
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective amination of 4-alkylisoquinoline-1,3(2H,4H)-dione derivatives†
Cheng Chenga,
Ying-Xian Lia,
Xue-Min Jiaa,
Ji-Quan Zhanga,
Yong-Long Zhao a,
Wei Fengb,
Lei Tang*ac and
Yuan-Yong Yang*ac
a,
Wei Fengb,
Lei Tang*ac and
Yuan-Yong Yang*ac
aState Key Laboratory of Functions and Applications of Medicinal Plants, Guizhou Provincial Engineering Technology Research Center for Chemical Drug R&D, School of Pharmacy, Guizhou Medical University, Guiyang 550004, China. E-mail: yangyuanyong@gmc.edu.cn; tlei1974@hotmail.com
bBGI-Shenzhen, Building 11, Beishan Industrial Zone, Yantian, Shenzhen, 518083, China
cGuizhou Provincial Key Laboratory of Pathogenesis and Drug Research on Common Chronic Diseases, Guizhou Medical University, Guiyang 550004, China
First published on 25th November 2020
Abstract
A mild and efficient enantioselective amination of 4-alkylisoquinoline-1,3(2H,4H)-dione derivatives was established, which is compatible with a broad range of substrates and delivers the final products in excellent yields (up to 99%) and ee values (up to 99%) with low catalyst loading (down to 1 mol%). The synthetic potential of this methodology was also demonstrated in the gram scale level.
Isoquinolinedione, bearing the carbon skeleton of tetrahydroisoquinoline (THIQ), is an important structural motif present in bioactive compounds and natural products with a broad array of biological properties.1 However, the construction of isoquinolinedione, particularly the chiral version, is currently underdeveloped,2 and the reported methods heavily rely on the radical-initiated addition–cyclization of activated alkenes to prepare this structural motif that hard to be further diversified.3 From a pharmaceutical point of view, the presence of heteroatoms (such as nitrogen) is essential for their biological activity (Fig. 1).4 Therefore, the introduction of other functional groups or heteroatoms into this framework is a pressing issue to be addressed.
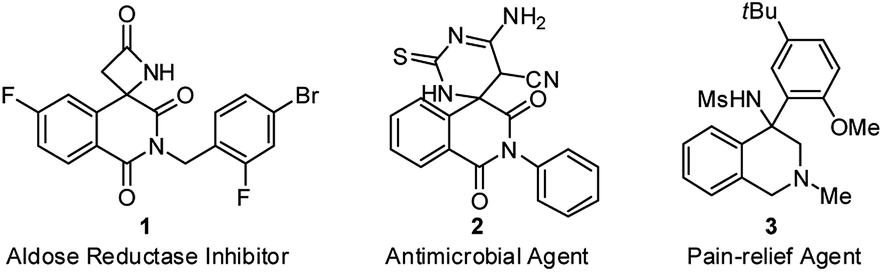 | ||
| Fig. 1 Bioactive compounds bearing isoquinolinedione or tetrahydroisoquinoline core structure. | ||
On a different note, amine attached to a stereogenic center is a ubiquitous structure in natural products and bioactive compounds and becomes impetus for continuous exploration.5 Using azodicarboxylates or nitrosoarenes as electrophilic amine sources,6 activated substrates such as 1,3-dicarbonyl compounds and pyrazolones could be readily transformed into the corresponding amination products in high ee and yields.7 With the pioneering work of List and Jørgensen, the α-amination of aldehydes could be realized through enamine activation.8 The α-amination of less activated substrates such as nitroisoxazole derivatives could be realized via phase-transfer catalysis.9 Recently, cyclic ketones or vinyl ketones were transformed into the corresponding amination products via organo- or metal catalysis.10 Surprisingly, reports on the amination of heterocyclic compounds are very limited, and they majorly focus on the oxindole scaffold.11 Therefore, the construction of other pharmaceutical relevant α-amination heterocyclic compounds would be a meaningful work.12 In addition, the organo-catalyzed asymmetric amination reactions generally require relatively high catalyst loading to achieve the optimal yields and enantioselectivities; for this reason, the development of an efficient amination protocol would still be highly desirable.
