
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCircular and linear: a tale of aptamer selection for the activation of SIRT1 to induce death in cancer cells†
Basma Al-Sudaniab,
Abby H. Ragazzon-Smithc,
Athar Azizb,
Rania Alansarid,
Natalie Ferryb,
Marija Krstic-Demonacosb and
Patricia A. Ragazzon *d
*d
aCollege of Pharmacy, Branch of Clinical Laboratory Sciences, University of Mustansiriya, UK
bBiomedical Research Centre, School of Environment and Life Sciences, University of Salford, UK
cSchool of Earth and Environmental Sciences, The University of Manchester, UK
dSchool of Pharmacy and Bioengineering, Keele University, Hornbeam Building (2.26), Keele ST5 5BG, UK. E-mail: p.ragazzon@keele.ac.uk
First published on 21st December 2020
Abstract
It is a challenge to select the right target to treat conditions without affecting non-diseased cells. Cancer belongs to the top 10 causes of death in the world and it remains difficult to treat. Amongst cancer emerging targets, silent information regulator 1 (SIRT1) – a histone deacetylase – has shown many roles in cancer, ageing and metabolism. Here we report novel SIRT1 ligands that bind and modulate the activity of SIRT1 within cells and enhance its enzymatic activity. We developed a modified aptamer capable of binding to and forming a complex with SIRT1. Our ligands are aptamers, they can be made of DNA or RNA oligonucleotides, their binding domain can recognise a target with very high affinity and specificity. We used the systematic evolution of ligands by exponential enrichment (SELEX) technique to develop circular and linear aptamers selectively binding to SIRT1. Cellular consequences of the interaction were monitored by fluorescence microscopy, cell viability assay, stability and enzymatic assays. Our results indicate that from our pool of aptamers, circular AC3 penetrates cancerous cells and is recruited to modulate the SIRT1 activity. This modulation of SIRT1 resulted in anticancer activity on different cancer cell lines. Furthermore, this modified aptamer showed no toxicity on one non-cancerous cell line and was stable in human plasma. We have demonstrated that aptamers are efficient tools for localisation of internal cell targets, and in this particular case, anticancer activity through modulation of SIRT1.
1 Introduction
Histone deacetylases (HDACs) remove the acetyl group from the ε-amino lysine residues on histones, making the interaction between histones and DNA stronger. This tight interaction reduces the chances of RNA polymerase contacting DNA.1–3 This interaction regulates gene expression affecting the development of multiple diseases including cancer.2 Human HDACs are classified into four classes: I, II III and IV.3 The class III enzymes are known as sirtuins,4 these enzymes are NAD+ -dependent consisting of SIRT1 to SIRT7.5,6 SIRT1 is the most studied; it can be present in the nucleus or the cytoplasm depending on the cell type or tissue evaluated.7 While the functions of SIRT1 are still largely unknown, a link between SIRT1 activity and the metabolic status of the cell has been suggested.7 SIRT1 targets multiple proteins such as p53, p73, E2F, HIC1 and Ku70, all of which are responsible for major cellular processes.8 SIRT1 expression has been shown to be significantly elevated in various cancers.6,9–12 However, the mechanism of dysregulation of SIRT1 is still unclear. Overexpression of SIRT1 could be a consequence of cancer rather than a cause.2 Under physiological conditions in response to cellular stress or to DNA damage, SIRT1 appears to promote cell survival via cell cycle arrest, DNA repair, or inhibition of apoptosis. In case of persistent stress signal or significantly high levels of cellular damage SIRT1 induces cell senescence.2Macromolecules, like DNA, can offer a more selective and effective approach in inhibition of enzymes. Nucleic acid–based aptamers rely on their 3D structure to recognise and bind their selective targets through a combination of quadruplexes, triplexes, pseudoknots, hairpins, bulges, loops and stems.13 Aptamers have many advantages over small molecules and to a certain extent over antibodies, for example, they present high specificity and affinity, present low immunogenicity,14,15 and can be selected in vitro with reduced costs. Aptamers are stable upon modifications presenting a longer half-life while avoiding bio-transformation and have a wide range of potential targets.16 The approach of combinatorial chemistry is used to generate and select aptamers, they are generally obtained through a process known as SELEX, where a library of oligonucleotides is incubated with the target. The bound sequences are separated from unbound ones, and they are subsequently amplified for further iterative cycles. After 8–12 rounds a small number of sequences are obtained and taken for further studies. They have applications in the environmental, pharmaceutical and analytical industries.17,18 Nucleic acids are prone to degradation by nucleases. Due to this, linear aptamers present stability issues in vivo. Chemically modified counterparts have shown improvements, for example, 3′-end capping with inverted thymidine,19 biotin conjugates,20 2′-fluoro (2′-F) or 2′-amino (2′-NH2) ribose groups,21 phosphodiester linkage of DNA replaced with methylphosphonate or phosphorothioate analogue,22 oligonucleotide phosphodiester linkage replaced with triazole linkages,23 L-DNA,24 5′-end with cholesterol,25 dialkyls lipids26 PEGylation27 and base modifications like phosphorodithioate (PS2) substitution28 or 5-(N-benzylcarboxyamide)-2′-deoxyuridine modification that produces Slow Off-rate Modified Aptamers (SOMAmers).29 Locked nucleic acids are analogues of ribonucleotides with a methylene linkage between 2′-O and 4′-C of the sugar ring; they are resistant to nuclease activity.30 Their secondary structure which is responsible for functionality might be affected due to post-selection modifications.
The SELEX methodology can be used employing a variety of methods, such as capillary electrophoresis (CE-SELEX),31 microfluidic (M-SELEX),32 cell (cell-SELEX),33 in vivo from a tumour inside an animal (in vivo-SELEX)34 and high-throughput sequencing (HTS-SELEX).35 Molecular entrance into the cells is favoured by non-charged molecules, hence many aptamers are designed for extracellular targets.19,36,37 Circular nucleic acids have also shown to be stable since they have no 3′ or 5′ ends exposed to be acted upon by exonuclease digestion.19 Circularisation of aptamers is an attractive alternative to chemical modification for improving aptamer stability as well as assisting with aptamer internalisation through the cell membrane.38 These aptamers can demonstrate good stability and internalisation potential. Circularisation of aptamers permits the use of unmodified nucleotides thus avoiding potential toxicity associated with chemical modification.
SIRT1 was chosen for its dual activity during cell cycle and its potential role in carcinogenesis. Our initial hypothesis stipulated that a circular aptamer would be able to penetrate a living cell due to its non-charged conformation and this could be monitored via fluorescence microscopy. Our aptamer selection used a DNA library in circular and linear forms. The circular selection produced an aptamer capable of selective binding to SIRT1 modulating its activity with further implications in biological activity studies.
2 Experimental
2.1 Materials and methods
Chemicals were purchased from ThermoFisher Scientific, Sigma-Aldrich or SLS (Scientific Laboratory Supplies). A549, MCF7, MDA-MB468 and U2OS cells were purchased from Sigma-Aldrich. Caco-2, Beas2B and HepG2 were purchased from the American Type Culture Collection (ATCC). BEGM BulletKit™ was purchased from Lonza. Recombinant human SIRT1-GST-tagged [SIRT1-462H] was purchased from Creative BioMart. PreScission Protease on-column GSTrap FF and NHS-HP SpinTrap column were purchased from GE HealthCare. DNA library 5′-TTCGGAAGAGATGGCGAC-N40–CGAGCTGATCCTGATGGAA-3′ was purchased from TriLink Bio Technologies. Phosphorylation reaction kit (EK00310), T4DNA (EL0012) and Exonuclease I (EN0581) were purchased from ThermoFisher Scientific. BAS-P1, BAS-P3 and BAS-P3- no tail primers (BAS-P1: 5′-TTCGGAAGAGATGGCGAC-3′, BAS-P3: 5′-ATGTCGTGCGTGCTA-SP18-TTCCATCAGGATCAGCTCG-3′, BASP3-notail: 5′-TTCCATCAGGATCAGCTCG-3′), C1: 5′-TTGCGGCATTTTGCCTTCCTGTTTTTGCTCACCCAGAAAC-3′; C2: 5′-TTCGGAAGAGATGGCGAC TTGCGGCATTTTGCCTTCCTGTTTTTGCTCACCCAGAAAC CGAGCTGATCCTGATGGAA-3′; AC3: 5′-CGAGTGGGTTACATCGAAACTGGATCTCAACAGCGGTAAC-3′; C4: 5′-TTCGGAAGAGATGGCGAC CGAGTGGGTTACATCGAAACTGGATCTCAACAGCGGTAAC CGAGCTGATCCTGATGGAA-3′; C5: 5′-CACTCCCTCTGCGTGCGAATTTTGCCTATGGCGCATATTC-3′; C6: 5′-TTCGGAAGAGATGGCGAC CACTCCCTCTGCGTGCGAATTTTGCCTATGGCGCATATTC CGAGCTGATCCTGATGGAA-3′; L1: 5′-CGGACTGCAACCTATGCTATCGTTGATGTCTGTCCAAGCA-3′; L2: 5′-TTCGGAAGAGATGGCGAC CGGACTGCAACCTATGCTATCGTTGATGTCTGTCCAAGCA CGAGCTGATCCTGATGGAA-3′; L3: 5′-CACTTTTCGGGGAAATGTGCGCGGAACCCCTATTTGTTTA-3′; L4: 5′-TTCGGAAGAGATGGCGAC CACTTTTCGGGGAAATGTGCGCGGAACCCCTATTTGTTTA CGAGCTGATCCTGATGGAA-3′; and AC3-FAM (5′-CGAG-i6-FAMK-GGGTTACATCGAAACTGGATCTCAACAGCGGTAAC-3′) were purchased from IDTDNA (FAM is an NHS ester); Scrambled: 5′-ACGCGATACAGTCCTAAGTGGATCAAGTGGCGATACCTGA-3′.ISOLATE II PCR and Gel kit, VENT DNA polymerase, Vector Ptz57R/T and E. coli DH5α were purchased from BioLabs. Nucleospin® Extract II kit and QIAprep Spin Miniprep Kit were purchased from Qiagen. Anti-SIRT1 antibody (ab104833) was purchased from Abcam. Alexa Fluor 594 goat anti-mouse IgG (AF594, Texas Red dye) was purchased from Life Technologies. Clear bottom imaging tissue culture plates were purchased from Corning. Human plasma was purchased from Seralab and used immediately.
