DOI:
10.1039/D0RA07587F
(Paper)
RSC Adv., 2020,
10, 40068-40083
Stimuli-responsive gelators from carbamoyl sugar derivatives and their responses to metal ions and tetrabutylammonium salts†
Received
4th September 2020
, Accepted 7th October 2020
First published on 4th November 2020
Abstract
Carbohydrate-based low molecular weight gelators (LMWGs) are interesting compounds with a variety of applications. In this research, a library of nineteen carbamate derivatives of N-acetyl-D-glucosamine were synthesized and characterized, and several derivatives were found to be effective LMWGs. They formed gels in pump oils as well as mixtures of water with ethanol or water with DMSO. The structures of the carbamoyl chains played an important role in the gelation properties, short chain aliphatic derivatives and phenyl carbamates formed gels in more solvents than certain aromatic and dimeric carbamates. The phenyl carbamate gelator was also selected for the encapsulation of naproxen sodium, and the drug slowly diffused from the gel to the aqueous phase as indicated by UV-vis spectroscopy. In addition, we also found that the p-methoxyl benzyl carbamate derivative showed interesting stimuli-responsive gelation properties in the presence of metal salts and tetrabutylammonium salts. The gels were characterized using optical microscopy, scanning electron microscopy, rheology and other methods. The self-assembling mechanisms of the gelators were studied using 1H NMR spectroscopy. The preparation, characterization, and molecular assembling properties of these compounds are reported. The results obtained from this study are useful for the design of other LMWGs and the sugar derivatives can be explored for different biological applications. The formation of spontaneous ionic gels can be applicable for a plethora of applications including catalysis and environmental remediation.
Introduction
Supramolecular gelation through low molecular weight gelators (LMWGs) has gained much attention over recent years. Several classes of LMWGs have been reported, depending on the solvents used, these include both hydrogelators and organogelators.1–6 They have been explored for applications in many research fields including drug delivery and tissue engineering,7–9 as catalysts for organic reactions,10 as well as in environmental applications to remove pollutants.11,12 The structure-based approach can allow for the design and discovery of supramolecular gelators with desirable functions. For instance, stimuli-responsive gelators have been studied for their unique and controllable properties.13–18 Metallogels and ionogels are useful soft materials with a variety of applications ranging from anion sensing and catalysis to electrochemical applications.19–22 Ionogels formed by gelators in ionic liquids or salts have been investigated for their potential applications in dye sensitized solar cells. Anion binding and anion-responsive molecular gelators can function as host–guest receptors, making them useful for anion sensing.23–26 Gels responsive to anions and cations may have distinctive properties to function as sensors and may also be beneficial for catalysis.20,27–32
Carbohydrate derived low molecular weight gelators are intriguing compounds with many potential applications.5,33–35 Several organogelators and hydrogelators have been obtained by selectively functionalizing the different hydroxyl groups from common monosaccharides such as D-glucose and D-glucosamine.36 Among the functional groups that are effective for molecular gelators, amide, carbamate, and urea functions have been shown to be efficient in the formation of supramolecular gels.37–40 Some bis-arylcarbamate derivatives38 as well as carbamoyl functionalized glucose and glucosamine derivatives were reported as LMWGs.39,40 Amides based gelators have been used as anion binding agents and they played a significant role in molecular recognition.19 Several amide based derivatives have shown susceptibility towards anion stimuli.41 A glucose-based gelator formed gels in tetramethyl ammonium bromide electrolyte solution, the resulting ionogels showed thermal reversibility and enhanced conductivity.42 Two glycolipid derived gelators were shown to be fluoride responsive.43 Organic carbamate or urethane derivatives have played important roles in the design and study of low molecular weight gelators (LMWGs).38,40,44–46 The carbamate functional group contains one hydrogen bonding donor (NH) and two hydrogen bonding acceptors (N and O), therefore they can form intermolecular hydrogen bonding networks among the gelators and also with solvent molecules. In comparison to amides, carbamates contain an extra hydrogen bond acceptor which may play an important role in molecular self-assemblies and the interaction with other atoms.
During the course of the functionalization of monosaccharide derivatives to produce effective supramolecular assemblies and advanced functional biomaterials; certain correlations between the structures of the compounds and their gelation properties have been established.40,47–50 As shown in Fig. 1, the various C-2 acylated derivatives of 4,6-O-benzylidene acetal protected D-glucosamine derivatives have proven to be effective low molecular weight gelators (LMWGs). These include the D-glucose derived carbamate 1, D-glucosamine derived carbamate 2,40 and D-glucosamine derived amide 3. Recently, we demonstrated that the 3-O-esters of glucosamine derivatives 4 were also effective gelators.47 These compounds were typically synthesized in a few steps from N-acetyl-D-glucosamine (NAG).49 Based on the interesting gelation results from the 3-ester derivatives, we envisioned that the isosteric carbamate derivatives at the 3-OH position may result in another class of effective gelators with the general structure 5. Previously, we have demonstrated that the replacement of an ester with a carbamate functional group by introducing the NH hydrogen bonding donor enhanced gelation properties of the sugar derivatives.40 The new carbamate derivatives 5 have several desirable features that will lead to a new class of organogelators. It contains an acetamide functional group at the C-2 position, which can also contribute to intermolecular hydrogen bonding. The carbamate derivatives are more stable at basic conditions in comparison to the corresponding ester analogs, therefore, for certain applications where stable gelators are desirable, the carbamates would be suitable. In this research, in order to probe the structural influence of sugar derivatives towards molecular self-assemblies and discover novel functional supramolecular gelators, we introduced carbamoyl functional groups to the 3-OH position of a 4,6-O-benzylidene acetal protected N-acetyl-D-glucosamine derivative. A series of carbamate derivatives with the general structures 5 and the dimeric carbamates 6 were synthesized and analyzed.
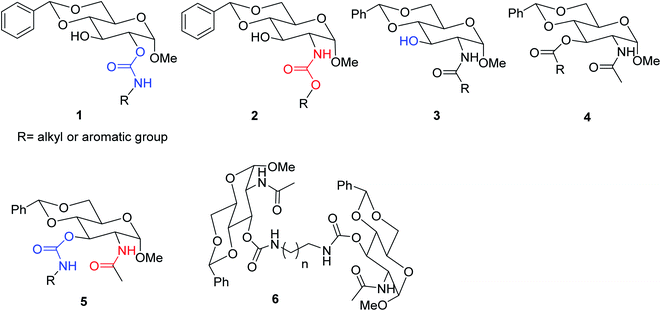 |
| | Fig. 1 Structures of several classes of sugar derivatives that can function as effective LMWGs (1–4) and the compounds synthesized in this study (5–6). | |
Results and discussions
As shown in Scheme 1, carbamate derivatives 9–27 were synthesized and characterized. Two methods were used for the preparation of the compound libraries. Compounds 12–14, 16–17 and 20–23, were prepared by following the methods reported previously using commercially available isocyanates.51 The others, including compounds 9–11, 15, 18–19, and the bis-carbamates 24–27 were synthesized using the imidazole carbonate intermediate 8 by reacting with different amines. Carbonyl di-imidazole (CDI) has been used often for the formation of cyclic carbamates from vicinal amino alcohol or linear carbamate by stepwise acylation reactions. The sugar alcohol 7 was treated with CDI first to form the corresponding imidazole carboxylate 8 effectively, this intermediate reacted readily with primary amines to produce the desired carbamates. When using α, ω-diamines, four different di-carbamate derivatives were synthesized successfully as shown in Scheme 1.
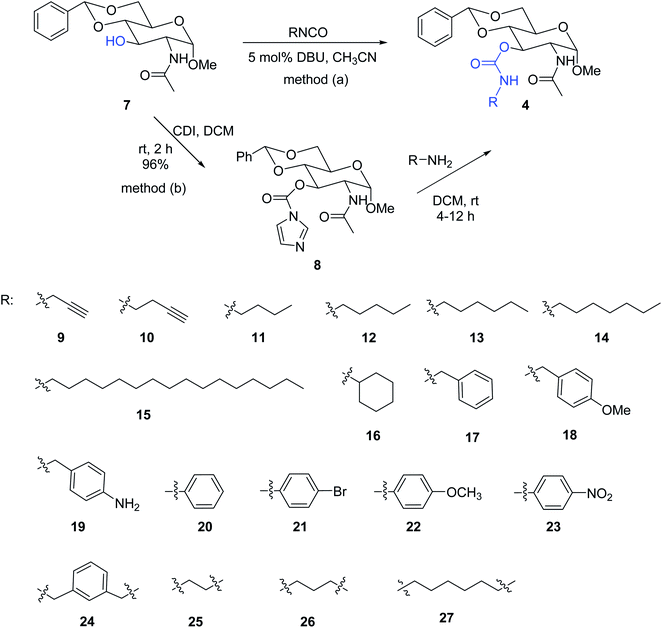 |
| | Scheme 1 Synthesis of monomeric 3-O-carbamate derivatives 9–23 and dimeric derivatives 24–27. | |
Gelation properties
After these compounds were synthesized and purified, their gelation properties were tested in a panel of organic solvents and aqueous solutions, the results are shown in Table 1. All compounds were insoluble in hexane, whilst most were insoluble in water except compounds 10 and 19, which formed partial gels at 20.0 and 6.7 mg mL−1, respectively. The short chain aliphatic carbamate derivatives showed similar gelation tendencies to the corresponding ester derivatives with the same alkyl chain. These compounds showed effective gelation properties for DMSO/water or ethanol/water mixtures. However, the aromatic derivatives were less effective gelators.
