
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe structure-directing role of graphene in composites with porous FeOOH nanorods for Li ion batteries
Hongliang Zhaoac,
Zhifan Songa,
Hanxi Gaoa,
Biqian Lia,
Tao Hu*b,
Fengqin Liu *a and
Hong Yong Sohnc
*a and
Hong Yong Sohnc
aSchool of Metallurgical and Ecological Engineering, University of Science and Technology Beijing, Beijing 100083, China. E-mail: liufq@ustb.edu.cn
bShanghai Energy New Materials Technology Co., Ltd., Shanghai 201399, China. E-mail: hutao@SEMCORPGlobal.com
cDepartment of Materials Science and Engineering, The University of Utah, Salt Lake City, Utah 84112, USA
First published on 12th November 2020
Abstract
Graphene sheets that contain porous iron oxides including Fe2O3 and FeOOH nanorods were synthesized via a one-step hydrothermal route. A novel mechanism for controlling the structure of graphene-based composites was developed. Porous FeOOH nanorods with a high capacity for electron- and ion-transport were synthesized by controlling the composition of GO dispersion. The synthesized graphene/FeOOH composite anode exhibited an excellent electrochemical performance in which a reversible capacity of 304 mA h g−1 was reached with nearly 100% coulombic efficiency after 1000 cycles of charge and discharge under a high current rate of 5 A g−1.
1. Introduction
Rechargeable lithium ion batteries (LIBs) have been widely utilized as secondary energy storage devices for portable electronic devices, electric vehicles and industrial power sources. Tao et al.1 reviewed the recent significant progress on high performance electrode materials for flexible lithium-ion batteries (LIBs). Wang et al.2 pointed out the recent progress of micro-battery materials with high energy density for micro-electronic devices. Zeng et al.3 reviewed the latest development of active electrode materials and battery chemistry for automotive batteries. In the above reports, graphene which has good conductivity and rate performance has great application potential for lithium-ion battery anode materials. However, the conventional graphite anode electrode for the commercial LIBs cannot meet the increasing demands for batteries with high energy and power densities due to its low theoretical specific capacity (372 mA h g−1).4–6 Therefore, there is a critical challenge to develop electrode materials with high energy densities, prolong lifetimes, and low costs for commercial LIBs.The metal oxides (MOs) can significantly improve the electrical performances of batteries and capacitors. The supercapacitors with nano metal oxides have high specific capacitance values, fast charge–discharge properties and have been widely used in industrial fields.7–9 Transition metal oxides with high theoretical capacities (>600 mA h g−1) have been extensively investigated as candidate negative electrode materials for LIBs. Guo et al.10 presented a simple wet chemical method to prepare graphite–SnO2 composite electrode which has excellent cyclability at high current density. Sun et al.11 prepared the RGO–CoO–Co3O4 composite using a facile solvothermal and sintering method. The hybrid composite anode delivered a high reversible capacity of 994 mA h g−1 at 100 mA g−1 after 200 cycles. Zhang et al.12 synthesized graphene–α-MnO2 composite (GMC) as the LIB anode by a conventional hydrothermal approach. The GMC anode showed a good reversible capacity that maintained 998 mA h g−1 at 60 mA g−1 after 30 cycles. Zhao et al.13 proposed an atomic layer deposition (ALD) method for preparing the NG–V2O5 composite electrode which exhibited a good electrochemical performance when used as anodes for LIBs. At 200 and 1000 mA g−1, the NG–V2O5 electrode showed good capacity retention and high discharge capacities of 608 (after 50 cycles) and 365 mA h g−1 (after 500 cycles), respectively. Especially, the iron oxides of Fe2O3, Fe3O4 and FeOOH also showed excellent discharge/charge performance, as the LIB anodes.14–16 Moreover, a variety of nanostructures, including uniform nanoribbons, nanotubes, nanocubes, nanowires, and other hierarchical nanostructures with large interfaces between the electrolyte and the electrode with shortened pathways for ion and electron transport, have been developed. These nanostructures avoid the pulverization of the electrodes and the rapid capacity decay that present serious drawbacks of bulk counterparts during Li+ insertion/extraction.
Among these MOs used for LIBs, iron oxides are considered as the most promising candidate materials for lithium storage owing to their high theoretical capacities of 1007 mA h g−1, low costs, abundance, and nontoxic properties. [In this paper, the term ‘iron oxides’ is used to represent the compounds of iron and oxide elements including ferric oxyhydroxide (FeOOH) and ferric oxide (Fe2O3).] However, in order to overcome their inherently poor conductivities and capacities that fade from the large volume change during cycling, a series of nanocomposites based on iron oxide nanostructures were fabricated by mixing various carbon additives that improve their electronic transport and also buffer the huge volume expansion of iron oxides.