2.2 Procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g at 4 °C for 30 min. The pellet was washed with 70% ethanol and repeated the centrifugation step then dried and re-suspended in 20 μl water and set up PCR in 100 μl volumes. To determine if the library could bind to the column in absence of SIRT1, 1.0 ml of BAS library in binding buffer was introduced in the column, with an OD260nm: 0.1155. After incubation of 2 hours at 37 °C, 3 ml were flushed through the column; the eluted was collected and the absorbance at 260 nm was recorded. The results showed an OD260nm: 0.0339 (equivalent to OD260nm: 0.1017 in 1 ml), demonstrating that no association was found between the oligonucleotides and the column matrix. Polymerase Chain Reaction (PCR): 1 μM of both BAS-P1 and BAS-P3 primers plus recovered sequences from the previous step, dissolved in 20 μl were used for the of PCR amplification process. The 18 atom hexaethylene-glycol spacer in the antisense primer BAS-P3 connects the complementary primer region to a 15 nt random sequence helping with strand separation of the PCR products by PAGE. The PCR program 20 cycles run as: denaturation 95 °C 30 s, annealing: 50 °C 45 s and extension: 72 °C 10 s, on a RoboCycler® Gradient 96 (Stratagene). The product was purified by urea PAGE and ISOLATE II PCR and Gel kit (Bioline BIO52058). In the case of linear selection, the product was used straight away. In the case of circular selection, phosphorylation and circularisation was performed on the pooled PCR products. Around 8 rounds were repeated for all the steps of selection circular aptamers. Cloning, plasmid isolation and sequencing: the final population of the selection step, dissolved in 2 μl was used as template, it was added 200 μM dNTP, 1 μM each of BAS-P1 and BASP3- no tail primers, buffer and VENT DNA polymerase. PCR products were purified for the cloning using the NucleoSpin® Extract II kit following manufacturer's instructions (Clontech, cat 636971). The ligation reaction was mixed with 0.52 pmol of purified PCR product and added to 6 μl of 5X ligation buffer, 0.17 pmol of Vector Ptz57R/T and 3U T4 DNA ligase in a final 30 μl volume, stirred for 5 min and then kept on 4 °C overnight. E. coli DH5α high efficiency competent cells were used for transformation following manufacturer's recommendation. For transformation, 100 μl cell cultures were plated onto duplicate LB/100 μg ml−1 ampicillin 50 μg ml−1/IPTG/X-Gal plates. Colonies were incubated overnight at 37 °C with gentle shaking, picked and inoculated into LB media supplemented with 50 μg ml−1 ampicillin and incubated overnight at 37 °C with gentle shaking. QIAprep Spin Miniprep Kit manufacturer's recommendations (Qiagen, cat 27104) were used for plasmid preparation, for each clone, 0.6 μg purified plasmid DNA was mixed with 20 pmol forward primers in a 100 μl PCR tube. All aptamer samples were outsourced for sequencing to Source Bio Science. The Data of sequencing were analysed by DNAMAN 5.29 software (Lynnon corp).
000 g at 4 °C for 30 min. The pellet was washed with 70% ethanol and repeated the centrifugation step then dried and re-suspended in 20 μl water and set up PCR in 100 μl volumes. To determine if the library could bind to the column in absence of SIRT1, 1.0 ml of BAS library in binding buffer was introduced in the column, with an OD260nm: 0.1155. After incubation of 2 hours at 37 °C, 3 ml were flushed through the column; the eluted was collected and the absorbance at 260 nm was recorded. The results showed an OD260nm: 0.0339 (equivalent to OD260nm: 0.1017 in 1 ml), demonstrating that no association was found between the oligonucleotides and the column matrix. Polymerase Chain Reaction (PCR): 1 μM of both BAS-P1 and BAS-P3 primers plus recovered sequences from the previous step, dissolved in 20 μl were used for the of PCR amplification process. The 18 atom hexaethylene-glycol spacer in the antisense primer BAS-P3 connects the complementary primer region to a 15 nt random sequence helping with strand separation of the PCR products by PAGE. The PCR program 20 cycles run as: denaturation 95 °C 30 s, annealing: 50 °C 45 s and extension: 72 °C 10 s, on a RoboCycler® Gradient 96 (Stratagene). The product was purified by urea PAGE and ISOLATE II PCR and Gel kit (Bioline BIO52058). In the case of linear selection, the product was used straight away. In the case of circular selection, phosphorylation and circularisation was performed on the pooled PCR products. Around 8 rounds were repeated for all the steps of selection circular aptamers. Cloning, plasmid isolation and sequencing: the final population of the selection step, dissolved in 2 μl was used as template, it was added 200 μM dNTP, 1 μM each of BAS-P1 and BASP3- no tail primers, buffer and VENT DNA polymerase. PCR products were purified for the cloning using the NucleoSpin® Extract II kit following manufacturer's instructions (Clontech, cat 636971). The ligation reaction was mixed with 0.52 pmol of purified PCR product and added to 6 μl of 5X ligation buffer, 0.17 pmol of Vector Ptz57R/T and 3U T4 DNA ligase in a final 30 μl volume, stirred for 5 min and then kept on 4 °C overnight. E. coli DH5α high efficiency competent cells were used for transformation following manufacturer's recommendation. For transformation, 100 μl cell cultures were plated onto duplicate LB/100 μg ml−1 ampicillin 50 μg ml−1/IPTG/X-Gal plates. Colonies were incubated overnight at 37 °C with gentle shaking, picked and inoculated into LB media supplemented with 50 μg ml−1 ampicillin and incubated overnight at 37 °C with gentle shaking. QIAprep Spin Miniprep Kit manufacturer's recommendations (Qiagen, cat 27104) were used for plasmid preparation, for each clone, 0.6 μg purified plasmid DNA was mixed with 20 pmol forward primers in a 100 μl PCR tube. All aptamer samples were outsourced for sequencing to Source Bio Science. The Data of sequencing were analysed by DNAMAN 5.29 software (Lynnon corp).
