
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mild hydrothermally treated brewer's spent grain for efficient removal of uranyl and rare earth metal ions†
Yi Su a,
Wendelin Böhmb,
Marco Wenzela,
Silvia Paaschc,
Margret Ackerd,
Thomas Doerte,
Eike Brunnerc,
Thomas Henleb and
Jan J. Weigand*a
a,
Wendelin Böhmb,
Marco Wenzela,
Silvia Paaschc,
Margret Ackerd,
Thomas Doerte,
Eike Brunnerc,
Thomas Henleb and
Jan J. Weigand*a
aChair of Inorganic Molecular Chemistry, TU Dresden, 01062 Dresden, Germany. E-mail: jan.weigand@tu-dresden.de
bChair of Food Chemistry, TU Dresden, 01062 Dresden, Germany
cChair of Bioanalytical Chemistry, TU Dresden, 01062 Dresden, Germany
dCentral Radionuclide Laboratory, TU Dresden, 01062 Dresden, Germany
eChair of Inorganic Chemistry II, TU Dresden, 01062 Dresden, Germany
First published on 22nd December 2020
Abstract
The increasing concerns on uranium and rare earth metal ion pollution in the environment require sustainable strategies to remove them from wastewater. The present study reports an eco-friendly approach to convert a kind of protein-rich biomass, brewer's spent grain (BSG), into effective biosorbents for uranyl and rare earth metal ions. The employed method reduces the energy consumption by performing the hydrothermal treatment at a significantly lower temperature (150 °C) than conventional hydrothermal carbonization. In addition, with the aid of the Maillard reaction between carbohydrates and proteins forming melanoidins, further activation processes are not required. Treatment at 150 °C for 16 h results in an altered biosorbent (ABSG) with increased content of carboxyl groups (1.46 mmol g−1) and a maximum adsorption capacity for La3+, Eu3+, Yb3+ (pH = 5.7) and UO22+ (pH = 4.7) of 38, 68, 46 and 221 mg g−1, respectively. Various characterization methods such as FT-IR, 13C CP/MAS NMR, SEM-EDX and STA-GC-MS analysis were performed to characterize the obtained material and to disclose the adsorption mechanisms. Aside from oxygen-containing functional groups, nitrogen-containing functional groups also contribute to the adsorption. These results strongly indicate that mild hydrothermal treatment of BSG could be applied as a greener, low-cost method to produce effective adsorbents for uranyl and rare earth metal ion removal.
Introduction
Uranium is one of the most widely used radioactive elements and an unreplaceable raw material for nuclear energy. In recent years, the concentration of uranium in aqueous environments has increased significantly as a result of human activities, such as mining and nuclear industry.1 These unwanted releases of uranium cause severe environmental pollution and health risks due to the chemical and radiologic toxicity of uranium.2 Owing to this, the World Health Organization (WHO) has recommended that the maximum concentration of uranium in drinking water should not exceed 30 μg L−1.3 Therefore, the removal of uranium from wastewater is necessary to protect the environment and health. In addition, rare earth elements are often found associated with uranium especially in wastewater from mining and spent nuclear fuel.4,5 It is reported that rare earth elements also can be tissue-specific bioaccumulated, thus causing damage to the lungs, liver and brain.6 Considering the health and environment hazards of uranyl and rare earth metal ions, it would be efficient and economical to remove both from wastewater simultaneously.Recently, adsorption process has risen more and more interests in both academic and industry fields to remove uranyl and rare earth metal ions from wastewater. This is because adsorption demonstrates obvious advantages e.g. fast kinetics, high adsorption capacity, remarkable stability and the avoidance of organic solvent. Furthermore, adsorption is considered to be inexpensive and easier to use than other methods.4 Different adsorbents have been developed and tested for the adsorption of uranyl and rare earth metal ions, such as graphene oxide-based adsorbent,7,8 nanomaterials,9 cationic resin,10 mesoporous silica-based adsorbent11 and biosorbent.12 On the base of cost-to-performance ratio, biosorbents are most preferable and economically viable among all the adsorbents, as they are convenient, readily available and cheap.13 They also present great potential as an eco-friendly alternative to synthetic adsorbents.14 Various kinds of biopolymers have been reported as suitable biosorbents for uranium and rare earth metal ions, such as fungi (yeast (Saccharomyces cerevisiae) embedded cellulose, 25.9 mg g−1 for Eu3+),15 bacteria (Pseudomonas aeruginosa, 208 μmol g−1 dry biomass for La3+)16 and cellulose nanofiber (167 mg g−1 for UO22+).17 However, the adsorption capacity of natural biomass is normally lower than that of synthetic adsorbents (e.g. graphene oxide-activated carbon felt composite, 298 mg g−1 for UO22+),18 which requires proper modification methods, either by chemical functionalization or thermal conversion.12
Comparing to traditional chemical modification methods for biosorbents, hydrothermal treatment presents some unique benefits as a greener and economical approach. It is highly flexible on the choice of feedstock, and there is no need of pre-drying of the biomass feeds and no toxic chemicals are required.19 In this context, hydrothermal carbonization (HTC) is the most commonly used hydrothermal process for biosorbent production, and is typically performed at high temperatures of up to 250 °C during which the biomass is submerged in water and heated under pressure.20 The resulting solid product, named hydrochar, possesses abundant surface functional groups21 and has a high affinity towards uranium and rare earth metal ions. For example, hydrochar prepared from pine needles22 and carbonaceous spheres from glucose with AlCl3 catalyst23 present adsorption capacity for UO22+ of 62.7 mg g−1 and 163 mg g−1, respectively. However, the application of hydrochar is limited by some drawbacks, like high operation temperature and pressure, low porosity, and low specific surface area. Especially, physical or chemical activation is usually required to obtain satisfying adsorption capacity of hydrochars.24
The goal of our studies is to develop an adsorbent for UO22+ and trivalent rare earth metal ions removal employing brewer's spent grain (BSG), a large-scale produced by-product of the beer production, as a low-cost and convenient manageable raw material. Literatures have reported the application of BSG as a biosorbent for metal ions, such as Mn2+, Zn2+, Ni2+, Cd2+, Cu2+ and Pb2+.25–28 Recently, activated hydrochar produced from BSG is reported for the adsorption of acetaminophen.29 Nevertheless, it is worth to notice that the activated hydrochar was obtained at 220 °C for 16 h with a chemical activation process using KOH and high temperature (800 °C). Both, high temperature and the strong alkaline condition inevitably undermine the environmental benefits of the hydrochar.
The common starting materials for HTC processes are lignocellulosic materials, while BSG could be described as a biomass rich in lignocellulose and protein.30 It is thus possible that the Maillard reaction takes place at an elevated temperature and the resulting products contribute to enhance the adsorption capacity.31 Based on this hypothesis, mild hydrothermal conditions (temperature of 150 °C) are evaluated in the present work to enhance the adsorption capacity of BSG with the aid of Maillard reaction. The results show that Maillard reaction between carbohydrates and proteins with the formation of melanoidins plays an important role in the enhanced presence of functional groups suitable for metal coordination and yields in high adsorption capacity of the altered biosorbent (ABSG) towards UO22+ without further treatment. This minimizes energy consumption and makes it possible to dispense additional activation processes, so that current disadvantages of the HTC process are overcome, indicating a simpler and eco-friendly route for the production of biosorbents. Effects of reaction temperature and time on the adsorption capacity, the properties of products and the Maillard reaction were investigated to shed light on the involved reaction mechanisms. Furthermore, spectroscopic and thermogravimetric methods were employed to fully characterize the obtained biosorbents. The adsorption properties towards UO22+ and La3+, Eu3+ and Yb3+ as representatives of the early, middle and late lanthanides onto BSG-based biosorbents were examined including the adsorption mechanisms.