Two-dimensional graphene sheets with large specific surface areas, excellent electronic conductivities and high mechanical properties have been extensively investigated as electrode materials or conductive substrates and ideal building blocks to grow MO nanostructures, thereby leading to improved electrochemical capacity and cycling stability.
However, there still exists a challenge to obtain well-dispersed graphene dispersions to grow composites of graphene with MO precursors by using a hydrothermal route based on its aggressive properties arising from the strong van der Waals force between the graphene sheets. Therefore, graphene oxide (GO), a precursor of chemically converted graphene (CCG), has taken the place of graphene in fabricating the composites because it exhibits hydrophilicity and exfoliates to single layer GO sheets which feature hydroxyl and epoxy groups on the basal plane along with carboxyl and carbonyl moieties located at the nanosheet edge. The precursor of MOs can bind with these oxygen-containing groups during the synthesize process when GO is the precursor chemical to fabricate graphene-based composites. Furthermore, Dimiev et al.17 reported that an aqueous GO dispersion is a stronger acid than commonly known carboxylic acids with a single functional group in a molecule that creates the potential of acting as an MO nanostructure by adjusting the pH of the reaction system.
On the other hand, iron oxide porous nanostructures such as hollow spheres, tubes, rings, and porous rods have been fabricated and used as LIB anode materials. With these structures, a higher power capability can be reached owing to the large surface area of these porous structures providing more active interfaces, which provides fast Li+ diffusion and electron transport. Additionally, the size and shape-dependent properties of the iron oxide porous nanomaterials can be well controlled by choosing different types of inorganic salts.18,19
Here, we report a facile, a one-step hydrothermal route for the synthesis of porous iron oxides including Fe2O3 and FeOOH nanorods on graphene sheets, in which low cost urea is used as the source of nitrogen while a GO dispersion is used to adjust the pH of the reaction system.
Under the synergistic effects of GO sheets and by adjusting the reaction time, one dimensional (1-D) porous Fe2O3 and FeOOH nanorods with diameters of 20–30 nm and lengths of 50–100 nm were obtained. Moreover, the porosity could be fine-tuned by the weight ratio of GO dispersion and reaction time. In addition, the electrochemical performances of graphene/FeOOH composite anodes were investigated. A reversible capacity of 304 mA h g−1 was reached with nearly 100% coulombic efficiency after 1000 cycles of charge and discharge at a high current rate of 5 A g−1.
2. Experimental
2.1 Preparation
Firstly, GO powder was produced by the modified Hummer method described in previous work.20Graphene–iron oxide composites were synthesized by a simultaneous hydrothermal synthesis and assembly procedure. Gr–Fe-3 was obtained by the following steps: firstly, 30 mL of 2 mg mL−1 GO dispersion was mixed with 5.4 g of urea by sonication for 30 min. Then, 278 mg FeSO4·7H2O was added with a 30 min ultra-sonication. The dispersion was transferred to a polytetrafluoroethylene (Teflon)-lined autoclave and hydrothermally treated at 160 °C for 5 h. Then, Gr–Fe-3 was obtained after washing several times, followed by vacuum drying overnight. Finally, microwave treatment was applied for two minutes to remove the oxygen functional groups of reduced graphene in Gr–Fe-3 using a household microwave oven (700 W, Galanz), which produced Gr–Fe-3-M.
2.2 Structural and electrochemical characterization
The structural, morphological, and microstructural properties of the graphene and iron oxide nanocomposites were characterized using a variety of characterization techniques such as X-ray diffraction (XRD), Raman analysis, scanning electron microscopy (SEM), transmission electron microscopy (TEM), and thermogravimetric analysis (TGA). More specifically, XRD measurements were done using a PANanalytical X-ray diffraction system. Raman analysis was performed by a Jobin-Yvon HR300 Raman spectrometer using a 532 nm green laser source. SEM images were taken on a Carl Zeiss Ultra 1540 Dual Beam FIB/SEM System. TEM images were obtained using a JEOL JEM-2010 instrument with an operating voltage of 200 kV. TGA measurements were performed in air atmosphere from 30 to 800 °C at a heating rate of 10 °C min−1 in a TA Instruments TGA-Q50. The XPS were recorded on a PHI 5000 Versa Probe system.2.3 Electrochemical measurements
Electrochemical performance was investigated by a coin cell configuration where Li foil was used as the negative electrode. The working electrode was fabricated by a slurry coating procedure that involved mixing the composites, carbon black and PVDF at a weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 in N-methylpyrrolidinone (NMP) and coating the slurry on the Cu foil and drying under 80 °C overnight in vacuum.
:10:10 in N-methylpyrrolidinone (NMP) and coating the slurry on the Cu foil and drying under 80 °C overnight in vacuum.
