
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInteraction of synthetic and lignin-based sulfonated polymers with hydrophilic, hydrophobic, and charged self-assembled monolayers†
Armin Eraghi Kazzaz and
Pedram Fatehi *
*
Biorefining Research Institute, Green Processes Research Centre, Chemical Engineering Department, Lakehead University, 955 Oliver Road, Thunder Bay, ON P7B5E1, Canada. E-mail: pfatehi@lakeheadu.ca
First published on 6th October 2020
Abstract
There is a need to understand the role of polymer structure on its interaction with surfaces to produce effective functional surfaces. In this work, we produced two anionic polymers of lignin-3-sulfopropyl methacrylate (L-S) and poly(vinyl alcohol-co-vinyl acetate)-3-sulfopropyl methacrylate (PVA-S) with similar charge densities and molecular weights. On the gold-coated surface, we deposited self-assembled monolayers (SAM) bearing different terminal moieties namely, hydroxyl, carboxyl, methyl, and amine groups of alkanethiols. This study highlighted the difference between the interaction of L-S and PVA-S and functionalized self-assembled surfaces. The information was generated using advanced tools, such as an X-ray photoelectron spectroscopy (XPS), and a quartz crystal microbalance with dissipation (QCM-D), which facilitated the correlation development between polymer properties and deposition performance on the functionalized surfaces. The higher deposition of PVA-S than L-S onto OH and COOH surfaces was observed due to its greater hydrogen bonding development and higher solubility. The solubility and structure of PVA-S were also beneficial for its higher adsorption than L-S onto CH3 and NH2 surfaces. However, the variation in pH, temperature, and salt significantly affected the adsorption of the macromolecules.
1. Introduction
Polymeric films are ubiquitous in applications ranging from automobiles to construction. The majority of polymer films are multilayer polymeric materials with varied functionalities. To generate multilayer films, the interaction of polymers with different surfaces is critical. Polymer adsorption on different surfaces and at the solid/water interface can happen as a result of hydrophobic, hydrogen-bonding, and electrostatic interactions between polymer segments and surfaces. Polymer adsorption is influenced by the properties of the polymer and the interaction between the polymer, surface, and solvent. In this context, the difference in the structure of the polymer, e.g., linear or three-dimensional, has shown to have a crucial impact on the adsorption behavior of the polymer on surfaces.1,2Poly(vinyl alcohol-co-vinyl acetate) (PVA) is known as an odorless, whitish or creamy, nontoxic, biocompatible, thermostable linear synthetic polymer used widely in different applications. PVA polymer is used vastly in textile, papermaking, coating industry, 3D printing, optical gas and humidity sensors, emission sensors for vehicles and oral drug delivery, and solar cells.3–7 PVA, by having a linear structure,2 has been functionalized with anionic and cationic monomers to improve its adsorption on fibrous pulp and broaden its application as adhesives and emulsifiers.6,8
Lignin, an abundant phenolic polymer, is one of the alternatives to petroleum feedstocks,9 which recently attracted tremendous attention and applications.10–15 Lignin, by having a complex three-dimensional structure, has been reported to show a distinct interaction behavior compared to linear polymeric materials.16,17 However, as lignin has a complicated structure, the interaction mechanisms of lignin derivatives with different functionalized surfaces are still unclear. Revealing these mechanisms is believed to highly affect its end-used applications in wastewater treatment, surface coating, and biological applications, for instance.
Polymers could adsorb following altered mechanisms.18–20 Thus, interaction mechanisms between the polymer and adsorbing surface include, but are not limited to, charge interaction, hydrogen bonding, van der Waals forces, and hydrophobic interactions. Although the mechanisms behind the adsorption of various polymers on different surfaces in contrastingly charged systems have been studied, information on the adsorption in the absence of electrostatic interaction for lignin and PVA based polymers is limited. To address this, two types of branched anionic polymers of poly(vinyl alcohol-co-vinyl acetate)-3-sulfopropyl methacrylate (PVA-S) and lignin-3-sulfopropyl methacrylate (L-S) were produced. To eliminate the effect of molecular weight and charged group in comparing lignin and PVA, the reaction condition was controlled to produce polymers with a similar molecular weight and charge density. This will elucidate the role of the structure and surface chemistry of polymers in adsorption.
Self-assembled monolayers (SAMs) with different surface chemistries were used to study the adsorption of polymers and the adsorption kinetics.21–24 These surfaces are extensively used in electrochemical sensors25 and also as model surfaces to study the adsorption of polymers, such as proteins.26,27 Various SAMs with various combinations of moieties can be used to elucidate the different contributions of the driving force for polymer adsorption.23 In studying polymer adsorption on SAM surfaces, it was found that polyelectrolytes and the surface with a like-charges were shown adsorption.23 In other words, although these systems had an interaction barrier of electrostatic origins, the adsorption of the polymer was fast.23 In another study, the adhesion forces between a hydrophobic surface (CH3-SAM) and alkali lignin was analyzed and the results revealed the importance of hydrophobic interaction between the CH3 surface and in non-modified lignin.24 As another objective of this work, the comparison of lignin and poly(vinyl alcohol-co-vinyl acetate) adsorption on different SAM surfaces could provide insights into the impact of polymer properties on the adsorption performance of polymers on altered surfaces. To the best of the authors' knowledge, the interaction of sulfonated PVA and lignin-based compounds on SAMs have not been studied.
In the present study, the adsorbed amount of PVA-S and L-S on OH, COOH, CH3, and NH2 functionalized surfaces were studied for the first time using QCM-D to provide information on their altered adsorption behavior. The selected SAM surfaces carried distinct terminal functional groups relevant to the surfaces used in different industries, such as surface coatings, mining, pharmaceutical, and cosmetics.4,7,9 Also, conducting adsorption studies under different saline and pH conditions would reveal the performance of these sulfonate-based polymers on altered surfaces in different environments and the interaction mechanisms of the adsorption processes on the surfaces.
The main goal of this paper was to identify how the interaction of lignin derivatives, i.e., highly branched materials, is different from their synthetic linear equivalents when their molecular weights and charge densities are similar. This paper provides fundamental insights into quantitative adsorption fundamentals of lignin and synthetic macromolecules. Demonstrating this difference would help establish methods to improve the characteristics of lignin for creating valorized lignin derivatives with desired functionality.
2. Experimental section
2.1 Materials
In this work, 11-mercapto-1-undecanol (–OH, 97%), 12-mercaptododecanoic acid (–COOH, 96%), 1-dodecanethiol (–CH3, ≥98%), ammonium hydroxide, 6-amino-1-hexanethiol hydrochloride (–NH2), vinyl acetate (99%), 3-trimethylsilyl-(2,2,3,3-D4)-propionic acid sodium salt (TMSP) (99.8%), hydrochloric acid (37%), sodium hydroxide (99.0%), poly diallyl dimethylammonium chloride (PDADMAC) with the molecular weight of 100–200 kg mol−1, potassium chloride (KCl), D2O (with the isotopic purity of 99.8%), 3-sulfopropyl methacrylate potassium salt (98%) (S), sodium persulfate (Na2S2O8), sodium chloride (99%), ethanol (99.8%), methanol (99.8%) and dimethyl sulfoxide-D6 ([D6]DMSO) (99.9%), and hydrogen peroxide (30 wt%) were all purchased from Sigma-Aldrich company. Cellulose acetate membrane with the molecular weight cut-off of 1000 g mol−1 was purchased from Spectrum Labs. Inc., USA. Also, softwood kraft lignin (L) was produced via the LignoForce technology of FPInnovations in Thunder Bay, ON, and received as a raw material. High-performance liquid chromatography grade water was produced using a Milli-Q water purifier with the resistivity of less than 18 MΩ cm−1 and used throughout this work. AT-cut gold-coated piezoelectric quartz crystal sensors (5 MHz resonant frequency) were purchased from Biolin Scientific Inc. Nylon filters with a pore size of 0.22 μm were purchased from the Celltreat Scientific company. Additionally, all of the chemicals utilized in this work were of analytical grades.2.2 Synthesis of L-S and PVA-S polymers
Softwood kraft lignin (L) was polymerized with 3-sulfopropyl methacrylate potassium salt (S) based on the methodology described in a previous study.25 The reaction was conducted at a molar ratio of 1.2 S/L, 10 wt% HCl, 90 wt% of the water in the presence of 1.5 wt% of potassium persulfate (as initiator) at 80 °C for 90 min. The produced polymer was purified with membrane dialysis for three days to remove any unreacted monomers, and the purified lignin-3-sulfopropyl methacrylate sample was denoted as L-S.In another set of reactions, vinyl acetate was used, instead of lignin, in polymerizing with 3-sulfopropyl methacrylate potassium salt (S). The reaction was conducted by using vinyl acetate (VA) and S in a molar ratio of 0.5 S/VA, 10% NaOH, 9/1 v/v of methanol/water in the presence of 1.5 wt% of potassium persulfate (as initiator) at 80 °C for 90 min.6,28 The produced polymer was precipitated by methanol precipitation and centrifugation at 3500 rpm for 10 min and then purified using membrane dialysis for three days. The poly(vinyl alcohol-co-vinyl acetate)-3-sulfopropyl methacrylate sample was donated as PVA-S. Polymeric solutions were prepared in the concentration of 1 g L−1 for different analyses in this study.
2.3 Static and dynamic light scattering
The light scattering analysis of produced L-S and PVA-S polymers were performed by a static light scattering instrument, Brookhaven BI-200SM, equipped with a goniometer. The laser polarized light was set at 633 nm.29 The cell was set at different temperatures ranging from 15 to 65 °C. The samples were passed through a 0.22 μm diameter porous filter. The second virial coefficient (A2), and the average radius of gyration (Rg) were obtained from the concentration dependence and slope of the angle based on Zimm plot eqn (1), respectively:30–32
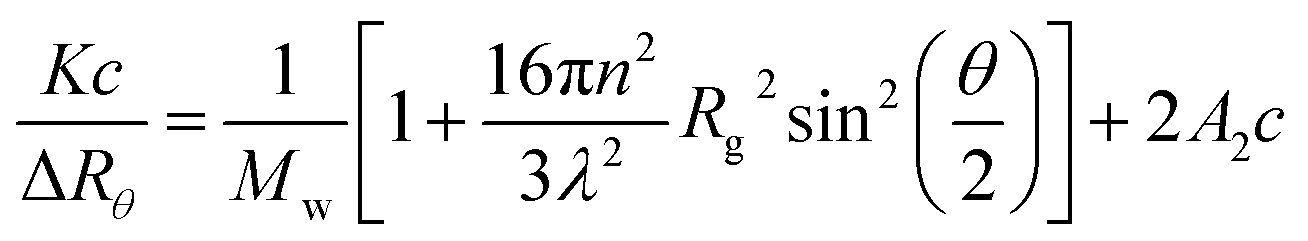 | (1) |
The hydrodynamic radius (Rh) of the polymers was determined using a dynamic light scattering (DLS) instrument, NanoBrook, ZetaPALS, Brookhaven Instruments Corp., USA, which was equipped with a 35 mW power laser (λ = 637 nm, wavelength). Measurements were made at 90° at different temperatures (15–65 °C), and sample solutions were filtered using 0.2 μm pore size filters for light scattering measurements. The hydrodynamic radius (Rh) was measured based on the diffusion coefficient (D) by using the Stoke–Einstein eqn (2):25,33,34
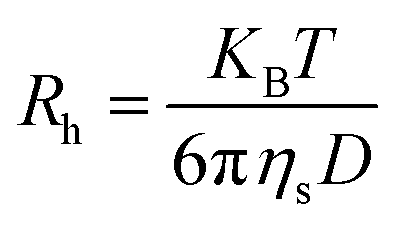 | (2) |
Differential refractometry (DR) technique was used to determine the macromolecular solutions' specific refractive index increments (dn/dc). It is essential to measure the dn/dc precisely to obtain an accurate Mw value. The refractive index increment of each polymer solution (dn/dc) was measured using a Brookhaven BI-DNDC instrument. Although many studies have reported dn/dc values for homopolymers, limited reports are found on the dn/dc values for heteropolymers.29,35
The second virial coefficient (A2), the average radius of gyration (Rg), hydrodynamic radius (Rh), and refractive index increments (dn/dc) were conducted three times and the average values were reported.