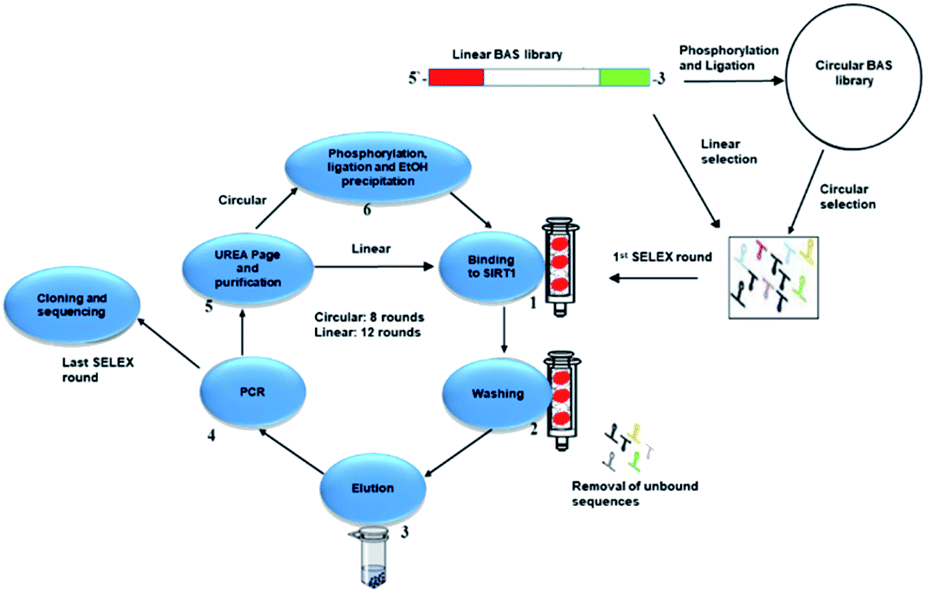 | ||
| Fig. 1 Workflow representation of the selection process. In the case of the circular library, the oligonucleotides were phosphorylated and ligated. In the case of the linear library, the oligonucleotides were unmodified. First stage of the cycle starts with the SIRT1 interacting with either library. Unbound sequences were eluted using different NaCl ionic concentration. The bound sequences were then amplified by PCR. In the case of circular library, the sequences were phosphorylated and ligated before entering the second round. The linear library sequences were used from the purification step after the PCR one. Once eight cycles (circular) and twelve (linear) were performed, the sequences were cloned, analysed and studied for biological activity. | ||
000 Caco-2, HepG2, U2OS, A549, MCF7, MBA-MB468 and Beas2B cells were seeded in 100 μl per well of complete medium in clear bottom imaging tissue culture plates for 18 hours. On the next day, cells were treated with AC3 as 0.1 μM for U2OS, and MDA-MB468, 0.2 μM for MCF-7 and HepG2, 0.3 μM for A549 and Caco2, and 1 μM to Beas-2B and incubated at 5% CO2/95% humidified air at 37 °C for 72 hours. At the end of the incubation period, the cells were washed three times in PBS, fixed for 5 min at room temperature with formalin 4%, washed twice in PBS, then permeabilised by 0.5% Triton X-100 for 5 min, washed thrice with PBS; nonspecific binding was blocked with 3% FBS for 1 h at room temperature, then the blocking solution was removed, 100 μl per well of anti-SIRT1 antibody 1:500 were added to cells and incubated overnight at 4 °C. On the next day, cells were washed three times in PBS, and incubated for 2 hours in the dark at room temperature with 1:2000 Alexa Fluor 594 goat anti-mouse IgG/1% FBS in PBS, then the cells were washed three times in PBS. Cells were treated with 10 μl of PBS containing DAPI, left for 1 hour before the microscopic examination and image capture in a Cytation™ 3 Cell Imaging Multi-Mode Reader. Fluorescence intensity was measured by LUMIStar Luminometer Microplate Reader. Filters were used as: GFP filter was used for FAM (carboxyfluorescein, green) exc. 495 nm, em. 509 nm; DAPI (4′,6-diamidino-2-phenylindole-dihydrochloride, blue) exc. 359 nm, em. 461 nm and Texas Red (sulforhodamine 101 acid chloride, red) exc. 596 nm, em. 615 nm. The results represent the mean ± SEM of duplicates.3 Results and discussion
Modulation of SIRT1 activity is beneficial against several diseases including neurodegeneration, cancer, inflammation and metabolic diseases.2,3,8,39,40 This manuscript describes two approaches to obtain novel DNA aptamers for selective targeting of intracellular SIRT1. Two different types of aptamers, linear and circular were compared. A comprehensive set of experiments were performed in order to select the optimal sequence from our pool of aptamers.As shown in Fig. 1, the selection steps involved binding, partition, elution, amplification (this section is known as SELEX rounds13) and cloning. This is an iterative process and can take up to 20 rounds of repeated cycles.16,41–43
Enrichment of selective sequences can be monitored after each round until a plateau of intensity for each band is reached as shown via polyacrylamide gels (Fig. 1A and B, ESI†).
3.1 Circularisation and preparation of SIRT1
Aptamers are generally developed through linear selection, in our case, for the linear selection, the library was used without any modifications. Circular aptamers obtained from a circular library is labouring and time consuming, requiring several steps of phosphorylation and ligation. A random DNA library contains various sequence motifs ranging from 1013 to 1015 in number44 of a central random sequence of 20–80 nucleotides with flanking regions of 18–21 nucleotides that will form the binding site for primers during PCR. This randomisation allows a wide diversity of possible sequences. For example, four different nucleotides at 40 randomised positions gives a theoretical possibility of 1024 different sequences.45 Our library named BAS has a random sequence of 40 nucleotides with flanking primer sequences forming a total of 77 nucleotides. It was used as precursor pool for the selection of aptamers for SIRT1. The library was 3′-phosphorylated and circularised. Circularisation of all the sequences in the library might not be possible, so a mix between inter-sequences and intra-sequences might form. To avoid this, the circularisation step was performed in a dilute solution to circumvent intra-strands association, and a step of gel purification was added after the circularisation. Gel assays showed no evidence of double aptamer (Fig. 2, ESI†). The circular form of our library was confirmed by digesting it with the enzyme Exonuclease I.Our target enzyme, SIRT1, was commercially obtained and purified to remove the GST-tag through cleavage of the GST-tag in a GSTrap FF column. The purification stage was evaluated by SDS-PAGE using 4–20% stacking polyacrylamide gel and stained with Instant Blue stain (Fig. 3, ESI†). This purified SIRT1 was later immobilised on a NHS HP SpinTrap column before proceeding to the SELEX step.
3.2 SELEX, DNA cloning and sequencing
Two pools of ssDNA libraries were employed. One pool was used for the linear selection and the second pool was phosphorylated and circularised producing the circular library.Oligonucleotides were dissolved in binding buffer, they interacted with the immobilised SIRT1 and any weak interactions were eluted by a range of buffers with increasing ionic strength. The buffer selection scheme was designed to wash away unbound sequences upon different ionic concentrations. This scheme ensures there is an enrichment of selective aptamers in direct relation to higher ionic strength. Different concentrations of NaCl were used to elute the unbound and bound oligonucleotides. A lower concentration of NaCl was used to elute unbound sequences and slightly higher ones were used to release mildly bound ones. Tightly bound oligonucleotides were recovered using a high salt concentration step. Once the sequences were recovered, the samples were desalted and amplified through PCR. In the case of the circular selection, prior to each new cycle, the library was again phosphorylated, circularised and purified.
After eight rounds of selection for the circular aptamer and twelve ones for the linear selection of aptamers, the PCR products were then cloned and 144 clones (72 for each linear and circular) were obtained for consensus sequence family analysis. From them, 50 complete sequences from each linear and circular set were used in class analysis sorted as shown in Table 1.
| Clone | Sequence (5′→3′) (N40)a | Frequency% |
|---|---|---|
| a TTCGGAAGAGATGGCGAC*-N40–CGAGCTGATCCTGATGGAA, primers in red.b Circular selection.c L: linear selection. | ||
| 1b | CAAGTGTAGCGGTCACGCTGCGCGTAACCACCACACCCGC | 12 |
| 2b | TTGCGGCATTTTGCCTTCCTGTTTTTGCTCACCCAGAAAC | 24 |
| 3b | CGAGTGGGTTACATCGAAACTGGATCTCAACAGCGGTAAC | 28 |
| 4b | CACTCCCTCTGCGTGCGAATTTTGCCTATGGCGCATATTC | 36 |
| 5c | ACAAGAGTCCACTATTAAAGAACGTGGACTCCAACGTGCC | 18 |
| 6c | CCAATAGGCCGAAATCGGCAAAATCCCTTATAAATCCTGC | 14 |
| 7c | CGGACTGCAACCTATGCTATCGTTGATGTCTGTCCAAGCA | 38 |
| 8c | CACTTTTCGGGGAAATGTGCGCGGAACCCCTATTTGTTTA | 30 |
In Table 1 the clones, without primer sequences, are showed as clones 1 to 4 from the circular selection and 5 to 8 from the linear selection. Clones 1, 5 and 6 were removed from further studies as the frequency was very low. For our further studies we re-named them as C or L denomination for circular or linear and added -P for the sequences tested with their primers. Clone 2 became C1, clone 3 is AC3, clone 4 is C5, clone 7 is L1 and clone 8 is L2.
3.3 SIRT1 and aptamer interaction
Aptamers are known for selective binding to their targets. Our selected aptamers were tested for activation and inhibition activities. We monitored the in vitro activity of the aptamers with and without the primers (aptamers with and without primers are presented in Table 1, ESI.† Circularity for aptamers is presented in Fig. 4, ESI†).The Fluor de Lys SIRT1 kit is a fluorescence-based assay, which uses a small lysine-acetylated peptide, corresponding to K382 of human p53, as a substrate. It has been extensively used in literature to study SIRT1 activators and inhibitors. The kit also recommends the use of resveratrol as positive control. This polyphenolic compound has been widely used due to its positive stimulation of SIRT1 activity.46,47 The assay monitors the fluorescence intensity of the Fluor de Lys de-acetylated product in a direct linear relationship to its concentration. To obtain fold induction values, the results at different concentrations of aptamers and resveratrol were normalised to only-vehicle samples.
The primers are part of the sequence during selection therefore, we tested if they had any effect. The aptamers with primers did not present any enhanced activity in relation to the counterpart without primers, confirming the activity was attributed to the target sequence and not due to the primer sequence (expanded results are shown Table 2, ESI†).
