
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMononuclear gold(III) complexes with diazanaphthalenes: the influence of the position of nitrogen atoms in the aromatic rings on the complex crystalline properties†
Biljana Đ. Glišić *a,
Beata Warżajtisb,
Marcin Hoffmannb,
Urszula Rychlewskab and
Miloš I. Djuranc
*a,
Beata Warżajtisb,
Marcin Hoffmannb,
Urszula Rychlewskab and
Miloš I. Djuranc
aUniversity of Kragujevac, Faculty of Science, Department of Chemistry, R. Domanovića 12, 34000 Kragujevac, Serbia. E-mail: biljana.glisic@pmf.kg.ac.rs
bFaculty of Chemistry, Adam Mickiewicz University in Poznań, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland
cSerbian Academy of Sciences and Arts, Knez Mihailova 35, 11000 Belgrade, Serbia
First published on 16th December 2020
Abstract
A series of mononuclear gold(III) complexes of the general formula [AuCl3(diazanaphthalene)], where diazanaphthalene is quinazoline (qz, 1), phthalazine (phtz, 2), 1,5-naphthyridine (1,5-naph, 3), 1,6-naphthyridine (1,6-naph, 4) or 1,8-naphthyridine (1,8-naph, 5), were prepared and fully characterized. The complexes 1–5 consist of discrete monomeric species with the Au(III) cation in a square planar coordination geometry surrounded by three chloride anions and one diazanaphthalene ligand. Crystallographic studies indicate the presence of an extended 4 + 1 or 4 + 2 geometry around the square planar [AuCl3(diazanaphthalene)] center due to Au⋯Cl and Au⋯N interactions. The crystal structures of these complexes are controlled by a variety of intermolecular interactions that utilize the amphiphilic properties of the coordinated chloride anions and involve C–H groups, π-electrons, and an uncoordinated nitrogen atom of the diazanaphthalene ligand. The usual offset π-stacking between the N-heteroaromatic ligands appears to be completely hindered between the 1,5-naph fragments and significantly weakened between the 1,6-naph and 1,8-naph in their respective complexes 3, 4 and 5, for which the average molecular polarizability (α) values are the lowest in the series. It is remarkable that the [AuCl3(benzodiazine)] complexes 1 and 2 form centrosymmetric crystals, but the [AuCl3(naphthyridine)] complexes 3–5 assemble into non-centrosymmetric aggregates, making them potential alternatives to the previously studied systems for application in various fields by taking advantage of their polarity.
Introduction
The observed interest in the coordination chemistry of gold(III) with aromatic nitrogen-containing heterocyclic ligands (N-heterocycles) has several reasons. N-Heterocycles are of great biological significance, since they can be found in many bioactive natural products, as well as medicinally and biologically important compounds.1,2 For example, one can mention a porphyrin ring in haemoglobin, which consists of four pyrrole molecules cyclically linked together, a corrin ring in vitamin B12, and purine and pyrimidine bases which are found in DNA and RNA structures. Besides that, N-heterocycles containing donor–acceptor substitution have been shown to exhibit unique photophysical and photochemical properties owing to the charge transfer. For example, 1,6-naphthyridine derivatives were reported as a new class of compounds for nonlinear optics.3 Due to their structural properties, N-heterocycles provide rigid building blocks for molecular architectures with various transition metals,4–6 so they have been extensively utilized for the formation of numerous mononuclear and polynuclear metal complexes,7–12 metallosupramolecular assemblies with desirable structures and properties,13 as well as for the construction of structural and functional models of metalloenzyme active sites.14–16Another important reason for investigation of gold(III) chemistry with N-heterocycles stems from the finding that use of these ligands in the synthesis of gold(III) complexes enhances the stability of Au(III) ion under physiological conditions, allowing evaluation of gold(III) complexes as potential antitumor17 and antimicrobial agents.18 Thus, different N-heterocycles such as pyridine, bipyridine, terpyridine, phenanthroline and porphyrins have been used and a number of mononuclear and dinuclear gold(III) complexes have been synthesized showing cytotoxic activity toward different human tumor cell lines,19–28 as well as significant activity against the microbes29 and the parasites which are causative agents of tropical diseases, such as malaria,30 leishmaniasis31,32 and trypanosomiasis.33,34
In the light of the abovementioned, we have already reported the synthesis, NMR and X-ray structural characterization of mononuclear gold(III) complexes with some diazaaromatics, namely pyridazine, pyrimidine, pyrazine, quinoxaline, phenazine, 1,7-phenanthroline and 4,7-phenanthroline, [AuCl3(N-heterocycle)].35–37 All complexes were obtained by reacting K[AuCl4] and the corresponding N-heterocycle in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in the mixture of water and ethanol or in the pure ethanol. Furthermore, we have synthesized a new class of N-heterocycles based on camphorquinoxaline core and the corresponding mononuclear gold(III) complexes.38 By fusing the quinoxaline with camphor moiety, we have made the two nitrogen binding sites nonequivalent in order to investigate which of the two nitrogen atoms will be involved in the formation of mononuclear complexes, and found out that the investigated camphoquinoxaline derivatives had encoded information, both spatial and directional, that led to the formation of single coordination products, in which Au(III) ion is bound to the less sterically hindered of the two nonequivalent nitrogen atoms.38
:1 molar ratio in the mixture of water and ethanol or in the pure ethanol. Furthermore, we have synthesized a new class of N-heterocycles based on camphorquinoxaline core and the corresponding mononuclear gold(III) complexes.38 By fusing the quinoxaline with camphor moiety, we have made the two nitrogen binding sites nonequivalent in order to investigate which of the two nitrogen atoms will be involved in the formation of mononuclear complexes, and found out that the investigated camphoquinoxaline derivatives had encoded information, both spatial and directional, that led to the formation of single coordination products, in which Au(III) ion is bound to the less sterically hindered of the two nonequivalent nitrogen atoms.38
In the course of the above investigations35–38 and in order to test the possible changes in the approach of nitrogen heterocycles with extended annelated π systems, we report herein the synthesis, spectroscopic and X-ray crystallographic characterization of gold(III) complexes with five diazanaphthalene ligands (Scheme 1). Density functional theory (DFT) calculations were performed in order to gain more insight into the coordination mode of diazanaphthalenes toward Au(III) ion.
 | ||
| Scheme 1 Schematic drawing of diazanaphthalene ligands used in the present study. The numbering of atoms used for NMR assignation follows the IUPAC rules and does not match the one applied in the X-ray and theoretical studies of gold(III) complexes with these ligands. The α positions are numbered 1, 4, 5 and 8, while the β positions are 2, 3, 6 and 7. | ||
Results and discussion
In the present study, we have investigated the coordination ability of five diazanaphthalenes, namely quinazoline (qz), phthalazine (phtz), 1,5-naphthyridine (1,5-naph), 1,6-naphthyridine (1,6-naph) and 1,8-naphthyridine (1,8-naph) (Scheme 1) toward Au(III) ion. Two of the five presently investigated diazanaphthalene ligands, qz and phtz, contain nitrogen atoms within the same ring, hence they belong to a group of benzodiazines, while the remaining three, i.e. 1,5-naph, 1,6-naph and 1,8-naph, contain nitrogen atoms in different rings and are classified to naphthyridines. As in naphthalene, in the diazanaphthalene isomers, one can distinguish two nonequivalent sites α and β at which a nitrogen atom can be placed, and to either of which the Au–N bond may be formed. As a consequence, in the diazanaphthalene ligands one can have the two nitrogen atoms either placed at different sites α and β (qz and 1,6-naph), or at the same site, β (phtz) or α (1,5-naph and 1,8-naph). It appears that, despite of the different steric and electronic properties of the investigated diazanaphthalenes, their reactions with [AuCl4]− invariably lead to the formation of mononuclear [AuCl3(qz)] (1), [AuCl3(phtz)] (2), [AuCl3(1,5-naph)] (3), [AuCl3(1,6-naph)] (4) and [AuCl3(1,8-naph)] (5) complexes having one monodentatedly coordinated diazanaphthalene ligand, regardless the different ratio of the used substrates. In this respect, they behave similarly to the previously investigated quinoxaline, another isomer of diazanaphthalene,36 and to camphorquinoxaline derivatives.38 In two of the five ligands, namely qz and 1,6-naph, the potential metal binding nitrogen atoms are nonequivalent (α and β), therefore it is legitimate to pose a question which of the two nitrogen atoms will be subjected to the attack of the Au(III) electrophile.Structural characterization of gold(III) complexes 1–5
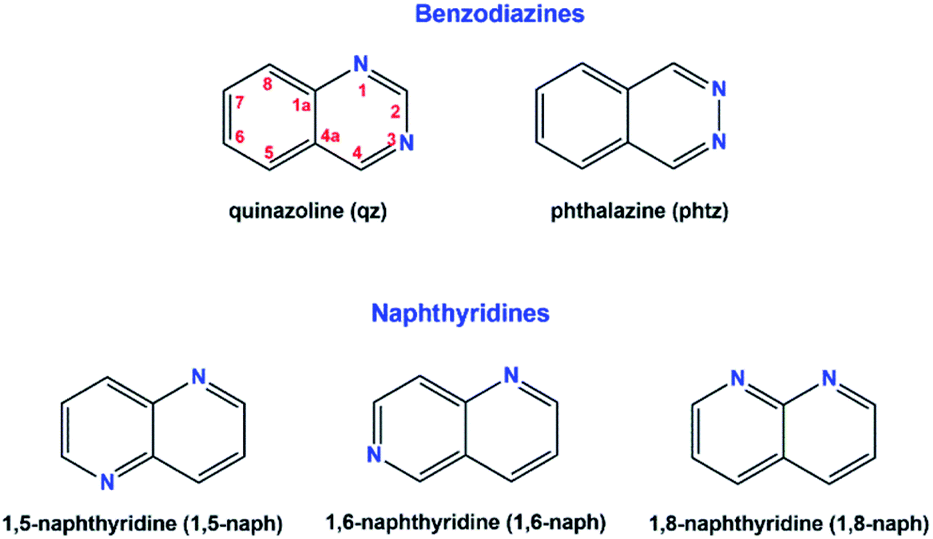
 | ||
| Fig. 1 Molecular structures of five [AuCl3(diazanaphthalene)] complexes showing atomic displacement parameters at 50% probability, except for the hydrogen atoms which were drawn in an arbitrary scale. | ||
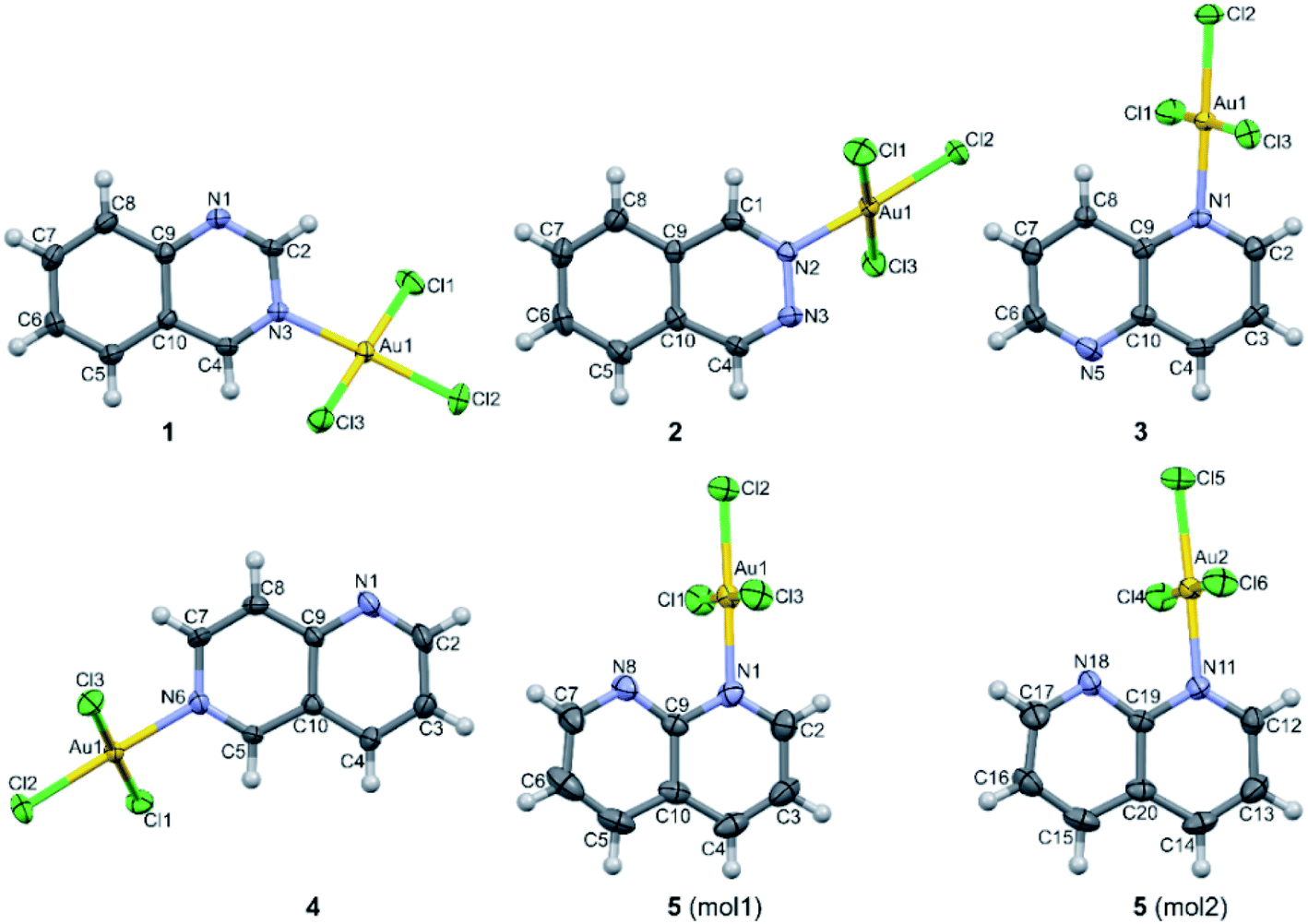
The crystal structure of all these complexes consists of discrete monomeric species with the Au(III) in a square planar coordination geometry surrounded by three chloride anions and only one diazanaphthalene ligand. In this respect, 3 constitutes the first example of the crystal structure, where only one nitrogen atom of the 1,5-naph ligand has been involved in the coordination to the metal ion. As the search of the CSD (Cambridge Structural Database) reveals,39 in the previously reported metal complexes, in which the 1,5-naph acts as a ligand, it always utilizes both nitrogen atoms to form coordination bonds in a bidentate-bridging mode, which results in the formation of 1D coordination polymers. As a consequence of the coordination of only one of the two diazanaphthalene nitrogen atoms, in each of the investigated complexes the second nitrogen atom of diazanaphthalene remains uncoordinated and is preferably utilized as an acceptor of C–H⋯N intermolecular hydrogen bond, with the exception of 2, in which the uncoordinated nitrogen atom N3 makes a close contact of 3.389(4) Å with the neighboring Au(III) ion (symmetry transformation 1 − x, −y, 1 − z). The qz and 1,6-naph ligands possess two alternative and distinguishable nitrogen atoms, to either of which a coordination bond may be formed during the reaction with K[AuCl4]. It appears that in the respective complexes 1 and 4, the nitrogen atom at the β site coordinates to the metal center. This allows the long axis of the ligand to point outward from the coordination plane and get involved in columnar stacking interactions. Fig. 2, which is an overlap of the molecules 3 and 4, illustrates the relative orientation of the heterocyclic rings with respect to the coordination plane in the complexes, where the ligand is coordinated via the α or β nitrogen, respectively.
 | ||
| Fig. 2 Overlap of the molecules 3 (red) and 4 (blue) illustrates the relative orientation of the heterocyclic rings with respect to the coordination plane in complexes, where the ligand is coordinated via α and β nitrogen, respectively. | ||
In all investigated crystal structures, the Au–Cl2 bond which is trans with respect to the Au–N bond, is consistently shorter than the remaining Au–Cl1 and Au–Cl3 bonds (for numbering scheme see Fig. 1), which might be considered as an indirect consequence of the higher trans effect of the chloride compared to the diazanaphthalene nitrogen atom. The corresponding Au–N bond lengths vary from 2.027(4) to 2.057(8) Å, with the shortest one being in the complex 2. The values of the valence angles around the coordinated nitrogen atom spread a wide range from 115.8(6) to 124.4(7)° in complexes 2 and 5, become more equally distributed in complexes 1 and 3 and almost equal in complex 4 (Table S2†). The mean value of the valence angle at the uncoordinated nitrogen atom equal to 116.8(1.3)° is characteristic for the heterocyclic nitrogen possessing free electron pair.
The complexes 1–5 differ in the orientation of the azaaromatic ring plane with respect to the plane defined by the four donor atoms surrounding the Au(III) ion. In the crystals of 5, the two planes are perpendicular (89.98(10) and 89.30(9)° for two independent molecules), they are somewhat inclined in the crystals of 3 (68.84(7)°), and achieve the values of 62.76(7) and 59.46(9)° in the crystals of 2 and 4, respectively, while in the crystals of 1, this interplanar angle is only 39.47(5)°.
Noticeable inclination of the N-heteroaromatic ring with respect to the coordination plane is a source of molecular helicity, which becomes meaningful in the chiral crystals formed by the molecules of 4. The complexes 1 and 2 that contain benzodiazines as ligands, form triclinic centrosymmetric crystals, in which the dominant intermolecular interactions operate between molecules related by inversion. In contrast, the complexes containing the naphthyridine ligands crystallize either in polar (3 and 5) or chiral (4) space group. The amphiphilic nature of the charge distribution on the chloride anion coordinated to the Au(III) cation allows it to serve as a Lewis base in Cl⋯Au interactions (Table S3†), as both a donor and acceptor in Cl⋯Cl halogen bond (Table S3†) and also as a hydrogen bond acceptor (Table S4†). As a result, in each of the investigated crystals (1–5), the square planar coordination around Au(III) is extended to square pyramidal or tetragonal bipyramidal due to the Au⋯Cl intermolecular interactions and the molecular packing is dominated by the intermolecular C–H⋯Cl hydrogen bonds, which outnumber the other types of the specific intermolecular interactions. Notably, the π⋯π stacking interactions play a significant structural role only in the centrosymmetric crystals of [AuCl3(benzodiazine)] complexes (1 and 2) and become either much weaker or completely absent in a series of [AuCl3(naphthyridine)] crystals (3–5). Geometrical parameters describing the π⋯π stacking interactions are listed in Table S5.†
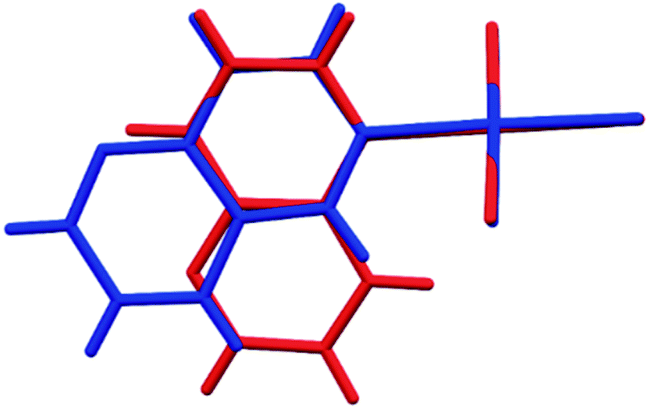
In the crystals of 1, the quinazoline ligand is coordinated to the Au(III) ion via the β nitrogen atom. This allows for the columnar stacking interactions. In the two-ring π-systems, such as benzodiazines, the offset π⋯π stacking interaction may involve two nitrogen-containing aromatic rings, two arene rings or the arene and the N-heteroaromatic rings. And indeed, in the crystals of 1, there are two types of stacking in each column: one involves the pyrimidine and arene moieties of the inversion related quinazoline ligands, while the other encompasses solely the arene rings generated by another set of the inversion centers (Fig. 3a and b). There is a slight preference for the pyrimidine⋯arene approach, followed by the arene⋯arene approach (Table S5†), while pyrimidine⋯pyrimidine type of interactions is nonexistent. Moreover, in the crystals of 1, one chlorido ligand approaches the Au(III) center at a distance of only 3.295(2) Å, while the sum of the van der Waals radii of the two atoms amounts to 3.41 Å. The approach of the chloride ion from the other side appears at far longer distance of 3.665(2) Å. Pairs of Au⋯Cl interactions form centrosymmetric dimer (Fig. 3). On both sides of this dimer, there are the quinazoline ligands that are engaged in columnar π⋯π stacking interactions. Another supramolecular motif joining molecules related by inversion is the R22(6) ring (graph set notation40), formed by a pair of inversion related C2–H2⋯N1 (2 − x, −y, 1 − z) hydrogen bonds involving the uncoordinated nitrogen atom as an acceptor (Table S4†). The appearance of such type of hydrogen bond has been previously noted in gold(III) complexes with pyridazine and pyrimidine monodentatedly coordinated to the metal center35 and in the crystal structure of gold(III) complex with quinoxaline.36 Moreover, the Au1–Cl1⋯Cl3 (x + 1, y, z) and Au1–Cl3⋯Cl3 (−x + 1, −y + 1, −z + 2) contacts indicate the presence of type I halogen bonding (see Table S3†).41
 | ||
| Fig. 3 Packing of the molecules of 1. The figure illustrates pairs of the centrosymmetric dimers formed via C–H⋯N hydrogen bonds and Au⋯Cl interactions (a); columnar π⋯π stacking, and two different modes of molecular overlap within the column (b). | ||
The packing of 2 is dominated by two features. First, the pairs of complex molecules related by inversion stack off-face at a distance 3.447 Å between the arene and pyridazine moieties (Fig. 4 and Table S5†). Second, the connection between the dimers takes place by means of pairs of inversion-related Au⋯Cl3 and Au⋯N3 interactions at distances of 3.530(2) and 3.389(4) Å, respectively. In this way the coordination sphere around Au(III) ion is supplemented to tetragonal bipyramidal (4 + 2). The presence of Au⋯N3 interactions is unique in the group of [AuCl3(N-heterocycle)] complexes. Moreover, the Au1–Cl3⋯Cl1–Au1 contact of 3.444(2) Å indicates the presence of type I halogen bonding.41
 | ||
| Fig. 4 Packing of the molecules of 2 demonstrating pairs of inversion related Au⋯N and Au⋯Cl as well as dimeric π⋯π stacking interactions (a); view perpendicular to phtz plane displaying a degree of the ring overlap (b). | ||
As was previously noted,42 aromatic nitrogen-containing heterocycles are electron poor systems and a metal which is coordinated to a nitrogen heteroatom will further enhance the electron-withdrawing effect through its positive charge. Hence, aromatic nitrogen-containing heterocycles should in principle be well suited for π⋯π interactions, because of their low π-electron density. In spite of this expectation, the crystals of 3 are deprived of any off-face stacking interactions. The complex molecules interact via C–H⋯N hydrogen bonds and the Au⋯Cl3 interactions, as illustrated in Fig. 5. The azaarene rings, rather than forming the π⋯π stacking, are engaged in (Cl2)⋯π interactions. The distance between Cl2 and the center of gravity (Cg) of the C9–C10 bond (symmetry operation −½ + x, ½ − y, z) is 3.386 Å and the Au–Cl⋯Cg angle amounts to 113.1°. There are also several Cl⋯Cl interactions operating along the polar z-axis, but geometrical parameters describing these interactions do not allow their straightforward classification into one of the two types of halogen bonds (Table S3†).43 The complete lack of π⋯π stacking interactions in crystals containing 1,5-naph as a ligand has been observed by us in 1D polymeric crystals formed by [ZnCl2(1,5-naph)]n complex, in which the ligand acts in a bidentate-bridging mode and is involved in anion⋯π interactions.44
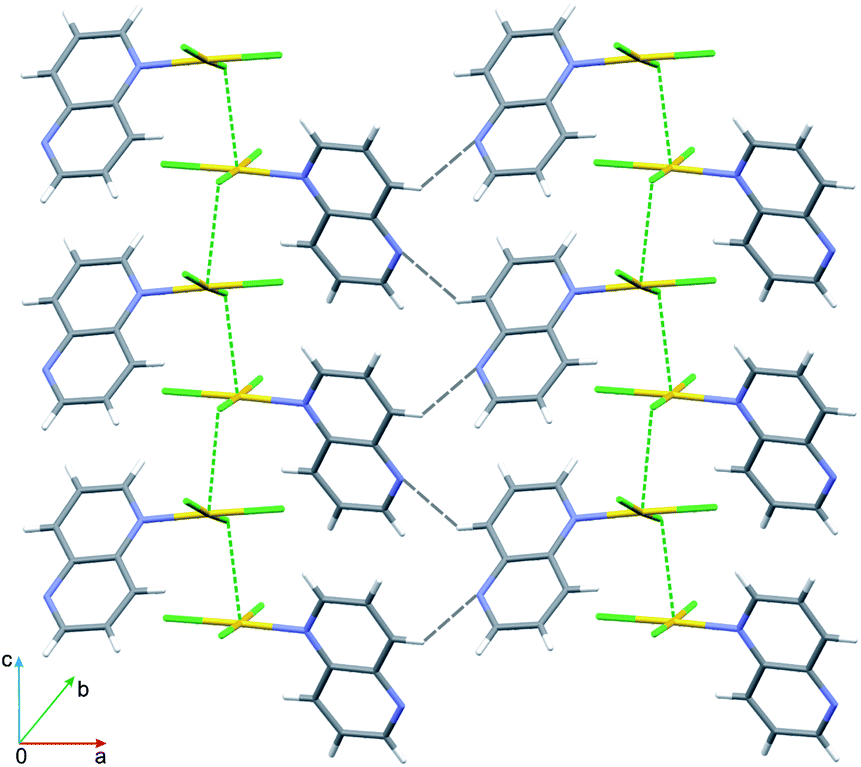 | ||
| Fig. 5 View of the aggregation in crystals of complex 3 in non-centrosymmetric space group Pna21. Note the arrangement of molecules along the polar c-direction held together by C–H⋯N and Au⋯Cl interactions. | ||
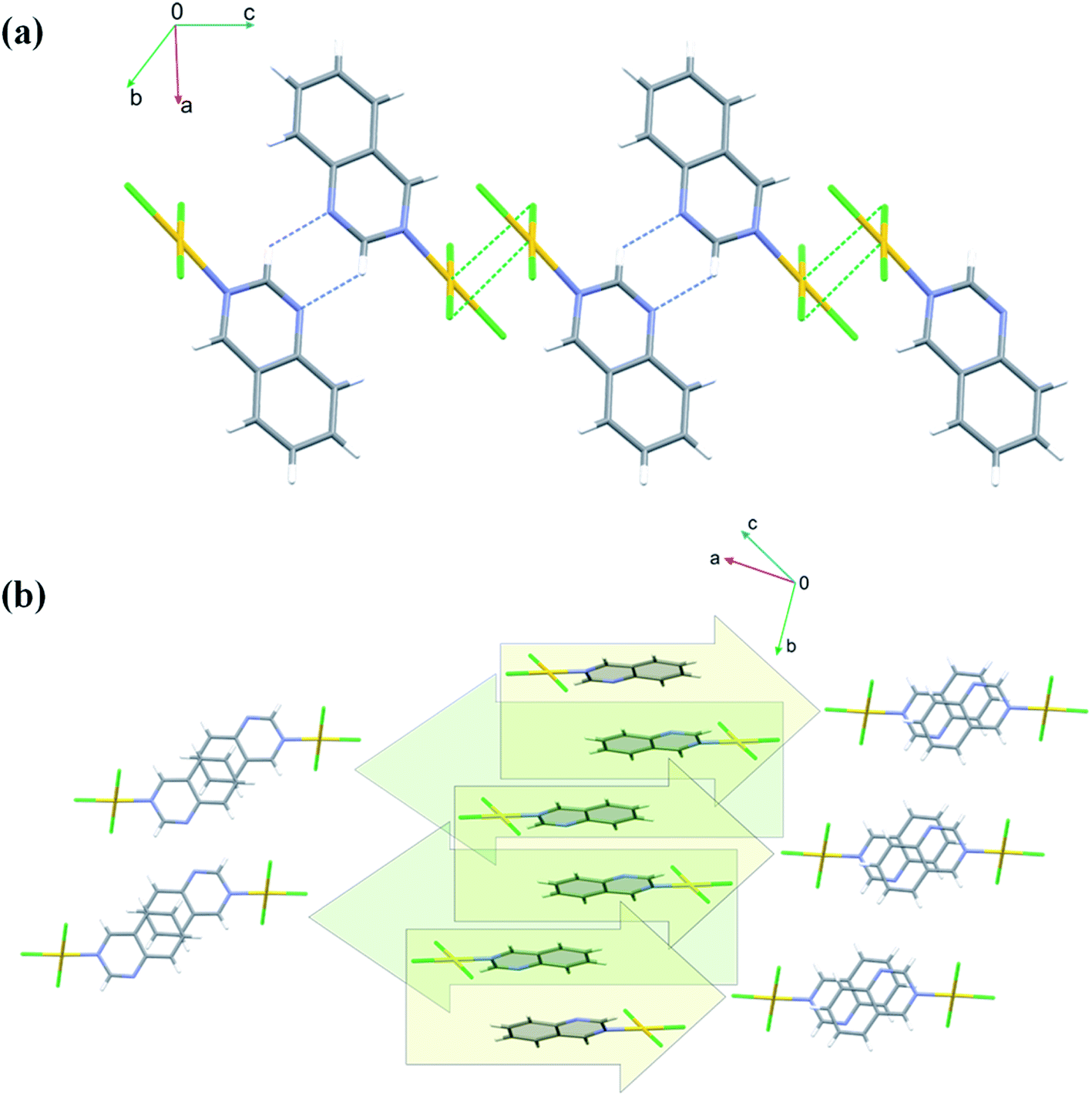
Crystals of 4 are both polar and chiral. Contrary to the anti-parallel arrangement of the molecules involved in the π⋯π stacking interactions in the crystals of 1 and 2, in the crystals of 4 the molecules engaged in stacking are related by single lattice translation along the a-direction, so they stack parallel to each other (Fig. 6). Geometrical parameters describing this type of interactions are less favorable than in the previous two cases (Fig. 6 and Table S5†). The Au(III) ion is approached by two chloride ions at distances 3.427(3) and 3.378(3) Å, extending its coordination sphere to tetragonal bipyramid (4 + 2). Pairwise Au⋯Cl1 and Au⋯Cl3 interactions lead to the formation of dimers that mimic those formed around an inversion center in the centrosymmetric crystals of 1 and 2 (Fig. 6).
 | ||
| Fig. 6 Arrangement of the molecules of 4 in chiral and polar space group P21 by means of C–H⋯N, pairwise Au⋯Cl1 and Au⋯Cl3 interactions and columnar stacking along the a-direction (a); view perpendicular to the 1,6-naph plane displaying a small degree of the ring overlap (b). | ||
The molecules of 5 crystallize in the non-centrosymmetric space group Cc with two symmetry independent molecules. The coordination of 1,8-naph molecule is monodentate and the interaction with the most distant nitrogen atom is minimized, as the angle between the plane of the naph moiety and the coordination plane is close to 90° (vide supra). In spite of that, there is probably some interaction between the distant nitrogen and the central Au atom (the Au⋯N distances are 2.942(6) and 2.901(6) Å in the two symmetry independent molecules) which is of steric importance, because it prevents the approach of another ligand from this side, thus hindering a possibility of extension of the square planar coordination around the Au(III) cation. However, the approach of the chloride anion is still possible from the other side of the coordination plane, which supplements the coordination around Au(III) to square pyramidal (4 + 1).
Two independent molecules of complex 5 display the same geometry and are related by a pseudo-center of symmetry having the approximate fractional x, y, z coordinates of 0.28, 0.39 and 0.35. The two molecules are held together by relatively weak π⋯π interactions (Table S5†). The dimeric motif is further extended along b-direction by means of Au⋯Cl interactions (Fig. 7) and propagates along a-direction by means of C–H⋯N hydrogen bonds, but overwhelming are the C–H⋯Cl interactions which operate in all three-dimensions (Table S4†). Using the standard settings inserted to the Mercury program,45 we were able to identify small voids between the molecular layers parallel to the (001) lattice planes. Multiplication of the number of symmetry independent molecules and the appearance of the structural voids are the signs that the molecule of 5 exhibits difficulties with packing in the crystal. This is also reflected in the values of the crystal density and the Kitajgorodskij packing index,46 which are the smallest within the investigated series (Table S1†).
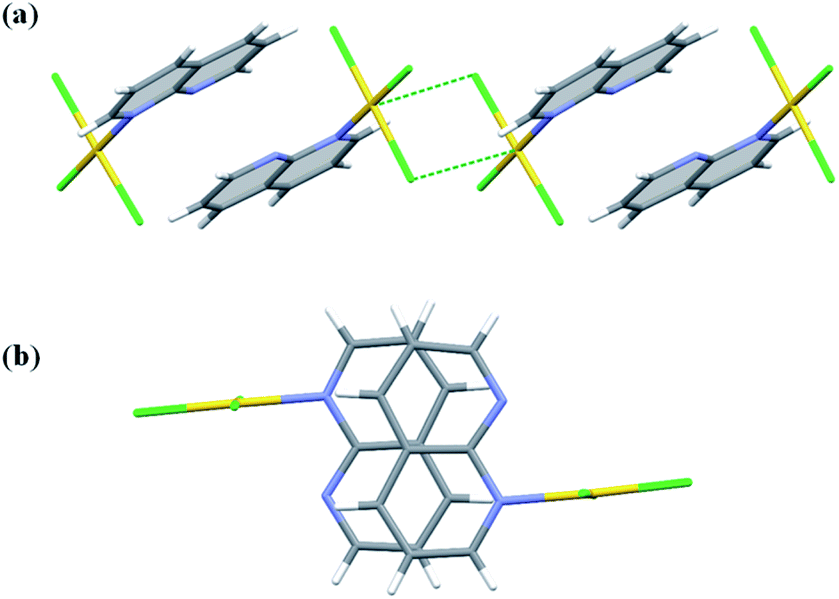 | ||
| Fig. 7 Pairwise π⋯π and Au⋯Cl interactions between two symmetry independent molecules of complex 5 in non-centrosymmetric space group Cc (a); view perpendicular to the 1,8-naph plane displaying a degree of the ring overlap (b). | ||
Since the work of Kitaigorodskij,46 it has been recognized that inversion centers in space groups are ‘‘good’’ for the close-packing of molecules. This does not seem to be reflected in the present study. Except for the molecule of 5, which crystallizes in polar space group Cc and exhibits some packing problems in crystal, the remaining crystals display similar values of the calculated density and packing index (Table S1†), despite the fact that some of them form centrosymmetric crystals but some do not. Our observations seem to indicate that the presence of an inversion center is needed for good stacking rather than good packing: the π⋯π stacking interactions in non-centrosymmetric crystals of 4 and 5 are relatively weak compared with those operating in centrosymmetric crystals of 1 and 2. Despite the fact that the centrosymmetric space groups are more favored than non-centrosymmetric, the naphthyridine ligands, for some reason, are well suited for the formation of the non-centrosymmetric aggregates. This observation might perhaps be of interest to the non-linear optics community, where the non-centrosymmetric space groups are a requirement.
The proton chemical shifts of the diazanaphthalene ligands in deuteroacetone generally agree with the literature values for the other solvents.47–49 The 1H NMR spectra of the gold(III) complexes 1–5 were noticeably different from those of the uncoordinated diazanaphthalene ligands; addition of the free ligand resulted in another set of 1H signals with the expected chemical shifts. The auration of all ligands resulted in 1H deshielding, with a mean value ΔH1–H8: +0.45–0.65 ppm, as deduced from the unassigned 1H NMR data. The observed downfield shifts for complexes 1–5 with respect to the free diazanaphthalene ligands were a consequence of the strong electron-withdrawing effect of Au(III) ion; i.e. the overall electron density is shifted toward the AuCl3 fragment, making diazanaphthalene ring electron-deficient compared to the uncoordinated ligand. Further evidence for the reduced nucleophilicity of the uncoordinated nitrogen atom in the investigated complexes is provided by DFT calculations (vide infra).
Coordination to Au(III) of phtz, 1,5-naph and 1,8-naph ligands resulted in the appearance of six sets of proton signals in the spectra of complexes 2, 3 and 5, respectively (three sets of proton signals are present in the spectra of free ligands). As an illustration of these findings, 1H NMR spectra of free 1,8-naph ligand and the corresponding gold(III) complex 5 are presented in Fig. S1a.† Moreover, from the obtained NMR spectroscopic results, it can be concluded that qz and 1,6-naph ligands in the complexes 1 and 4 are exclusively coordinated through the N3 and N6 nitrogen atoms, respectively. This is clearly illustrated in Fig. S1b,† which shows the 1H NMR spectra of the uncoordinated 1,6-naph and its complex 4.
The Au(III) complexation of the diazanaphthalene ligands resulted in an overall deshielding of ring carbons, in some cases up to 7.8 ppm (mean ΔC1–C8a: +1.2–3.4 ppm). In the case of complex 2, all carbon atoms were deshielded (up to 5.5 ppm). This is in accordance with downfield shifting of carbon signals previously found for [AuCl3(pydz)] (pydz is pyridazine) complex; both these complexes contain two adjacent nitrogen atoms.35 On the other hand, in the remaining four gold(III) complexes, nitrogen-adjacent carbons were shielded (up to −13.5 ppm for C8a, which is positioned between N1 and N8 atoms, in complex 5). These spectral characteristics were also previously noted for the mononuclear gold(III) complexes with monodentatedly coordinated pyrimidine,35 pyrazine,35 quinoxaline36 and phenazine,36 as well as for [Au(AMP)Cl2]Cl·H2O complex (AMP is bidentatedly coordinated 2-(aminomethyl)pyridine)50 and some pyridinium cations.51–53
The UV-Vis spectra of the presently investigated gold(III) complexes 1–5 were recorded in acetone. The wavelengths of maximum absorption for 1–5 (λmax, nm) and molar extinction coefficients (ε, M−1 cm−1) determined right after dissolution of the complexes, are listed in the Experimental section. As can be expected, the shape of the spectra and values of λmax are almost identical for all complexes indicating the same monodentate coordination mode of N-heterocycles. The UV-Vis data show absorption around 320.0 nm, which corresponds to LMCT (ligand-to-metal charge transfer) transitions and could be assigned to Cl → Au charge transfer by analogy to auric acid absorption spectra.54–56
Theoretical studies
In order to gain better understanding on the coordination mode of the investigated diazanaphthalenes toward the Au(III) ion, the structures of the ligands and corresponding gold(III) complexes 1–5 were optimized for the isolated molecules by means of DFT methods. Of the three experimental approaches (B3LYP, M06 and M06-2X, see Experimental section), the M06-2X method showed the best agreement with the corresponding crystallographic data. Consequently, this method was chosen for the structure optimization of the gold(III) complexes 1–5 and the investigated diazanaphthalenes. The calculated bond lengths (Å) and angles (°) for 1–5 are given in Table S2† and show a good agreement with the corresponding crystallographic data.The donor ability of diazanaphthalenes toward Au(III) has been evaluated by observing the composition of the highest occupied molecular orbital (HOMO) and the molecular charge distribution, by using NBO analysis. The calculated NBO charges (e) of donor nitrogen atoms and eigenvalues (hartree) of HOMOs and LUMOs of diazanaphthalenes are presented in Table 1. The results summarized in this table pointed out that electron-donating ability of diazanaphthalenes increases with increasing of the HOMOs eigenvalues57–59 in the following order: 1,8-naph ≈ 1,5-naph < phtz < qz ≈ 1,6-naph. Furthermore, the nitrogen N3 and N6 atoms in asymmetrical qz and 1,6-naph ligands, respectively, are slightly more negatively charged than the corresponding N1 nitrogen.
| Ligand/gold(III) complex | qN1 | qN2 | qN3 | qN5 | qN6 | qN8 | EHOMO | ELUMO |
|---|---|---|---|---|---|---|---|---|
| a Compound is symmetric. | ||||||||
| qz | −0.446 | −0.460 | −0.3041 | −0.0426 | ||||
| 1 | −0.414 | −0.499 | −0.3395 | −0.1071 | ||||
| phtza | −0.232 | −0.232 | −0.3091 | −0.0403 | ||||
| 2 | −0.259 | −0.191 | −0.3271 | −0.09283 | ||||
| 1,5-napha | −0.420 | −0.420 | −0.3150 | −0.0421 | ||||
| 3 | −0.434 | −0.404 | −0.3377 | −0.1036 | ||||
| 1,6-naph | −0.423 | −0.426 | −0.3037 | −0.0430 | ||||
| 4 | −0.417 | −0.461 | −0.3372 | −0.1047 | ||||
| 1,8-napha | −0.404 | −0.404 | −0.3155 | −0.0426 | ||||
| 5 | −0.430 | −0.408 | −0.3240 | −0.0954 | ||||
To obtain more information about the favorable site of Au(III) electrophilic attack in these two ligands, we have examined their molecular electrostatic potential (MEP) mapped on the electron density surface generated at M06-2X/cc-pVTZ level of theory (Fig. 8a). The MEP has been shown so far as a very useful tool for prediction of the sites of electrophilic and nucleophilic attacks, as well as hydrogen bonding interactions.38,60,61 The regions of the negative potentials, colored in red, are related to the possible sites of an electrophilic attack, while the regions of positive potential (blue) may indicate the sites of a nucleophilic attack.
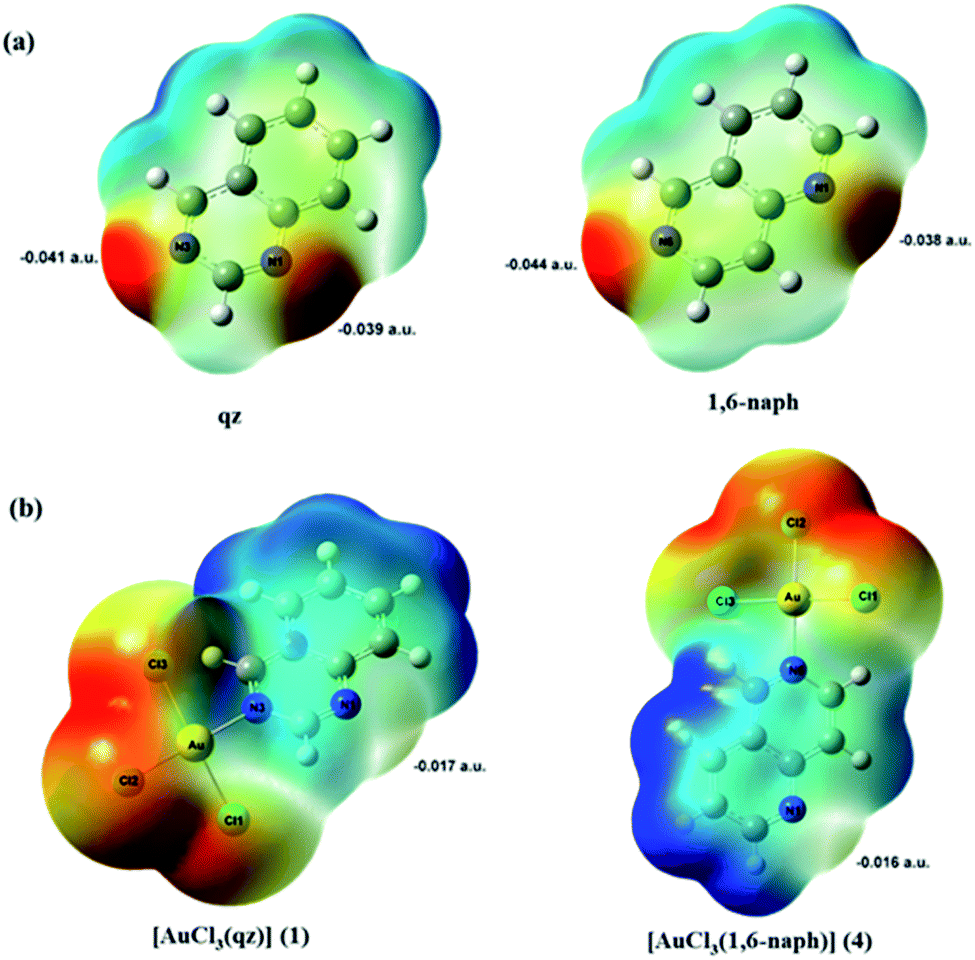 | ||
| Fig. 8 Electrostatic potential mapped on electron density surfaces for (a) asymmetrical qz and 1,6-naph ligands and (b) complexes 1 and 4 generated at DFT M06-2X/cc-pVTZ and M06-2X/(LanL2TZ(f)+cc-pVTZ) levels of theory, respectively (isoval 0.0004 a.u.). Color change from red most negative to blue most positive. | ||
As can be expected for the diazanaphthalene ligands, the negative potential regions are on the electronegative nitrogen atoms. The negative electrostatic potential values are −0.039 a.u. for N1 and −0.041 a.u. for N3 in qz, and −0.038 a.u. for N1 and −0.044 a.u. for N6 in 1,6-naph (Fig. 8a). Due to the fact that in both qz and 1,6-naph ligands the difference between the values of the electrostatic potential for the two nitrogens is not very large, we have optimized the structures and calculated the energies for the four distinct mononuclear gold(III) complexes, in which qz and 1,6-naph ligands are coordinated via either of the two nitrogen atoms. The structures of these complexes in the gas phase, calculated for the reaction of qz and 1,6-naph with an equimolar amount of [AuCl4]−, are presented in Fig. 9 along with their relative energies. Gold(III) complexes with N1 coordinated nitrogen were calculated to be higher in energy by 1.1 and 2.6 kcal mol−1, respectively, in comparison with the complexes 1 and 4 having N3 and N6 coordinated qz and 1,6-naph ligands, respectively. This undoubtedly confirms that the latter two complexes are energetically more favorable, being in agreement with our spectroscopic and crystallographic results.
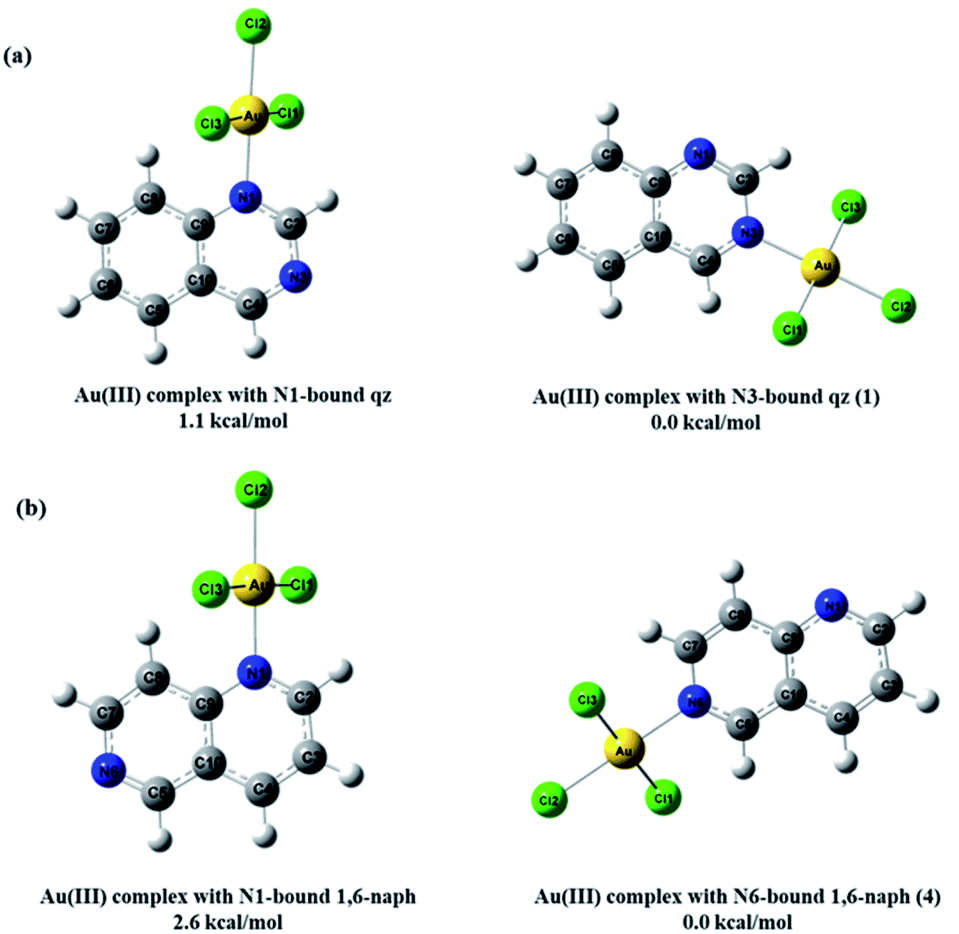 | ||
| Fig. 9 The calculated structures of the four mononuclear gold(III) complexes that could supposedly be formed as the final product of the reactions of (a) qz and (b) 1,6-naph with an equimolar amount of [AuCl4]−, as well as their relative energies in the gas phase. | ||
DFT calculations have been also used to provide an evidence that the presence of an electron-pulling AuCl3 group bound to the nitrogen atom of diazanaphthalene ring makes it electron-deficient and reduces the nucleophilicity of the second nitrogen atom, preventing the formation of dinuclear gold(III) species. As can be seen from Table 1, coordination of all N-heterocycles to the Au(III) caused a decrease in the eigenvalues of the HOMOs, which indicates that the obtained mononuclear gold(III) complexes have lower electron-donating ability than the corresponding ligands. To obtain information about the potential sites in [AuCl3(N-heterocycle)] complexes, which can be a target of Au(III) electrophilic attack, their MEPs have been generated at M06-2X/(LanL2TZ(f)+cc-pVTZ) level of theory. The MEP maps for all investigated gold(III) complexes have shown that Au(III) ion bound to the heteroaromatic ring made it electron-deficient compared to the uncoordinated ligands. This has caused, as a consequence, an increase of the electrostatic potential value on the uncoordinated nitrogen atom; accordingly, this nitrogen is no longer a preferable site for the electrophilic attack of the subsequent AuCl3 fragment. As an illustration of this finding, the MEPs for complexes 1 and 4 are presented in Fig. 8b.
Nonlinear optical (NLO) properties of complexes 1–5
In the last decades, many transition metal complexes have been gaining much attention as potential building blocks for second order nonlinear optical (NLO) materials.62–64 Compared to organic molecules, metal complexes offer a larger variety of molecular structures in relation to metal nd configuration, oxidation and spin state, and can satisfy the different demands of second order NLO materials, such as switchable, tunable and multidimensional optical properties.64It is well known that the nonlinear optical response of an isolated molecule in an electric field Ei(ω) can be presented as a Taylor series expansion of the total dipole moment, μ, induced by the field:
| μ = μ0 + αijEj + βijkEjEk +… |
The polar properties of the gold(III) complexes 1–5 were calculated at M06-2X/(LanL2TZ(f)+cc-pVTZ) level of theory in the gas phase and the obtained results are presented in Table 2. According to this table, the calculated α values for the investigated gold(III) complexes increase in the order: 5 < 3 < 4 < 2 < 1, and the calculated β values follow the similar trend, i.e. 5 < 4 < 3 < 2 < 1. Urea is one of the prototypical compounds used in the study of NLO properties of the molecules.67 Therefore, urea was selected as a reference, and its μ, α and β values were calculated at M06-2X/cc-pVTZ level of theory. Almost all presently investigated gold(III) complexes have the greater α and β values than the corresponding values of urea (up to 6.5 and 4.3 times, respectively). The exception is the complex 5, which has a slightly lower β value than that of urea (1.2 times). This indicates that gold(III) complexes with diazanaphthalenes 1–5 might be good candidates for NLO materials.
| Compound | Dipole moment μ (D) | Polarizability α (×10−24 esu) | Hyperpolarizability β (×10−30 esu) |
|---|---|---|---|
| a The presented values for urea are calculated at M06-2X/cc-pVTZ level of theory in the gas phase. | |||
| Ureaa | 4.3852 | 4.39 | 1.10 |
| 1 | 10.4442 | 28.36 | 4.73 |
| 2 | 13.7133 | 28.31 | 2.82 |
| 3 | 8.0700 | 27.51 | 2.29 |
| 4 | 10.1137 | 28.20 | 1.36 |
| 5 | 12.5106 | 27.46 | 0.94 |
As revealed by the X-ray analysis (vide supra), the complexes that display the lowest values of the polarizability (α) and hyperpolarizability (β), namely 3, 4 and 5, form polar crystals. Polar crystals are of great interest because of their physical properties, such as pyroelectricity, ferroelectricity, piezoelectricity, second harmonic generation, which are only allowed in polar systems (crystalline or amorphous)68 and are therefore highly requested in materials for advanced applications. Moreover, there is also a nice correlation between the value of the average polarizability (α) for the studied complex and its involvement in π⋯π stacking interactions in crystals. Namely, the highest α values are associated with the presence, while the lowest with the absence of π⋯π stacking. Indeed, the calculated correlation coefficient between the calculated polarizability (α) and the shortest distance between the planes defined by the diazanaphthalene fragments (h) (see Table S5†) is equal to −0.818. It means that the determination coefficient equal to 0.669 and the calculated polarizability (α) explain 66.9% of observed variation in the values of distances between planes defined by diazanaphthalene fragments (h). Unfortunately, due to the lack of the material, we were not able to perform the second-harmonic generation (SHG) measurements.
Experimental
Materials and measurements
Potassium tetrachloridoaurate(III) (K[AuCl4]), quinazoline (qz), dichloromethane, ethanol, acetone and deuteroacetone were purchased from the Sigma-Aldrich Chemical Co. Phthalazine (phtz), 1,5-naphthyridine (1,5-naph), 1,6-naphthyridine (1,6-naph) and 1,8-naphthyridine (1,8-naph) were obtained from abcr GmbH & Co. KG. All the employed chemicals were of analytical reagent grade and used without further purification.Elemental microanalyses for carbon, hydrogen and nitrogen were performed on a Vario EL instrument by the Microanalytical Laboratory, Department of Organic Chemistry, University of Heidelberg. All NMR spectra were recorded at 25 °C in acetone-d6 on a Bruker Avance II 400 MHz spectrometer. Chemical shifts are reported in ppm (δ) and scalar couplings are reported in Hertz. 5.0 mg of each compound was dissolved in 0.6 mL of acetone-d6 and transferred into a 5 mm NMR tube. The splittings are designated as: s, singlet; d, doublet; m, multiplet; dd, doublet of doublets; dt, doublet of triplets; td, triplet of doublets; ddd, doublet of doublet of doublets. The UV-Vis spectra were recorded on a Jasco V570 spectrophotometer after dissolving the corresponding gold(III) complex in acetone, over the wavelength range 600–200 nm. The concentration of the gold(III) complexes was 2.5 × 10−4 M.
Synthesis of the gold(III) complexes 1–5
Gold(III) complexes with five isomeric dizanaphthalenes were synthesized according to the modified procedure published in the literature for the preparation of [AuCl3(N-heterocycle)] complexes (N-heterocycle is pyridazine, pyrimidine, pyrazine, quinoxaline, phenazine, 1,7- and 4,7-phenanthroline, and methylcamphorquinoxaline derivatives).35–38The solution of 0.5 mmol of the corresponding diazanaphthalene ligand (65.1 mg) in 2.0 mL of ethanol was added slowly under stirring to the solution containing an equimolar amount of K[AuCl4] (188.9 mg) in 10.0 mL of water. The yellow precipitate, formed immediately after addition of the N-heterocyclic ligand, was filtered off, washed with water, and recrystallized in dichloromethane to form yellow crystals of gold(III) complexes 1–5. The crystals were collected from the solution and dried in the dark at ambient temperature. The yield was 69% for [AuCl3(qz)] (1) (149.5 mg), 65% for [AuCl3(phtz)] (2) (140.9 mg), 54% for [AuCl3(1,5-naph)] (3) (117.0 mg), 43% for [AuCl3(1,6-naph)] (4) (93.2 mg) and 58% for [AuCl3(1,8-naph)] (5) (125.7 mg).
Anal. calcd for 1 = C8H6AuCl3N2 (MW = 433.46): C, 22.17; H, 1.40; N, 6.46. Found: C, 22.13; H, 1.46; N, 6.51%. 1H NMR (400 MHz, CD3COCD3): 8.13 (ddd, J = 8.1, 7.1, 1.0 Hz, H6), 8.29 (dd, J = 8.5, 0.6 Hz, H7), 8.47 (ddd, J = 8.5, 7.0, 1.4 Hz, H5), 8.59 (dd, J = 8.3, 0.7 Hz, H8), 9.64 (s, H4), 10.32 (s, H2) ppm. 13C NMR (101 MHz, CD3COCD3): 125.4 (C8a), 128.2 (C7), 130.4 (C8), 131.2 (C6), 140.0 (C5), 149.8 (C4a), 150.6 (C4), 162.2 (C2) ppm. UV-Vis (acetone, λmax, nm): 323.0 (ε = 4.0 × 103 M−1 cm−1).
Quinazoline (qz)*: 1H NMR (400 MHz, CD3COCD3): 7.76 (ddd, J = 8.1, 4.5, 3.5 Hz, H6), 8.02 (dd, J = 4.7, 1.1 Hz, H5 and H7), 8.13 (dt, J = 8.2, 1.0 Hz, H8), 9.26 (s, H4), 9.54 (s, H2) ppm. 13C NMR (101 MHz, CD3COCD3): 125.2 (C8a), 127.6 (C6), 127.9 (C8), 128.1 (C7), 134.1 (C5), 149.9 (C4a), 155.2 (C4), 160.4 (C2) ppm.
Anal. calcd for 2 = C8H6AuCl3N2 (MW = 433.46): C, 22.17; H, 1.40; N, 6.46. Found: C, 22.10; H, 1.53; N, 6.56%. 1H NMR (400 MHz, CD3COCD3): 8.45 (ddd, J = 8.2, 7.1, 1.3 Hz, H6), 8.53 (ddd, J = 8.2, 7.1, 1.2 Hz, H7), 8.61 (dd, J = 8.2, 1.0 Hz, H5), 8.69 (dd, J = 8.0, 0.8 Hz, H8), 9.90 (s, H4), 10.47 (s, H1) ppm. 13C NMR (101 MHz, CD3COCD3): 127.6 (C8a), 127.9 (C5), 129.2 (C8), 129.7 (C4a), 135.2 (C6), 138.1 (C7), 155.0 (C4), 156.2 (C1) ppm. UV-Vis (acetone, λmax, nm): 323.0 (ε = 3.7 103 M−1 cm−1).
Phthalazine (phtz): 1H NMR (400 MHz, CD3COCD3): 8.05 (dd, J = 6.0, 3.2 Hz, H6 and H7), 8.18 (dd, J = 6.0, 3.6 Hz, H5 and H8), 9.65 (s, H1 and H4) ppm. 13C NMR (101 MHz, CD3COCD3): 126.3 (C5 and C8), 126.4 (C4a and C8a), 132.6 (C6 and C7), 150.8 (C1 and C4) ppm.
Anal. calcd for 3 = C8H6AuCl3N2 (MW = 433.46): C, 22.17; H, 1.40; N, 6.46. Found: C, 22.02; H, 1.58; N, 6.58%. 1H NMR (400 MHz, CD3COCD3): 8.28 (dd, J = 8.8, 4.0 Hz, H7), 8.44 (dd, J = 8.5, 5.5 Hz, H3), 9.13 (d, J = 8.4 Hz, H4), 9.31 (d, J = 8.8 Hz, H8), 9.36 (dd, J = 4.0, 1.2 Hz, H6), 9.86 (dd, J = 5.6, 1.2 Hz, H2) ppm. 13C NMR (101 MHz, CD3COCD3): 127.6 (C3), 128.1 (C7), 134.1 (C8), 143.1 (C8a), 144.9 (C4), 145.4 (C4a), 154.1 (C2), 154.2 (C6) ppm. UV-Vis (acetone, λmax, nm): 323.0 (ε = 4.3 × 103 M−1 cm−1).
1,5-Naphthyridine (1,5-naph): 1H NMR (400 MHz, CD3COCD3): 7.79 (dd, J = 8.4, 4.0 Hz, H3 and H7), 8.43 (m), 9.02 (dd, J = 4.0, 1.5 Hz, H2 and H6) ppm. 13C NMR (101 MHz, CD3COCD3): 124.5 (C3 and C7), 137.1 (C4 and C8), 143.9 (C4a and C8a), 151.3 (C2 and C6) ppm.
Anal. calcd for 4 = C8H6AuCl3N2 (MW = 433.46): C, 22.17; H, 1.40; N, 6.46. Found: C, 22.29; H, 1.54; N, 6.34%. 1H NMR (400 MHz, CD3COCD3): 8.05 (dd, J = 8.5, 4.3 Hz, H3), 8.46 (d, J = 6.9 Hz, H8), 8.98 (d, J = 8.5 Hz, H4), 9.18 (d, J = 6.1 Hz, H7), 9.50 (dd, J = 4.3, 1.7 Hz, H2), 10.13 (s, H5) ppm. 13C NMR (101 MHz, CD3COCD3): 124.8 (C4a), 125.6 (C3), 127.0 (C8), 138.4 (C4), 144.9 (C7), 156.4 (C5 and C8a), 160.0 (C2) ppm. UV-Vis (acetone, λmax, nm): 322.0 (ε = 2.3 × 103 M−1 cm−1).
1,6-Naphthyridine (1,6-naph): 1H NMR (400 MHz, CD3COCD3): 7.71 (dd, J = 8.4, 4.4 Hz, H3), 7.92 (d, J = 6.0 Hz, H8), 8.56 (ddd, J = 8.3, 1.7, 0.8 Hz, H4), 8.77 (d, J = 6.0 Hz, H7), 9.15 (dd, J = 4.2, 1.7 Hz, H2), 9.39 (s, H5) ppm. 13C NMR (101 MHz, CD3COCD3): 122.7 (C8), 123.8 (C3), 124.7 (C4a), 136.6 (C4), 147.6 (C7), 151.3 (C8a), 154.1 (C5), 155.9 (C2) ppm.
Anal. calcd for 5 = C8H6AuCl3N2 (MW = 433.46): C, 22.17; H, 1.40; N, 6.46. Found: C, 22.20; H, 1.58; N, 6.49%. 1H NMR (400 MHz, CD3COCD3): 8.09 (dd, J = 8.2, 4.2 Hz, H5), 8.29 (dd, J = 8.0, 5.6 Hz, H4), 8.90 (dd, J = 8.0, 1.2 Hz, H6), 9.20 (d, J = 8.0 Hz, H3), 9.43 (d, J = 2.8 Hz, H7), 9.81 (d, J = 5.2 Hz, H2) ppm. 13C NMR (101 MHz, CD3COCD3): 125.6 (C5), 126.2 (C4), 127.2 (C4a), 140.0 (C6), 143.8 (C8a), 146.0 (C3), 156.1 (C2), 157.4 (C7) ppm. UV-Vis (acetone, λmax, nm): 323.0 (ε = 3.4 × 103 M−1 cm−1).
1,8-Naphthyridine (1,8-naph): 1H NMR (400 MHz, CD3COCD3): 7.64 (dd, J = 8.2, 4.2 Hz, H4 and H5), 8.46 (dd, J = 8.0, 2.0 Hz, H3 and H6), 9.13 (dd, J = 4.0, 2.0 Hz, H2 and H7) ppm. 13C NMR (101 MHz, CD3COCD3): 123.1 (C4 and C5), 123.9 (C4a), 138.2 (C3 and C6), 154.3 (C2 and C7), 157.3 (C8a) ppm.
* NMR data for the free ligands are given for comparative purposes.
Crystallographic data collection and refinement of the structures
X-ray measurements were carried out at 295 K with Mo Kα radiation (λ = 0.71073 Å) on Xcalibur kappa-geometry diffractometer equipped with Eos CCD detector using CrysAlisPro software.69 Analytical numeric absorption correction using a multifaceted crystal model based on expressions derived by Clark & Reid70 was applied. Further crystallographic and refinement data can be found in Table S1.† The structures were solved by direct methods using SHELXT71 and refined by full-matrix least-squares calculations on F2 with SHELXL.72 Anisotropic displacement parameters were refined for all non-hydrogen atoms. The positions of the hydrogen atoms attached to the carbon atoms were calculated at standardized distances (0.93 Å) and refined using a riding model with isotropic displacement parameters 20% higher than the isotropic equivalent of their carriers. Crystals of 3 and 5 were refined as racemic twins with a ratio of twin components 0.608:0.392 and 0.442:558, respectively. MERCURY45 computer graphics program was used to prepare drawings.
Quantum-mechanical calculations
The M06, M06-2X and B3LYP density functional theory methods were used in our calculations. The M06 and M06-2X methods are the hybrid meta-GGA functionals, where the Hartree–Fock exchange energy (X) is equal to 27 and 54% respectively.73 The B3LYP consists of Becke three-parameter hybrid exchange functional (B3)74 and the Lee, Yang, and Parr correlation functional (LYP) at the gradient-corrected DFT level.75 The B3LYP and M06 functionals are known to give good descriptions of transition metal complexes,73,76–78 while the M06-2X are recommended for the aromatic nitrogen-containing heterocycles.79 The basis set for calculations on diazanaphthalenes was Dunning's correlation consistent polarized valence double-ζ (cc-pVDZ) basis set,80 while for calculations for gold(III) complexes the basis set was composite: cc-pVDZ basis set for the hydrogen, nitrogen, carbon and chlorine80 and LanL2TZ(f) basis set for gold atom.81 The latter contains the LanL2 relativistic effective core potential (RECP) of Hay and Wadt71 and a flexible triple-ζ basis set augmented with f polarization functions in the treatment of valence shell on the Au atom. All structures were fully optimized without any geometric constraints. To check the influence of basis set effects for each optimized geometry of the diazanaphthalenes and gold(III) complexes, the additional single-point calculations were completed at the M06-2X/cc-pVTZ and M06-2X/(LanL2TZ(f)+cc-pVTZ) levels of theory, respectively (cc-pVTZ is correlation consistent polarized valence triple-ζ basis set80). The optimized structures were confirmed to be potential energy minima by vibrational frequency calculations at the same level of theory because no imaginary frequency was found. The natural bond analyses (NBO)82 were performed at M06-2X/cc-pVTZ and M06-2X/(LanL2TZ(f)+cc-pVTZ) levels of theory in order to provide indices of the diazanaphthalenes and gold(III) complexes, respectively. Finally, polarizability and hyperpolarizability calculations for the gold(III) complexes were performed at M06-2X/(LanL2TZ(f)+cc-pVTZ) level of theory. All DFT calculations were performed using the Gaussian 09 program package.83 The M06, M06-2X and B3LYP methods, as well as cc-pVDZ and cc-pVTZ basis sets were employed as implemented in the software package, while the LanL2TZ(f) basis set for gold was obtained from EMSL Basis set Exchange.84Conclusions
Although ligands with hypodentate coordination mode are known to increase their denticity when a coligand can be expelled from the metal coordination sphere, this is not taking place in [AuCl3(L)] complexes with aromatic diazaheterocycles (L). In the investigated series of Au(III) complexes, the dipodal diazanaphthalene ligands always act as monodentate. This seems to be a consequence of an increase of the electrostatic potential value on the uncoordinated nitrogen atom upon coordination of diazanaphthalene. Whenever in the diazaaromatic ligand the two sites available for coordination are nonequivalent (as in qz and 1,6-naph ligands), the Au–N bond is formed to the nitrogen atom in the naphthalene β position. This coordination mode eases the columnar π⋯π stacking interactions between the heteroaromatic rings in crystals. Crystallographic studies indicate the presence of the extended 4 + 1 or 4 + 2 geometry around the Au(III) center, mainly due to Au⋯Cl, but also due to Au⋯N interactions, and the importance of Cl⋯Cl contacts and C–H⋯Cl, and C–H⋯N hydrogen bonds as specific tools for organizing all these molecules in the solid state. On the other hand, the structure-determining role of π⋯π stacking between diazaaromatic ligands is unambiguous only in [AuCl3(benzodiazine)] complexes, which form centrosymmetric crystals and appears vague in the crystals formed by [AuCl3(naphthyridine)] complexes, which arrange into non-centrosymmetric polar aggregates. Interestingly, the molecules belonging to the latter group display the lowest values of polarizability (α) and hyperpolarizability (β). These results suggest that, in the investigated series of complexes, the dispersion contributions to the aromatic π⋯π interaction are significant.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded in part by the Ministry of Education, Science and Technological Development of the Republic of Serbia (Agreement no. 451-03-68/2020-14/200122) and was also supported in part by PL-Grid infrastructure and by the Serbian Academy of Sciences and Arts under strategic projects programme – grant agreement no. 01-2019-F65. B. Đ .G. gratefully acknowledges financial support from the Ministry of Education, Science and Technological Development of the Republic of Serbia during postdoctoral stay at the Adam Mickiewicz University in Poznań, Poland and Prof. Peter Comba (Heidelberg University, Germany) for the hospitality and support during postdoctoral stay in his group financed by DAAD.Notes and references
- M. S. Butler, J. Nat. Prod., 2004, 67, 2141 Search PubMed.
- J. A. Joule and K. Mills, Heterocyclic Chemistry, Blackwell Science, Oxford, 2000 Search PubMed.
- V. K. Indirapriyadharshini, P. Ramamurthy, V. Raghukumar and V. T. Ramakrishnan, Spectrochim. Acta, Part A, 2002, 58, 1535 Search PubMed.
- S. Derossi, M. Casanova, E. Iengo, E. Zangrando, M. Stener and E. Alessio, Inorg. Chem., 2007, 46, 11243 Search PubMed.
- M. Schweiger, S. R. Seidel, A. M. Arif and P. J. Stang, Angew. Chem., Int. Ed., 2001, 113, 3575 Search PubMed.
- M. Willermann, C. Mulcahy, R. K. O. Sigel, M. M. Cerdà, E. Freisinger, P. J. Sanz Miguel, M. Roitzsch and B. Lippert, Inorg. Chem., 2006, 45, 2093 Search PubMed.
- C. J. Sumby, Coord. Chem. Rev., 2011, 255, 1937 Search PubMed.
- C. Kaes, A. Katz and M. W. Hosseini, Chem. Rev., 2000, 100, 3553 Search PubMed.
- P. J. Steel, Coord. Chem. Rev., 1990, 106, 227 Search PubMed.
- D. P. Ašanin, M. D. Živković, S. Rajković, B. Warżajtis, U. Rychlewska and M. I. Djuran, Polyhedron, 2013, 51, 255 Search PubMed.
- S. Rajković, D. P. Ašanin, M. D. Živković and M. I. Djuran, Polyhedron, 2013, 65, 42 Search PubMed.
- S. Rajković, U. Rychlewska, B. Warżajtis, D. P. Ašanin, M. D. Živković and M. I. Djuran, Polyhedron, 2014, 67, 279 Search PubMed.
- V. Balzani, A. Juris, M. Venturi, S. Campagna and S. Serroni, Chem. Rev., 1996, 96, 759 Search PubMed.
- D. E. Fenton, Chem. Soc. Rev., 1999, 28, 159 Search PubMed.
- A. M. Barrios and S. J. Lippard, Inorg. Chem., 2001, 40, 1250 Search PubMed.
- A. M. Barrios and S. J. Lippard, Inorg. Chem., 2001, 40, 1060 Search PubMed.
- B. Bertrand and A. Casini, Dalton Trans., 2014, 43, 4209 Search PubMed.
- B. Đ. Glišić and M. I. Djuran, Dalton Trans., 2014, 43, 5950 Search PubMed.
- K. Palanichamy, N. Sreejayan and A. C. Ontko, J. Inorg. Biochem., 2012, 106, 32 Search PubMed.
- L. Messori, F. Abbate, G. Marcon, P. Orioli, M. Fontani, E. Mini, T. Mazzei, S. Carotti, T. O'Connell and P. Zanello, J. Med. Chem., 2000, 43, 3541 Search PubMed.
- G. Marcon, S. Carotti, M. Coronnello, L. Messori, E. Mini, P. Orioli, T. Mazzei, M. A. Cinellu and G. Minghetti, J. Med. Chem., 2002, 45, 1672 Search PubMed.
- L. Messori, G. Marcon, M. A. Cinellu, M. Coronnelo, E. Mini, C. Gabbiani and P. Orioli, Bioorg. Med. Chem., 2004, 12, 6039 Search PubMed.
- A. Casini, M. A. Cinellu, G. Minghetti, C. Gabbiani, M. Coronnello, E. Mini and L. Messori, J. Med. Chem., 2006, 49, 5524 Search PubMed.
- C. Gabbiani, A. Casini, L. Messori, A. Guerri, M. A. Cinellu, G. Minghetti, M. Corsini, C. Rosani, P. Zanello and M. Arca, Inorg. Chem., 2008, 47, 2368 Search PubMed.
- A. N. Wein, A. T. Stockhausen, K. I. Hardcastle, M. R. Saadein, S. Peng, D. Wang, D. M. Shin, Z. Chen and J. F. Eichler, J. Inorg. Biochem., 2011, 105, 663 Search PubMed.
- R. W.-Y. Sun and C.-M. Che, Coord. Chem. Rev., 2009, 253, 1682 Search PubMed.
- C. T. Lum, A. S.-T. Wong, M. Lin, C.-M. Che and R. W.-Y. Sun, Chem. Commun., 2013, 49, 4364 Search PubMed.
- R. D. Teo, H. B. Gray, P. Lim, J. Termini, E. Domeshek and Z. Gross, Chem. Commun., 2014, 50, 13789 Search PubMed.
- N. D. Savić, D. R. Milivojevic, B. Đ. Glišić, T. Ilic-Tomic, J. Veselinovic, A. Pavic, B. Vasiljevic, J. Nikodinovic-Runic and M. I. Djuran, RSC Adv., 2016, 6, 13193 Search PubMed.
- C. Gabbiani, L. Messori, M. A. Cinellu, A. Casini, P. Mura, A. R. Sannella, C. Severini, G. Majori, A. R. Bilia and F. F. Vincieri, J. Inorg. Biochem., 2009, 103, 310 Search PubMed.
- M. Navarro, C. Hernández, I. Colmenares, P. Hernández, M. Fernández, A. Sierraalta and E. Marchán, J. Inorg. Biochem., 2007, 101, 111 Search PubMed.
- S. P. Fricker, R. M. Mosi, B. R. Cameron, I. Baird, Y. Zhu, V. Anastassov, J. Cox, P. S. Doyle, E. Hansell, G. Lau, J. Langille, M. Olsen, L. Qin, R. Skerlj, R. S. Y. Wong, Z. Santucci and J. H. McKerrow, J. Inorg. Biochem., 2008, 102, 1839 Search PubMed.
- R. A. Sánchez-Delgado, M. Navarro, K. Lazardi, R. Atencio, M. Capparelli, F. Vargas, J. A. Urbina, A. Bouillez, A. F. Noels and D. Masi, Inorg. Chim. Acta, 1998, 275–276, 528 Search PubMed.
- E. Nyarko, T. Hara, D. J. Grab, A. Habib, Y. Kim, O. Nikolskaia, T. Fukuma and M. Tabata, Chem.-Biol. Interact., 2004, 148, 19 Search PubMed.
- B. Warżajtis, B. Đ. Glišić, N. S. Radulović, U. Rychlewska and M. I. Djuran, Polyhedron, 2014, 79, 221 Search PubMed.
- B. Đ. Glišić, B. Warżajtis, N. S. Radulović, U. Rychlewska and M. I. Djuran, Polyhedron, 2015, 87, 208 Search PubMed.
- A. Pavic, B. Đ. Glišić, S. Vojnovic, B. Warżajtis, N. D. Savić, M. Antić, S. Radenković, G. V. Janjić, J. Nikodinovic-Runic, U. Rychlewska and M. I. Djuran, J. Inorg. Biochem., 2017, 174, 156 Search PubMed.
- B. Ð. Glišić, M. Hoffmann, B. Warżajtis, M. S. Genčić, P. D. Blagojević, N. S. Radulović, U. Rychlewska and M. I. Djuran, Polyhedron, 2016, 105, 137 Search PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 Search PubMed.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed., 1995, 34, 1555 Search PubMed.
- G. R. Desiraju, P. Shing Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711 Search PubMed.
- Ch. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 Search PubMed.
- G. R. Desiraju and R. J. Parthasarathy, J. Am. Chem. Soc., 1989, 111, 8725 Search PubMed.
- T. P. Andrejević, B. Warżajtis, B. Đ. Glišić, S. Vojnovic, M. Mojicevic, N. Lj. Stevanović, J. Nikodinovic-Runic, U. Rychlewska and M. I. Djuran, J. Inorg. Biochem., 2020, 208, 111089 Search PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453 Search PubMed.
- A. I. Kitaigorodskii, Organic Chemical Crystallography, Consultant’s Bureau, New York, 1961 Search PubMed.
- S. Zhu, K. X. Moreno, R. M. Jenkins and J. A. Walmsley, Dalton Trans., 2008, 6401 Search PubMed.
- P. J. Black and M. L. Heffernan, Aust. J. Chem., 1965, 18, 707 Search PubMed.
- W. W. Paudler and T. J. Kress, J. Heterocyclic Chem., 1965, 2, 393 Search PubMed.
- B. Warżajtis, B. Đ. Glišić, M. D. Živković, S. Rajković, M. I. Djuran and U. Rychlewska, Polyhedron, 2015, 91, 35 Search PubMed.
- D. Niedzielska, T. Pawlak, M. Bozejewicz, A. Wojtczak, L. Pazderski and E. Szłyk, J. Mol. Struct., 2013, 1032, 195 Search PubMed.
- A. Pevec and A. Demšar, J. Fluorine Chem., 2008, 129, 707 Search PubMed.
- A. Pevec, M. Tekavec and A. Demšar, Polyhedron, 2011, 30, 549 Search PubMed.
- K. Esumi, M. Nawa, N. Aihara and K. Usui, New J. Chem., 1998, 22, 719 Search PubMed.
- N. Pantelić, B. B. Zmejkovski, J. Trifunović-Macedoljan, A. Savić, D. Stanković, A. Damjanović, Z. Juranić, G. N. Kaluđerović and T. J. Sabo, J. Inorg. Biochem., 2013, 128, 146 Search PubMed.
- N. Pantelić, T. P. Stanojković, B. B. Zmejkovski, T. J. Sabo and G. N. Kaluđerović, Eur. J. Med. Chem., 2015, 90, 766 Search PubMed.
- P. Tyagi, S. Chandra, B. S. Saraswat and D. Sharma, Spectrochim. Acta, Part A, 2015, 143, 1 Search PubMed.
- H. Hu, L. Du, X. Li, H. Zhao, X. Zhang, S. Shi, H. Li, X. Tang and J. Yang, Int. J. Electrochem. Sci., 2013, 8, 11228 Search PubMed.
- Y.-H. Zhou, J. Tao, D.-L. Sun, L.-Q. Chen, W.-G. Ji and Y. Cheng, Polyhedron, 2015, 85, 849 Search PubMed.
- F. J. Luque, J. M. López and M. Orozco, Theor. Chem. Acc., 2000, 103, 343 Search PubMed.
- R. V. Pinjari and S. P. Gejji, J. Phys. Chem. A, 2008, 112, 12679 Search PubMed.
- Y.-X. Sun, Q.-L. Hao, W.-X. Wei, Z.-X. Yu, L.-D. Lu, X. Wang and Y.-S. Wang, J. Mol. Struct.: THEOCHEM, 2009, 904, 74 Search PubMed.
- L. Pilia, M. Pizzotti, F. Tessore and N. Robertson, Inorg. Chem., 2014, 53, 4517 Search PubMed.
- S. Di Bella, Chem. Soc. Rev., 2001, 30, 355 Search PubMed.
- K. S. Thanthiriwatte and K. M. N. de Silva, J. Mol. Struct.: THEOCHEM, 2002, 617, 169 Search PubMed.
- A. Esme and S. G. Sagdinc, J. Mol. Struct., 2013, 1048, 185 Search PubMed.
- K. Sayin and D. Karakaş, Spectrochim. Acta, Part A, 2015, 144, 176 Search PubMed.
- T. Hahn and H. Klapper, Point groups and crystal classes, in International Tables for Crystallography, ed. T. Hahn and D. Reidel, Publishing Company, Dordrecht, Holland, 1983, vol. A, p. 746 Search PubMed.
- CrysAlis PRO, Agilent Technologies, Yarnton, Oxfordshire, England, 2014 Search PubMed.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 Search PubMed.
- S. Q. Niu and M. B. Hall, Chem. Rev., 2000, 100, 353 Search PubMed.
- A. P. Martins, A. Marrone, A. Ciancetta, A. G. Cobo, M. Echevarría, T. F. Moura, N. Re, A. Casini and G. Soveral, PLoS One, 2012, 7, e37435 Search PubMed.
- Y. Zhao and D. G. Truhlar, Chem. Phys. Lett., 2011, 502, 1 Search PubMed.
- R. G. A. R. Maclagan, S. Gronert and M. Meot-Ner, J. Phys. Chem. A, 2015, 119, 127 Search PubMed.
- (a) T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007 Search PubMed; (b) D. E. Woon and T. H. Dunning Jr, J. Chem. Phys., 1993, 98, 1358 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 Search PubMed.
- (a) A. E. Reed and F. Weinhold, J. Chem. Phys., 1983, 78, 4066 Search PubMed; (b) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 Search PubMed; (c) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- (a) D. Feller, J. Comp. Chem., 1996, 17, 1571 Search PubMed; (b) K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and Tables S1–S5. CCDC 2017216–2017220 (1–5). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra08731a |
| This journal is © The Royal Society of Chemistry 2020 |