Experimental
Materials
NaOH (97%, VWR chemicals), La(NO3)3·6H2O (99.9%, Thermo Fisher GmbH), Eu(NO3)3·5H2O (99.9%, Sigma-Aldrich), Yb(NO3)3·5H2O (99.9%, Sigma-Aldrich), UO2(CH3COO)2·2H2O (Merck), HNO3 (supra pure, 69%, Carl Roth GmbH), Na2CO3 (99.5%, Grüssing GmbH), NaHCO3 (99%, Grüssing GmbH) and potassium hydrogen phthalate (Laborchemie Apolda GmbH) were applied as purchased. Ultra-pure water (18.2 MΩ cm−1, arium® pro, Sartorius) was used in all experiments.Preparation of standardized brewer's spent grain
Brewer's spent grain (BSG, water content 78 wt%) was obtained from our laboratory-scale brewery plant (Technical University of Dresden, Germany) during the production of a Pilsner beer directly after the mashing process. 14.6 kg Pilsner malt (Weyermann) was used in this brewing. During mashing process, 53 L of water was poured initially, with a replenishment volume of 58 L. The temperature and time of different mashing procedures are summarized in Table S1 in the ESI.† The fresh BSG was then stored at −16 °C until further processing. For the preparation of standardized BSG, the material was defrosted at room temperature and dried at 60 °C under reduced pressure (<70 mbar) for 72 h (water content 3.0 wt%). Afterwards the BSG was milled using a coffee grinder (MayOcean) for 30 s, left to rest for 10 s and milled again for 20 s. The milled BSG was passed through a 315 μm sieve, characterized and used for the adsorption studies.Hydrothermal treatment of brewer's spent grain (BSG) to yield altered biosorbent (ABSG)
Hydrothermal treatment of BSG was conducted in an autoclave reactor (DAB-3, Berghof Products + Instruments GmbH) with 250 mL volume. Typically, 15 g fresh, defrosted BSG was mixed manually inside the reactor with 10 mL ultra-pure water and the pH was adjusted to 10 using 0.01 M NaOH. The reactor was heated in an oven (Dry-line 115, VWR International) at different temperatures (100 °C, 125 °C, 150 °C, 175 °C) for various time periods (1 h, 4 h, 8 h, 16 h, 24 h). After reaction, the solid product was collected and dried at 60 °C under reduced pressure (<70 mbar) overnight to reduce the water content to less than 5 wt%, crushed and passed through 315 μm sieve to obtain a homogeneous fraction. The hydrothermal treated BSG was designated with ABSG-θ, t, where θ is the reaction temperature and t is the reaction time, in order to investigate the influence of the hydrothermal treatment parameters. For general characterization (13C solid state NMR, oxygen functional groups and STA-GC-MS analysis) and adsorption study, ABSG (ABSG-150 °C, 16 h) was used with a water content of 4.3 wt%, N content of 5.0 wt% and an estimate protein content of 29.2 wt%.Adsorption study
Batch adsorption experiments for BSG and ABSG were performed by suspending 2 mg dry adsorbent in 2 mL solution containing the metal ions of the required concentration in micro centrifuge tubes (2 cm3, Safe-Lock, Eppendorf) with an overhead shaker (Reax 2, Heidolph) at room temperature. For experiments using 169YbCl3 radiotracer, 1 mg adsorbent and 1 mL metal solution was used. The kinetic studies were conducted under constant initial concentration at pH = 5.7 for La3+, Eu3+ and Yb3+ and at pH = 4.7 for UO22+ with a series of time intervals ranging from 0 to 180 min. 0.1 M or 1.0 M HNO3 was used to adjust the initial pH in the range of 1–6 for La3+, Eu3+ and Yb3+ and 1–5 for UO22+. The pH range was carefully chosen in order to prevent precipitation. The equilibrium pH was measured with an InLab micro pH electrode (Mettler Toledo). Adsorption isotherms were obtained with different initial metal concentrations ranging from 100 mg L−1 to 1000 mg L−1 at pH = 5.7 for La3+, Eu3+ and Yb3+ and at pH = 4.7 for UO22+. After adsorption the solution was filtered through a 13 mm syringe filter with a 0.22 μm PTFE film (Fisher Scientific), and the metal concentration before and after adsorption was determined by ICP-OES (OPTIMA 2000DV, PerkinElmer). The adsorption capacity (qe, mg g−1) was calculated using the following equation (eqn (1)):
 | (1) |
Adsorption experiments using 169Yb radiotracer (YbCl3, Polatom) were also performed. In these experiments, the metal ions distribution between the solution and adsorbent was determined radiometrically employing radiation from 169Yb with a NaI (TI) scintillation counter (Hidex AMG, Hidex GmbH). The count rate (CPM, counts per minute) of 0.5 mL of the supernatant liquid and the remaining 0.5 mL supernatant liquid with the adsorbent were determined. The adsorption capacity (qe, mg g−1) is calculated as eqn (2):
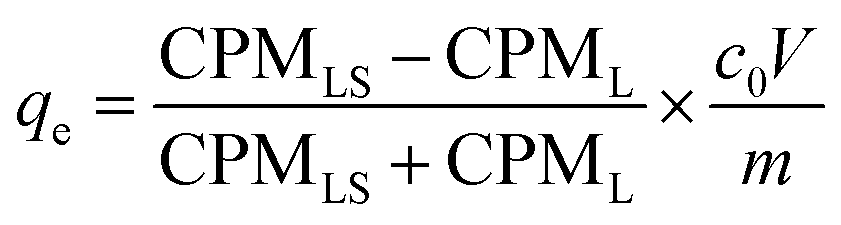 | (2) |
Effects of temperature on the adsorption capacity of the biosorbents were examined by performing adsorption isotherms at three different temperatures, 25 °C, 45 °C and 65 °C. Generally, 2 mg adsorbent was mixed with 2 mL metal ions solution with different initial concentrations (100–600 mg L−1) at pH = 4.7 (UO22+) and pH = 5.7 (La3+, Eu3+ and Yb3+) in 10 mL test tube using magnetic stirrer (IKA®, RCT basic) at a stirrer speed of 180 rpm. The temperature was controlled by a circulation thermostat (UH 4, MLW-Medingen). Thermodynamic parameters were calculated according to the following equations (eqn (3) and (4)):
| ΔG = ΔH0 − TΔS0 | (3) |
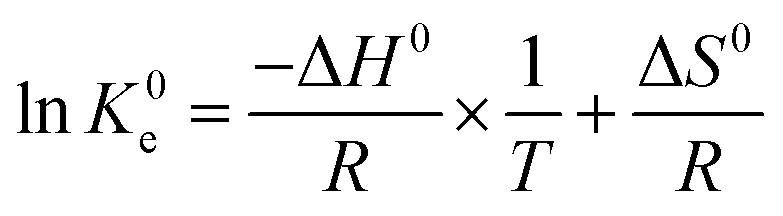 | (4) |
Characterization
BSG and ABSG with a water content of less than 5 wt% were used in all characterizations. Infrared (FT-IR) spectra of adsorbents before and after the metal adsorption were obtained with a single beam Fourier transform infrared VERTEX 70 spectrometer (Bruker). An ATR (attenuated total reflectance) unit (diamond) with a single reflection optics at an interaction angle of 45° was used. The spectra were recorded over the range of 4500 to 400 cm−1 with a resolution of 4 cm−1, and averaged over 32 scans. In order to investigate the detailed changes of chemical structures, the obtained spectra were processed using the OPUS software package as provided by Bruker to compare the intensity of certain bands. Baseline corrections were applied at 3014 cm−1, 2807 cm−1, 1783 cm−1, 1532 cm−1, 1086 cm−1, 900 cm−1 and 400 cm−1. Then the spectra were normalized respecting to –CH2– antisymmetric stretching vibration bands (≈2924 cm−1). For normalization, the absorbance value of this band was set to 1.0 and the complete spectrum was multiplied by a ratio factor. The raw spectra are provided in Fig. S1 in the ESI† to demonstrate that no obvious intensity changes occurred during the spectra manipulation.13C solid state NMR spectra were recorded on a Bruker Ascend 800 MHz spectrometer using a commercial 3.2 mm MAS NMR probe and operating at a resonance frequency of 201.2 MHz. The MAS frequency was 15 kHz. Adamantane was used as external standard. Ramped 1H–13C cross-polarization (CP, contact time: 4 ms) and SPINAL 1H-decoupling during the signal acquisition was applied. The recycle delay was 3 s. 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 scans (BSG)/28000 scans (ABSG) were accumulated for signal-to-noise improvement.
000 scans (BSG)/28000 scans (ABSG) were accumulated for signal-to-noise improvement.
The specific surface area of BSG and ABSG was determined by the Brunauer–Emmett–Teller (BET) method (SA 9600, Horiba Scientific). The sample was subjected to a pre-treatment at 110 °C for 90 min. The analysis was performed using 8% N2 in He. The adsorption took place at −196 °C and the desorption was at room temperature.
Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX) analysis were performed on a scanning electron microscope (SU8020, HITACHI) equipped with an energy-dispersive X-ray spectrometer X-MaxN (OXFORD Instrument) at an electron beam voltage of 20 kV. The metal-loaded ABSG samples were dried at 60 °C under reduced pressure (<70 mbar) for 48 h and coated with a Au layer by a rotary pumped coater (Q150R ES, Quorum) at 5 mA. The surface morphology images of metal loaded ABSG were taken at magnification of 1000 times, and the EDX mapping was taken at 20 kV/10 μA, magnification of 1000–5000 times for 25 frames.
The thermal stability of adsorbents and volatile products produced during decomposition were analyzed by simultaneous thermal analyzer (STA 8000, PerkinElmer) coupled with a GC-MS (GC Clarus 680, MS Clarus SQ 8S, PerkinElmer). The samples were heated from 40 °C to 600 °C with a heating rate of 20 °C min−1 under helium atmosphere. The volatile products generated at 375–385 °C (BSG, ABSG) and at 340–360 °C (Yb-ABSG) were detected using GC-MS. The GC temperature was initially set to 35 °C with a hold time of 3 min, and then increased with a heat rate of 5 °C min−1 until 220 °C, and hold at 220 °C for 3 min. The obtained MS spectra were interpreted using NIST mass spectral search program provided by PerkinElmer.