Recently, our group reported the amination of 4-arylisoquinolinedione via organo-catalysis.13 However, due to the attenuated reactivity at low temperatures, high catalyst loading is required for satisfactory yields and enantioselectivities, and the substrate scope is limited to 4-aryl substituents. To further expand the scope of this reaction, we tried to extend this amination methodology to 4-alkylisoquinolinedione derivatives.
Our study commenced with 2-benzyl-4-butylisoquinoline-1,3(2H,4H)-dione 4a and di-tert-butyl azodicarboxylate 5 as model substrates for condition optimization (Table 1). With previously optimized bifunctional catalyst 7, the reaction proceeds smoothly and delivers the amination product in moderate yields and excellent enantioselectivity after 24 h (Table 1, entry 1). Gratifyingly, the chemical yield could be increased by raising the temperature and maintaining the ee value (Table 1, entry 2). Further solvent optimization reveals that the polarity of solvents poses a positive effect on the chemical yield but negative effect on the ee value (Table 1, entries 3–5), indicating that the polar solvent may contribute to the stabilization of the enolate intermediate via dipole–dipole interactions but also interrupting the efficient interaction of the substrate with the catalyst.
|
||||
|---|---|---|---|---|
| Entrya | 7 (mol%) | Solvent | Yield 6ab (%) | eec (%) |
| a All reaction was conducted with 0.2 mmol compound 4a, 0.44 mmol compound 5, in 0.5 mL solvent and reacted at 25 °C for 24 h.b Isolated yield.c The ee was determined by HPLC analysis.d Reaction was conducted at 5 °C and reacted for 24 h.e Reaction was run for 35 h.f Reaction was reacted for 72 h.g Reaction was conducted at 40 °C. | ||||
| 1d | 10 | CHCl3 | 50 | 98 |
| 2 | 10 | CHCl3 | 71 | 97 |
| 3 | 10 | Toluene | 82 | 74 |
| 4 | 10 | Ether | 95 | 80 |
| 5 | 10 | THF | 99 | 21 |
| 6 | 10 | DCM | 99 | 90 |
| 7 | 10 | Chlorobenzene | 85 | 94 |
| 8 | 10 | CH2ClCH2Cl | 99 | 98 |
| 9 | 5 | CH2ClCH2Cl | 97 | 97 |
| 10e | 2 | CH2ClCH2Cl | 99 | 97 |
| 11f,g | 1 | CH2ClCH2Cl | 83 | 95 |
| 12f,g | 1 | CH2ClCH2Cl | 88 | 93 |
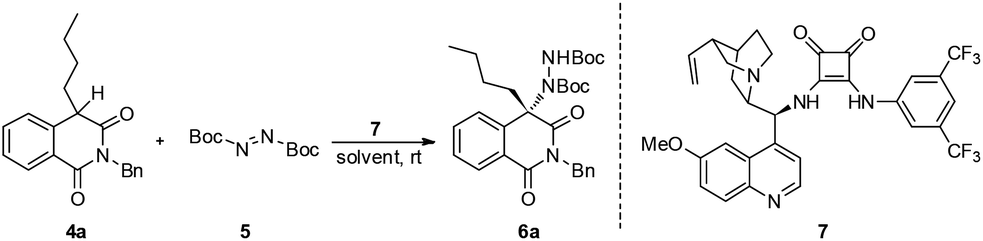
Further solvent optimization reveals that DCM gives the best yield along with very good ee (Table 1, entry 6). Then, another chlorinated solvent was tested and found that 1,2-dichloroethane gives the best results both in ee and yield (Table 1, entry 7 and 8). At this point, we try to study the catalyst loading effect on this amination reaction. At lower catalyst loadings, the ee dropped marginally and also the chemical yield, while the decrease in the chemical yield could be compensated by longer reaction time (Table 1, entries 9 and 10). We also tried to further decrease the catalyst loading to 1 mol%, but a much longer reaction time was required to get a satisfactory yield (Table 1, entry 11). The chemical yield could be increased slightly when the reaction was conducted at 40 °C; however, at the expense of ee (Table 1, entry 12). Therefore, taking account of the yield and ee of the final product, the 2 mol% catalyst at room temperature in 1,2-dichloroethane was established as under optimal reaction conditions for further exploration.
