), 7.54 (m, 5H), 7.40 (m, 9H), 7.20 (m, 4H), 4.88 (m, 1H), 4.18 (m, 1H), 4.05 (m, 1H), 3.87 (m, 1H), 1.30 (m, 3H), 0.91 (s, 1H). 31P{1H} NMR (CDCl3): δ 18.5 (d, JPP 82 Hz, 1P), −0.8 (d, JPP 82 Hz, 1P).2.9. Crystal structure determinations
Single crystals of 1–5 suitable for X-ray diffraction analysis were grown by slow diffusion of n-hexane into a CH2Cl2 solution containing each product. Suitable crystals were mounted on a Bruker D8 Venture diffractometer equipped with a PHOTON II CPAD detector using a Nylon loop and Paratone oil, and the diffraction data were collected at low temperatures (see Table 1) using Mo-Kα radiation (λ = 0.71073). Data reduction and integration were carried out with SAINT+,17 and absorption corrections were applied using the program SADABS.18 The structures were solved with the ShelXS19 structure solution program by direct methods and refined by full-matrix least-squares based on F2 using ShelXL20 (for 1–3) or XL19 (for 4–5) within the OLEX2 (ref. 21) graphical user interface. All non-hydrogen atoms were refined anisotropically, and the hydrogen atoms were included using a riding model. Cluster 4 co-crystallizes with a disordered molecule of MeOH whose carbon atom was disordered over two sites (with an occupancy ratio 63:37) and refined isotropically. The hydrogen atoms for this disordered solvent molecule were not included in the refinement. Pertinent crystallographic parameters are given in Table 1.
| Compound | 1 | 2 | 3 | 4 | 5 |
| CCDC | 2036482 | 2036483 | 2036484 | 2036485 | 2036486 |
| Empirical formula | C33H24O9Os3P2 | C33H23O9Os3P2 | C33H22O9Os3P2 | C36H25O12Os3P2 | C36H28O11Os3P2 |
| Formula weight | 1197.06 | 1196.05 | 1195.04 | 1282.10 | 1269.12 |
| Temperature (K) | 193(1) | 193(1) | 150(1) | 150(1) | 150(1) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group | P21/n | ![[P with combining macron]](https://www.rsc.org/images/entities/i_char_0050_0304.gif) 1 1 |
1 |
1 |
1 |
|
|||||
| Unit cell dimensions: | |||||
| a (Å) | 12.9006(4) | 10.642(7) | 12.403(5) | 10.8434(8) | 10.9749(8) |
| b (Å) | 11.7117(4) | 12.082(9) | 13.586(7) | 11.4530(9) | 12.9394(10) |
| c (Å) | 23.7556(8) | 16.209(13) | 14.093(6) | 16.7944(12) | 14.3221(11) |
| α (°) | 90 | 76.185(19) | 63.446(14) | 70.658(3) | 74.898(4) |
| β (°) | 105.5930(10) | 71.21(3) | 81.056(13) | 87.463(3) | 89.703(3) |
| γ (°) | 90 | 73.02(2) | 68.94(2) | 76.322(3) | 72.437(3) |
| Volume (Å3) | 3457.1(2) | 1863(2) | 1982.5(16) | 1910.8(3) | 1866.1(2) |
| Z | 4 | 2 | 2 | 2 | 2 |
| Density (calculated) (Mg m−3) | 2.300 | 2.132 | 2.002 | 2.228 | 2.259 |
| Absorption coefficient (mm−1) | 11.143 | 10.340 | 9.716 | 10.094 | 10.333 |
| F(000) | 2208 | 1102 | 1100 | 1190 | 1180 |
| Crystal size (mm3) | 0.215 × 0.101 × 0.068 | 0.187 × 0.122 × 0.018 | 0.265 × 0.211 × 0.111 | 0.435 × 0.381 × 0.097 | 0.197 × 0.134 × 0.031 |
| 2θ range for data collection (°) | 5.4 to 56.702 | 4.602 to 56.68 | 5.808 to 54.34 | 4.87 to 54.418 | 5.576 to 50.866 |
| Reflections collected | 133154 | 49680 |
53938 |
62539 |
60422 |
| Independent reflections [Rint] | 8608 [Rint = 0.0404] | 9245 [Rint = 0.0907] | 8764 [Rint = 0.0490] | 8495 [Rint = 0.0430] | 6874 [Rint = 0.0416] |
| Data/restraints/parameters | 8608/0/432 | 9245/1/308 | 8764/0/425 | 8495/0/480 | 6874/0/475 |
| Goodness-of-fit on F2 | 1.046 | 1.035 | 1.085 | 1.051 | 1.040 |
| Final R indices [I > 2σ(I)] | R1 = 0.0186, wR2 = 0.0360 | R1 = 0.0504, wR2 = 0.0909 | R1 = 0.0212, wR2 = 0.0567 | R1 = 0.0347, wR2 = 0.1112 | R1 = 0.0176, wR2 = 0.0363 |
| R Indices (all data) | R1 = 0.0245, wR2 = 0.0376 | R1 = 0.0888, wR2 = 0.1013 | R1 = 0.0226, wR2 = 0.0573 | R1 = 0.0362, wR2 = 0.1128 | R1 = 0.0221, wR2 = 0.0377 |
| Largest diff. peak and hole (e Å−3) | 2.69 and −1.35 | 2.64 and −1.53 | 1.87 and −1.44 | 2.71 and −2.95 | 0.94 and −0.70 |
2.10. Computational methodology
All calculations were performed with the hybrid meta exchange-correlation functional M06,22 as implemented by the Gaussian 09 program package.23 The osmium atoms were described by Stuttgart–Dresden effective core potentials (ECP) and an SDD basis set,24 while a 6-31G(d′) basis set was employed for the remaining atoms.25The input data for the unrestricted optimizations were taken from the coordinates of the experimental structures. The reported geometries represent fully optimized ground states (positive eigenvalues) based on the Hessian matrix, and the natural charges (Q) and Wiberg bond indices were computed using Weinhold's natural bond orbital (NBO) program (NBO version 3.1).26,27 The geometry-optimized structures presented here have been drawn with the JIMP2 molecular visualization and manipulation program.28
3. Results and discussion
3.1. Preparation of [Os3(CO)10(μ-OH)(μ-H)]
Part of our contribution describes the synthesis of the known doubly bridged cluster [Os3(CO)10(μ-OH)(μ-H)] on a semi-preparative scale (Scheme 1). This particular cluster serves as a useful precursor to various polynuclear osmium clusters.29 Starting from [Os3(CO)12], the labile cluster [Os3(CO)10(NCMe)2] is easily prepared and used in the hydrolysis reaction that gives [Os3(CO)10(μ-OH)(μ-H)]. The method described here using [Os3(CO)10(NCMe)2] furnishes [Os3(CO)10(μ-OH)(μ-H)] in yields > 70%, and this is comparable (percent yield) to the heterogeneous procedure that employs the silica-supported cluster [Os3(CO)10(μ-H)(μ-OSi![[triple bond, length half m-dash]](https://www.rsc.org/images/entities/char_e007.gif) )].7,30
)].7,30
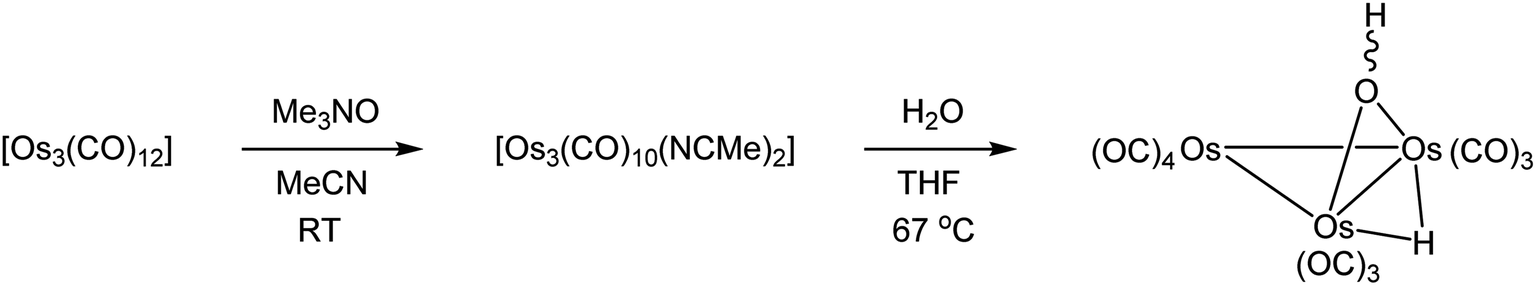 | ||
| Scheme 1 Synthesis of [Os3(CO)10(μ-OH)(μ-H)]. | ||
The first structural reports on [Os3(CO)10(μ-OH)(μ-H)] appeared in 1995,31–33 and all three structures contained hydrogens that were placed in fixed positions. The hydroxyl hydrogen in [Os3(CO)10(μ-OH)(μ-H)] is diastereotopic and can exist as a mixture of cis and trans stereoisomers depending on the disposition of the hydrogen with respect to an appropriate reference point, which we will define here as the axial CO in the Os(CO)4 moiety that is located on the same face of the Os3 plane as the bridging hydroxyl group. The two possible orientations for the hydroxyl hydrogen are depicted in Scheme 2. Lin and Leong published the structure of the trans stereoisomer where an intermolecular O–H⋯O bond between the hydroxyl group in [Os3(CO)10(μ-OH)(μ-H)] and α-pyrone controls the stereochemistry.34 The ability of such hydrogen bonding to control the molecular architecture of organometallic compounds has been discussed.35 The disposition of the ancillary ligands in the structure reported by Lin and Leong is similar to the ligands in the earlier report from Churchill on the related methoxy-bridged cluster [Os3(CO)10(μ-OMe)(μ-H)].36
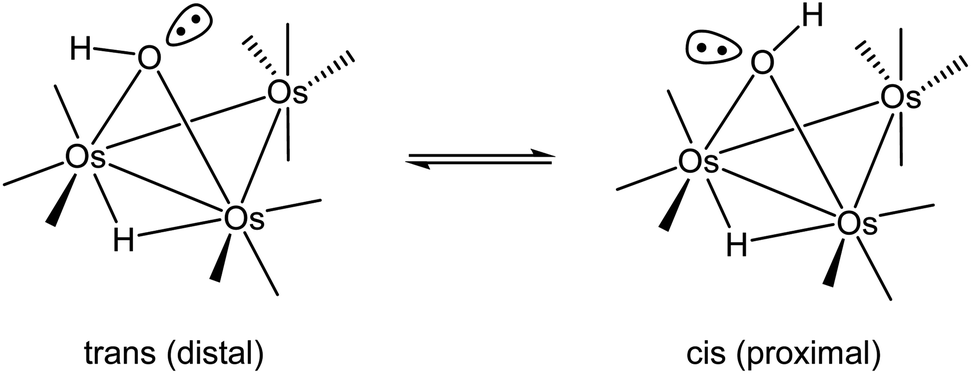 | ||
| Scheme 2 Cis and trans stereoisomers for [Os3(CO)10(μ-OH)(μ-H)]. | ||
The bonding in the trans and cis isomers of [Os3(CO)10(μ-OH)(μ-H)] was analyzed by electronic structure calculations. The trans isomer A1 is computed to be 0.6 kcal mol−1 (ΔG) more stable than the cis isomer A2, with Fig. 1 showing the optimized structures of the isomeric pair. The barrier for trans–cis interconversion (A1 ⇌ A2) was also evaluated to establish the role this process plays in the stereochemical equilibration of the isomers. If the barrier is low, the composition of the isomers in solution will reflect a thermally equilibrated mixture. The two isomers readily invert through a pyramidalization process at the oxygen through TSA1A2 that lies 4.6 kcal mol−1 above the thermodynamically preferred isomer A1. The pyramidalization barrier is sufficiently low in the gas phase at room temperature. The rapid equilibration of the two isomers will result in a time-averaged environment for the hydroxyl hydrogen in the 1H NMR spectrum of [Os3(CO)10(μ-OH)(μ-H)], along with a single resonance for the edge-bridging hydride ligand. This NMR behavior is exactly the result found by Aime and coworkers in their NMR relaxation study of [Os3(CO)10(μ-OH)(μ-H)] when CDCl3 was employed as the solvent.37 The appearance of a single hydride resonance in the 1H NMR spectrum of [Os3(CO)10(μ-OH)(μ-H)] in CDCl3 does not allow one to determine if a single isomer is present or if there is a rapid equilibration of isomers. Changing the solvent from CDCl3 to acetone allowed Aime and coworkers to sufficiently slow down the isomer equilibration and directly observe both isomers in solution. The increased dipolar electrostatic interactions between the O–H⋯acetone entities help raise the pyramidalization barrier through favorable electrostatic bonding. The unequivocal identity of each isomer was established using the spin-lattice relaxation (T1) data. These data allowed the determination of the rH–H distance between the hydride and hydroxyl hydrogen in the two species with the trans isomer exhibiting an rH–H distance of 2.90 Å and the cis isomer an rH–H distance of 3.26 Å. The NMR-based distances compare well with the DFT distances of 2.97 Å and 3.49 Å computed for A1 and A2, respectively.
