
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transaminase-mediated synthesis of enantiopure drug-like 1-(3′,4′-disubstituted phenyl)propan-2-amines†
Ágnes Lakóab,
Zsófia Molnára,
Ricardo Mendonçab and
László Poppe *ac
*ac
aDepartment of Organic Chemistry and Technology, Budapest University of Technology and Economics, Műegyetem rkp. 3, 1111 Budapest, Hungary. E-mail: poppe@mail.bme.hu; Tel: +36-1-463-3299
bHovione Farmaciência, S.A., Campus do Lumiar, Edifício R, Estrada do Paço do Lumiar, 1649-038 Lisboa, Portugal
cBiocatalysis and Biotransformation Research Center, Faculty of Chemistry and Chemical Engineering, Babes-Bolyai University of Cluj-Napoca, Arany János Str. 11, 400028 Cluj-Napoca, Romania
First published on 10th November 2020
Abstract
Transaminases (TAs) offer an environmentally and economically attractive method for the direct synthesis of pharmaceutically relevant disubstituted 1-phenylpropan-2-amine derivatives starting from prochiral ketones. In this work, we report the application of immobilised whole-cell biocatalysts with (R)-transaminase activity for the synthesis of novel disubstituted 1-phenylpropan-2-amines. After optimisation of the asymmetric synthesis, the (R)-enantiomers could be produced with 88–89% conversion and >99% ee, while the (S)-enantiomers could be selectively obtained as the unreacted fraction of the corresponding racemic amines in kinetic resolution with >48% conversion and >95% ee.
Introduction
Chiral amines can be found as constituents in approximately 40% of all active pharmaceutical ingredients, but are also used as resolving agents for the separation of enantiomers,1 thus, there is a high demand for the synthesis of enantiomerically pure amines. For the synthesis of chiral amines, several solutions have been developed; however, these methods usually operate with chiral auxiliaries or metal complexes with chiral ligands and sometimes suffer from low enantioselectivity and low atom efficiency.2 Biocatalytic approaches via kinetic resolution (employing lipases3 monoamine oxidases4 or transaminases5), dynamic kinetic resolution with lipases,6 deracemisation with monoamine oxidases,7 or asymmetric synthesis (employing transaminases,8 amine dehydrogenases9 or imine reductases10) represent appealing alternatives for the synthesis of enantiopure amines.Commercial drugs (Fig. 1) containing both enantiomers of amphetamine and related compounds (1-arylpropan-2-amines) exist for the treatment of obesity (benzphetamine, 1),11,12 Parkinson's disease (L-DOPA,13 2; selegiline,14 3), narcolepsy and attention deficit hyperactivity disorder (dextroamphetamine, 4)15,16 and benign prostatic hyperplasia (tamsulosin, 5).17 Furthermore, L-amphetamine [6, (2R)-1-phenylpropan-2-amine] has been reported to improve cognitive function in multiple sclerosis patients.18
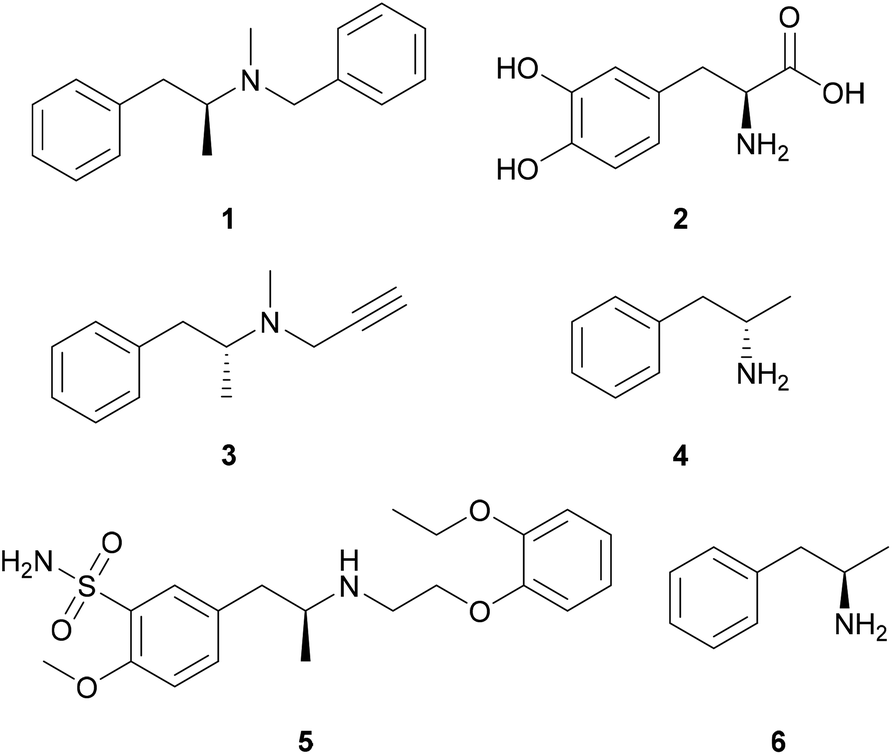 | ||
| Fig. 1 APIs containing the 1-phenylpropan-2-amine building block. | ||
The synthesis of enantiopure (S)-1-phenylpropan-2-amine derivatives is more explored than of the (R)-amines; however, chemical and biocatalytic approaches both are reported for the enantioselective synthesis of L-amphetamine as well.
Some chemical methods (Scheme 1) for synthesis of L-amphetamine (6) include diastereoselective organocerium additions to (S)-1-amino-2-ethoxymethyl-pyrrolidine hydrazone derivatives followed by hydrogenolysis (A),19 proline-catalysed α-aminooxylation (9 steps) and α-amination (4 steps) strategies (B),20 hydrogenation of α,β-disubstituted nitroalkenes with rhodium and chiral phosphorus ligands (C),21 and Sharpless asymmetric dihydroxylation of olefins (D).22
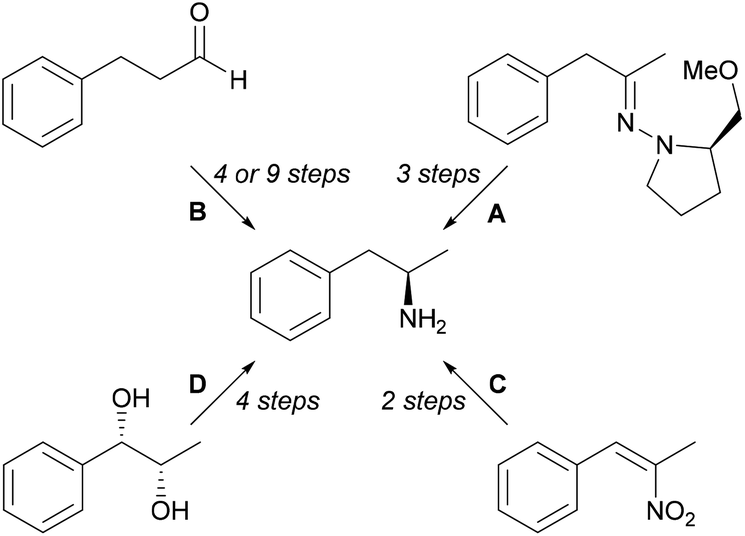 | ||
| Scheme 1 Chemical methods for the synthesis of (R)-1-phenylpropan-2-amine. | ||
Multiple biocatalytic strategies have been developed employing different (coupled) enzymes from various starting compounds (Scheme 2). Racemic 1-phenylpropan-2-amine can be enzymatically resolved via lipases (E);23–26 however, kinetic resolution only allows a maximum of 50% yield. The asymmetric reductive amination of carbonyl compounds with TAs (F) has been one of the main research areas of biocatalytic methods leading to chiral amines.27,28 TA-mediated amine syntheses have been widely reported as a one-enzyme system using a variety of amine donors without further transformation of the ketone co-product.29–33 Furthermore, there were reports on TAs coupled in cascades with pyruvate-reducing enzyme lactate dehydrogenase (LDH) and NADH-regenerating glucose dehydrogenase (GDH).34,35 Amine dehydrogenases (G, AmDH) can utilise ammonia for the synthesis of the desired amines by reductive amination of the prochiral ketone.36 Enzyme cascades further widen the possibilities. Starting from racemic alcohols, a five-enzyme system applying two enzymes for the synthesis [alcohol dehydrogenase (H, ADH), AmDH] and three enzymes [NADP-oxidase, catalase, and formate dehydrogenase (FDH)] for cofactor regeneration allowed the synthesis of the desired (R)-amines in near quantitative yields.37 An in vitro hydrogen-borrowing amination combining whole-cells with ADH and AmDH activity were applied in tandem operation,38 and was further developed to operate in E. coli cells co-expressing the two enzymes as well.39 This approach was also improved by employing a mutant AmDH,36 or by a sustainable immobilized system containing AmDH and GDH.40 Furthermore, ketoximes in a one-pot cascade consisting of laccases (I) and TAs (F) gave the desired compounds as well, although in this case, the starting materials were prepared from the ketone derivatives.41
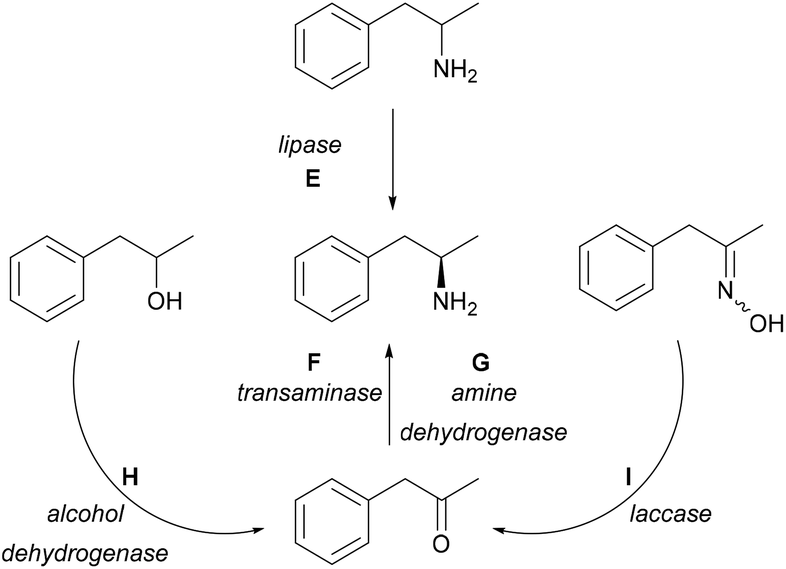 | ||
| Scheme 2 Biocatalytic syntheses of (R)-1-phenylpropan-2-amine. | ||
Immobilisation of the biocatalysts can improve productivity, as the immobilised forms can have enhanced stability and can be reused in multiple catalytic cycles.42–44 Furthermore, whole-cell immobilisation combines the benefits of elimination of the high costs of enzyme purification and the possibility of reuse, thus representing a cost-efficient way of employing biocatalysts.45
Herein, we report the application of immobilized E. coli whole-cells overexpressing transaminases from Arthrobacter sp. (ArR-TA and ArRm-TA, natural and engineered, respectively) and Aspergillus terreus (AtR-TA), the optimisation of the asymmetric synthesis of (R)-arylpropan-2-amines using 1-phenylpropan-2-one as model compound, and the kinetic resolution of the racemic amines providing access to the (S)-enantiomers.
Results and discussion
Our aim was to first optimise the asymmetric biocatalysis providing (R)-1-phenylpropan-2-amine (8a) from prochiral 1-phenylpropan-2-one (7a) with the aid of our sol–gel immobilised whole-cell TA biocatalysts.46,47 Additional goal was to extend the transamination with (R)-selective TAs to three further disubstituted derivatives (7b–d) as well (Scheme 3). Finally, to gain access to the (S)-enantiomers of the four amines by the (R)-selective TAs, we also investigated the kinetic resolution of all the racemic amines.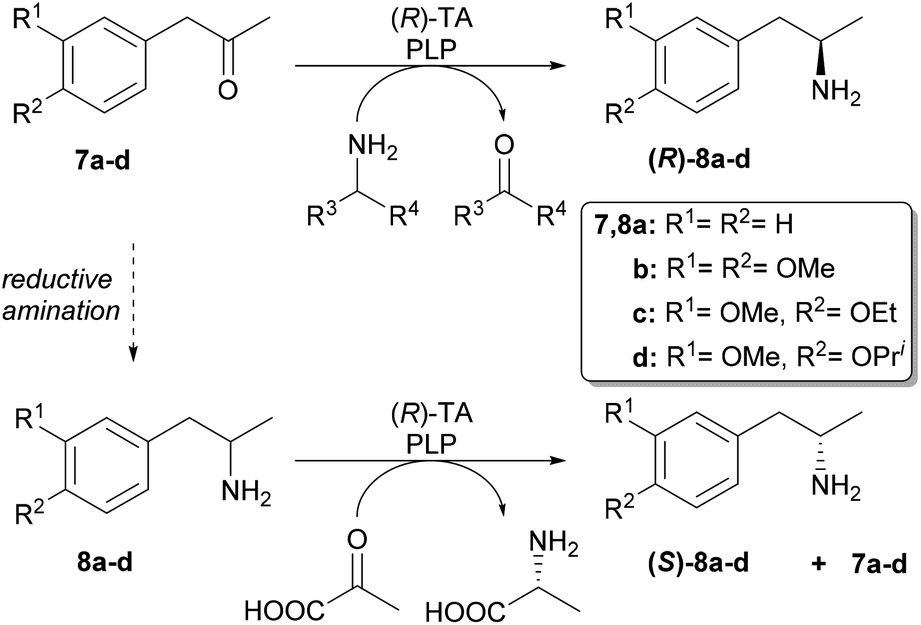 | ||
| Scheme 3 Transaminase-catalysed biotransformations leading to the (R)- and (S)-enantiomers of 1-arylpropan-2-amines 8a–d. | ||
Chemical synthesis of the prochiral 3,4-disubstituted 1-phenylpropan-2-ones (7b–d) and the corresponding racemic amines 8a–d
Aromatic aldehydes 9b–d bearing substituents of various bulkiness were obtained from vanillin (10) by introducing three different alkyl groups (methyl, ethyl and isopropyl) with O-alkylation (Scheme 4). Darzens condensation from the O-alkylated aldehydes 9b–d resulted in the ketones 7b–d. Notably, efficient condensation required the use of freshly prepared sodium methoxide. Finally, Pd-catalysed transfer hydrogenation from the ketones 7a–d resulted in the racemic amines (8a–d) of sufficient purity enabling the further operations without purification.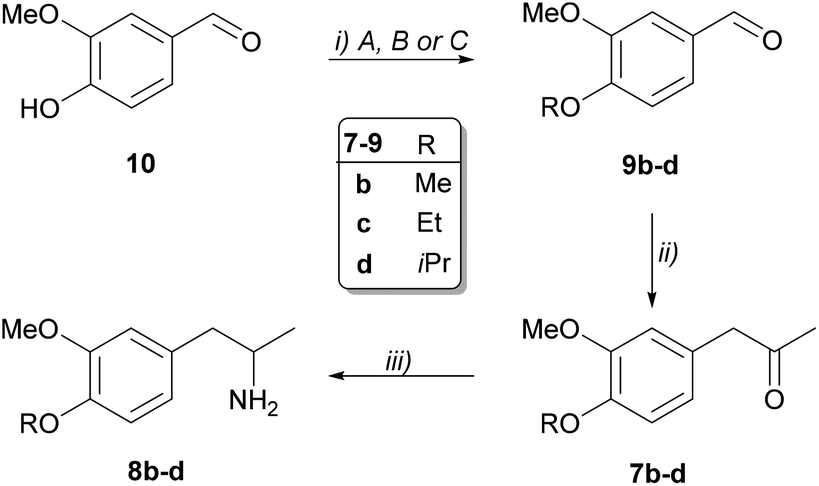 | ||
| Scheme 4 Synthesis of racemic disubstituted 1-phenylpropan-2-amines (8b–d). Reaction conditions: (i) A: 10 (20 mmol), K2CO3 (1 equiv.), Me2SO4 (2 equiv.) in 75 mL acetone, reflux; B: 10 (9.9 mmol), K2CO3 (1.5 equiv.), EtI (1.25 equiv.) in 15 mL DMF; C: 10 (6.6 mmol), K2CO3 (1.5 equiv.), iPrBr (1.5 equiv.) in 6.6 mL DMF, 80 °C; (ii) 9b–d (6 mmol), (±)-2-chloropropionic acid methyl ester (1.2 equiv.), NaOMe/MeOH (1.15 mmol, 25 wt%) in toluene (10 mL); 1N NaOH in toluene at 50 °C; 5N HCl in toluene, reflux; (iii) 7b–d (1 mmol), ammonium formate (10 equiv.), 10% Pd/C (0.04 equiv.) in 5 mL methanol. | ||
Reductive amination of 1-phenylpropan-2-one (7a) with (R)-selective whole-cell TA biocatalysts
The TA-mediated asymmetric synthesis of the (R)-amines was optimised in the biotransformations of the unsubstituted ketone 7a. Three immobilised TA-biocatalysts (ArR-TA, ArRm-TA, and AtR-TA) were screened in the reductive amination of 7a with seven different amine donors including investigation of the effect of DMSO as cosolvent. To enhance productivity, substrate concentration was varied as well.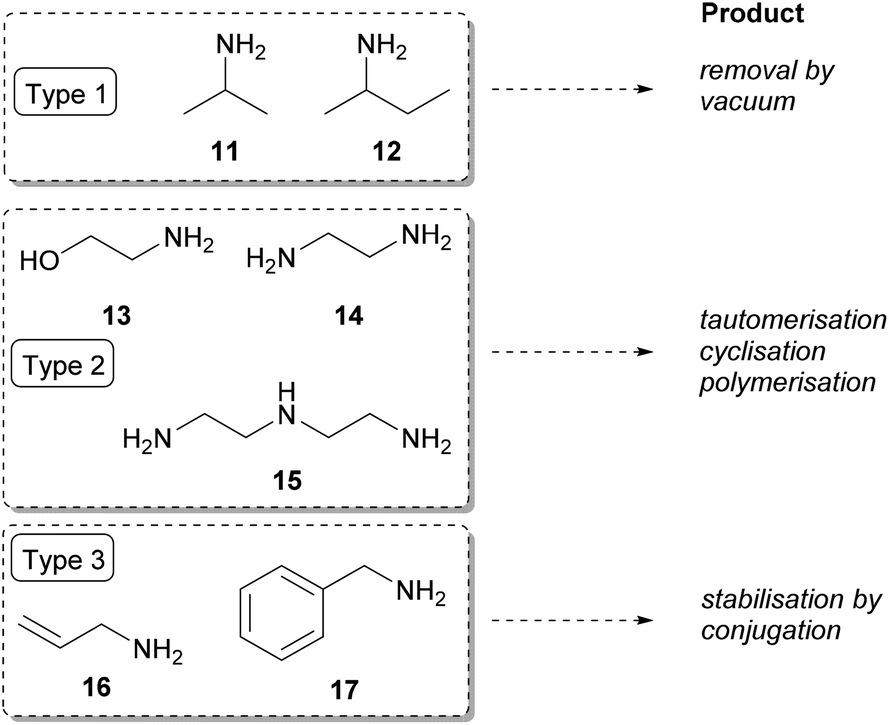 | ||
| Fig. 2 Various amine donors facilitating the displacement of the equilibrium of transamination. | ||