Coin cells were assembled in an argon-filled glove box. The electrolyte was 1 M LiPF6 dissolved in the solution with 1:1 ethylene carbonate and diethyl carbonate by volume (Novolyte Technologies). The electrochemical behavior of the electrodes was characterized using an Arbin BT 2000 testing station. The electrochemical test was performed between 0.01–3.0 V vs. Li+/Li. The cyclic voltammetry (CV) and Nyquist plots were analyzed by using a potentiostat VersaSTAT 4 (Princeton Applied Research). All the electrochemical measurements were carried out at room temperature.
3. Results and discussion
3.1 Synthesis
GO powder can be well dispersed in deionized water (DIW) to form GO dispersion (2 g L−1) owing to its hydrophilic. Then, a urea solution was introduced to create an alkaline system due to the fact that urea can slowly release hydroxyl and ammonium ions. NH4+ binds with the oxygen-containing groups of GO platelets simultaneously with the hydroxyl ions. Cong et al.21 pointed the ferrous (Fe2+) ions of FeSO4 diffuse toward GO sheets by electrostatic interactions. Because most of the oxygen-containing functional groups on GO surface were occupied by NH4+, ferrous ions initially hydrolyzed to Fe(OH)2. Moreover, the pH of reaction system can be tuned by the initial weight ratio between GO and FeSO4 (W(GO/Fe2+)), pH value being much lower at higher W(GO/Fe2+) as applied in our reaction.Qu et al.22 and Xue et al.23 found the resultant Fe(OH)2 can convert to FeOOH and Fe2O3 anchored on the graphene sheet surface with different OH− ions in low and high pH values, respectively, similar to the formation of graphene and other iron composites. GO can be reduced to graphene when its oxygen containing functional groups are removed by one of the following mechanisms: the reduction by nitrogen-based groups reported by Tu et al.,24 reaction with Fe2+ reported by Wang et al.25 and self-assembly of GO groups in hydrothermal reaction reported by Xu et al.26 As suggested by the following reaction:
| CO(NH2)2 + 3H2O → 2NH4+ + CO2 + 2OH− | (1) |
| Fe2+ + 2OH− → Fe(OH)2 | (2) |
| 2Fe(OH)2 + 2OH− → Fe2O3 + 3H2O + 2e (low W(GO/Fe2+)) | (3) |
| Fe(OH)2 + OH− → FeOOH + H2O + e (high W(GO/Fe2+)) | (4) |
The experimental conditions and the structures of the composites are summarized in Table 1.
| Sample name | GO dispersion (2 g L−1, mL) | Urea (mg) | FeSO4 (mg) | Reaction T (°C) | Reaction time (h) | Crystal type | Morphology |
|---|---|---|---|---|---|---|---|
| Gr–Fe-2 | 30 | 5.4 | 834 | 160 | 5 | Fe2O3 | Nanorod |
| Gr–Fe-4 | 30 | 5.4 | 417 | 160 | 5 | Fe2O3 | Nanorod |
| Gr–Fe-3 | 30 | 5.4 | 278 | 160 | 5 | FeOOH | Porous, nanorod |
| Gr–Fe-1 | 30 | 5.4 | 834 | 160 | 15 | Fe2O3 | Porous, nanorod |
| Gr–Fe-5 | 0 | 5.4 | 278 | 160 | 5 | Fe2O3 | Nanoparticle |
| Gr-6 | 30 | 5.4 | 0 | 160 | 5 | Graphene | Graphene |
Generally, FeOOH as a precursor in the preparation of Fe2O3, was formed via olation and oxolation of the FeO6 units. Moreover, Musić et al.27 and Song et al.28 revealed that chloride ions (Cl−) play a significant role in tuning the channel type porous structure of FeOOH because small chloride ions can occupy the tunnel sites during the formation of the FeOOH nanorods.
However, in this work, at high W(GO/Fe2+), porous FeOOH nanorods were obtained. GO plays an important role in the formation of FeOOH nanorods. To further demonstrate this, particles of Fe2O3 with 50–100 nm diameter (Gr–Fe-5) was produced without GO, which is consistent with our understanding that Fe2O3 crystals tend to form under a high pH value. Therefore, under increased presence of GO in the reaction system, FeOOH nanorods adsorb on the GO surface and the functional groups of GO control the growth and shape of nanorods. Comparison of Gr–Fe-3 and Gr–Fe-5 is shown in Table 1, without the addition of graphene oxide, only Fe2O3 was crystallized. FeOOH did was not observed without graphene oxide.
In previous investigations, Chaudhari and Yu29 reported that porous Fe2O3 was formed by annealing at 250 °C for 2 h and FeOOH was obtained in the presence of Cl−, while in this work Fe2O3 was obtained at 160 °C only by controlling the W(GO/Fe2+) and porous FeOOH crystals less than 20 nm in diameter was obtained without the presence of Cl− ions.
It is worth noting that the crystal types of iron oxides changed from FeOOH to Fe2O3 as the reaction time and temperature remained constant while the W(GO/Fe2+) was increased from 60/278 to 60/417 and 60/834. The results indicated that GO can act as a structure-directing template in influencing the crystal growth of Fe2O3.