Table 1 Gelation properties of the carbamate derivativesa
| No. |
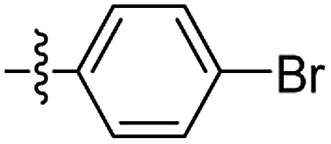
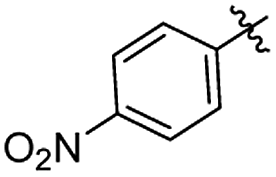
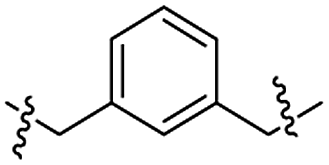
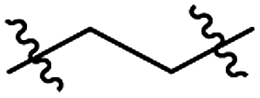
Structures of R: |
Pump oil |
Tol |
i-PrOH |
EtOH |
EG |
Glycerol |
H2O |
EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1) :H2O (1:1) |
EtOH:H2O (1:2) |
DMSO:H2O (1:1) |
DMSO:H2O (1:2) |
| G, gel at room temperature, the numbers are the corresponding minimum gelation concentrations (MGCs) in mg mL−1; GC, clear or transparent gel; GT, translucent gel; GO, opaque gel; GP, partial gel at 20 mg mL−1 unless otherwise specified; I, insoluble; P, crystallize or precipitate; S, soluble at ∼20 mg mL−1. |
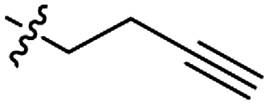
| 9 |
 |
P |
GP |
P |
P |
P |
GO 20.0 |
I |
P |
P |
P |
GO 20.0 |
| 10 |
 |
GT 20.0 |
S |
P |
P |
P |
P |
GP |
GP |
GP |
P |
GP |
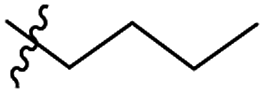
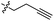
| 11 |
 |
GT 20.0 |
S |
P |
S |
GO 20.0 |
GT 20.0 |
P |
GP |
GP |
GO 20.0 |
GO 20.0 |
| 12 |
 |
GO 10.0 |
Go 20.0 |
S |
S |
P |
I |
I |
GT 20.0 |
GO 20.0 |
GO 10.0 |
P |
| 13 |
 |
GO 5.0 |
S |
S |
S |
P |
GO 20.0 |
I |
GO 10.0 |
GO 10.0 |
GO 20.0 |
GO 20.0 |
| 14 |
 |
GO 5.0 |
S |
S |
S |
GO 10.0 |
I |
I |
GO 10.0 |
GO 5.0 |
GO 10.0 |
I |
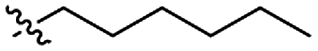
| 15 |
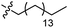 |
GT 5.0 |
P |
P |
P |
GP |
S |
P |
P |
P |
P |
I |
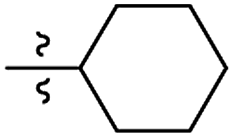
| 16 |
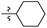 |
I |
S |
S |
S |
I |
S |
I |
GO 5.0 |
GO 3.3 |
GO 6.7 |
I |
| 17 |
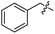 |
I |
GC 20.0 |
P |
P |
S |
P |
I |
GO 2.5 |
GO 2.5 |
GO 10.0 |
GO 10.0 |
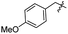
| 18 |
 |
GT 20.0 |
P |
GO 10.0 |
P |
GO 10.0 |
GC 6.7 |
I |
GO 4.0 |
GO 10.0 |
GO 2.5 |
GO 3.3 |
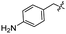
| 19 |
 |
P |
I |
S |
P |
S |
S |
GP 6.7 |
S |
GP |
S |
S |
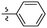
| 20 |
 |
I |
GO 2.8 |
GC 10.0 |
GC 20.0 |
P |
GO 20.0 |
I |
GO 3.3 |
GO 2.5 |
Go 4.0 |
GO 2.8 |
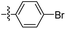
| 21 |
 |
I |
GC 20.0 |
P |
P |
P |
P |
I |
I |
I |
P |
P |
| 22 |
 |
I |
P |
P |
P |
P |
I |
I |
I |
P |
P |
P |
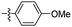
| 23 |
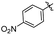 |
I |
P |
S |
S |
P |
I |
I |
P |
P |
P |
P |
| 24 |
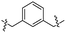 |
GT 20.0 |
I |
I |
GP |
P |
S |
I |
GP |
GP |
P |
GT 6.7 |
| 25 |
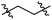 |
I |
I |
GP |
GP |
GT 20.0 |
I |
I |
GP |
P |
I |
GP |
| 26 |
 |
GT 10.0 |
GP |
P |
P |
P |
S |
I |
I |
I |
P |
P |
| 27 |
 |
I |
GP |
GP |
P |
P |
S |
I |
I |
I |
P |
I |
The linear aliphatic carbamate derivatives formed gels in pump oil, with the five carbon to seven carbon (compounds 12–14) being the most efficient; forming gels at lower MGCs in pump oil in comparison to the others. The hexyl carbamate derivative 13 was the most versatile gelator among the alkyl derivatives, forming gels in six of the selected solvents. The very long chain derivative 15 only formed gels in pump oil but not in the other selected solvents, the compound contains a very long chain fatty acyl group which increases the hydrophobic interactions and van der Waals forces significantly. The cyclohexyl derivative 16 formed gels in ethanol/water mixtures and DMSO/water (1:1) mixture, similarly to the short chain linear carbamates but at lower MGCs. For the aromatic derivatives, the benzyl derivative 17 showed good gelation properties in ethanol/water mixtures and DMSO/water mixtures, the p-methoxy benzyl carbamate 18 and the phenyl carbamate 20 were the most versatile gelators among all the compounds synthesized in this study, forming gels in eight different solvents. Interestingly, the substituted phenyl derivatives were not effective gelators as most of them were insoluble in the tested solvents. Several dimeric bis-polar carbamate derivatives were also synthesized but these compounds did not form gels in most of the tested solvents, except that compounds 24 and 26 formed gels in pump oil and 24 formed a gel in DMSO/H2O (v/v 1:2). The dimeric carbamates with two opposing sugar headgroups have intense intermolecular interactions which result in stronger packing therefore becoming insoluble or forming a precipitate.
Many carbamate derivatives formed stable gels in the DMSO and water mixture although not in pure water. To find out the minimum amount of DMSO required for gelation in aqueous solutions, the gelation properties of compounds 18 and 20 were tested in different ratios of DMSO and water, the results are shown in Table 2. These two most versatile gelators formed gels at different ratios of DMSO with water from 1:1 up to 1:9. Most of the gels appeared opaque, while some were translucent or transparent. A few representative examples are included in Fig. 2.
Table 2 Gelation test in DMSO water mixture for compounds 18 and 20a
| Compound |
DMSO:H2O volume ratio |
| 1:0 |
1:1 |
1:2 |
1:3 |
1:4 |
1:5 |
1:6 |
1:7 |
1:8 |
1:9 |
| G, gel formed after heating; S, soluble; P, precipitation. GT, translucent gel, GO, opaque gel. The numbers following the letters are the concentrations in mg mL−1 of the solvent mixture. |
| 18 |
S |
GT 10.0 |
GT 6.7 |
GT 5.0 |
GT 4.0 |
GT 3.3 |
GT 2.8 |
GT 2.5 |
GT 2.2 |
P |
| 20 |
S |
GO 10.0 |
GO 6.7 |
GO 5.0 |
GO 4.0 |
GO 3.3 |
GO 2.8 |
GO 2.5 |
GO 2.2 |
GO 2.0 |
 |
| | Fig. 2 (a) An opaque gel formed by compound 13 in EtOH/H2O (v/v 1:1) at 10 mg mL−1; (b) an opaque gel formed by compound 17 in EtOH/H2O (v/v 1:2) at 2.5 mg mL−1; (c) a transparent gel formed by compound 20 in i-PrOH at 10.0 mg mL−1. | |
Further gelation properties of compound 18 for different metal ions
To further explore the potential applications of these sugar based gelators, the properties of these compounds were further evaluated. The p-methoxyl benzyl compound 18 was selected as the sample gelator since it formed stable gels in most solvents. This compound was tested for gelation in the presence of several earth-abundant metals including Hg(OAc)2, Ni(OAc)2, Zn(OAc)2·2H2O, Cu(OAc)2, Pb(OAc)4, and FeCl2. The gelation test was performed by the standard heating and cooling method and several salts were able to form homogenous stable gels. The tests were then repeated by mixing the metal ion solutions with the gelator solution to observe whether the gels can form spontaneously without heating. The compound 18 was dissolved in DMSO first, addition of an equal volume of the aqueous metal salt solution to the gelator solution resulted in spontaneous gelation for several metal salts. The gel photos are shown in Fig. 3.
 |
| | Fig. 3 Photograph of spontaneous gels containing Cu2+, Zn2+, Fe2+, Ni2+, Pb4+. The mercury salt mixture only formed a precipitate. The solvent is DMSO:H2O (v/v 1:1) and the molar ratio of gelator 18 to the metal ions is 1:2 for all samples. | |
For samples in biomimetic environments and for potential biomedical applications, it is desirable to form hydrogels with minimum amount of DMSO. We analyzed the gelation properties of compound 18 for several biologically relevant metal ions such as Ca2+, Zn2+, Fe3+, Cu2+, and vitamin B2. Spontaneous gels were formed by simply adding the aqueous solutions of the metal ions to the compound 18 solution at room temperature, these are shown in Fig. 4. The metal salts used are (a) Cu(OAc)2·H2O, (b) FeCl3, (c) Zn(OAc)2·2H2O, (d) CaO3PO(CH2)2N(Cl)(CH3)3·4H2O. Vitamin B2 co-gels are shown in Fig. 4e and f.