Type 1 amine donors are cheap and volatile; thus, these amines can be used in high excess to shift the reaction equilibrium towards product formation. Isopropylamine (11) and racemic sec-butylamine (12) and the corresponding ketones are low boiling-compounds that can be easily removed by vacuum.8,49,50 Although the pool of TAs which accept isopropylamine (11) as amine donor is limited, in case of acceptance industrial use of 11 is preferred compared to L-alanine, as the forming acetone can be easily removed.51 However, due to its unfavoured Keq, 11 has to be used in high excess and the forming acetone shows severe inhibitory effect. As an alternative, homologous sec-butylamine (12) was shown to be accepted in racemic form by TAs from Arthrobacter sp.32 and Aspergillus terreus29 as well. The more favourable Keq with 12 allows use of the amine donor in smaller excess (e.g. 10 equiv. compared to 50 equiv.52), while the forming methyl ethyl ketone co-product is still volatile and doesn't show severe reaction inhibition.
The Type 2 amine donors [ethanolamine (13), ethylenediamine (14), diethylenetriamine (15)] can be considered as ‘smart’ co-substrates, where a coupled tautomerisation, dimerisation, or cyclisation of the carbonyl co-product to non-inhibiting co-products cause the equilibrium displacement.53
In case of Type 3 amine donors [allylamine (16) and benzylamine (17)], the forming carbonyl-compounds are energetically favoured due to conjugation of the carbonyl moiety to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond or to the phenyl group.
C double bond or to the phenyl group.
Initially, the activity of all amine donors with the three TAs were screened in 10-fold excess (Table 1); however, using amines 13, 14 and 15 the conversion did not exceed 5% in any case (not shown in Table 1). The highest conversions could be achieved with all three (R)-TAs using sec-butylamine as amine donor, followed by isopropylamine being the second best. While ArR-TA and AtR-TA resulted in excellent ee in all cases (>99%), the highly mutated ArRm-TA evolved for efficient transformation of a bulky substrate did not show perfect enantioselectivity.8
| Entry | Enzyme | Amine donor | Conversion [%] | ee [%] |
|---|---|---|---|---|
| a Reaction conditions: immobilised whole-cell TA biocatalyst (20 mg), 7a (10 mM), amine donor (100 mM), PLP (1 mM), sodium phosphate buffer (100 mM, pH 7.5), DMSO (5 v/v%), 30 °C, 24 h. | ||||
| 1 | ArR-TA | Isopropylamine | 27.8 | >99 (R) |
| 2 | sec-Butylamine | 83.0 | >99 (R) | |
| 3 | Allylamine | 8.7 | >99 (R) | |
| 4 | Benzylamine | 3.1 | >99 (R) | |
| 5 | ArRm-TA | Isopropylamine | 25.4 | 62.2 (R) |
| 6 | sec-Butylamine | 51.6 | 82.4 (R) | |
| 7 | Allylamine | 14.4 | 91.5 (R) | |
| 8 | Benzylamine | 8.0 | 82.4 (R) | |
| 9 | AtR-TA | Isopropylamine | 8.5 | >99 (R) |
| 10 | sec-Butylamine | 68.5 | >99 (R) | |
| 11 | Allylamine | 0.8 | >99 (R) | |
| 12 | Benzylamine | 2.8 | >99 (R) | |
Next, the amine donors in lower equivalencies were tested with the most active enzyme ArR-TA, resulting in the transamination with excellent ee (>99%) but various conversions (Fig. 3). In this series of investigation, the reactions with 10-fold excess of amine donors (the usual minimal requirement of Type 1 amine donors, such as isopropylamine 11 or sec-butylamine 12) were compared to the reactions with no excess of amine donors (since smart co-substrates can drive the reaction to completion even in small excess), and to the ones with 5-fold excess of amine donors as well. Since in the lower than 10-fold equivalent amine donor cases the pH of the reaction remained within the operational range of the ArR-TA, no pH correction was necessary. The results indicated that reactions with amine donors 13, 14, 15 and 16 were not sufficient for effective synthetic processes, thus they were not investigated any further. Unsurprisingly, with benzylamine 17 higher conversion could be achieved than with isopropylamine 11, reaching 75% conversion at 10-fold excess compared to that of 50% with isopropylamine. Because sec-butylamine 12 provided the highest conversion at any substrate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :amine donor ratio (a reasonable 30.3% conversion could be achieved even with 1 equiv. of 12), the further optimisation was performed with 12 as amine donor. The significant difference between IPA (11) and SBA (12) can be rationalized by the more beneficial Keq, with 12 as amine donor due to the better affinity of the larger 12 to TAs as compared to 11.
