
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOpening the internal structure for transport of ions: improvement of the structural and chemical properties of single-walled carbon nanohorns for supercapacitor electrodes†
Wojciech Zieba a,
Piotr Olejnikb,
Stanislaw Koterc,
Piotr Kowalczykd,
Marta E. Plonska-Brzezinska*b and
Artur P. Terzyk*a
a,
Piotr Olejnikb,
Stanislaw Koterc,
Piotr Kowalczykd,
Marta E. Plonska-Brzezinska*b and
Artur P. Terzyk*a
aFaculty of Chemistry, Physicochemistry of Carbon Materials Research Group, Nicolaus Copernicus University in Toruń, Gagarin Street 7, 87-100 Toruń, Poland. E-mail: aterzyk@chem.uni.torun.pl
bDepartment of Organic Chemistry, Faculty of Pharmacy with the Division of Laboratory Medicine, Medical University of Bialystok, Mickiewicza 2A, 15-222 Bialystok, Poland. E-mail: marta.plonska-brzezinska@umb.edu.pl
cFaculty of Chemistry, Department of Physical Chemistry, Nicolaus Copernicus University in Toruń, Gagarin Street 7, 87-100 Toruń, Poland
dCollege of Science, Health, Engineering and Education, Murdoch University, WA, 6150, Australia
First published on 19th October 2020
Abstract
We investigated the electrochemical performance of single-walled carbon nanohorns (SWCNHs) for use as supercapacitor electrodes. For the first time, we used acid-treatment for oxidation of SWCNHs and hole creation in their structure. A detailed study was performed on the correlation between the oxidation of SWCNHs via acid treatment and variable acid treatment times, the structural properties of the oxidized carbon nanostructures, and the specific capacitance of the SWCNH electrodes. We showed that simple functionalization of carbon nanostructures under controlled conditions leads to an almost 3-fold increase in their specific capacitance (from 65 to 180 F g−1 in 0.1 M H2SO4). This phenomenon indicates higher accessibility of the surface area of the electrodes by electrolyte ions as a result of gradual opening of the SWCNH internal channels.
1 Introduction
Recently, great interest has been focused on the application of nanostructured carbons for electrocatalysis1 and energy storage applications,2–5 particularly in electrochemical capacitors or supercapacitors (SCs).6–10 Due to a large specific surface area connected with high mesoporosity, nanostructured carbons have a high capability for charge accumulation at the electrode/electrolyte interface (known as an electric double layer).11–13 Accumulation of an ionically conducting electrolyte solution on an electronically conducting electrode surface leads to the establishment of electric double-layer capacitance (EDLC). In brief, the electrochemical performance of these systems depends on the control of mass and charge transport (i.e., electronic and ionic mobility) and electron transfer kinetics in multiphase boundaries,14 and these are size dependent processes. To this end, much attention is focused on preparation of nanostructured carbon materials with controlled properties, including internal and external porosity, electrical conductivity and rich functional chemistry.15–17In literature, we find many reports of the use of nanostructured carbon materials as electrode materials in SCs,4,5,11 and the most popular are carbon nanotubes (CNT) and graphene electrodes.18 For example, multiwalled CNTs were heated to enhance their specific surface area and pore size distribution, and after thermal treatment, the CNT electrodes reached a maximum specific capacitance of 180 F g−1 with a power density of 20 kW kg−1.19 Even with special preparation of CNTs, their specific surface area is still much lower than that of mesoporous carbons, ordered mesoporous carbons or activated carbon electrodes (specific surface areas in the range of 600 and 3000 m2 g−1, respectively).20–22 Surface modification of carbon nanostructures (CNs) via acidic or alkaline treatment, which introduces oxygen functional groups, has an additional effect and improves the electrochemical properties of SCs.13,23,24 It should also be emphasized that the gravimetric capacity of carbon materials does not increase linearly with the specific surface area, and based on the density functional theory model, it reaches a plateau for values of ca. 1200 m2 g−1.22 The authors suggested that this limitation of the gravimetric capacity can be ascribed to space constriction for charge accommodation inside the pore walls.22 In this context, a further increase in the specific surface of carbonaceous materials does not affect the increase in gravimetric capacity.
Single-walled carbon nanohorns (SWCNH) belong to the class of novel carbon nanomaterials. Thousands of cone-end shaped tubules with diameters of 2–5 nm are agglomerated in spheres with a diameter in the range of 60–120 nm. The spheres can occur in few different forms: dahlia, petal-dahlia, bud and seed. Dahlia and petal-dahlia shapes have long cone endings on a spherical surface in contrast to bud and seed shapes.25 The material is produced by CO2 laser ablation of graphite and is catalyst-free with notably high purity of up to 90%. To obtain access to the internal volume of SWCNH, various oxidation reactions have been applied, e.g., heat treatment under different atmospheres, such as air,26,27 CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 and O2.29–32 Less popular methods are the so-called “wet methods” using, e.g., H2O2,33–36 concentrated acids or their mixtures, e.g., nitric acid37–41 or “piranha” solution.42,43 Oxidation changes selected chemical and physical properties of the material, thus increasing the specific surface area and pore volume. Surface oxygen groups increase hydrophilicity, enabling the formation of stable colloids in water.39 Generally, these functionalization approaches increase the specific effective surface of the SWCNHs while optimizing the structural parameters that influence the capacitive properties of the SWCNH electrodes.27,29,31,44 Therefore, due to a large surface area, homogeneous structure and capacity for modifications, SWCNHs have found application in many electrochemical areas, such as Li-ion batteries,30,45–48 SCs,26,27,29,31,44,49,50 sensors41,51–53 and/or fuel cells.54–56 Moreover, SWCNHs could also be used as composites to construct complicated and hierarchical systems that exhibit optimized targeted properties.33
28 and O2.29–32 Less popular methods are the so-called “wet methods” using, e.g., H2O2,33–36 concentrated acids or their mixtures, e.g., nitric acid37–41 or “piranha” solution.42,43 Oxidation changes selected chemical and physical properties of the material, thus increasing the specific surface area and pore volume. Surface oxygen groups increase hydrophilicity, enabling the formation of stable colloids in water.39 Generally, these functionalization approaches increase the specific effective surface of the SWCNHs while optimizing the structural parameters that influence the capacitive properties of the SWCNH electrodes.27,29,31,44 Therefore, due to a large surface area, homogeneous structure and capacity for modifications, SWCNHs have found application in many electrochemical areas, such as Li-ion batteries,30,45–48 SCs,26,27,29,31,44,49,50 sensors41,51–53 and/or fuel cells.54–56 Moreover, SWCNHs could also be used as composites to construct complicated and hierarchical systems that exhibit optimized targeted properties.33