 |
| | Fig. 4 Gels formed by compound 18 with different metal ions (a–d) and vitamin B2 (e–f) in DMSO:H2O (v/v 1:8). The molar ratio of gelator 18 to the metal ions are 1:5 for (a–d). The vitamin B2 to gelator molar ratio was 1:19 in (e) and 1:2 in (f). | |
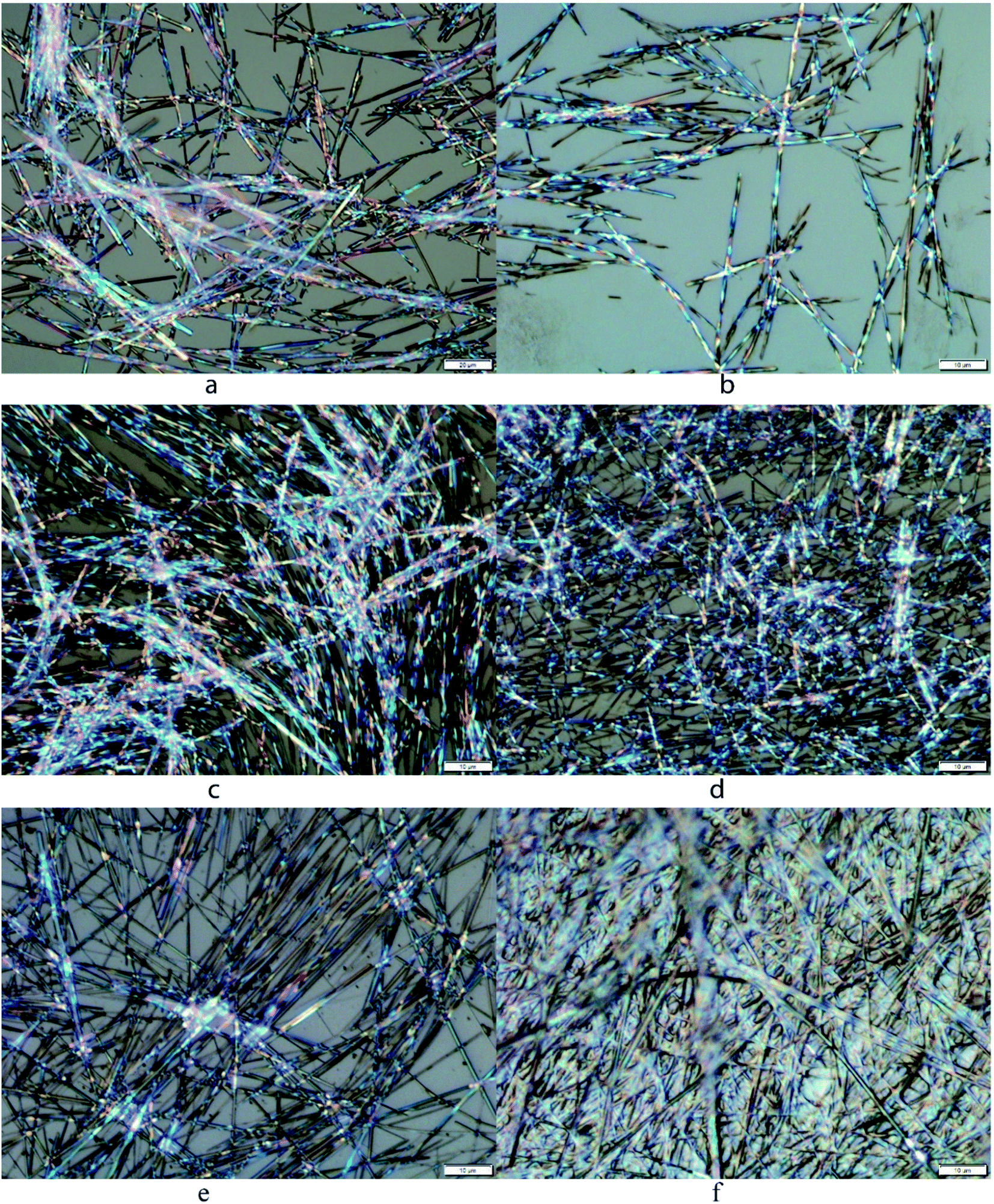
Gel characterizations
The gel morphology was characterized using optical microscopy and scanning electron microscopy. Some examples of these micrographs are included in Fig. 5 and 6. The gel formed by the p-methoxyl benzyl carbamate 18 in EtOH:H2O (v/v 1:1) showed bright birefringent fibers or tubular networks (Fig. 5a). The phenyl derivative 20 in DMSO:H2O (v/v 1:1) formed smaller and shorter fibers (Fig. 5b). The gel of compound 18 in DMSO:H2O (v/v 1:1) formed very long and uniform fibrous assemblies with smaller diameters (Fig. 5c). Compound 18 also formed gels in the presence of metal salts, the gel morphology showed some differences with the addition of various metals in comparison to the gel morphology without the metal salts. The Zn2+-containing gel showed shorter but uniform fibrous networks (Fig. 5d), and the nickel ion containing gel apparently led to birefringent fibers with larger diameters and longer length than the zinc gel (Fig. 5e). Finally, in the presence of Fe2+, the gels formed very long and straight fibers but less birefringent, and more densely packed fibrous assemblies (Fig. 5f). The optical micrograph also showed the fibers emerging and cross-linking within the solvent, forming the bulk of the gel. The presence of the metal ions seems to affect the molecular assembling process by forming extended uniform fibrous networks. The interaction of the compound 18 with metal ions led to enhanced intermolecular electrostatic interactions.
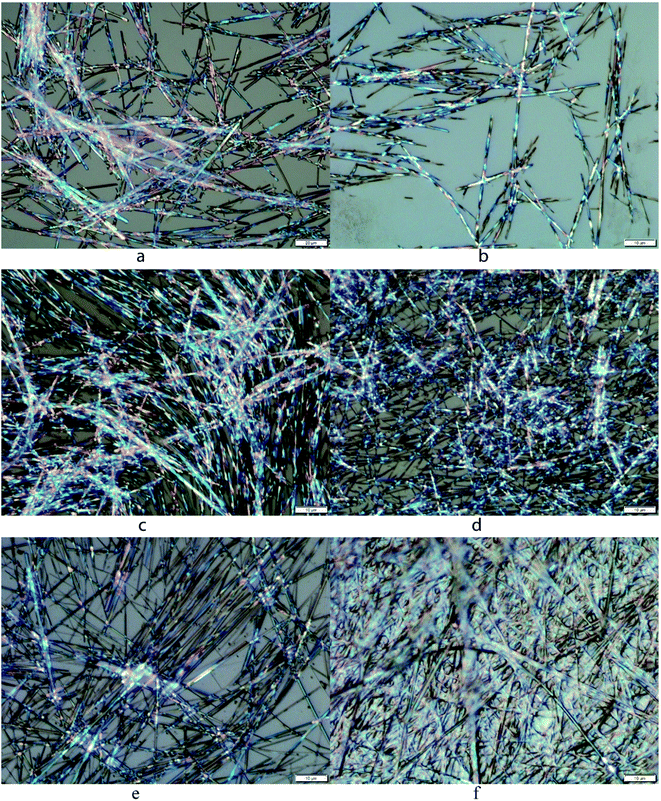 |
| | Fig. 5 Optical micrographs for several gels: (a) compound 18 in EtOH:H2O (v/v 1:1) at 4.0 mg mL−1, (b) compound 20 in DMSO:H2O (v/v 1:1) at 4.0 mg mL−1, (c) compound 18 in DMSO:H2O (v/v 1:1) at 2.5 mg mL−1, (d–f) compound 18 in DMSO:H2O (v/v 1:1) and with added 2 equiv. of metal salts: (d) Zn(OAc)2, (e) Ni(OAc)2, (f) FeCl2, the gelator concentration was 2.5 mg mL−1, 5.1 mM. | |
 |
| | Fig. 6 Scanning electron micrographs for several gels: (a and b) compound 17 in EtOH:H2O (v/v 1:1) at 2.5 mg mL−1, (c and d) compound 18 in EtOH:H2O (v/v 1:1) at 4.0 mg mL−1. | |
The SEM images for the dried gels of compounds 17 and 18 in EtOH:H2O (v/v 1:1) are shown in Fig. 6. The benzyl derivative 17 formed very small and densely packed fibrous networks (Fig. 6a and b), however compound 18 formed wide planar sheet like assemblies (Fig. 6c and d), the individual sheets have an average diameter of 2.5 μm. Compound 17 had a relatively lower gelation concentration and it formed a smaller fibrous network with tiny fibers packing into more stacked smooth films. On the other hand, compound 18 had higher MGCs and it formed large planar sheets (Fig. 6d). The morphology of the dried gels has more local crystallinity order; a similar trend was observed from the optical micrographs (OMs) whereby the more efficient gelators typically formed smaller fibrous networks and the less efficient ones formed more rigid planar sheets or stacking ribbons.
The melting point ranges of some of the gels formed by hexyl carbamate 13, heptyl carbamate 14, and cyclohexyl carbamate 16 in 1:1 DMSO:H2O were measured (Table 2). The three temperatures recorded were the temperature at which liquid was first seen, the temperature at which the gel was halfway melted, and the temperature at which the gel was fully melted to liquid. The gels started melting at 41.5–50.8 °C and completely melted at 77.2–122.2 °C. The melting point range for the gels increased as gel concentration decreased. All the gels measured in this experiment showed a large melting point range, which suggests that the gels are relatively stable to heat (Table 3).
Table 3 The melting point range for some of the gels in DMSO:H2O (v/v 1:1)
| Compound |
Gelator conc. (mM) |
mg mL−1 |
T1 (°C) |
T2 (°C) |
T3 (°C) |
| 13 |
44.4 |
20.0 |
48.0 |
71.3 |
77.2 |
| 14 |
21.5 |
10.0 |
50.8 |
85.1 |
95.6 |
| 16 |
14.9 |
6.7 |
41.5 |
91.3 |
122.2 |
The rheological properties of the gels formed by compounds 17 and 20 in several solvents were evaluated, these are shown in Fig. 7 and S5. For all gels, their storage moduli G′ were greater than the loss moduli G′′ in the full range of angular frequency from 0.1 to 100 rad S−1, indicating the stability of the gels. The ethanol/water (1:2) gel of compound 17 had smaller G′ and G′′ compared to the gel of compound 20 in the same solvent. The gel formed by compound 20 in (EtOH:H2O, 1:2) had the largest G′ and G′′ values among all the tested samples, and the toluene gel of compound 20 had the smallest G′ and G′′ values among all the tested samples.
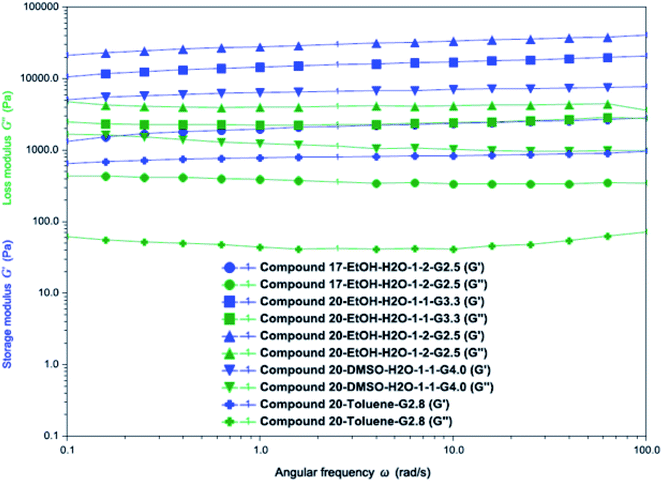 |
| | Fig. 7 Rheological properties of the gels formed by compound 17 (EtOH:H2O, 1:2, 2.5 mg mL−1), compound 20 (EtOH:H2O, 1:1, 3.3 mg mL−1), compound 20 (EtOH:H2O, 1:2, 2.5 mg mL−1), compound 20 (DMSO:H2O, 1:1, 4.0 mg mL−1) and compound 20 (toluene, 2.8 mg mL−1). Applied strain was 1% for all the samples, all solvent ratios are volume ratios. | |
NMR analysis at different temperatures to probe intermolecular forces
To analyze the structural impact on gelation, the 1H NMR spectra at different temperatures for two representative compounds, the hexyl derivative 13 and phenyl derivative 20 were studied and these are shown in Fig. 8 and 9. Upon increasing temperatures from 30 to 60 °C in compound 13, the NHAc chemical shift changed from 7.80 to 7.56 ppm, the amide proton shifted upfield by 0.24 ppm; and the carbamate NH signal also had an upfield change of 0.16 ppm. Compound 20 showed a similar upfield change for the two NH signals, the amide showed an upfield shift of 0.23 ppm and the carbamate moved upfield by 0.14 ppm. These indicate that both NH functional groups participated in the intermolecular hydrogen bonding interactions. The acetamide showed a bigger shift which perhaps is due to less steric hindrance comparing to the carbamate at the C-3 position.