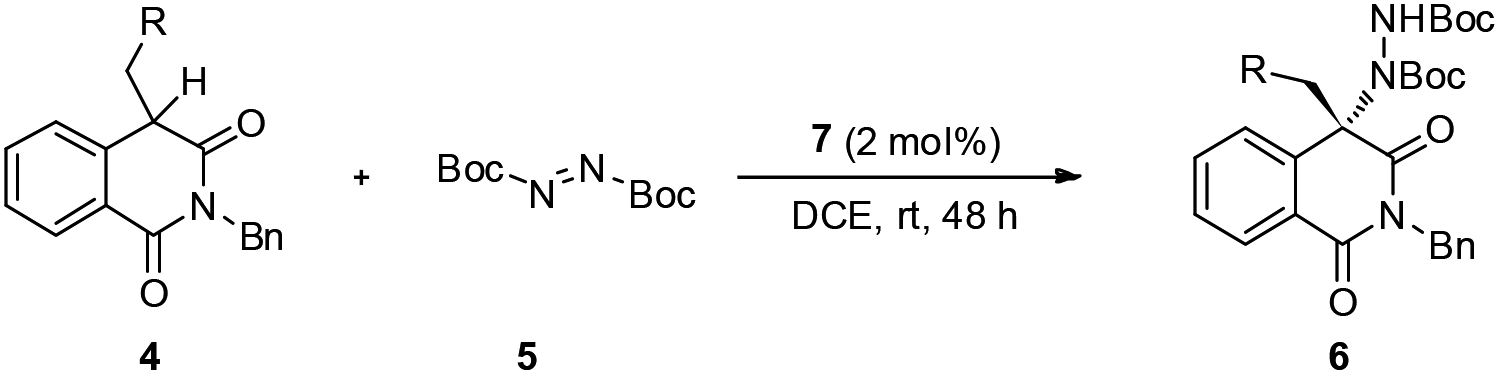
With a set of optimal reaction conditions in hand, the substrate scope for this amination reaction was explored. By changing the linear n-butyl to branched or substituted alkyl groups, the final products were obtained in very good yields and ee values (Table 2, entries 1–4). Except the meta-substituted benzyl groups, other benzyl groups generally give excellent yields and ee values regardless of the electronic or steric properties of the aromatic rings (Table 2, entries 5–16). Moreover, this methodology is also compatible with other steric or heteroaromatic substrates and excellent results were obtained (Table 2, entries 17–21). The absolute configuration of 6i determined via single crystal X-ray diffraction was S, and the absolute configurations of other products 6 were assigned by analogy.14
|
||||
|---|---|---|---|---|
| Entrya | R | Product | Yieldb (%) | eec (%) |
| a Reactions were run on a 0.03 mmol 1 and 0.036 mmol 2 with the 2 mol% catalyst in 500 μL solvent at 25 °C for 48 h.b Yield was based on the isolated product of 3.c The ee was determined via HPLC analysis. | ||||
| 1 | n-Propyl | 6a | 99 | 97 |
| 2 | i-Butyl | 6b | 96 | 94 |
| 3 | i-Propyl | 6c | 99 | 81 |
| 4 | PhCH2CH2 | 6d | 94 | 93 |
| 5 | Ph | 6e | 99 | 97 |
| 6 | 4-MeC6H4 | 6f | 99 | 92 |
| 7 | 4-OMeC6H4 | 6g | 90 | 97 |
| 8 | 4-FC6H4 | 6h | 96 | 96 |
| 9 | 4-ClC6H4 | 6i | 99 | 99 |
| 10 | 4-BrC6H4 | 6j | 99 | 99 |
| 11 | 3-MeC6H4 | 6k | 99 | 76 |
| 12 | 3-BrC6H4 | 6l | 99 | 89 |
| 13 | 2-OmeC6H4 | 6m | 99 | 93 |