For the first time, we report the structural changes of SWCNHs that were performed using oxidation by nitric acid. A variable acidic treatment time was applied, and the correlations between the structural changes and the electrochemical properties of the obtained CNs were examined. We showed that simple functionalization of CNs under controlled conditions leads to an almost 3-fold increase in their specific capacitance due to an ‘opening structure’ for ion mobility. Additionally, the resulting SWCNH electrode has excellent electrochemical stability.
2 Results and discussion
2.1. Structural and textural characterization of SWCNHs
The physicochemical properties of CNs are closely related to their structure, type of carbon atoms, number of defects on the surface, type of functional groups, etc. Therefore, we performed systematic experimental and theoretical studies of the structure and surface of the SWCNHs modified in the oxidation process. Fig. 1A presents the N2 adsorption–desorption isotherms measured for the pristine and the oxidized SWCNHs. The shape of the isotherm measured for the pristine SWCNHs (Fig. 1A) suggests the presence of mesopores. The BET surface area (SBET) was calculated using the Brunauer–Emmett–Teller57 adsorption isotherm equation. However, to make the porosity parameters more realistic, we used a series of local isotherms for cylinders calculated using grand canonical Monte Carlo (GCMC) simulations, and this procedure is described in detail in the Experimental section (fit quality is shown in Fig. S1 and S2†). The SBET value for SWCNHs (Table 1) is in good agreement with the values reported for this system by other authors58 and supplied by the producer.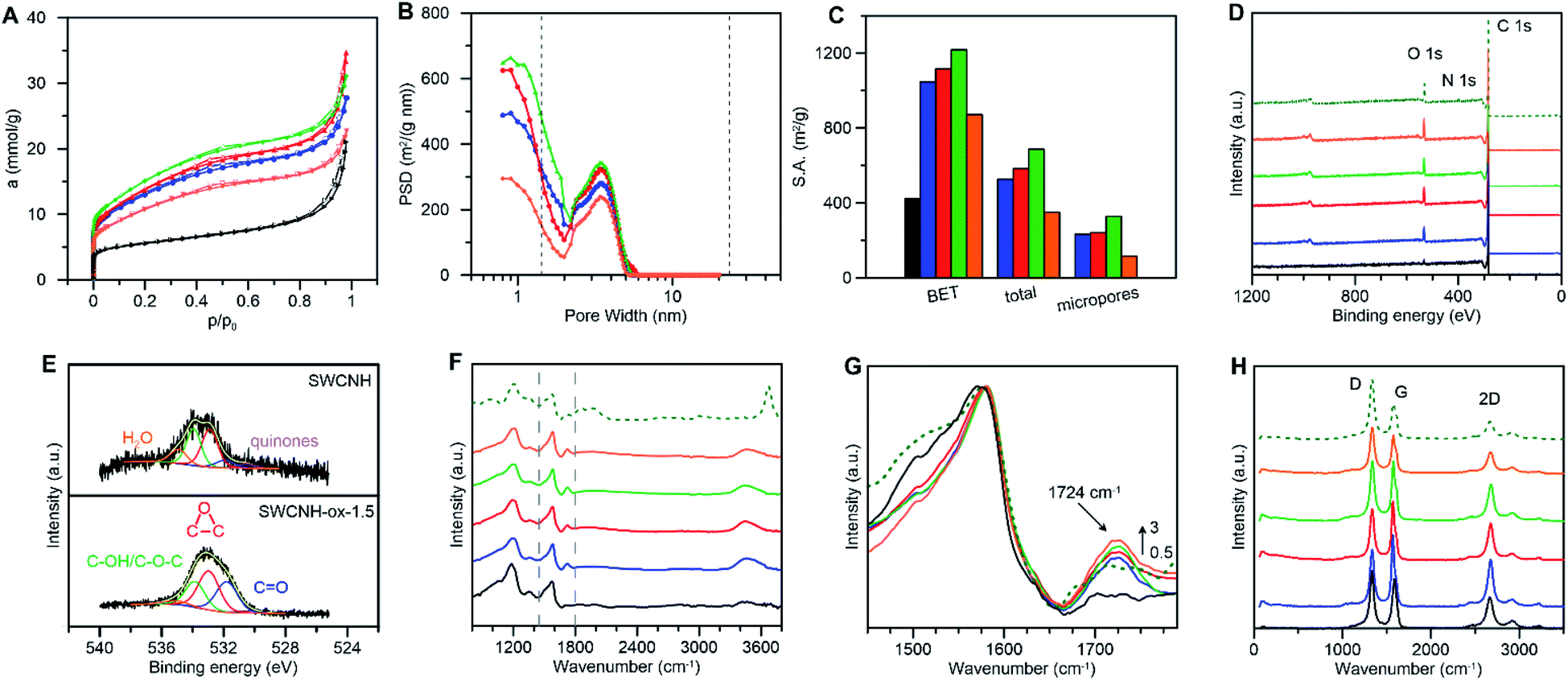 | ||
| Fig. 1 (A) Low temperature N2 adsorption–desorption isotherms, (B) pore size distributions, (C) pore surface areas, (D) XPS survey spectra, (E) high-resolution O 1s XPS spectra, (F and G) FTIR and (H) Raman spectra for SWCNH (black), SWCNH-ox-0.5 (blue), SWCNH-ox-1.0 (red), SWCNH-ox-1.5 (green), SWCNH-ox-3 (orange) and r-SWCNH-ox-1.5 (dotted dark green). For r-SWCNH-ox-1.5, practically the same adsorption isotherms and porosity parameters as for SWCNH-ox-1.5 are observed, and thus the adsorption and pore size distribution results for this sample are omitted for clarity. | ||
| Materials | SBET (m2 g−1) | Stotal (m2 g−1) | Smeso (m2 g−1) | Smicro (m2 g−1) | vtotal (cm3 g−1) | vmeso (cm3 g−1) | vmicro (cm3 g−1) | Dmicro (nm) | Dmeso (nm) |
|---|---|---|---|---|---|---|---|---|---|
| a SBET – the apparent BET surface area, equal to 421 m2 g−1 for pristine SWCNH, Stotal – total internal surface area, Smeso – internal surface area of mesopores, Smicro – internal surface area of micropores, vtotal – total internal pore volume, vmeso – internal volume of mesopores, vmicro – internal volume of micropores, Dmicro – average diameter of micropores, and Dmeso – average diameter of mesopores. For r-SWCNH-ox-1.5 (not shown), practically the same isotherms and porosity parameters as for SWCNH-ox-1.5 were observed. | |||||||||
| SWCNH-ox-0.5 | 1047 | 527 | 294 | 233 | 0.33 | 0.25 | 0.08 | 0.69 | 3.4 |
| SWCNH-ox-1 | 1116 | 584 | 342 | 242 | 0.37 | 0.30 | 0.07 | 0.83 | 3.3 |
| SWCNH-ox-1.5 | 1219 | 687 | 358 | 328 | 0.42 | 0.31 | 0.11 | 0.90 | 3.4 |
| SWCNH-ox-3 | 872 | 350 | 234 | 116 | 0.24 | 0.20 | 0.04 | 0.86 | 3.4 |
From the data collected in Fig. 1A, B, S1 and S2† it can be concluded that increasing the oxidation time of the SWCNHs led to increasing N2 adsorption, which consequently led to increasing SBET, Stotal and total pore volumes (vtotal) with the creation of micropores on the SWCNH surface (Table 1). This observation is schematically presented in Fig. 1C. The SBET values are between 872 (SWCNH-ox-3) and 1219 m2 g−1 (SWCNH-ox-1.5) for the oxidized materials. The maximum values for all of these parameters were recorded for SWCNH-ox-1.5 (Table 1). Therefore, the reduction reaction of the SWCNH-ox-1.5 material was performed to produce r-SWCNH-ox-1.5, which was used in further studies as a comparison.
Oxidation treatment of SWCNHs causes gradual opening of the internal channels of the carbon nanotubes, as confirmed by the HR-TEM images collected in Fig. 2B. However, for oxidation time longer than 1.5 h, burn-off of tubules and creation of amorphous carbon around the walls were observed (Fig. 2C). These conclusions confirm the decrease in N2 adsorption (Fig. 1A) and the consequent decrease of the SBET and the vtotal values that were observed for SWCNH-ox-3 (Table 1). However, for electrochemical application of CNs, the most commonly used parameter is SBET, a more justified approach is the use of internal surfaces, the parameters connected with the presence of meso and micropores. The Smeso and the Smicro values for the oxidized SWCNHs were increased in the order SWCNH-ox-3 < SWCNH-ox-0.5 < SWCNH-ox-1 < SWCNH-ox-1.5 (Table 1). The sharp drop in the Smeso and the Smicro values with a long oxidation time (SWCNH-ox-3) is related to the adsorption of amorphous carbon on the SWCNH surface, as mentioned previously, that was observed by HR-TEM studies (Fig. 2C). An analogous relationship for the SWCNH structures and oxidation time was observed for the volume of meso and micropores (Table 1). It must be emphasized that gradual treatment of the SWCNHs with HNO3 shows minimal change in the average diameters of mesopores (Dmeso) and slightly changes the diameters of micropores (Dmicro) (Table 1). A longer oxidation process led to an increase in the average diameter of micropores from approximately 0.7 to 0.9 nm.
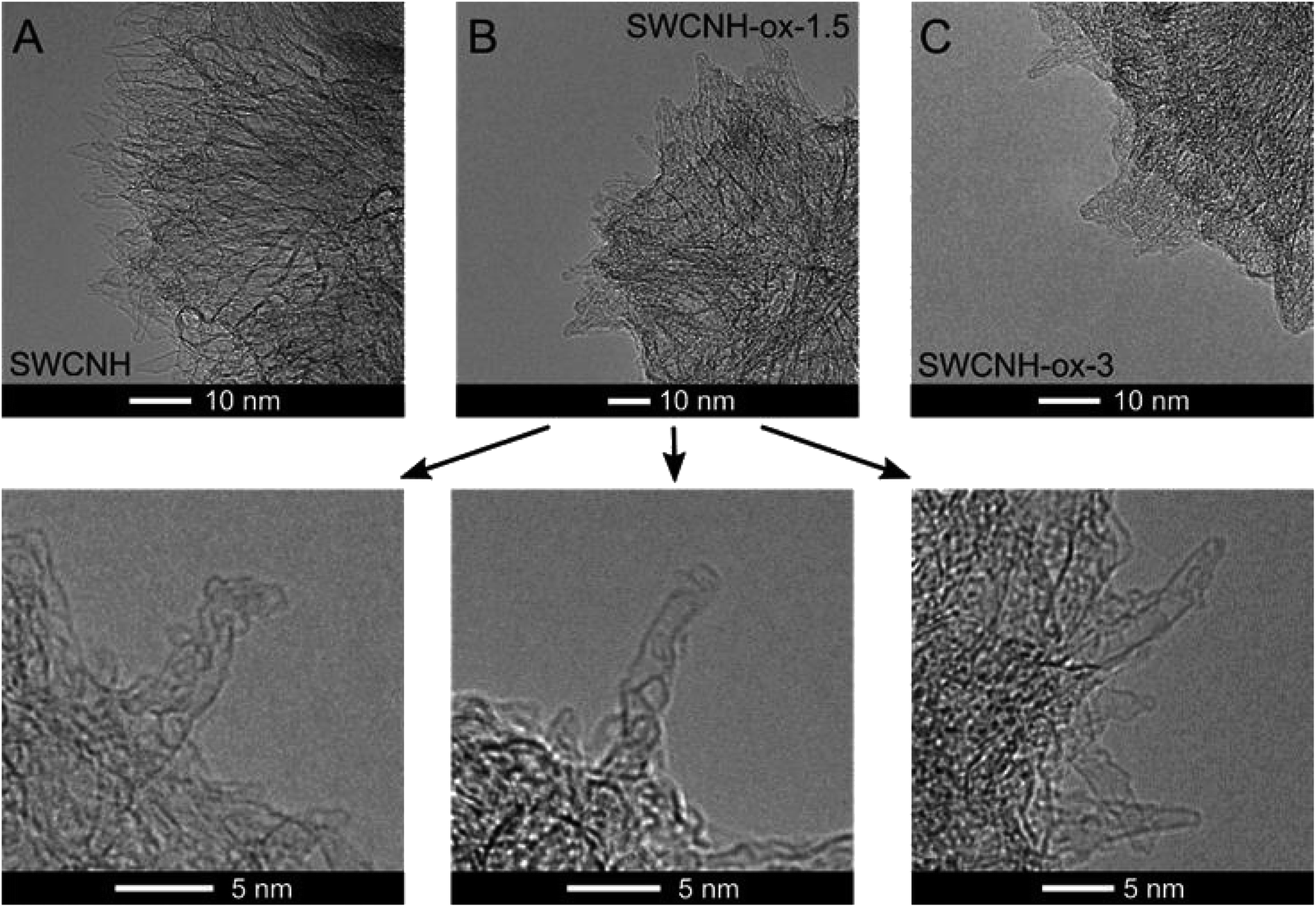 | ||
| Fig. 2 HR-TEM images of (A) SWCNHs and selected oxidized samples, (B) SWCNH-ox-1.5 and (C) SWCNH-ox-3.0 (bottom panel shows the holes in the horns of the SWCNH-ox-1.5 sample). | ||
X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared spectroscopy (FTIR) were performed to confirm the oxidation of the SWCNH surface. Quantitative and qualitative studies were conducted using XPS (Fig. 1D and E).
The survey spectra for the SWCNHs and the oxidized materials are compared in Fig. 1D, and the elemental quantification is shown in Fig. S3–S5.† The XPS spectra for the pristine SWCNHs contain one peak (of C 1s) and two peaks (of C 1s and O 1s) for SWCNH-ox (Fig. 1D). A small N 1s peak is also observed for the reduced sample (Fig. 1D).