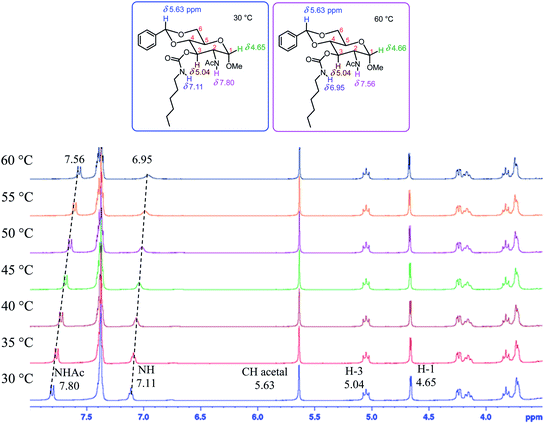 |
| | Fig. 8 The 1H NMR spectra (3.5 to 8.0 ppm) of compound 13 from 30 °C to 60 °C in DMSO-d6 (10.0 mg mL−1). | |
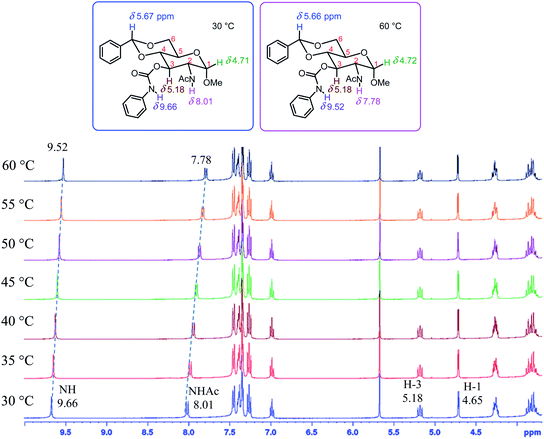 |
| | Fig. 9 The 1H NMR spectra (3.7 to 10.0 ppm) of compound 20 from 30 °C to 60 °C in DMSO-d6 (10.0 mg mL−1). | |
Naproxen entrapment and timed release studies
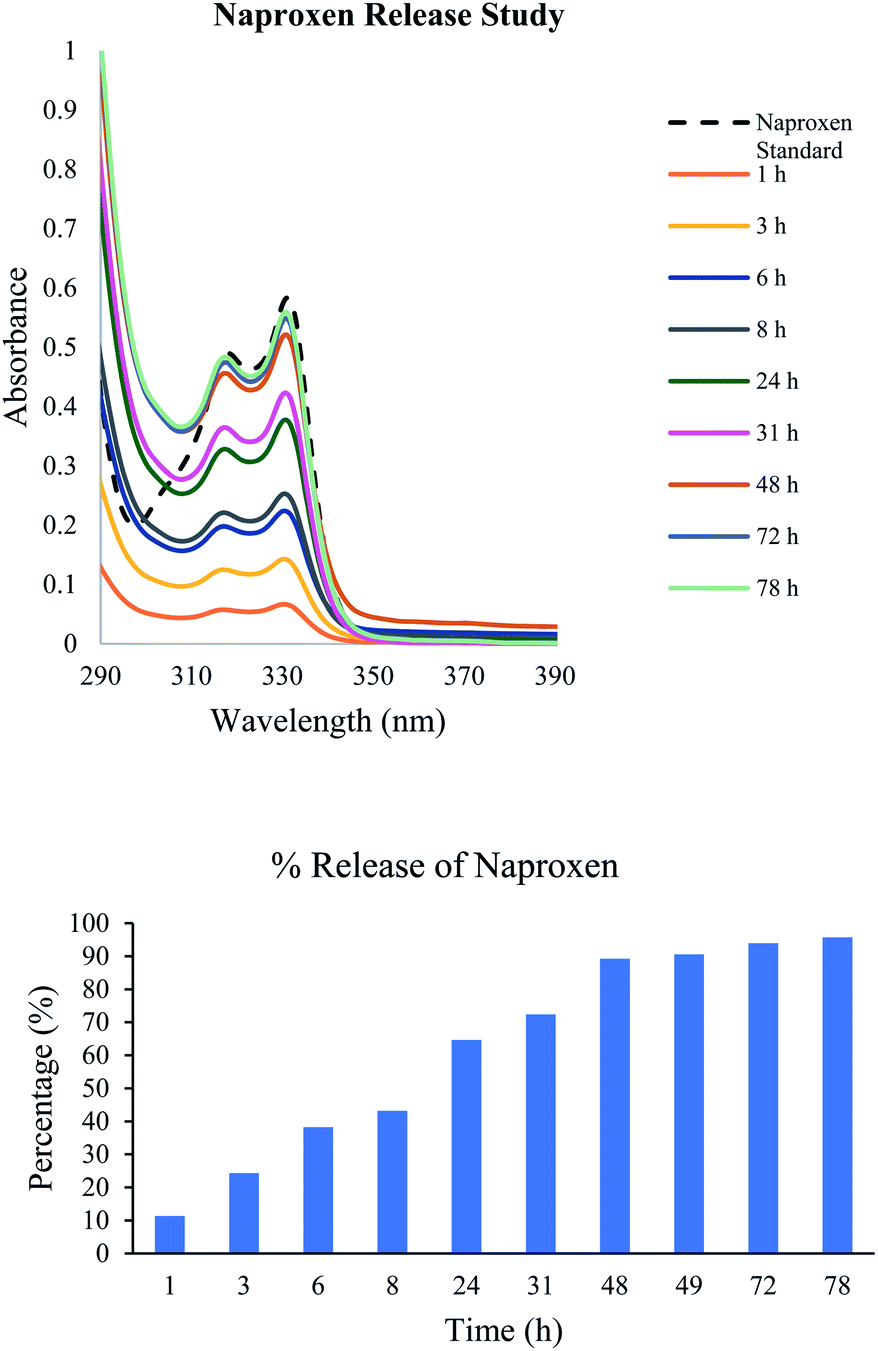
Due to the ease of syntheses of these carbamate derivatives, their applications for various other systems would be important. Compound 20 was utilized for the analysis of the encapsulation of naproxen as well as the transfer rate of the naproxen from the gel to the aqueous phase. This was monitored using UV-vis spectroscopy and the results are shown in Fig. 10. About 50% of the naproxen was released to the aqueous phase after 8 hours, and reached full equilibrium at about 2 days. The gels were stable during these measurements and no visible changes were noted. The gel photos are included in Fig. S15.†
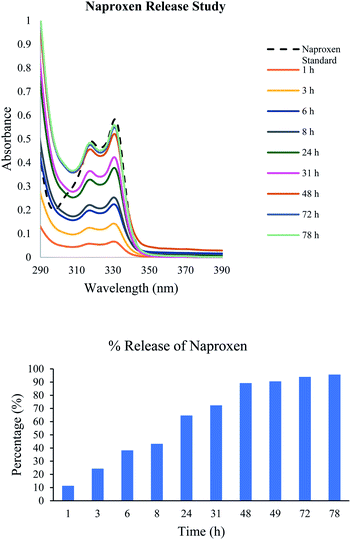 |
| | Fig. 10 Release of naproxen from the gel to the aqueous phase (a) UV spectra of naproxen release at different times from the gel formed by compound 20 (5.6 mg) in 2.0 mL of DMSO:H2O (v/v 1:7) with naproxen (0.25 mg) in the presence of phosphate buffered saline at pH 7.47 (2.0 mL) on top of the gel. (b) The % release at different times based on the UV absorbance at 331 nm. | |
Stability of the carbamates under basic conditions
These carbamates are interesting compounds that should be more stable in basic conditions in comparison to the corresponding esters derivatives. The stability of several compounds in alkaline conditions was analyzed and the results are summarized in ESI Fig. S1.† The gels formed by the hexyl carbamate 13 and the benzyl derivative 17 in EtOH:H2O (v/v 1:1) were chosen for the analysis. An equal volume of aqueous base (NaOH) at pH 10 or 12 solution was added on top of the gels. The stability of the gels in the presence of the basic solutions was observed by visual inspection and the gel stability was checked by tilting the vials at certain time intervals (14 h, 36 h, 144 h). All gels were stable at 14 h, the gels formed by 13 were stable in the presence of both basic solutions for 6 days, and the gel formed by 17 was stable in pH 10 solution for 6 days, but became unstable in pH 12 solution after 14 h. The mixture was analyzed after 6 days in pH 12 solution for compound 17, the extracted compounds showed no decomposition under this condition (ESI Fig. S1 and S2†). Besides basic solutions, an organic base (DBU) was also used to analyze the stability of the p-methoxyl benzyl carbamate derivative 18, the compound was dissolved in DMSO/H2O (v/v 1:1), 5 equivalents of DBU was added to the solution, the mixture was heated and left standing at rt for 1 h, no decomposition was observed by TLC and 1H NMR analysis (ESI Fig. S1–S3†). From this study, we showed that the carbamate gelators were resistant to base degradation, this property could be important with certain applications when the stability of gelators is required.
Gelation for electrolytes and response to anions or chemical stimuli
The carbamate derivative 18 also formed stable gels in the presence of tetramethylammonium bromide (TMABr) at 0.5, 5.0 and 10.0 equivalents. This indicates that the gelator can form stable co-gels with the ammonium salts, which are electrolytes.42,52 The gelation response towards several tetrabutylammonium salts were also studied for compound 18. In DMSO:H2O (v/v 1:1) solvent, compound 18 formed stable gels with several tetrabutylammonium salts (TBAX). These include tetrabutylammonium salts of iodide, bromide, bisulfate, and fluoride; abbreviated as TBAI, TBABr, TBAHS, and TBAF. The initial TBAX amount (2.1 mM) was 0.5 equivalent to compound 18 (2.5 mg mL−1, or 4.1 mM), stable gels were obtained for all four salts. To the same gels, addition of another 4.5 equivalents of TBA salts also resulted in stable gels as shown in Fig. S4a.† However upon further addition of another 5 equivalents of TBAX to the corresponding gels, the gels containing TBA salts of bromide and bisulfate became unstable, but the vials containing TBAI and TBAF were able to reform stable gels (Fig. S4b†). To the precipitated mixtures of the samples containing TBABr and TBAHS, further addition of 10 equivalents of acetic acid led to the restoration of gels for TBABr, but not for TBAHS (Fig. S4c†). Fig. 11 shows the responses towards the salt and acid for samples with addition of TBABr salt.
 |
| | Fig. 11 Gelation properties of compound 18 with TBABr, (a) gel formed with 5 equiv. of TBABr, (b) precipitation with 10 equiv. of TBABr, (c) resumed gelation after adding acetic acid to the vial in (b). | |
NMR studies of gelators with different amounts of TBABr
The NMR spectra of gelator 18 with different amounts of TBABr were obtained in two different solvents, d-chloroform and d6-acetone. Deuterated chloroform is a non-hydrogen bonding solvent and the interaction of gelator with the anions should be more pronounced. We observed a significant amount of chemical shift change for the gelator upon the addition of different amounts of the tetrabutylammonium salt (Fig. 12, S10 and S11†). However in d6-acetone (Fig. S12†), not many chemical shift changes were observed with the added TBABr salt, which indicates that the gelator self-assembling mechanisms are very different in different solvents, the molecules will interact with the solvents significantly, therefore the effect of the ammonium salt was not important.
Compound 18 showed the greatest chemical shift changes in deuterated chloroform. The 1H NMR spectra of gelator 18 with different amounts of TBABr are shown in Fig. 12. From 0 to 10 equivalents of TBABr, the NHAc chemical shift changed from 5.99 to 5.92 ppm, with an upfield shift of 0.07 ppm. Interestingly, the carbamate NH signal showed almost no change, and all other C–H signals moved about 0.1 ppm upfield. Consequently, with 15 equiv. of TBABr, nearly all C–H signals moved upfield by 0.14 ppm from 0 to 15 equivalents of the salt (Fig. S11†). This trend is the same for all hydrogens except the carbamate hydrogen NHCO, which stayed the same with more Br-ions and only showed 0.01 ppm upfield change from 5.09 to 5.08 ppm, this trend is very different compared to the rest of the compounds. The amide NHAc (δN–H) signal shifted upfield by 0.12 ppm, and most of other C–H signals showed an upfield chemical shift change of 0.14 ppm. The observed upfield change of δAr–H upon addition of more TBABr indicated that π–π stacking interactions of the aromatic functions are involved in the gelation process.41,53,54 With increasing amount of TBABr, possible anion–π interaction is also present. On the other hand, other C–H signals moving upfield were probably due to CH–π interactions and enhanced van der Waals forces, these indicate that the sugar ring was also involved in gel formation. The NH signal of the carbamate participated in hydrogen bonding with the bromide, this effect does not change upon further addition of the salt, one equivalent of the salt was sufficient in forming the hydrogen bonds.
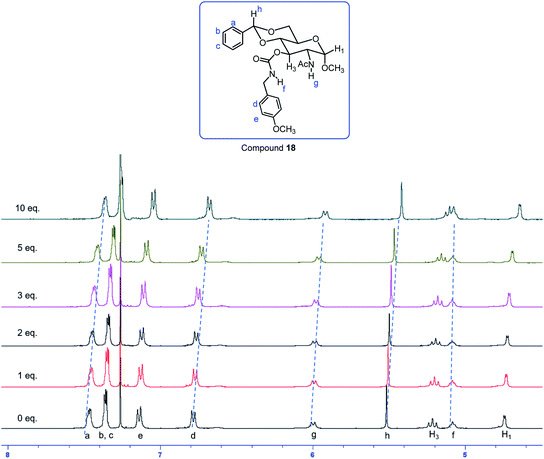 |
| | Fig. 12 1H NMR spectra of gelator 18 in CDCl3 with different amount of tetrabutylammonium bromide (TBABr). | |
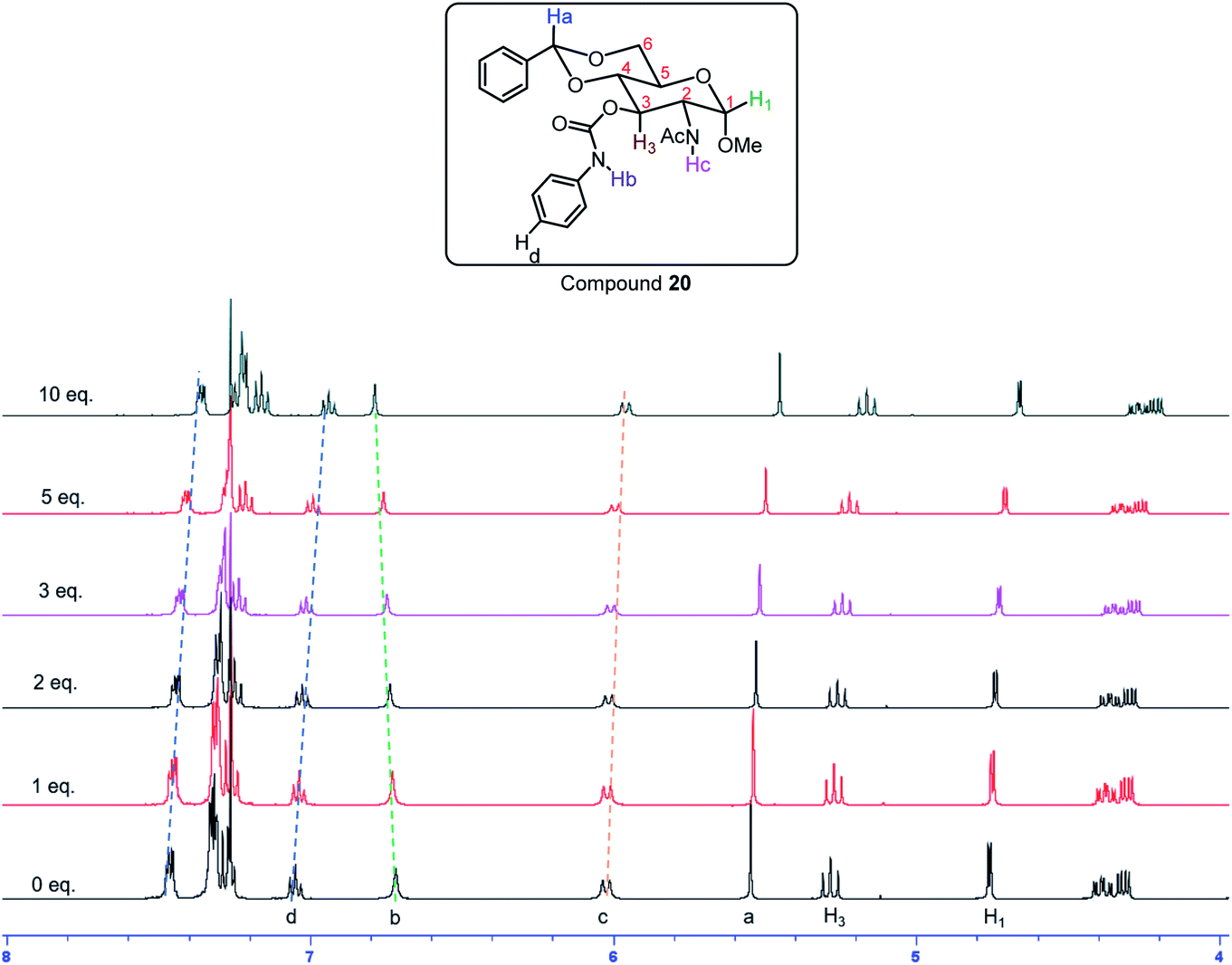
The same trend was observed for the aromatic carbamate 20, in different solvents the chemical shift showed significantly different trends. In CDCl3, with different amounts of TBABr from 0 to 10 equivalents, most of the C–H signals in compound 20 moved upfield by 0.10 ppm, with the C–H3 displaying a 0.12 ppm change (Fig. 13 and S13†). The amide NHAc only showed a 0.06 ppm upfield shift from δ 6.01 to 5.95 ppm; and the carbamate NHb showed 0.06 ppm downfield change from δ 6.72 to 6.78 ppm, which is opposite to the others. This indicates that both the amide and carbamate NH participates in hydrogen bonding strongly and contributed to gelation. With the addition of more bromide ions, the C–H signals in the NMR spectra showed significant upfield changes, including both the aromatic peaks and the sugar ring C–H signals, which signified that at higher TBABr salt concentrations the gelator showed enhanced π–π stacking interactions and C–H–π interactions. In the DMSO solvent, the influence of the anion binding is much less than that of chloroform. In the DMSO-d6 solvent (Fig. S14†) the signals did not show much change upon the titration of TBABr from 0 to 10 equivalents, the carbamate NHCO signal remained mostly unchanged (from 9.65 to 9.66 ppm), the NH signal showed slightly more upfield change of 0.05 ppm from δ 8.01 to 7.96 ppm. The acetal CH showed a small downfield change of 0.04 ppm from 5.66 to 5.70 ppm; whilst the C–H3, and C–H1 showed small upfield changes of 0.02 and 0.03 ppm, respectively.
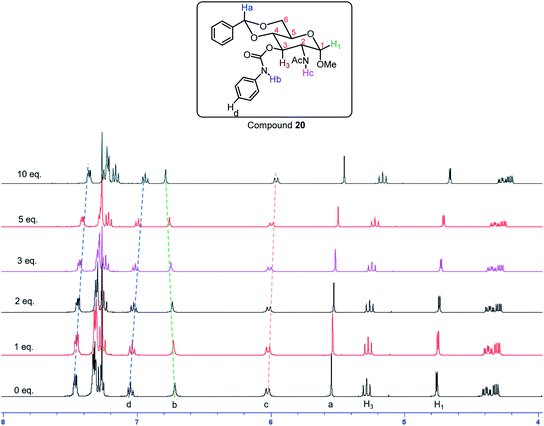 |
| | Fig. 13 1H NMR spectra of gelator 20 in CDCl3 with different amount of tetrabutylammonium bromide (TBABr). | |
Conclusions
A series of nineteen 3-O-carbamate derivatives of the N-acetyl-D-glucosamine headgroup has been synthesized and their self-assembling mechanisms in a variety of solvents were studied. The structure of the functional group attached to the carbamoyl moiety plays an important role in the gelation properties. The aliphatic carbamates were more effective than the aromatic derivatives with an exception of the phenyl carbamate. Interestingly, the dimeric sugar carbamate derivatives were not as effective gelators in comparison to the monomeric carbamate derivatives. Among these carbamate derivatives, most of the alkyl carbamates formed gels in pump oil and several also formed gels in mixed solvents of water with ethanol or water with DMSO. Certain aryl carbamates were also effective gelators for a few of the tested solvents. The best performing compounds are the p-methoxybenzyl derivative 18 and the phenyl carbamate 20 which formed gels in eight of the tested solvents and with low MGCs of up to 2.5 mg mL−1. The compound 18 was also an effective gelator for metal ions and tetrabutylammonium salts. It formed spontaneous gels in the presence of several metal ions including Ca2+, Cu2+, Fe2+, Zn2+, and Ni2+. Besides metal ions, the compound also formed stable gels in the presence of tetramethyl or tetrabutyl ammonium salts. The gels were characterized using optical microscopy, rheology and scanning electron microscopy. The gelators formed long and uniform fibrous networks as indicated by the optical micrographs and SEMs. The gelation properties of some representative compounds were further studied and analyzed using 1H NMR spectroscopy at different temperatures and different amounts of the TBABr salts. The phenyl carbamate derivative was also used for entrapping and releasing naproxen. The structure and gelation properties obtained from this study will be used for the design of other relevant molecular gelators, these compounds will also be further studied for other applications such as sensors for bromide ions, etc.
Experimental section
General methods
Reagents and solvents were used as they were received from the suppliers. All purification was conducted by flash chromatography using 230–400 mesh silica gel obtained from Natland International Corporation. 1H NMR and proton-decoupled 13C NMR spectra were obtained with a Bruker 400 MHz spectrometer in DMSO-d6 or CDCl3. The chemical shifts were reported using CDCl3/DMSO-d6 as internal standard at 7.26/2.50 ppm and at 77.00/39.50 ppm, respectively. 2D NMR experiments (HSQC, COSY) were also conducted using a 400 MHz Bruker NMR spectrometer to assist the proton and carbon signal assignment. Melting point measurements were carried out using a Fisher Jones melting point apparatus. The molecular mass was measured using LCMS on an Agilent 6120B Single Quad Mass Spectrometer and LC1260 system or Shimadzu LCMS-2020 with ESI in positive ionization mode.
Optical microscopy. A thin slice of the gel was transferred onto a clean glass slide and then left to air-dry for a day or so. The gel was then observed under an Olympus BX60M optical microscope using an Olympus DP73-1-51 high-performance 17MP digital camera with pixel shifting and Peltier cooling. The program used to acquire and store the images is CellSens Dimension 1.11.
Gelation test. Approximately 2 mg of the desired compound was placed in a one-dram vial and 0.1 mL of the gelation solvent or solution was transferred to the vial to attain a concentration of 20 mg mL−1. The vial was then heated until the gelator dissolved fully; sometimes, the mixture was sonicated to help with dissolving the compound and the mixture was left to cool for approximately 15 minutes or longer for the gel to form. After this period, if the solution was clear, this was recorded as soluble; if the solid reappeared, this was recorded as a precipitate; if the sample formed a gel, then the vial was inverted; if no solvent was flowing, this indicated that a stable gel was formed; otherwise, this was recorded as an unstable gel. If gelation occurs, another 0.1 mL solvent is added and the method is repeated until an unstable gel is formed. The minimum gelation concentration (MGC) which represents the concentration prior to unstable gelation, was recorded.For the gelation screening in DMSO and water mixtures, typically, about 2.0 mg of the compound was first dissolved in small amount of DMSO (0.1 mL), followed by addition of water (0.1 mL) incrementally until a stable gel can no longer form. The concentration of the stable gelation was recorded as the gelation concentration (Table 2).
Gelation test for metal ions. Compound 18 (2.0 mg, 4.11 × 10−3 mmol, 1 equivalent) was dissolved in 0.4 mL of DMSO in a 1 dram vial. Then the aqueous solution (2.05 × 10−2 mol L−1) of Cu(OAc)2, Zn(OAc)2·2H2O, FeCl2, Ni(OAc)2, Pb(OAc)4, Hg(OAc)2, (0.4 mL, 8.22 × 10−3 mmol, 2.0 equivalents to compound 18) was added to the vials. The samples were kept at room temperature for 20 min and the vials were inverted to examine if gelation occurred. The photos of the vials which exhibited spontaneous gels in 20 min are shown in Fig. 3.
Gelation tests in DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :H2O (v/v 1:8). Compound 18 (2.0 mg, 4.11 × 10−3 mmol, 1 equivalent) was dissolved in 0.09 mL of DMSO in a 1 dram vial. Aqueous solutions (0.71 mL, 2.06 × 10−2 mol L−1, 5 equivalent) of phosphocholine chloride calcium salt tetrahydrate, Zn(OAc)2·2H2O, Cu(OAc)2·H2O and FeCl3 were added to the above DMSO solution at room temperature, respectively. For the vitamin B2 gelation test, compound 18 (2.0 mg, 4.11 × 10−3 mmol, 1 equivalent) was dissolved in 0.09 mL of DMSO in a 1 dram vial, then an aqueous solution (0.71 mL, 2.99 × 10−4 mol L−1 and 2.48 × 10−3 mol L−1) of riboflavin B2 was added to the above DMSO solution at room temperature, respectively. The gel photos are shown in Fig. 4.
:H2O (v/v 1:8). Compound 18 (2.0 mg, 4.11 × 10−3 mmol, 1 equivalent) was dissolved in 0.09 mL of DMSO in a 1 dram vial. Aqueous solutions (0.71 mL, 2.06 × 10−2 mol L−1, 5 equivalent) of phosphocholine chloride calcium salt tetrahydrate, Zn(OAc)2·2H2O, Cu(OAc)2·H2O and FeCl3 were added to the above DMSO solution at room temperature, respectively. For the vitamin B2 gelation test, compound 18 (2.0 mg, 4.11 × 10−3 mmol, 1 equivalent) was dissolved in 0.09 mL of DMSO in a 1 dram vial, then an aqueous solution (0.71 mL, 2.99 × 10−4 mol L−1 and 2.48 × 10−3 mol L−1) of riboflavin B2 was added to the above DMSO solution at room temperature, respectively. The gel photos are shown in Fig. 4.
Scanning electron microscopy (SEM). A thin slice of the gel was transferred on to a clean glass slide (1.0 mm × 1.0 mm) and then left to air-dry for at least one day, afterwards it was placed in a desiccator for additional two days. The sample was then coated with a thin layer of gold (∼50 nm) using a sputter coater (BOC Edwards 306 Evaporator, <10−6 torr). The sample was observed, and images were taken using SU8010 Hitachi field emission scanning electron microscope.
Naproxen trapping and release studies. Naproxen sodium was dissolved in the desired solvent, and this was used to prepare the gel first. Naproxen sodium (2.5 mg) was dissolved in 10.0 mL of DMSO:H2O (v/v 1:7). This solution (2 mL) was used to prepare the gel with compound 20 (5.6 mg). The gelator concentration was 2.8 mg mL−1, and the initial naproxen sodium concentration was 0.25 mg mL−1. The gel was left at room temperature for 14 hours, and then phosphate buffered saline (pH 7.47) was placed on top of the gel. The UV absorbance of the aqueous phase was taken at different time intervals by transferring the solution to a cuvette and then cautiously returning it to the vial after the measurement. The final naproxen concentration should be 0.125 mg mL−1 in both aqueous and gel phases if equilibrium has been achieved. Phosphate buffered saline was made by dissolving 8.01 of sodium chloride, 0.2 g of potassium chloride, 2.72 g of di-sodium hydrogen phosphate heptahydrate and 0.27 g of potassium dihydrogen phosphate in 1.0 L of DI water. This was autoclaved for 20 minutes at 121 °C or filtered using a 0.45 μm filter to sterilize.
Synthesis of carbamate derivatives
Compounds 12–14, 16, 17, 20–23 were prepared using the methods described previously.51 The other compounds 9–11, 15, 18–19, 24–27 were synthesized using the CDI method. The detailed procedures are given for one mono-carbamate and one di-carbamate. Only the characterization data of other compounds were given unless the method used was different.
Synthesis of imidazole carboxylate compound 8. Compound 7 (1.00 g, 3.13 mmol, 1 equiv.) and CDI (1.02 g, 6.26 mmol, 2 equiv.) were dissolved in anhydrous DCM (10 mL). The reaction mixture was stirred at room temperature for 2 hours under N2 at which 1H NMR spectrum showed full conversion. The mixture was washed with water (2 mL × 2) and the combined organic phases were dried over Na2SO4. The solvent was removed to give the crude, which was purified by column chromatography using eluent from 0.5% to 5% MeOH/DCM to afford a white solid (1.25 g, 2.99 mmol, 96%) as the desired product. Rf = 0.3 in 2% MeOH/DCM; mp 245.0–246.0 °C. 1H NMR (400 MHz, CDCl3) δ 8.12 (s, 1H), 7.42–7.40 (m, 3H), 7.34–7.31 (m, 3H), 7.04 (s, 1H), 5.94 (d, J = 9.8 Hz, 1H), 5.54 (s, 1H), 5.39 (t, J = 9.9 Hz, 1H), 4.76 (d, J = 3.7 Hz, 1H), 4.58 (dt, J = 10.2, 3.7 Hz, 1H), 4.33 (dd, J = 10.2, 4.7 Hz, 1H), 3.97–3.91 (m, 1H), 3.89–3.80 (m, 2H), 3.44 (s, 3H), 1.91 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.0, 148.8, 137.3, 136.6, 130.8, 129.2, 128.2, 126.1, 117.3, 101.7, 99.0, 78.8, 75.0, 68.8, 62.8, 55.4, 51.8, 23.1. LC-MS (ESI+) calcd for C20H24N3O7 [M + H]+ 418.2, found 418.2.
General procedure for carbamate synthesis using CDI method
For mono-carbamate synthesis, the imidazole carboxylate 8 (75 mg, 0.18 mmol, 1.0 equiv.), amine (1.2 or 1.5 equiv.) and 2 mL of anhydrous DCM were added to a 50 mL round bottomed flask. For bis-carbamate synthesis, compound 8 (120 mg, 0.29 mmol, 2.1 equiv.), di-amine (1.0 equiv.) and 2 mL of anhydrous DCM were added to a 50 mL round bottom flask. The mixture was stirred at room temperature until full conversion was confirmed by 1H NMR analysis. The solvent was removed to give the crude product, which was further purified by column chromatography using eluent from 0.5% MeOH/DCM to 5% MeOH/DCM to afford the purified carbamate derivatives.
Synthesis of compound 9. Compound 8 (100 mg, 0.24 mmol, 1.0 equiv.) and propargylamine (0.023 mL, 0.36 mmol, 1.5 equiv.) were added to a 50 mL round bottom flask. The mixture was stirred for 8 hours at room temperature. A white solid (81 mg, 84%) was obtained as the desired product (Rf = 0.5 in 5% MeOH/DCM). mp 220.1–223.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.49–7.43 (m, 2H), 7.37–7.31 (m, 3H), 6.00 (d, J = 9.3 Hz, 1H), 5.52 (s, 1H), 5.19 (t, J = 10.0 Hz, 1H), 5.04 (br s, 1H), 4.73 (d, J = 3.4 Hz, 1H), 4.36–4.25 (m, 2H), 4.02–3.81 (m, 3H), 3.79 (t, J = 10.2 Hz, 1H), 3.68 (t, J = 9.5 Hz, 1H), 3.40 (s, 3H), 2.18 (br s, 1H), 1.98 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 169.3, 155.0, 136.0, 128.1, 127.2, 125.3, 100.7, 98.1, 78.4, 78.2, 70.6, 70.3, 67.9, 61.9, 54.3, 51.9, 29.8, 22.3. LC-MS (ESI+) calcd for C20H25N2O7 [M + H]+ 405, found 405.
Synthesis of compound 10. Compound 8 (75 mg, 0.18 mmol, 1.0 equiv.) and 3-butynylamine (0.022 mL, 0.27 mmol, 1.5 equiv.) were added to a 50 mL round bottom flask. The mixture was stirred for 5 hours at room temperature. A white solid (65 mg, 87%) was obtained as the desired product (Rf = 0.36 in 5% MeOH/DCM). mp 192.0–194.2 °C; 1H NMR (400 MHz, CDCl3) δ 7.50–7.43 (m, 2H), 7.38–7.31 (m, 3H), 6.03 (d, J = 8.9 Hz, 1H), 5.52 (s, 1H), 5.24–5.09 (m, 2H), 4.74 (d, J = 3.5 Hz, 1H), 4.34–4.23 (m, 2H), 3.92–3.84 (m, 1H), 3.82–3.73 (m, 1H), 3.67 (t, J = 9.4 Hz, 1H), 3.40 (s, 3H), 3.32–3.24 (m, 2H), 2.38–2.32 (m, 2H), 1.97 (s, 3H), 1.96 (t, J = 2.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 170.2, 156.4, 137.1, 129.1, 128.2, 126.3, 101.6, 99.1, 81.0, 79.3, 70.9, 70.2, 68.9, 62.9, 55.3, 53.0, 39.8, 23.2, 19.6. LC-MS (ESI+) calcd for C21H27N2O7 [M + H]+ 419, found 419.
Synthesis of compound 11. Compound 8 (75 mg, 0.18 mmol, 1.0 equiv.) and 1-butylamine (0.027 mL, 0.27 mmol, 1.5 equiv.) were added. The mixture was stirred for 11 hours at room temperature. A white solid (65 mg, 86%) was obtained as the desired product (Rf = 0.42 in 5% MeOH/DCM). mp 209.0–211.1 °C; 1H NMR (400 MHz, CDCl3) δ 7.47–7.44 (m, 2H), 7.36–7.33 (m, 3H), 6.07 (d, J = 9.1 Hz, 1H), 5.52 (s, 1H), 5.17 (t, J = 10.1 Hz, 1H), 4.78 (t, J = 5.4 Hz, 1H), 4.74 (d, J = 3.3 Hz, 1H), 4.30–4.23 (m, 2H), 3.91–3.84 (m, 1H), 3.78 (t, J = 10.1 Hz, 1H), 3.66 (t, J = 9.5 Hz, 1H), 3.39 (s, 3H), 3.20–3.07 (m, 2H), 1.97 (s, 3H), 1.46–1.39 (m, 2H), 1.34–1.26 (m, 2H), 0.88 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.2, 156.5, 137.1, 129.1, 128.2, 126.3, 101.6, 99.1, 79.3, 70.6, 69.0, 62.9, 55.3, 53.2, 40.8, 31.9, 23.2, 19.8, 13.7. LC-MS (ESI+) calcd for C21H31N2O7 [M + H]+ 423, found 423.
Synthesis of compound 16. Compound 8 (100 mg, 0.24 mmol, 1.0 equiv.) and cyclohexylamine (35.7 mg, 0.36 mmol, 1.5 equiv.) were added to a 50 mL round bottom flask. The mixture was stirred for 12 hours at room temperature. A white solid (78 mg, 89%) was obtained as the desired product (Rf = 0.3 in 5% MeOH/DCM). The characterization data are the same as reported previously.51
Synthesis of compound 18. Compound 8 (100 mg, 0.24 mmol, 1 equiv.) and 4-methoxybenzylamine (0.037 mL, 0.29 mmol, 1.2 equiv.) were added to a 50 mL round bottom flask. The mixture was stirred for 4 hours at room temperature. A white solid (105 mg, 90%) was obtained as the desired product (Rf = 0.3 in 2% MeOH/DCM); mp 251.0–252.5 °C; 1H NMR (400 MHz, CDCl3) δ 7.47–7.45 (m, 2H), 7.36–7.35 (m, 3H), 7.13 (d, J = 8.4 Hz, 2H), 6.78 (d, J = 8.4 Hz, 2H), 6.03 (d, J = 9.0 Hz, 1H), 5.51 (s, 1H), 5.21 (t, J = 10.0 Hz, 1H), 5.11 (t, J = 5.7 Hz, 1H), 4.74 (d, J = 3.4 Hz, 1H), 4.30–4.23 (m, 4H), 3.91–3.85 (m, 1H), 3.82–3.72 (m, 4H), 3.66 (t, J = 9.4 Hz, 1H), 3.40 (s, 3H), 1.90 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.3, 159.0, 156.5, 137.1, 130.3, 129.1, 128.6, 128.2, 126.3, 114.0, 101.7, 99.1, 79.3, 70.9, 69.0, 62.9, 55.3, 53.0, 44.5, 23.2. LC-MS (ESI+) calcd for C25H31N2O8 [M + H]+ 487.2, found 487.2.
Synthesis of compound 19. Compound 8 (75 mg, 0.18 mmol, 1.0 equiv.) and 4-aminobenzylamine (0.031 mL, 0.27 mmol, 1.5 equiv.) were added to a 50 mL round bottom flask. The mixture was stirred for 12 hours at room temperature. A white solid (66 mg, 78%) was obtained as the desired product (Rf = 0.35 in 5% MeOH/DCM). mp 208.1–210.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.51–7.43 (m, 2H), 7.40–7.31 (m, 3H), 7.00 (d, J = 7.9 Hz, 2H), 6.56 (d, J = 7.9 Hz, 2H), 6.07 (d, J = 8.9 Hz, 1H), 5.51 (s, 1H), 5.21 (t, J = 10.1 Hz, 1H), 5.07 (br s, 1H), 4.73 (d, J = 2.7 Hz, 1H), 4.33–4.11 (m, 4H), 3.93–3.84 (m, 1H), 3.79 (t, J = 10.2 Hz, 1H), 3.70–3.54 (m, 3H), 3.39 (s, 3H), 1.91 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.3, 156.5, 145.8, 137.1, 129.1, 128.7, 128.2, 128.0, 126.3, 115.2, 101.7, 99.1, 79.3, 70.8, 69.0, 62.9, 55.3 (2 OCH3), 53.1, 44.7, 23.2. LC-MS (ESI+) calcd for C24H30N3O7 [M + H]+ 472, found 472.
Synthesis of compound 24. Compound 8 (120 mg, 0.288 mmol, 2.1 equiv.), m-xylylene diamine (18.6 mg, 0.137 mmol, 1.0 equiv.) were added to a 50 mL round bottom flask. The mixture was stirred for 6 hours at room temperature. A white solid (91 mg, 80%) was obtained as the desired product. Rf = 0.3 in 5% MeOH/DCM. mp > 275.0 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.84 (d, J = 9.4 Hz, 2H), 7.68 (t, J = 6.1 Hz, 2H), 7.40–7.35 (m, 10H), 7.00–6.93 (m, 4H), 5.65 (s, 2H), 5.10 (t, J = 9.8 Hz, 2H), 4.66 (d, J = 3.5 Hz, 2H), 4.25–3.97 (m, 8H), 3.84–3.72 (m, 6H), 3.36 (s, 6H), 1.77 (s, 6H); 13C NMR (100 MHz, DMSO-d6) δ 170.1, 156.6, 140.1, 137.9, 129.3, 128.6, 128.5, 126.6, 125.8, 125.3, 101.0, 99.5, 79.8, 69.9, 68.4, 63.2, 55.5, 52.3, 44.0, 22.8. LC-MS (ESI+) calcd for C42H51N4O14 [M + H]+ 835.3, found 835.3.
Synthesis of compound 25. Compound 8 (120 mg, 0.288 mmol, 2.1 equiv.) and ethylenediamine (8.2 mg, 0.137 mmol, 1 equiv.) were added to a 50 mL round bottom flask. The mixture was stirred for 4 hours at room temperature. A white solid (76.8 mg, 74%) was obtained as the desired product. Rf = 0.3 in 5% MeOH/DCM. mp > 275.0 °C; 1H NMR (400 MHz, CDCl3 + d4-MeOH) δ 7.38–7.37 (m, 4H), 7.28–7.26 (m, 6H), 6.71 (d, J = 9.5 Hz, 2H), 5.42 (s, 2H), 5.03 (t, J = 9.9 Hz, 2H), 4.64 (d, J = 3.5 Hz, 2H), 4.23–4.16 (m, 4H), 3.84–3.78 (m, 2H), 3.71 (t, J = 10.2 Hz, 2H), 3.60 (t, J = 9.5 Hz, 2H), 3.35 (s, 6H), 3.19–3.08 (m, 4H), 1.85 (s, 6H); 13C NMR (100 MHz, CDCl3 + d4-MeOH) δ 171.5, 156.9, 136.9, 129.1, 128.2, 126.2, 101.7, 99.0, 79.2, 70.9, 68.8, 62.7, 55.3, 52.7, 40.7, 22.7. LC-MS (ESI+) calcd for C36H47N4O14 [M + H]+ 759.3, found 759.3.
Synthesis of compound 26. Compound 8 (120 mg, 0.288 mmol, 2.1 equiv.), 1, 3-diaminopropane (10.1 mg, 0.137 mmol, 1 equiv.) were added to a 50 mL round bottom flask. The mixture was stirred for 6 hours at room temperature. A white solid (77 mg, 73%) was obtained as the desired product. Rf = 0.3 in 5% MeOH/DCM. mp > 275.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.44–7.43 (m, 4H), 7.31–7.29 (m, 6H), 6.09 (d, J = 8.9 Hz, 2H), 5.52 (s, 2H), 5.20–5.12 (m, 4H), 4.74 (d, J = 3.6 Hz, 2H), 4.32–4.27 (m, 4H), 3.91–3.85 (m, 2H), 3.79 (t, J = 10.2 Hz, 2H), 3.67 (t, J = 9.5 Hz, 2H), 3.41 (s, 6H), 3.05 (m, 4H), 1.95 (s, 6H); 1.56–1.47 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 170.2, 156.9, 137.0, 129.1, 128.2, 126.3, 101.7, 99.1, 79.4, 70.9, 68.9, 62.9, 55.4, 52.8, 37.3, 29.8, 23.2. LC-MS (ESI+) calcd for C37H49N4O14 [M + H]+ 773.3, found 773.3.