:amine donor ratio (a reasonable 30.3% conversion could be achieved even with 1 equiv. of 12), the further optimisation was performed with 12 as amine donor. The significant difference between IPA (11) and SBA (12) can be rationalized by the more beneficial Keq, with 12 as amine donor due to the better affinity of the larger 12 to TAs as compared to 11.
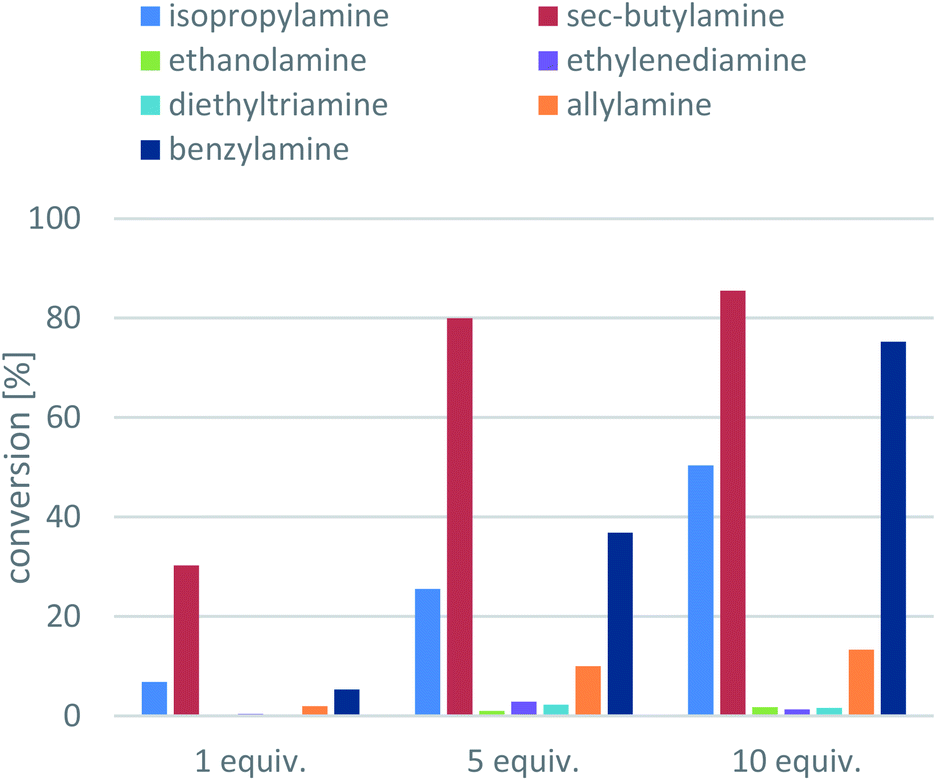 | ||
| Fig. 3 Screening the amine donors in ArR-TA-mediated transamination of 1-phenylpropan-2-one (7a). Reaction conditions: immobilised whole-cell TA biocatalyst (20 mg), 7a (10 mM), amine donor (10–100 mM), PLP (1 mM), sodium phosphate buffer (100 mM, pH 7.5), DMSO (5 v/v%), 30 °C, 24 h. | ||
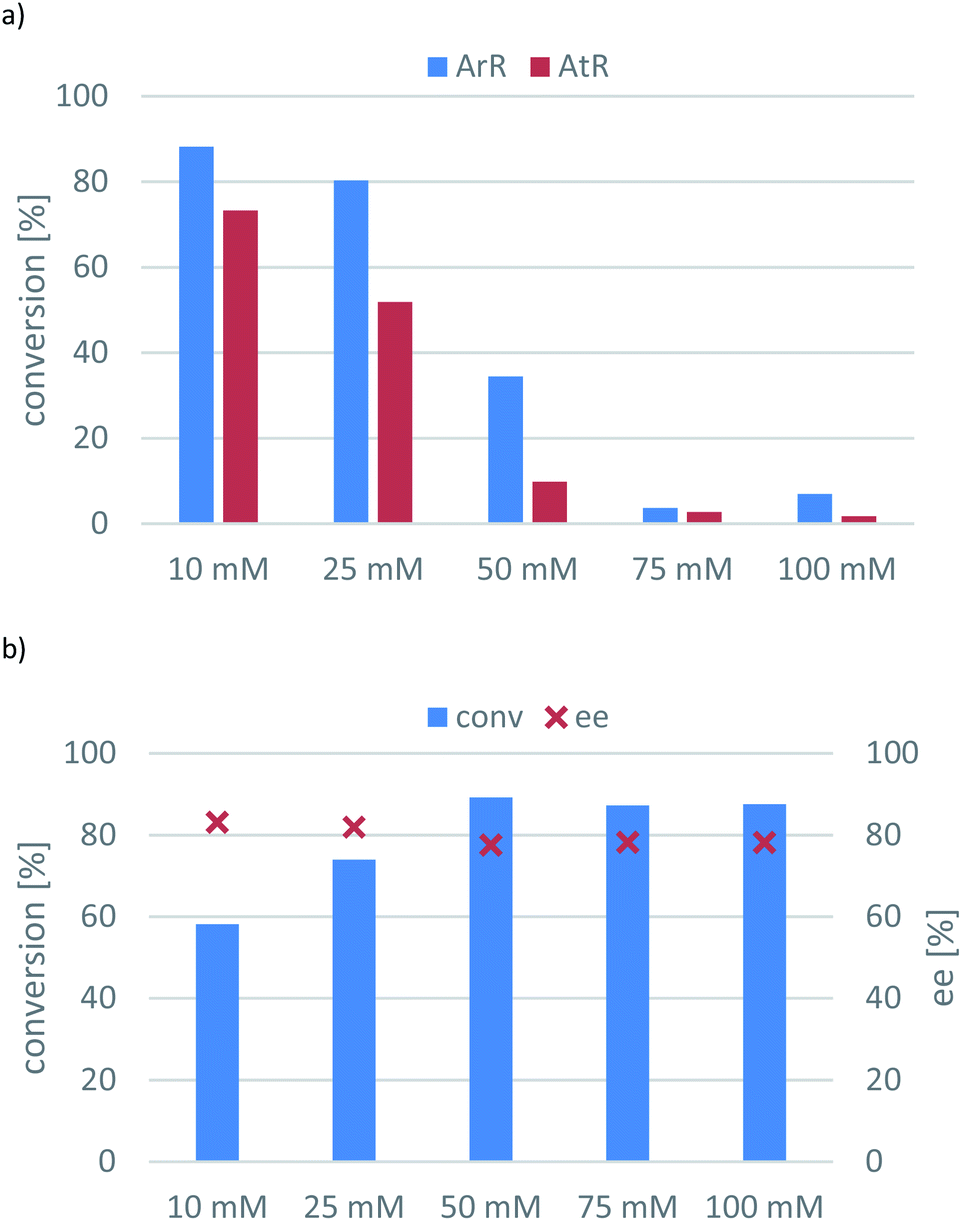 | ||
| Fig. 4 The effect of initial substrate concentration on the conversion of the transamination of 1-phenylpropan-2-one (7a) mediated by ArR-TA or AtR-TA (panel (a)); or ArRm-TA (panel (b)). Reaction conditions: immobilised whole-cell TA biocatalyst (20 mg), 7a (10–100 mM), sec-butylamine 12 (0.1–1 M), PLP (1 mM), sodium phosphate buffer (100 mM, pH 7.5), DMSO (5 v/v%), 30 °C, 24 h. | ||
Inspection of the substrate loading effect for 7a (Fig. 4) answered the question whether sol–gel immobilization could have protecting effect on the substrate/product inhibition of the transaminase-catalysed reaction. Because the conversions of the ArR-TA- and AtR-TA-catalysed reactions dropped significantly – especially with AtR-TA – (Fig. 4a), it is apparent that sol–gel whole cell immobilization could not protect against substrate/product and this phenomena seems to be an intrinsic property of these two TAs (similarly to many other TAs). On the other hand, substrate inhibition was not a serious issue for ArRm-TA (Fig. 4b), because this evolved TA retained its improved property of avoiding the substrate inhibition in sitagliptin intermediate production in the transamination of ketone 7a as well.