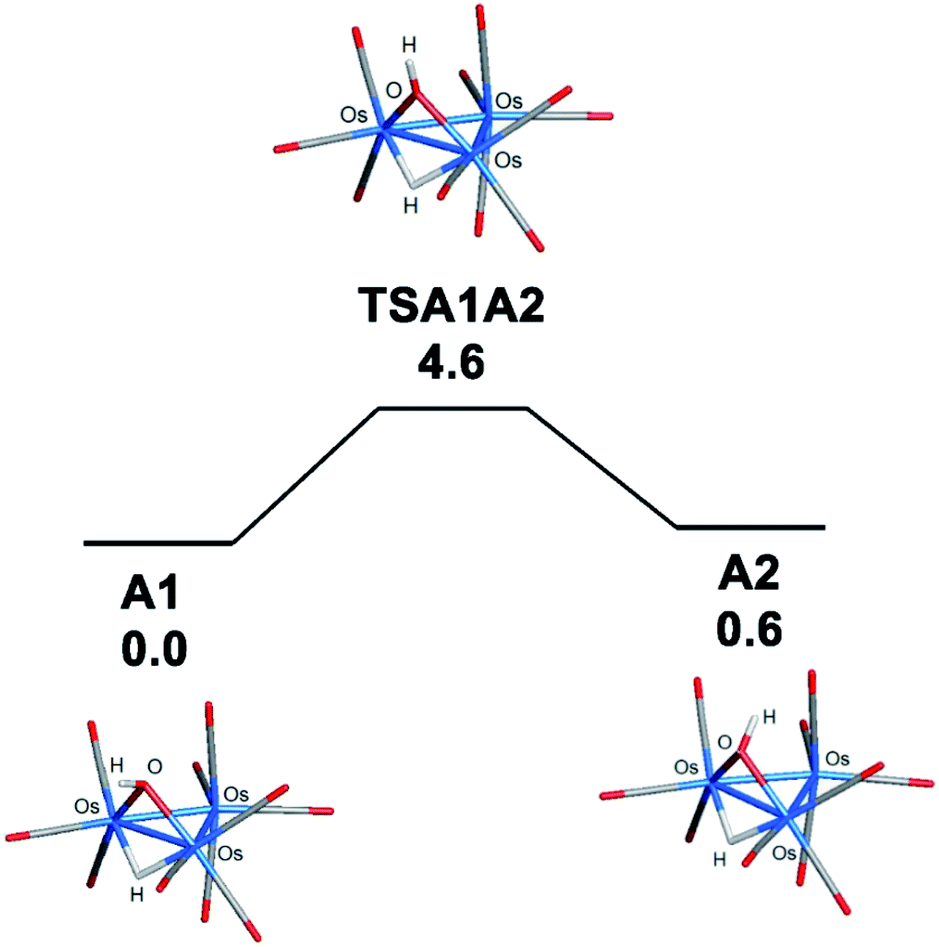 | ||
| Fig. 1 DFT-optimized structures of the trans (A1) and cis (A2) isomers of [Os3(CO)10(μ-OH)(μ-H)] and the free-energy surface for pyramidal inversion via TSA1A2. Energy values are ΔG in kcal mol−1 relative to A1. | ||
3.2. Reaction of [Os3(CO)10(μ-OH)(μ-H)] with dppm
As mentioned earlier, we needed [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)] (1) for external studies and utilized the procedure reported by Johnson and Lewis10 in our synthesis. Thermolysis of [Os3(CO)10(μ-OH)(μ-H)] with a slight excess of dppm in refluxing cyclohexane afforded two products after chromatographic separation. The major product (62% yield) was confirmed as [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)] (1) based on IR and NMR spectroscopy, and X-ray diffraction analysis. The minor product (15% yield) was subsequently established as the isomeric cluster [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)] (2) based on solution spectroscopic data and X-ray crystallography (Scheme 3). The major difference between clusters 1 and 2 resides in the disposition of bridging ligands. In 1, all three bridging ligands share a common Os–Os edge while in 2 the dppm ligand ligates an adjacent Os–Os edge. A total of four stereoisomers are possible when the stereochemistry of the hydroxyl hydrogen (trans/cis forms) is taken into account.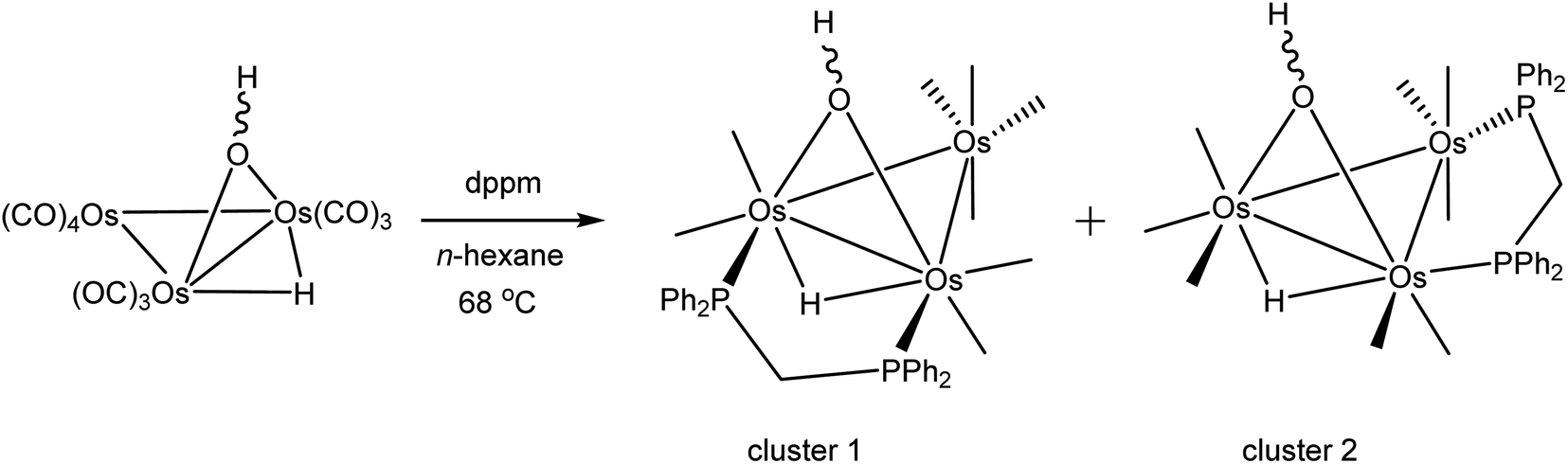 | ||
| Scheme 3 Reaction of [Os3(CO)10(μ-OH)(μ-H)] with dppm. | ||
Control experiments confirmed that 1 slowly transforms into 2 in cyclohexane at 81 °C. This transformation likely proceeds through a dissociative release of one of the coordinated phosphine moieties of the dppm ligand (κ2 → κ1 process). Ligand rearrangement, followed by phosphine ring closure, completes the isomerization sequence. Heating 2 in cyclohexane under comparable conditions returned the starting cluster in quantitative yield when the reaction was monitored by TLC. Both isomers undergo oxidative-addition of the O–H bond with concomitant elimination of H2 to produce the oxo-capped [Os3(CO)7(μ3-CO)(μ3-O)(μ-dppm)] (3) in refluxing xylene (vide infra). The stability of cluster 3 at 139 °C is limited, and the product yield in refluxing xylene has not been maximized. We have also observed that 1 furnishes 2 and 3 in 13 and 43% yield, respectively, when refluxed in xylene, and 2 furnishes 1 and 3 in 13% and 27% yield, respectively, under comparable conditions. All reactions are accompanied by considerable material loss (Scheme 4). Cluster 1 was produced as the sole product when a mixture of [Os3(CO)10(μ-OH)(μ-H)] and dppm in CH2Cl2 was treated with a methanolic solution of Me3NO. Finally, we also examined the reaction between [Os3(CO)10(μ-dppm)] and H2O under different conditions in an effort to synthesize clusters 1 and 2 but did not observe either of these products, paralleling the earlier results reported by Johnson and Lewis.10
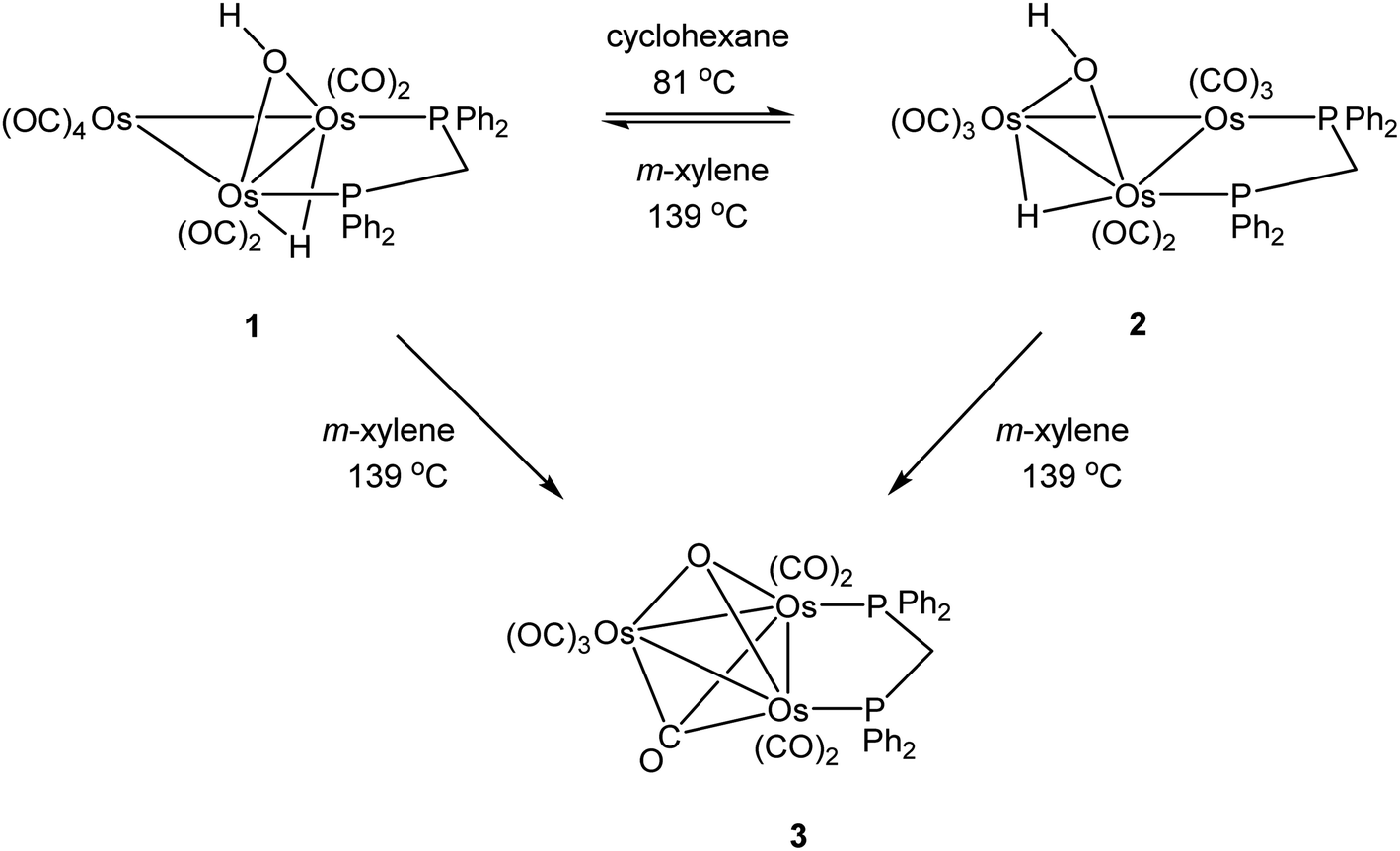 | ||
| Scheme 4 Interconversion of 1 and 2 and their transformation paths to 3. | ||
Initially, cluster 1 was structurally characterized by Johnson and Lewis, and the solid-state was consistent with the reported spectroscopic data.10 Although the dataset for the diffraction data was quite good, the location of hydride and hydroxyl hydrogen could not be established. The locus of the former group was assigned based on the distribution of CO ligands about the cluster polyhedron. Since we were able to grow single crystals of 1, we collected a new data set at low temperature, and the results are shown in Fig. 2. Selected bond distances and angles for 1 are given in the figure caption. We obtained the same polymorph with metric parameters very similar to those reported earlier by Johnson and Lewis and were able to find the positions for the hydride and hydroxyl hydrogen atom from the difference maps. The hydroxyl hydrogen is oriented over the metallic plane and exhibits an Os(3)–O(9)–H(9) angle of 79.03(3)° in keeping with the cis stereochemistry displayed by the hydroxyl group in A2.
