DOI:
10.1039/D0RA07567A
(Paper)
RSC Adv., 2020,
10, 41393-41402
Visible light responsive TiO2 photocatalysts for degradation of indoor acetaldehyde†
Received
3rd September 2020
, Accepted 5th November 2020
First published on 12th November 2020
Abstract
Photocatalysis is a promising technique for developing sustainable and environmentally friendly materials to improve indoor air quality. Visible-light-responsive TiO2 has been widely investigated but there are inconsistent results because photocatalytic properties depend strongly on synthetic methods. Herein, we synthesize TiO2 doped with 10 different metal ions (M–TiO2) by conducting a dialysis in a sol–gel method to obtain the best photocatalyst for the degradation of acetaldehyde under LED irradiation. Purification of a sol by dialysis enables us to discuss pure effects of dopants on the photocatalytic activity because impurities such as counter ions of metal salts are removed before sintering. Only Cr–, Pt–, V–, and Fe–TiO2 show photocatalytic activity and the optimal doping amounts are 0.50–1.7, 0.10, 1.0, and 0.10 atom%, respectively. Such differences in the optimal amounts can be explained in terms of the dopant ions having different valence states, suggesting the formation of oxygen vacancies. The Cr–TiO2 powder exhibits high activity even at the doping amount of 4.2 atom%. We also demonstrate that the Cr–TiO2 film prepared on a glass substrate can be used to degrade acetaldehyde by changing the film thickness and the LED intensity depending on the degree of the indoor contamination.
Introduction
Indoor air quality has become a major concern since people spend more than 80% of their time in indoor environments.1,2 Many volatile organic compounds (VOCs) including formaldehyde, acetaldehyde (CH3CHO), and toluene are detected as indoor pollutants.3 These chemicals are emitted continuously to the indoor air from household products such as paints, cleaning agents, constructive materials, and furniture.4–6 Many technologies have been investigated to reduce the concentration of indoor VOCs by using mainly CH3CHO as a probe molecule. Adsorption on carbon-based materials is the most used technology for the removal of VOCs because of its easy operation.7,8 However, regeneration of adsorbents is needed for the use of adsorption process. Photocatalytic oxidation is the most promising technique for developing sustainable and environmentally friendly materials to improve indoor air quality9,10 because it promotes the decomposition of VOCs to CO2 and H2O. Especially, titanium dioxide (TiO2) has been widely investigated for outdoor and indoor air purification.5,6,11 However, TiO2 can be activated only under ultraviolet (UV) irradiation because of the wide band gap (3.2 eV). Such UV light is scarce in indoor lighting since white light emitting diode (LED) has been widely utilized instead of fluorescent lamps for energy saving. Doping of TiO2 with metal or non-metal elements is a well-known strategy for promoting the absorption of visible light (VL) by creating new energy levels in the band gap of TiO2. TiO2 doped with Pt,12 Mn,1 Ir,13 or C4 and co-doped with C and V14 or with N and Fe2 have been reported as active photocatalysts for the degradation of CH3CHO under VL irradiation. There are many studies on photocatalysis of TiO2 doped with other elements. However, photocatalytic properties depend strongly on the synthetic method. This fact seems to be reasonable because the location and the energy level of the dopants in TiO2 might be affected by the presence of impurities (e.g. counter ions of metal salts) during the sintering process and might be varied depending on whether the doping is conducted by using commercially available TiO2 powder or by preparing TiO2 nanoparticles in a sol–gel method. On the other hand, understanding the factors affecting photocatalytic activity is needed to develop more excellent materials for indoor air purification. In this study, TiO2 doped with 10 different metal ions (M–TiO2) are synthesized by conducting a dialysis in the sol–gel method using Ti(OC3H7)4 (TTIP) as a staring material. By conducting the dialysis, proton, 2-propanol which is formed by hydrolysis of TTIP, and counter ions of the metal salts used for doping are removed before the sintering. Furthermore, we have already found that during dialysis, V(III), Pt(IV) or Ru(IV) remained in the TiO2 sol but in the case of a larger metal ion such as Cu(II) or Co(II), most of them moved out of the TiO2 sol through the dialysis tube.15,16 This finding indicates that metal ion dopants can be divided into two groups, i.e. with and without some interaction with TiO2 nanoparticles in the sol. By sintering the purified sol after the dialysis, only metal ions which interact with TiO2 nanoparticles in the sol can be doped. This might be useful to elucidate the factors affecting the photocatalysis because we do not have to consider two different situations, with and without the interaction between metal ions and TiO2 nanoparticles. The purpose of this study is clarifying the best choice of M–TiO2 for the degradation of CH3CHO under LED irradiation and understanding the factors affecting the photocatalysis. Furthermore, the photocatalytic activity of the M–TiO2 film is evaluated because the film is more appropriate than powders to the practical application for indoor air purification.
Experimental
Synthesis of photocatalysts
Approximately 15 ml of TTIP was added dropwise to 180 ml of aqueous solutions containing 1.3 ml of HNO3. The obtained suspension was stirred at room temperature with a magnetic stirrer for 6 days to get a highly dispersed colloidal solution. This sol was dialyzed in 2750 ml of water by using a precleaned molecularly porous dialysis tubing (Spectra/Por® 3, molecular weight cut-off: 3500) for 2 days by changing water once a day to get pH 2. The cleaning method of the dialysis tubing was described in ESI.† Then, the obtained transparent sol was placed in a beaker and mixed with 2 ml of aqueous solutions containing CrCl3·6H2O, H2PtCl6·6H2O, VCl3, FeCl3·6H2O, CuCl2, MnCl2·4H2O, Na2MoO4·2H2O, Na2WO4·2H2O, NbCl5, or RuCl3·nH2O to get the concentration of metal ion of 0.1–10 atom% against the total of Ti and the metal ion. After stirring, this solution was dialyzed for 1 day to remove counter ions of metal salts. Removal of the counter ions was confirmed by using ion chromatograph (Shimadzu, PIA-1000, column: Shim-pack IC-A3(S)). For example, 0.1036 g of CrCl3·6H2O was used for the synthesis of 0.8 atom% Cr–TiO2. The concentration of metal ion which was moved out of the dialysis tube during the dialysis was evaluated by inductively coupled plasma atomic emission spectroscopy (Varian, ICP-AES Liberty Series II and Agilent Technologies, 5110 ICP-OES). A finally obtained solution (M–TiO2 sol) was dried, crushed into powder, and sintered at 500 °C for 3 h with ramping rate at 3 °C min−1. For comparison, pure TiO2 was synthesized by conducting the dialysis for 3 days without the addition of the metal ions. Table S1† lists the amounts of metal ions added in the synthesis and those doped in the finally obtained samples.