The content of Ca, Fe, Mn, Mg, Zn, K, Na, P and Si (mineral elements) of BSG and ABSG were determined by ICP-OES after microwave-assisted digestion. In general, 10 mL HNO3 (supra pure, 69%) were added to 0.1 g adsorbent and reacted for 15 min at room temperature before heating for 15 min at 210 °C (MARS 6, CEM GmbH). Elemental analysis were performed on a Vario MICRO cube (Elementar Analysensysteme GmbH) in CHNS mode to determine the content of carbon, nitrogen, hydrogen and sulfur. The oxygen content was calculated by mass balance considering the content of carbon, nitrogen, hydrogen, sulfur and the mineral elements determined by ICP-OES. The results of elemental analysis and mineral elements analysis are provided in Tables S2 and S3 in the ESI.† Protein content was estimated according to nitrogen content by multiplying factor 5.83.33
Maillard reaction products (MRPs) were analyzed according to literature with slight modifications.34 Typically, the samples were enzymatically hydrolyzed, cleaned up through a solid phase extraction cartridge, and analyzed via HPLC-MS/MS (Agilent Technologies). For separation, a HPLC-column (Kinetex-C-18 column, 1.7 μm, 100 Å, 50 × 2.1 mm) was used. Amino acid analysis (ion-exchange chromatography with ninhydrin detection) was performed by the method proposed by Hellwig et al..35 Point of zero charge (pHpzc) of BSG and ABSG was determined by solid addition method,36 using 0.2 g adsorbent suspended in 10 mL 0.1 M NaNO3 solution. The initial pH of the latter was adjusted to 1–10 using 0.1 M HNO3 or 0.1 M NaOH. The content of oxygen functional groups (OFGs) were quantified using Boehm titration.37 In general, a mixture of 0.9 g adsorbents and 50.00 mL of one of the three reaction bases, NaHCO3, Na2CO3 and NaOH, in a concentration of 0.05 M was shaken for 24 h. The mixtures were filtered and three 10.00 mL aliquots were taken for titration. The NaHCO3 and NaOH samples were acidified with 20.00 mL 0.05 M HCl, whereas for Na2CO3 samples 30.00 mL of 0.05 M HCl was added. The acidified solutions were then put into ultrasonic bath (Sonorex RK 52H, Bandelin electronic GmbH & Co. KG) for 20 min to expel dissolved CO2 and titrated with 0.05 M NaOH using phenolphthalein indicator.
Results and discussion
Hydrothermal treatment of brewer's spent grain
 | ||
| Fig. 1 Effects of (a) reaction temperature (for 16 h) and (b) time (at 150 °C) on the adsorption capacity and chemical composition of ABSG. For adsorption experiments: 2 mg ABSG/2 mL solution, pH = 5.7, c0(La3+) = 100 mg L−1, t = 2 h, room temperature. | ||
Fig. 1(a) and (b) also show the changes of the element composition of ABSG as the reaction temperature and time varies. The carbon content increases as the reaction temperature increases and time prolongs, along with the decreasing of hydrogen and oxygen content. This may be due to the condensation polymerization mechanism which causes a loss of O-containing functional groups and low polarity of ABSG.38 This explains the decrease of adsorption capacity when the temperature and time exceed certain values. The nitrogen content of ABSG remains nearly unchanged as the temperature and time varies, which illustrates that N-containing groups have a higher persistence than O-containing functional groups. In this case, the N-containing groups may be incorporated into ABSG via Maillard reaction.39 In order to further investigate the reaction type during hydrothermal treatment, H/C atomic ratios versus O/C atomic ratios of different samples and reference materials are plotted in van Krevelen diagram40 as shown in Fig. 2. BSG is depicted as a kind of lignocellulose material with a chemical composition more similar to cellulose than pure lignin. The direct vector points at a dehydration pathway as the temperature increases, while both the dehydration and decarboxylation reaction occur with increasing reaction time. Both reaction pathways contribute to the lower H/C and O/C ratios of ABSG.40 In addition, the decrease of the H/C ratio also indicates aromatization mechanism during the hydrothermal conversion.41 It is noteworthy that all the reaction mechanisms, dehydration, decarboxylation, aromatization as well as Maillard reaction do not take place in consecutive steps, but rather in a parallel network of different pathways leading to the properties of ABSG.
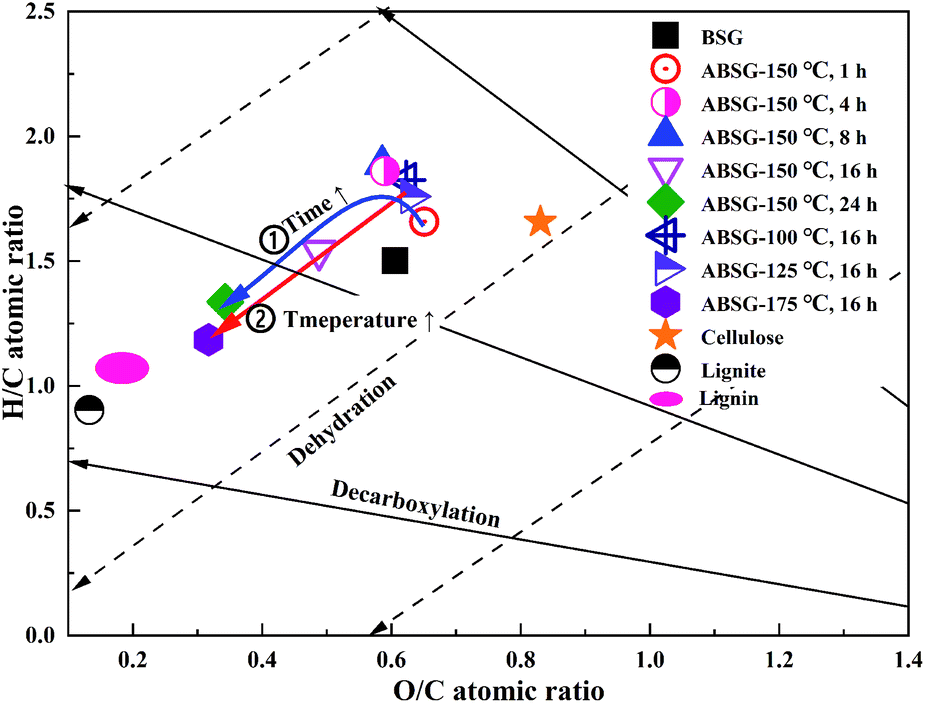 | ||
| Fig. 2 van Krevelen diagram of BSG-based biosorbents, typical biomass and coal. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration) increases when a temperature over 150 °C and a time over 8 h is applied, while the intensity of C–O stretching vibration at 1021 cm−1 decreases continuously with increasing temperature and time. These changes indicate the transformation of C–O bonds in the glucose units of cellulose and hemicellulose to CO bonds,41 resulting in the increase of adsorption capacity when the temperature and time increase. When a temperature of 175 °C is applied, all above mentioned absorption bands decrease in intensity, indicating the loss of functional groups and thus decreasing adsorption capacity.
O stretching vibration) increases when a temperature over 150 °C and a time over 8 h is applied, while the intensity of C–O stretching vibration at 1021 cm−1 decreases continuously with increasing temperature and time. These changes indicate the transformation of C–O bonds in the glucose units of cellulose and hemicellulose to CO bonds,41 resulting in the increase of adsorption capacity when the temperature and time increase. When a temperature of 175 °C is applied, all above mentioned absorption bands decrease in intensity, indicating the loss of functional groups and thus decreasing adsorption capacity.