| 14 | 2-MeC6H4 | 6n | 92 | 93 |
| 15 | 2-ClC6H4 | 6o | 95 | 98 |
| 16 | 3,4,5-OmeC6H2 | 6p | 99 | 87 |
| 17 | 2-Naphthyl | 6q | 99 | 99 |
| 18 | 2-Indolyl | 6r | 90 | 82 |
| 19 | 3-Indolyl | 6s | 99 | 99 |
| 20 | 2-Me-3-indolyl | 6t | 99 | 96 |
| 21 | 2-Fural | 6u | 99 | 97 |
To demonstrate the practical synthetic application of current protocol, the gram scale synthesis of chiral 6i has been demonstrated (Scheme 1). The product was produced in excellent yield and ee value at the 2 mmol scale. Moreover, a synthetically desirable amino product could be obtained from the cleavage of the N–N bond and deprotection of the Boc group in two steps with very good yield and ee value (Scheme 2).15
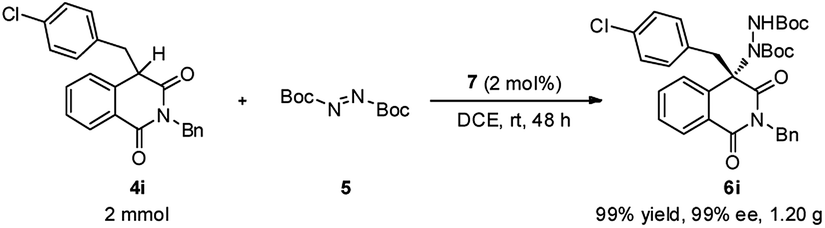 | ||
| Scheme 1 Gram scale preparation of 6i. | ||
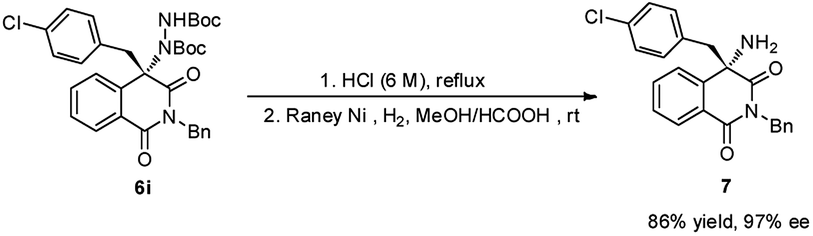 | ||
| Scheme 2 Transform the product into amino product. | ||
In an effort to account for the observed stereocontrol of the reaction, a plausible reaction mechanism is proposed in Scheme 3. With the previously established bifunctional catalyst by Rawal et al.,16 the isoquinolinedione was activated by the alkyl amine moiety to attack the azodicarboxylate that was activated by the squaramide moiety via hydrogen bonding interactions in a well-defined manner to deliver the final product in S configuration.17 The outcome in this study is in accordance with our previous reports13 as the benzyl group alleviates the steric hindrance of the substituted phenyl ring from the reaction center and delivers the product in a high ee value (Table 2, entry 14).