3.2 Structural and chemical properties
To investigate the crystallinity and structural properties of graphene–iron oxide composites, X-ray powder diffraction (XRD) pattern was collected for each sample. As shown in the Fig. 1, all iron samples are the complex of two different phases, and but Gr–Fe-3 clearly exhibits the absence of Fe2O3 peaks. For Gr–Fe-1, Gr–Fe-2, Gr–Fe-4, and Gr–Fe-5, the Fe2O3 phases of hematite and maghemite (Joint Committee on Powder Diffraction Standards (JCPDS) JCPDS no. 13-053 and 40-755, respectively) are displayed, and the diffraction peaks at 2θ = 24.8 and 32.1 correspond to the (012) and (300) planes of hematite α-Fe2O3 and maghemite γ-Fe2O3, respectively. In contrast, the dominant peaks of Gr–Fe-3 indicate that goethite and lepidocrocite FeOOH (JCPDS no. 30-251 and 00-136, respectively) were formed under high W(GO/Fe2+).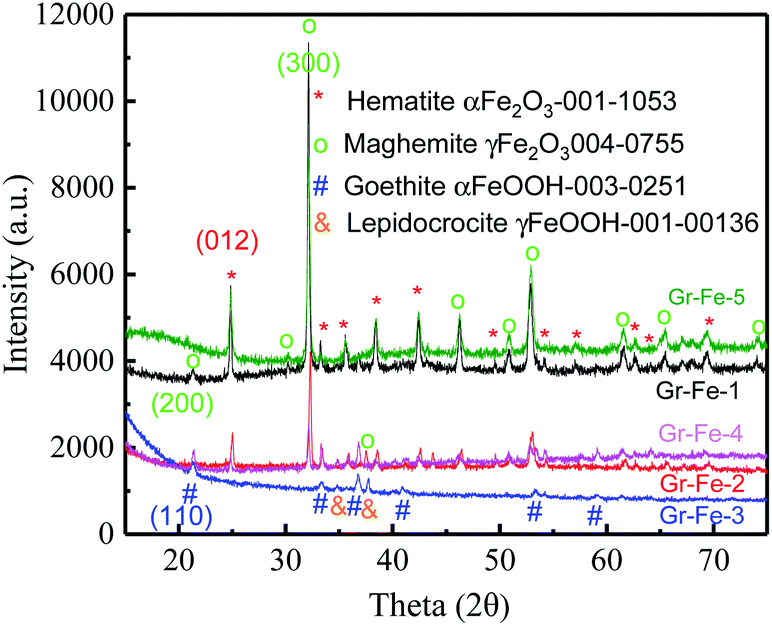 | ||
| Fig. 1 XRD patterns of the graphene and iron oxide composites. | ||
Compared with Fe2O3, the Fe in Gr–Fe-3-M (graphene/porous FeOOH nanorods) has an intermediate valence with more oxygen vacancies that promote the reaction of Li+. In addition, the porous structure of FeOOH nanorods provides a large reaction area. Thus, Gr–Fe-3-M was selected for further evaluation.
Although the XRD patterns (see Fig. 2) reveal no obvious changes caused by microwave treatment. The main phase before and after microwave treatment was FeOOH, and a small amount of Fe2O3 was detected after the microwave treatment. Due to low concentration, no obvious XRD diffraction peak of graphene was detected. Fig. 3 shows the Raman spectra before and after the microwave treatment and the intensity ratios I(D)/I(G) were compared by calculating the areas of D and G peaks. I(D)/I(G) decreased from about 1.32 to 1.19 after the microwave treatment. This indicates that microwave rapidly removes the residual oxygen-containing groups of the composites.
 | ||
| Fig. 2 XRD patterns before (bottom) and after (top) microwave treatment. | ||
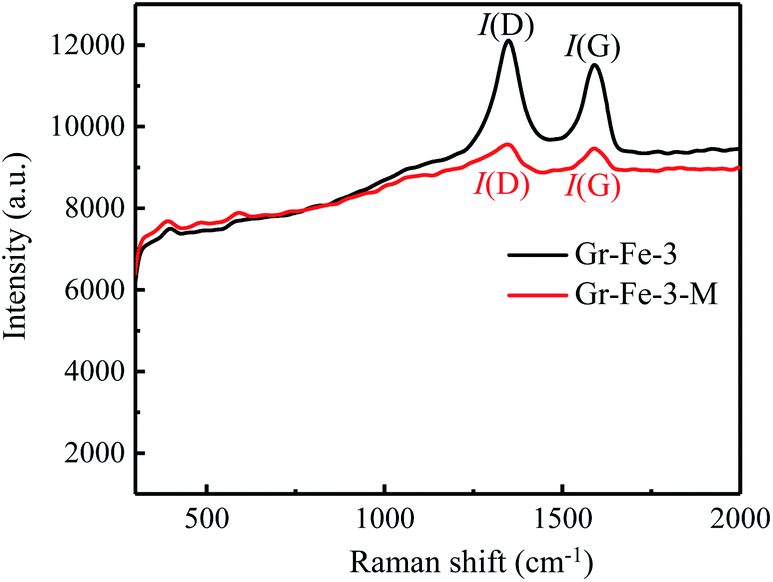 | ||
| Fig. 3 Raman spectra before and after microwave treatment. | ||
As shown in Fig. 4, for both Gr–Fe-3 and Gr–Fe-3-M, 9% and 6% weight loss can be attributed to the decomposition of FeOOH between 250 °C and 350 °C, respectively. Furthermore, 56–63% of iron oxide weight is left after heating to 500 °C, indicating that microwave treatment promotes reduction of the composites of GO and metal oxides.