Deconvolution of the high-resolution C 1s core level spectra for the pristine SWCNHs shows three main peaks centered at 284.7 eV (56.4% at. concentration of C; C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C sp2; graphitic carbon), 284.9 eV (13.6% at. concentration of C; C–C sp3) and 285.2 eV (8.2% at. concentration of C; high C–C sp3/C–H sp3), (Fig. S3†).59–61 After the oxidation reaction, the C 1s XP spectrum exhibits additional energy regions that confirm the presence of oxygen-containing groups on the SWCNH surface and could be deconvoluted into six peaks at 285.7 eV (ether groups, C–O–C), 286.1 eV (hydroxyl groups, C–OH), 286.7 eV (epoxy groups, (CC)
C sp2; graphitic carbon), 284.9 eV (13.6% at. concentration of C; C–C sp3) and 285.2 eV (8.2% at. concentration of C; high C–C sp3/C–H sp3), (Fig. S3†).59–61 After the oxidation reaction, the C 1s XP spectrum exhibits additional energy regions that confirm the presence of oxygen-containing groups on the SWCNH surface and could be deconvoluted into six peaks at 285.7 eV (ether groups, C–O–C), 286.1 eV (hydroxyl groups, C–OH), 286.7 eV (epoxy groups, (CC)![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) O), 287.3 eV (carbonyl groups, CO), 289.2 eV (carboxyl groups, COOH, very weak signal), (Fig. S3†),62,63 and an additional peak centered at 284.2 eV related to the vacancy defects of the graphene lattice.64 The level of oxidation of the SWCNHs was determined from the areas under the C 1s and O 1s peaks (Table S1†). As expected, the amount of functional groups (C–O–C, C–OH, (CC)O, CO and COOH) increased with increasing oxidation reaction time, and after reaching the reaction time of one and half hours, their amounts on the surface of the CN decrease (a progressive decrease in the C 1s/O 1s ratio, Table 2). However, it should be emphasized that this observation is correct in considering the total number of oxygen-containing groups (Table S1†). The amount of COOH groups on the SWCNH surface increased proportionally with the increase in the oxidation time (the value for 0.5 h was not determined; in the range of 1–3 h oxidation reaction the values are between 1.0%–2.3% at. concentration of C).
O), 287.3 eV (carbonyl groups, CO), 289.2 eV (carboxyl groups, COOH, very weak signal), (Fig. S3†),62,63 and an additional peak centered at 284.2 eV related to the vacancy defects of the graphene lattice.64 The level of oxidation of the SWCNHs was determined from the areas under the C 1s and O 1s peaks (Table S1†). As expected, the amount of functional groups (C–O–C, C–OH, (CC)O, CO and COOH) increased with increasing oxidation reaction time, and after reaching the reaction time of one and half hours, their amounts on the surface of the CN decrease (a progressive decrease in the C 1s/O 1s ratio, Table 2). However, it should be emphasized that this observation is correct in considering the total number of oxygen-containing groups (Table S1†). The amount of COOH groups on the SWCNH surface increased proportionally with the increase in the oxidation time (the value for 0.5 h was not determined; in the range of 1–3 h oxidation reaction the values are between 1.0%–2.3% at. concentration of C).
| Materials | C 1s/O 1s | νD (cm−1) | νG (cm−1) | ID/IG | ζ potential (mV) |
|---|---|---|---|---|---|
| SWCNH | 15.110 | 1332 | 1580 | 1.20 | n.a. |
| SWCNH-ox-0.5 | 4.143 | 1335 | 1575 | 1.28 | −6.65 |
| SWCNH-ox-1 | 3.087 | 1335 | 1571 | 1.37 | −17.91 |
| SWCNH-ox-1.5 | 3.172 | 1338 | 1575 | 1.50 | −22.35 |
| SWCNH-ox-3 | 2.603 | 1336 | 1574 | 1.68 | −29.65 |
| r-SWCNH-ox-1.5 | 4.050 | 1334 | 1577 | 1.66 | +2.45 |
Deconvolution of the O 1s XPS peaks produced additional information on the nature of the surface-containing groups. However, for the SWCNHs, selected oxygen functionalities were found with the contribution of chemisorbed water. The calculated percentages of these groups are quite low, and they are shown in Fig. S4† for comparison purposes only. As shown in Fig. 1E, the O 1s core signal for the oxidized CNs could be deconvoluted into peaks at 530.5 eV (quinones),65 531.8 eV (CO),66 532.9 eV ((CC)O),67 533.8 eV (C–OH, C–O–C) and 535.0 eV (chemisorbed water).68,69 The percentage of these functional groups for all oxidized SWCNH materials is shown in Fig. S4.† After reduction of the SWCNH-ox-1.5 by hydrazine, the amount of oxygen-containing groups on the SWCNH surface decreases significantly, with the exception of the quinone groups, which proves that the groups containing O were successfully reduced.
This observation is confirmed by high-resolution N 1s XPS studies, which were decomposed into four identified components (Fig. S5†). For r-SWCNH-ox-1.5, we observed the appearance of N–H bonds at 399.8 eV, pyrrolic or amine moieties at 400.2 and 400.6 eV, and quaternary N at 401.8 eV.64,70,71 We observed the hydrophilization of the SWCNH surface by the creation of surface oxides. The ratio of C 1s/O 1s peaks decreases rapidly after a short oxidation process (after 0.5 h), from 15.110 to 4.143, and subsequently changes during a longer oxidation time, reaching a value of 2.603 for SWCNH-ox-3 (Table 2) and a value of 4.05 for r-SWCNH-ox-1.5.
This observation is also strongly supported by the decrease in the ζ potential values (from −6.65 to −29.65) determined at pH = 2 (Table 2). As confirmed by the XPS data (Table S1†) the presence of nitrogen-containing groups for the r-SWCNH-ox-1.5 sample is manifested by the positive ζ potential value (ζ potential = +2.45) caused by the basic nature of selected surface nitrogen groups.
The FTIR-HATR technique was used to determine the nature of the functional groups on the SWCNH surface. Fig. 1F and G show the FTIR spectra of the pristine, oxidized and reduced SWCNHs. Three clearly distinct bands appear for the pristine SWCNHs at 1573, 1356 and 1185 cm−1, with small shoulders at 1059 cm−1. Thus, the band at 1573 cm−1 might be assigned to stretching vibrations of the CC aromatic groups.72–74 The presence of the peaks at 1356 and 1185 cm−1 are strongly affected by the defective structure of CNs and was also observed for ultradispersed diamond powders.75 However, it should be emphasized that these peaks are found in the area of several overlapping peaks that occur in the range from 1400 to 900 cm−1, therefore, the determination of their intensities is often ambiguous. In the literature, this phenomenon is often translated as the occurrence of ‘dangling bonds’ on the surface of CNs, which are sensitive to chemisorption of foreign elements.75 These surface carbon atoms have free valances that can be saturated by chemisorption or that might interact or react with foreign elements (H2O, H, N, O, etc.) to form bonded surface functional groups.76 These absorption features still exist in the region of 1600 to 900 cm−1 in the reduced form of the SWCNHs, but they are somewhat different. Therefore, this wavenumber region might be assigned to edge carbon atoms on the SWCNH surface, which are sensitive to chemical reactions (oxidation or reduction), that in successive stages have changed the chemical nature.
Successful oxidation of the SWCNH surface is strongly supported by the peak at 1724 cm−1, which can be assigned to CO stretching vibrations (Fig. 1G),75 and these vibrations are not observed for the pristine SWCNHs. The spectra of the oxidized SWCNH materials clearly indicate that a longer oxidation process of the CNs leads to an increase in the intensity of the CO stretching vibrations (from 0.5 h to 3 h), which consequently proves the increase in the amount of oxygen-containing groups on the SWCNH surface. For the reduced form of the SWCNHs, the intensity of this peak decreased, but an additional shoulder is observed at 1716 cm−1 (dashed line on Fig. 1G). This peak might be assigned to CO stretching vibrations in ketones or aldehydes.74 Based on the FTIR spectroscopic characterization of functionalized single-walled carbon nanotubes, the band detected at approximately 1570 cm−1 was attributed to CC stretching vibration of functionalized carbons.72,77 The increased intensity in the high frequency region between 3700 and 3200 cm−1 could also indicate successful oxidation of the SWCNH surface.72 This band is the result of the O–H stretching vibration and can be observed in the spectra in Fig. 1F as a broad band with a maximum at 3440 cm−1. Upon surface reduction with hydrazine, the spectrum for the r-SWCNH-ox-1.5 shows a strong and broad band in the 3500–3800 cm−1 region.75 The maximum falls near 3650 cm−1, and the band might be assigned to N–H asymmetric stretching vibrations.74 Two additional broad peaks are observed. The first peak is found in the 2100–1830 cm−1 region with two maxima at 1980 and 1860 cm−1. These peaks might be assigned to C–NH3+ and CO stretching vibrations of anhydride or cyclic acid anhydride.74 In the 1050–900 cm−1 region with a maximum near 970 cm−1, the peak observed might be assigned to C–C–C bending vibrations or C–O–C symmetric stretching vibrations.74
The Raman spectra of the pristine and the oxidized SWCNH samples registered at 532 nm are shown in Fig. 1H and S6† and are summarized in Table 2. The most distinctive signal at approximately 1580 cm−1 is known as the G band, which corresponds to the in-plane optical mode of vibration for two adjacent sp2 C atoms on an ideal hexagonal ring of graphite.78–80 The spectra are also dominated by the D band at 1332 cm−1.81,82 The presence of the D band is due to defects in the carbon crystalline curved structure. Additional combined tones for the peaks are located at 2665 cm−1 (2D) and 2919 cm−1 (D + G) (Fig. S6†) that are observed for all investigated materials and correspond to the second-order Raman spectrum.83 The D band for all oxidized SWCNH materials and the reduced sample is shifted compared with that of the pristine SWCNHs (from 1332 to 1338 cm−1). The origin and dispersive behavior of the D band can be explained by the double resonance Raman mechanism84 that was also observed for nanographite.85