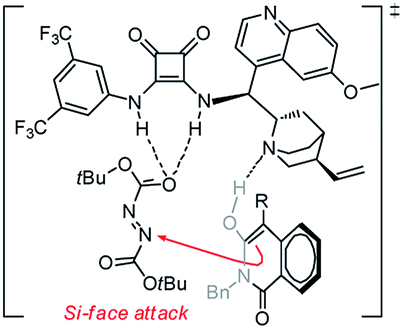 | ||
| Scheme 3 Proposed mechanism for the amination reaction. | ||
To summarize, a highly enantioselective amination methodology with low catalyst loading was established (down to 1 mol%), which is compatible with a broad range of substrates and delivers the final products in excellent yields (up to 99%) and ee values (up to 99%). Moreover, the maintaining of yield and ee in up-scale preparation clearly demonstrates the synthetic potential of this methodology. Most importantly, this reaction is mild and operationally simple and could be performed without the exclusion of air or moisture at room temperature.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the financial support from the National Natural Science Foundation of China (22061012, 21807120), the Office of Science & Technology of Guizhou Province ([2020]4Y208, [2018]5779-62), and the National-Local Joint Engineering Research Center for Innovative & Generic Chemical Drug.Notes and references
- (a) J. Kankanala, C. Marchand, M. Abdelmalak, H. Aihara, Y. Pommier and Z. Wang, J. Med. Chem., 2016, 59, 2734–2746 CrossRef CAS; (b) S. K. V. Vernekar, Z. Liu, E. Nagy, L. Miller, K. A. Kirby, D. J. Wilson, J. Kankanala, S. T. Sarafianos, M. A. Parniak and Z. Wang, J. Med. Chem., 2015, 58, 651–664 CrossRef CAS; (c) Y.-L. Chen, J. Tang, M. J. Kesler, Y. Y. Sham, R. Vince, R. J. Geraghty and Z. Wang, Bioorg. Med. Chem., 2012, 20, 467–479 CrossRef CAS.
- Y. You, W.-Y. Lu, K.-X. Xie, J.-Q. Zhao, Z.-H. Wang and W.-C. Yuan, Chem. Commun., 2019, 55, 8478–8481 RSC.
- (a) Z.-Z. Li, J. Yu, L.-N. Wang, S.-L. Chen, R.-L. Sheng and S. Tang, Tetrahedron, 2018, 74, 6558–6568 CrossRef CAS; (b) X.-F. Xia, S.-L. Zhu, D. Wang and Y.-M. Liang, Adv. Synth. Catal., 2017, 359, 859–865 CrossRef CAS; (c) M. Lu, Z. Liu, J. Zhang, Y. Tian, H. Qin, M. Huang, S. Hu and S. Cai, Org. Biomol. Chem., 2018, 16, 6564–6568 RSC; (d) T. Zhang, X. Guo, Y. Shi, C. He and C. Duan, Nat. Commun., 2018, 9, 4024 CrossRef; (e) X. Li, S. Zhuang, X. Fang, P. Liu and P. Sun, Org. Biomol. Chem., 2017, 15, 1821–1827 RSC; (f) S. Huang, P. Niu, Y. Su, D. Hu and C. Huo, Org. Biomol. Chem., 2018, 16, 7748–7752 RSC; (g) Y.-O. Yuan, P. S. Kumar, C.-N. Zhang, M.-H. Yang and S.-R. Guo, Org. Biomol. Chem., 2017, 15, 7330–7338 RSC.
- (a) M. S. Malamas, J. Heterocycl. Chem., 1994, 31, 565–568 CrossRef CAS; (b) M. M. Youssef and M. A. Amin, Molecules, 2010, 15, 8827–8840 CrossRef CAS; (c) Y. Besidski and A. Claesson, Int. Pat. WO 2009005459, 2009.
- (a) Chiral Amine Synthesis: Methods, Developments and Applications, ed. T. C. Nugent, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010 Search PubMed; (b) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS.
- (a) F. Zhou, F.-M. Liao, J.-S. Yu and J. Zhou, Synthesis, 2014, 46, 2983–3003 CrossRef CAS; (b) M. G. Memeo and P. Quadrelli, Chem. Rev., 2017, 117, 2108–2200 CrossRef CAS.