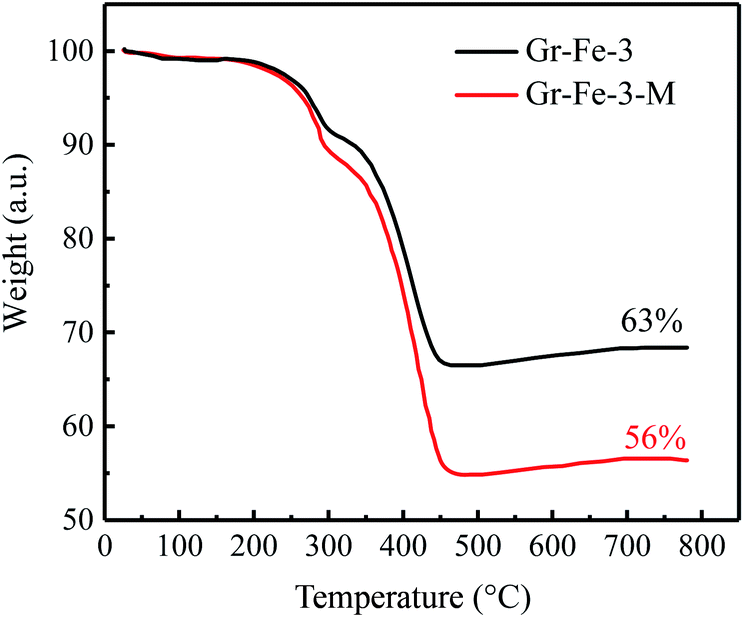 | ||
| Fig. 4 TGA curves of Gr–Fe-3 and Gr–Fe-3-M. | ||
To further examine the morphology and microstructure of the composites, a series of scanning electron microscope (SEM) and transmission electron microscope (TEM) analyses were carried out. In the presence of GO, iron oxide nanorods were dispersed uniformly on the surface of graphene layers (Fig. 5a and b), whereas without GO, the shapes of metal oxides had hierarchical morphology with nano-scale diameters.
 | ||
| Fig. 5 Morphology and microstructure of the composite electrodes: (a and b) SEM and (c and d) TEM images of Gr–Fe-3-M nanostructures. | ||
TEM images of the composites (Fig. 5c and d) show that flexible graphene sheets were covered by a uniform layer of iron oxide nanostructure with length and diameter in the range of 50–100 nm and 20–30 nm, respectively. The composites surface was concave and convex which showed a porous structure indicating that long reaction time and high W(GO/Fe2+) produce a nanoporous structure on the surface of crystalline iron oxides. Moreover, the 0.269 nm lattice fringe spacing in the high-resolution TEM (HRTEM) image (Fig. 5d) corresponds to the (130) lattice plane of FeOOH, consistent with the XRD results.