Preparation of films
Three coating methods were employed to prepare the Cr–TiO2, Pt–TiO2 and V–TiO2 films on slide glasses (4.0 × 1.5 cm2) which were precleaned with sonication in water and acetone. A suspension of 0.10 g of the Cr–TiO2, Pt–TiO2 or V–TiO2 powder in 10 ml of ethanol was used for making films by using a spin coater (Kyowariken, K359S1) with a rotation at 500 rpm for 15 s followed by that at 2000 rpm for 30 s. This process was repeated 5 times as a standard condition before the sintering. The obtained film was denoted as M–TiO2_SC. To make a thick Cr–TiO2 film (Cr–TiO2_TK), 0.30 g of Cr–TiO2 was mixed with 0.015 g of polyethylene glycol and 1.5 ml of ethanol by using a mortar and a pestle and was spread in a recess which was made by using adhesive tapes with a height of 53 μm as spacers on the slide glass. The obtained surface was flattened by moving a glass rod, dried and sintered after removing the tapes. The Cr–TiO2_DC film was prepared by using the Cr–TiO2 sol just after the dialysis and a dip-coater (SDI, DT-0001-S1) under the condition of withdrawal/immersion rate of 9.0 mm s−1 and dip-duration of 10 min followed by the sintering. This procedure was repeated three times. Sintering conditions for making all films were the same as those for the synthesis of the M–TiO2 powder.
Characterization
X-ray diffraction (XRD, Rigaku, Miniflex 600) analysis was performed with Cu Kα radiation (40 kV, 15 mA) at 2θ angles from 10° to 90° with a scan speed of 10° min−1. The Brunauer–Emmett–Teller (BET) specific surface area was measured with nitrogen as the adsorptive gas by automatic surface area analyzer (Shimadzu, Tristar II 3020). Diffuse reflectance UV-vis absorption spectra of the powder samples were obtained by using a spectrophotometer (JASCO, V-670). The valence state of Cr, Pt or V ion near the TiO2 surface was analyzed by X-ray photoelectron spectroscopy (XPS, Thermo SCIENTIFIC, K-Alpha). The binding energies were calibrated with reference to the C 1s peak (284.6 eV) originating from the surface impurity carbons. Electrochemical Impedance Spectroscopy (EIS) was measured to evaluate a flat band potential of TiO2 or M–TiO2 electrode by using potentiostat/galvanostat (Solartron Analytical, SI 1287) and frequency response analyzer (Solartron Analytical, SI 1260). These electrodes were prepared on transparent conductive substrate (fluorine-doped tin oxide (FTO) on glass, GEOMATEC, 10 Ω sq−1) in the same way as Cr–TiO2_DC film although the coating process was not repeated. In the electrochemical cell, a platinum electrode was used as the counter electrode, Ag/AgCl (in saturated KCl) as a reference, and 1.0 mol dm−3 NaOH solution as an electrolyte and bubbling with argon gas was conducted to remove oxygen before the measurements.
Photocatalytic degradation of gaseous CH3CHO
The synthesized M–TiO2 powder (0.03 g) or film was placed in a transparent Pyrex vial (40 ml, capped with a Mininert valve) with two small branches, each of which was connected with a two-way valve by Fluran tubing. Reactant gas stream containing CH3CHO, oxygen, and water vapor was prepared by adjusting the flowrate of the gas stream from gas cylinders containing CH3CHO (199 ppmv, N2 balance, Taiyo Nippon Sanso Co.), oxygen (>99.9%, Iwatani Co.) and nitrogen (>99.9%, Taiyo Nippon Sanso Co.) and by passing partly through the saturator containing water. After the reactant gas was passed through the vial for 1 h in the dark to achieve an adsorption–desorption equilibrium on M–TiO2, the two-way valves were closed. The gas of 300 μl was taken by using a gas-tight syringe through the Mininert valve and analyzed by gas-chromatography (Shimadzu, GC-2014, equipped with flame ionization and thermal conductivity detectors). It is noted that 300 μl of nitrogen was injected in the vial just before the sampling to suppress the pressure decrease through the experiments. After confirming the constant concentration of CH3CHO by GC, the vial was irradiated with 16 W LED (LDR100-220 V 16N–H, Mitsubishi) or 4.4 W LED (LDA4N-G/40 W, Toshiba) which did not emit light with wavelength shorter than 400 nm. The light intensity was measured with a solar power meter (MK-Scientific, TM-207) to be 1.0 or 0.10 mW cm−2 for 16 W or 4.4 W LED, respectively. The photocatalytic experiments were performed at 25 ± 1 °C by circulating the surrounding air and the sampling was conducted every 20 min. Initial reaction rate (r0) was evaluated from the slope of the plots of the concentration of CH3CHO vs. irradiation time (r0 = −d[CH3CHO]t=0/dt). In this paper, r0 is expressed in the unit of ppmv min−1 where 1 ppmv min−1 corresponds to 1.64 × 10−9 mol min−1. Unless otherwise noted, the photocatalytic experiments were performed in the reactant gas containing CH3CHO (mole fraction: 1.0 × 10−4), oxygen (0.20), and water vapor (6.0 × 10−3).
Result and discussion
Effect of amounts of metal ion dopants in M–TiO2
Fig. 1 indicates dependence of r0 on the amounts of metal ion in M–TiO2 sintered at 500 °C. Only Cr–, Pt–, V–, and Fe–TiO2 show higher activity than TiO2 which possesses r0 of 0.42 ppmv min−1 as shown by a dashed line in Fig. 1. The Cr–TiO2 powder exhibits the highest r0 value (1.17 ppmv min−1) at 0.5 atom% but the r0 value does not change significantly at the doping amount of 0.5–1.7 atom% (r0 = 1.12 ± 0.05 ppmv min−1). Thus, we select 0.8 atom% Cr–TiO2 as a standard sample in later discussion. In the case of Pt–TiO2 powder, the r0 value is 1.11 ppmv min−1 at 0.10 atom% and decreases drastically with an increase in the doping amount. A similar behavior is observed with Fe–TiO2 although the highest r0 value is about half of that of Pt–TiO2. The optimal doping amount of V–TiO2 is 1.0 atom% (r0 = 1.08 ppmv min−1). Doping of Pt ≥ 1.0 atom%, Fe ≥ 0.5 atom% or other metal ions lowers the photocatalytic activity of TiO2, suggesting that these metal ions might act as the recombination center of the photogenerated holes and electrons. Hereafter, characterization of the Cr–TiO2, Pt–TiO2, and V–TiO2 powder was compared to explain their differences in the photocatalytic performance.