Although ArRm-TA could operate with 88% conversion at 100 mM substrate concentration (10-fold increase compared to the initial tests), the enantiomeric excess remained around 80%. Since ArRm-TA could not be used in highly enantiotope selective mode, the optimal substrate concentration was defined at 10 mM for the other two (R)-TAs.
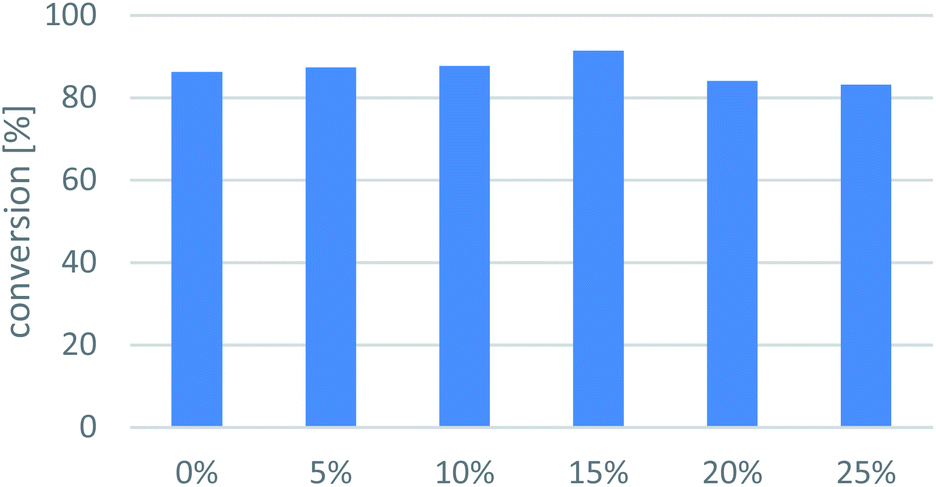 | ||
| Fig. 5 Cosolvent effect on conversion of the ArR-TA-mediated transamination of 1-phenylpropan-2-one (7a). Reaction conditions: immobilised whole-cell TA biocatalyst (20 mg), 7a (10 mM), sec-butylamine 12 (100 mM), PLP (1 mM), sodium phosphate buffer (100 mM, pH 7.5), DMSO (0–25 v/v%), 30 °C, 24 h. | ||
Transamination of 1-phenylpropan-2-one derivatives 7a–d
The optimized conditions were applied for screening the synthesis of the disubstituted (R)-phenylpropan-2-amines (8b–d) mediated by the three selected immobilized whole-cell (R)-TA biocatalysts (Table 2). With ArR-TA and AtR-TA all the four 1-phenylpropan-2-ones 7a–d were transformed with similar efficiency (the conversions were 88–89% for ArR-TA and 69–76% for AtR-TA). Interestingly, although ArRm-TA was evolved to transform a bulky substrate, this TA variant transformed the substituted derivatives 7b–d with significantly lower conversion (<36%) than the non-substituted ketone 7a (62%). Enantiotope selectivity of the ArRm-TA-mediated amination of the smaller ketones 7a–c was moderate (ee = 84.9–88.5%). Expectedly, the highest enantiotope selectivity could be achieved in amination of 7d (ee = 92.7%) due to the increased bulkiness of this substrate.| Entry | Substrate | Enzyme | Conversion [%] | ee [%] |
|---|---|---|---|---|
| a Reaction conditions: immobilised whole-cell TA biocatalyst (20 mg), 7a–d (10 mM), sec-butylamine 12 (100 mM), PLP (1 mM), sodium phosphate buffer (100 mM, pH 7.5), DMSO (5 v/v%), 30 °C, 24 h. | ||||
| 1 | 7a | ArR-TA | 88.9 | >99 (R) |
| 2 | AtR-TA | 76.2 | >99 (R) | |
| 3 | ArRm-TA | 62.2 | 84.9 (R) | |
| 4 | 7b | ArR-TA | 88.0 | >99 (R) |
| 5 | AtR-TA | 69.1 | >99 (R) | |
| 6 | ArRm-TA | 23.4 | 88.5 (R) | |
| 7 | 7c | ArR-TA | 88.5 | >99 (R) |
| 8 | AtR-TA | 68.7 | >99 (R) | |
| 9 | ArRm-TA | 21.1 | 85.4 (R) | |
| 10 | 7d | ArR-TA | 89.0 | >99 (R) |
| 11 | AtR-TA | 70.5 | >99 (R) | |
| 12 | ArRm-TA | 35.7 | 92.7 (R) | |
To compare the efficiency of our TA biocatalysts, an estimated ∼0.1 recombinant TA-content per mg immobilized biocatalyst can be considered (for details, see Experimental section). However, comparison with previously reported methods can only made in a qualitative and not quantitative way. The various methods used the TA biocatalysts in different forms and applied various amine donors as well.
For the transamination of 7a, commercially available purified TAs,30,60 lyophilized whole cells41 or cell-free extracts (CFEs) directly29 were applied as TA-biocatalysts. In case of lyophilized whole cells, 250% w/w TA-biocatalyst was used for transforming 17 mM of 7a with 1 M IPA (∼60 equiv.) and 1 mM PLP for 24 h at 30 °C.41 TA as CFE (∼2 mg protein) was also employed for conversion of 7a (10 mM) in presence of 5–10 equiv. amine donor (L-alanine, IPA, SBA, methylbenzylamine) and 0.1 mM PLP for 24 h at 30 °C.29
The early reports on transamination of molecule 7b applied cells from fermentation broth and defined the quantity of catalyst only as whole cells harvested from 100 mL culture broth.31,32,61 Only one report described the production of (R)-8b using commercially available TAs,60 using 2 mg of purified (R)-selective TA in to transform 7b (20 mM) with IPA (11, 50 equiv.) and PLP (1 mM) for 24 h at 30 °C in 91% conversion. When the enzyme load was increased to 1:1 w/w TA:substrate ratio, the amine donor equivalent could be lowered to 16.
Thus, in this work the TA-mediated reactions with already investigated substrates 7a,b proceeded with similar results as previously disclosed, and data on novel substrates 7c,d extended the applicability of the immobilized whole-cell TA form.