Resveratrol is the most widely studied SIRT1 activator, in our studies it produced a 2.8 fold activity at 200 μM. Table 2 shows the results for the most active aptamers with and without their primer sequences. At 0.8 μM, 2.7, 2.7 and 2.9 fold induction activity were produced by L3, L3-P and AC3 respectively. These aptamers have demonstrated extremely high performance. Though C3-P produced a very remarkable fold induction activity, its cell viability was quite modest when compared to AC3 alongside the difficulty in producing vast quantities for further studies.
A time course study up to 45 minutes using the same kit was employed to study the behaviour of AC3, C3-P, L3 and L3-P in respect to resveratrol, at different concentrations. Our results (Fig. 5, ESI†) showed all our aptamers yielded more product than resveratrol at 30 and 45 minutes of incubation, even at 0.1 μM concentration.
In general SIRT1 allosteric activators decrease the acylated protein equilibrium dissociation constant for the substrate.48 In a possible mechanism of action the aptamer would bind to an allosteric site of SIRT1 inducing conformational changes in the substrate binding pocket increasing the activity.
Surface Plasmon Resonance (SPR) provides information about the interaction of a protein and its substrate (extensive explanation can be found in SPR Section in ESI†). L3, L3-P, AC3 and C4 were the best performing aptamers in our activity tests therefore they were taken for SPR studies. The sensor surface was built upon by immobilisation of aptamers on the sensor chip, and SIRT1 was injected at different concentrations up to 800 nM. An aptamer level of 1000–1200 RU on the sensor surface was maintained during the immobilisation of the aptamer. SPR sensorgram results are shown in Table 3 (Fig. 6A to D, ESI†). Our results revealed a very tight and stable binding behaviour of SIRT1 enzyme to AC3 presenting an equilibrium dissociation constant (KD) of 0.27 ± 0.01 nM.
| Aptamer | KD (nM) |
|---|---|
| AC3 | 0.27 ± 0.01 |
| C3-P | 0.66 ± 0.01 |
| L3 | 0.48 ± 0.01 |
| L3-P | 1.03 ± 0.01 |
The SPR data have demonstrated the high affinity of AC3 for SIRT1, with KD values in the sub-nanomolar range. These data are consistent with published results of other aptamers including aptamers against thrombin (KD = 25–200 nmol l−1),49 PSMA (KD = 2 nmol l−1),50 tenascin C (KD = 5 nmol l−1)51 and c-di-GMP Vibrio cholerae (KD in the range 10 pM).52
3.4 Cell viability
L3, L3-P, AC3 and C3-P were moved forward for studies on a range of different cancer cell lines relevant to the most common types of cancer. The selected cancer cell lines presented different expression patterns of SIRT1. TBHP (tert-butyl hydroperoxide (tBuOOH)), an organic peroxide was used as positive control as it is widely known to produce cell stress followed by apoptosis.53 Results, as presented in Fig. 2, demonstrate excellent activity by the aptamer AC3.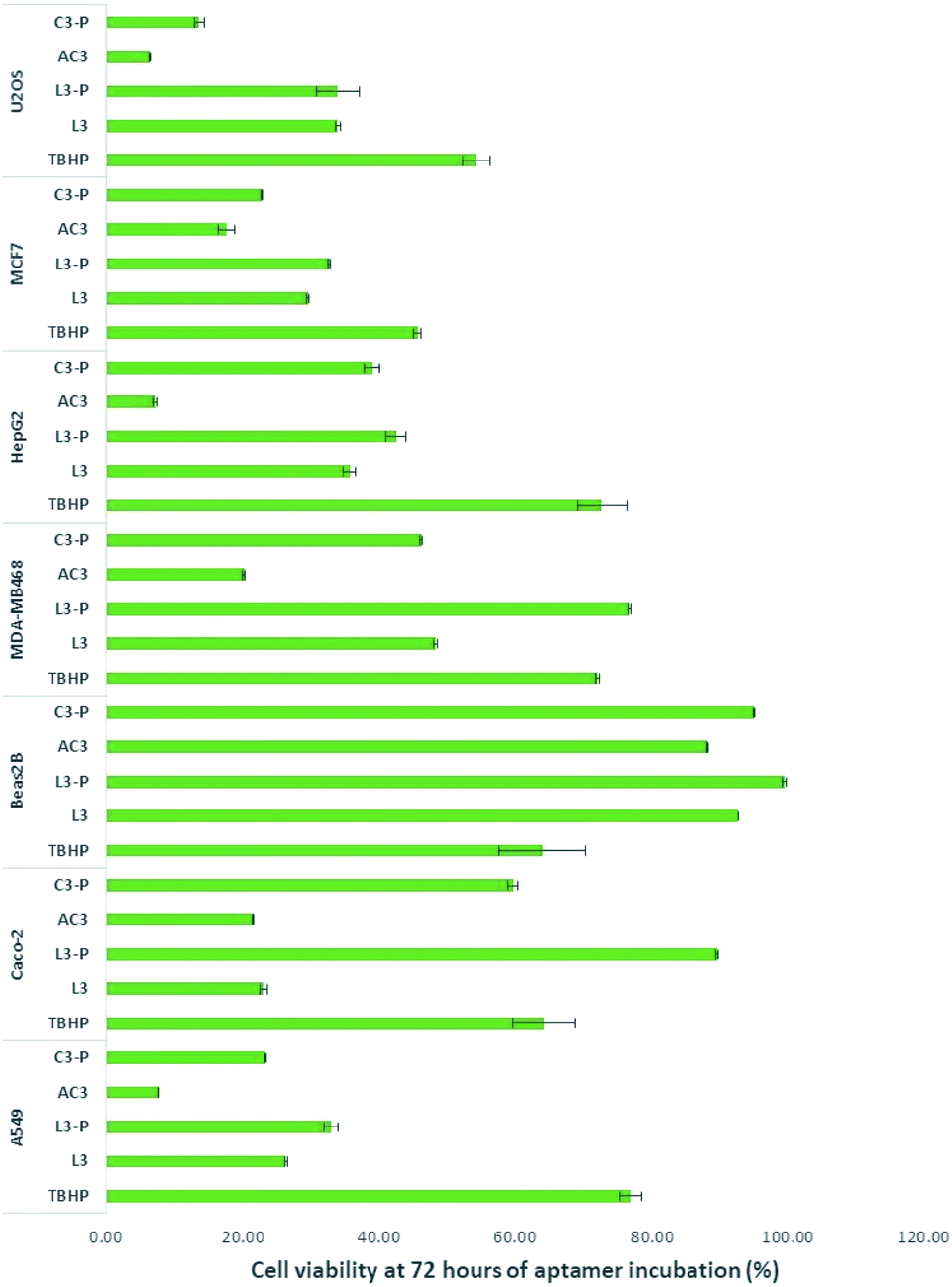 | ||
| Fig. 2 Cell viability results at 72 hours of incubation with aptamers at 2.5 μM final concentration. The results represent the mean ± SEM of 2 independent experiments. The p-value for AC3 treatment relative to TBHP show for A549: p < 0.00005; Hepg2: p < 0.0005; MCF7: p < 0.0001; MDA-MB468: p < 0.005; U2OS: p < 0.0005; Caco-2: p < 0.001 and Beas2B: p < 0.05. | ||
The human osteosarcoma U2OS cell line is an example of SIRT1 overexpression.54–56 L3 and L3-P were the least active with their toxicity remaining stable. C3-P and AC3 were more toxic to U2OS, especially AC3.
SIRT1 expression on breast cancer cells appears to be mixed, with over57,58 and under expression.59,60 In this work we employed two breast cancer cell lines, MCF-7 as breast cancer cell line oestrogen positive61 and MDA-MB468 as oestrogen negative.58,62 Aptamers L3 and L3-P were the least active on MCF7 though their toxicity increased towards the 72 hours period of incubation. AC3 was the most toxic to the cells. On MDA-MB468, L3-P had no effect on this cell line. L3, AC3 and C3-P showed an increase activity on this cell line, especially AC3.
HepG2 is a hepatocellular cancer cell line documented to overexpress SIRT1. In HepG2 all aptamers showed activity on this cell line, especially AC3.
Beas2B cells are immortalised human bronchial epithelium normal cells.63,64 No over or under expression of SIRT1 has been reported in this cell line. The aptamers did not affect significantly this cell line; implying there is a relationship between SIRT1 stimulation and cancer death.
SIRT1 expression appears to be decreased in human ColoRectal Cancer (CRC) including Caco-2.65 The opposite is found on human non-small cell lung cancer cell lines such as A549.66 On Caco-2 L3, AC3 and C3-P were toxic to the cells. On A549, all aptamers were toxic to A549 cells mainly AC3.
As shown in Fig. 2, at 72 hours AC3 left 6.3, 17.6, 20.1, 7.1, 21.6, 7.6 and 88.2% of viable cells for U2OS, MCF7, MDA-MB468, HepG2, Caco-2, A549 and Beas2B respectively (results at 24 and 48 hours of incubation are presented in Fig. 7, ESI†). In all cases the aptamers without the primer sequence demonstrated to be more toxic to the cells.