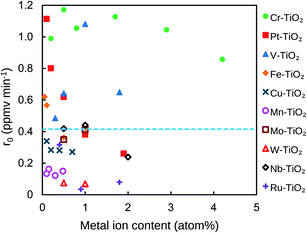 |
| | Fig. 1 Effect of metal ion content in M–TiO2 on the initial degradation rate of CH3CHO. The broken line indicates the initial degradation rate of CH3CHO on TiO2 powder. | |
Comparison of Cr–TiO2, Pt–TiO2, and V–TiO2
Fig. S1† shows XRD patterns of Cr–TiO2, Pt–TiO2, V–TiO2, and TiO2 sintered at 500 °C, indicating that the former three samples exhibit the peaks attributable to anatase, brookite, and rutile but TiO2 shows only rutile which is the most thermodynamically stable phase. Fig. 2 indicates the composition ratio of anatase, rutile, and brookite, which was evaluated by Rietveld analysis for the XRD patterns. We also examined the effect of the sintering temperature on the r0 values in the range of 200–600 °C by using 0.8 atom% Cr–TiO2, 0.1 atom% Pt–TiO2, and 1.0 atom% V–TiO2. Both Pt–TiO2 and V–TiO2 exhibited the highest r0 values when being sintered at 500 °C although the r0 value for Cr–TiO2 sintered at 400 °C was slightly higher than that at 500 °C (Fig. S2†). Besides, for the preparation of the FTO electrode and the slide glass which were coated with M–TiO2 as discussed later, sintering at 500 °C was needed to prevent the film from being peeled off from the substrates. Thus, we selected the sintering temperature to be 500 °C for the comparison of the r0 values for Cr–TiO2, Pt–TiO2, and V–TiO2 powder.
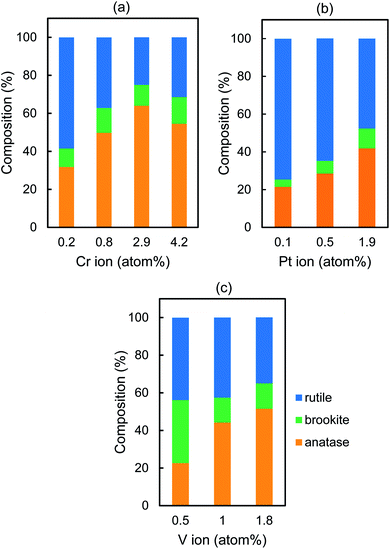 |
| | Fig. 2 Crystal phase composition of (a) Cr–TiO2, (b) Pt–TiO2, and (c) V–TiO2 powder. | |
Fig. S3† shows the UV-vis diffuse reflectance spectra, indicating that absorption edge is shifted to the longer wavelength with increasing the doping amount. The band gap energy (Eg) was calculated by Tauc plots in which indirect transition band gap was postulated (Table 1).17–19 The Cr–TiO2, Pt–TiO2, and V–TiO2 samples possess a similar tendency that the transformation to rutile is suppressed and Eg decreases as the doping amount increases. Table 1 also lists the BET specific surface area (SSA) of these samples, which does not change systematically except for Cr–TiO2. These findings cannot explain the different behavior observed with Cr–TiO2, Pt–TiO2, and V–TiO2 as shown in Fig. 1.
Table 1 Band gap energy (Eg) and BET specific surface area (SSA) of Cr–TiO2, Pt–TiO2, and V–TiO2 powder
| Cr content (atom%) |
Cr–TiO2 |
Pt content (atom%) |
Pt–TiO2 |
V content (atom%) |
V–TiO2 |
| Eg (eV) |
SSA (m2 g−1) |
Eg (eV) |
SSA (m2 g−1) |
Eg (eV) |
SSA (m2 g−1) |
| 0.20 |
2.82 |
75.5 |
0.10 |
2.90 |
34.4 |
0.50 |
3.01 |
56.9 |
| 0.80 |
2.80 |
50.5 |
0.50 |
2.70 |
51.9 |
1.0 |
2.86 |
38.8 |
| 1.7 |
2.44 |
40.0 |
1.0 |
2.69 |
43.0 |
1.8 |
2.71 |
65.4 |
| 2.9 |
2.40 |
— |
1.9 |
2.47 |
— |
|
|
|
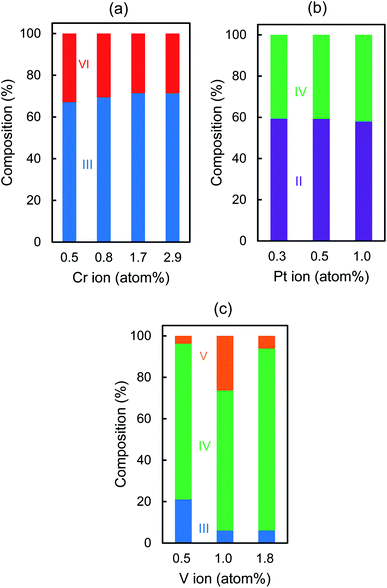
Fig. 3 shows the valence states of the metal ion dopants which are evaluated from the peak area after deconvolution in the XPS spectra (Fig. S4†). In the case of Cr–TiO2 or Pt–TiO2, Cr(VI) or Pt(II) is formed during the synthesis and the ratio of Cr(III)/Cr(VI) or Pt(II)/Pt(IV) keeps almost constant, i.e. 2.3 ± 0.3 or 1.4 ± 0.1, respectively, regardless of the doping amounts. On the other hand, V(III), V(IV), and V(V) are detected and V(IV) exists predominantly in all V–TiO2 samples. Flat band potential of TiO2 is affected by the doping of metal ions. Fig. S5† indicates Mott–Schottky plots of the TiO2 film prepared on FTO under various frequencies in the range of 250–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz, indicating positive slopes which are typical for n-type semiconductors. A carrier density (ND) and a flat band potential (VFB) of TiO2 were estimated to be 3.6 × 1018 cm−3 and −0.76 V vs. Ag/AgCl, respectively, at 1000 Hz.