The ratio of the intensities of the D and G bands (ID/IG) is used as a convenient measure of crystalline order and could confirm the functionalization process of the pristine SWCNHs (Table 2). The effectiveness of the functionalization of CNs increased, and the heights of the “disorder” deconvoluted peaks (D′, D) also increased as the G peak decreased.86 Based on this relationship, the Raman spectrum for the pristine SWCNHs shows the lowest D/G intensity ratio (ID/IG = 1.2), and the highest ID/IG value was determined for SWCNH-ox-3 (ID/IG = 1.68). Oxidation with strong acids causes the creation of pores with different dimensionality. Therefore, the hybridization of the C atoms changes from sp2 to sp3, which is recognized as formation of the defect in the CN structure.87 The larger D band intensity is also caused by the presence of oxygen functional groups on the SWCNH surface that are recognized as a defect in the structure of carbonaceous material. It must also be noted that the G band in the SWCNH-ox spectra exhibits an apparent shoulder, which corresponds to the D′ band, a second-order band, at ∼1604 cm−1. This phenomenon is commonly associated with disorder in graphite and could be associated with oxidation treatment of CNs (Fig. S6†).86,88
2.2. Electrochemical performance of SWCNH electrodes
To explore the potential application of these materials in energy storage devices, the samples were used as supercapacitor electrodes and characterized using cyclic voltammetry (CV) in 0.1 mol dm−3 aqueous solution. Fig. 3A shows a comparison of the electrochemical properties of the glassy carbon electrode (GCE) modified with SWCNH, SWCNH-ox-1 and r-SWCNH-ox-1 layers. Modification of the electrode was performed by the drop-casting method using an ethanol dispersion of SWCNH, SWCNH-ox-1 or r-SWCNH-ox-1 with the same concentration of CNs (please see Experimental section, where modification of the GCE is described in the details).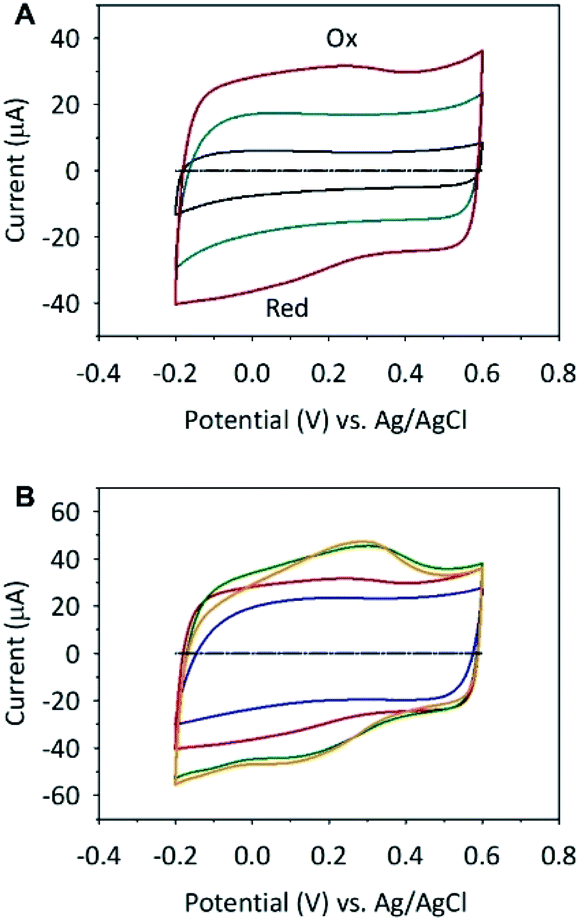 | ||
| Fig. 3 CVs in 0.1 mol dm−3 NaCl of (A) nonmodified GCE (black dashed line) and covered with SWCNH (black line), SWCNH-ox-1 (red line) or r-SWCNH-ox-1 (blue line); and (B) nonmodified GCE (black dashed line) and SWCNH-ox prepared with different oxidation time: SWCNH-ox-0.5 (blue line), SWCNH-ox-1 (red line), SWCNH-ox-1.5 (green line), and SWCNH-ox-3 (orange line). The sweep rate was 10 mV s−1. | ||
The potential was cycled between −0.2 and +0.6 V vs. Ag/AgCl without any noticeable change in the shape of the CVs. The electrodes modified with SWCNH and r-SWCNH-ox-1 exhibit CV curves with the typical capacitance behaviour and almost pseudorectangular anodic and cathodic profiles. The symmetry of both CVs indicates that the capacitance of the electrodes is reversible in the anodic and cathodic directions.
A small departure from the ideal rectangular shape is observed for the oxidized SWCNH materials (Fig. 3). The broad and poorly defined peaks Ox and Red were recorded on CVs in anodic and cathodic cycles, respectively (Fig. 3A, red line). The difference between the observed capacitances of the nonmodified SWCNH electrodes and their oxygen-containing derivatives could be due to the more microporous structure of SWCNH-ox and the pseudocapacitive faradaic reactions (reversible oxidation/reduction) of oxygen functionalities (mainly C–OH, CO and COOH) generated during chemical oxidation.89,90 The characteristic weak shoulders in both cycles is characteristic of the oxidized CNs due to the low redox kinetics of the surface functional groups compared with the charging kinetics of the electrochemical double layer.89,91 This effect also depends on the small fraction of oxygen-containing groups relative to the whole surface of the CNs. Moreover, all CVs are stable for these films, which proves their electrochemical stability in the studied potential range.89,91
A previous study showed that an increase in surface oxides on the CNs,92 which are used as an electrode material, results in increasing intensities of both redox processes (Fig. 3B). The amount of oxygen-containing groups on the oxidized SWCNH surface depends on the oxidation time of this nanostructure (please see the spectroscopic results in Fig. 1G). A previous study also showed that surface oxides prefer proton adsorption in the micropores of the carbon materials.89,91 Therefore, the specific adsorption results in local changes to the charge density in the double-layer capacitance. The pseudocapacitive effect is observed for all SWCNH-ox (Fig. 3B).
The specific capacitances (Cs) of the SWCNH and their oxidized and reduced forms (SWCNH-ox and r-SWCNH-ox, respectively) were determined using the CV method based on the following eqn (1):
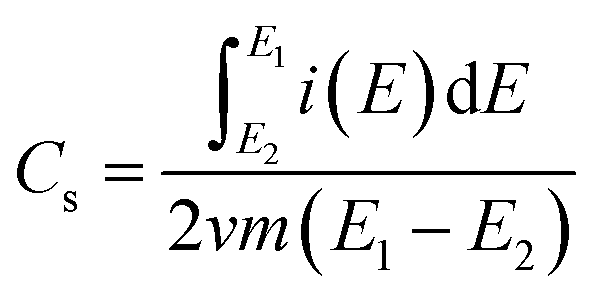 | (1) |
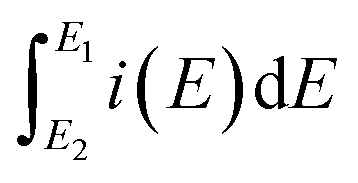 is the integrated current in the potential window, v is the sweep rate (V s−1), and m is the mass of the active material. The calculated Cs are summarized in Table 3. A clear correlation exists between the concentration of the oxygen functionalities in the SWCNH structure and the Cs values (Fig. 3B). The first interesting observation is an enhancement of the Cs of the SWCNH materials, from 65 F g−1 found for the pristine SWCNH to 180 F g−1 for the oxidized SWCNHs (SWCNH-ox-1.5 in acidic media, H2SO4). This value is in good agreement with a previous report on the oxidized SWCNHs exhibiting higher capacitance than their pristine form due to modification of the micropore surface area and micropore volume due to hole-opening of the SWCNHs.26,29,31,44
is the integrated current in the potential window, v is the sweep rate (V s−1), and m is the mass of the active material. The calculated Cs are summarized in Table 3. A clear correlation exists between the concentration of the oxygen functionalities in the SWCNH structure and the Cs values (Fig. 3B). The first interesting observation is an enhancement of the Cs of the SWCNH materials, from 65 F g−1 found for the pristine SWCNH to 180 F g−1 for the oxidized SWCNHs (SWCNH-ox-1.5 in acidic media, H2SO4). This value is in good agreement with a previous report on the oxidized SWCNHs exhibiting higher capacitance than their pristine form due to modification of the micropore surface area and micropore volume due to hole-opening of the SWCNHs.26,29,31,44
Generally, increasing the SBET value of materials leads to an increasing overall capacitance due to a higher electrode/electrolyte contact area (Table 3). However, it should be noted that the number of pores and also their size (micro and mesopores, and outer surfaces of the particles) affect the capacity of the layers (Smicro and Smeso) (Table 1). These parameters determine the possibility of ion mobility in the electrode layer and, consequently, the formation of electric double-layer capacitance.13,93 In addition, the method of measuring the electrode surface has an influence on the capacitance of porous carbons,92 considering the Cs relative to the morphology of the carbon material and surface chemistry (e.g., wettability and type of oxygen-containing group). As presented above, the capacitance of materials is a highly complex process, and therefore, for each newly obtained or modified CN, the structure and surface properties must be analyzed in detail in relation to many parameters.
Even for SWCNHs oxidized via the heat-treatment method,26,27,29,31,44 for which the literature is quite scarce, we find many discrepancies and no simple relationship between SBET and Cs. For example, oxidation of the SWCNHs via heat-treatment in an air atmosphere at different temperatures in the range of 400 to 580 °C results in an increasing specific surface area, from 400 to 1420 m2 g−1 (limit values for the pristine SWCNHs and the oxidized SWCNHs, respectively), and consequently leads to increasing Cs, from 66 to 114 F g−1.31 Using the same type of modification (at 550 °C), other authors have produced materials with the following parameters: SBET of 1690 m2 g−1 and Cs of 100 F g−1.26 However, Sano et al. used heat treatment of pristine SWCNHs in an air atmosphere at the same temperature (550 °C), and they obtained an oxidized material with a much lower SBET value of 964 m2 g−1, although the Cs was quite high 365 F g−1 (at sweep rates of 10 mV s−1). As noted many times, no simple correlation is found between the SBET and Cs of the CNs because the values presented above show that the same method of oxygenation and nearly the same experimental conditions can lead to different electrochemical properties of the materials. Although there is a general tendency that more efficient oxidation of CNs leads to an increase of SBET and consequently an increase in the Cs of SWCNHs, the resulting Cs values differ quite significantly. It seems that a key to increasing Cs, as Kaneko reports, is the type and the size of the pores created in the SWCNHs during the oxidation treatment, known as ‘tip opening’ and/or ‘sidewall opening’.29,31
For the first time, we used acid treatment for oxidation of SWCNHs and hole-creation in their structure. Because the pore size of an electrode material should be suitable for the ion size of the electrolyte, a systematic study of the effects of SWCNH porosity on the ion–electrode interactions was performed.
Additionally, the surface area of porous carbons are measured by gas adsorption, which can penetrate the open pores that are available for the molecular size of the adsorbate. Electrolyte penetration into pores is more restricted,92 and thus a correlation between the electrolyte accessibility and carbon structure and their surface properties was studied.
Herein, the effect of the supporting electrolyte on the capacitive properties of the GCE modified with SWCNH (Fig. 4, panel (I)), SWCNH-ox-1.5 (Fig. 4, panel (II)) or r-SWCNH-ox-1.5 (Fig. 4, panel (III)) films was studied. This investigation was performed in an aqueous solution containing different supporting electrolytes of H2SO4, Na2SO4, NaCl or (Et)4NCl. The Cs values were determined using eqn (1) and are summarized in Table 3.
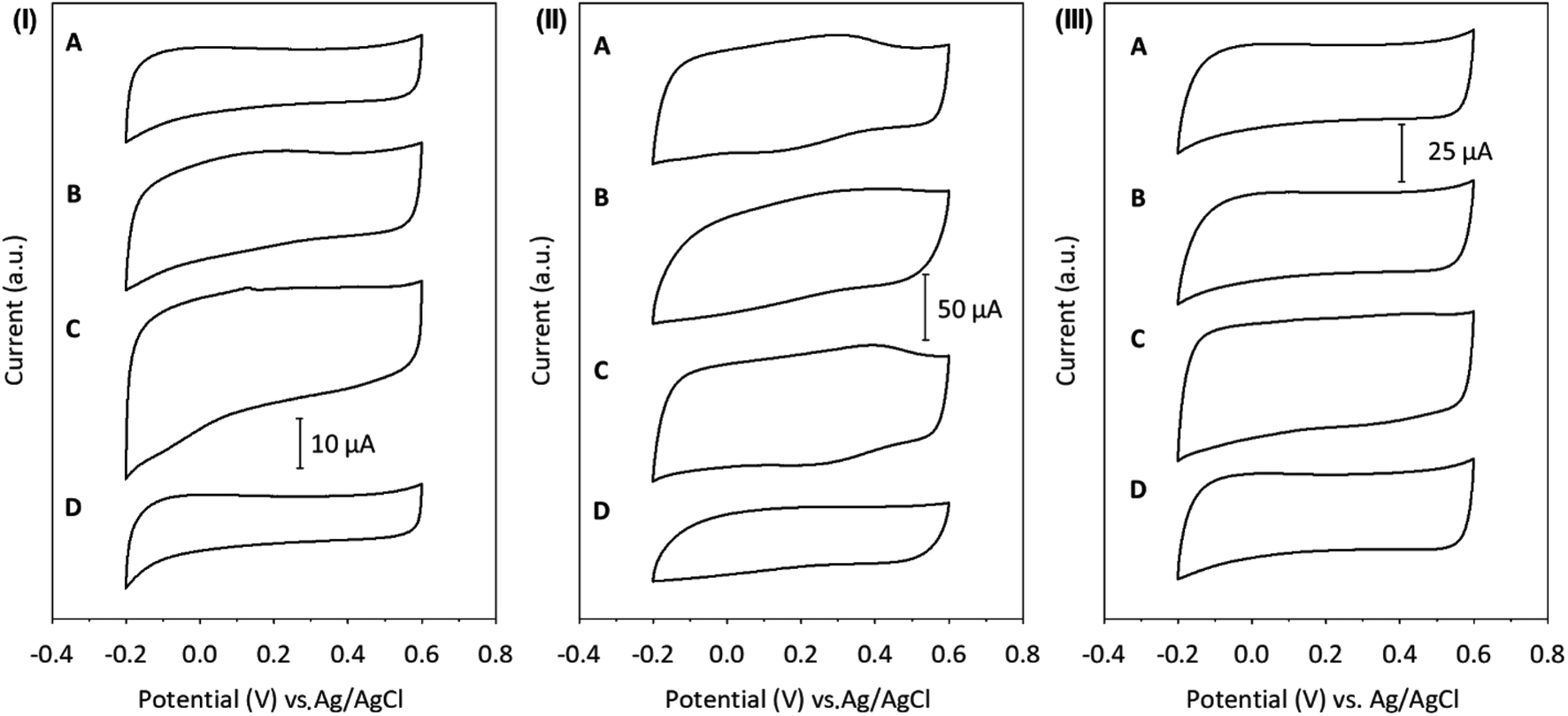 | ||
| Fig. 4 CVs of (I) SWCNH, (II) SWCNH-ox-1.5 and (III) r-SWCNH-ox-1.5 films in 0.1 M (A) NaCl, (B) Na2SO4, (C) H2SO4, and (D) (Et)4NCl. The sweep rate was 10 mV s−1. | ||
There are two effects responsible for the influence of the supporting electrolyte on Cs, the degree of the film penetration of the counter ions and the structure of the double layer on the CNO surface and electrolyte solution.24 A few parameters might have an influence on these effects: the hydrated radius of the ions (rh), the chemistry of the CN surface, the number of pores in the CNs and their size. According to Stokes' law, the migration velocity is inversely proportional to the effective rh.94 The Stokes radius (rs), rh and standard molar enthalpy of hydration (ΔhydH°) for the ions of the supporting electrolytes are summarized in Table S2.† However, the size of rh is almost the same, and therefore, the supporting electrolyte affected the electrochemical properties of the films (Table 3). The SWCNH-ox might interact more strongly with cations than with anions. Additionally, the interaction of water molecules with the oxidized surface of CNs can influence the wettability of the carbon surface in aqueous solution and affect their electrochemical properties. The observed value of the Cs of r-SWCNH-ox-1.5 is higher than for unmodified nanostructures in all supporting electrolytes (Fig. 4, panel (III)), but the tendency of the supporting electrolyte to influence the electrochemical properties remains as in the oxidized form of SWCNHs. However, it should be emphasized, that reversible oxidation and reduction of oxygen functionalities generated during chemical oxidation are not visible on CVs of r-SWCNH-ox-1.5. Therefore, it may be assumed, that the chemical reduction of SWCNH-ox-1.5 with the use of hydrazine led to the reduction of carboxylic groups, creating quinone or alcoholic groups. The higher values of Cs of the reduced nanostructures (r-SWCNH-ox-1.5) compared to SWCNHs may also result from incomplete reduction of SWCNH-ox-1.5. It can be assumed that the surface of CNs has not been completely reduced and is still hydrophilic in nature (Table 3).
To investigate the rate performance of the CNs, the electrodes were cycled between −0.2 and 0.6 V (vs. Ag/AgCl) at different potential sweep rates. Fig. 5 and S7† show the CV curves of the SWCNH (Fig. 5) and SWCNH-ox-1.5 (Fig. S7†) layers on the GCE surface in an aqueous solution of 0.1 mol dm−3 in the different supporting electrolytes recorded at different sweep rates.
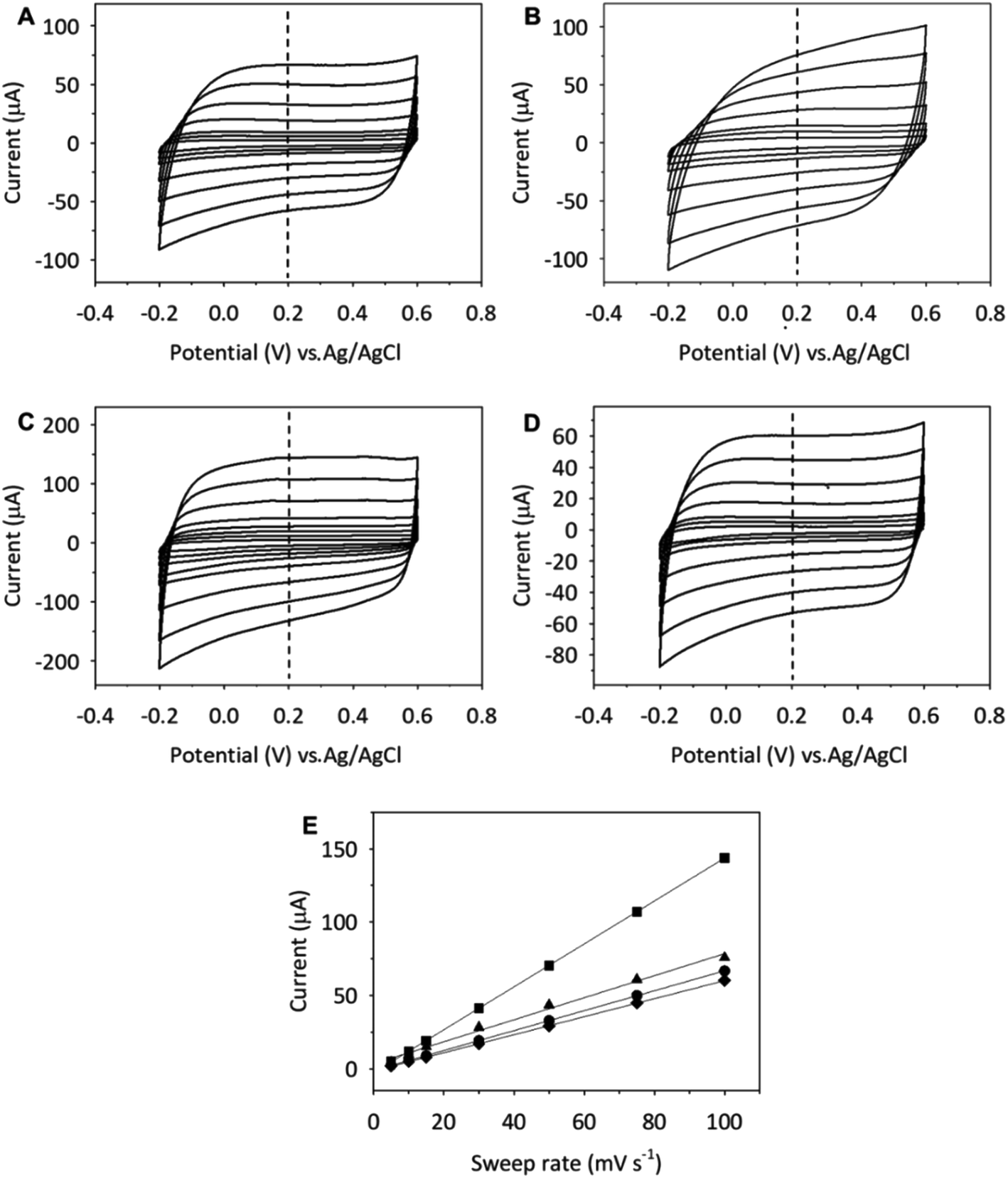 | ||
| Fig. 5 CVs of SWCNH in different supporting electrolytes (0.1 M): (A) NaCl, (B) Na2SO4, (C) H2SO4, and (D) (Et)4NCl at different sweep rates: 5, 10, 15, 30, 50, 75, and 100 mV s−1. (E) Dependence of the current of SWCNH at 0.2 V (vs. Ag/AgCl) for (●) NaCl, (▲) Na2SO4, (■) H2SO4, and (♦) (Et)4NCl. | ||
The CV curves were obtained for each nanomaterial at 5, 10, 15, 30, 50, 75 and 100 mV s−1, showing an increase in the current peak with increasing v. A linear relationship between the capacitive current (I) and the sweep rate (v) is observed for all forms of CNs (Fig. 5 and S7†). In this case, the capacitive current is expressed by the following equation:
| I = Csvm | (2) |
Given the existence of a linear correlation between the current peak and the scan rate, the Cs can be calculated from the slope of the linear fit using eqn (2).95 The Cs values are summarized in Table S3,† which compares these values obtained by both methods (eqn (1) and (2)). The values of Cs obtained by integration of the I–E curves are slightly higher than those calculated from the slope of the I–v plots. Generally, the Cs of the SWCNH, SWCNH-ox-1.5 and r-SWCNH-ox-1.5 electrodes calculated by these two methods in the presence of the inorganic electrolytes are highly similar.