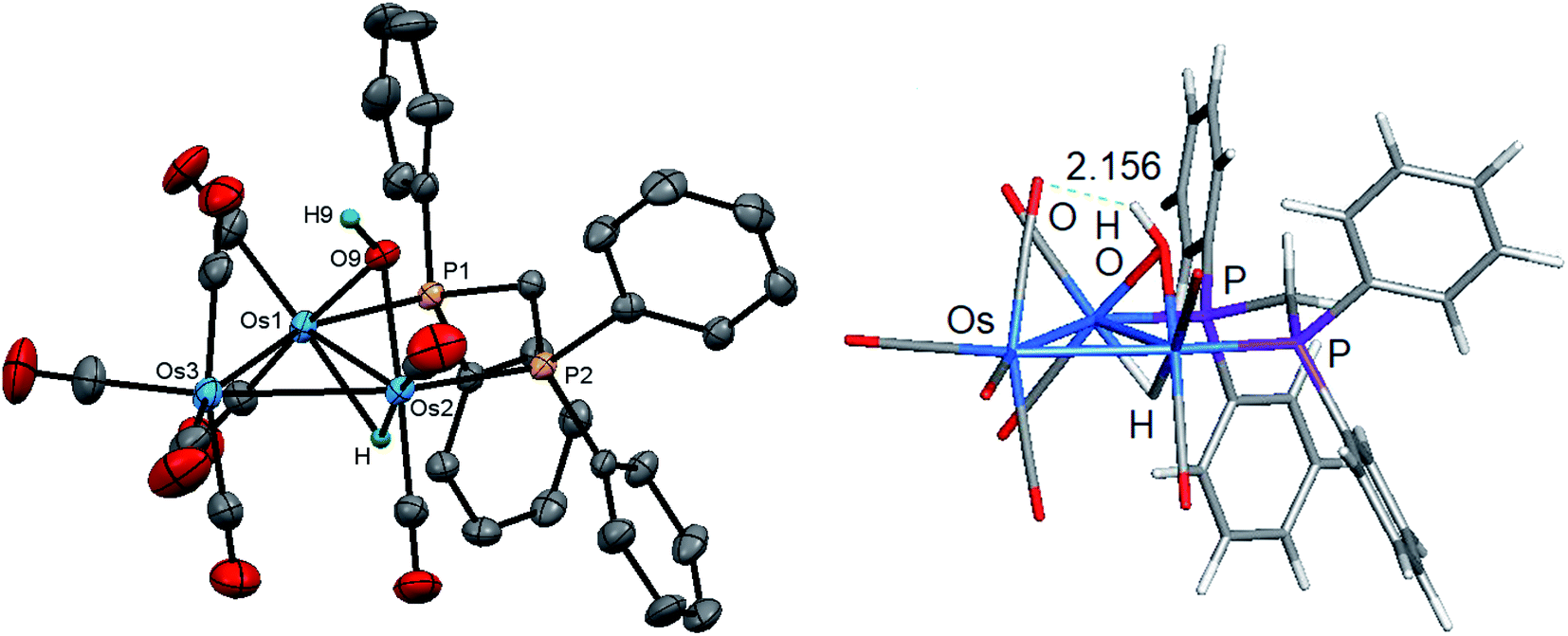 | ||
| Fig. 2 Molecular structure of [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)] (1, left) and DFT-optimized structure B1 (right). The atomic displacement ellipsoids are shown at the 50% probability and the hydrogen atoms (except for the hydride and hydroxyl proton) are omitted for clarity. Selected bond lengths (Å) and bond angles (°) for the experimental structure: Os(1)–Os(2) 2.77027(18), Os(1)–Os(3) 2.82141(18), Os(2)–Os(3) 2.85356(18), Os(1)–P(1) 2.3350(7), Os(2)–P(2) 2.3589(8), Os(1)–O(9) 2.149(2), Os(2)–O(9) 2.144(2), P(1)–Os(1)–Os(2) 94.627(19), P(1)–Os(1)–Os(3) 155.817(19), P(1)–Os(1)–O(9) 80.38(6), O(9)–Os(1)–Os(2) 49.73(6), O(9)–Os(1)–Os(3) 85.24(6), Os(1)–O(9)–Os(2) 80.37(8), Os(3)–O(9)–H(9) 79.03(3). | ||
The optimized structure of B1 is shown alongside the solid-state structure. Excellent agreement between the two structures exists. The adopted cis disposition of the H(9)–O(9) vector gives rise to an attractive 3c,4e interaction with the O(5) atom of the axial C(5)O(5) group with the internuclear O(9)–H(9)⋯O(5) distance of 2.18(5) Å well within the van der Waals radii for the oxygen and hydrogens atoms involved in the formation of this hydrogen bond.38 The computed hydrogen bond distance for this interaction is 2.156 Å, and the magnitude closely matches the distance in the experimental structure. The natural charges (Q) and the Wiberg bond indices in B1 were evaluated, and the three osmium atoms are all negatively charged. The unique Os(CO)4 site exhibits a Q value of −1.44 and the bridged osmium atoms exhibit a mean charge of −1.06. The Q value associated with the oxygen and hydride bridging groups is −0.81 and 0.08, respectively. The hydroxyl hydrogen atom is considerably more electrophilic than the bridging hydride whose charge is 0.53. The two phosphorus atoms exhibit a mean Q of 0.75. The charge on the carbon and oxygen atoms of the CO ligands are uniform in magnitude with mean Q values of 0.76 (C) and −0.47 (O). The two unbridged Os–Os bonds display a mean Wiberg bond index (WBI) of 0.47 and the ligand-bridged Os–Os vector exhibits a WBI of 0.29. The former WBI is consistent with the existence of an Os–Os single bond while the latter WBI is best interpreted within a 4c,4e framework involving bond-pair donation to the osmium centers from the bridging hydroxyl and hydride groups.39,40
Coordination of the dppm ligand in 1 leads to steric crowding about the triply bridged Os–Os vector, and this accounts for B1 as the thermodynamically favored isomer where such contacts are minimized. The assumption concerning the steric environment was corroborated by DFT calculations on several different isomeric forms based on 1. The energy ordering for the isomers explored is shown in Fig. 3 (top portion), where the trans isomer B2 lies 3.2 kcal mol−1 higher in energy than B1. The lower portion of the figure illustrates the destabilizing interactions in B2 involving unfavorable van der Waals contacts between the hydrogen on the OH group and an ortho hydrogen on one of the aryl groups (2.141 Å) and the axial hydrogen of the methylene spacer (2.039 Å). The ortho hydrogen associated with the aryl group on the adjacent phosphorus atom exhibits an internuclear OH⋯H distance of 2.557 Å that is well outside the accepted range for repulsion. Isomer B3 is the product of a “cyclopentane-like” ring flip involving the methylene spacer. While the initial OH⋯Hax interaction involving the methylene spacer has been eliminated, new conformational crowding between the aryls groups and the methylene spacer help to destabilize B3. Species B4, a stereoisomer of B3, contains a cis OH group, and it suffers from similar aryl congestion as in B3. Finally, coordination of the dppm ligand through available axial sites adjacent to the bridging hydride gives B5, which is the least stable of the computed isomers, lying nearly 30 kcal mol−1 above B1.