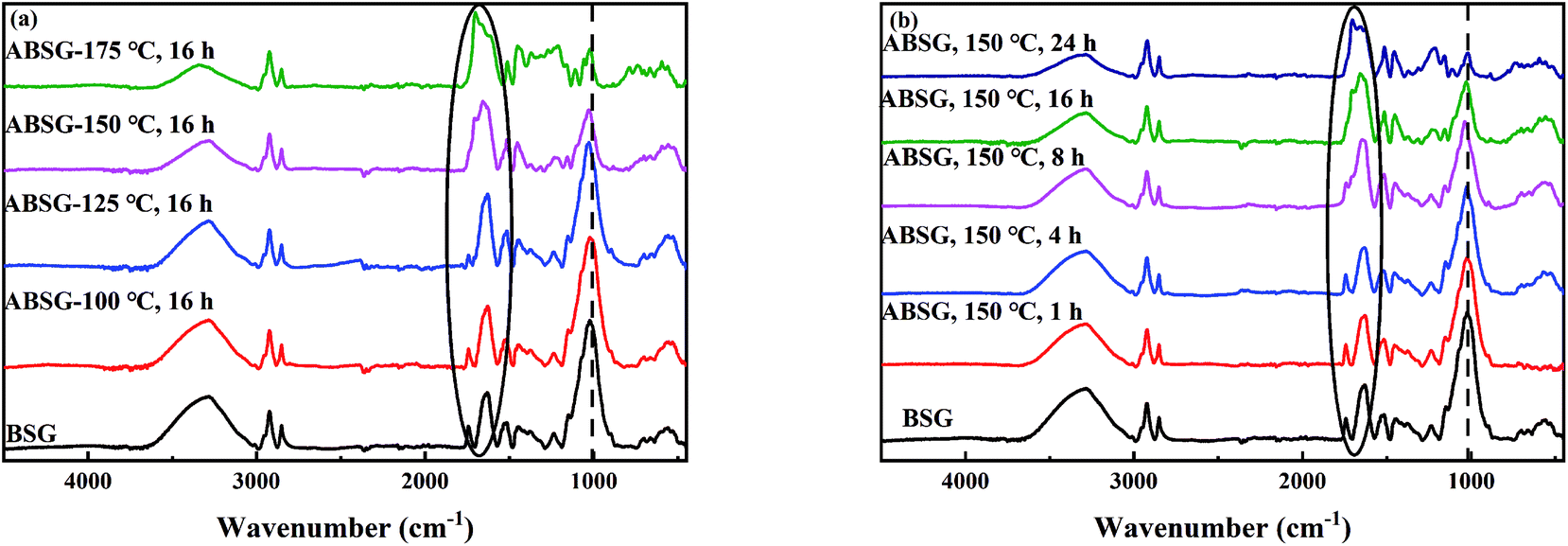 | ||
| Fig. 3 FT-IR spectra of BSG and ABSG samples with different (a) reaction temperature (for 16 h) and (b) time (at 150 °C). For adsorption experiments: 2 mg ABSG/2 mL solution, pH = 5.7, c0(La3+) = 100 mg L−1, t = 2 h, room temperature. | ||
 | ||
| Fig. 4 Effects of reaction temperature (for 16 h) on the (a) content of MRPs, (b) adsorption capacity and the content of amino acids, and effects of reaction time (at 150 °C) on the (c) content of MRPs, (d) adsorption capacity and the content of amino acids. For adsorption experiments: 2 mg ABSG/2 mL solution, pH = 5.7, c0(La3+) = 100 mg L−1, t = 2 h, room temperature. | ||
Characterization
In order to gain information about structure changes of BSG after hydrothermal treatment, 13C CP/MAS solid state NMR spectra (Fig. 5(a)) were recorded. The spectra show that after hydrothermal treatment the main chemical structure of BSG is conserved. The resonances in both spectra can be divided into four groups: aliphatic carbons (10–40 ppm), typical cellulose carbons C1 (105 ppm), C4 (80–90 ppm), C2,3,5 (72–75 ppm), C6 (62–65 ppm), lignin aromatic carbons (130 ppm) and carboxyl carbons (174 ppm).46,47 The emerging resonance between 110–160 ppm after hydrothermal treatment points at an increase content of unsaturated or aromatic carbons in ABSG. This is in accord with the increased intensity of the absorption band at 1650 cm−1 attributed to the stretching vibration of the CO bond in the FT-IR spectra and the increase of the H/C ratio obtained in the elemental analysis. Compared to BSG, the 13C NMR spectrum of ABSG shows a more intense signal at 56 ppm due to methoxyl carbons, presumably bound to an aromatic ring,47 which also results from the aromatization during hydrothermal treatment. However, it must be noted that the quantitative analysis of CP spectra is limited by the different polarization transfer efficiencies of the various signals and depends on the water content of the sample. Probably, the extremely restricted mobility of the protons in BSG and/or their chemical inhomogeneity results in broad and hence practically invisible signals. In contrast, the slightly higher water content of ABSG may lead to better resolved signals in the spectrum. Note that a higher mobility of the protons can also cause a weakening or even suppression of the CP signals.
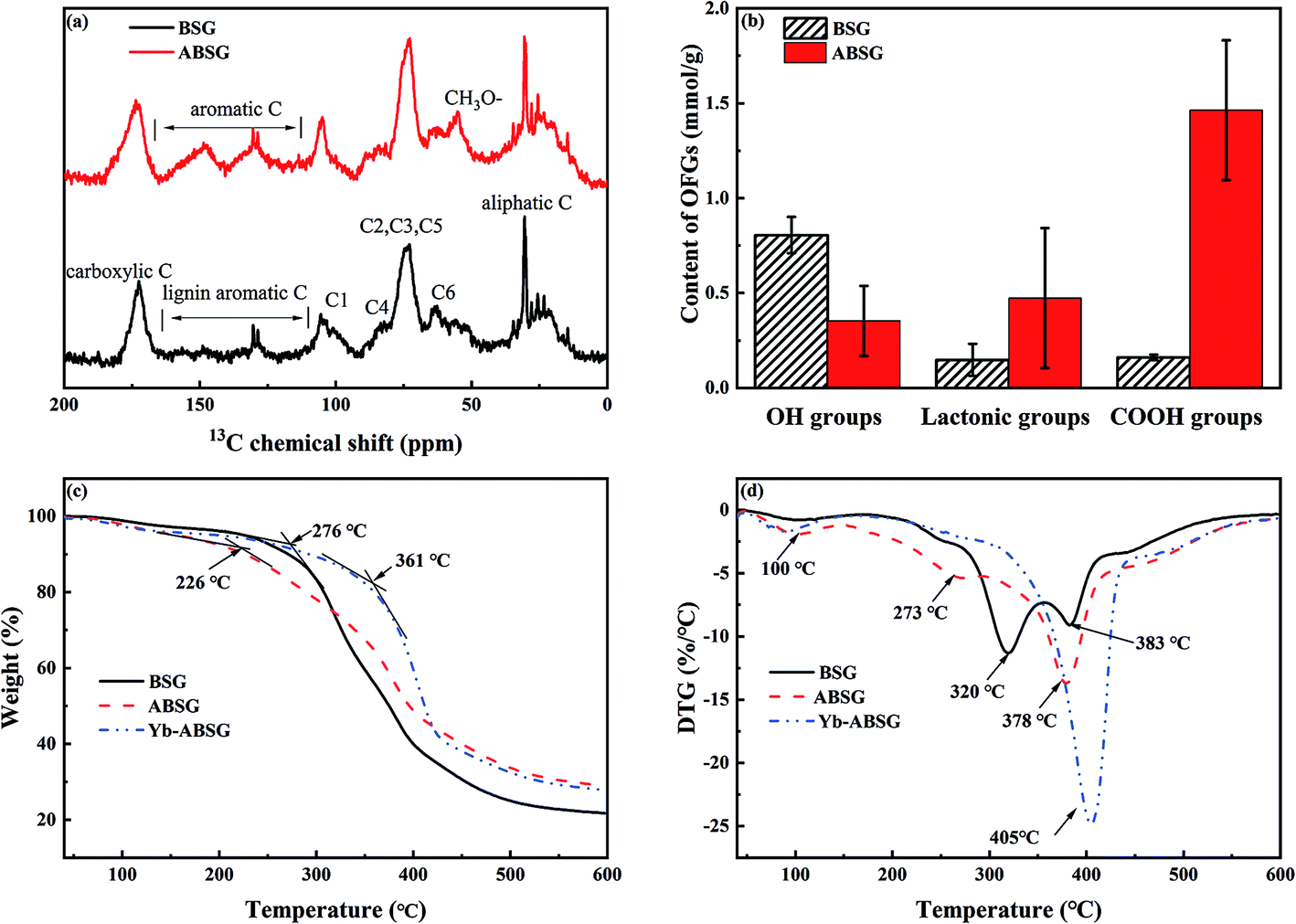 | ||
| Fig. 5 (a) 13C CP/MAS solid state NMR spectra of BSG and ABSG, (b) oxygen functional groups (OFGs) of BSG and ABSG, (c) TG and (d) DTG of BSG, ABSG and Yb-ABSG (20 °C min−1, He atmosphere). | ||
The content of the oxygen functional groups (OFG) of BSG and ABSG were quantified by Boehm titration. The results are depicted in Fig. 5(b) and show a decrease in the content of the hydroxyl groups for ABSG (0.48 mmol g−1) compared to BSG (0.8 mmol g−1). In contrast, the content of the lactonic groups increases from 0.15 mmol g−1 for BSG to 0.47 mmol g−1 for ABSG. The increase of the content of the carboxyl groups is even more pronounced from 0.15 mmol g−1 for BSG to 1.46 mmol g−1 for ABSG, which is comparable to the promoted oxidization of cellulosic materials (1.4 mmol g−1).17 However, this is rather unexpected as the decarboxylation and dehydration during the hydrothermal treatment of BSG would cause a loss of O-containing functional groups. Presumable three reasons can be assigned for this behavior: first, the emergence of carboxyl groups could be due to small fraction acids that form during the degradation of cellulose and hemicellulose, which are incorporated within the matrix as terminal groups.48 Second, the Maillard reaction produces various products with abundant functional groups, especially carboxyl groups. And third, it is possible that wet oxidation occurs producing carboxyl groups at the employed temperature (150 °C).49 The latter is supported by experiments employing an argon atmosphere during the hydrothermal treatment (150 °C, 16 h) yielding in an adsorbent (ABSG-Ar) with a reduced adsorption capacity (see Fig. S5 in the ESI†).
The thermal decomposition of BSG, ABSG and Yb3+ loaded ABSG (Yb-ABSG) was explored by STA-GC-MS analysis in order to obtain further information about the thermal stability and structural changes during the hydrothermal treatment and adsorption. Both BSG and ABSG show three derived thermogravimetric (DTG) peaks (maximum decomposition temperature) while Yb-ABSG shows only two peaks (Fig. 5(d)). The first DTG peak at around 100 °C could be assigned to the evaporation of water with a mass loss of 3.0 wt% (BSG), 4.3 wt% (ABSG) and 4.6 wt% (Yb-ABSG) in thermogravimetric curves (TG, Fig. 5(c)). The extrapolated onset temperatures in the TG of BSG, ABSG and Yb-ABSG are 276 °C, 226 °C and 361 °C, respectively. The lower of onset temperature after hydrothermal treatment is probably due to the increase of carboxyl groups on the surface of ABSG with low thermal stability.50 In contrast, an increase of the onset temperature to 361 °C is obtained for Yb-ABSG. Presumable, the coordination of the functional groups towards the metal ions may result in an increase in the thermal stability.51 The DTG-2 peaks could be attributed to the decomposition of hemicellulose and cellulose, while the DTG-3 peaks could be assigned to the continuous decomposition of cellulose materials and lignin.52 The DTG-3 peak area of ABSG is larger than that of BSG, which results from the degradation of intermediate hydrothermal products, e.g. advanced Maillard reaction products as well as polymerized melanoidins.53 For Yb-ABSG, DTG-2 and DTG-3 peaks overlap and form one large decomposition peak at higher temperature (405 °C). At even higher temperature, all samples undergo a moderate decomposition and carbonization stage with small change of sample mass. In this stage, some molecules in the hydrothermal samples (ABSG and Yb-ABSG) crosslink within the matrix,54 increasing the thermal stability and the residue mass from 21.7 wt% (BSG) to 29.0 wt% (ABSG) and 27.8 wt% (Yb-ABSG).
The volatile products at 375–385 °C (BSG, ABSG) and at 340–360 °C (Yb-ABSG) are analyzed by GC-MS (Table S4 in the ESI†). CO2 and water are the most abundant products, of which CO2 counting for 67.7% (BSG), 48.6% (ABSG) and 42.1% (Yb-ABSG) and water for 25.4% (BSG), 43.8% (ABSG) and 39.7% (Yb-ABSG) of the total analyzed products. The release of CO2 results from the decomposition of O-functional groups, while the source of water could be ascribed to the dehydration and fragmentation of the hydroxyl substituents in lignin, hemicellulose and cellulose.55 A group of furan-derivatives e.g. 3-methylfuran and 2,5-dimethylfuran are detected with over 0.9% among the products, and small molecules such as acetic anhydride and 1-hydroxypropan-2-one are also noticed. These products are evolved from cellulose degradation under helium atmosphere.42 It is noteworthy that acetic acid is not detected during the decomposition of BSG. However, approximately 1–3% among the decomposition products of ABSG (1.3%) and Yb-ABSG (2.7%) are detected as acetic acid. This increase is in agreement with the abovementioned enhanced presence of carboxyl groups on the surface of ABSG.
Adsorption study
The preliminary adsorption experiments employing La3+ show that the highest adsorption of 34.1 mg g−1 is obtained for ABSG treated at 150 °C and 16 h (see Fig. 1(a) and (b)). Thus, the biosorbents BSG and ABSG (150 °C, 16 h) were used for the batch adsorption experiments using UO22+, La3+, Eu3+ and Yb3+. ICP-OES was used to determine the concentration of the metal ions before and after the adsorption. In addition, adsorption experiments were also performed using the radiotracer technique employing the radiation from 169Yb. The results are depicted in Fig. S6 in the ESI† and show comparable adsorption capacities for BSG (21 mg g−1 for ICP method and 22 mg g−1 for radiotracer method) and ABSG (42 mg g−1 for ICP method and 49 mg g−1 for radiotracer method), indicating both methods can be used.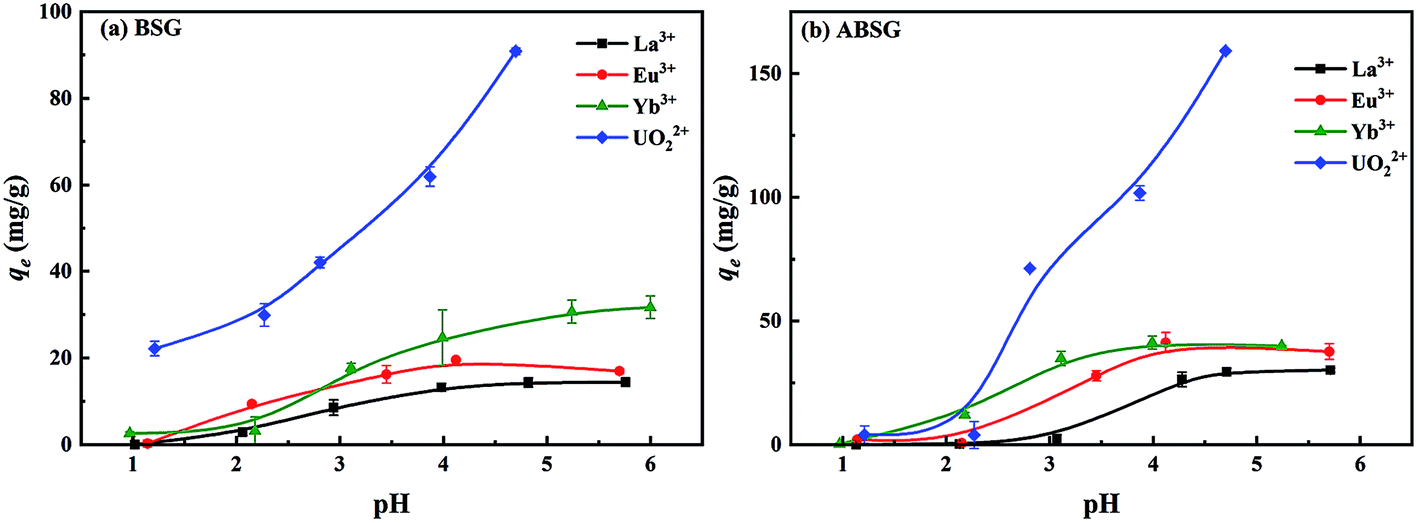 | ||
| Fig. 6 Effect of initial pH on the adsorption capacity of (a) BSG and (b) ABSG. For adsorption: 2 mg adsorbent/2 mL solution, c0(La3+, Eu3+, Yb3+) = 100 mg L−1, c0(UO22+) = 300 mg L−1, 2 h, room temperature. | ||
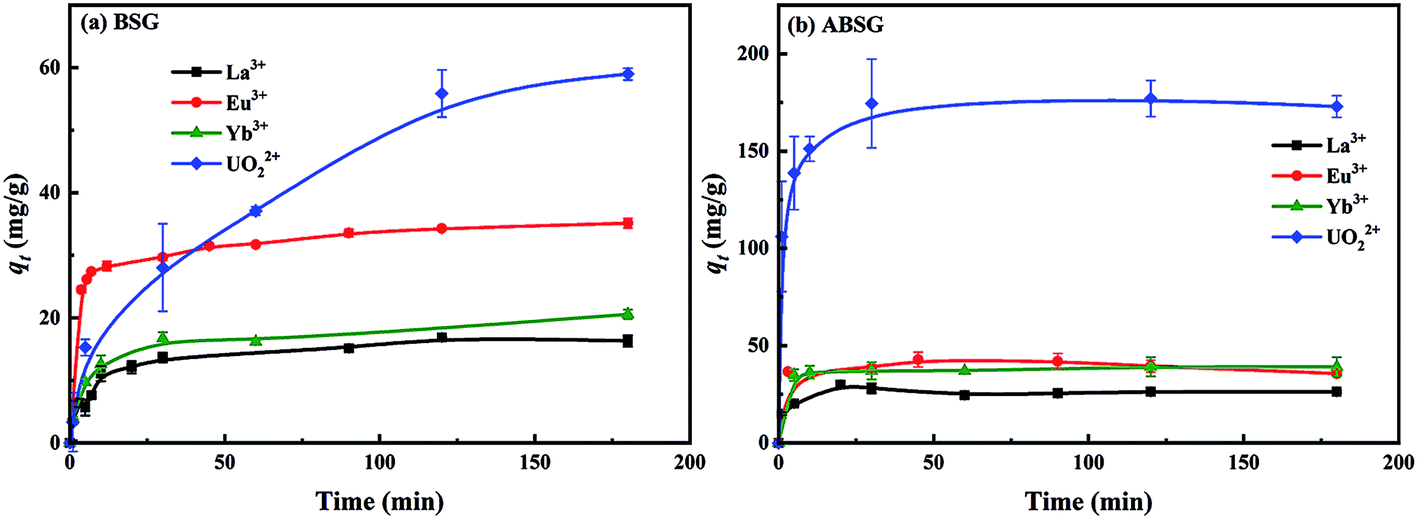 | ||
| Fig. 7 Effect of contact time on the adsorption capacity of (a) BSG and (b) ABSG. For adsorption, 2 mg adsorbent/2 mL solution, c0(La3+, Eu3+, Yb3+) = 100 mg L−1, c0(UO22+) = 300 mg L−1, pH(La3+, Eu3+, Yb3+) = 5.7, pH(UO22+) = 4.7, room temperature. | ||
| Adsorbent | Adsorbate | Kinetic model | R2 | k (g (mg−1 min−1)) | qe,cal (mg g−1) | qe,exp (mg g−1) |
|---|---|---|---|---|---|---|
| BSG | La3+ | PFO | 0.3078 | 0.0011 | 4.1 | 16.9 |
| PSO | 0.9982 | 0.0055 | 16.5 | |||
| Eu3+ | PFO | 0.9835 | 0.0203 | 9.8 | 35.1 | |
| PSO | 0.9987 | 0.0032 | 35.4 | |||
| Yb3+ | PFO | 0.5726 | 0.0206 | 11.6 | 20.6 | |