Frequently, the presence of functional groups on the CN's surface can contribute to capacitor instability of these electrodes.92 Therefore, we checked the electrochemical stability of the oxidized SWCNH electrodes in two supporting electrolytes, an inert and an acidic solution. Fig. 6 shows the CVs for up to 1000 cycles of the SWCNH-ox-1.5 electrode. As shown in Fig. 6, the Cs of the SWCNH-ox-1.5 electrode showed a slightly different electrochemical stability in the two supporting electrolytes. The Cs decreases rapidly during the first 100 cycles but remained nearly constant thereafter up to 1000 cycles in H2SO4 solution.
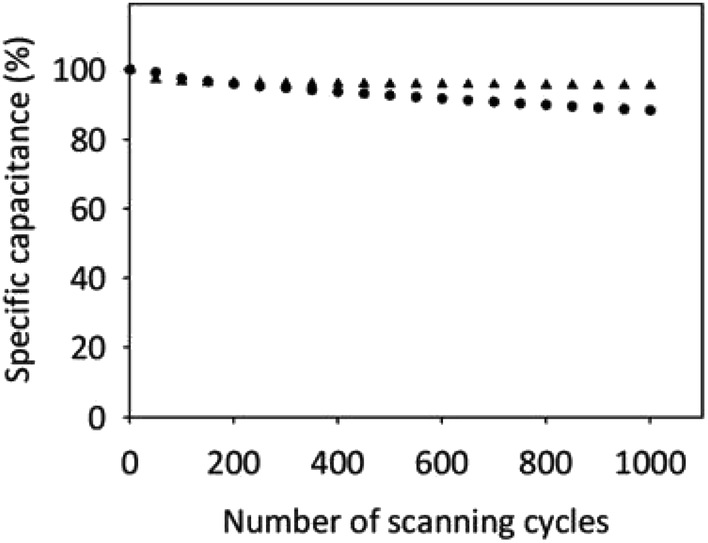 | ||
| Fig. 6 Cyclic life test of SWCNH-ox-1.5 in 0.1 mol dm−3 NaCl (●) and H2SO4 (▲) measured by CVs at +0.2 V (vs. Ag/AgCl). The sweep rate was 50 mV s−1. | ||
In the case of NaCl solution, the Cs for this electrode decreases gradually throughout the cycling potential window. The value of Cs decreased by approximately 12% (from 230 to 202 F g−1) for NaCl solution and approximately 5% (from 233 to 221 F g−1) for H2SO4 solution after 1000 cycles. The SWCNH-ox-1.5 electrode in H2SO4 solution was retained approximately 95% of the initial capacitive current after 1000 cycles. The study confirmed long-term cycling stability of the SWCNH-ox-1.5 electrode in acidic solution.
3 Experimental
3.1. Modification of SWCNHs
An amount of 0.5 g of SWCNH (NEC, Minato, Tokyo, Japan) was oxidized using 250 mL of 65% HNO3 (Chempur, Piekary Śląskie, Poland) at 80 °C for different times (0.5, 1, 1.5 or 3 h). The samples were labeled as SWCNH for the initial material and SWCNH-ox-y, where ‘ox’ denotes the oxidation and ‘y’ oxidation time, respectively. After the oxidation reaction, the mixture was cooled and diluted to 1000 mL, filtered under vacuum on a 0.45 μm nylon membrane (Whatman) and washed with hot distilled water until the conductivity of the mixture was no larger than 10 μS. The obtained material was collected, dispersed in distilled water, centrifuged and freeze-dried. The SWCNH-ox-1.5 was reduced. To the reduction reaction, 50 mg of SWCNH-ox-1.5 was added to 100 mL of 5% hydrazine aqueous solution. The mixture was refluxed for two hours, and in the last step, the mixture was centrifuged, washed with water until low conductivity was reached, and freeze-dried. The material was labeled as r-SWCNH-ox-1.5.3.2. Methods
Nitrogen adsorption–desorption isotherms (77 K) were measured using an ASAP 2010 adsorption apparatus (Micromeritics, Norcross, USA). We computed the BET apparent surface areas (SBET) from the classical Brunauer–Emmett–Teller57 adsorption isotherm equation. Porosity parameters were calculated from of the kernel of local N2 isotherms (77 K) simulated in infinitely long structureless graphene cylinders and the in-house grand canonical Monte Carlo (GCMC) simulation code. We used a single-site (12, 6) Lennard-Jones potential model (σff = 0.3615 nm and εff/kB = 101.5 K) for computing N2–N2 interactions. The cylindrical sections of SWNHs were modeled by a structureless graphene layers. The solid-fluid potential was computed from:
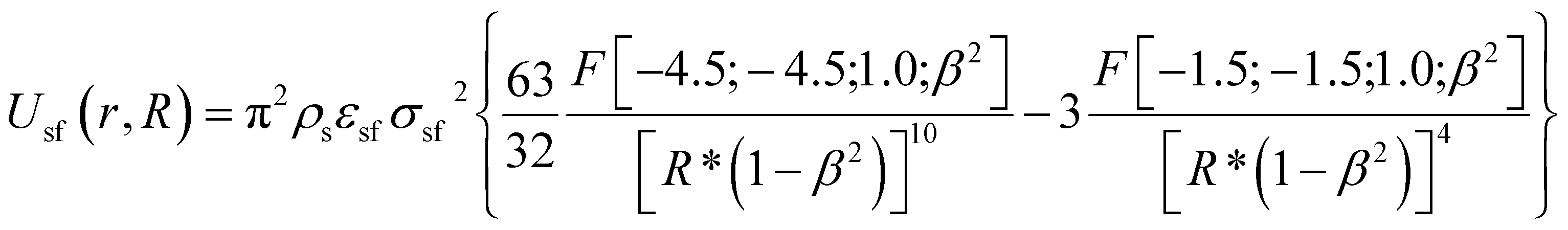 | (3) |
For GCMC simulations of N2 adsorption in graphene cylinders, we used a periodic unit with a length of 10σff. The periodic boundary conditions and the minimum image convention was used for computing N2–N2 interactions in the longitudinal direction. Interactions between N2 molecules were truncated at 5σff without applying the long-range correction. The relationship between pressure and chemical potential for N2 at 77 K was computed from canonical Monte Carlo simulations and the Widom particle insertion method.96 At each point along the N2 adsorption isotherm, the GCMC simulations were equilibrated using 4 × 107 Monte Carlo steps. The ensemble averages were computed from additional 2 × 107 Monte Carlo steps. In all GCMC simulations, we used an equal probability of 1/3 for the insertion, deletion and displacement perturbation step.
The absolute value of N2 adsorption per surface area of graphene wall, Scyl, was computed from:
| nabs(D;h) = N(h)/Scyl | (4) |
The pore size distribution function f(D) was computed from the general adsorption integral equation:
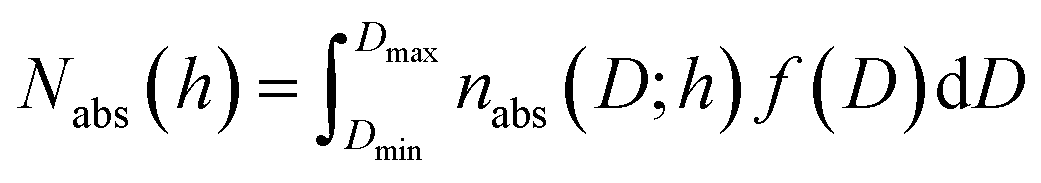 | (5) |
The experimental value of the absolute N2 adsorption inside SWNHs was estimated from the following equation:
| Nabs(h) = Nopened(h) − Nclosed(h) | (6) |
For X-ray photoelectron spectroscopy (XPS), an UHV analytical system (Prevac, Rogow, Poland) equipped with a nonmonochromatic Al K (1486.7 eV) radiation source (VG Scienta SAX 100) and a monochromator (VG Scienta XM 780) was used. The Al K source was operated at 12 kV and 30 mA. A low-resolution survey scan (0–1200 eV) was performed for all samples. The pristine SWCNH and SWCNH-ox powder samples were placed on naturally oxidized Si wafers (ITME, Warsaw, Poland) that were precoated with approximately 50 nm of Au (purity 99.999%) using molecular beam epitaxy (MBE, Prevac). The measurements were performed at room temperature. The collected data were described using Casa XPS software (Casa Software Ltd., Teignmouth, USA).
The materials were also characterized using high-resolution transmission electron microscopy (HR-TEM). The images were collected using a transmission electron microscope F20X-TWIN (FEI-Tecnai, Hillsboro, USA) operated at 200 kV. The drop of sample dispersed in methanol (99.8% p.a.) was placed on a Cu grid coated with an ultrathin amorphous carbon film and dried under ambient conditions.
The ζ-potential (25 °C, Particulate Systems, NanoPlus HD, Micromeritics, Norcross, USA) was measured using solutions with a concentration of carbon materials of 0.01 mg mL−1 and an ionic strength of 0.1 M. Each solution was prepared using HCl and KCl (analytical grade, Chempur, Piekary Śląskie, Poland) to adjust the ionic strength. After 24 h, the pH value was measured again, and the ζ-potential was measured.
The infrared (IR) spectra were recorded using a NICOLET IN10 MX infrared microscope (Thermo Scientific, Waltham, Massachusetts, USA). The microscope was operated primarily in reflectance mode, and the mercury–cadmium–telluride (MCT) detector was cooled with liquid N2. A zinc selenide (ZnSe) crystal (θ = 45°, np = 2.4) was used in the FTIR-HATR (Fourier transform infrared spectroscopy-horizontal attenuated total reflectance) technique. The solution of the analyzed samples was placed in HATR cells, and a beam of infrared radiation entering a crystal underwent multiple internal reflections. The resultant radiation was measured and plotted as a function of wavelength. The spectral resolution was 4 cm−1, and 256 scans were averaged to obtain a single spectrum.