Kinetic resolution of the racemic amines 8a–d
Due to thermodynamic equilibrium reasons in the TA-mediated processes, kinetic resolution (KR) of racemic amines is more preferred to perform than asymmetric synthesis, however, at maximum conversion of the racemic substrate only 50% yield of the unconsumed enantiomer that can be achieved.62 Because in our preliminary screens none of the (S)-selective TAs in our hands (ArS-TA, VfS-TA, CvS-TAm)47 showed sufficient activity in the asymmetric synthesis of (S)-8a–d, these (S)-amines were accessed by the (R)-selective TAs also, using KR of the corresponding racemic amines 8a–d (Scheme 3, Fig. 6). In all cases, AtR-TA showed the highest activity and resulted in almost full conversions (>48%). Three substrates (8a, 8c and 8d) could be resolved efficiently with high selectivity (ee > 95%, c > 49%) while KR of 8b proceeded with lower conversion (c = 48.2%) reaching only 91% ee within 24 h.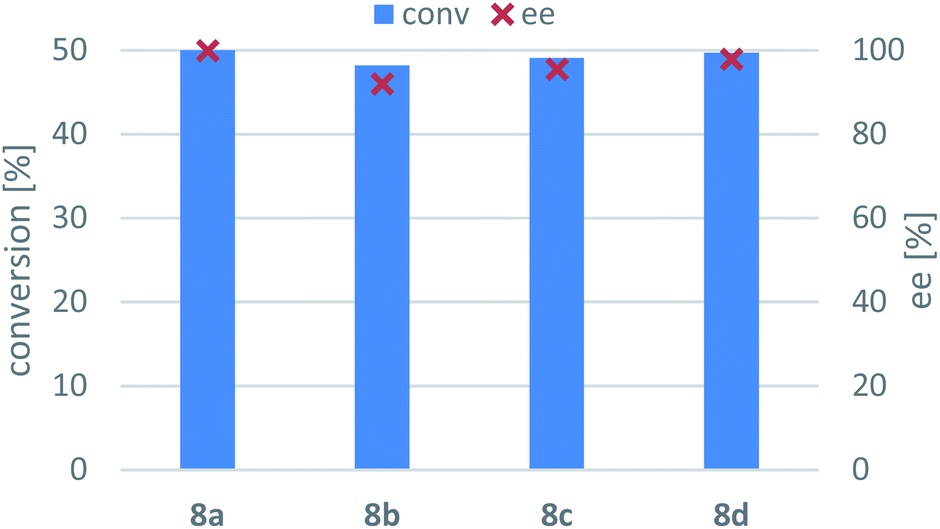 | ||
| Fig. 6 AtR-TA-mediated kinetic resolution of the racemic amines 8a–d. Reaction conditions: immobilised whole-cell TA biocatalyst (20 mg), 8a–d (10 mM), sodium pyruvate (5 mM), PLP (1 mM), sodium phosphate buffer (100 mM, pH 7.5), DMSO (5 v/v%), 30 °C, 24 h. | ||
Experimental
Methods and materials
The TAs used had high expression level (∼40% of all proteins in the cell, with expressions plasmids using T7 promoter, even 50% recombinant protein content could be achieved).64 On average, wet E. coli cell mass contains 200–320 mg mL−1 protein,65 and the density of the cells is ∼1.1 g cm−3.66 Furthermore, it is known from our previous studies that ∼0.9 g of such supported whole-cell TA biocatalyst can be produced from 1 g of wet E. coli cells.46,47 Thus, the estimated recombinant TA content of 1 g immobilized E. coli cells is ∼90–140 mg. Consequently, our supported whole-cell TA-biocatalysts contained approximately 8–13% w/w recombinant TA (as a part of the ∼20–33% w/w total protein content).
:1) and Hydrodex β-6TBDM column [25 m × 0.25 mm × 0.25 μm film with heptakis-(2,3-di-O-methyl-6-O-t-butyldimethylsilyl)-β-cyclodextrine; Macherey & Nagel].where eeR is the enantiomeric excess of the (R)-amine, PR and PS indicate the peak area of the corresponding enantiomer of the investigated amine 8a–d.
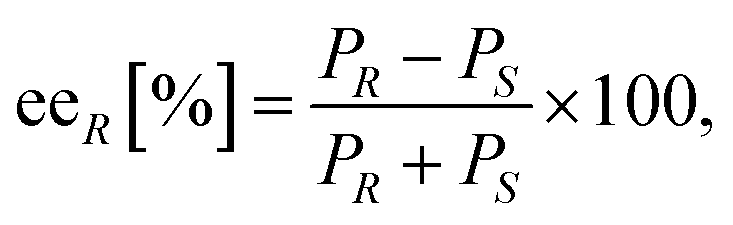
The conversion of the racemic amines 8a–d in the highly selective kinetic resolution (ckr) was calculated by the following equation:
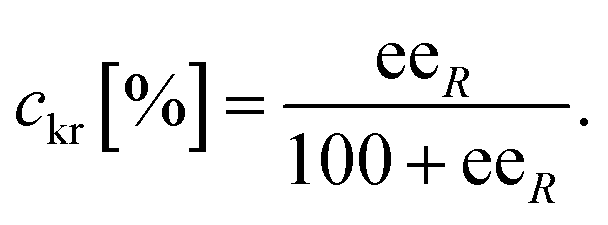
The molar response factor (f) of the amine 7a–d related to the corresponding starting ketone 8a–d was also determined from KR data as follows:
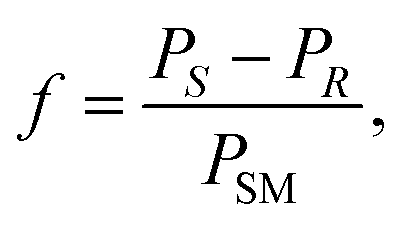
The conversion of the ketones 7a–d in the asymmetric synthesis (cas) was calculated using the response factor f as follows:
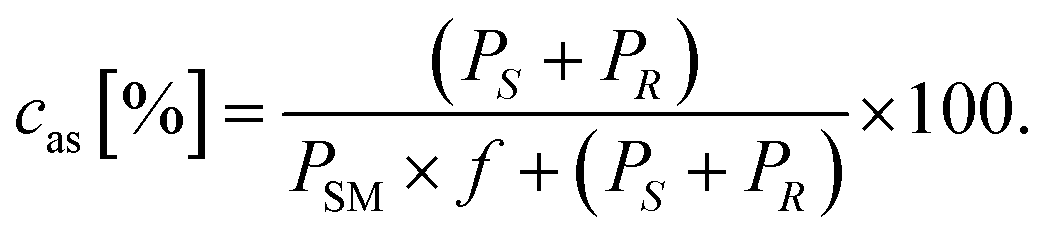
Synthesis of 3,4-disubstituted benzaldehydes (9b–d)
:4, TLC: Rf = 0.4) to give aldehyde 9b (2.42 g, 73%). White crystals, mp: 42–43 °C. 1H-NMR (500 MHz, CDCl3) 9.85 (s, 1H), 7.46 (dd, J = 8.2, 1.9 Hz, 1H), 7.41 (d, J = 2.0 Hz, 1H), 6.98 (d, J = 8.1 Hz, 1H), 3.97 (s, 4H), 3.94 (s, 3H); IR (cm−1) 2844, 1685, 1587, 1514, 1271, 1138. Physical and spectral data are in accordance with ref. 67.Production of ketones 7b–d by Darzens reaction70
:3) to give the desired ketone (7b–d).Synthesis of racemic amines 8a–d by reductive amination71,72
Biotransformations
Statistical analysis on the conversion of the unsubstituted ketone 7a in transamination reaction could be performed from 5 points: 83.0% (Table 1), 85.5% (Fig. 3), 88.2% (Fig. 4), 88.9% (Table 2), 87.4% (Fig. 5). Based on these data, standard deviation of the population could be calculated as 2.1% for μ = 86.6% as the mean value. Since the SD of conversion was quite moderate in biotransformation of 7a, the further investigations were performed as single sets of experiments.General method. To a 4 mL screw-capped vial containing KPi buffer (890 μL, 100 mM, pH 7.5) were added immobilized whole-cell TA (20 mg), PLP solution (10 μL, 100 mM in KPi buffer), DMSO solution of the racemic amine (50 μL, 200 mM of 8a–d in DMSO), and sodium pyruvate solution (50 μL, 100 mM in KPi buffer) and the resulted mixture was shaken at 450 rpm at 30 °C for 24 h. Then, the reaction was terminated by the addition of 1 M aqueous NaOH solution (100 μL) and EtOAc (500 μL). The aqueous phase was separated, and the organic phases was dried over anhydrous sodium sulphate. The conversion from the racemic amine (8a–d) to ketone (7a–d) and the enantiomeric excess of the unreacted amine [(S)-8a–d] were determined by chiral GC [50 μL of extracted reaction sample was derivatised to the corresponding acetamides (by addition of 10 μL acetic anhydride and shaking at 60 °C by 750 rpm for 1 h)]. The results are summarized in Fig. 6.