To confirm the observed cell activity was only due to AC3 and not any external DNA, a cell viability assay was performed on Caco-2. With a final concentration of 2.5 μM at 24 hours of incubation, the activities of the library, alongside AC3 in linear form and a scrambled oligonucleotide were explored. The results presented in Table 3 (in ESI†), demonstrated that both the library and scrambled oligonucleotide are not active. The linear form of AC3 is mildly active (79% viable cells) when compared to the high activity presented by the circular form (29% of viable cells). This indicates the observed activity is due to AC3 structure and not to the presence of external DNA.
SPR studies showed AC3 and L3 had lower KD in respect to (C3-P and L3-P). AC3 achieved the lowest KD indicating the strongest binding.
From these studies, aptamer AC3 has demonstrated to be the most active. AC3 is a circular aptamer therefore the 5′ and 3′ ends are ligated, potential conformation could involve the formation of 2 loops. A few questions arose: would AC3 stand plasma degradation? Would AC3 enter the cells and modulate SIRT1 producing an effect? Our initial cell-based studies indicate a possible yes to all these questions. A set of assays were performed to study in more detail the characteristics and effect of AC3.
3.5 Plasma stability of AC3
DNA oligonucleotides have an in vitro half-life in plasma of up to 60 minutes.45 Circularisation can potentially protect an aptamer from degradation in plasma. AC3 was tested in human plasma for degradation. Stability assays were carried out by incubating 1 μM of AC3 with human plasma for up to 24 hours. We analysed the outcome via HPLC-UV. The test samples were run on a C18 column with absorption monitored at 260 nm. Our studies indicate that human plasma eluted between 0.50 to 2.50 minutes. AC3 eluted at 4.07 minutes as showed in Fig. 3A. The samples of human plasma plus AC3 showed there is no matrix interference between the analytes. We used the retention time of 4.07 minutes for monitoring the degradation of the aptamer. As showed in Fig. 3B, 96.3% of AC3 is still remaining at 15 minutes. This lowers to 69.0% at 2 hours and 62.7% at 24 hours. This nearly 63% of intact circular AC3 indicates the circular form is very stable.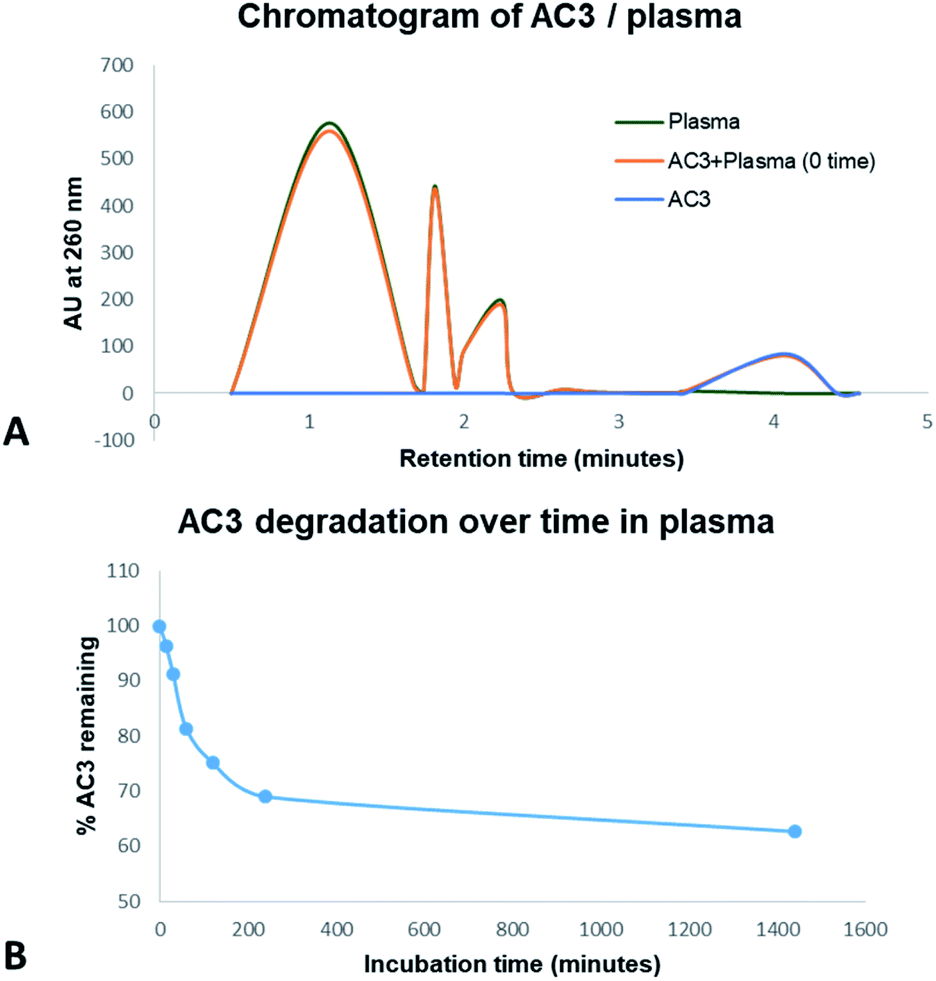 | ||
| Fig. 3 Stability of AC3 in human plasma up to 24 hours monitored by HPLC-UV. (A) Comparison of human plasma elution time with AC3. (B) Degradation of AC3 in human plasma as a function of time. | ||
3.6 Cell internalisation of AC3
To study the location of AC3's recognition and binding to SIRT1, fluorescence microscopy was used. The cells were incubated with AC3 labelled with FAM for a period of 72 hours. The cells were dosed with a low concentration of AC3 to avoid cell death, this depended on their IC50. To localise SIRT1 in the cell, a primary antibody was used coupled with a Texas Red labelled secondary antibody. The DNA in the nuclei was localised using DAPI (AC3 untreated cells are presented in Fig. 16 and 17, ESI†). In Fig. 4, the overlap of the emissions of AC3, SIRT1 and the nuclei are presented (AC3 treated cell images for only DAPI and SIRT1 localisation are presented in Fig. 18 to 21, ESI†). The cells treated with AC3 showed co-localisation with SIRT1 confirming the binding. Interestingly, there was no observable fluorescence signal on the control cells Beas2B when treated with AC3. This could be due to the difference in expression levels of SIRT1 in respect to the malignant cell lines or to the intrinsic nature of this cell line. Fluorescence microscopy studies showed AC3 was internalised and was co-localised with SIRT1 in our cancerous cell panel. This demonstrates that AC3 has crossed the lipid bilayer of the living cells and was recruited to its target.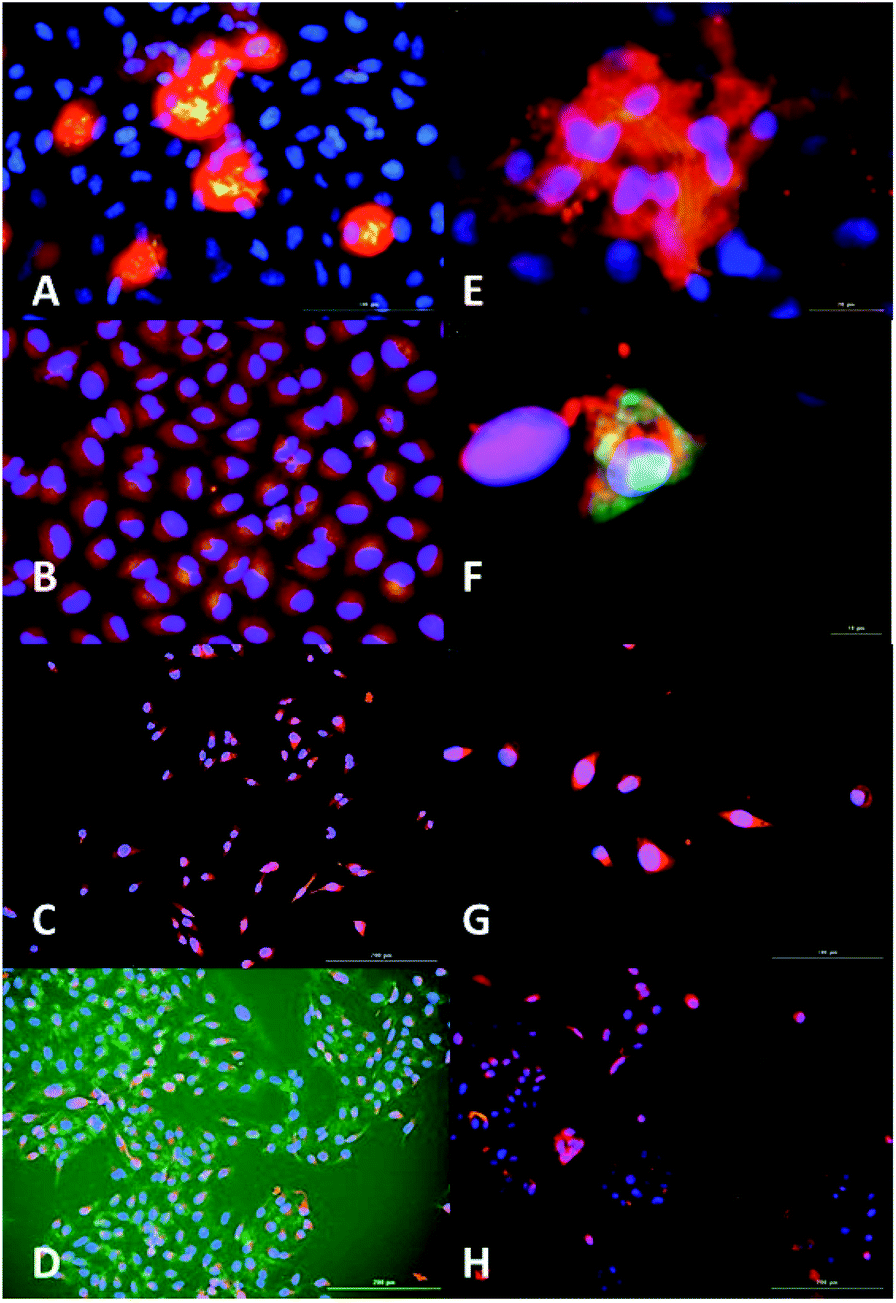 | ||
| Fig. 4 Fluorescence microscopy analysis of AC3 binding to SIRT1 enzyme and nuclei localisation. AC3-FAM (green-yellow), nuclei (DAPI, blue) and SIRT1 (Texas Red, red). Overlap of images can produced alterations in the colour results such as yellow produced by overlap of red and green, magenta produced by red and blue and cyan produced by green and blue. (A) A549, scale 100 μm. (B) HepG2 (augmentation in Fig. 20, ESI.† (C) Caco2, scale 200 μm). (D) U2OS, scale 200 μm. (E) MCF7, scale 20 μm. (F) MCF7 zoomed. (G) MDAMB-468, scale 100 μm. (H) Beas2B, scale 200 μm. Fluorescence intensity was measured by LUMIStar luminometer microplate readers as: green ex. 495 nm, em. 509 nm; blue ex. 359 nm, em. 461 nm and red ex. 596 nm, em. 615 nm. The results represent the mean ± SEM of two different experiments performed twice. Images were taken at a magnification of 100× in a Cytation 3. | ||