| PSO | 0.9988 | 0.0140 | 20.9 | |||
| UO22+ | PFO | 0.9439 | 0.0230 | 59.2 | 59.0 | |
| PSO | 0.9604 | 0.0140 | 67.0 | |||
| ABSG | La3+ | PFO | 0.9510 | 0.0267 | 9.0 | 30.0 |
| PSO | 0.9968 | 0.7062 | 26.0 | |||
| Eu3+ | PFO | 0.8576 | 0.0158 | 4.1 | 40.2 | |
| PSO | 0.9986 | 0.0213 | 36.9 | |||
| Yb3+ | PFO | 0.6214 | 0.0074 | 2.8 | 39.2 | |
| PSO | 0.9983 | 0.0181 | 39.2 | |||
| UO22+ | PFO | 0.9955 | 0.1126 | 75.2 | 177.0 | |
| PSO | 0.9996 | 0.0077 | 175.1 |
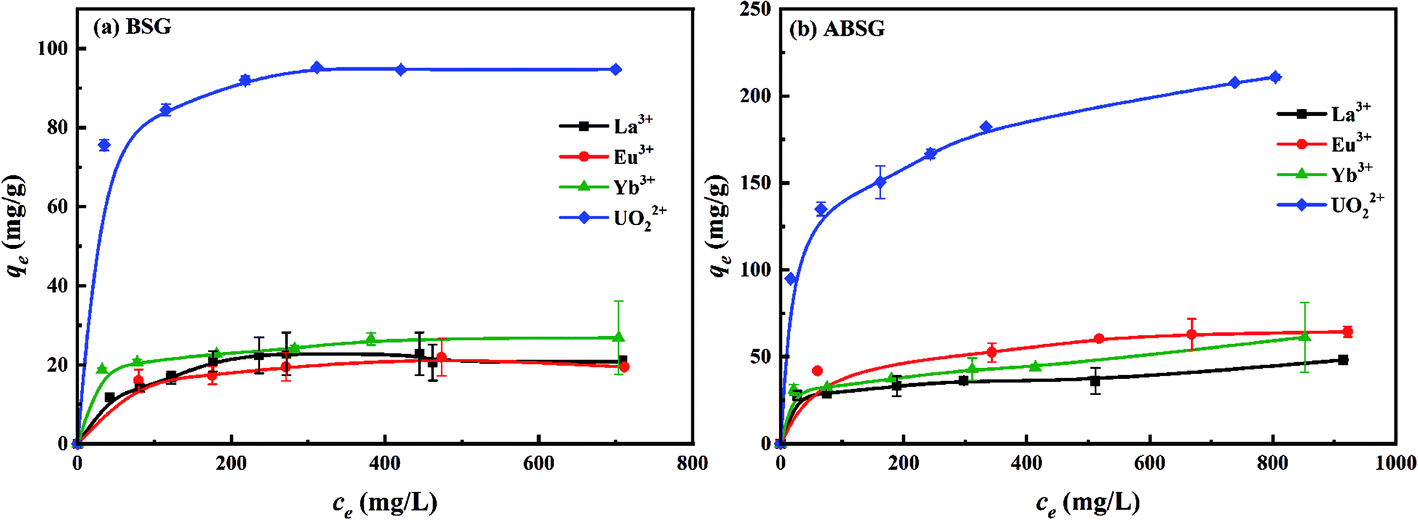 | ||
| Fig. 8 Effect of initial concentration on the adsorption capacity of (a) BSG and (b) ABSG. For adsorption, 2 mg adsorbent/2 mL solution, t(BSG) = 2 h, t(ABSG) = 1 h, pH(La3+, Eu3+, Yb3+) = 5.7, pH(UO22+) = 4.7, room temperature. | ||
| Adsorbate | Adsorbent | Langmuir model | Freundlich model | |||
|---|---|---|---|---|---|---|
| R2 | qm (mg g−1) | RL | R2 | n | ||
| La3+ | BSG | 0.9823 | 29.4 | 0.2073–0.6108 | 0.9775 | 2.60 |
| ABSG | 0.9981 | 37.4 | 0.0837–0.4773 | 0.8975 | 9.74 | |
| Eu3+ | BSG | 0.9981 | 25.1 | 0.0630–0.4022 | 0.9738 | 6.03 |
| ABSG | 0.9926 | 68.3 | 0.0596–0.3880 | 0.9809 | 6.12 | |
| Yb3+ | BSG | 0.9972 | 27.8 | 0.0286–0.5706 | 0.9631 | 8.00 |
| ABSG | 0.9930 | 46.0 | 0.0258–0.3164 | 0.8654 | 8.03 | |
| UO22+ | BSG | 0.9997 | 96.0 | 0.0092–0.0915 | 0.9907 | 9.42 |
| ABSG | 0.9914 | 220.6 | 0.0541–0.5411 | 0.9872 | 4.97 | |
K0e versus 1/T for thermodynamic parameter calculations (Fig. S11, ESI†) are given in the ESI.† Table 3 summarizes the thermodynamic parameters of the different adsorption process. The negative values of ΔG0 of all studied cases indicate a spontaneous adsorption process. In addition, the positive values of ΔS0 in all cases could be explained as the increase of randomness at the solid/liquid interface during the adsorption.66 Comparing the ΔH0 values with −TΔS0 values shows that the thermodynamic driving force of the uranyl and rare earth metal ions onto the biosorbents is the change of entropy.67 The adsorption of the studied rare earth metal ions onto ABSG and BSG presents positive ΔH0, indicating an endothermic process as reported in most of the literature.68 The adsorption of UO22+ onto BSG is also endothermic (ΔH0 = 55.8 kJ mol−1), which is consistent with the uranyl complexation by anionic organic ligands, producing endothermic enthalpies and large positive entropies of complexation.18 Nevertheless, the adsorption of UO22+ onto ABSG is exothermic (ΔH0 = −12.6 kJ mol−1). Literature has reported that different surface properties of biochar would result in different thermodynamic mechanisms of uranyl adsorption.67 Therefore, variations of the surface characters of BSG and ABSG such as the content of carboxyl groups (0.15 mmol g−1 for BSG and 1.46 mmol g−1 for ABSG) and pHPZC (5.7 for BSG and 4.1 for ABSG) may result in the obtained difference in ΔH0.
| Adsorbent | Adsorbate | ΔH0 (kJ mol−1) | ΔS0 (J (mol−1 K−1)) | ΔG0 (kJ mol−1) | −TΔS0 (kJ mol−1) | ||||
|---|---|---|---|---|---|---|---|---|---|
| 298 K | 318 K | 338 K | 298 K | 318 K | 338 K | ||||
| ABSG | UO22+ | −12.6 | 39.6 | −24.4 | −25.2 | −25.9 | −11.8 | −12.6 | −13.4 |
| Yb3+ | 14.4 | 113.1 | −19.3 | −21.5 | −23.8 | −33.7 | −36.0 | −38.2 | |
| Eu3+ | 18.0 | 124.3 | −19.1 | −21.6 | −24.0 | −37.0 | −39.5 | −42.0 | |
| La3+ | 12.2 | 109.5 | −20.4 | −22.6 | −24.8 | −32.6 | −34.8 | −37.0 | |
| BSG | UO22+ | 55.8 | 261.1 | −22.0 | −27.2 | −32.5 | −77.8 | −83.0 | −88.3 |
| Yb3+ | 27.0 | 146.3 | −16.6 | −19.5 | −22.4 | −43.6 | −46.5 | −49.4 | |
| Eu3+ | 12.6 | 108.1 | −19.6 | −21.8 | −24.0 | −32.2 | −34.4 | −36.5 | |
| La3+ | 7.5 | 89.8 | −19.3 | −21.1 | −22.9 | −26.8 | −28.6 | −30.4 | |
Investigation of adsorption mechanism
The determined BET surface area of BSG and ABSG is <2 m2 g−1, which is consistent with the literature reporting a BET surface of BSG of 0.48 m2 g−1.69 Thus, the surface area of the biosorbent is very small and has little effect on the adsorption process.SEM/EDX analysis prove the metal ion adsorption onto to surface of ABSG, as the SEM image, the EDX element mapping and the uranium distribution of uranyl ions-loaded ABSG depicted in Fig. 9 indicate. Similar results are obtained for the examined rare earth metal ions and are displayed in Fig. S12 in the ESI.† The SEM images show an irregular and rough surface of ABSG with no apparent pore structure (Fig. 9(a)), which is consistent with the low BET surface area. Therefore, the adsorption of the metal ions by ABSG is due to interactions of the functional groups on the surface. This is supported by the EDX element mapping (Fig. 9(b)) and the distribution of uranium (Fig. 9(c)). The irregular shape of the particles leads to noticeable shading so that no uniform recording of the X-ray signal can be expected. However, besides some small areas with uranium accumulation the uranyl ions seem to be more or less evenly distributed on the surface of ABSG.