2.4 Characterization
The charge density of samples was determined with a Particle Charge Detector (PCD 04, BTG Mütek GmbH) using a 0.005 mol L−1 PDADMAC or 0.005 mol L−1 PVSK solution as the titrant, as explained elsewhere.14,36 The elemental analysis of the polymers was performed using an elemental analyzer (Vario EL Cube, Elemental Analyzer, Germany) as explained in detail elsewhere.37 The molecular weight of PVA-S and L-S polymers was measured by a gel permeation chromatography (GPC, Malvern GPCmax VE2001 with multi-detectors) after treating the samples at different pH values (4.0, 6.7, and 11.0) for 12 h and purifying the samples using membrane dialysis for 3 days.38 The details of this analysis can be found in the ESI† file.The phenolic hydroxyl and carboxylate group contents of L and L-S samples were determined using an automatic potentiometric titrator (785 DMP Titrino, Metrohm, Switzerland) with the HCl standard solution as a titrant (more information can be found in the ESI†).
The contact angle of water–air (θW/A) of PVA-S and L-S polymers at different pH were conducted using Theta Lite contact angle analyzer (Biolin Scientific, Finland) associated with a camera-based on our previous experiment.25 PVA-S and L-S were coated on glass slides using a spin coater (WS-650, Laurell Technologies Corp) and dried overnight. Then, the contact angle of 1.5 μL of a droplet on coated surfaces was determined.
The structure of L, PVA-S, and L-S was analyzed by a 1H NMR spectroscopy with 32 scans. Samples were dissolved in D2O or [D6]DMSO and stirred until fully dissolved (i.e., for 12 h).9,39 Trimethylsilyl propionic acid (TMSP) was used as the internal standard.40 More information about this experiment is available in the ESI† file.
The zeta potential of lignin (L), L-S, and PVA-S polymers were analyzed using a NanoBrook Zeta PALS (Brookhaven Instruments Corp, USA) at pH ranging from 3.0 to 11.0. The analysis was performed three times, and the average values were reported in this study.
2.5 Self-assembled monolayers (SAM)s preparation
In the QCM-D measurements, AT-cut piezoelectric quartz crystals (Biolin Scientific Inc), which were covered with gold and had a fundamental frequency of 5 MHz, were used. The crystals were modified according to the procedure described by Hedin et al.41 The surfaces were initially cleaned for 10 min in a UV/ozone chamber, which was followed by immersing in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:6 mixture of hydrogen peroxide (25%), ammonium hydroxide (25%), and Milli-Q water for 8 min at 78 °C. To obtain chemically well-defined and electrically inert SAM surfaces, the crystals were immersed overnight (>15 h) in 20 mL of 2 mM of alkanethiol solution in degassed ethanol (99.8%) and an amber bottle, and stirred at 50 rpm and 25 °C in a water bath. Then, 11-mercapto-1-undecanol (–OH), 1-dodecanethiol (–CH), 12-mercaptododecanoic acid (–COOH), and 6-amino-1-hexanethiol hydrochloride (–NH2) chemicals were used to form hydroxyl (OH), methyl (CH3), (COOH) and (NH2) terminated SAMs on the surfaces, respectively. The thiol group of n-alkanethiols bonds to Au surfaces by metal–sulfur bonds (i.e., chemisorption) and forms close-packed SAMs (i.e., self-assembled monolayers), which leaves –OH, –COOH, CH3, and NH2 (i.e., the other end of the functional groups) available on the surface.23,41 To remove excess thiols from the treated surfaces, the surfaces were washed and ultrasonicated in ethanol five times, each time for 5 min. The SAM surfaces were dried under N2 gas.
:1:6 mixture of hydrogen peroxide (25%), ammonium hydroxide (25%), and Milli-Q water for 8 min at 78 °C. To obtain chemically well-defined and electrically inert SAM surfaces, the crystals were immersed overnight (>15 h) in 20 mL of 2 mM of alkanethiol solution in degassed ethanol (99.8%) and an amber bottle, and stirred at 50 rpm and 25 °C in a water bath. Then, 11-mercapto-1-undecanol (–OH), 1-dodecanethiol (–CH), 12-mercaptododecanoic acid (–COOH), and 6-amino-1-hexanethiol hydrochloride (–NH2) chemicals were used to form hydroxyl (OH), methyl (CH3), (COOH) and (NH2) terminated SAMs on the surfaces, respectively. The thiol group of n-alkanethiols bonds to Au surfaces by metal–sulfur bonds (i.e., chemisorption) and forms close-packed SAMs (i.e., self-assembled monolayers), which leaves –OH, –COOH, CH3, and NH2 (i.e., the other end of the functional groups) available on the surface.23,41 To remove excess thiols from the treated surfaces, the surfaces were washed and ultrasonicated in ethanol five times, each time for 5 min. The SAM surfaces were dried under N2 gas.
2.6 SAM characterization
Theta Lite Contact Angle analyzer (Biolin Scientific, Finland) associated with a camera was employed to quantitatively analyze the wettability of the Au surfaces before and after SAM deposition. The contact angle analysis of water droplets (1.5 μL) on the bare and modified QCM sensors was conducted using the sessile drop method based on Young's equation with three independent measurements. The values of the water contact angle were evaluated for the four types of SAM are reported in Table 2 and are consistent with reports in the literature.23,42,43The X-ray photoelectron spectroscopy (XPS) measurements were performed on the bare and SAM-coated sensors by using a Kratos Axis Supra with a monochromatic Al Kα radiation (1486.6 eV) with 1 mm spot diameter at a base pressure of about 3 × 10−10 mbar and 20 eV pass energy. Based on the surface plane, the take-off angle for the detected photoelectrons was adjusted to 60°. For energy referencing, spectra were calibrated to the C1 line peak at 284.6 eV.44 Data analysis and peak fitting were conducted using ESCApe software (V1.2.0.1325). The thickness of SAM-terminated surfaces on Au substrates was estimated by means of the attenuation of the Au 4f signal using the following eqn (3):
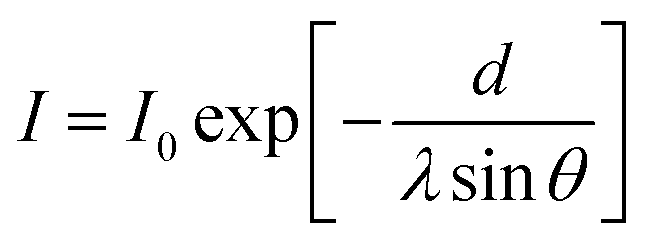 | (3) |
2.7 Quartz crystal microbalance with dissipation (QCM-D)
The detailed description of the QCM-D technique has been explained by Li et al.46 and Pensini et al.47 The adsorption studies of L-S and PVA-S were conducted on the above mentioned SAM-coated gold sensors by using a QCM-D, E1, 401, instrument (Q-Sense Inc., Gothenburg, Sweden). A peristaltic pump was used to pump solutions at the flow rate of 0.15 mL min−1 through the chamber of the QCM instrument. The temperature was set to room temperature (25 °C) for all experiments. The adsorption experiment was initially conducted with a buffer solution of the desired aqueous pH and electrolytes, until achieving a baseline signal in the analysis. Then, the adsorption experiment was initiated by replacing the buffer solution with L-S or PVA-S polymer solutions with the same pH and electrolyte concentration. After reaching saturation in adsorption, the solutions were changed to the buffer solutions for removing unadsorbed components from the SAM coated surfaces. The solutions were degassed prior to the experiment. This analysis was conducted at different salt concentrations (1–1000 mM) and pH (3.0–11.0) with PVA-S and L-S solutions. It is worth noting that the PVA-S, L-S, and buffer solutions had the same pH and salinity in all experiments.In this analysis, Sauerbrey and Voigt equations were used to evaluate the properties of the adsorbed layers and analyse the changes in the adsorbed mass on the sensor (more information is available in the ESI†). The Sauerbrey equation was used when ΔD ≤ 1 × 10−6, which would be an indication of an elastic and rigid layer. However, data points were fitted into the Voigt model for layers with the higher dissipation using three harmonic overtones of 5, 7, and 9 of the Q-Tools software. The 9th overtone was used for the better representation of data (details available in the ESI†). Also, the fluid density and viscosity were considered to be 0.99 g cm−3 and 1.05 mPa, respectively. The error bars related to the adsorbed mass present the standard deviation of the repeated experiments.
3. Results and discussion
3.1 Characterization of produced polymers
The 1H-NMR spectra of the produced samples are depicted in Fig. S1 and S2.† The appearance of a new peak at 0.96 and 1.10 ppm is assigned to the methyl group of S monomer in the 1H-NMR spectra.25,48,49 The three peaks at 4.7, 4.5, and 4.2 ppm are assigned to the proton in the (OH) group of PVA (Fig. S2†).39,50 The peak at 1.97 ppm is also assigned to the methyl (CH3) group of PVA.39,51 More information could be found in the ESI.† The results in Fig. S1 and S2† confirm the successful production of L-S and PVA-S. Also, FTIR analysis was performed on the obtained polymers and the spectroscopy, as well as the peak data, are shown in Fig. S3 and Table S1 in the ESI† file, respectively. The results of this analysis also confirmed the successful polymerization and the production of L-S and PVA-S. In addition, the reaction yield for the production of L-S (65.32%) was measured to be lower than the reaction yield for the production of PVA-S (74.21%). The properties of produced polymers are shown in Table 1. Based on the elemental analysis (Table 1), L (unmodified lignin), L-S, and PVA-S had 0.76, 8.03, and 8.17 wt% sulfur element, respectively, and no traceable nitrogen. The charge density (CD) of L, L-S, and PVA-S were −0.85, −3.17, and −3.20 meq. g−1, respectively, which would indicate that the amount of introduced sulfur content to polyvinyl acetate and lignin was equal. The GPC analysis demonstrated the molecular weight of 1.8 × 104, 11.4 × 104, and 11.3 × 104 g mol−1 for L, L-S, and PVA-S, respectively. Since the adsorption of the produced polymers is going to be analyzed on different surfaces at different pH, it is crucial to analyze the stability of the polymers at different pH. To do so, the molecular weight analysis was performed using GPC for the samples pretreated in both acidic and alkaline pH for 12 h. As seen in Table S2,† compared to neutral pH, the PVA-S, and L-S samples showed 7.6–8.9, and 4.0–0.2% reduction in their molecular weights, respectively. These results reveal that both polymers are relatively stable over the examined pH range. The charge of produced polymers at various pH was determined through zeta potential analysis and presented in Fig. S4 in the ESI† file. By changing the pH, the zeta potential of L was changed from about −7.5 to −17.6 mV. The zeta potential increment was observed more drastically for both PVA-S and L-S polymers than L, which could be due to their sulfonate groups.| Samples name | L | L-S | PVA-S |
|---|---|---|---|
| a Error was <0.05%.b Method sensitivity <0.09. | |||
| Nitrogen content,a wt% | <0.09b | <0.09b | <0.09b |
| Sulfur content,a wt% | 0.76 | 8.03 | 8.17 |
| Charge density,a μeq. g−1 | −0.85 | −3.17 | −3.20 |
| Carboxylate content,a mmol g−1 | 0.17 | 0.16 | — |
| Phenolic hydroxyl group content,a mmol g−1 | 1.86 | 0.18 | — |
| Mw (GPC), g mol−1 | 1.8 × 104 | 11.4 × 104 | 11.3 × 104 |
| Mn (GPC), g mol−1 | 0.48 × 104 | 6.38 × 104 | 6.05 × 104 |
| dn/dc,a mL g−1 | 0.1320 | 0.1601 | 0.0660 |
| Reaction yield, % | 100 | 65.32 | 74.21 |
3.2 Characterization of self-assembled monolayers (SAM)
A series of SAM-modified surfaces were produced by reacting thiols with different end groups on the QCM sensor. Fig. 1 demonstrates the X-ray photoelectron spectroscopy (XPS) of SAM with –OH, –NH2, –CH3, and –COOH end groups on the sensors. The presence of a peak at 162 eV is attributed to the Au–S energy binding, which indicates the successful modification of Au with SAMs. | ||
| Fig. 1 SAMs with different terminated groups on gold: XPS S 2p spectra of (a) –OH, (b) –COOH, (c) –CH3, and (d) –NH2. | ||
Table S3 (in the ESI† file) includes the relative atomic concentrations of SAM-modified surfaces (C, O, S, and N), which confirms the successful formation of SAM. The excess amount of O in –CH3 could be attributed to the atmospheric contamination. In the case of –NH2 modified surface, the excess amount of O could be attributed to the atmospheric contamination (3.8%) and partial oxidation of the S–Au bond to sulfonate (7.3%). The S2p level of –OH, –COOH, –CH3, and –NH2 SAM-terminated surfaces were studied, and it has been found that the partial oxidation of –SH to sulfonate happened only in NH2 terminated SAM, which could be attributed to the HCl used to stabilize amino compound.52
Table 2 demonstrates the properties of the four types of SAMs. The sessile drop method was used to measure the deionized water contact angle on SAM coated sensors.42,43 The water contact angle of the gold sensor was 75 ± 2°. The contact angle of –OH, –COOH, –CH3, and –NH2 surfaces were measured to be <10°, 30°, 110°, and 41°, respectively. At neutral pH (pH 6.7), –OH and –CH3 carry no charges while –NH2 is positively charged and –COOH is negatively charged.53 The contact angle measurements of SAM films suggested that the surface coated with the –CH3 group was highly hydrophobic, while the surfaces coated with –OH, –COOH, and –NH2 groups were wettable. SAMs functionalized with –OH could be considered as super-hydrophilic as the water droplet spread on the SAM surface (a contact angle of <10°). According to the literature, the contact angle for –OH functionalized SAM was reported to be as low as 9.5°23,52 and as high as 40–50°.54,55 The monolayer surface containing OH has been reported to be unstable.56 The rate of contact angle variation was also revealed to be dependent on temperature. Also, freshly prepared surfaces were suggested to be used for more credibility of this analysis, which was performed accordingly in this work. These SAMs (–CH3, –OH, –COOH, and –NH2) were well characterized and used extensively as model surfaces.57,58
| Surface | Chemical formula | SAMs name | Water contact angle, ° | Features | Charge at pH 6.7 | Theoretical length, Å | Thickness, Å | Tilt angle, ° |
|---|---|---|---|---|---|---|---|---|
| OH | HS(CH2)11OH | 11-Mercapto-1-undecanol | <10 | Hydrophilic | Neutral | 17.1 | 12.97 | 40.6 |
| COOH | HS(CH2)11COOH | 12-Mercaptododecanoic acid | 30 ± 3 | Hydrophilic | − | 18.3 | 15.11 | 34.3 |
| CH3 | HS(CH2)11CH3 | 1-Dodecanethiol | 110 ± 2 | Hydrophobic | Neutral | 17.3 | 15.00 | 29.9 |
| NH2 | HS(CH2)6NH2·HCl | 6-Amino-1-hexanethiol hydrochloride | 41 ± 1 | Hydrophilic | + | 10.8 | 9.55 | 27.8 |
The theoretical lengths of SAMs were obtained from computational modeling, Avogadro software (V1.2.0), are reported in Table 2. By considering the estimated values achieved from the Au 4f (Fig. S5†) attenuation analysis, the SAMs show an atilt angle in the range of 27.8–40.6° for the surface (Table 2 and Fig. S5†). If SAMs were to be defective, isolated domains would have been formed on the surfaces, which would have further resulted in single molecules to generate a lying-down configuration.52 Thus, our results reveal a defect-free and tightly packed SAMs. In other words, all SAMs had significant high-packing densities while exhibiting differential end groups.52,58
Also, to analyze the interaction of SAM surfaces with water molecules, water adsorption on different SAM surfaces was conducted at different temperatures (Fig. S6–S9†) and pH (Fig. S10–S13†). Based on the obtained results, no significant difference (i.e., within 10%) was observed between SAM surfaces. This indicates that the chemically modified SAM surfaces are stable under the examined conditions. Also, these conditions (different temperatures and pH) showed no specific effect on the swelling of SAMs (Fig. S6–S13†).