Raman spectra were recorded using a SENTERRA (Bruker Optics, Billerica, MA, USA) micro-Raman system. Spectra were investigated in the range of 60–4500 cm−1. A green laser (532 nm) was used as the excitation source, and the beam was focused on samples through a 20× microscope objective. Spectra were collected with laser powers at 2 mW to avoid sample heating and damage. Each sample was measured at least three times with a total integration time of 60 s (2 × 30 s). The obtained spectra were averaged and normalized to the G band intensity.
Voltammetric experiments were performed using an AUTOLAB (Utrecht, The Netherlands) computerized electrochemistry system equipped with a PGSTAT 12 potentiostat. The AUTOLAB system was controlled with Nova 2.0 software from the same manufacturer. A three-electrode cell was selected. A glassy carbon disk electrode (GCE, Bioanalytical Systems Inc. West Lafayette, USA) with a diameter of 2 mm was used as the working electrode. Before each experiment, the surface of the GCE electrode was polished using extra fine carborundum paper (Buehler) followed by 0.3 μm Al2O3 and 0.25 μm diamond polishing compounds (Metadi II, Buehler). The GCE electrode was sonicated in water to remove traces of Al2O3 from the metal surface, washed with water, and dried. The film on the GCE was prepared as follows. An amount of 4 mg of active material (SWCNH, oxidized or reduced form) was suspended in ethanol containing conductive carbon paint (v/v; 10:1) and sonicated for 15 minutes. An amount of 10 μL of the SWCNH dispersion was transferred to the GCE surface, and the solvent was evaporated under an Ar atmosphere. The counter electrode was a platinum flag with an area of approximately 0.5 cm2. A silver wire immersed in 0.1 mol L−1 AgCl and separated from the working electrode by a ceramic tip (Bioanalytical Systems Inc. West Lafayette, USA) served as the reference electrode. All experiments were performed in water purified through a Millipore apparatus. Oxygen was removed from the solution by purging with argon. The studied solutions were not buffered.
4 Conclusions
Nitric acid treatment for oxidation of SWCNHs leading to hole creation in their structure was used for the first time. The obtained materials were characterized using several physicochemical methods and experimental and theoretical studies. The N2 adsorption technique, which was supported by GCMC simulations, led to the porosity parameters. XPS, HR-TEM, FTIR and Raman spectroscopy and ζ-potential proved the successful oxidation of the SWCNH structures and their chemical composition. Electrochemical measurements were performed, supplying the correlation between the structural properties of oxidized CNs and the specific capacitance of the SWCNH electrodes. The most important conclusion from our study is that use of simple functionalization of CNs under controlled conditions leads to an almost 3-fold increase of their specific capacitance. Thus, nitric acid treatment is a simple method for enhancement of the specific capacitance of SWCNHs and facilitates application of this material for supercapacitor electrodes. Additionally, the resulting SWCNH electrode has excellent electrochemical stability, indicating the possibility of potential future applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Science Center (NSC), Poland for financial support via grant number OPUS 13 UMO-2017/25/B/ST5/00975.References
- Y. Liu, H. Zhang, P. K. Behara, X. Wang, D. Zhu, S. Ding, S. P. Ganesh, M. Dupuis, G. Wu and M. T. Swihart, ACS Appl. Mater. Interfaces, 2018, 10, 42417–42426 CrossRef CAS.
- A. B. Papandrew, R. A. Elgammal, M. Tian, W. D. Tennyson, C. M. Rouleau, A. A. Puretzky, G. M. Veith, D. B. Geohegan and T. A. Zawodzinski, J. Power Sources, 2017, 337, 145–151 CrossRef CAS.
- D. Mirabile Gattia, M. V. Antisari, L. Giorgi, R. Marazzi, E. Piscopiello, A. Montone, S. Bellitto, S. Licoccia and E. Traversa, J. Power Sources, 2009, 194, 243–251 CrossRef CAS.
- G. G. Wallace, J. Chen, D. Li, S. E. Moulton and J. M. Razal, J. Mater. Chem., 2010, 20, 3553 RSC.
- S. L. Candelaria, Y. Shao, W. Zhou, X. Li, J. Xiao, J.-G. Zhang, Y. Wang, J. Liu, J. Li and G. Cao, Nano Energy, 2012, 1, 195–220 CrossRef CAS.
- W. Xu, Z. Wang, Z. Guo, Y. Liu, N. Zhou, B. Niu, Z. Shi and H. Zhang, J. Power Sources, 2013, 232, 193–198 CrossRef CAS.
- S. He, H. Hou and W. Chen, J. Power Sources, 2015, 280, 678–686 CrossRef CAS.
- Z. Zhou and X.-F. Wu, J. Power Sources, 2014, 262, 44–49 CrossRef CAS.
- X. Li and B. Wei, Nano Energy, 2013, 2, 159–173 CrossRef CAS.
- E. Frackowiak and F. Béguin, Carbon, 2001, 39, 937–950 CrossRef CAS.
- S. R. S. Prabaharan, R. Vimala and Z. Zainal, J. Power Sources, 2006, 161, 730–736 CrossRef CAS.
- S. He and W. Chen, J. Power Sources, 2014, 262, 391–400 CrossRef CAS.
- E. Frackowiak and F. Beguin, Carbon, 2002, 40, 1775–1787 CrossRef CAS.
- G. Centi and S. Perathoner, Catal. Today, 2010, 150, 151–162 CrossRef CAS.
- Z. Zhao, S. Liu, J. Zhu, J. Xu, L. Li, Z. Huang, C. Zhang and T. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 19871–19880 CrossRef CAS.
- Y. Yang, S. Jin, Z. Zhang, Z. Du, H. Liu, J. Yang, H. Xu and H. Ji, ACS Appl. Mater. Interfaces, 2017, 9, 14180–14186 CrossRef CAS.
- J. Cao, C. J. Jafta, J. Gong, Q. Ran, X. Lin, R. Félix, R. G. Wilks, M. Bär, J. Yuan, M. Ballauff and Y. Lu, ACS Appl. Mater. Interfaces, 2016, 8, 29628–29636 CrossRef CAS.
- Y. S. Kim, J. Y. Oh, J. H. Kim, M. H. Shin, Y. C. Jeong, S. J. Sung, J. Park, S. J. Yang and C. R. Park, ACS Appl. Mater. Interfaces, 2017, 9, 17552–17564 CrossRef CAS.
- K. H. An, W. S. Kim, Y. S. Park, Y. C. Choi, S. M. Lee, D. C. Chung, D. J. Bae, S. C. Lim and Y. H. Lee, Adv. Mater., 2001, 13, 497–500 CrossRef CAS.
- R. Ryoo, S. H. Joo, M. Kruk and M. Jaroniec, Adv. Mater., 2001, 13, 677–681 CrossRef CAS.
- M. Kruk, K. M. Kohlhaas, B. Dufour, E. B. Celer, M. Jaroniec, K. Matyjaszewski, R. S. Ruoff and T. Kowalewski, Microporous Mesoporous Mater., 2007, 102, 178–187 CrossRef CAS.
- O. Barbieri, M. Hahn, A. Herzog and R. Kötz, Carbon, 2005, 43, 1303–1310 CrossRef CAS.
- V. V. N. Obreja, Physica E Low Dimens. Syst. Nanostruct., 2008, 40, 2596–2605 CrossRef CAS.
- M. E. Plonska-Brzezinska, A. Lapinski, A. Z. Wilczewska, A. T. Dubis, A. Villalta-Cerdas, K. Winkler and L. Echegoyen, Carbon, 2011, 49, 5079–5089 CrossRef CAS.
- T. Azami, D. Kasuya, R. Yuge, M. Yudasaka, S. Iijima, T. Yoshitake and Y. Kubo, J. Phys. Chem. C, 2008, 112, 1330–1334 CrossRef CAS.
- R. Yuge, T. Manako, K. Nakahara, M. Yasui, S. Iwasa and T. Yoshitake, Carbon, 2012, 50, 5569–5573 CrossRef CAS.
- Y. Nan, B. Li, X. Song and N. Sano, Carbon, 2019, 142, 150–155 CrossRef CAS.
- E. Bekyarova, K. Kaneko, M. Yudasaka, D. Kasuya, S. Iijima, A. Huidobro and F. Rodriguez-Reinoso, J. Phys. Chem. B, 2003, 107, 4479–4484 CrossRef CAS.
- C.-M. Yang, Y.-J. Kim, J. Miyawaki, Y. A. Kim, M. Yudasaka, S. Iijima and K. Kaneko, J. Phys. Chem. C, 2015, 119, 2935–2940 CrossRef CAS.
- W. Wu, Y. Zhao, C. Wu and L. Guan, RSC Adv., 2014, 4, 28636–28639 RSC.
- C.-M. Yang, Y.-J. Kim, M. Endo, H. Kanoh, M. Yudasaka, S. Iijima and K. Kaneko, J. Am. Chem. Soc., 2007, 129, 20–21 CrossRef CAS.
- E. Bekyarova, K. Kaneko, D. Kasuya, K. Murata, M. Yudasaka and S. Iijima, Langmuir, 2002, 18, 4138–4141 CrossRef CAS.
- S. M. Unni, R. Illathvalappil, S. N. Bhange, H. Puthenpediakkal and S. Kurungot, ACS Appl. Mater. Interfaces, 2015, 7, 24256–24264 CrossRef CAS.
- S. M. Unni, S. Ramadas, R. Illathvalappil, S. N. Bhange and S. Kurungot, J. Mater. Chem. A, 2015, 3, 4361–4367 RSC.
- E. Sánchez-Tirado, A. González-Cortés, M. Yudasaka, S. Iijima, F. Langa, P. Yáñez-Sedeño and J. M. Pingarrón, J. Electroanal. Chem., 2017, 793, 197–202 CrossRef.
- M. Zhang, M. Yudasaka, K. Ajima, J. Miyawaki and S. Iijima, ACS Nano, 2007, 1, 265–272 CrossRef CAS.
- C.-M. Yang, H. Noguchi, K. Murata, M. Yudasaka, A. Hashimoto, S. Iijima and K. Kaneko, Adv. Mater., 2005, 17, 866–870 CrossRef CAS.
- E. Aryee, A. K. Dalai and J. Adjaye, Top. Catal., 2014, 57, 796–805 CrossRef CAS.
- F. Agresti, S. Barison, A. Famengo, C. Pagura, L. Fedele, S. Rossi, S. Bobbo, M. Rancan and M. Fabrizio, J. Colloid Interface Sci., 2018, 514, 528–533 CrossRef CAS.
- S. Zhu, X.-E. Zhao, J. You, G. Xu and H. Wang, Analyst, 2015, 140, 6398–6403 RSC.