 | ||
| Fig. 9 (a) SEM image (magnification of 1000 times) of uranyl ions loaded-ABSG, (b) EDX element mapping of uranyl ions loaded-ABSG (20 kV/10 μA, magnification of 1000 times, 25 frames) and (c) distribution of uranium on uranyl ions loaded-ABSG. For ion-loading: 50 mg ABSG/50 mL solution, pH(UO22+) = 4.7, c0(UO22+) = 300 mg L−1, t = 1 h, room temperature. | ||
FT-IR spectra of ABSG before and after adsorption are displayed in Fig. 10. For all ion-loaded samples the strengths of the absorption bands at 1654 cm−1 are decreased, which could be attributed to the antisymmetric stretching vibration of –COO− groups. This indicates the involvement of carboxyl groups in the adsorption process. In addition, the chelate model is calculated based on the differences between the antisymmetric and symmetric stretching bands of the –COO− groups (Δνas–νs). As shown in Table 4, Δνas–νs values of 197 cm−1 and 201 cm−1 are obtained for La3+ and Yb3+ loaded samples, respectively. This minor difference is negligibly small considering the resolution of the recorded spectra of 4 cm−1. In general, value of Δνas–νs > 200 cm−1 indicates a monodentate binding of metal ions, which involves an interaction with the metal ions through an electrostatic effect and ion exchange.70 In contrast, Δνas–νs of 179 cm−1 and 187 cm−1 are obtained for the Eu3+ and UO22+ loaded samples, pointing at a different binding mode. In the case of UO22+, this is presumable due to the presence of hydrated uranyl species and a more complex coordination occurs. This is confirmed by a new absorption band occurring at 922 cm−1 which is assigned to the antisymmetric stretching vibration of UO in (UO2)3(OH)5+.71 In addition, new absorption bands are obtained for all studied metal ions after adsorption at 530 to 540 cm−1 and 440 to 450 cm−1, which could be ascribed to metal–N vibration and metal–O vibration, respectively.72 This indicates that N-containing functional groups of the ABSG also contribute to the adsorption of the studied ions. And is consistent with the literature reporting the involvement of O and N-containing functional groups for the adsorption of uranyl73 and rare earth metal ions.8 Thus, the adsorption of La3+, Eu3+, Yb3+ and UO22+ onto ABSG is predominated by the electrostatic effect between negative charged functional groups and cations. However, in case of Eu3+ and UO22+, a coordination of the cation with O and N-containing functional groups also plays an important role, thus resulting in higher adsorption capacity than that of La3+ and Yb3+ as observed in the isotherms study.
 | ||
| Fig. 10 FT-IR spectra of ABSG and ion-loaded ABSG. For ion-loading: 50 mg ABSG/50 mL solution, pH(La3+, Eu3+, Yb3+) = 5.7, pH(UO22+) = 4.7, c0(La3+, Eu3+, Yb3+) = 100 mg L−1, c0(UO22+) = 300 mg L−1, t = 1 h, room temperature. | ||
| ABSG | La-ABSG | Eu-ABSG | Yb-ABSG | UO2-ABSG | |
|---|---|---|---|---|---|
| Antisymmetric stretching of –COO− (νas, −COO−), cm−1 | 1654 | 1650 | 1631 | 1655 | 1639 |
| Symmetric stretching of –COO− (νs, −COO−), cm−1 | 1452 | 1453 | 1453 | 1454 | 1452 |
| Δνas–νs, cm−1 | 202 | 197 | 179 | 201 | 187 |
Conclusions
In present study, BSG is successfully transformed into an effective biosorbent ABSG via a mild hydrothermal treatment approach (150 °C, 16 h) in line with the principles of green chemistry. This is performed at a significantly lower temperature than traditional HTC method without additional activation process, which minimizes the energy consumption and environmental impact during the treatment. Maillard reaction with the formation of melanoidins plays an important role in the increase of adsorption capacity, along with other pathways like dehydration, decarboxylation, aromatization and oxidation. Thus, the ABSG obtained has an increase content of carboxyl groups from 0.15 mmol g−1 (BSG) to 1.46 mmol g−1 with increasing adsorption capacity. The Langmuir maximum adsorption capacity of La3+, Yb3+, Eu3+ and UO22+ onto ABSG obtained at optimal pH of 5.7 for La3+, Eu3+ and Yb3+ and at 4.7 for UO22+ is 38, 46, 68 and 221 mg g−1, respectively. In addition, FT-IR spectra show that both O-containing functional groups and N-containing functional groups are involved in the adsorption of studied ions. Overall, the present study demonstrates the potential of hydrothermal treated BSG for uranyl and rare earth metal ions removal from wastewater with low cost and high sustainability. Furthermore, the results about Maillard reaction provide information regarding the hydrothermal treatment of protein-rich biomass at low temperature, which may give inspirations on the exploration of similar biomass as biosorbents after treatment at low temperatures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the German Federal Ministry of Education and Research (FENABIUM project 02NUK046A) and China Scholarship Council (CSC No. 201804910464) for financial support. We also thank Andrea Brünner for the SEM/EDX measurements and the Publication Fund of the TU Dresden for Open Access Funding.Notes and references
- A. Waseem, H. Ullah, M. K. Rauf and I. Ahmad, Crit. Rev. Environ. Sci. Technol., 2015, 45, 2391–2423 CrossRef CAS.
- D. Brugge and V. Buchner, Rev. Environ. Health, 2011, 26, 231–249 CAS.
- R. W. Herschy, in Encyclopedia of Lakes and Reservoirs, ed. L. Bengtsson, R. W. Herschy and R. W. Fairbridge, Springer Netherlands, Dordrecht, 2012, pp. 876–883 Search PubMed.
- J. Veliscek-Carolan, J. Hazard. Mater., 2016, 318, 266–281 CrossRef CAS.
- C. Tunsu, M. Petranikova, M. Gergorić, C. Ekberg and T. Retegan, Hydrometallurgy, 2015, 156, 239–258 CrossRef CAS.
- G. Pagano, M. Guida, F. Tommasi and R. Oral, Ecotoxicol. Environ. Saf., 2015, 115, 40–48 CrossRef CAS.
- C. Liao, X. R. Zhao, X. Y. Jiang, J. Teng and J. G. Yu, Microchem. J., 2020, 152, 104288 CrossRef CAS.
- X. Zhao, X. Jiang, D. Peng, J. Teng and J. Yu, J. Rare Earths, 2021, 39, 90–97 CrossRef CAS.
- T. Kegl, A. Košak, A. Lobnik, Z. Novak, A. K. Kralj and I. Ban, J. Hazard. Mater., 2020, 386, 121632 CrossRef CAS.
- A. Kumari, S. Singh, K. Parmar, D. D. Pathak and M. Kumar Jha, J. Ind. Eng. Chem., 2020, 83, 421–429 CrossRef CAS.
- T. Takei, M. Takehara, T. Takabayashi, S. Yanagida and N. Kumada, Colloids Surf., A, 2020, 125664 CrossRef.
- N. K. Gupta, A. Sengupta, A. Gupta, J. R. Sonawane and H. Sahoo, J. Environ. Chem. Eng., 2018, 6, 2159–2175 CrossRef CAS.
- P. Rajasulochana and V. Preethy, Resour. Technol., 2016, 2, 175–184 Search PubMed.
- N. R. Nicomel, L. Otero-Gonzalez, L. Arashiro, M. Garfí, I. Ferrer, P. Van Der Voort, K. Verbeken, T. Hennebel and G. Du Laing, Green Chem., 2020, 22, 1985–1995 RSC.
- B. Arunraj, T. Sathvika, V. Rajesh and N. Rajesh, ACS Omega, 2019, 4, 940–952 CrossRef CAS.
- A. C. Texier, Y. Andrès, C. Faur-Brasquet and P. Le Cloirec, Chemosphere, 2002, 47, 333–342 CrossRef CAS.
- H. Ma, B. S. Hsiao and B. Chu, ACS Macro Lett., 2012, 1, 213–216 CrossRef CAS.
- S. Chen, J. Hong, H. Yang and J. Yang, J. Environ. Radioact., 2013, 126, 253–258 CrossRef CAS.
- S. A. Nicolae, H. Au, P. Modugno, H. Luo, A. E. Szego, M. Qiao, L. Li, W. Yin, H. J. Heeres, N. Berge and M.-M. Titirici, Green Chem., 2020, 22, 4747–4800 RSC.
- M. Heidari, A. Dutta, B. Acharya and S. Mahmud, J. Energy Inst., 2019, 92, 1779–1799 CrossRef CAS.
- Z. Zhang, Z. Zhu, B. Shen and L. Liu, Energy, 2019, 171, 581–598 CrossRef CAS.
- Z. B. Zhang, X. H. Cao, P. Liang and Y. H. Liu, J. Radioanal. Nucl. Chem., 2013, 295, 1201–1208 CrossRef CAS.
- H. Cai, X. Lin, L. Tian and X. Luo, Ind. Eng. Chem. Res., 2016, 55, 9648–9656 CrossRef CAS.
- H. S. Kambo and A. Dutta, Renewable Sustainable Energy Rev., 2015, 45, 359–378 CrossRef CAS.
- S. Lu and S. W. Gibb, Bioresour. Technol., 2008, 99, 1509–1517 CrossRef CAS.
- K. S. Low, C. K. Lee and C. H. Low, J. Appl. Polym. Sci., 2001, 82, 2128–2134 CrossRef CAS.
- S. Wierzba and A. Kłos, J. Cleaner Prod., 2019, 225, 112–120 CrossRef CAS.
- S. Wierzba, M. Rajfur, M. Nabrdalik and A. Kłos, Microchem. J., 2019, 145, 196–203 CrossRef CAS.
- T. P. de Araújo, H. B. Quesada, R. Bergamasco, D. T. Vareschini and M. A. S. D. de Barros, Bioresour. Technol., 2020, 310, 123399 CrossRef.
- S. I. Mussatto, G. Dragone and I. C. Roberto, J. Cereal Sci., 2006, 43, 1–14 CrossRef CAS.
- M. Hellwig and T. Henle, Angew. Chem., Int. Ed., 2014, 53, 10316–10329 CrossRef CAS.
- T. Kegl, I. Ban, A. Lobnik and A. Košak, J. Hazard. Mater., 2019, 378, 120764 CrossRef CAS.
- A. L. Merrill and B. K. Watt, Energy value of foods: basis and derivation, ARS United States Department of Agriculture, Washington DC, 1973 Search PubMed.
- U. Schwarzenbolz, T. Hofmann, N. Sparmann and T. Henle, J. Agric. Food Chem., 2016, 64, 5071–5078 CrossRef CAS.
- M. Hellwig, S. Witte and T. Henle, J. Agric. Food Chem., 2016, 64, 7234–7243 CrossRef CAS.
- L. S. Balistrierl and J. W. Murray, Am. J. Sci., 1981, 281, 788–806 CrossRef.
- A. M. Oickle, S. L. Goertzen, K. R. Hopper, Y. O. Abdalla and H. A. Andreas, Carbon, 2010, 48, 3313–3322 CrossRef CAS.
- A. Funke and F. Ziegler, Biofuels, Bioprod. Biorefin., 2010, 4, 160–177 CrossRef CAS.
- P. J. Arauzo, L. Du, M. P. Olszewski, M. F. Meza Zavala, M. J. Alhnidi and A. Kruse, Bioresour. Technol., 2019, 293, 122117 CrossRef CAS.
- X. Xu, R. Tu, Y. Sun, Y. Wu, E. Jiang and J. Zhen, Bioresour. Technol., 2019, 271, 427–438 CrossRef CAS.
- H. Simsir, N. Eltugral and S. Karagoz, Bioresour. Technol., 2017, 246, 82–87 CrossRef CAS.
- A. O. Balogun, F. Sotoudehniakarani and A. G. McDonald, J. Anal. Appl. Pyrolysis, 2017, 127, 8–16 CrossRef CAS.
- Q. Li, L. Chai, Q. Wang, Z. Yang, H. Yan and Y. Wang, Bioresour. Technol., 2010, 101, 3796–3799 CrossRef CAS.
- M. N. Lund and C. A. Ray, J. Agric. Food Chem., 2017, 65, 4537–4552 CrossRef CAS.
- J. A. Rufián-Henares and S. P. De La Cueva, J. Agric. Food Chem., 2009, 57, 432–438 CrossRef.
- Y. Li, D. Cui, Y. Tong and L. Xu, Int. J. Biol. Macromol., 2013, 62, 663–669 CrossRef CAS.
- B. Weber, E. A. Stadlbauer, S. Eichenauer, C. Koch, K. Albert, M. Kramer and D. Steffens, J. Biobased Mater. Bioenergy, 2013, 7, 367–375 CrossRef CAS.
- X. Qi, L. Li, T. Tan, W. Chen and R. L. Smith, Environ. Sci. Technol., 2013, 47, 2792–2798 CrossRef CAS.
- A. Yousefifar, S. Baroutian, M. M. Farid, D. J. Gapes and B. R. Young, Bioresour. Technol., 2017, 226, 229–237 CrossRef CAS.
- D. M. Dos Santos, A. De Lacerda Bukzem, D. P. R. Ascheri, R. Signini and G. L. B. De Aquino, Carbohydr. Polym., 2015, 131, 125–133 CrossRef.
- A. Hardy, G. Vanhoyland, E. Geuzens, M. K. Van Bael, J. Mullens, L. C. Van Poucke and J. D'Haen, J. Sol-Gel Sci. Technol., 2005, 33, 283–298 CrossRef CAS.
- A. I. Osman, E. O'Connor, G. McSpadden, J. K. Abu-Dahrieh, C. Farrell, A. H. Al-Muhtaseb, J. Harrison and D. W. Rooney, J. Chem. Technol. Biotechnol., 2020, 95, 183–195 CrossRef CAS.
- A. Kruse and T. Zevaco, Energies, 2018, 11, 674 CrossRef.
- M. P. Olszewski, P. J. Arauzo, P. A. Maziarka, F. Ronsse and A. Kruse, Catalysts, 2019, 9, 625 CrossRef CAS.
- G. Özsin and A. E. Pütün, Energy Convers. Manage., 2017, 149, 675–685 CrossRef.
- J. Qu, X. Meng, Y. Zhang, Q. Meng, Y. Tao, Q. Hu, X. Jiang, H. You and C. A. Shoemaker, J. Hazard. Mater., 2019, 379, 120804 CrossRef CAS.
- P. Misaelides, A. Godelitsas, A. Filippidis, D. Charistos and I. Anousis, Sci. Total Environ., 1995, 173–174, 237–246 CrossRef.
- W. Zhao, X. Lin, H. Cai, T. Mu and X. Luo, Ind. Eng. Chem. Res., 2017, 56, 12745–12754 CrossRef CAS.
- Y. Zhang, X. Yue, W. Xu, H. Zhang and F. Li, J. Hazard. Mater., 2019, 379, 120783 CrossRef CAS.
- K. Y. Foo and B. H. Hameed, Chem. Eng. J., 2010, 156, 2–10 CrossRef CAS.
- S. J. Allen, G. Mckay and J. F. Porter, J. Colloid Interface Sci., 2004, 280, 322–333 CrossRef CAS.
- T. W. Weber and R. K. Chakravorti, AIChE J., 1974, 20, 228–238 CrossRef CAS.
- E. Kusrini, W. Wicaksono, C. Gunawan, N. Z. A. Daud and A. Usman, J. Environ. Chem. Eng., 2018, 6, 6580–6588 CrossRef CAS.
- X. R. Zhao, X. Xu, X. Y. Jiang, J. Teng and J. G. Yu, Desalin. Water Treat., 2019, 148, 188–194 CrossRef CAS.
- R. Bai, F. Yang, Y. Zhang, Z. Zhao, Q. Liao, P. Chen, P. Zhao, W. Guo and C. Cai, Carbohydr. Polym., 2018, 190, 255–261 CrossRef CAS.
- L. Tan, Q. Liu, D. Song, X. Jing, J. Liu, R. Li, S. Hu, L. Liu and J. Wang, New J. Chem., 2015, 39, 2832–2838 RSC.
- M. S. Alam, D. Gorman-Lewis, N. Chen, S. Safari, K. Baek, K. O. Konhauser and D. S. Alessi, Environ. Sci. Technol., 2018, 52, 13057–13067 CrossRef CAS.
- S. Iftekhar, D. L. Ramasamy, V. Srivastava, M. B. Asif and M. Sillanpää, Chemosphere, 2018, 204, 413–430 CrossRef CAS.
- J. Zhang and Q. Wang, J. Cleaner Prod., 2016, 112, 3927–3934 CrossRef CAS.
- O. W. Duckworth and S. T. Martin, Geochim. Cosmochim. Acta, 2001, 65, 4289–4301 CrossRef CAS.
- F. Quilès and A. Burneau, Vib. Spectrosc., 2000, 23, 231–241 CrossRef.
- H. F. G. Barbosa and É. T. G. Cavalheiro, Int. J. Biol. Macromol., 2019, 121, 1179–1185 CrossRef CAS.
- G. Tian, J. Geng, Y. Jin, C. Wang, S. Li, Z. Chen, H. Wang, Y. Zhao and S. Li, J. Hazard. Mater., 2011, 190, 442–450 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08164g |
| This journal is © The Royal Society of Chemistry 2020 |