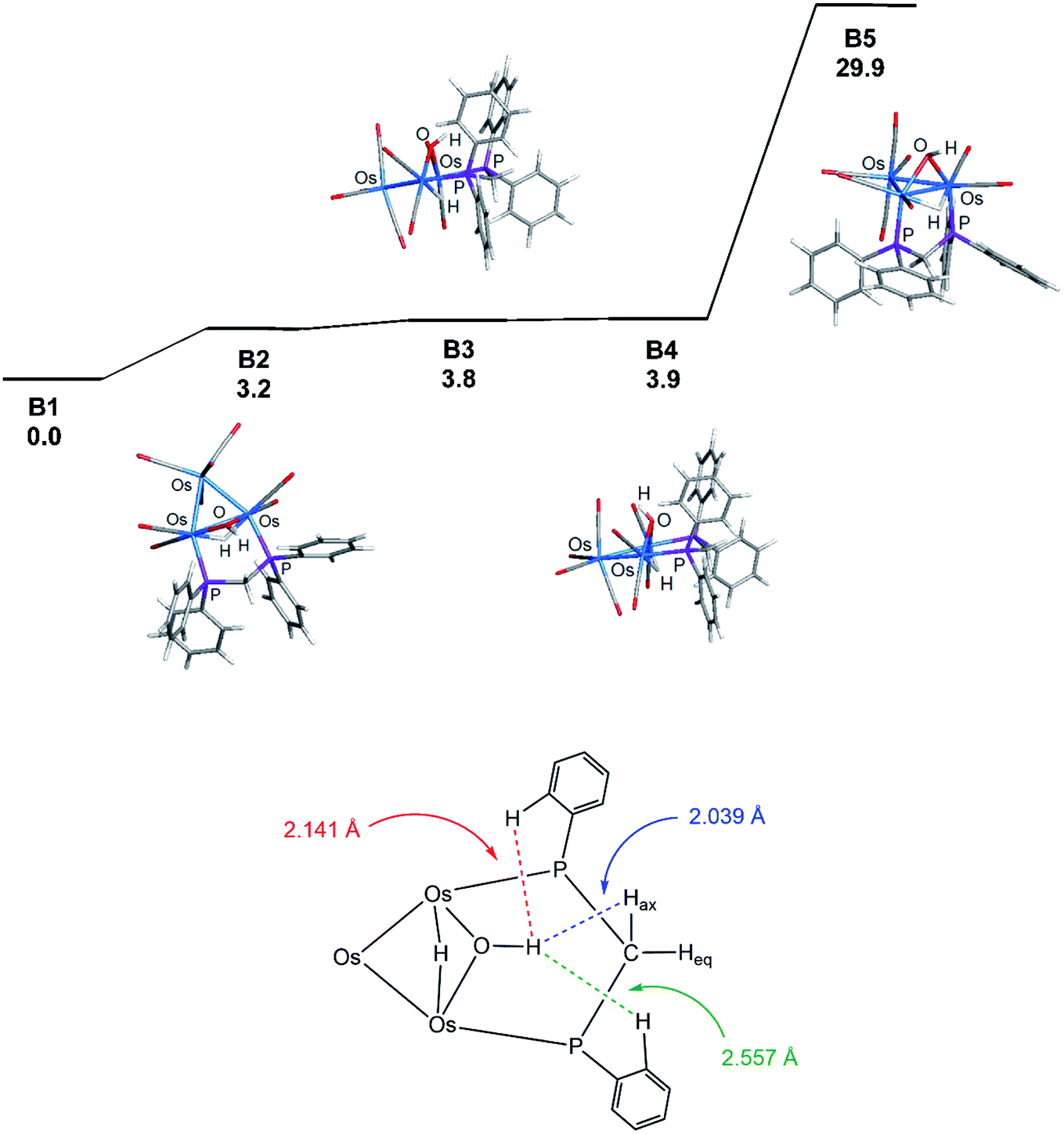 | ||
| Fig. 3 Optimization and energy ordering for the isomeric clusters based on 1 (top) and important van der Waals contacts in B2 involving the hydroxyl hydrogen and the dppm ligand (bottom). The energy difference (ΔG) on the upper potential energy surface is relative to B1 in kcal mol−1 and is not drawn to scale. The ancillary CO ligands and distal phenyl groups in the bottom representation of B2 have been omitted for clarity. | ||
The molecular structure of 2 is shown in Fig. 4 with the caption containing selected bond distances and angles. The molecule contains a triosmium core ligated by eight carbonyls, a bridging dppm ligand, a bridging hydroxyl ligand, and an edge bridging hydride. The hydride was located structurally, but we could not locate the hydroxyl hydrogen from structural analysis. The difference between isomeric 1 and 2 lies in the arrangement of the bridging ligands around Os–Os edges. In 1, all three bridging ligands span the same Os–Os edge, whereas in 2 the hydroxyl and hydride ligands function as edge-bridging groups at the Os(2)–Os(3) vector while the dppm ligand bridges the adjacent Os(1)–Os(2) vector. The P(2) atom coordinated at Os(2) occupies one of the two coordination sites that reside outside the plane defined by the three osmium atoms. Here we define these two sites relative to the bridging hydroxyl group with the P(2) atom and the C(4)O(4) ligand residing at the proximal and distal sites, respectively. The metric parameters for the Os–Os, Os–P, and Os–O bond distances are very similar to those observed in 1. The spectroscopic data indicate that the solid-state structure persists in solution. The 1H NMR spectrum displays an upfield doublet at δ −11.12 (J 35.6 Hz) for the bridging hydride consistent with coupling to only one phosphorus atom of the dppm ligand. The singlet at δ 0.12, which disappears upon the addition of D2O, is assigned to the hydroxyl proton. The 1H NMR spectrum also shows two multiplets at δ 6.13 and 4.38, each integrating to one proton, for the methylene moiety, and a series of aryl multiplets from δ 7.69–7.18 associated with the dppm ligand. The 31P{1H} NMR spectrum shows two equal intensity doublets at δ −19.2 and −22.8 (J 44 Hz) in accord with the solid-state structure.