- (a) M. Terada, M. Nakano and H. Ube, J. Am. Chem. Soc., 2006, 128, 12044–12045 CrossRef; (b) X. Xiao, L. Lin, X. Lian, X. Liu and X. Feng, Org. Chem. Front., 2016, 3, 809–812 RSC; (c) H. Konishi, T. Y. Lam, J. P. Malerich and V. H. Rawal, Org. Lett., 2010, 12, 2028–2031 CrossRef CAS; (d) S. Yarlagadda, B. Ramesh, C. R. Reddy, L. Srinivas, B. Sridhar and B. V. S. Reddy, Org. Lett., 2017, 19, 170–173 CrossRef CAS; (e) C. Xu, L. Zhang and S. Luo, J. Org. Chem., 2014, 79, 11517–11526 CrossRef CAS; (f) Y. Wei, H.-X. Zhang, J.-L. Zeng, J. Nie and J.-A. Ma, Org. Lett., 2017, 19, 2162–2165 CrossRef CAS; (g) A. Kumar, S. K. Ghosh and J. A. Gladysz, Org. Lett., 2016, 18, 760–763 CrossRef CAS.
- (a) B. List, J. Am. Chem. Soc., 2002, 124, 5656–5657 CrossRef CAS; (b) A. Bøgevig, K. Juhl, N. Kumaragurubaran, W. Zhuang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2002, 41, 1790–1793 CrossRef.
- B. Zhu, R. Lee, Y. Yin, F. Li, M. L. Coote and Z. Jiang, Org. Lett., 2018, 20, 429–432 CrossRef CAS.
- (a) T. Takeda and M. Terada, J. Am. Chem. Soc., 2013, 135, 15306–15309 CrossRef CAS; (b) X. Yang and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 3205–3208 CrossRef CAS; (c) G. A. Shevchenko, G. Pupo and B. List, Synlett, 2015, 26, 1413–1416 CrossRef CAS; (d) B. M. Trost, J. S. Tracy and T. Saget, Chem. Sci., 2018, 9, 2975–2980 RSC; (e) Y. Han and E. J. Corey, Org. Lett., 2019, 21, 283–286 CrossRef CAS; (f) B. M. Trost, J. S. Tracy and E. Y. Lin, ACS Catal., 2019, 9, 11082–11087 CrossRef CAS.
- (a) T. Bui, M. Borregan and C. F. Barbas III, J. Org. Chem., 2009, 74, 8935–8938 CrossRef CAS; (b) L. Cheng, L. Liu, D. Wang and Y.-J. Chen, Org. Lett., 2009, 11, 3874–3877 CrossRef CAS; (c) F. Zhou, M. Ding, Y.-L. Liu, C.-H. Wang, C.-B. Ji, Y.-Y. Zhang and J. Zhou, Adv. Synth. Catal., 2011, 353, 2945–2952 CrossRef CAS; (d) S. Paria, Q.-K. Kang, M. Hatanaka and K. Maruoka, ACS Catal., 2019, 9, 78–82 CrossRef CAS; (e) S. Yarlagadda, B. Ramesh, C. R. Reddy, L. Srinivas, B. Sridhar and B. V. S. Reddy, Org. Lett., 2017, 19, 170–173 CrossRef CAS.
- (a) T. Mashiko, K. Hara, D. Tanaka, Y. Fujiwara, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 11342–11343 CrossRef CAS; (b) T. Mashiko, N. Kumagai and M. Shibasaki, Org. Lett., 2008, 10, 2725–2728 CrossRef CAS; (c) T. Mashiko, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 14990–14999 CrossRef CAS.
- C. Cheng, Y.-X. Li, J.-Q. Zhang, Y.-L. Zhao, L. hang, C. Li, Y.-S. Yang, L. Tang and Y.-Y. Yang, Adv. Synth. Catal., 2019, 361, 5317–5321 CrossRef CAS.
- CCDC 2009628 (6i) contains the supplementary crystallographic data for this paper.
- Y. Huang, X. Fu, H.-Y. Bai, D.-G. Zhu and S.-Y. Zhang, Org. Lett., 2018, 20, 3469–3472 CrossRef.
- J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416–14417 CrossRef CAS.
- (a) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS; (b) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390–2431 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2009628. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra07806a |
| This journal is © The Royal Society of Chemistry 2020 |