General method. To a 4 mL screw-capped vial containing KPi buffer (930 μL, 100 mM, pH 7.5) were added immobilized whole-cell TA (20 mg), PLP solution (10 μL, 100 mM in KPi buffer), DMSO solution of the ketone (50 μL, 200 mM of 7a–d in DMSO), and sec-butylamine 12 (10.1 μL, equal to 100 mM in the final volume), and the pH 8 was set with 1 M aqueous HCl solution. Then, the resulted mixture was shaken at 450 rpm and 30 °C for 24 h, and the reactions were terminated by the addition of 1 M aqueous NaOH solution (100 μL) and EtOAc (500 μL). The aqueous phase was separated, and the organic phases was dried over anhydrous sodium sulphate. The conversion from the ketone (7a–d) and the enantiomeric excess of the formed amine [(R)-8a–d] were determined by chiral GC [50 μL of extracted reaction sample was derivatised to the corresponding acetamides (by addition of 10 μL acetic anhydride and shaking at 60 °C by 750 rpm for 1 h)]. The results are summarized in Table 2.
Conclusions
This study with three (R)-selective transaminases of Arthrobacter sp. and Aspergillus terreus applied as immobilized whole-cell biocatalysts of the overexpressing E. coli cells showed that enantiopure drug-like (R)-1-(3′,4′-disubstituted phenyl)propan-2-amines (R)-8a–d could be obtained from the corresponding 1-arylpropan-2-ones (7a–d) by asymmetric transamination, while kinetic resolution with these (R)-selective TAs could provide the (S)-amines (S)-8a–d from the corresponding racemic amines 8a–d.The 3′,4′-disubstituted 1-phenylpropan-2-ones (7b–d) have been synthesised conveniently from vanillin (10) in two steps. The high-yield O-alkylations of 10 followed by Darzens reaction gave the desired ketones 7b–d in good yields. The racemic amines (8b–d) were obtained by Pd-catalysed reductive amination of 7b–d.
The reaction optimisation on model compound 7a probing seven different amine donors, and the effect of the substrate and co-solvent DMSO concentration revealed sec-butylamine as the most suitable amine donor. While high conversions with ArR-TA and AtR-TA with excellent selectivity (ee > 99%) could be achieved only up to 10 mM substrate concentration, the highly engineered ArRm-TA provided sufficiently high conversion even at 100 mM, albeit with insufficient selectivity (ee ∼ 80%).
With ArR-TA as the most versatile biocatalyst in this study, the (R)-1-phenylpropan-2-amine (8a) was obtained with 92% conversion and >99% ee employing sec-butylamine as amine donor under the optimised reaction conditions in 24 h. ArR-TA catalysed the transamination of all the 3,4-disubstituted ketones 7b–d as well with equally high conversion (88–89%) and excellent selectivity (ee > 99%). In addition, the kinetic resolution of 8a,c,d with AtR-TA could provide the (S)-amines (S)-8a–d with perfect conversion (>49% in KR) and good selectivity (ee > 95%), only the KR of 8b proceeded with lower conversion (c = 48.2%) and moderate selectivity (ee = 92%).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was partially funded by Higher Education Excellence Program of the Ministry of Human Capacities in the frame of Biotechnology Research Area of Budapest University of Technology and Economics (BME FIKP-BIO). Authors thank Prof. Beáta G. Vértessy (BME, Hungary) for providing access to protein expression facilities of her laboratories. Á. Lakó thanks the PhD fellowship (SPAC/BDE/08) from Hovione Farmaciência. The authors also acknowledge the Gedeon Richter Talentum Foundation for the PhD fellowship of Z. Molnár.Notes and references
- D. Ghislieri and N. J. Turner, Top. Catal., 2014, 57, 284–300 CrossRef CAS.
- T. C. Nugent, Chiral Amine Synthesis: Methods, Developments and Applications, Wiley-VCH, 2010 Search PubMed.
- Y. J. Kim, Y. S. Choi, S. Yang, W. R. Yang and J. H. Jeong, Synlett, 2015, 26, 1981–1984 CrossRef CAS.
- Z. Chen, Y. Ma, M. He, H. Ren, S. Zhou, D. Lai, Z. Wang and L. Jiang, Appl. Biochem. Biotechnol., 2015, 176, 2267–2278 CrossRef CAS.
- R. L. Hanson, B. L. Davis, Y. Chen, S. L. Goldberg, W. L. Parker, T. P. Tully, M. A. Montana and R. N. Patel, Adv. Synth. Catal., 2008, 350, 1367–1375 CrossRef CAS.
- E. Farkas, M. Oláh, A. Földi, J. Kóti, J. Éles, J. Nagy, C. A. Gal, C. Paizs, G. Hornyánszky and L. Poppe, Org. Lett., 2018, 20, 8052–8056 CrossRef CAS.
- K. R. Bailey, A. J. Ellis, R. Reiss, T. J. Snape and N. J. Turner, Chem. Commun., 2007, 44, 3640–3642 RSC.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS.
- M. J. Abrahamson, E. Vázquez-Figueroa, N. B. Woodall, J. C. Moore and A. S. Bommarius, Angew. Chem., Int. Ed., 2012, 51, 3969–3972 CrossRef CAS.
- F. Leipold, S. Hussain, D. Ghislieri and N. J. Turner, ChemCatChem, 2013, 5, 3505–3508 CrossRef CAS.
- Northeastern University, Methods And Compositions for Preventing Or Treating Obesity, WO 2012/027695 A1, 2012.
- K. Stemmer, T. D. Müller, R. D. DiMarchi, P. T. Pfluger and M. H. Tschöp, J. Clin. Invest., 2019, 129, 4058–4071 CrossRef.
- E. L. Lane, Eur. J. Neurosci., 2019, 49, 384–398 CrossRef.
- M. Naoi, W. Maruyama and M. Shamoto-Nagai, J. Neural Transm., 2020, 127, 131–147 CrossRef CAS.
- T. Winston, Methods For Treating Attention Deficit Hyperactivity Disorder Using A Combination Of Bupropion ((±)-2-(Tert-Butylamino)-1-(3-Chlorophenyl)Propan-1-One) And Phentermine (2-Methyl-1-Phenylpropan-2-Amine), US Pat., US 2016/0022659 A1, 2016.
- S. Kotagal, Paediatr. Respir. Rev., 2018, 25, 19–24 Search PubMed.
- S. Pattanaik, R. S. Mavuduru, A. Panda, J. L. Mathew, M. M. Agarwal, E. C. Hwang, J. A. Lyon, S. K. Singh and A. K. Mandal, BJU Int., 2019, 124, 27–34 CrossRef CAS.
- J. F. Sumowski, N. Chiaravalloti, D. Erlanger, T. Kaushik, R. H. B. Benedict and J. Deluca, Mult. Scler. J., 2011, 17, 1141–1145 CrossRef CAS.
- S. E. Denmark, T. Weber and D. W. Piotrowski, J. Am. Chem. Soc., 1987, 109, 2224–2225 CrossRef CAS.
- S. K. Talluri and A. Sudalai, Tetrahedron, 2007, 63, 9758–9763 CrossRef CAS.
- S. Li, K. Huang and X. Zhang, Chem. Commun., 2014, 50, 8878 RSC.
- I. A. Sayyed and A. Sudalai, Tetrahedron: Asymmetry, 2004, 15, 3111–3116 CrossRef CAS.
- L. Muñoz, A. M. Rodriguez, G. Rosell, M. P. Bosch and A. Guerrero, Org. Biomol. Chem., 2011, 9, 8171–8177 RSC.
- J. González-Sabín, V. Gotor and F. Rebolledo, Tetrahedron: Asymmetry, 2002, 13, 1315–1320 CrossRef.
- F. Poulhès, N. Vanthuyne, M. P. Bertrand, S. Gastaldi and G. Gil, J. Org. Chem., 2011, 76, 7281–7286 CrossRef.
- B. A. Davis and D. A. Durden, Synth. Commun., 2001, 31, 569–578 CrossRef CAS.
- D. Zhu and L. Hua, Biotechnol. J., 2009, 4, 1420–1431 CrossRef CAS.
- J. Lalonde, Curr. Opin. Biotechnol., 2016, 42, 152–158 CrossRef CAS.
- K. Fesko, K. Steiner, R. Breinbauer, H. Schwab, M. Schürmann and G. A. Strohmeier, J. Mol. Catal. B: Enzym., 2013, 96, 103–110 CrossRef CAS.