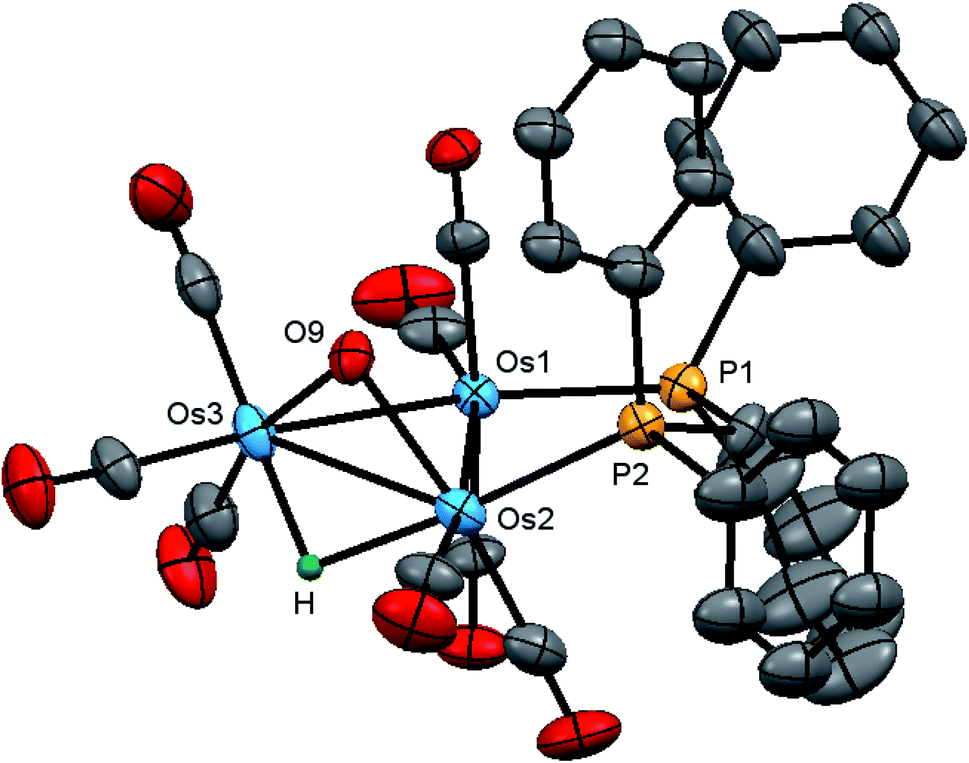 | ||
| Fig. 4 Molecular structure of [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)] (2) showing 50% probability atomic displacement ellipsoids. Hydrogen atoms (except the hydride) are omitted for clarity. Selected bond lengths (Å) and bond angles (°): Os(1)–Os(2) 2.830(2), Os(1)–Os(3) 2.8079(17), Os(2)–Os(3) 2.7896(17), Os(1)–P(1) 2.326(3), Os(2)–P(2) 2.305(3), Os(2)–O(9) 2.147(6), Os(3)–O(9) 2.144(6), P(1)–Os(1)–Os(2) 94.93(6), P(1)–Os(1)–Os(3) 153.52(6), P(2)–Os(2)–Os(1) 86.69(6), P(2)–Os(2)–Os(3) 134.27(7), P(2)–Os(2)–O(9) 97.54(18), O(9)–Os(2)–Os(3) 49.41(16), O(9)–Os(2)–Os(1) 79.42(15), Os(2)–O(9)–Os(3) 81.1(2). | ||
The stereochemistry of the hydroxyl hydrogen in 2 was addressed through electronic structure calculations. Fig. 5 shows the optimized structures of the trans (left) and cis (right) stereoisomers C2 and C1, respectively. The energy difference (ΔG) between the two species is only 0.3 kcal mol−1 and favors C1, whose hydroxyl hydrogen is located over the metallic plane. Given the low barrier computed for the pyramidalization of the hydroxyl group in [Os3(CO)10(μ-OH)(μ-H)], both C1 and C2 likely exist in solution as a rapidly equilibrating mixture of stereoisomers. The solid-state structure of 2 corresponds to C2 based on the disposition of the hydroxyl oxygen atom relative to the internal anchor points Os(1) and C(3). The Os(1)–O(9) [3.223(7) Å] and C(3)–O(9) [2.67(1) Å] distances in 2 closely match the bond lengths of 2.672 Å and 3.216 Å for the same pair of bonds in C2. In contrast, the same bond distances in C1 are substantially longer at 3.448 Å and 2.895 Å for the corresponding bonds. Relative to B1, C2 is thermodynamically more stable by 1.7 kcal mol−1.
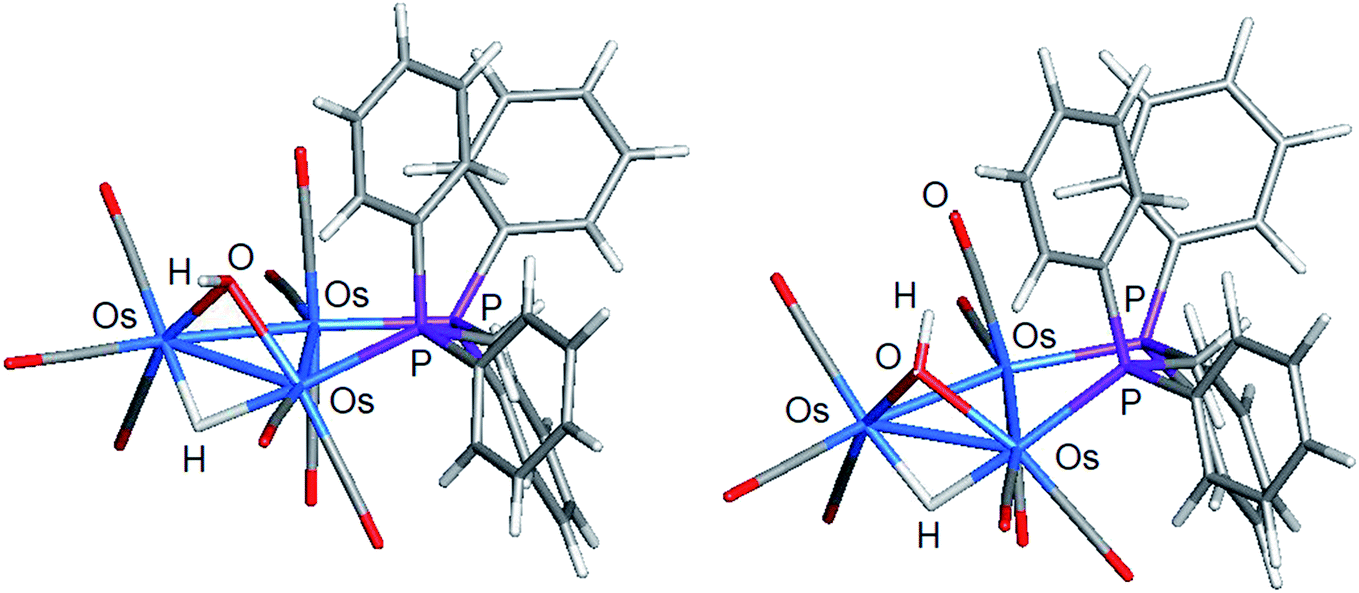 | ||
| Fig. 5 DFT-optimized structures of C2 (left) and C1 (right) based on the minor product isolated from the reaction of [Os3(CO)10(μ-OH)(μ-H)] and dppm. | ||
Switching the P(2) and C(4)O(4) ligands in 2 would furnish a new isomer where the P(2) occupies the distal site relative to the bridging hydroxyl ligand. Depending on the orientation of the hydroxyl hydrogen in this new isomer, both cis and trans forms are expected, as depicted in Scheme 5. The pair of stereoisomers C3 (trans) and C4 (cis) was evaluated by DFT (not shown) and found to lie 7.6 and 7.9 kcal mol−1 above C2, confirming the thermodynamic site preference (C1/C2 versus C3/C4) for the disposition of the bridging dppm ligand in cluster 2.
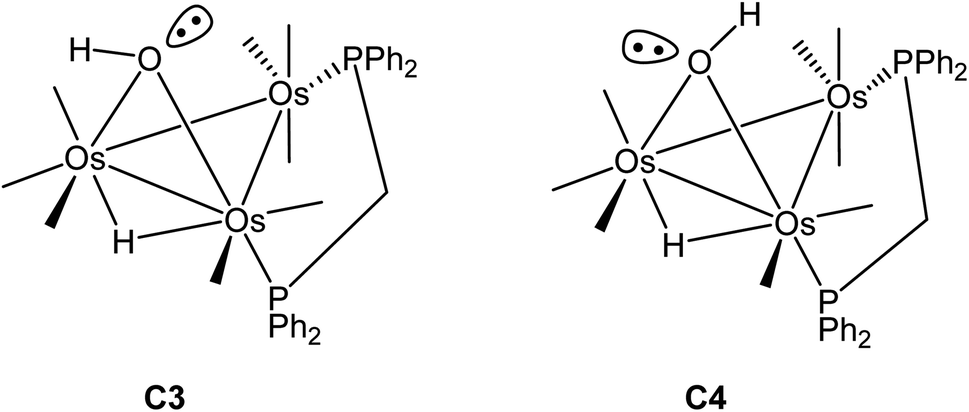 | ||
| Scheme 5 Alternative isomers for cluster 2. | ||
3.3. Thermolysis reactions of [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)]
Refluxing samples of either 1 or 2 in xylene afforded the oxo-capped cluster 3, which was characterized in solution by traditional spectroscopic methods, and its molecular structure established by single-crystal X-ray diffraction analysis. Fig. 6 shows the molecular structure of 3 with selected bond distances and angles reported in the caption. The molecule possesses a non-crystallographic mirror plane of symmetry that contains both capping ligands, the methylene carbon and its protons, the Os(3) atom, and the apical C(6)O(6) ligand. The molecule consists of an approximately equilateral triangle of osmium atoms whose Os–Os distances range between 2.7472(8)–2.7754(11) Å and exhibits a mean Os–Os distance of 2.7603 Å. The μ3-oxo and a triply bridging carbonyl ligand cap opposite polyhedral faces of the Os3 triangle. The Os–O(9) and Os–C(8) bond distances associated with the two face-capping ligands are non-symmetrical. The mean distance of the Os–C(8) vectors [2.145 Å] at the dppm-substituted osmium sites [Os(1)–P(1) and Os(2)–P(2)] is 0.086 Å shorter than the Os(3)–C(8) bond [2.231(4) Å] at the Os(CO)3 moiety. This trend is reversed for the Os–O(oxo) bonds where the mean Os–O(9) distance of 2.088 Å is found for the Os–O(9) bonds involving the Os(1) and Os(2) sites, which are ca. 0.02 Å longer