- M. V. Bracamonte, M. Melchionna, A. Giuliani, L. Nasi, C. Tavagnacco, M. Prato and P. Fornasiero, Sens. Actuators, B, 2017, 239, 923–932 CrossRef CAS.
- F. Valentini, E. Ciambella, A. Boaretto, G. Rizzitelli, M. Carbone, V. Conte, F. Cataldo, V. Russo, C. S. Casari, D. F. Chillura-Martino, E. Caponetti, M. Bonchio, F. Giacalone, Z. Syrgiannis and M. Prato, Electroanalysis, 2016, 28, 2489–2499 CrossRef CAS.
- F. Valentini, E. Ciambella, V. Conte, L. Sabatini, N. Ditaranto, F. Cataldo, G. Palleschi, M. Bonchio, F. Giacalone, Z. Syrgiannis and M. Prato, Biosens. Bioelectron., 2014, 59, 94–98 CrossRef CAS.
- H. J. Jung, Y.-J. Kim, J. H. Han, M. Yudasaka, S. Iijima, H. Kanoh, Y. A. Kim, K. Kaneko and C.-M. Yang, J. Phys. Chem. C, 2013, 117, 25877–25883 CrossRef CAS.
- Y. Zhao, J. Li, Y. Ding and L. Guan, Chem. Commun., 2011, 47, 7416–7418 RSC.
- Y. Zhao, J. Li, Y. Ding and L. Guan, RSC Adv., 2011, 1, 852–856 RSC.
- H. Lai, J. Li, Z. Chen and Z. Huang, ACS Appl. Mater. Interfaces, 2012, 4, 2325–2328 CrossRef CAS.
- R. Yuge, N. Tamura, T. Manako, K. Nakano and K. Nakahara, J. Power Sources, 2014, 266, 471–474 CrossRef CAS.
- Q.-F. Lü, S. Wang, J. Zhou, F.-F. Duan, H. Yang and R. Liu, ChemistrySelect, 2019, 4, 7270–7277 CrossRef.
- H. Hwang, C. H. Kim, J.-H. Wee, J. H. Han and C.-M. Yang, Appl. Surf. Sci., 2019, 489, 708–716 CrossRef CAS.
- L. Lv, C. Cui, C. Liang, W. Quan, S. Wang and Z. Guo, Food Control, 2016, 60, 296–301 CrossRef CAS.
- G. Zhang, P. He, W. Feng, S. Ding, J. Chen, L. Li, H. He, S. Zhang and F. Dong, J. Electroanal. Chem., 2016, 760, 24–31 CrossRef CAS.
- L. Yang, X. Ran, L. Cai, Y. Li, H. Zhao and C.-P. Li, Biosens. Bioelectron., 2016, 83, 347–352 CrossRef CAS.
- E. Antolini, Appl. Catal., B, 2009, 88, 1–24 CrossRef CAS.
- L. Zhang, N. Zheng, A. Gao, C. Zhu, Z. Wang, Y. Wang, Z. Shi and Y. Liu, J. Power Sources, 2012, 220, 449–454 CrossRef CAS.
- C. Zhu, D. Liu, Z. Chen, L. Li and T. You, J. Colloid Interface Sci., 2018, 511, 77–83 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- K. Murata, K. Kaneko, F. Kokai, K. Takahashi, M. Yudasaka and S. Iijima, Chem. Phys. Lett., 2000, 331, 14–20 CrossRef CAS.
- D. Wei, Y. Liu, Y. Wang, H. Zhang, L. Huang and G. Yu, Nano Lett., 2009, 9, 1752–1758 CrossRef CAS.
- A. Dimiev, D. V. Kosynkin, A. Sinitskii, A. Slesarev, Z. Sun and J. M. Tour, Science, 2011, 331, 1168–1172 CrossRef CAS.
- R. Blume, P. R. Kidambi, B. C. Bayer, R. S. Weatherup, Z.-J. Wang, G. Weinberg, M.-G. Willinger, M. Greiner, S. Hofmann, A. Knop-Gericke and R. Schlögl, Phys. Chem. Chem. Phys., 2014, 16, 25989–26003 RSC.
- M. Koinuma, H. Tateishi, K. Hatakeyama, S. Miyamoto, C. Ogata, A. Funatsu, T. Taniguchi and Y. Matsumoto, Chem. Lett., 2013, 42, 924–926 CrossRef CAS.
- Y.-C. Chiang, Y.-J. Chen and C.-Y. Wu, Materials, 2017, 10, 1296 CrossRef.
- M. K. Rabchinskii, S. A. Ryzhkov, D. A. Kirilenko, N. V. Ulin, M. V. Baidakova, V. V. Shnitov, S. I. Pavlov, R. G. Chumakov, D. Yu. Stolyarova, N. A. Besedina, A. V. Shvidchenko, D. V. Potorochin, F. Roth, D. A. Smirnov, M. V. Gudkov, M. Brzhezinskaya, O. I. Lebedev, V. P. Melnikov and P. N. Brunkov, Sci. Rep., 2020, 10, 6902–6914 CrossRef CAS.
- A. Barinov, O. B. Malcioǧlu, S. Fabris, T. Sun, L. Gregoratti, M. Dalmiglio and M. Kiskinova, J. Phys. Chem. C, 2009, 113, 9009–9013 CrossRef CAS.
- A. Fujimoto, Y. Yamada, M. Koinuma and S. Sato, Anal. Chem., 2016, 88, 6110–6114 CrossRef CAS.
- B. Lesiak, L. Kövér, J. Tóth, J. Zemek, P. Jiricek, A. Kromka and N. Rangam, Appl. Surf. Sci., 2018, 452, 223–231 CrossRef CAS.
- C. Petit, M. Seredych and T. J. Bandosz, J. Mater. Chem., 2009, 19, 9176 RSC.
- E. Desimoni, G. I. Casella, A. M. Salvi, T. R. I. Cataldi and A. Morone, Carbon, 1992, 30, 527–531 CrossRef CAS.
- N. K. Gupta, B. Peng, G. L. Haller, E. E. Ember and J. A. Lercher, ACS Catal., 2016, 6, 5843–5855 CrossRef CAS.
- R. Arrigo, M. E. Schuster, Z. Xie, Y. Yi, G. Wowsnick, L. L. Sun, K. E. Hermann, M. Friedrich, P. Kast, M. Hävecker, A. Knop-Gericke and R. Schlögl, ACS Catal., 2015, 5, 2740–2753 CrossRef CAS.
- J. Zhang, H. Zou, Q. Qing, Y. Yang, Q. Li, Z. Liu, X. Guo and Z. Du, J. Phys. Chem. B, 2003, 107, 3712–3718 CrossRef CAS.
- J. Kastner, T. Pichler, H. Kuzmany, S. Curran, W. Blau, D. N. Weldon, M. Delamesiere, S. Draper and H. Zandbergen, Chem. Phys. Lett., 1994, 221, 53–58 CrossRef CAS.
- T. Petit and L. Puskar, Diamond Relat. Mater., 2018, 89, 52–66 CrossRef CAS.
- T. Jiang and K. Xu, Carbon, 1995, 33, 1663–1671 CrossRef CAS.
- R. Sappok and H. P. Boehm, Carbon, 1968, 6, 283–295 CrossRef CAS.
- M. X. Pulikkathara, O. V. Kuznetsov and V. N. Khabashesku, Chem. Mater., 2008, 20, 2685–2695 CrossRef CAS.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- R. Al-Jishi and G. Dresselhaus, Phys. Rev. B, 1982, 26, 4514–4522 CrossRef CAS.
- R. J. Nemanich and S. A. Solin, Phys. Rev. B, 1979, 20, 392–401 CrossRef CAS.
- R. Tsu, J. H. González and I. C. Hernández, Solid State Commun., 1978, 27, 507–510 CrossRef CAS.
- V. Mochalin, S. Osswald and Y. Gogotsi, Chem. Mater., 2009, 21, 273–279 CrossRef CAS.
- E. D. Obraztsova, M. Fujii, S. Hayashi, V. L. Kuznetsov, Yu. V. Butenko and A. L. Chuvilin, Carbon, 1998, 36, 821–826 CrossRef CAS.
- C. Thomsen and S. Reich, Phys. Rev. Lett., 2000, 85, 5214–5217 CrossRef CAS.
- L. G. Cançado, K. Takai, T. Enoki, M. Endo, Y. A. Kim, H. Mizusaki, A. Jorio, L. N. Coelho, R. Magalhães-Paniago and M. A. Pimenta, Appl. Phys. Lett., 2006, 88, 163106 CrossRef.
- D. Torres, J. L. Pinilla, R. Moliner and I. Suelves, Carbon, 2015, 81, 405–417 CrossRef CAS.
- M. E. Plonska-Brzezinska, A. T. Dubis, A. Lapinski, A. Villalta-Cerdas and L. Echegoyen, ChemPhysChem, 2011, 12, 2659–2668 CrossRef CAS.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51–87 CrossRef CAS.
- M. A. McArthur, N. Hordy, S. Coulombe and S. Omanovic, Electrochim. Acta, 2015, 162, 245–253 CrossRef CAS.
- D. Torres, S. Pérez-Rodríguez, D. Sebastián, J. L. Pinilla, M. J. Lázaro and I. Suelves, Appl. Surf. Sci., 2020, 509, 144774 CrossRef CAS.
- S. K. Park, Q. Mahmood and H. S. Park, Nanoscale, 2013, 5, 12304 RSC.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS.
- C. Portet, G. Yushin and Y. Gogotsi, Carbon, 2007, 45, 2511–2518 CrossRef CAS.
- K. D. Collins and M. W. Washabaugh, Q. Rev. Biophys., 1985, 18, 323–422 CrossRef CAS.
- M. Nasibi, M. A. Golozar and G. Rashed, J. Power Sources, 2012, 206, 108–110 CrossRef CAS.
- B. Widom, J. Chem. Phys., 1963, 39, 2808–2812 CrossRef CAS.
- P. Kowalczyk, A. P. Terzyk, P. A. Gauden, R. Leboda, E. Szmechtig-Gauden, G. Rychlicki, Z. Ryu and H. Rong, Carbon, 2003, 41, 1113–1125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Selected GCMC simulated local N2 (77 K) adsorption isotherms and the fit to experimental data, high-resolution C 1s XPS and N 1s spectra, deconvolution of Raman spectra, CVs of SWCNH-ox-1.5 in 0.1 M different supporting electrolytes, specific capacitance of SWCNH, SWCNH-ox and r-SWCNH-ox in aqueous solutions of different supporting electrolytes. See DOI: 10.1039/d0ra07748h |
| This journal is © The Royal Society of Chemistry 2020 |