3.3 Battery performance
The electrochemical properties of graphene–iron composites Gr–FeOOH were investigated as the anode electrode of LIBs.Fig. 6 shows the CVs of Gr–Fe-3-M (graphene/porous FeOOH nanorod) electrodes. When compared to that of Fe2O3,21 the additional peak at 1.41 V can be assigned to the lithium intercalation before the reduction reaction since crystallized α-FeOOH has a layered structure. However, it disappears in the subsequent cycles, implying that this lithium intercalation process is irreversible reported by Lou et al.30
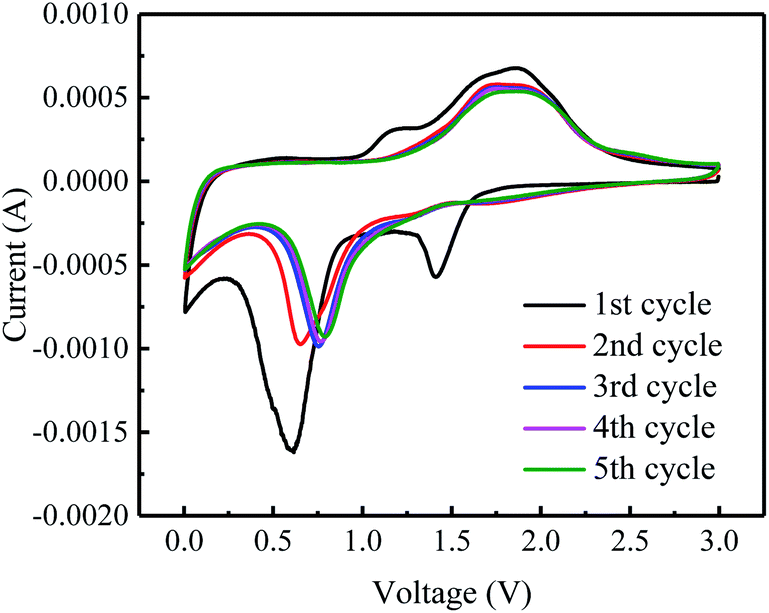 | ||
| Fig. 6 CVs curves of the Gr–Fe-3-M electrode at a scan rate of 0.5 mV s−1 for five cycles. | ||
In addition, the peak at 0.59 V can be attributed to the reduction of Fe3+ to Fe0. It shifts to the higher potential in the following cycles. It can also be speculated that there are two small oxidation peaks at 1.62 and 1.94 V corresponding to the oxidation of Fe0 to Fe2+ and Fe3+. These oxidation peaks remain stable during the subsequently discharge–charge processes, indicating reversibility of the Fe0 to Fe3+ reaction, similar to Fe2O3 anode.
The main text of the article should appear here with headings as appropriate. Fig. 7 shows the charge–discharge capacities of the electrodes before and after the microwave treatment. The microwave treatment significantly improves the electrode capacity. For example, at a current density of 0.1 A g−1, the capacity of the electrode without microwave treatment (see Fig. 7a) is only about 750 mA h g−1, which is much lower than that after microwave treatment (about 1100 mA h g−1, as seen in Fig. 7b). After the microwave treatment, as the current density increases from 0.1 to 1, 10, 30, 50, and 80 A g−1, all of the electrodes exhibit good capacity retention, and it is worth to note that at 30 A g−1, a capacity of 196 mA h g−1 is obtained. Gr–Fe-3-M shows the best capacity at high current rates indicating that a porous structure with large surface area increases the electrochemical reaction area especially under low current densities, and more graphene sheets can accommodate the strain of Li insertion–extraction at high current rates.
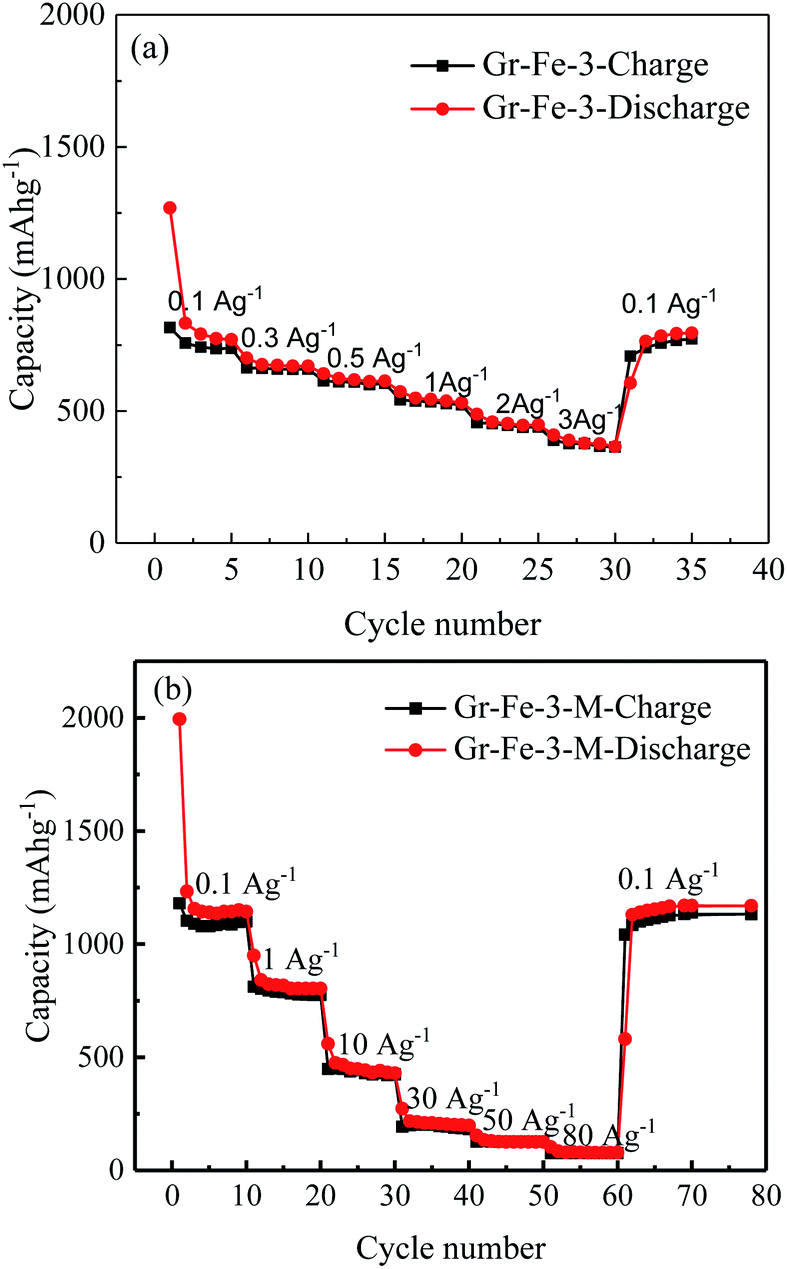 | ||
| Fig. 7 Rate capacities of the electrode at various current densities (a) before and (b) after microwave treatment. | ||
To investigated the electrochemical performance of Gr–Fe-3-M porous nanorods, an electrochemical impedance spectroscopy (EIS) measurement was performed. Based on an appropriate electric equivalent circuit, which was selected for testing the electrochemical reaction and diffusion processes in the battery,31,32 the electrochemical impedance spectroscopy (EIS) measurement was performed. The Nyquist plots in Fig. 8 reveal that the Gr–Fe-3-M electrode has a lower charge transfer resistance (RCT = 203.4 Ω) than that of the Gr–Fe-3 electrode (RCT = 365.1 Ω), accompanied by a lower Zw due to the faster diffusion of Li+.