than the Os(3)–O(9) vector [2.069 Å]. These trends reflect the donor properties of the dppm ligand and the pi-accepting properties of the μ3-O and μ3-CO ligands. The Os–O and Os–C distances involving these capping ligands are within the range reported for related complexes.41–43 The DFT-optimized structure of D is shown alongside the experimental structure, and good agreement between the two structures is noted. The metal triangle is also coordinated by seven terminal carbonyls and a dppm ligand that bridges the Os(1) and Os(2) atoms. The 31P{1H} NMR spectrum shows two equal intensity doublets centered at δ 16.7 and −4.1 (J 58 Hz) for the dppm ligand that argue against a molecule with idealized Cs symmetry in solution. In addition to ν(CO) bands due to the terminal carbonyls, the IR spectrum also shows a weak absorption at 1643 cm−1 consistent with the presence of a μ3-CO ligand.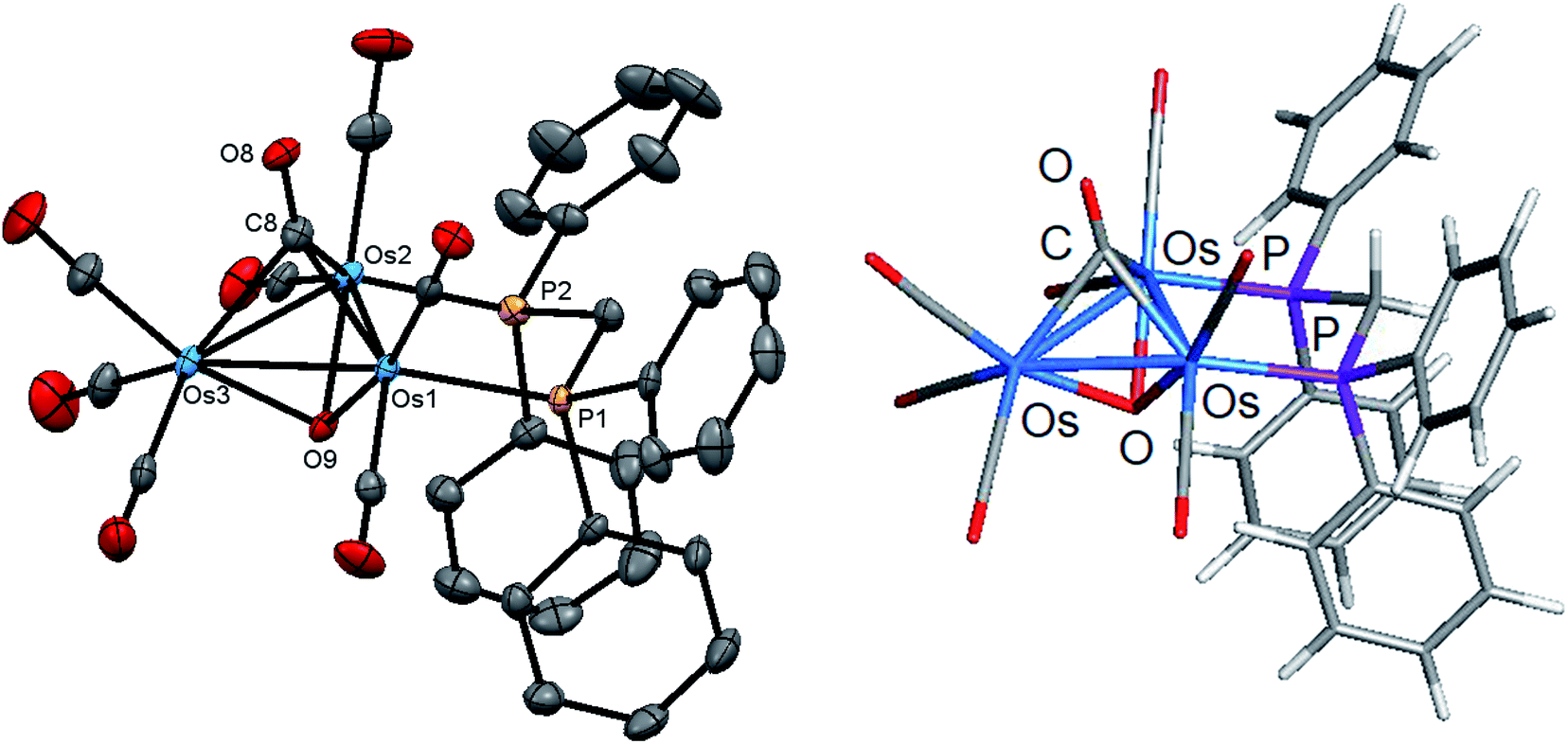 | ||
| Fig. 6 Molecular structure of [Os3(CO)7(μ3-CO)(μ3-O)(μ-dppm)] (3, left) and DFT-optimized structure D (right). The atomic displacement ellipsoids are shown at the 50% probability and the hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond angles (°) for the experimental structure: Os(1)–Os(2) 2.7754(11), Os(1)–Os(3) 2.7583(11), Os(2)–Os(3) 2.7472(8), Os(1)–P(1) 2.3301(13), Os(2)–P(2) 2.3233(13), Os(1)–O(9) 2.087(3),Os(2)–O(9) 2.088(3), Os(3)–O(9) 2.069(3), Os(1)–C(8) 2.143(4), Os(2)–C(8) 2.147(4), Os(3)–C(8) 2.231(4), P(1)–Os(1)–Os(2) 92.72(3), P(1)–Os(1)–Os(3) 144.24(3), P(1)–Os(1)–O(9) 97.03(8), O(9)–Os(1)–Os(2) 48.34(7), Os(1)–O(9)–Os(2) 83.33(10), C(8)–Os(1)–Os(2) 49.76(10), Os(1)–C(8)–Os(2) 80.63(14). | ||
3.4. Reactions of [Os3(CO)10(μ-dppm)] with ROH: formation of [Os3(CO)8(μ-OH)(μ,η1,κ1-OCOR)(μ-dppm)] using Me3NO
Related to the current study is our earlier work on the reactivity of methoxide-bridged cluster [Os3(CO)10(μ-OMe)(μ-H)] with different diphosphine ligands.44,45 Thermolysis of dppm with [Os3(CO)10(μ-OMe)(μ-H)] afforded the known cluster [Os3(CO)10(μ-dppm)] along with [Os3(CO)8(μ-OMe)(μ-H)(μ-dppm)] as the exclusive products. The latter product is the methoxy derivative of 1, and its synthesis employing [Os3(CO)10(μ-dppm)] as a starting material instead of [Os3(CO)10(μ-OMe)(μ-H)] was explored under different reaction conditions.Thermolysis of [Os3(CO)10(μ-dppm)] and ROH (R = Me, Et) in refluxing toluene did not afford alcohol-based products but rather the known cyclometalated cluster [HOs3(CO)8{μ3-Ph2PCH2P(μ-C6H4)Ph}]46 and unreacted starting cluster [Os3(CO)10(μ-dppm)]. Repeating the reaction in refluxing benzene in the presence of Me3NO furnished the new clusters [Os3(CO)8(μ-OH)(μ,η1,κ1-OCOR)(μ-dppm)] (4, R = Me; 5, R = Et) in low yields, after the usual workup and chromatographic separation (Scheme 6).
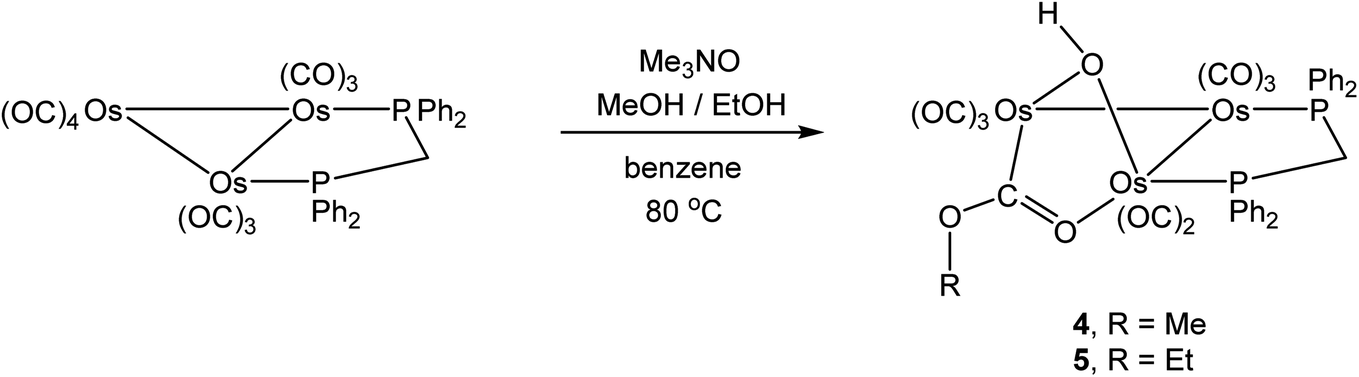 | ||
| Scheme 6 Synthesis of hydroxyl-bridged clusters [Os3(CO)8(μ-OH)(μ,η1,κ1-OCOR)(μ-dppm)]. | ||
Both 4 and 5 require a nucleophilic attack of an alcoholic oxygen atom at one of the carbonyl carbons at the remote osmium Os(CO)4 site in [Os3(CO)10(μ-dppm)]. While the source of the hydroxyl group is unclear at this time, the equilibrium depicted in Scheme 7 provides a potential source of the alkoxide anion that is needed for the reduction of the CO ligand. An equilibrium mixture produced by proton transfer from the alcohol to Me3NO would yield hydroxytrimethylammonium and methoxide. The alkoxide generated in this equilibrium can directly attack a carbon atom of a CO ligand in [Os3(CO)10(μ-dppm)] to give an anionic metalloester [Os3(CO)9(κ1-CO2R)(μ-dppm)]−, which could then capture the hydroxyl group in hydroxytrimethylammonium cation to furnish the observed product [Os3(CO)8(μ-OH)(μ,η1,κ1-OCOR)(μ-dppm)]. While the direct attack of the anionic metalloester [Os3(CO)9(κ1-CO2R)(μ-dppm)]− on the tetrahedral nitrogen center in [Me3NOH]+ cation is unlikely, a multistep sequence involving an electron-transfer process cannot be ruled out at this time.
 | ||
| Scheme 7 Potential source of alkoxide anion from Me3NO and alcohol. | ||
The new clusters 4 and 5 have been characterized by a combination of analytical and spectroscopic methods and the solid-state structure of each established by single-crystal X-ray diffraction analysis. The molecular structures of 4 and 5 are depicted in Fig. 7 and 8 and the captions contain selected bond distances and angles. Both molecules contain an opened cluster of three osmium atoms with the opened edge simultaneously bridged by the μ-OH and a μ-OCOR ligands. The bonding Os–Os distances are comparable in magnitude [Os(1)–Os(2) 2.8772(3) and Os(1)–Os(3) 2.8762(3) Å in 4; Os(1)–Os(2) 2.8683(3) and Os(1)–Os(3) 2.8716(3) Å in 5]. The dppm ligand acts in a bridging capacity and spans one of the Os–Os edges. The non-bonding distance between the Os(2) and Os(3) atoms [3.3787(4) Å in 4 and 3.3700(4) Å in 5] precludes any significant bonding between these metallic centers. The μ-OH ligand “symmetrically” bridges this expanded edge based on bond distances of 2.121(4) Å [Os(2)–O(9)] and 2.115(4)Å [Os(3)–O(9)] in 4, and 2.113(3) Å [Os(2)–O(9)] and 2.107(3) Å [Os(3)–O(9)] in 5; these Os–O bond lengths are similar to those distances found in 2 and 3. While we could not locate the hydroxyl hydrogen in 4, it was located crystallographically for 5, oriented over the bridging ester moiety and away from the plane defined by the three osmium atoms. The μ-OCOR ligand bridges the same Os atoms as the μ-OH ligand, and here the Os–C and Os–O distances involving this bridging ester ligand [Os(3)–C(9) 2.083(6) and Os(2)–O(10) 2.145(4) Å in 4; Os(3)–C(9) 2.073(4) and Os(2)–O(10) 2.148(2) Å in 5] are unremarkable in comparison to those bond distances reported in related clusters.47,48
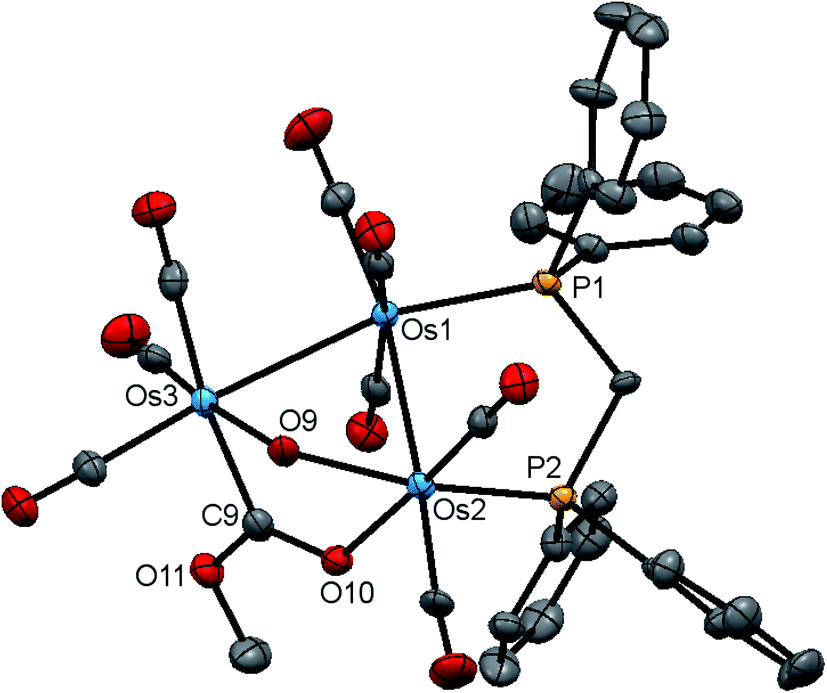 | ||
| Fig. 7 Molecular structure of [Os3(CO)8(μ-OH)(μ,η1,κ1-OCOMe)(μ-dppm)] (4) showing 50% probability atomic displacement ellipsoids. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond angles (°): Os(1)–Os(2) 2.8772(3), Os(1)–Os(3) 2.8762(3), Os(1)–P(1) 2.3201(14), Os(2)–P(2) 2.2957(14), Os(2)–O(9) 2.121(4), Os(3)–O(9) 2.115(4), Os(2)–O(10) 2.145(4), Os(3)–C(9) 2.083(6), Os(2)–Os(1)–Os(3) 71.926(9), Os(2)–O(9)–Os(3) 105.84(18), P(1)–Os(1)–Os(2) 92.23(4), P(1)–Os(1)–Os(3) 164.03(4), O(9)–Os(2)–Os(1) 80.54(11), O(10)–Os(2)–O(9) 81.73(16), O(10)–Os(2)–Os(1) 87.94(11), C(9)–Os(3)–O(9) 82.2(2), C(9)–Os(3)–Os(1) 87.24(16). | ||
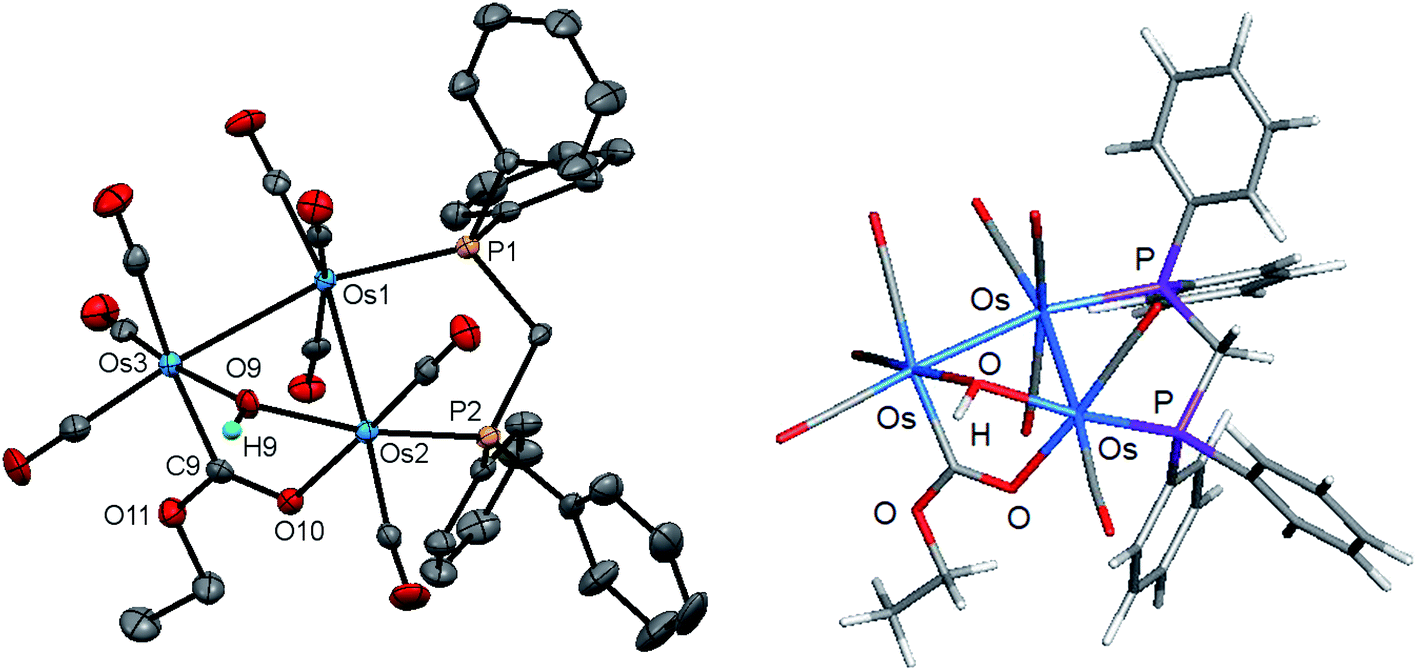 | ||
| Fig. 8 Molecular structure of [Os3(CO)8(μ-OH)(μ,η1,κ1-OCOEt)(μ-dppm)] (5) and DFT-optimized structure E (right). The atomic displacement ellipsoids are shown at the 50% probability and the hydrogen atoms (except for the hydroxyl proton) are omitted for clarity. Selected bond lengths (Å) and bond angles (°) for the experimental structure: Os(1)–Os(2) 2.8683(3), Os(1)–Os(3) 2.8716(3), Os(1)–P(1) 2.3204(9), Os(2)–P(2) 2.2993(9), Os(2)–O(9) 2.113(3), Os(3)–O(9) 2.107(3), Os(2)–O(10) 2.148(2), Os(3)–C(9) 2.073(4), Os(2)–Os(1)–Os(3) 71.906(6), Os(2)–O(9)–Os(3) 105.98(12), P(1)–Os(1)–Os(2) 90.82(2), P(1)–Os(1)–Os(3) 162.73(2), O(9)–Os(2)–Os(1) 79.63(8), O(10)–Os(2)–O(9) 82.22(10), O(10)–Os(2)–Os(1) 89.00(6), C(9)–Os(3)–O(9) 82.35(12), C(9)–Os(3)–Os(1) 87.53(10). | ||
The DFT-optimized structure of E depicted in Fig. 8 closely mirrors the experimental structure. All three osmium exhibit a negative charge, with Q values ranging from −1.61 [Os(3)] to −0.79 [Os(1)]. A mean charge of 1.42 is computed for the two phosphorus atoms, and the bridging hydroxyl group exhibits a charge of −0.62 for the oxygen atom and 0.38 for the hydrogen atom. The bridging CO2Et moiety displays Q values of −0.60 [O(10)] and −0.56 [O(11)] for the oxygen atoms, and the ester carbon C(9) is electrophilic based on a charge of 0.79. Mean Wiberg bond indices of 0.44 and 0.82 are computed for the Os–Os and Os–P bonds, respectively, consistent with a single-bond designation for each of these vectors. The two Os–OH bonds reveal a mean WBI of 0.48 while the bridging ester moiety exhibits Wiberg indices of 0.76 [Os(3)–C(9)], 0.40 [Os(2)-O(10)], 1.40 [C(9)–O(10)], and 1.09 [C(9)–O(11)] consistent with the nature of this ligand that functions as a 3e donor.
The spectroscopic data for 4 and 5 indicate that both clusters retain their solid-state structure in solution. The 31P{1H} NMR spectrum of each product exhibits two equal intensity doublets [δ 18.1 and −0.9 (JPP 84 Hz) for 4; δ 18.5 and −0.8 (JPP 82 Hz) for 5] due to inequivalent 31P atoms in the bridging dppm ligand. In addition to the recorded aryl protons (20H), whose resonances appear over the range δ 7.70–7.20, the 1H NMR spectrum of both clusters displays two multiplets (1H each) in the aliphatic region [δ 4.97 and 4.08 for 4; δ 4.88 and 4.18 for 5] for the inequivalent methylene protons of the dppm ligand. Both products exhibit a sharp singlet for the hydroxyl hydrogen [δ −0.39 for 4; δ 0.91 for 5] and a methyl singlet at δ 3.64 for 4 and three multiplets centered at δ 4.05, 3.87, and 1.30 with an intensity ratio of 1:1:3 for the ethyl protons of μ-OCOEt ligand for 5. The appearance of two downfield resonances for the methylene moiety in the μ-OCOEt ligand supports the restricted rotation of the ethyl group, which renders these two hydrogens diastereotopic.