000 Hz, indicating positive slopes which are typical for n-type semiconductors. A carrier density (ND) and a flat band potential (VFB) of TiO2 were estimated to be 3.6 × 1018 cm−3 and −0.76 V vs. Ag/AgCl, respectively, at 1000 Hz.
 |
| | Fig. 3 Atomic valence composition of (a) Cr–TiO2, (b) Pt–TiO2, and (c) V–TiO2 powder. | |
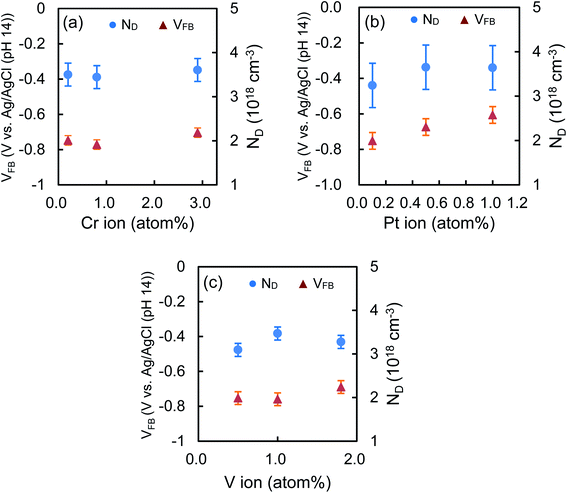
Fig. 4 shows the effect of the doping amounts on VFB and ND of the Cr–TiO2, Pt–TiO2 and V–TiO2 film, which were evaluated at 1000 Hz. The ND values of these samples are almost the same as that of TiO2 even if the doping amounts are changed. This fact is presumably due to the low doping amounts in this study. The VFB values of Cr–TiO2 and V–TiO2 are estimated to be −0.74 ± 0.04 and −0.73 ± 0.05 V, respectively. The position of the CB edge of n-type semiconductor is adjacent to VFB. Takle et al. reported that the doping of Cr(III) created a new energy level just above the VB edge of TiO2.20 Rossi et al. reported that the energy level induced by doping of V(IV) was located at ca. 2.2 eV below the CB edge of TiO2.21 Because of such deep levels of Cr(III) and V(IV) dopants, it is likely that the VFB values of Cr–TiO2 and V–TiO2 are not affected by the doping and are nearly equal to that of TiO2. On the other hand, the VFB of Pt–TiO2 increases from −0.75 to −0.61 V with increasing the doping amount from 0.1 to 1.0 atom%. Kim et al. also reported that VFB was shifted positively by 50 mV by doping Pt ion in TiO2, indicating the CB edge position was slightly lowered.22 The remarkable decrease in the photocatalytic degradation of CH3CHO on Pt–TiO2 with increasing the doping amounts is attributable to the lower CB edge which retards the transfer of the photogenerated electron to oxygen, resulting in the enhancement of the recombination. Table 2 lists the average values of the composition ratio of the dopant ions which are calculated from Fig. 3 with their ionic radii in parentheses. The ionic radius of Cr(III), Pt(IV), or V(III), whose compounds are used as the starting materials in the synthesis, is 0.755, 0.765, or 0.78 Å, respectively.23 Because their size is close to Ti(IV) (0.745 Å), they are easily substituted for Ti(IV) during the synthesis. After being sintered at 500 °C, Cr(III) still exists predominantly (69.9%) and is partially oxidized to Cr(VI) (30.1%).
 |
| | Fig. 4 VFB and ND of (a) Cr–TiO2, (b) Pt–TiO2, and (c) V–TiO2 powder. | |
Table 2 Ionic radii (Å) of metal ions and their average composition ratio (%)a
| Valence |
| 6+ |
5+ |
4+ |
3+ |
2+ |
| Ionic radii cited from literature23 are written in parentheses. |
| |
|
Ti (0.745) |
|
|
| Cr (0.58) 30.1 |
|
|
Cr (0.755) 69.9 |
|
| |
|
Pt (0.765) 41.2 |
|
Pt (0.94) 58.8 |
| |
V (0.68) 12.1 |
V (0.72) 76.8 |
V (0.78) 11.1 |
|
In the case of V–TiO2, V(IV) is formed predominantly (76.8%) together with V(III) (11.1%) and V(V) (12.1%), among which V(IV) and V(III) have a similar size as Ti(IV). Substitution of Ti(IV) with metal ions having a lower valence state, e.g. Cr(III) or V(III), induces the removal of oxygen atom from the TiO2 lattice to balance the charge, leading to the formation of oxygen vacancy. Some research groups reported that the presence of oxygen vacancy enhanced the photocatalytic activity by trapping the photogenerated electron to suppress the recombination.24–29 It is likely that more oxygen vacancies are created in Cr–TiO2 compared with V–TiO2 because the main component of Cr–TiO2 or V–TiO2 is Cr(III) or V(IV), respectively. Therefore, the high performance of Cr–TiO2 is attributable to the formation of more oxygen vacancies. In the case of V–TiO2, the optimal doping amount was 1.0 atom% where the highest amount of V(V) was detected (Fig. 3c). This finding suggests that the presence of V(V) enhances the photocatalytic activity. Ren et al. synthesized the V-doped TiO2 by using titanic acid and NH4VO3 and reported that a part of V(V) was reduced to V(IV) into TiO2 lattice and others existed as V2O5 on the surface where V(V) received the photogenerated electrons, resulting in the effective separation of the photogenerated carriers.30 In the case of Pt–TiO2, more Pt(II) are formed than Pt(IV). The ionic radius of Pt(II) (0.94 Å) is much larger than Pt(IV) or Ti(IV). Previously, we demonstrated by measuring XPS and XANES of Pt–TiO2 sintered at 200 °C that Pt(II) ion existed near the surface and only Pt(IV) was located in the bulk.31 This finding was easily explained in terms of their ionic radii. Mukri et al. reported that the presence of Pt(II) brought about its d states above the VB edge and oxygen vacancies.32 They demonstrated that Pt(II)-substituted TiO2 with oxygen vacancies exhibited higher photocatalytic activity for CO oxidation than Pt(IV)-substituted TiO2 without oxygen vacancies. Kim et al. mentioned that Pt dopants created energy levels above the VB edge where the trapped holes are highly localized and the oxygen vacancies were created near the Pt(II) sites.33 Indeed, we reported that the photocatalytic degradation of 4-chlorophenol was enhanced with increasing the ratio of Pt(II)/Pt(IV) in the range of 2.7–21.2 in Pt–TiO2 sintered at 200 °C.31 On the other hand, in this study, the ratio of Pt(II)/Pt(IV) was estimated to be 1.4 ± 0.1 for Pt–TiO2 sintered at 500 °C, indicating that the amounts of Pt(II) and Pt(IV) are almost comparable. Kim et al. mentioned that the presence of both Pt(II) and Pt(IV) might reduce the beneficial role of Pt(II) because the energy level created with Pt(II) and Pt(IV) lay close to each other, leading to the recombination of the photogenerated electron–hole pair.33 Therefore, lower photocatalytic activity observed with Pt–TiO2 is due to the less formation of effective oxygen vacancies near Pt(II) compared with the oxygen vacancies around Cr(III) in Cr–TiO2. There is another possibility that oxygen vacancies induced by Cr(III) in the TiO2 bulk and those by Pt(II) near the surface might be different. Our findings on the different dependence of r0 on the doping amounts of Cr–, Pt– and V–TiO2 can be explained in terms of the difference in their main valence states which are correlated with the formation of the oxygen vacancies. Further studies are needed to conclude the oxygen vacancies as the most important factor for the VL-responsive M–TiO2 photocatalyst. For that purpose, developing the method for a quantitative analysis of the oxygen vacancies is required. It is worthy to note that 0.8 atom% Cr–TiO2 prepared by conducting the dialysis shows much higher activity than that without the dialysis (Fig. S6†). Conducting dialysis is beneficial not only for the removal of impurities before the sintering process but also for the enhancement of the photocatalytic activity. Hereafter, the photocatalytic activity of 0.8 atom% Cr–TiO2 for the degradation of CH3CHO is discussed.
Photocatalytic degradation of CH3CHO on Cr–TiO2
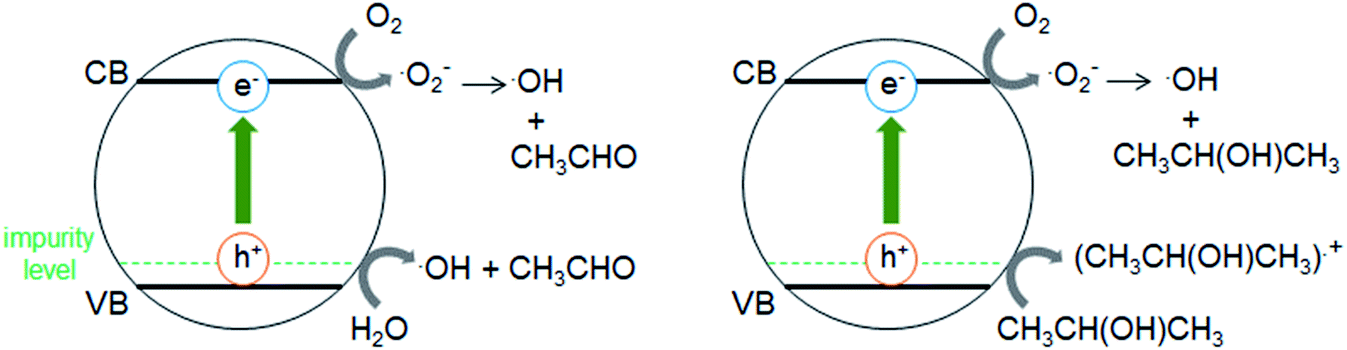
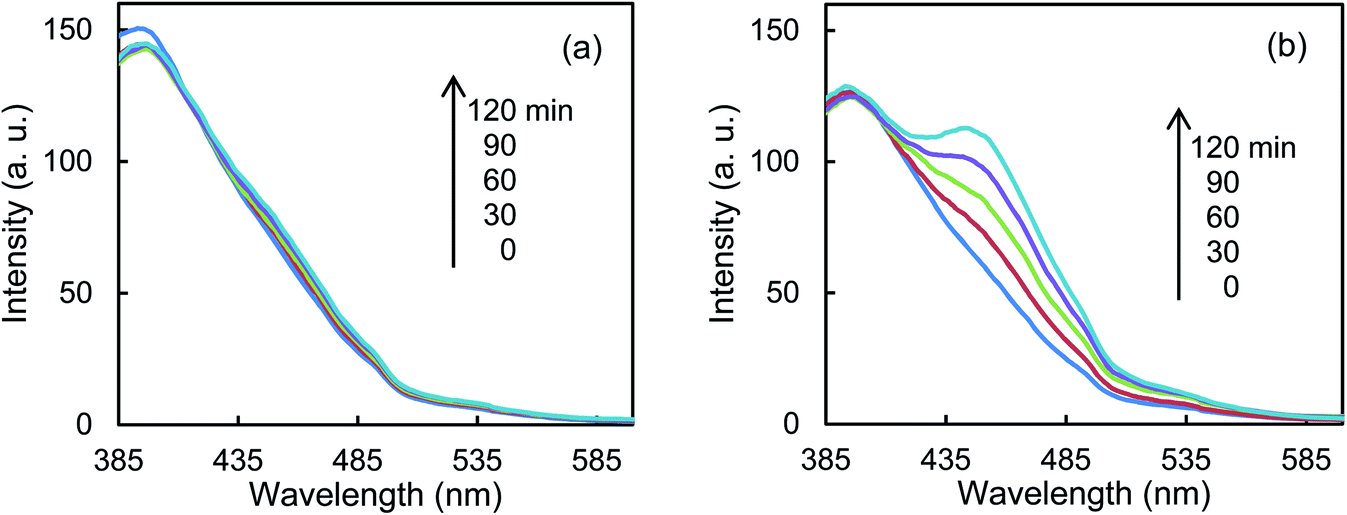
Fig. 5a shows that the r0 value is almost independent on the mole fraction of oxygen (5.0 × 10−6–0.50) when the experiments are performed in the reactant gas containing water vapor (mole fraction: 6.0 × 10−3) and CH3CHO (mole fraction: 1.0 × 10−4 or 5.0 × 10−4). In the absence of water vapor, CH3CHO is not degraded and the r0 value increases to reach a constant value of 1.09 ± 0.03 ppmv min−1 (Fig. 5b). Fig. 5c exhibits a linear relationship between r0−1 vs. [CH3CHO]−1, indicating that the reaction follows the Langmuir–Hinshelwood kinetic model where adsorption of the reactant is assumed prior to reaction. Generally, for the photocatalytic degradation of organic compounds in the gas phase, the degradation rate decreases with an increase in the mole fraction of water vapor due to the competitive adsorption on reactive sites. Indeed, as shown in Fig. 5b, when 2-propanol is used instead of CH3CHO in the experiments, the r0 value decreases with increasing mole fraction of water vapor. To understand such a different behavior, we measured the time course of the concentration of CH3CHO or 2-propanol in the dark by injecting 10 ml of 198 ppmv CH3CHO or 199 ppmv 2-propanol to the vial containing oxygen (mole fraction of 0.20), water vapor (0, 6.0 × 10−3, or 1.6 × 10−2) and 3.0 × 10−3 g of 0.8 atom% Cr–TiO2. In the absence of water vapor, 17.9 ± 1.4 ppmv of CH3CHO was detected but no 2-propanol was observed, indicating all of 2-propanol was adsorbed on the Cr–TiO2 surface (Fig. S7†). The adsorbed amounts of CH3CHO or 2-propanol was calculated by analyzing the data in Fig. S7.† Fig. 5d shows the adsorbed amount of CH3CHO decreases with an increase in the water vapor and is less than that of 2-propanol at any water vapor mole fraction. These results indicate that both CH3CHO and 2-propanol adsorb on Cr–TiO2 competitively with water but the adsorption of 2-propanol is more strongly than CH3CHO. The different dependence of r0 on the mole fraction of water vapor as shown in Fig. 5b suggests that 2-propanol is mainly degraded by the photogenerated holes whereas CH3CHO is degraded by OH radicals which are formed by the oxidation of water vapor by the photogenerated holes (Scheme 1). Therefore, the presence of water vapor is crucial for the degradation of CH3CHO. Takeuchi et al. reported that CH3CHO was oxidized by the OH radicals which were formed by the photooxidation of H2O molecules on TiO2 under UV irradiation.34 It is well-known that coumarin traps OH radical to generate 7-hydroxycoumarin (7-HC) which emits fluorescence at ca. 460 nm.35 We measured the fluorescence spectra of aqueous solutions containing coumarin (5.0 × 10−5 mol dm−3) and 0.2 wt% of TiO2 or Cr–TiO2 under VL irradiation for 120 min. Fig. 6 shows that the fluorescence at 460 nm increases slightly in the presence of TiO2 whereas it remarkably increases in the case of Cr–TiO2. This finding demonstrates the formation of OH radicals on Cr–TiO2 under VL irradiation. Fig. 7a shows the time course of CH3CHO and CO2 on Cr–TiO2, indicating that 80 ppmv of CH3CHO is degraded under VL irradiation for 80 min but the concentration of CO2 continues to increase. At the irradiation for 720 min, the stoichiometric ratio of [CO2]formed/[CH3CHO]degraded was estimated to be 2.39, which is above the theoretical value for the complete mineralization and can be ascribed to CH3CHO preadsorbed on Cr–TiO2 before the VL irradiation. Fig. 7b shows diffuse reflectance of FTIR on Cr–TiO2, indicating that new peaks appear at 1529 and 1440 cm−1 at the VL irradiation for 20 min and decrease gradually with increasing the irradiation time. These two peaks are coincident with those observed with TiO2 which was immersed in acetic acid (1 mol dm−3) followed by dryness (Fig. 7b). Acetic acid is formed by the photocatalytic oxidation of CH3CHO on TiO2 under UV irradiation.36 Therefore, the peaks at 1529 and 1440 cm−1 are attributable to νas(COO) and δas(CH3), respectively.37 It is noted that these peaks can be seen even at the VL irradiation for 720 min, indicating that a small quantity of acetic acid exists on the Cr–TiO2 surface.
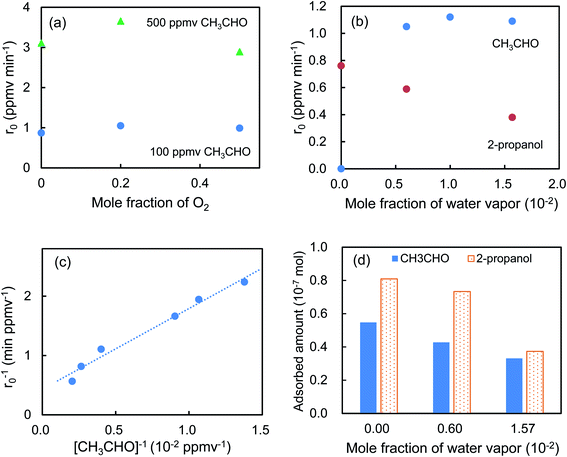 |
| | Fig. 5 Effect of mole fractions of (a) oxygen, (b) water vapor, and (c) CH3CHO on the r0 values. (d) Dependence of the adsorbed amount of CH3CHO on the mole fraction of water vapor. For comparison, the data obtained with 2-propanol were also depicted in (b) and (d). | |
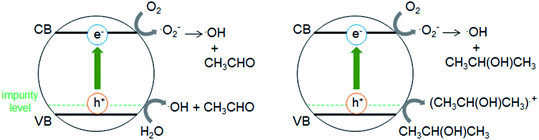 |
| | Scheme 1 Illustration of photocatalytic degradation of CH3CHO and 2-propanol on Cr–TiO2 under VL irradiation. | |
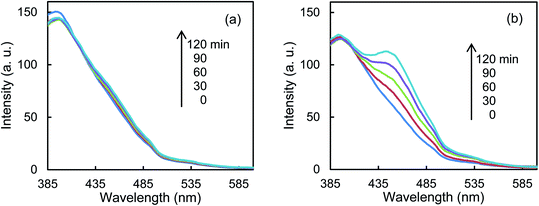 |
| | Fig. 6 Fluorescence spectra of an aqueous solution containing coumarin and (a) TiO2 or (b) Cr–TiO2 powder under VL irradiation for 120 min. | |
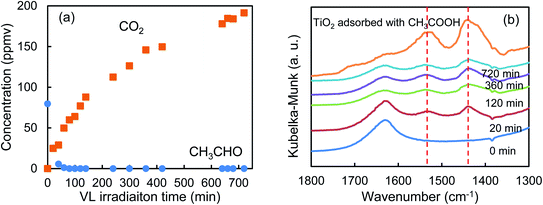 |
| | Fig. 7 Time course of (a) the concentrations of CH3CHO and CO2 and (b) diffuse reflectance FTIR spectra of Cr–TiO2 under VL irradiation for 720 min. For comparison, diffuse reflectance FTIR spectra of TiO2 adsorbed with acetic acid is depicted. | |
Photocatalytic degradation of CH3CHO on Cr–TiO2, Pt–TiO2 and V–TiO2 films for practical application
Effect of number of coating on the r0 value and on the transmittance of the 0.8 atom% Cr–TiO2_SC films were investigated. Fig. 8a and b indicate that r0 increases but the transmittance decreases with increasing the number of coating (n). These findings suggest that CH3CHO is degraded more effectively by increasing the amounts of the photocatalysts. Fig. 8c shows the effect of the doping amounts of the metal ions on the r0 values of M–TiO2_SC (n = 5) film. As the doping amount increases, the r0 value for Pt–TiO2 decreases, that for V–TiO2 shows the highest at 1.0 atom% and that for Cr–TiO2 slightly decreases, indicating a similar tendency as the M–TiO2 powder (Fig. 1). The amount of photocatalyst can be changed by using different coating methods. Fig. 9 shows the degradation of CH3CHO on Cr–TiO2_TK, Cr–TiO2_SC (n = 5), and Cr–TiO2_DC, in which the content of Cr ion is 0.8 atom%. Under the irradiation of LED of 6.5 mW cm−2, the most transparent Cr–TiO2_DC film shows the slowest degradation rate (r0 = 0.295 ppmv min−1) and the Cr–TiO2_SC (n = 5) exhibits 1.35 ppmv min−1 which is higher by a factor of 4.6. Such a high degradation rate is obtained with the Cr–TiO2_TK film even under the LED intensity of 0.1 mW cm−2. A tolerable concentration of CH3CHO in air has been evaluated to be 300 μg m−3 by World Health Organization,38 which is equivalent to 0.17 ppmv at 25 °C. The Cr–TiO2 film can be used to degrade CH3CHO in indoor air by changing the film thickness and the LED intensity depending on the degree of the indoor contamination.