 | ||
| Fig. 8 Nyquist plots of the Gr–Fe-3-M electrode at a discharged potential of 0.1 V (vs. Li/Li+) from 100 kHz to 10 mHz (inset: modelled equivalent circuit of EIS). | ||
The cycle retention and capacity performance of the Gr–Fe-3-M anode were further evaluated at 5 A g−1 for 1000 cycles. As shown in Fig. 9, capacity decay still occurs during the full charge/discharge cycles. It is possible that continuous SEI growth with cycling limits Li salt diffusion into the pores of the composite electrode and increases its polarization resistance, resulting in the electrode capacity fade. However, after 1000 cycles of charge and discharge, a reversible capacity of 304 mA h g−1 still remains with nearly 100% coulombic efficiency. These results demonstrate that graphene–FeOOH composite nanostructures with high porosity and large surface area effectively shorten the diffusion pathway of lithium ions and electrons, and increase the electrochemical reaction area, allowing the improved penetration of electrolyte and accommodating the strain of Li insertion–extraction.
 | ||
| Fig. 9 Cycle performance of the Gr–Fe-3-M electrode when performing full discharge–charge at 2 A g−1 for 1000 cycles. | ||
4. Conclusions
A facile one-step synthesis route was developed to synthesize graphene–iron oxides composites. It was determined in this work that:(1) FeOOH nanorods formed on the graphene surface at a high W(GO/Fe2+) system, and the functional groups of GO controlled the growth and shape of nanorods. FeOOH was crystallized instead of Fe2O3 when 30 mL of GO (2 g L−1) was added.
(2) The graphene/FeOOH composite anode showed a higher charge–discharge capacity after microwave treatment than without it.
(3) After 1000 cycles at 5 A g−1, the graphene/FeOOH composite anode exhibited a high reversible capacity of 304 mA h g−1 for which is attributed to the combination of porous FeOOH nanorods and highly conductive 2D graphene nanosheets.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the China Scholarship Council for providing Chinese Government Scholarship to Hongliang Zhao to study as a Visiting Scholar in Professor Hong Yong Sohn's laboratory at the University of Utah.References
- T. Tao, S. G. Lu and Y. Chen, Adv. Mater. Technol., 2018, 3, 1700375 CrossRef.
- Y. Wang, B. Liu, Q. Li, S. Cartmell, S. Ferrara, Z. D. Deng and J. Xiao, J. Power Sources, 2015, 286, 330–345 CrossRef CAS.
- X. Q. Zeng, M. Li, D. Abd El-Hady, W. Alshitari, A. S. Al-Bogami, J. Lu and K. Amine, Adv. Energy Mater., 2019, 9, 1900161 CrossRef.
- D. H. Jiao, Z. W. Xie, Q. Wan, Z. K. Wei, X. X. Yan, F. K. Butt and M. Z. Qu, Int. J. Electrochem. Sci., 2018, 14, 848 Search PubMed.
- J. Kang, H. V. Kim, S. A. Chae and K. H. Kim, Small, 2018, 14, 1704394 CrossRef.