4. Conclusions
In summary, this article describes a convenient synthetic method for the preparation of the hydroxy-bridged triosmium cluster [Os3(CO)10(μ-OH)(μ-H)] along with the re-evaluation of the reaction of this cluster with dppm that has led to the isolation of a new isomer for [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)]. We have shown that [Os3(CO)10(μ-OH)(μ-H)] may be obtained in ca. 75% yield from the hydrolysis of [Os3(CO)10(NCMe)2] at 67 °C; the latter precursor is easily synthesized from commercially available [Os3(CO)12] and Me3NO/MeCN at room temperature in ca. 80% yield.11 Thus, this method furnishes [Os3(CO)10(μ-OH)(μ-H)] in 56% overall yield from [Os3(CO)12]. Reaction between [Os3(CO)10(μ-OH)(μ-H)] and dppm furnished a pair of isomers (1 and 2) with the general formula [Os3(CO)8(μ-OH)(μ-H)(μ-dppm)] that differ by the disposition of the bridging dppm ligand. Control experiments showed that 1 and 2 are inter-convertible, and both isomers may be employed as starting materials for the synthesis of the oxo-capped [Os3(CO)7(μ3-CO)(μ3-O)(μ-dppm)] (3). Treatment of [Os3(CO)10(μ-dppm)] with the alcohols MeOH and EtOH in the presence of added Me3NO gave the unexpected clusters [Os3(CO)8(μ-OH)(μ,η1,κ1-OCOMe)(μ-dppm)] (4) and [Os3(CO)8(μ-OH)(μ,η1,κ1-OCOEt)(μ-dppm)] (5). The generality of this last reaction and the mechanism responsible for the formation of 4 and 5 are under active investigation by our groups.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Ministry of Science and Technology, the Government of the People's Republic of Bangladesh (SG and SEK) and the Robert A. Welch Foundation (Grant B-1093-MGR) is acknowledged. The DFT calculations were performed at UNT through CASCaM, which is an NSF-supported facility (CHE-1531468).References
- B. F. G. Johnson, J. Lewis and P. A. Kilty, J. Chem. Soc. A, 1968, 2859 Search PubMed.
- A. J. Arce, A. J. Deeming, S. Donovan-Mtunzi and S. E. Kabir, J. Chem. Soc., Dalton Trans., 1985, 2479 Search PubMed.
- J. Banford, M. J. Mays and P. A. Raithby, J. Chem. Soc., Dalton Trans., 1985, 1355 Search PubMed.
- E. G. Bryan, B. F. G. Johnson and J. Lewis, J. Chem. Soc., Dalton Trans., 1977, 1328 Search PubMed.
- D. Roberto, E. Lucenti, C. Roveda and R. Ugo, Organometallics, 1997, 16, 5974 Search PubMed.
- C. Dossi, A. Fusi, M. Pizzotti and R. Psaro, Organometallics, 1990, 9, 1994 Search PubMed.
- C. E. Anson, E. J. Ditzel, M. Fajardo, H. D. Holden, B. F. G. Johnson, J. Lewis, J. Puga and P. R. Raithby, J. Chem. Soc., Dalton Trans., 1984, 2723 Search PubMed.
- E. D. Kamiets, V. A. Maksakov, L. K. Kedrova, N. I. Shakot’ko and S. P. Gupin, Izv. Akad. Nauk. SSR, Ser. Khim., 1983, 2, 435 Search PubMed.
- M. W. Lum and W. K. Leong, J. Organomet. Chem., 2003, 687, 203 Search PubMed.
- S. R. Hodge, B. F. G. Johnson, J. Lewis and P. R. Raithby, J. Chem. Soc., Dalton Trans., 1987, 931 Search PubMed.
- B. F. G. Johnson, J. Lewis and D. A. Pippard, J. Chem. Soc., Dalton Trans., 1981, 407 Search PubMed.
- J. J. B. Lidasan, J. A. D. Del Rosario and J. D. Ocon, Catalysts, 2020, 10, 740 Search PubMed.
- G. L. Elizarova, L. G. Matvienko, A. O. Kuzmin, E. R. Savinova and V. N. Parmon, Mendeleev Comm, 2001, 11, 15 Search PubMed.
-
(a) B. Besson, A. Choplin, L. D'Ornelas and J. M. Basset, Chem. Comm., 1982, 843 Search PubMed;
(b) A. Choplin, B. Besson, L. D'Ornelas, R. Sanchez-Delgado and J. M. Basset, J. Am. Chem. Soc., 1988, 110, 2783 Search PubMed;
(c) For an example of hydrogen-transfer reduction using [Os3(CO)10(μ-H)(μ-OSi)], see: J. Kaspar, A. Trovarelli, M. Graziani, C. Dossi, A. Fusi, R. Psaro, R. Ugo, R. Ganzeria and M. Lenarda, J. Mol. Catal., 1989, 51, 181 Search PubMed.
- S. E. Kabir and G. Hogarth, Coord. Chem. Rev., 2009, 253, 1285 Search PubMed.
- H. Begum and S. E. Kabir, J. Bangl. Acad. Sci., 1992, 16, 169 Search PubMed.
- Bruker, SAINT (8.37A), Bruker AXS Inc., Madison, Wisconsin, USA, 2015 Search PubMed.
- Bruker, SADABS-2014/5, Bruker AXS Inc., Madison, Wisconsin, USA, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr. C, 2015, 71, 3 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, 2016, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 Search PubMed.
- (a) G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193 Search PubMed; (b) G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081 Search PubMed.
- (a) NBO Version 3.1,E. D. Glendening, A. E. Reed, J. E. Carpenter, and F. Weinhold Search PubMed; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 Search PubMed.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083 Search PubMed.
- (a) JIMP2, version 0.091, a free program for the visualization and manipulation of molecules: M. B. Hall and R. F. Fenske, Inorg. Chem., 1972, 11, 768 Search PubMed; (b) J. Manson, C. E. Webster, M. B. Hall, JIMP2 molecular visualization and manipulation program, Texas A&M University, College Station, TX, 2006, http://www.chem.tamu.edu/jimp2/index.html Search PubMed.
- E. J. Ditzel, M. Gómez-Sal, B. F. G. Johnson, J. Lewis and P. R. Raithby, J. Chem. Soc., Dalton Trans., 1987, 1623 Search PubMed.
- C. Dragonetti, E. Lucenti, D. Roberto, W. K. Leong and Q. Lin, Inorg. Synth., 2004, 34, 215 Search PubMed.
- D. W. Knoeppel, J. H. Chung and S. G. Shore, Acta Crystallogr. C, 1995, 51, 42 Search PubMed.
- M. G. Karpov, S. P. Tunik, V. R. Denisov, G. L. Starova, A. B. Nikol’skii, F. M. Dolgushin, A. I. Yanovsky and Y. T. Struchkov, J. Organomet. Chem., 1995, 485, 219 Search PubMed.
- W. G.-Y. Ho and W.-T. Wong, Polyhedron, 1995, 14, 2855 Search PubMed.
- Q. Lin and W. K. Leong, Organometallics, 2003, 22, 3639 Search PubMed.
- D. Braga, F. Grepioni and G. R. Desiraju, Chem. Rev., 1998, 98, 1375 Search PubMed.
- M. R. Churchill and H. J. Wasserman, Inorg. Chem., 1980, 19, 2391 Search PubMed.
- S. Aime, M. Cisero, R. Gobetto, D. Osella and A. J. Arce, Inorg. Chem., 1991, 30, 1614 Search PubMed.
- The atomic radius for the hydrogen (1.20 Å) and oxygen (1.52 Å) atoms were taken from the available periodic table of the elements on the Los Alamos National Laboratory (LANL) site: http://periodic.lanl.gov/1.shtml.
- J. F. van der Maelen, S. García-Granda and J. A. Cabeza, Comp. Theor. Chem., 2011, 968, 55 Search PubMed.
- G. Parkin, Struct. Bond., 2010, 136, 113 Search PubMed.
- J. S.-Y. Wong and W.-T. Wong, New J. Chem., 2002, 26, 94 Search PubMed.
- (a) K. A. Azam, G. M. G. Hossain, S. E. Kabir, K. M. A. Malik, M. A. Mottalib, S. Perven and N. C. Sarker, Polyhedron, 2002, 21, 381 Search PubMed; (b) H. Akter, A. J. Deeming, G. M. G. Hossain, S. E. Kabir, D. N. Mondol, E. Nordlander, A. Sharmin and D. A. Tocher, J. Organomet. Chem., 2005, 690, 4628 Search PubMed.
- R. J. Goudsmit, B. F. G. Johnson, J. Lewis, P. R. Raithby and K. H. Whitmire, Chem. Comm., 1983, 246 Search PubMed.
- S. E. Kabir, M. A. Mottalib, G. M. G. Hossaon, E. Nordlander and E. Rosenberg, Polyhedron, 2006, 25, 95 Search PubMed.
- For a report on the reactivity of [Os3(CO)10(μ-OR)(μ-H)] with monodentate phosphine ligands, see: E. J. Ditzel, M. P. Gómez-Sal, B. F. G. Johnson, J. Lewis and P. R. Raithby, J. Chem. Soc., Dalton Trans., 1987, 1623 Search PubMed.
- J. A. Clucas, D. F. Foster, M. M. Harding and A. K. Smith, J. Chem. Soc., Chem. Comm., 1984, 949 Search PubMed.
- N. C. Bhoumik, T. K. Saha, S. Ghosh, V. N. Nesterov, M. G. Richmond and S. E. Kabir, J. Organomet. Chem., 2020, 911, 121133 Search PubMed.
- (a) Y. Chi, J.-W. Lan, S.-M. Peng and G.-H. Lee, J. Clust. Sci., 2001, 12, 421 Search PubMed; (b) B. F. G. Johnson, J. Lewis, P. R. Raithby and W. T. Wong, J. Organomet. Chem., 1991, 401, C50 Search PubMed; (c) G. R. John, B. F. G. Johnson, J. Lewis and K. C. Wong, J. Organomet. Chem., 1979, 169, C23 Search PubMed; (d) C. R. Eady, B. F. G. Johnson, J. Lewis and M. C. Malatesta, J. Chem. Soc., Dalton Trans., 1978, 1358 Search PubMed.
Footnote |
| † CCDC 2036482 (for 1), 2036483 (for 2), 2036484 (for 3), 2036485 (for 4), and 2036486 (for 5). Atomic coordinates and energies for all DFT-optimized structures are available upon request (MGR). For crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra08783a |
| This journal is © The Royal Society of Chemistry 2020 |