3.7 Catalytic activity characterisation of AC3 towards SIRT1 and effect on cell viability
The activity of AC3 was tested on the same panel of cells as described before using Fluor de Lys SIRT1 kit. Our results, as showed in Table 4, indicate that AC3 increases the activity of SIRT1 in all the tested cancerous cell lines. At a concentration of 1 μM, it showed an increase fold activity of 1.7 for A549, 2.1 for HepG2, 1.4 for both MDA-MB468 and MCF7, 2.7 for Caco-2 and 1.8 for U2OS. It is noticeable that AC3 has not increased activity of SIRT1 in Beas2B. Thus, AC3 appears to discriminate between cancer cells and non-cancerous cells, Beas2B. This is an interesting feature on Beas2B, AC3 seemed to not interact with these cells, maybe due to the lack of SIRT1 expression, which is also accompanied by a lack of SIRT1 activity and cell death. Resveratrol entered this cell line, produced cell death and increased SIRT'1 activity. Resveratrol has been shown to have several targets within a cell67 and maybe both effects on enhancing catalytic and cell death activities could be due to an earlier and/or alternative pathways. Nevertheless it is remarkable AC3 has demonstrating activity only on the cancer cells of our panel; and that stimulation of SIRT1 activity is relevant to induce cancer death.68| SIRT1 fold activitya | |||||||
|---|---|---|---|---|---|---|---|
| A549 | HepG2 | MDAMB-468 | MCF7 | Caco-2 | U2OS | Beas2B | |
| a Fluor de Lys SIRT1 kit assay; values represent mean values with standard deviation () of two experiments and data normalised to control (SIRT1/cell + water). A549: p < 0.0005. Hepg2: p < 0.0001. MCF7: p < 0.001. MDA-MB468: p < 0.001. U2OS: p < 0.001. Caco-2: p < 0.00001. Beas2B: p > 0.05. Nicotinamide was used as negative control as it is an inhibitor of SIRT1 therefore no increment in SIRT1 catalytic activity was observed. | |||||||
| AC3 1.0 μM | 1.70 ± 0.08 | 2.07 ± 0.01 | 1.42 ± 0.05 | 1.44 ± 0.05 | 2.66 ± 0.19 | 1.83 ± 0.14 | 1.05 ± 0.05 |
| AC3 0.5 μM | 1.39 ± 0.00 | 1.41 ± 0.04 | 1.18 ± 0.05 | 0.98 ± 0.06 | 1.48 ± 0.15 | 1.26 ± 0.02 | 1.09 ± 0.02 |
| AC3 0.25 μM | 1.19 ± 0.01 | 1.16 ± 0.01 | 1.03 ± 0.05 | 0.87 ± 0.06 | 1.15 ± 0.13 | 0.87 ± 0.05 | 1.01 ± 0.00 |
| Resveratrol 100 μM | 1.74 ± 0.09 | 1.90 ± 0.08 | 1.40 ± 0.05 | 1.04 ± 0.11 | 2.86 ± 0.10 | 1.78 ± 0.12 | 1.78 ± 0.07 |
| Nicotinamide 100 μM | 0.81 ± 0.11 | 0.82 ± 0.01 | 0.79 ± 0.10 | 0.75 ± 0.01 | 0.93 ± 0.02 | 0.79 ± 0.04 | 0.70 ± 0.03 |
Though these values are very similar to the ones obtained by resveratrol at 100 μM, the concentration of this polyphenolic compound proved to be toxic on all cell lines used in this study, demonstrating no discrimination between cancer and non-cancerous cells. This incremental activity of SIRT1 in the assessed cancer cells, was also accompanied by a reduction of the cell viability. Further analyses to determine the effect of AC3 at lower concentrations during the first 24 hours demonstrated the stimulation of SIRT1 induced cell death on cancer cells, as shown in Table 5.
| Cell line | % cell viability ata 24 h | |||
|---|---|---|---|---|
| AC3 | Resveratrol | |||
| 0.25 μM | 0.50 μM | 1.00 μM | 100 μM | |
| a Cell viability, MTT assay; values represent mean values with standard deviation () of two experiments and data normalised to control (cell + water). A549: p < 0.0005. Hepg2: p < 0.0001. MCF7: p < 0.001. MDA-MB468: p < 0.001. U2OS: p < 0.001. Caco-2: p < 0.00001. Beas2B: p > 0.05. | ||||
| A549 | 78.06 ± 0.11 | 73.01 ± 0.06 | 22.51 ± 0.03 | 6.79 ± 0.01 |
| HepG2 | 66.96 ± 0.01 | 46.04 ± 0.01 | 8.70 ± 0.05 | 6.04 ± 0.01 |
| MCF-7 | 69.10 ± 0.01 | 62.93 ± 0.02 | 14.07 ± 0.01 | 4.41 ± 0.01 |
| U2OS | 80.48 ± 0.05 | 54.56 ± 0.01 | 16.42 ± 0.07 | 8.50 ± 0.02 |
| MDA-MB468 | 76.51 ± 0.02 | 35.77 ± 0.04 | 13.21 ± 0.04 | 7.04 ± 0.01 |
| Caco-2 | 83.63 ± 0.08 | 49.23 ± 0.05 | 11.55 ± 0.01 | 12.07 ± 0.01 |
| Beas2B | 88.31 ± 0.03 | 77.72 ± 0.02 | 86.89 ± 0.06 | 8.86 ± 0.01 |
At 1.0 μM of AC3, the cell death on A549, MCF7, U2OS, MDA-MB468, A549, HepG2 and Caco-2 was in the range of 75–91%. On the non-cancerous cells Beas2B, AC3 did not increase SIRT1's catalytic activity for more than 10% and did not substantially affect viability of these cells either as 90% of the cell population was still viable after 24 hours. It is important to notice that resveratrol at 100 μM showed to be toxic on the cancerous and non-cancerous cells. Further studies to obtain inhibition of growth, provided IC50 values as: 0.32 μM (SD: 0.05) for A549, 0.20 μM (SD: 0.01) for HepG2, 0.14 μM (SD: 0.02) for MCF7, 0.10 μM (SD: 0.01) for MDA-MB468, 0.06 μM (SD: 0.01) for U2OS, and 0.26 μM (SD: 0.09) for Caco-2 (Fig. 8 to 15, ESI†).