Synthesis of compound 27. Compound 8 (120 mg, 0.288 mmol, 2.1 equiv.), 1,6-hexanediamine (15.9 mg, 0.137 mmol, 1 equiv.) were added to a 50 mL round bottom flask. The mixture was stirred for 12 hours at room temperature. A white solid (90.3 mg, 0.11 mmol, 81%) was obtained as the desired product (Rf = 0.4 in 5% MeOH/DCM). mp > 275.0 °C; 1H NMR (400 MHz, CDCl3 + d4-MeOH) δ 7.47–7.40 (m, 4H), 7.35–7.28 (m, 6H), 6.28 (d, J = 8.8 Hz, 2H), 5.53 (s, 2H), 5.19 (t, J = 10.1 Hz, 2H), 4.89–4.81 (m, 2H), 4.76 (d, J = 3.4 Hz, 2H), 4.37–4.25 (m, 4H), 3.96–3.86 (m, 2H), 3.81 (t, J = 10.2 Hz, 2H), 3.71 (t, J = 9.6 Hz, 2H), 3.42 (s, 6H), 3.02–2.81 (m, 4H), 1.97 (s, 6H), 1.39–1.28 (m, 4H), 1.23–1.16 (m, 4H); 13C NMR (100 MHz, CDCl3 + d4-MeOH) δ 171.1, 156.7, 137.0, 129.0, 128.1, 126.2, 101.6, 99.1, 79.5, 70.3, 68.8, 62.8, 55.3, 52.7, 40.3, 29.1, 25.7, 22.8. LC-MS (ESI+) calcd for C40H55N4O14 [M + H]+ 815.4, found 815.4.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for financial support from the National Science Foundation grant CHE# 1808609. We thank Dr Lalith Samankumara and Ms. Consuelo Garcia for their assistance with synthesis and analysis of some compounds. We also thank Dr Harold O. Lee III for his assistance with using scanning electron microscope.
References
- D. Yuan and B. Xu, J. Mater. Chem. B, 2016, 4, 5638–5649 RSC.
- M. D. Segarra-Maset, V. J. Nebot, J. F. Miravet and B. Escuder, Chem. Soc. Rev., 2013, 42, 7086–7098 RSC.
- A. H. Karoyo and L. D. Wilson, Gels, 2017, 3, 1 CrossRef.
- M. Liu, G. Ouyang, D. Niu and Y. Sang, Org. Chem. Front., 2018, 5, 2885–2900 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS.
- A. Tanaka, Y. Fukuoka, Y. Morimoto, T. Honjo, D. Koda, M. Goto and T. Maruyama, J. Am. Chem. Soc., 2015, 137, 770–775 CrossRef CAS.
- J. Zeng, Y. Yin, L. Zhang, W. Hu, C. Zhang and W. Chen, Macromol. Biosci., 2016, 16, 363–370 CrossRef CAS.
- K. Lalitha, Y. S. Prasad, C. U. Maheswari, V. Sridharan, G. John and S. Nagarajan, J. Mater. Chem. B, 2015, 3, 5560–5568 RSC.
- B. Escuder, F. Rodriguez-Llansola and J. F. Miravet, New J. Chem., 2010, 34, 1044–1054 RSC.
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- H. Oh, N. Yaraghi and S. R. Raghavan, Langmuir, 2015, 31, 5259–5264 CrossRef CAS.
- R. Balamurugan, Y. S. Zhang, S. Fitriyani and J. H. Liu, Soft Matter, 2016, 12, 5214–5223 RSC.
- M. Piccinno, C. A. Angulo-Pachon, P. Ballester, B. Escuder and A. D. Cort, RSC Adv., 2016, 6, 57306–57309 RSC.
- P. Byrne, G. O. Lloyd, L. Applegarth, K. M. Anderson, N. Clarke and J. W. Steed, New J. Chem., 2010, 34, 2261–2274 RSC.
- L. Viau, C. Tourne-Peteilh, J.-M. Devoisselle and A. Vioux, Chem. Commun., 2010, 46, 228–230 RSC.
- T. Xiao and L. Wang, Tetrahedron Lett., 2018, 59, 1172–1182 CrossRef CAS.
- T. Xiao, L. Xu, L. Zhou, X.-Q. Sun, C. Lin and L. Wang, J. Mater. Chem. B, 2019, 7, 1526–1540 RSC.
- H. Maeda, Chem.–Eur. J., 2008, 14, 11274–11282 CrossRef CAS.
- H. Wu, J. Zheng, A. L. Kjoniksen, W. Wang, Y. Zhang and J. Ma, Adv. Mater., 2019, 31, e1806204 CrossRef.
- M. O. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS.
- J. Le Bideau, L. Viau and A. Vioux, Chem. Soc. Rev., 2011, 40, 907–925 RSC.
- A. Ghosh, P. Das, R. Kaushik, K. K. Damodaran and D. A. Jose, RSC Adv., 2016, 6, 83303–83311 RSC.
- N. Malviya, M. Das, P. Mandal and S. Mukhopadhyay, Soft
Matter, 2017, 13, 6243–6249 RSC.
- Y. Zhai, W. Chai, W. Cao, Z. Sun and Y. Huang, Front. Chem. Sci. Eng., 2015, 9, 488–493 CrossRef CAS.
- Q. Lin, X. Zhu, Y.-P. Fu, Y.-M. Zhang, R. Fang, L.-Z. Yang and T.-B. Wei, Soft Matter, 2014, 10, 5715–5723 RSC.
- N. Malviya, C. Sonkar, B. K. Kundu and S. Mukhopadhyay, Langmuir, 2018, 34, 11575–11585 CrossRef CAS.
- P. Xue, B. Yao, P. Wang, P. Gong, Z. Zhang and R. Lu, Chem.–Eur. J., 2015, 21, 17508–17515 CrossRef CAS.
- S. Sarkar, S. Dutta, S. Chakrabarti, P. Bairi and T. Pal, ACS Appl. Mater. Interfaces, 2014, 6, 6308–6316 CrossRef CAS.
- L. Li, N. Zhou, H. Kong and X. He, Polym. Chem., 2019, 10, 5465–5472 RSC.
- H. Yao, J. Wang, Q. Zhou, X.-W. Guan, Y.-Q. Fan, Y.-M. Zhang, T.-B. Wei and Q. Lin, Soft Matter, 2018, 14, 8390–8394 RSC.
- L. He, Z. W. Peng, Z. W. Jiang, X. Q. Tang, C. Z. Huang and Y. F. Li, ACS Appl. Mater. Interfaces, 2017, 9, 31834–31840 CrossRef CAS.
- J. Zhou, J. Li, X. Du and B. Xu, Biomaterials, 2017, 129, 1–27 CrossRef CAS.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS.
- F. Ono, H. Watanabe and S. Shinkai, RSC Adv., 2014, 4, 25940–25947 RSC.
- S. Datta and S. Bhattacharya, Chem. Soc. Rev., 2015, 44, 5596–5637 RSC.
- J. G. Hardy, A. R. Hirst, I. Ashworth, C. Brennan and D. K. Smith, Tetrahedron, 2007, 63, 7397–7406 CrossRef CAS.
- O. Demir-Ordu, H. Simsir and K. Alper, Tetrahedron, 2015, 71, 1529–1539 CrossRef CAS.
- G. Wang, S. Cheuk, K. Williams, V. Sharma, L. Dakessian and Z. Thorton, Carbohydr. Res., 2006, 341, 705–716 CrossRef CAS.
- G. Wang, S. Cheuk, H. Yang, N. Goyal, P. V. N. Reddy and B. Hopkinson, Langmuir, 2009, 25, 8696–8705 CrossRef CAS.
- J. E. Webb, M. J. Crossley, P. Turner and P. Thordarson, J. Am. Chem. Soc., 2007, 129, 7155–7162 CrossRef CAS.
- M. Bielejewski, K. Nowicka, N. Bielejewska and J. Tritt-Goc, J. Electrochem. Soc., 2016, 163, G187–G195 CrossRef CAS.
- C.-C. Tsai, W.-T. Chuang, Y.-F. Tsai, J.-T. Li, Y.-F. Wu and C.-C. Liao, J. Mater. Chem. B, 2013, 1, 819–827 RSC.
- S. Baddi, D. S. Sarma and A. Palanisamy, J. Sol-Gel Sci. Technol., 2016, 79, 637–649 CrossRef CAS.
- L. Lascialfari, G. Pescitelli, A. Brandi, M. Mannini, D. Berti and S. Cicchi, Soft Matter, 2015, 11, 8333–8341 RSC.
- A. D'Aleo, J.-L. Pozzo, K. Heuze, F. Voegtle and F. Fages, Tetrahedron, 2007, 63, 7482–7488 CrossRef.
- A. Chen, L. P. Samankumara, C. Garcia, K. Bashaw and G. Wang, New J. Chem., 2019, 43, 7950–7961 RSC.
- G. Wang, N. Goyal, H. P. R. Mangunuru, H. Yang, S. Cheuk and P. V. N. Reddy, J. Org. Chem., 2015, 80, 733–743 CrossRef CAS.
- N. Goyal, S. Cheuk and G. Wang, Tetrahedron, 2010, 66, 5962–5971 CrossRef CAS.
- G. Wang, A. Chen, H. P. R. Mangunuru and J. R. Yerabolu, RSC Adv., 2017, 7, 40887–40895 RSC.
- A. Chen, D. Wang, L. P. Samankumara and G. Wang, Synthesis, 2019, 51, 2897–2908 CrossRef CAS.
- M. Bielejewski, A. Lapinski and O. Demchuk, J. Colloid Interface Sci., 2017, 490, 279–286 CrossRef CAS.
- C. Po, Z. Ke, A. Y.-Y. Tam, H.-F. Chow and V. W.-W. Yam, Chem.–Eur. J., 2013, 19, 15735–15744 CrossRef CAS.
- Q. Lin, P.-P. Mao, Y.-Q. Fan, P.-P. Jia, J. Liu, Y.-M. Zhang, H. Yao and T.-B. Wei, Soft Matter, 2017, 13, 7360–7364 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Gelation properties in tetramethylammonium bromide and tetrabutylammonium salts, stability of the gels in basic conditions, NMR spectra for compounds 13 and 20 at different temperatures; NMR studies of compounds 18 and 20 with different amount of TBABr, additional gel photos for naproxen. The 1H and 13C NMR spectra of all intermediates and final products are provided. See DOI: 10.1039/d0ra07587f |
| ‡ Current address: TCG GreenChem, Inc. 737 North 5th Street, Suite 501, Richmond, VA 23219, USA. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Joedian Morris and
Guijun Wang
,
Joedian Morris and
Guijun Wang