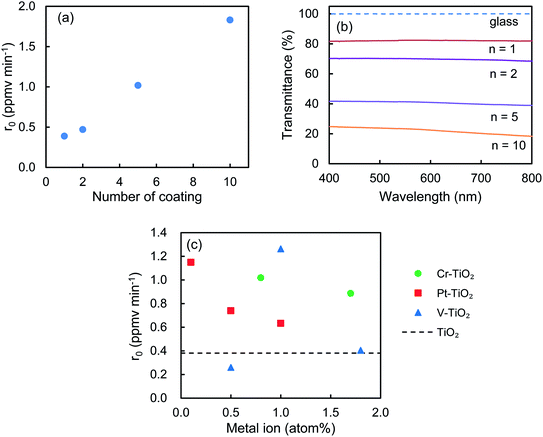 |
| | Fig. 8 Effect of number of coating (n) on (a) the r0 value and (b) the transmittance of 0.8 atom% Cr–TiO2_SC films. (c) Effect of metal ion content on the r0 value of M–TiO2_SC (n = 5) film (M = Cr, Pt, V). | |
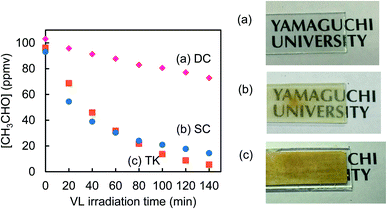 |
| | Fig. 9 Photocatalytic degradation of CH3CHO on (a) Cr–TiO2_DC and (b) Cr–TiO2_SC (n = 5) films under the irradiation of LED (6.5 mW cm−2) and on (c) Cr–TiO2_TK film under LED (0.1 mW cm−2). | |
Conclusions
This study has demonstrated that chromium ion is the best dopant among ten different metal ions for the degradation of CH3CHO under white LED irradiation because Cr–TiO2 exhibits high photocatalytic activity over a wide range of the doping amount. Furthermore, the physicochemical property and the photocatalytic activity of Cr–TiO2, Pt–TiO2 and V–TiO2 were compared. We prepared these photocatalysts by conducting the dialysis in the sol–gel synthesis. This synthetic method enables us to elucidate the true effect of the metal ion dopants because a homogeneous aqueous sol containing TiO2 nanoparticles and metal ions is dried and sintered after all impurities are removed by the dialysis. Furthermore, metal ions having similar ionic size as Ti(IV) interact with TiO2 nanoparticles because they remain but larger metal ions such as Cu(II) are lost in the TiO2 sol during the dialysis. Our findings indicate that the different dependence of the degradation rate of CH3CHO on the doping amounts of Cr–TiO2, Pt–TiO2 and V–TiO2 is attributable to their main valence states which are correlated with the formation of the oxygen vacancies. Recently, the oxygen vacancies have attracted much attention because they are believed to play an important role to improve the photocatalytic activity. They can be created by sintering TiO2 at high temperatures in an oxygen-poor atmosphere but disappear slowly when being exposed to air.28 However, it is very difficult to discuss the oxygen vacancies in TiO2 doped with other elements because crystal lattice might be distorted by forming several point defects such as Ti interstitial, oxygen vacancies, and interstitial or substitutional impurities including dopant ions. The present results suggest that Cr(III) in Cr–TiO2 stabilizes the formation of oxygen vacancies. The Cr–TiO2 powder synthesized in this study might be promising to elucidate the role of oxygen vacancies in photocatalysis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI Grant 18K05298. We thank Yamaguchi University Science Research Center for the ICP-OES and XRD measurements. We appreciate Mr T. Tonosaki for the technical support on the XPS measurements at Collaborative Center for Engineering Research Equipment, Faculty of Engineering, Yamaguchi University.
Notes and references
- E. S. Karafas, M. N. Romanias, V. S. Stefanopoulos, V. Binas, A. Zachopoulos, G. Kiriakidis and P. Papagiannakopoulos, J. Photochem. Photobiol., A, 2019, 371, 255–263 CrossRef CAS.
- S. Izadyar and S. Fatemi, Ind. Eng. Chem. Res., 2013, 52, 10961–10968 CrossRef CAS.
- T. Takigawa, B. Wang, Y. Saijo, K. Morimoto, K. Nakayama, M. Tanaka, E. Shibata, T. Yoshimura, H. Chikara, K. Ogino and R. Kishi, Int. Arch. Occup. Environ. Health, 2010, 83, 225–235 CrossRef CAS.
- F. Salvadores, R. I. Minen, J. Carballada, O. M. Alfano and M. M. Ballari, Chem. Eng. Technol., 2016, 39, 166–174 CrossRef CAS.
- H. Ichimura, T. Seike and A. Kozu, Chemosphere, 2020, 256, 127143 CrossRef.
- S. Weon and W. Choi, Environ. Sci. Technol., 2016, 50, 2556–2563 CrossRef CAS.
- V. Kumar, Y. Lee, J. Shin, K. Kim, D. Kukkar and Y. F. Tsang, Environ. Int., 2020, 135, 105356 CrossRef CAS.
- I. Bravo, F. Figueroa, M. I. Swasy, M. F. Attia, M. Ateia, D. Encalada, K. Vizuete, S. Galeas, V. H. Guerrero, A. Debut, D. C. Whitehead and F. Alexis, RSC Adv., 2020, 10, 7967–7975 RSC.
- W. Qu, P. Wang, M. Gao, J. Hasegawa, Z. Shen, Q. Wang, R. Li and D. Zhang, Environ. Sci. Technol., 2020, 54, 9693–9701 CrossRef CAS.
- R. Magudieshwaran, J. Ishii, K. Chandar, N. Raja, C. Terashima, R. Venkatachalam, A. Fujishima and S. Pitchaimuthu, Mater. Lett., 2019, 239, 40–44 CrossRef CAS.
- A. H. Mamaghani, F. Haghighat and C. Lee, Chemosphere, 2019, 219, 804–825 CrossRef CAS.
- I. Wysocka, A. Markowska-Szczupak, P. Szweda, J. Ryl, M. Endo-Kimura, E. Kowalska, G. Nowaczyk and A. Zielinska-Jurek, Indoor Air, 2019, 29, 979–992 CrossRef CAS.
- V. Menendez-Flores and T. Ohno, Catal. Today, 2014, 230, 214–220 CrossRef CAS.
- X. Yang, C. Cao, K. Hohn, L. Erickson, R. Maghirang, D. Hamal and K. Klabunde, J. Catal., 2007, 252, 296–302 CrossRef CAS.
- N. Nishiyama, Y. Fujiwara, K. Adachi, K. Inumaru and S. Yamazaki, Appl. Catal., B, 2015, 176–177, 347–353 CrossRef CAS.
- S. Yamazaki, D. Takaki, N. Nishiyama, and Y. Yamazaki, in Current Developments in Photocatalysis and Photocatalytic Materials, ed. X. Wang, M. Anpo, and X. Fu, Elsevier, 2019, pp. 23–38 Search PubMed.
- P. Makula, M. Pacia and W. Macyk, J. Phys. Chem. Lett., 2018, 9, 6814–6817 CrossRef CAS.
- T. T. Loan, V. H. Huong, V. T. Tham and N. N. Long, Phys. B, 2018, 532, 210–215 CrossRef CAS.
- X. Yu, J. Xie, H. Dong, Q. Liu and Y. Li, Chem. Phys. Lett., 2020, 754, 137732 CrossRef CAS.
- S. P. Takle, O. A. Apine, J. D. Ambekar, S. L. Landge, N. N. Bhujbal, B. B. Kale and R. S. Sonawane, RSC Adv., 2019, 9, 4226–4238 RSC.
- G. Rossi, L. Pasquini, D. Catone, A. Piccioni, N. Patelli, A. Paladini, A. Molinari, S. Caramori, P. O'Keeffe and F. Boscherini, Appl. Catal., B, 2018, 237, 603–612 CrossRef CAS.
- S. Kim, S. Hwang and W. Choi, J. Phys. Chem. B, 2005, 109, 24260–24267 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- X. Chen, C. Meng, Y. Wang, Q. Zhao, Y. Li, X. Chen, D. Yang, Y. Li and Y. Zhou, ACS Sustainable Chem. Eng., 2020, 8, 1095–1101 CrossRef CAS.
- F. Yang, R. Yang, L. Yan, X. Liu, X. Luo and L. Zhang, Catal. Sci. Technol., 2020, 10, 5659–5665 RSC.
- J. Li, H. Zhuo, Z. Wei, G. Zhuang, X. Zhong, S. Deng, X. Li and J. Wang, J. Mater. Chem. A, 2018, 6, 2264–2272 RSC.
- B. B. Adormaa, W. K. Darkwah and Y. Ao, RSC Adv., 2018, 8, 33551–33563 RSC.
- Q. Wu, Q. Zheng and R. Krol, J. Phys. Chem. C, 2012, 116, 7219–7226 CrossRef CAS.
- A. Naldoni, M. Altomare, G. Zoppellaro, N. Liu, S. Kment, R. Zboril and P. Schmuki, ACS Catal., 2019, 9, 345–364 CrossRef CAS.
- F. Ren, H. Li, Y. Wang and J. Yang, Appl. Catal., B, 2015, 176–177, 160–172 CrossRef CAS.
- N. Nishiyama and S. Yamazaki, ACS Omega, 2017, 2, 9033–9039 CrossRef CAS.
- B. D. Mukri, U. V. Waghmare and M. S. Hegde, Chem. Mater., 2013, 25, 3822–3833 CrossRef CAS.
- W. Kim, T. Tachikawa, H. Kim, N. Lakshminarasimhan, P. Murugan, H. Park, T. Majima and W. Choi, Appl. Catal., B, 2014, 147, 642–650 CrossRef CAS.
- M. Takeuchi, J. Deguchi, S. Sakai and M. Anpo, Appl. Catal., B, 2010, 96, 218–223 CrossRef CAS.
- K. Ishibashi, A. Fujishima, T. Watanabe and K. Hashimoto, Electrochem. Commun., 2000, 2, 207–210 CrossRef CAS.
- H. Li, S. Yin, Y. Wang and T. Sato, Environ. Sci. Technol., 2012, 46, 7741–7745 CrossRef CAS.
- B. Hauchecorne, D. Terrens, S. Verbruggen, J. A. Martens, H. V. Langenhove, K. Demeestere and S. Lenaerts, Appl. Catal., B, 2011, 106, 630–638 CrossRef CAS.
- World Health Organization, IPCS Environmental Health Criteria 167, ACETALDEHYDE, Geneva, 1996 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07567a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Keisuke Kozasa,
Kohshiro Okimura and
Kensuke Honda
*,
Keisuke Kozasa,
Kohshiro Okimura and
Kensuke Honda