4 Conclusions
SIRT1 has many functions; the molecular pathway for SIRT1's anticancer activity might be extremely complex and dependent on the cell type. A study on BRCA1 mutant cancer cells showed the use of resveratrol restored the activity of SIRT1 and produced inhibition of surviving stimulating apoptosis.69 A study on APC min ± mice by Firestein et al.70 showed that overexpression of SIRT1 reduces colon cancer formation, this appeared to be the result of SIRT1 deacetylating β-catenin and promoting the oncogenic form to be localised in the cytoplasm.p53 regulates positively SIRT1 transcription though in a negative loop p53's inactivation could reduce the expression of SIRT1, which would then increase p53's activity.71 It appears this response would depend on the catalytic activity of SIRT1, as only overexpression of catalytically active SIRT1 removed p53 acetylation. Another form of regulation on p53 by SIRT1, relies on deacetylating p300, a histone acetyltransferase of p53.72 Similar negative feedback loops have been found, in vitro, on FOXO3a, FOXL2 and E2F1, that can act as tumour suppressors in some situations.71,72 Other mechanisms of action might be possible for SIRT1, as it interacts with many autophagy proteins regulating this process.65,66,70–73
SIRT1 plays an important role in DNA damage response and genome integrity by maintaining proper chromatin structure and DNA damage repair foci formation. Given SIRT1 activity is NAD+ dependent, which itself is metabolism dependent, over activity of SIRT1 can deplete or skew the energy resources of cancer cells abrogating the energy metabolism of malignant cells leading to cell death.
In our results, AC3 did not over-activate SIRT1 in non-malignant cells indicating that in physiological conditions of normal cells, energy metabolism will remain unaffected. It is likely that difference in metabolic demands in different types of malignant cells can account for the difference in SIRT1 activation and suppression. In order to dissect the mechanism of SIRT1 induced cell death, energy metabolism of malignant cells, DNA damage and NFκB pathway will need to be analysed with AC3 treatment of control and malignant cells as further work.
SIRT1 is the most studied of the seven mammalian sirtuins.74 Modulation of SIRT1 by small ligands such as resveratrol, which might not be sufficiently selective, produce unwanted cell death when tested in vitro. Using SELEX we developed an extremely selective ligand against SIRT1.
In this manuscript, we demonstrate two types of aptamer selection, linear and circular. From our results, AC3 a circular aptamer, stood up as the best performing aptamer for inducing SIRT1 activity and performing anticancer action.
Our aptamer, was produced in vitro using a modified library, we characterised the biophysical interactions between AC3 and SIRT1. AC3 was recruited to intracellular SIRT1, confirmed by co-localisation. AC3 has shown anticancer activity for several models of cancer including lung (A549), colon (Caco-2), the liver (HepG2), osteosarcoma (U2OS) and breast cancer models (MCF-7 and MDA-MB468). The most interesting feature is that AC3 was non-toxic on the non-cancerous cell line Beas2B, implying it might be safe to non-cancerous tissue. Stability studies also showed AC3 to resist the degradation of plasma enzymes up to 24 hours.
In conclusion, using SELEX methodology we have produced an aptamer which is: (a) selective for SIRT1, (b) enters the living cells and interacts with SIRT1, (c) is stable upon enzymatic attack, (d) active on cancer cells, (e) the mode of action is through activation of SIRT1 and (f) is safe on non-cancerous cells. A pharmacological activation of SIRT1 enhanced cell death suggesting a tumour suppressive function. The complex SIRT1-aptamer identified in this study may be used in the future for cancer treatment or biological evaluation of SIRT1.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would also like to express our gratitude to Dr Nanda Puspita for advice and training in the cell culture room, the Biomolecular Analysis Core Facility at the University of Manchester for their assistance with SPR studies, Dr Caroline Topham for her advice with the image analyses and Dr John Hadfield for his advice on the writing of this manuscript. This work was supported by the Iraqi Ministry of Higher Education and Scientific Research [MOHESR].References
- Y. Olmos, J. J. Brosens and E. W. F. Lam, Drug Resist. Updates, 2011, 14, 35–44 Search PubMed.
- L. Bosch-Presegué and A. Vaquero, Genes Cancer, 2011, 2, 648–662 Search PubMed.
- Z. Wang and W. Chen, Genes Cancer, 2013, 4, 82–90 Search PubMed.
- C.-X. Deng, Int. J. Biol. Sci., 2009, 5, 147–152 Search PubMed.
- C. C. S. Chini, J. M. Espindola-Netto, G. Mondal, A. M. G. Guerrico, V. Nin, C. Escande, M. Sola-Penna, J.-S. Zhang, D. D. Billadeau and E. N. Chini, Clin. Cancer Res., 2016, 22, 2496 Search PubMed.
- T. F. Liu and C. E. McCall, Genes Cancer, 2013, 4, 135–147 Search PubMed.
- C. Cantó and J. Auwerx, Pharmacol. Rev., 2012, 64, 166–187 Search PubMed.
- J. Yi and J. Luo, Biochim. Biophys. Acta, 2010, 1804, 1684–1689 Search PubMed.
- L. Bosch-Presegué and A. Vaquero, Oncogene, 2014, 33, 3764–3775 Search PubMed.
- K. Li and J. Luo, N. Am. J. Med. Sci., 2011, 4, 104–106 Search PubMed.
- J. M. Solomon, R. Pasupuleti, L. Xu, T. McDonagh, R. Curtis, P. S. DiStefano and L. J. Huber, Mol. Cell. Biol., 2006, 26, 28–38 Search PubMed.
- C.-S. Lim, Med. Hypotheses, 2006, 67, 341–344 Search PubMed.
- M. Blind and M. Blank, Mol. Ther.--Nucleic Acids, 2015, 4, e223 Search PubMed.
- M. Avci-Adali, H. Steinle, T. Michel, C. Schlensak and H. P. Wendel, PLoS One, 2013, 8, e68810 Search PubMed.
- J. G. Bruno, Pharmaceuticals, 2018, 11, 62 Search PubMed.
- Z. Zhuo, Y. Yu, M. Wang, J. Li, Z. Zhang, J. Liu, X. Wu, A. Lu, G. Zhang and B. Zhang, Int. J. Mol. Sci., 2017, 18, 2142 Search PubMed.
- K. Thiel, Nat. Biotechnol., 2004, 22, 649–651 Search PubMed.
- D. H. J. Bunka, O. Platonova and P. G. Stockley, Curr. Opin. Pharmacol., 2010, 10, 557–562 Search PubMed.
- J. Guo, X. Gao, L. Su, H. Xia, G. Gu, Z. Pang, X. Jiang, L. Yao, J. Chen and H. Chen, Biomaterials, 2011, 32, 8010–8020 Search PubMed.
- H. Dougan, D. M. Lyster, C. V. Vo, A. Stafford, J. I. Weitz and J. B. Hobbs, Nucl. Med. Biol., 2000, 27, 289–297 Search PubMed.
- S. Ni, H. Yao, L. Wang, J. Lu, F. Jiang, A. Lu and G. Zhang, Int. J. Mol. Sci., 2017, 18, 1683 Search PubMed.
- B. Saccà, L. Lacroix and J.-L. Mergny, Nucleic Acids Res., 2005, 33, 1182–1192 Search PubMed.
- A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388–1405 Search PubMed.
- C. Maasch, A. Vater, K. Buchner, W. G. Purschke, D. Eulberg, S. Vonhoff and S. Klussmann, J. Biol. Chem., 2010, 285, 40012–40018 Search PubMed.
- C. H. Lee, S.-H. Lee, J. H. Kim, Y.-H. Noh, G.-J. Noh and S.-W. Lee, Mol. Ther.--Nucleic Acids, 2015, 4, e254 Search PubMed.
- V. Ramaswamy, A. Monsalve, L. Sautina, M. S. Segal, J. Dobson and J. B. Allen, Nucleic Acid Ther., 2015, 25, 227–234 Search PubMed.
- S. Hoffmann, J. Hoos, S. Klussmann and S. Vonhoff, RNA Aptamers and Spiegelmers: Synthesis, Purification, and Post-Synthetic PEG Conjugation, 2011 Search PubMed.
- L. Zandarashvili, D. Nguyen, K. M. Anderson, M. A. White, D. G. Gorenstein and J. Iwahara, Biophys. J., 2015, 109, 1026–1037 Search PubMed.
- D. R. Davies, A. D. Gelinas, C. Zhang, J. C. Rohloff, J. D. Carter, D. O'Connell, S. M. Waugh, S. K. Wolk, W. S. Mayfield, A. B. Burgin, T. E. Edwards, L. J. Stewart, L. Gold, N. Janjic and T. C. Jarvis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19971–19976 Search PubMed.
- M. Kimoto, R. Yamashige, K.-i. Matsunaga, S. Yokoyama and I. Hirao, Nat. Biotechnol., 2013, 31, 453 Search PubMed.
- N. S. Hamedani and J. Müller, in Nucleic Acid Aptamers: Selection, Characterisation, and Application, ed. G. Mayer, Springer New York, New York, NY, 2016, pp. 61–75, DOI:10.1007/978-1-4939-3197-2_5.