3.3 Measurements of the hydrodynamic radius (Rh), the radius of gyration (Rg), and second virial coefficient (A2)
Fig. 2a depicts the hydrodynamic radius, Rh, of PVA-S, and L-S in pure water. As can be seen, the Rh of L-S increased by increasing the temperature from 15 to 65 °C. The increase in the overall size of L-S with temperature enhancement could be due to more hydration of lignin structure at a higher temperature. In the case of PVA-S, the insignificant change in the Rh might be attributed to the dehydration of PVA-S caused by polyvinyl acetate chains at a high temperature. Similar behavior was reported for poly(ethylene oxide) in water.59,60 Fig. 2b depicts the radius of gyration, Rg, defined as the root mean square distance of a particle's components from its center of mass. As seen, enhancing the temperature drastically increased the Rg of L-S, while it did not significantly affect the Rg of PVA-S. It is worth mentioning that Rg is sensitive to the refractive index distribution (mass distribution), while Rh is sensitive to hydrodynamics.33 It is well established that the ratio of Rg/Rh is a characteristic parameter related to the conformation of polymer chains in solutions. The values of Rg/Rh for uniform hard-sphere, random coil, and rod-like structures have been reported to be 0.778, 1.78, and ≥2, respectively.61–64 In the case of L-S, both Rg/Rh and Rg have increased with increasing the temperature. At 15 °C, the Rg/Rh ratio for L-S was about 0.77, while the Rg/Rh ratio increased to about 1.27 at 65 °C, revealing that the L-S present a swollen structure.32 An increment in the Rg/Rh ratio for L-S in the range of 0.77 to 1.27 (from 15 to 65 °C) is in agreement with the intermediate structure from a hard-sphere to a looser structure due to the water swelling effect.65 The respective Rg/Rh ratio of PVA-S changed from ∼0.60 at 15 °C to ∼0.76 at 65 °C. Thus, the spherical shape of the PVA-S polymer was insignificantly altered via temperature alteration. The higher Rg/Rh ratio at a high temperature also ascribed to the dehydration of PVA-S.66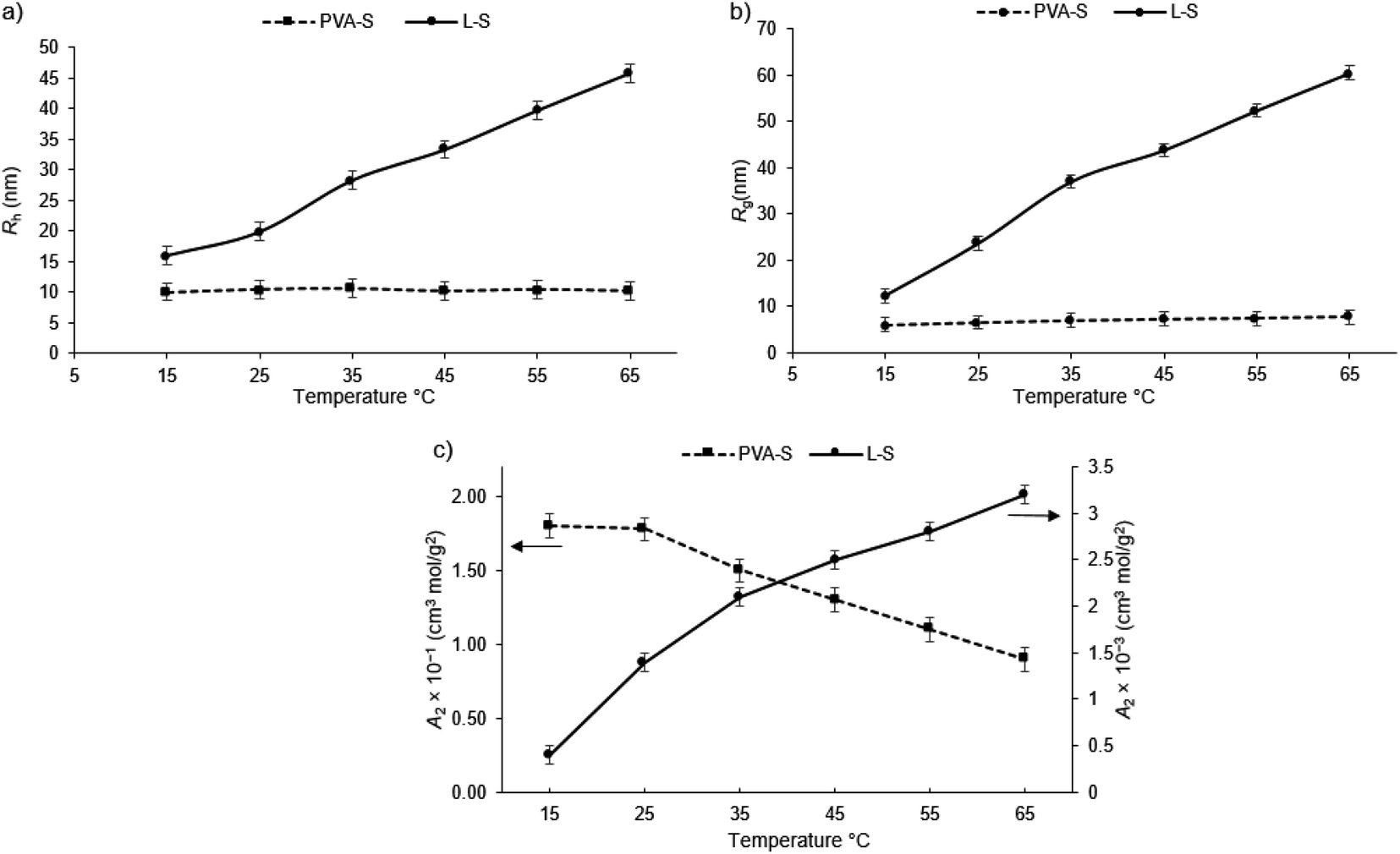 | ||
| Fig. 2 Temperature dependence of (a) hydrodynamic radius (Rh), (b) radius of gyration (Rg), and (c) second virial coefficient (A2) of PVA-S and L-S polymers. | ||
Fig. 2c indicates the second virial coefficient (A2) of the polymers, which signifies the polymer–solvent interactions. A low value of A2 means strong interactions among solute particles, indicating that the solute is in a poor solvent, which means that the polymers are partially or completely insoluble in the solvent. The magnitude of A2 values denotes the strength of such interactions. The variation in A2 with temperature depends on the hydrophilic/hydrophobic structure of polymers.67–69 Based on Fig. 2c, the A2 value for L-S polymer increases as the temperature rises from 15 to 65 °C, implying that high temperature improved the interaction of L-S polymer and water. The intensive interactions between polymer segments could lead to a more compact structure with the hard-sphere configuration at low temperature, while the more efficient interaction between L-S and water loosened the structure of L-S polymer at a higher temperature, which is supported by results depicted in Fig. 2a and b. In the case of PVA-S (Fig. 2c), A2 values decreased by increasing the temperature implying that water has become a poor solvent for this polymer at a higher temperature. Similar behavior has been reported for poly(vinyl alcohol).70,71 As reported in the literature, the PVA macromolecule chain becomes dehydrated above critical solution temperature (25 °C in our case).72,73
The lower solubility of L-S than PVA-S, based on the second virial coefficient (A2), could be due to two reasons: its aromatic structure,74,75 and, its pKa value (phenolic groups), which drops at higher temperatures and leads to an increase in its solubility.76,77
3.4 Effect of temperature on the adsorption of L-S and PVA-S on SAMs
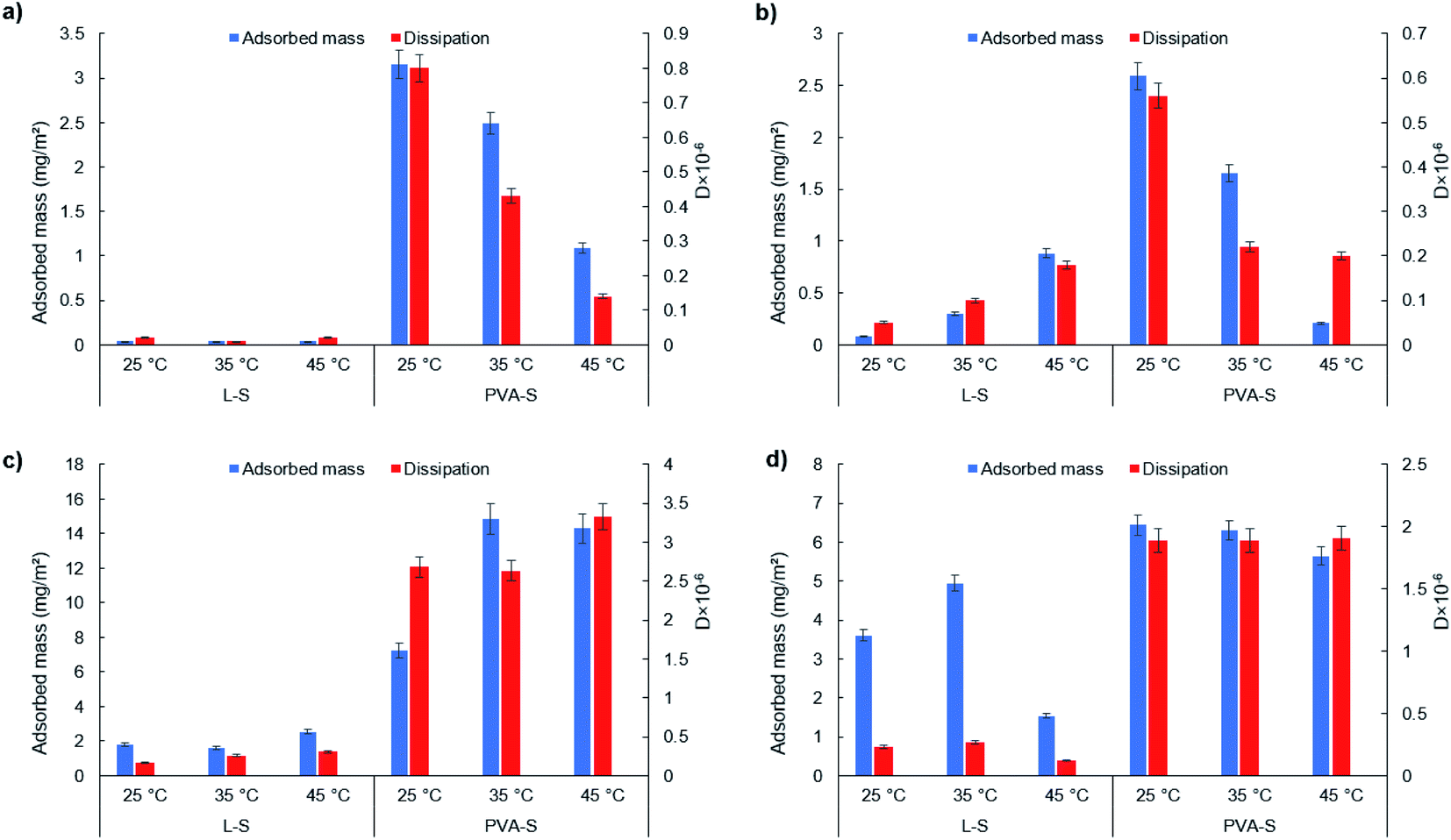 | ||
| Fig. 3 Adsorbed mass and dissipation of L-S and PVA-S on (a) OH, (b) COOH, (c) CH3, and (d) NH2 functionalized surfaces at different temperatures. | ||
The adsorbed amount of PVA-S is observed to decrease sharply from 2.59 mg m−2 at 25 °C to 0.21 mg m−2 at 45 °C. As explained earlier, the solubility of PVA-S was reduced at higher temperatures (Fig. 2c), and thus its driving force for adsorption dropped. By increasing the temperature, the interaction between hydrophilic groups of PVA-S would decrease. The reason for this could be the denser structure of PVA at higher temperatures, which would result in dehydration in the structure of PVA72 and a lower adsorption amount on the –OH functionalized surface. On the other hand, by lowering the hydrogen bonding capability of PVA-S, more hydrophobic parts of PVA-S (i.e., CH) would be exposed and interact with –CH3 functionalized surface.
Overall, comparing the adsorption of L-S on the NH2 (Fig. 3d) and CH3 (Fig. 3c) surfaces, the higher adsorption of L-S was found on the NH2-functionalized surfaces. This implies that the electrostatic interaction could be more dominant than hydrophobic interactions for L-S. In other words, temperature increment could improve the electrostatic interaction development of L-S with surfaces more than its hydrophobic interaction. The similar trend observed for PVA-S adsorption on –NH2 and –CH3-functionalized surfaces could indicate that the electrostatic attraction and hydrophobic interaction were both playing important roles in PVA-S adsorption onto the functionalized surfaces. In addition, comparing L-S and PVA-S, the highest adsorption capacity of L-S (4.95 mg m−2) was achieved at 35 °C on –NH2 functionalized surface (Fig. 3d). Meanwhile, the highest adsorption amount of PVA-S (14.83 mg m−2) was obtained at 35 °C on the –CH3 functionalized surface (Fig. 3c). The unmodified lignin (L) polymer contains different functional groups of phenolic hydroxyl (1.86 mmol g−1 (Table 1), carboxylate (0.17 mmol g−1 (Table 1), carbonyl, and methoxy groups, which could provide the macromolecule with hydrogen bonding capability. However, the rigid and three-dimensional structure of lignin11 would limit the chance of available functional groups on L-S to interact with the solid surfaces containing –OH and COOH groups and lead the sites on lignin to prefer developing hydrogen bonds with the solution rather than the surfaces.
3.5 Effect of pH on the adsorption of L-S and PVA-S on SAMs
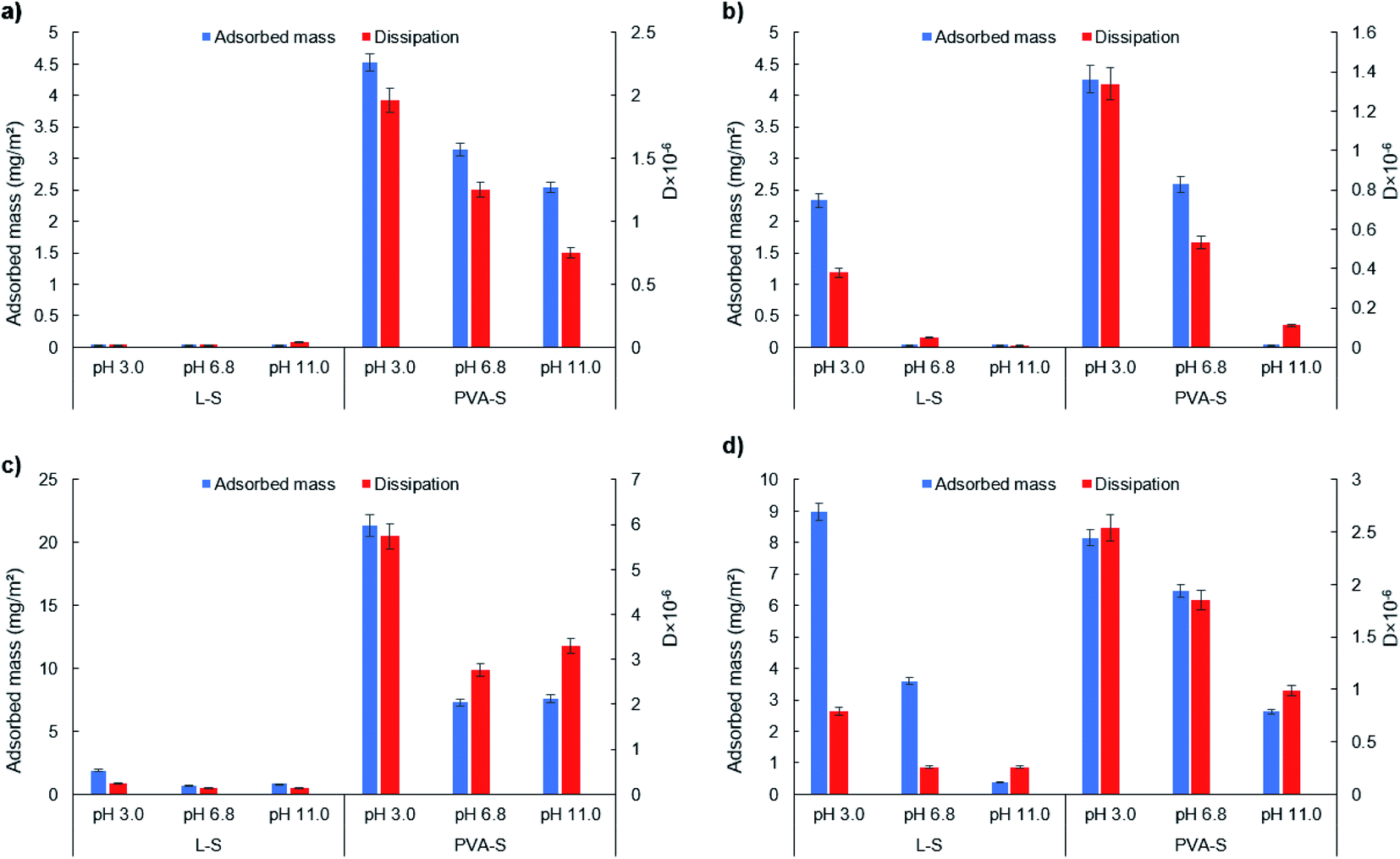 | ||
| Fig. 4 Adsorbed mass and dissipation of L-S and PVA-S on (a) OH, (b) COOH, (c) CH3, and (d) NH2 functionalized surfaces at different pH. | ||
A similar trend of reduction in the PVA-S adsorption to –COOH surface was observed by increasing the pH, which is due to the enhancement in the repulsion force between the polymer and the surface. While electrostatic interactions play a critical role in the adsorption of like-charged polymers (i.e., electrostatic repulsion), the interesting PVA-S adsorption on the negatively charged –COOH surface indicates the sensible development of nonelectrostatic interactions between PVA-S and COOH surface (e.g., van der Waals and hydrogen bonding).