- D. González-Martínez, V. Gotor and V. Gotor-Fernández, Adv. Synth. Catal., 2019, 361, 2582–2593 Search PubMed.
- A. Iwasaki, Y. Yamada, Y. Ikenaka and J. Hasegawa, Biotechnol. Lett., 2003, 25, 1843–1846 CrossRef CAS.
- A. Iwasaki, Y. Yamada, N. Kizaki, Y. Ikenaka and J. Hasegawa, Appl. Microbiol. Biotechnol., 2006, 69, 499–505 CrossRef CAS.
- J. Paris, A. Telzerow, N. Ríos-Lombardía, K. Steiner, H. Schwab, F. Morís, H. Gröger and J. González-Sabín, ACS Sustainable Chem. Eng., 2019, 7, 5486–5493 CrossRef CAS.
- A. Telzerow, J. Paris, M. Håkansson, J. González-Sabín, N. Ríos-Lombardía, M. Schürmann, H. Gröger, F. Morís, R. Kourist, H. Schwab and K. Steiner, ACS Catal., 2019, 9, 1140–1148 CrossRef CAS.
- D. Koszelewski, I. Lavandera, D. Clay, D. Rozzell and W. Kroutil, Adv. Synth. Catal., 2008, 350, 2761–2766 CrossRef CAS.
- L. J. Ye, H. H. Toh, Y. Yang, J. P. Adams, R. Snajdrova and Z. Li, ACS Catal., 2015, 5, 1119–1122 CrossRef CAS.
- T. Knaus, L. Cariati, M. F. Masman and F. G. Mutti, Org. Biomol. Chem., 2017, 15, 8313–8325 RSC.
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 CrossRef CAS.
- J. A. Houwman, T. Knaus, M. Costa and F. G. Mutti, Green Chem., 2019, 21, 3846–3857 RSC.
- J. Liu, B. Q. W. Pang, J. P. Adams, R. Snajdrova and Z. Li, ChemCatChem, 2017, 9, 425–431 CrossRef CAS.
- R. S. Correia Cordeiro, N. Ríos-Lombardía, F. Morís, R. Kourist and J. González-Sabín, ChemCatChem, 2019, 11, 1272–1277 CrossRef CAS.
- Y. Zhang, J. Ge and Z. Liu, ACS Catal., 2015, 5, 4503–4513 CrossRef.
- P. Tufvesson, J. Lima-Ramos, J. S. Jensen, N. Al-Haque, W. Neto and J. M. Woodley, Biotechnol. Bioeng., 2011, 108, 1479–1493 CrossRef CAS.
- A. I. Benítez-Mateos, M. L. Contente, S. Velasco-Lozano, F. Paradisi and F. López-Gallego, ACS Sustainable Chem. Eng., 2018, 6, 13151–13159 CrossRef.
- P. Tufvesson, J. Lima-Ramos, M. Nordblad and J. M. Woodley, Org. Process Res. Dev., 2011, 15, 266–274 CrossRef CAS.
- L. Nagy-Győr, E. Abaházi, V. Bódai, P. Sátorhelyi, B. Erdélyi, D. Balogh-Weiser, C. Paizs, G. Hornyánszky and L. Poppe, ChemBioChem, 2018, 19, 1845–1848 CrossRef.
- Z. Molnár, E. Farkas, Á. Lakó, B. Erdélyi, W. Kroutil, B. G. Vértessy, C. Paizs and L. Poppe, Catalysts, 2019, 9, 1–11 CrossRef.
- D. Koszelewski, K. Tauber, K. Faber and W. Kroutil, Trends Biotechnol., 2010, 28, 324–332 CrossRef CAS.
- A. W. H. Dawood, M. S. Weiß, C. Schulz, I. V. Pavlidis, H. Iding, R. O. M. A. de Souza and U. T. Bornscheuer, ChemCatChem, 2018, 10, 3943–3949 CrossRef CAS.
- H. Yun, B.-K. Cho and B.-G. Kim, Biotechnol. Bioeng., 2004, 87, 772–778 CrossRef CAS.
- P. Kelefiotis-Stratidakis, T. Tyrikos-Ergas and I. V. Pavlidis, Org. Biomol. Chem., 2019, 17, 1634–1642 RSC.
- C. E. Paul, M. Rodríguez-Mata, E. Busto, I. Lavandera, V. Gotor-Fernández, V. Gotor, S. García-Cerrada, J. Mendiola, Ó. de Frutos and I. Collado, Org. Process Res. Dev., 2014, 18, 788–792 CrossRef CAS.
- A. Gomm, W. Lewis, A. P. Green and E. O’Reilly, Chem.–Eur. J., 2016, 22, 12692–12695 CrossRef CAS.
- M. D. Truppo, H. Strotman and G. Hughes, ChemCatChem, 2012, 4, 1071–1074 CrossRef CAS.
- Y. Satyawali, E. Ehimen, L. Cauwenberghs, M. Maesen, P. Vandezande and W. Dejonghe, Biochem. Eng. J., 2017, 117, 97–104 CrossRef CAS.
- L. H. Andrade, W. Kroutil and T. F. Jamison, Org. Lett., 2014, 16, 6092–6095 CrossRef CAS.
- C. S. Fuchs, R. C. Simon, W. Riethorst, F. Zepeck and W. Kroutil, Bioorg. Med. Chem., 2014, 22, 5558–5562 CrossRef CAS.
- F. G. Mutti and W. Kroutil, Adv. Synth. Catal., 2012, 354, 3409–3413 CrossRef CAS.
- S. Chen, H. Land, P. Berglund and M. S. Humble, J. Mol. Catal. B: Enzym., 2016, 124, 20–28 CrossRef CAS.
- L. Martínez-Montero, V. Gotor, V. Gotor-Fernández and I. Lavandera, Adv. Synth. Catal., 2016, 358, 1618–1624 CrossRef.
- K. Nakamichi, T. Shibatani, Y. Yamamoto and T. Sato, Appl. Microbiol. Biotechnol., 1990, 33, 637–640 CrossRef CAS.
- M. S. Malik, E.-S. Park and J.-S. Shin, Appl. Microbiol. Biotechnol., 2012, 94, 1163–1171 CrossRef CAS.
- F. G. Mutti, C. S. Fuchs, D. Pressnitz, J. H. Sattler and W. Kroutil, Adv. Synth. Catal., 2011, 353, 3227–3233 CrossRef CAS.
- G. L. Rosano and E. A. Ceccarelli, Front. Microbiol., 2014, 5, 1–17 Search PubMed.
- M. B. Elowitz, M. G. Surette, P. E. Wolf, J. B. Stock and S. Leibler, J. Bacteriol., 1999, 181, 197–203 CrossRef CAS.
- E. Martinez-Salas, J. A. Martin and M. Vicente, J. Bacteriol., 1981, 147, 97–100 CrossRef CAS.
- L. Gavara, T. Boisse, J. P. Hénichart, A. Daïch, B. Rigo and P. Gautret, Tetrahedron, 2010, 66, 7544–7561 CrossRef CAS.
- H. Doušová, Z. Růžičková and P. Šimůnek, J. Heterocycl. Chem., 2018, 55, 670–684 CrossRef.
- A. Younai, G. F. Chin and J. T. Shaw, J. Org. Chem., 2010, 75, 8333–8336 CrossRef CAS.
- J. Zimbron, M. Seeger-Weibel, H. Hirt and F. Gallou, Synthesis, 2008, 2008, 1221–1226 CrossRef.
- M. Vahermo, T. Suominen, A. Leinonen and J. Yli-Kauhaluoma, Arch. Pharm., 2009, 342, 201–209 CrossRef CAS.
- P. Falus, Z. Boros, G. Hornyánszky, J. Nagy, F. Darvas, L. Ürge and L. Poppe, Tetrahedron Lett., 2011, 52, 1310–1312 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08134e |
| This journal is © The Royal Society of Chemistry 2020 |