- G. Hybarger, J. Bynum, R. F. Williams, J. J. Valdes and J. P. Chambers, Anal. Bioanal. Chem., 2006, 384, 191–198 Search PubMed.
- D. A. Daniels, H. Chen, B. J. Hicke, K. M. Swiderek and L. Gold, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15416–15421 Search PubMed.
- J. Mi, Y. Liu, Z. N. Rabbani, Z. Yang, J. H. Urban, B. A. Sullenger and B. M. Clary, Nat. Chem. Biol., 2009, 6, 22 Search PubMed.
- N. Nguyen Quang, G. Perret and F. Ducongé, Pharmaceuticals, 2016, 9, 76 Search PubMed.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537 Search PubMed.
- H. Kuai, Z. Zhao, L. Mo, H. Liu, X. Hu, T. Fu, X. Zhang and W. Tan, J. Am. Chem. Soc., 2017, 139, 9128–9131 Search PubMed.
- D. A. Di Giusto, S. M. Knox, Y. Lai, G. D. Tyrelle, M. T. Aung and G. C. King, ChemBioChem, 2006, 7, 535–544 Search PubMed.
- Y. Hida, Y. Kubo, K. Murao and S. Arase, Arch. Dermatol. Res., 2007, 299, 103–106 Search PubMed.
- M. Dokmanovic, C. Clarke and P. A. Marks, Mol. Cancer Res., 2007, 5, 981 Search PubMed.
- S. Arnold, G. Pampalakis, K. Kantiotou, D. Silva, C. Cortez, S. Missailidis and G. Sotiropoulou, Biol. Chem., 2012, 393, 343 Search PubMed.
- K. Setlem, B. Mondal, S. Ramlal and J. Kingston, Front. Microbiol., 2016, 7, 1909 Search PubMed.
- R. Stoltenburg, T. Schubert and B. Strehlitz, PLoS One, 2015, 10, e0134403 Search PubMed.
- J. Zhou and J. Rossi, Nat. Rev. Drug Discovery, 2017, 16, 181–202 Search PubMed.
- S. M. Nimjee, R. R. White, R. C. Becker and B. A. Sullenger, Annu. Rev. Pharmacol. Toxicol., 2017, 57, 61–79 Search PubMed.
- K. T. Howitz, K. J. Bitterman, H. Y. Cohen, D. W. Lamming, S. Lavu, J. G. Wood, R. E. Zipkin, P. Chung, A. Kisielewski, L.-L. Zhang, B. Scherer and D. A. Sinclair, Nature, 2003, 425, 191–196 Search PubMed.
- V. C. J. de Boer, M. C. de Goffau, I. C. W. Arts, P. C. H. Hollman and J. Keijer, Mech. Ageing Dev., 2006, 127, 618–627 Search PubMed.
- D. Cao, M. Wang, X. Qiu, D. Liu, H. Jiang, N. Yang and R.-M. Xu, Genes Dev., 2015, 29, 1316–1325 Search PubMed.
- L. C. Griffin, J. J. Toole and L. L. K. Leung, Gene, 1993, 137, 25–31 Search PubMed.
- S. E. Lupold, B. J. Hicke, Y. Lin and D. S. Coffey, Cancer Res., 2002, 62, 4029 Search PubMed.
- B. J. Hicke, C. Marion, Y.-F. Chang, T. Gould, C. K. Lynott, D. Parma, P. G. Schmidt and S. Warren, J. Biol. Chem., 2001, 276, 48644–48654 Search PubMed.
- K. D. Smith, S. V. Lipchock, T. D. Ames, J. Wang, R. R. Breaker and S. A. Strobel, Nat. Struct. Mol. Biol., 2009, 16, 1218–1223 Search PubMed.
- C.-H. Lin, N.-T. Li, H.-S. Cheng and M.-L. Yen, J. Cell. Mol. Med., 2018, 22, 786–796 Search PubMed.
- R. Garva, C. Thepmalee, U. Yasamut, S. Sudsaward, A. Guazzelli, R. Rajendran, N. Tongmuang, S. Khunchai, P. Meysami, T. Limjindaporn, P.-t. Yenchitsomanus, L. Mutti, M. Krstic-Demonacos and C. Demonacos, Front. Oncol., 2019, 9, 49 Search PubMed.
- L. Peng, H. Ling, Z. Yuan, B. Fang, G. Bloom, K. Fukasawa, J. Koomen, J. Chen, W. S. Lane and E. Seto, Mol. Cell. Biol., 2012, 32, 2823–2836 Search PubMed.
- K. Furuya, T. Ozaki, T. Hanamoto, M. Hosoda, S. Hayashi, P. A. Barker, K. Takano, M. Matsumoto and A. Nakagawara, J. Biol. Chem., 2007, 282, 18365–18378 Search PubMed.
- Y. Zhang, M. Zhang, H. Dong, S. Yong, X. Li, N. Olashaw, P. A. Kruk, J. Q. Cheng, W. Bai, J. Chen, S. V. Nicosia and X. Zhang, Oncogene, 2008, 28, 445 Search PubMed.
- S. Elangovan, S. Ramachandran, N. Venkatesan, S. Ananth, J. P. Gnana-Prakasam, P. M. Martin, D. D. Browning, P. V. Schoenlein, P. D. Prasad, V. Ganapathy and M. Thangaraju, Cancer Res., 2011, 71, 6654–6664 Search PubMed.
- K. Rifaï, M. Idrissou, F. Penault-Llorca, Y.-J. Bignon and D. Bernard-Gallon, Cancers, 2018, 10, 409 Search PubMed.
- K. Rifaï, G. Judes, M. Idrissou, M. Daures, Y.-J. Bignon, F. Penault-Llorca and D. Bernard-Gallon, Oncotarget, 2017, 8, 110922–110930 Search PubMed.
- S. Liarte, J. L. Alonso-Romero and F. J. Nicolás, Front. Endocrinol., 2018, 9, 552 Search PubMed.
- Y. W. Yi, H. J. Kang, H. J. Kim, Y. Kong, M. L. Brown and I. Bae, Oncotarget, 2013, 4, 984–994 Search PubMed.
- R. R. Reddel, Y. Ke, B. I. Gerwin, M. G. McMenamin, J. F. Lechner, R. T. Su, D. E. Brash, J. B. Park, J. Rhim and C. Harris, Cancer Res., 1988, 48, 1904–1909 Search PubMed.
- C. Stewart, E. Torr, N. H. Mohd Jamili, C. Bosquillon and I. Sayers, Evaluation of Differentiated Human Bronchial Epithelial Cell Culture Systems for Asthma Research, 2012 Search PubMed.
- V. Pazienza, A. Piepoli, A. Panza, M. R. Valvano, G. Benegiamo, M. Vinciguerra, A. Andriulli and G. Mazzoccoli, SIRT1 and the Clock Gene Machinery in Colorectal Cancer, 2011 Search PubMed.
- I. Grbesa, M. J. Pajares, E. Martínez-Terroba, J. Agorreta, A.-M. Mikecin, M. Larráyoz, M. A. Idoate, K. Gall-Troselj, R. Pio and L. M. Montuenga, PLoS One, 2015, 10, e0124670 Search PubMed.
- S. S. Kulkarni and C. Cantó, Biochim. Biophys. Acta, 2015, 1852, 1114–1123 Search PubMed.
- S.-C. Chao, Y.-J. Chen, K.-H. Huang, K.-L. Kuo, T.-H. Yang, K.-Y. Huang, C.-C. Wang, C.-H. Tang, R.-S. Yang and S.-H. Liu, Sci. Rep., 2017, 7, 3180 Search PubMed.
- R.-H. Wang, Y. Zheng, H.-S. Kim, X. Xu, L. Cao, T. Luhasen, M.-H. Lee, C. Xiao, A. Vassilopoulos, W. Chen, K. Gardner, Y.-G. Man, M.-C. Hung, T. Finkel and C.-X. Deng, Mol. Cell., 2008, 32, 11–20 Search PubMed.
- R. Firestein, G. Blander, S. Michan, P. Oberdoerffer, S. Ogino, J. Campbell, A. Bhimavarapu, S. Luikenhuis, R. de Cabo, C. Fuchs, W. C. Hahn, L. P. Guarente and D. A. Sinclair, PLoS One, 2008, 3, e2020 Search PubMed.
- J.-E. Kim, Z. Lou and J. Chen, Cell Cycle, 2009, 8, 3784–3785 Search PubMed.
- G. Qiu, X. Li, C. Wei, X. Che, S. He, J. Lu, S. Wang, K. Pang and L. Fan, Dis. Markers, 2016, 2016, 6869415 Search PubMed.
- M. Roth and W. Y. Chen, Oncogene, 2014, 33, 1609–1620 Search PubMed.
- K. E. Maier and M. Levy, Mol. Ther.--Methods Clin. Dev., 2016, 3, 16014 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures and tables related to the aptamer selection, protein purification, clones obtained, SPR results, activity results, cell images and IC50 binding results. See DOI: 10.1039/d0ra07857c |
| This journal is © The Royal Society of Chemistry 2020 |