It is seen that L-S showed less hydrophobic interaction than PVA-S with the CH3 functionalized surface at all pH studied. This might be because of the linear structure of PVA-S, which facilitates the hydrophobic interaction of PVA-S with the surface, while the three-dimensional structure of L-S prohibits the hydrophobic interaction of the aromatic core of lignin with CH3 (i.e., steric hindrance).
Furthermore, increasing the pH (from 3.0 to 11.0) led to the enhancement in the hydrodynamic radius (Rh) of L-S and PVA-S polymers, and this change was more sensible for L-S (Fig. S30†), which can be due to its better solubility at higher pH. According to the contact angle analysis (Table S4†), PVA-S was more hydrophilic than L-S at all pH. This means that PVA-S could trap more water in its structure, which might lead to more changes in the adsorbed mass upon its adsorption to SAM surfaces. Although these two polymers have the same amount of sulfonate groups, their structural difference1 led to their altered interactions with varied surfaces. Also, its linear structure2 favored the adsorption of PVA-S on the functionalized surfaces.
In general, the maximum adsorption amount of L-S (8.99 mg m−2) was attained at pH 3.0 on the –NH2 functionalized surface. Meanwhile, the highest adsorption amount of PVA-S (21.33 mg m−2) was obtained at pH 3.0 on –CH3 functionalized surface.
3.6 Effect of salt concentration on the adsorption of L-S and PVA-S on SAMs
The adsorption of L-S and PVA-S on different functionalized surfaces at equilibrium is shown in Fig. 5 and 6 (the original data is provided as Fig. S31–S34 in the ESI† file). In saline systems, the long-range electrostatic double-layer interactions between the polymers and surfaces become remarkably small while the nonelectrostatic forces come into action.23 By increasing the salt concentration, the adsorption of L-S polymer increased on all surfaces (Fig. 5a). These results suggest that nonelectrostatic forces played a role in the adsorption of L-S on carboxyl and hydroxyl-coated surfaces. The more significant change for OH and COOH might be due to nonelectrostatic forces becoming relatively important compared to electrostatic interaction in salty systems.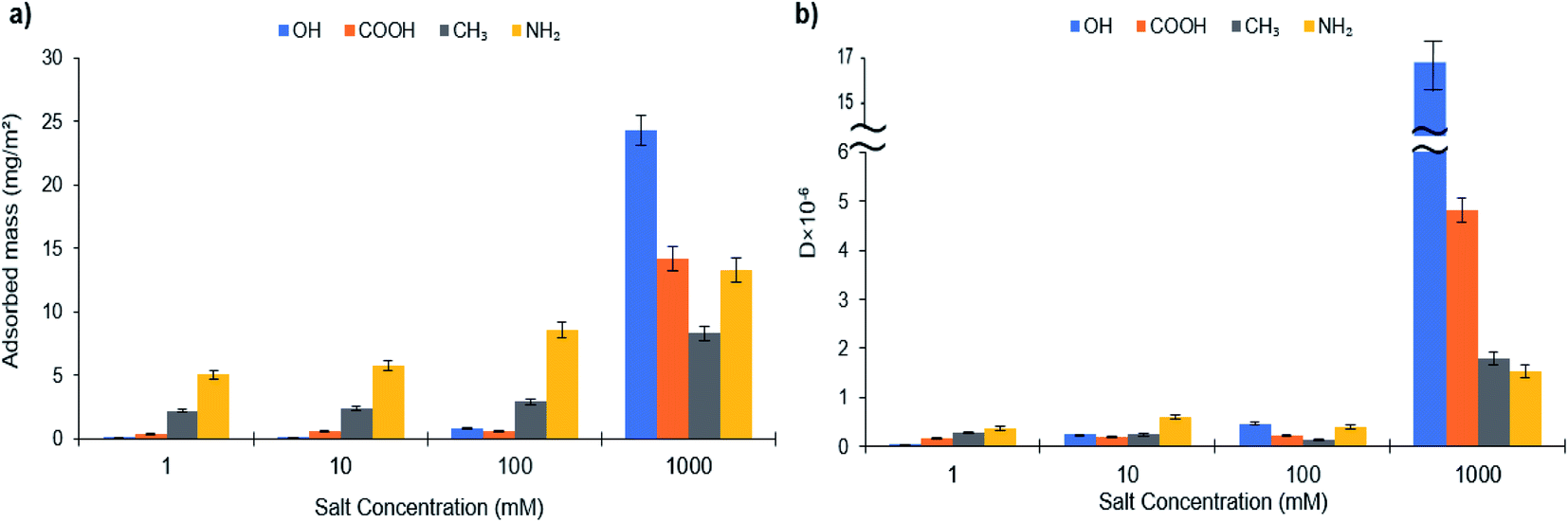 | ||
| Fig. 5 (a) Adsorbed mass and (b) dissipation of L-S on SAMs of different surfaces at different salt concentrations. | ||
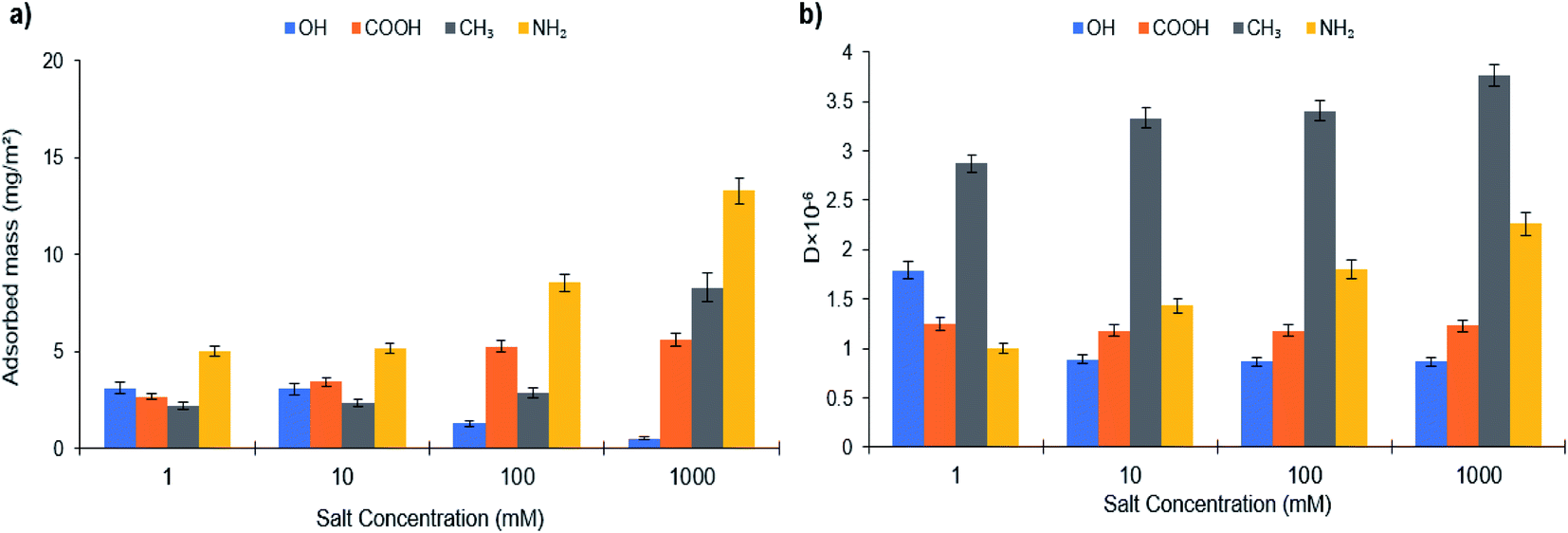 | ||
| Fig. 6 (a) Adsorbed mass and (b) dissipation of PVA-S on SAMs of different chemistry at different salt concentrations. | ||
Fig. 5b illustrates the dissipation changes on different alkanethiol surfaces. It is seen that the increase in the salt concentration (from 1 mM to 1000 mM) elevated the dissipation of L-S polymer on the –OH surface while depicting the maximum dissipation (16.48 × 106) at 1000 mM of salt concentration (Fig. 5b). Other surfaces also showed an increase in their dissipation when salt concentration increased from 100 mM to 1000 mM. This increase in dissipation might be raised by two phenomena; an increase in the adsorption of L-S on the surfaces and adaptation of an extended configuration in salty systems (Fig. S32†), which might be due to screening of the intramolecular interactions in the L-S.87 Increasing the salt concentration affected the hydrodynamic radius (Rh) of the polymer due to electrostatic shielding and attraction between sulfonate groups of L-S and cations available from salt. In this case, the Rh increment was observed to be more in L-S than in PVA-S (Fig. S35†). The addition of salt would increase the attraction between adjacent polymer cores by eliminating the electrostatic interaction and exposing the hydrophobic sites of the polymer. This would lead to more L-S particles to aggregate and thus enhance L-S adsorption onto surfaces. Similar behaviour was observed for the self-aggregation of CNC (cellulose nanocrystals) at 160 mM KCl.88
Fig. 6 depicts the adsorbed mass and dissipation of PVA-S adsorption on different functionalized surfaces as a function of salt concentration. If electrostatic interactions between the polymer and oppositely charged substrates were dominant, there would be a cutoff of adsorption with increased ionic strength, above which the adsorption would be decreased. Normally, the cutoff is abrupted when there are only electrostatic interactions deriving the adsorption.86,89–91 The adsorption of PVA-S polymer decreased on the –OH surface, while it was increased on –COOH, –CH3, and –NH2 modified surfaces (Fig. 6a). Compared to the results obtained from different surfaces, having constant adsorption indicates that nonelectrostatic forces dominated (rather than electrostatic interactions) when the polymer was not ionized.23 Also, a decrease in the adsorption of PVA-S on the –OH surface in the saline system has been reported in other studies.23,92 The fast adsorption kinetics (Fig. S33†) seen on positively charged NH2 terminated thiolates were observed to be unaffected by increasing the ionic strength (Fig. 6). This could be due to the electrostatic attraction force, which is always favorable and fast.88,92,93
In contrast, the adsorption kinetics for liked-charged systems (PVA-S and L-S on –COOH SAMs) was slow while a late saturation was attained (Fig. S32 and S33†). Salt reduced the kinetic barrier between like-charged polymers and surfaces, further favoring the adsorption.21,23,94 The noticeable change in the adsorbed mass of the –CH3 surface indicates that the hydrophobic interaction developed between the acetate group of the PVA-S and the surface, while all charges on the polymer were screened. The adsorbed mass was increased marginally for PVA-S on amine-rich monolayers by enhancing the ionic strength, which could be related to the higher adsorption of counterions from the solution, suppressing the polymer adsorption to this surface (Fig. 6).
Also, as mentioned earlier, the Rh of L-S and PVA-S increased as a result of an augment in salinity (Fig. S35†). This might be because increasing ionic strength causes the inter and intra-chain repulsion to develop a looser manner and expand polymer chains. These changes in the conformation of polymers lead to more sights become available for the interaction, which might have contributed to the increased adsorption of these polymers on most surfaces (Fig. 5 and 6). Polymerized kraft lignin-acrylic acid (KL-AA) with a molecular weight of 7.4 × 105 g mol−1 was reported to have the Rh value of 25.2 nm in salt-free solution.95 Moreover, the same behavior has been reported in the literature for poly(potassium-2-sulfopropylmethacrylate) when salt was added.96
In salinity, the highest adsorption amount for L-S polymer (24.32 mg m−2) was attained at 1000 mM of salt concentration on the –OH functionalized surface. Meanwhile, the maximum adsorption of PVA-S (13.3 mg m−2) was achieved at 1000 mM on the –NH2 surface.
Fig. 6b shows the dissipation changes on different alkanethiol surfaces with PVA-S adsorption concerning the ionic strength. It can be seen that the reduction in dissipation could be due to the decrease in the adsorption amount. In the case of –COOH, although the adsorbed mass was increased by the salt addition, the reduction in the dissipation could be due to PVA-S adopting a more compact structure, which was generated from nonelectrostatic forces in saline systems. In the case of –OH, –CH3 and NH2 surfaces, the dissipation value was changed proportionally to the adsorption of PVA-S on these surfaces.
3.7 Application
OH-coated surfaces interact with other materials, such as organic dyes polyesters,97 fatty acids, and alkyd resins.98 These surfaces are generally used in wastewater treatments, polymer blends, attachment of antibodies, and crystal engineering.9,99 The adsorption of L-S was less significant than PVA-S. This may make PVA-S a more effective coating material. Considering the temperature, salt, and pH variations, the results suggest that increasing the temperature, salt, and pH have an adverse impact on the adsorption of PVA-S onto the OH-functionalized surface.For COOH coated surfaces that interact with other materials, such as proteins, surfactants, antigen detection,100,101 PVA-S showed to be a more effective adsorbent than L-S, but its effectiveness was significantly affected by pH and temperature (Fig. 3 and 4).
For CH3 coated surfaces that interact with other materials, such as surfactants or surface-active agents,102 and have applications such as self-cleaning, anti-fogging, and anti-corrosion,103 PVA-S showed to be a better candidate. The temperature analysis showed the limited impact of PVA-S adsorption onto this surface. Also, the pH analysis confirmed that at acidic pH, the hydrophobic interaction of both PVA-S and L-S polymers with the surface is more significant. The salinity analysis confirmed that a higher salt concentration (1000 mM) could increase the adsorption for both PVA-S and L-S polymers.
In NH2 coated surfaces that interact with other materials, such as heavy metal, and have applications as flocculants, adsorbents, and dispersants, PVA-S observed to be a better candidate. The temperature analysis showed the limited impact of temperature variation on PVA-S adsorption and adverse effect on L-S adsorption. Also, the pH analysis confirmed that alkaline pH has a negative impact on the adsorption for both PVA-S and L-S polymers. The salt analysis confirmed that increasing the salt increases the adsorption for both PVA-S and L-S polymers.
Also, the contact angle analysis confirmed that PVA-S is more hydrophilic than the L-S polymer. Based on the second virial coefficient (A2) obtained from static light scattering, increasing the temperature adversely impacted the water solubility of PVA-S, while increasing that of L-S. Based on the hydrodynamic radius obtained from dynamic light scattering, pH enhancement has a more intense impact on the L-S polymer than PVA-S. On the other hand, PVA-S showed sensitivity to temperature, pH, and salt. Although the L-S adsorption was more limited, it was insensitive to temperature. Therefore, L-S could be used in applications that are sensitive to temperature but do not require a high level of adsorption. Also, the adsorption of L-S showed improvement by increasing the salt concentration. This demonstrates the affinity of lignin in developing different interaction mechanisms with different functionalized surfaces over PVA-S in saline systems. Thus, the contribution of nonelectrostatic forces seems to be more significant in the L-S polymer, illustrating practically an irreversible adsorption onto all surfaces, and its adsorption to the surfaces even when the electrostatic barrier exists. Therefore, it could be revealed that in applications that contain salts, e.g. wastewater treatment,15 additives for composites,104 lignin could be a better candidate to be used than PVA-S.
3.8 Future trend
Understanding the interaction of sulfonated lignin and PVA would help advance scenarios to improve the properties of lignin for generating super functional lignin derivatives. The analysis in this paper reveals that the inherent steric hindrance of lignin may need to be reduced, via depolymerization, oxidation, for instance, for elevating its adsorption on surfaces. Furthermore, raised from its folded and compact molecular structure, most of the functional groups on lignin were not exposed but need to be activated. Alternatively, lignin can be decorated with more functional groups to boost its interaction with surfaces. For example, a combination of sulfonation, carboxylation, and polymerization may induce lignin with desired properties.4. Conclusions
There has not been any systematic study in the literature on evaluating the performance of synthetic and lignin-based polymers having a similar charge density and molecular weight on well-defined self-assembled monolayers. In this study, we synthesized two types of anionic polymers of lignin-3-sulfopropyl methacrylate (L-S), and poly(vinyl alcohol-co-vinyl acetate)-3-sulfopropyl methacrylate (PVA-S) with similar charge densities and molecular weights. The Rh and Rg of PVA-S were smaller than those of L-S, while PVA-S had a larger A2. The A2 of PVA-S decreased with augment in the temperature whereas it enhanced for L-S, which was in agreement with the more compact structure of PVA-S. On the OH surface, PVA-S was adsorbed more than L-S, which could be due to its limited hydrogen bonding. Also, temperature, salt, and pH variations adversely affected the adsorption of PVA-S onto this surface. Although PVA-S was a more effective adsorbent on the COOH coated surface due to its higher solubility, pH and temperature were observed to remarkably impact its adsorption performance. The adsorption of both polymers was also increased with salinity enhancement on the COOH-surface. For the CH3 coated surface, although the PVA-S was observed to be a better candidate than L-S, increasing the temperature enhanced the L-S adsorption onto this surface due to more exposure of its hydrophobic parts. Interestingly, under acidic conditions, the hydrophobic interaction of both PVA-S and L-S polymers with the surface was increased. PVA-S also adsorbed more onto the NH2 coated surface than L-S due to its solubility and polymer structure. The temperature was more significantly affected the L-S adsorption onto this surface than did PVA-S, while the similar negative impact was observed for the adsorption of both polymers onto this surface at higher pH. Also, in saline systems, L-S adsorption was improved more significantly compared to PVA-S. Overall, the maximum adsorption of L-S (24.32 mg m−2) and PVA-S (21.33 mg m−2) polymers were observed on –OH functionalized surface at 1000 mM salt and on –CH3 functionalized surface at pH 3.0, respectively.Conflicts of interest
No competing financial interests are declared by authors.Acknowledgements
The authors would like to acknowledge NSERC, Canada Foundation for Innovation, Canada Research Chairs, Northern Ontario Heritage Fund Corporation, and Ontario Research Fund programs for supporting this research.References
- E. R. Van der Hage, M. M. Mulder and J. J. Boon, Structural characterization of lignin polymers by temperature-resolved in-source pyrolysis-mass spectrometry and Curie-point pyrolysis-gas chromatography/mass spectrometry, J. Anal. Appl. Pyrolysis, 1993, 25, 149–183 CrossRef CAS.
- F. Wang, P. Chandler, R. Oszust, E. Sowell, Z. Graham, W. Ardito and X. Hu, Thermal and structural analysis of silk–polyvinyl acetate blends, J. Therm. Anal. Calorim., 2017, 127, 923–929 CrossRef CAS.
- S. Jie, Y. Li, B. Jie, Z. Chu-Shu, L. Shao-Fang, Z. Feng and Y. Qing-Li, Peanut protein–polyvinyl alcohol composite fibers extruded from an ionic liquid, RSC Adv., 2013, 3, 10619–10622 RSC.
- A. Veerabhadraiah, S. Ramakrishna, G. Angadi, M. Venkatram, V. K. Ananthapadmanabha, K. N. M. H. NarayanaRao and K. Munishamaiah, Development of polyvinyl acetate thin films by electrospinning for sensor applications, Appl. Nanosci., 2017, 7, 355–363 CrossRef CAS.
- J. A. Gidigbi, S. A. Osemeahon, A. M. Ngoshe and A. Babanyaya, Modification of Polyvinyl Acetate with Hydroxylated Avocado Seed Oil as a Copolymer Binder for Possible Application in Coating Industry, International Journal of Recent Innovations in Academic Research, 2019, 3, 231–244 Search PubMed.
- T. Moritani and J. Yamauchi, Functional modification of poly(vinyl alcohol) by copolymerization III. Modification with cationic monomers, Polymer, 1998, 39, 559–572 CrossRef CAS.
- S. Ur-Rehman, M. Noman, A. D. Khan, A. Saboor, M. S. Ahmad and H. U. Khan, Synthesis of polyvinyl acetate/graphene nanocomposite and its application as an electrolyte in dye sensitized solar cells, Optik, 2020, 202, 163591 CrossRef.
- T. Moritani and J. Yamauchi, Functional modification of poly(vinyl alcohol) by copolymerization: II. Modification with a sulfonate monomer, Polymer, 1998, 39, 553–557 CrossRef CAS.
- A. E. Kazzaz, Z. H. Feizi and P. Fatehi, Grafting strategies for hydroxy groups of lignin for producing materials, Green Chem., 2019, 21, 5714–5752 RSC.
- M. Moradipour, E. K. Chase, M. A. Khan, S. O. Asare, B. C. Lynn, S. E. Rankin and B. L. Knutson, Interaction of lignin-derived dimer and eugenol-functionalized silica nanoparticles with supported lipid bilayers, Colloids Surf., B, 2020, 111028 CrossRef CAS.
- X. Luo, A. Mohanty and M. Misra, Lignin as a reactive reinforcing filler for water-blown rigid biofoam composites from soy oil-based polyurethane, Ind. Crops Prod., 2013, 47, 13–19 CrossRef CAS.
- Z. H. Feizi, A. E. Kazzaz, F. Kong and P. Fatehi, Evolving a flocculation process for isolating lignosulfonate from solution, Sep. Purif. Technol., 2019, 222, 254–263 CrossRef CAS.
- A. E. Kazzaz and P. Fatehi, Technical lignin and its potential modification routes: a mini-review, Ind. Crops Prod., 2020, 154, 112732 CrossRef.
- A. E. Kazzaz, Z. H. Feizi and P. Fatehi, Interaction of sulfomethylated lignin and aluminum oxide, Colloid Polym. Sci., 2018, 296, 1867–1878 CrossRef CAS.
- J. Chen, A. E. Kazzaz, N. AlipoorMazandarani, Z. H. Feizi and P. Fatehi, Production of flocculants, adsorbents, and dispersants from lignin, Molecules, 2018, 23, 868 CrossRef.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Lignin biosynthesis and structure, Plant Physiol., 2010, 153, 895–905 CrossRef CAS.
- T. M. Garver and P. T. Callaghan, Hydrodynamics of kraft lignins, Macromolecules, 1991, 24, 420–430 CrossRef CAS.
- S. L. Clark and P. T. Hammond, The role of secondary interactions in selective electrostatic multilayer deposition, Langmuir, 2000, 16, 10206–10214 CrossRef CAS.
- X. Jiang, C. Ortiz and P. T. Hammond, Exploring the rules for selective deposition: interactions of model polyamines on acid and oligoethylene oxide surfaces, Langmuir, 2002, 18, 1131–1143 CrossRef CAS.
- C. Friedsam, A. D. C. Bécares, U. Jonas, M. Seitz and H. E. Gaub, Adsorption of polyacrylic acid on self-assembled monolayers investigated by single-molecule force spectroscopy, New J. Phys., 2004, 6, 9 CrossRef.
- P. Maroni, F. J. M. Ruiz-Cabello and A. Tiraferri, Studying the role of surface chemistry on polyelectrolyte adsorption using gold–thiol self-assembled monolayer with optical reflectivity, Soft Matter, 2014, 10, 9220–9225 RSC.
- H. Eto, N. Soga, H. G. Franquelim, P. Glock, A. Khmelinskaia, L. Kai and P. Schwille, Design of sealable custom-shaped cell mimicries based on self-assembled monolayers on CYTOP polymer, ACS Appl. Mater. Interfaces, 2019, 11, 21372–21380 CrossRef CAS.
- P. Maroni, F. J. Montes Ruiz-Cabello, C. Cardoso and A. Tiraferri, Adsorbed mass of polymers on self-assembled monolayers: effect of surface chemistry and polymer charge, Langmuir, 2015, 31, 6045–6054 CrossRef CAS.
- Y. Song, J. Park, C. Lim and D. W. Lee, In-Depth Study of the Interaction Mechanism between the Lignin Nanofilms: Toward a Renewable and Organic Solvent-Free Binder, ACS Sustainable Chem. Eng., 2020, 8, 362–371 CrossRef CAS.
- A. E. Kazzaz and P. Fatehi, Fabrication of Amphoteric Lignin and its Hydrophilicity/Oleophilicity at Oil/Water Interface, J. Colloid Interface Sci., 2020, 561, 231–243 CrossRef.
- J. J. Gooding, F. Mearns, W. Yang and J. Liu, Self-assembled monolayers into the 21st century: recent advances and applications, Electroanalysis, 2003, 15, 81–96 CrossRef CAS.
- S. Huang, Q. Hou, D. Guo, H. Yang, T. Chen, F. Liu and J. Wang, Adsorption mechanism of mussel-derived adhesive proteins onto various self-assembled monolayers, RSC Adv., 2017, 7, 39530–39538 RSC.
- C. K. Haweel and S. H. Ammar, Preparation of polyvinyl alcohol from local raw material, J. Chem. Pet. Eng., 2008, 9, 15–21 Search PubMed.
- S. Contreras, A. R. Gaspar, A. Guerra, L. A. Lucia and D. S. Argyropoulos, Propensity of lignin to associate: light scattering photometry study with native lignins, Biomacromolecules, 2008, 9, 3362–3369 CrossRef CAS.
- S. M. Munzert, G. Schwarz and D. G. Kurth, Tailoring length and viscosity of dynamic metallo-supramolecular polymers in solution, RSC Adv., 2016, 6, 15441–15450 RSC.
- F. C. Giacomelli, I. C. Riegel, C. L. Petzhold, N. P. da Silveira and P. Štěpánek, Aggregation behavior of a new series of ABA triblock copolymers bearing short outer A blocks in B-selective solvent: from free chains to bridged micelles, Langmuir, 2008, 25, 731–738 CrossRef.
- P. Dimitrov, M. Jamróz-Piegza, B. Trzebicka and A. Dworak, The influence of hydrophobic substitution on self-association of poly(ethylene oxide)-b-poly(n-alkyl glycidyl carbamate) sb-poly(ethylene oxide) triblock copolymers in aqueous media, Polymer, 2007, 48, 1866–1874 CrossRef CAS.
- H. Hussain, B. H. Tan, G. L. Seah, Y. Liu, C. B. He and T. P. Davis, Micelle formation and gelation of (PEG-P (MA-POSS)) amphiphilic block copolymers via associative hydrophobic effects, Langmuir, 2010, 26, 11763–11773 CrossRef CAS.
- C. L. Berhaut, D. Lemordant, P. Porion, L. Timperman, G. Schmidt and M. Anouti, Ionic association analysis of LiTDI, LiFSI and LiPF6 in EC/DMC for better Li-ion battery performances, RSC Adv., 2019, 9, 4599–4608 RSC.
- A. Ishikubo, J. Mays and M. Tirrell, Behavior of cationic surfactants in poly(styrene sulfonate) brushes, Ind. Eng. Chem. Res., 2008, 47, 6426–6433 CrossRef CAS.
- Z. H. Feizi and P. Fatehi, Carboxymethylated cellulose nanocrystals as clay suspension dispersants: effect of size and surface functional groups, Cellulose, 2020, 27, 3759–3772 CrossRef.
- M. K. Konduri, F. Kong and P. Fatehi, Production of carboxymethylated lignin and its application as a dispersant, Eur. Polym. J., 2015, 70, 371–383 CrossRef CAS.
- A. E. Kazzaz, Z. H. Feizi, F. Kong and P. Fatehi, Interaction of poly(acrylic acid) and aluminum oxide particles in suspension: particle size effect, Colloids Surf., A, 2018, 556, 218–226 CrossRef CAS.
- C. J. Huang, F. S. Shieu, W. P. Hsieh and T. C. Chang, Acidic hydrolysis of a poly(vinyl acetate) matrix by the catalytic effect of Ag nanoparticles and the micellization of Ag-metal-containing polymer, J. Appl. Polym. Sci., 2006, 100, 1457–1464 CrossRef CAS.
- P. Maes, Y. B. Monakhova, T. Kuballa, H. Reusch and D. W. Lachenmeier, Qualitative and quantitative control of carbonated cola beverages using 1H NMR spectroscopy, J. Agric. Food Chem., 2012, 60, 2778–2784 CrossRef CAS.
- J. Hedin, J. E. Löfroth and M. Nydén, Adsorption behavior and crosL-Sinking of EHEC and HM-EHEC at hydrophilic and hydrophobic modified surfaces monitored by SPR and QCM-D, Langmuir, 2007, 23, 6148–6155 CrossRef CAS.
- H. Wang, S. Chen, L. Li and S. Jiang, Improved method for the preparation of carboxylic acid and amine terminated self-assembled monolayers of alkanethiolates, Langmuir, 2005, 21, 2633–2636 CrossRef CAS.
- M. L. Wallwork, D. A. Smith, J. Zhang, J. Kirkham and C. Robinson, Complex chemical force titration behavior of amine-terminated self-assembled monolayers, Langmuir, 2001, 17, 1126–1131 CrossRef CAS.
- L. Herrer, V. Sebastian, S. Martín, A. González-Orive, F. Pérez-Murano, P. J. Low and P. Cea, High surface coverage of a self-assembled monolayer by in situ synthesis of palladium nanodeposits, Nanoscale, 2017, 9, 13281–13290 RSC.
- C. D. Bain and G. M. Whitesides, Attenuation lengths of photoelectrons in hydrocarbon films, J. Phys. Chem., 1989, 93, 1670–1673 CrossRef CAS.
- Q. Li, R. Yuan and Y. Li, Study on the molecular behavior of hydrophobically modified poly(acrylic acid) in aqueous solution and its emulsion-stabilizing capacity, J. Appl. Polym. Sci., 2013, 128, 206–215 CrossRef CAS.
- E. Pensini, C. M. Yip, D. O'Carroll and B. E. Sleep, Carboxymethyl cellulose binding to mineral substrates: characterization by atomic force microscopy-based Force spectroscopy and quartz-crystal microbalance with dissipation monitoring, J. Colloid Interface Sci., 2013, 402, 58–67 CrossRef CAS.
- G. Masci, D. Bontempo, N. Tiso, M. Diociaiuti, L. Mannina, D. Capitani and V. Crescenzi, Atom transfer radical polymerization of potassium 3-sulfopropyl methacrylate: direct synthesis of amphiphilic block copolymers with methyl methacrylate, Macromolecules, 2004, 37, 4464–4473 CrossRef CAS.
- Y. Xu, A. Walther and A. H. Müller, Direct synthesis of poly(potassium 3-sulfopropyl methacrylate) cylindrical polymer brushes via ATRP using a supramolecular complex with crown ether, Macromol. Rapid Commun., 2010, 31, 1462–1466 CrossRef CAS.
- I. Korbag and S. Mohamed Saleh, Studies on the formation of intermolecular interactions and structural characterization of polyvinyl alcohol/lignin film, Int. J. Environ. Stud., 2016, 73, 226–235 CrossRef CAS.
- H. Lee, M. F. Rubner and R. E. Cohen, Coating, US Pat., 13215869, 2012.
- R. Hajiraissi, M. Hanke, Y. Yang, B. Duderija, A. Gonzalez Orive, G. Grundmeier and A. Keller, Adsorption and fibrillization of islet amyloid polypeptide at self-assembled monolayers studied by QCM-D, AFM, and PM-IRRAS, Langmuir, 2018, 34, 3517–3524 CrossRef CAS.
- H. T. Phan, S. Bartelt-Hunt, K. B. Rodenhausen, M. Schubert and J. C. Bartz, Investigation of bovine serum albumin (BSA) attachment onto self-assembled monolayers (SAMs) using combinatorial quartz crystal microbalance with dissipation (QCM-D) and spectroscopic ellipsometry (SE), PLoS One, 2015, 10, e0141282 CrossRef.
- J. A. Jones, L. A. Qin, H. Meyerson, I. K. Kwon, T. Matsuda and J. M. Anderson, Instability of self-assembled monolayers as a model material system for macrophage/FBGC cellular behavior, J. Biomed. Mater. Res., Part A, 2008, 86, 261–268 CrossRef.
- J. Li, T. Guan, C. Hao, L. Li and Y. Zhang, Effects of self-assembled monolayers with different chemical groups on ovarian cancer cell line behavior in vitro, J. Chem., 2015, 10, 784626 Search PubMed.
- S. D. Evans, R. Sharma and A. Ulman, Contact angle stability: reorganization of monolayer surfaces?, Langmuir, 1991, 7, 156–161 CrossRef CAS.
- K. S. Chong, S. Sun and G. J. Leggett, Measurement of the kinetics of photo-oxidation of self-assembled monolayers using friction force microscopy, Langmuir, 2005, 21, 3903–3909 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Self-assembled monolayers of thiolates on metals as a form of nanotechnology, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS.
- H. Hussain, K. Busse and J. Kressler, Poly(ethylene oxide)-and Poly(perfluorohexylethyl methacrylate)-Containing Amphiphilic Block Copolymers: Association Properties in Aqueous Solution, Macromol. Chem. Phys., 2003, 204, 936–946 CrossRef CAS.
- C. Booth and D. Attwood, Effects of block architecture and composition on the association properties of poly(oxyalkylene) copolymers in aqueous solution, Macromol. Rapid Commun., 2000, 21, 501–527 CrossRef CAS.
- Y. Kawata, S. Kozuka and S. I. Yusa, Thermo-responsive behavior of amphoteric diblock copolymers bearing sulfonate and quaternary amino pendant groups, Langmuir, 2018, 35, 1458–1464 CrossRef.
- K. Huber, S. Bantle, P. Lutz and W. Burchard, Hydrodynamic and thermodynamic behavior of short-chain polystyrene in toluene and cyclohexane at 34.5 degree. C, Macromolecules, 1985, 18, 1461–1467 CrossRef CAS.
- T. Konishi, T. Yoshizaki and H. Yamakawa, On the“ Universal Constants” ρ and Φ of flexible polymers, Macromolecules, 1991, 24, 5614–5622 CrossRef CAS.
- H. Hussain, K. Y. Mya and C. He, Self-assembly of brush-like poly [poly(ethylene glycol) methyl ether methacrylate] synthesized via aqueous atom transfer radical polymerization, Langmuir, 2008, 24, 13279–13286 CrossRef CAS.
- J. H. Yao, K. Y. Mya, X. Li, M. Parameswaran, Q. H. Xu, K. P. Loh and Z. K. Chen, Light scattering and luminescence studies on self-aggregation behavior of amphiphilic copolymer micelles, J. Phys. Chem. B, 2008, 112, 749–755 CrossRef CAS.
- H. Hussain, B. H. Tan, C. S. Gudipati, Y. Liu, C. B. He and T. P. Davis, Synthesis and self-assembly of poly(styrene)-b-poly(N-vinylpyrrolidone) amphiphilic diblock copolymers made via a combined ATRP and MADIX approach, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5604–5615 CrossRef CAS.
- T. Liu, Z. Zhou, C. Wu, B. Chu, D. K. Schneider and V. M. Nace, Self-Assembly of Poly(oxybutylene)-Poly(oxyethylene)-Poly(oxybutylene)(B6E46B6) Triblock Copolymer in Aqueous Solution, J. Phys. Chem. B, 1997, 101, 8808–8815 CrossRef CAS.
- B. Jeong, Y. H. Bae and S. W. Kim, Biodegradable thermosensitive micelles of PEG-PLGA-PEG triblock copolymers, Colloids Surf., B, 1999, 16, 185–193 CrossRef CAS.
- E. Castro, S. Barbosa, J. Juarez, P. Taboada, I. A. Katime and V. Mosquera, Influence of external factors on the micellization process and aggregate structure of poly(oxy) styrene-poly(oxy) ethylene block copolymers, J. Phys. Chem. B, 2008, 112, 5296–5304 CrossRef CAS.
- A. A. Tager, A. A. Anikeyeva, L. V. Adamova, V. M. Andreyeva, T. A. Kuz'mina and M. V. Tsilipotkina, The effect of temperature on the water solubility of polyvinyl alcohol, Polym. Sci., 1971, 13, 751–758 Search PubMed.
- V. T. Matsuo and H. Inagaki, Über den lösungszustand des polyvinylalkohols in wasser I. Mitt.: metastabiler zustand der lösung, Macromol. Chem. Phys., 1962, 53, 130–144 CrossRef.
- J. Liu, C. Detrembleur, M. Hurtgen, A. Debuigne, M. C. De Pauw-Gillet, S. Mornet and C. Jérôme, Thermo-responsive gold/poly(vinyl alcohol)-b-poly(N-vinylcaprolactam) core-corona nanoparticles as a drug delivery system, Polym. Chem., 2014, 5, 5289–5299 RSC.
- N. A. Cortez-Lemus and A. Licea-Claverie, Preparation of a mini-library of thermo-responsive star (NVCL/NVP-VAc) polymers with tailored properties using a hexafunctional xanthate RAFT agent, Polymers, 2018, 10, 20 CrossRef.
- J. P. Inwood, L. Pakzad and P. Fatehi, Production of sulfur containing kraft lignin products, BioResources, 2018, 13, 53–70 CAS.
- A. N. Evdokimov, A. V. Kurzin, O. V. Fedorova, P. V. Lukanin, V. G. Kazakov and A. D. Trifonova, Desulfurization of kraft lignin, Wood Sci. Technol., 2018, 52, 1165–1174 CrossRef CAS.
- M. Norgren and B. Lindström, Dissociation of phenolic groups in kraft lignin at elevated temperatures, Holzforschung, 2000, 54, 519–527 CAS.
- W. Zhu and H. Theliander, Precipitation of lignin from softwood black liquor: an investigation of the equilibrium and molecular properties of lignin, BioResources, 2015, 10, 1696–1714 Search PubMed.
- R. Ou, H. Zhang, J. Wei, S. Kim, L. Wan, N. S. Nguyen and H. Wang, Thermoresponsive amphoteric metal–organic frameworks for efficient and reversible adsorption of multiple salts from water, Adv. Mater., 2018, 30, 1802767 CrossRef.
- R. K. Sharma, J. B. Wooten, V. L. Baliga, X. Lin, W. G. Chan and M. R. Hajaligol, Characterization of chars from pyrolysis of lignin, Fuel, 2004, 83, 1469–1482 CrossRef CAS.
- A. A. M. Nada, M. A. Yousef, K. A. Shaffei and A. M. Salah, Infrared spectroscopy of some treated lignins, Polym. Degrad. Stab., 1998, 62, 157–163 CrossRef CAS.
- A. D. Phan and T. X. Hoang, The pH-dependent electrostatic interaction of a metal nanoparticle with the MS2 virus-like particles, Chem. Phys. Lett., 2019, 730, 84–88 CrossRef CAS.
- X. Zhang, Z. Zhao, G. Ran, Y. Liu, S. Liu, B. Zhou and Z. Wang, Synthesis of lignin-modified silica nanoparticles from black liquor of rice straw pulping, Powder Technol., 2013, 246, 664–668 CrossRef CAS.
- I. Popa, B. P. Cahill, P. Maroni, G. Papastavrou and M. Borkovec, Thin adsorbed films of a strong cationic polyelectrolyte on silica substrates, J. Colloid Interface Sci., 2007, 309, 28–35 CrossRef CAS.
- M. Porus, P. Maroni and M. Borkovec, Structure of adsorbed polyelectrolyte monolayers investigated by combining optical reflectometry and piezoelectric techniques, Langmuir, 2012, 28, 5642–5651 CrossRef CAS.
- D. Bauer, H. Buchhammer, A. Fuchs, W. Jaeger, E. Killmann, K. Lunkwitz, R. Rehmet and S. Schwarz, Stability of colloidal silica, sikron and polystyrene latex influenced by the adsorption of polycations of different charge density, Colloids Surf., A, 1999, 156, 291–305 CrossRef CAS.
- I. Szilagyi, G. Trefalt, A. Tiraferri, P. Maroni and M. Borkovec, Polyelectrolyte adsorption, interparticle forces, and colloidal aggregation, Soft Matter, 2014, 10, 2479–2502 RSC.
- A. Sweity, W. Ying, M. S. Ali-Shtayeh, F. Yang, A. Bick, G. Oron and M. Herzberg, Relation between EPS adherence, viscoelastic properties, and MBR operation: Biofouling study with QCM-D, Water Res., 2011, 45, 6430–6440 CrossRef CAS.
- T. Phan-Xuan, A. Thuresson, M. Skepö, A. Labrador, R. Bordes and A. Matic, Aggregation behavior of aqueous cellulose nanocrystals: the effect of inorganic salts, Cellulose, 2016, 23, 3653–3663 CrossRef.
- M. Jiang, I. Popa, P. Maroni and M. Borkovec, Adsorption of poly(l-lysine) on silica probed by optical reflectometry, Colloids Surf., A, 2010, 360, 20–25 CrossRef CAS.
- J. Forsman, Polyelectrolyte adsorption: electrostatic mechanisms and nonmonotonic responses to salt addition, Langmuir, 2012, 28, 5138–5150 CrossRef CAS.
- F. Xie, T. Nylander, L. Piculell, S. Utsel, L. Wågberg, T. Åkesson and J. Forsman, Polyelectrolyte adsorption on solid surfaces: theoretical predictions and experimental measurements, Langmuir, 2013, 29, 12421–12431 CrossRef CAS.
- S. J. De Carvalho, R. Metzler and A. G. Cherstvy, Critical adsorption of polyelectrolytes onto charged Janus nanospheres, Phys. Chem. Chem. Phys., 2014, 16, 15539–15550 RSC.
- N. Hansupalak and M. M. Santore, Sharp polyelectrolyte adsorption cutoff induced by a monovalent salt, Langmuir, 2003, 19, 7423–7426 CrossRef CAS.
- N. G. Hoogeveen, M. A. C. Stuart and G. J. Fleer, Polyelectrolyte adsorption on oxides: I. Kinetics and adsorbed amounts, J. Colloid Interface Sci., 1996, 182, 133–145 CrossRef CAS.
- F. Kong, S. Wang, W. Gao and P. Fatehi, Novel pathway to produce high molecular weight kraft lignin–acrylic acid polymers in acidic suspension systems, RSC Adv., 2018, 8, 12322–12336 RSC.
- D. J. Liaw and C. C. Huang, Dilute solution properties of anionic poly(potassium-2-sulfopropylmethacrylate), J. Appl. Polym. Sci., 1997, 63, 175–185 CrossRef CAS.
- K. J. D. MacKenzie, Innovative applications of inorganic polymers (geopolymers), in Handbook of Alkali-Activated Cements, Mortars and Concretes, Woodhead Publ., 2015, pp. 777–805 Search PubMed.
- D. H. Solomon and J. J. Hopwood, Reactivity of functional groups in surface coating polymers. Part I. Hydroxyl groups in alkyd resins, J. Appl. Polym. Sci., 1966, 10, 981–991 CrossRef.
- D. Andrews, G. Scholes and G. Wiederrecht, Comprehensive nanoscience and technology, Academic Press, 2010 Search PubMed.
- P. K. Ajikumar, J. K. Ng, Y. C. Tang, J. Y. Lee, G. Stephanopoulos and H. P. Too, Carboxyl-terminated dendrimer-coated bioactive interface for protein microarray: high-sensitivity detection of antigen in complex biological samples, Langmuir, 2007, 23, 5670–5677 CrossRef CAS.
- M. Gasser, B. Rothen-Rutishauser, H. F. Krug, P. Gehr, M. Nelle, B. Yan and P. Wick, The adsorption of biomolecules to multi-walled carbon nanotubes is influenced by both pulmonary surfactant lipids and surface chemistry, J. Nanobiotechnol., 2010, 8, 31 CrossRef CAS.
- M. Jawaid, S. Boufi and A. Khalil H. P. S., Cellulose-Reinforced nanofibre composites: Production, properties and applications, Woodhead Publ., 2017 Search PubMed.
- M. K. Meena, B. K. Tudu, A. Kumar and B. Bhushan, Development of polyurethane-based superhydrophobic coatings on steel surfaces, Philos. Trans. R. Soc., A, 2020, 378, 20190446 CrossRef.
- C. Salas, O. J. Rojas, L. A. Lucia, M. A. Hubbe and J. Genzer, On the surface interactions of proteins with lignin, ACS Appl. Mater. Interfaces, 2013, 5, 199–206 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectroscopy (Fig. S1 and S2), FTIR (Table S1 and Fig. S3), GPC (Table S2), zeta potential (Fig. S4), XPS (Table S3 and Fig. S5), water adsorption at a different temperature on different SAM surfaces (Fig. S6–S9), effect of temperature on the adsorption of L-S, and PVA-S on SAMs (Fig. S10–S17), effect of pH on the hydrodynamic radius (Rh) of L-S, and PVA-S (Fig. S18), water adsorption at different pH on different SAM surfaces (Fig. S19–S22), effect of pH on the adsorption of L-S and PVA-S on SAMs (Fig. S23–S30), contact angle of water–air (θW/A) interfaces of PVA-S and L-S polymers at different pH (Table S4), effect of salt concentration on the hydrodynamic radius (Rh) of L-S and PVA-S (Fig. S31), effect of salt concentration on the adsorption of L-S and PVA-S on SAMs (Fig. S32–S35). See DOI: 10.1039/d0ra07554j |
| This journal is © The Royal Society of Chemistry 2020 |