- Z. Y. Pan, H. Sun, J. Pan, J. Zhang, B. J. Wan and H. S. Peng, Carbon, 2018, 133, 384 CrossRef CAS.
- S. Najib and E. Erdem, Nanoscale Adv., 2019, 1, 2817 RSC.
- M. Toufani, S. Kasap, A. Tufani, F. Bakan, S. Weber and E. Erdem, Nanoscale, 2020, 12, 12790 RSC.
- S. Najib, F. Bakan, N. Abdullayeva, R. Bahariqushchi, S. Kasap, G. Franzò, M. Sankir, N. D. Sankir, S. Mirabella and E. Erdem, Nanoscale, 2020, 12, 16162 RSC.
- J. X. Guo, B. Jiang, X. Zhang and H. T. Liu, J. Power Sources, 2014, 262, 15 CrossRef CAS.
- L. N. Sun, Q. W. Deng, Y. L. Li, H. W. Mi, S. H. Wang, L. B. Deng, X. Z. Ren and P. X. Zhang, Electrochim. Acta, 2017, 241, 252 CrossRef CAS.
- Y. Zhang, H. Liu, Z. H. Zhu, K. W. Wong, R. Mi, J. Mei and W. M. Lau, Electrochim. Acta, 2013, 108, 465 CrossRef CAS.
- H. L. Zhao, H. X. Gao, B. Q. Li, Z. F. Song, T. Hu and F. Q. Liu, Mater. Lett., 2019, 252, 215 CrossRef CAS.
- M. S. Balogun, Z. P. Wu, Y. Luo, W. T. Qiu, X. L. Fan, B. Long, M. Huang, P. Liu and Y. X. Tong, J. Power Sources, 2016, 308, 7 CrossRef CAS.
- B. Jin, G. Y. Chen, X. B. Zhong, Y. Liu, K. Y. Zhou, P. Sun, P. Lu, W. X. Zhang and J. C. Liang, Ceram. Int., 2014, 40, 10359 CrossRef CAS.
- L. H. Yu, L. P. Wang, S. B. Xi, P. Yang, Y. H. Du, M. Srinivasan and J. Z. Xu, Chem. Mater., 2015, 27, 5340 CrossRef CAS.
- A. M. Dimiev, L. B. Alemany and J. M. Tour, ACS Nano, 2013, 7, 576 CrossRef CAS.
- X. L. Hu and C. Y. Yu, Adv. Funct. Mater., 2008, 18, 880 CrossRef CAS.
- C. Z. Wu, P. Yin, X. Zhu, C. Z. OuYang and Y. Xie, J. Phys. Chem. B, 2006, 110, 17806 CrossRef CAS.
- T. Hu, M. Xie, J. Zhong, H. T. Sun, X. Sun, S. Scott, S. M. George, C. S. Liu and J. Lian, Carbon, 2014, 76, 141 CrossRef CAS.
- H. P. Cong, X. C. Ren, P. Wang and S. H. Yu, ACS Nano, 2012, 6, 2693 CrossRef CAS.
- Q. T. Qu, S. B. Yang and X. L. Feng, Adv. Mater., 2011, 23, 5574 CrossRef CAS.
- Y. H. Xue, H. Chen, D. S. Yu, S. Y. Wang, M. Yardeni, Q. B. Dai, M. M. Guo, Y. Liu, F. Lu, J. Qu and L. M. Dai, Chem. Commun., 2011, 47, 11689 RSC.
- J. Tu, M. Zhao, X. J. Zhan, Z. J. Ruan, H. L. Zhang, Q. Q. Li and Z. Li, Polym. Chem., 2016, 7, 4054 RSC.
- F. F. Wang, F. Wang, G. D. Gao and W. Chen, Environ. Toxicol. Chem., 2015, 34, 1975 CrossRef CAS.
- Y. X. Xu, K. X. Sheng, C. Li and G. Q. Shi, ACS Nano, 2010, 4, 4324 CrossRef CAS.
- S. Musić, S. Krehula and S. Popović, Mater. Lett., 2004, 58, 2640 CrossRef.
- H. J. Song, X. H. Jia, N. Li, X. F. Yang and H. Tang, J. Mater. Chem., 2012, 22, 895 RSC.
- N. K. Chaudhari and J. S. Yu, J. Phys. Chem. C, 2008, 112, 19957 CrossRef CAS.
- X. M. Lou, X. Z. Wu and Y. X. Zhang, Electrochem. Commun., 2009, 11, 1696 CrossRef CAS.
- T. Hu, M. Xie, J. Zhong, H. T. Sun, X. Sun, S. Scott, S. M. George, C. S. Liu and J. Lian, Carbon, 2014, 76, 141 CrossRef CAS.
- Z. S. Wu, W. C. Ren, L. Xu, F. Li and H. M. Cheng, ACS Nano, 2011, 5, 5463 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |