
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel calix[4]arene p-benzazole derivatives and investigation of their DNA binding and cleavage activities with molecular docking and experimental studies†
Seyda Cigdem Ozkanab,
Fatma Aksakalc and
Aydan Yilmaz *b
*b
aDepartment of Chemical and Chemical Processing Technologies, Acigol Vocational School of Technical Sciences, Nevsehir Haci Bektas Veli University, Nevsehir, Turkey. E-mail: aydan@selcuk.edu.tr; Fax: +90 332 2412499; Tel: +90 332 2233866
bDepartment of Chemistry, Faculty of Science, Selcuk University, 42075, Konya, Turkey
cDepartment of Chemistry, Faculty of Science, Hacettepe University, Ankara, Turkey
First published on 21st October 2020
Abstract
In this study, novel p-benzimidazole-derived calix[4]arene compounds with different structures, and a benzothiazole-derived calix[4]arene compound, were synthesized by a microwave-assisted method and their structures were determined by FTIR, 1H NMR, 13C NMR, MALDI-TOF mass spectroscopy, and elemental analysis. The effects of functional calixarenes against bacterial (pBR322 plasmid DNA) and eukaryotic DNA (calf thymus DNA = CT-DNA) were investigated. The studies with plasmid DNA have shown that compounds 6 and 10 containing methyl and benzyl groups, respectively, have DNA cleavage activity at the highest concentrations (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μM). Interactions with plasmid DNA using some restriction enzymes (BamHI and HindIII) were also investigated. The binding ability of p-substituted calix[4]arene compounds towards CT-DNA was examined using UV-vis and fluorescence spectroscopy and it was determined that some compounds showed efficiency. In particular, it was observed that the functional compounds (10 and 5) containing benzyl and chloro-groups had higher activity (Kb binding constants were found to be 7.1 × 103 M−1 and 9.3 × 102 M−1 respectively) on DNA than other compounds. Competitive binding experiments using ethidium bromide also gave an idea about the binding properties. Docking studies of the synthesized compounds with DNA were performed to predict the binding modes, affinities and noncovalent interactions stabilizing the DNA–compound complexes at the molecular level. Docking results were in good agreement with the experimental findings on the DNA binding activities of compounds. Based on these results, this preliminary study could shed light on future experimental antibacterial and/or anticancer research.
000 μM). Interactions with plasmid DNA using some restriction enzymes (BamHI and HindIII) were also investigated. The binding ability of p-substituted calix[4]arene compounds towards CT-DNA was examined using UV-vis and fluorescence spectroscopy and it was determined that some compounds showed efficiency. In particular, it was observed that the functional compounds (10 and 5) containing benzyl and chloro-groups had higher activity (Kb binding constants were found to be 7.1 × 103 M−1 and 9.3 × 102 M−1 respectively) on DNA than other compounds. Competitive binding experiments using ethidium bromide also gave an idea about the binding properties. Docking studies of the synthesized compounds with DNA were performed to predict the binding modes, affinities and noncovalent interactions stabilizing the DNA–compound complexes at the molecular level. Docking results were in good agreement with the experimental findings on the DNA binding activities of compounds. Based on these results, this preliminary study could shed light on future experimental antibacterial and/or anticancer research.
Introduction
One of the developments in the field of chemistry in recent years has been the birth and progress of supramolecular chemistry. The discovery and many different features of macrostructures such as cyclodextrins,1 crown ethers2 and calixarenes3 have contributed to the advancement in this area.Calixarenes are excellent products of phenol-formaldehyde chemistry, which naturally act as sensors for many cations,4 anions5 and neutral molecules6 due to their inherent structural properties. However, the idea that calixarenes can be used in areas such as medicine and biology has led scientists to work on this subject due to the increase in the occurrence of diseases (such as cancer, Alzheimer's, and Parkinson's) that cannot be completely cured and pose daily threats to human life. Various studies on biological applications of calixarenes, such as antibacterial,7 antifungal,8 antiviral,9 anti-HIV,10 anti-inflammatory,11 anticancer,12 and biotechnological studies,13 have been carried out in recent years. Many studies on the interaction of calixarenes with DNA have been previously described;14 DNA cleavage studies were performed using plasmid DNA, and highly effective results were obtained.7b,15 However, there are also studies using eukaryotic DNA, and similar results have been obtained in these studies.16
Preliminary anticancer studies on interaction of candidate molecules with DNA are mostly examined. Most of the calixarene molecules that have been studied and those that bind to or interact with DNA are cationic structures.12a,b,16b However, as an alternative to these molecules, it is necessary to develop molecules that are uncharged but contain functional groups that can bind to DNA. For this purpose, the effects of calixarene compounds containing uncharged and functional groups on cancer cells were investigated and good results were obtained.12c–e In our study, we focus on synthesizing molecules that are uncharged but contain functional group(s) that can bind to DNA. We chose the benzimidazole groups and benzothiazole group, which have aromatic structures containing hetero groups as the functional groups.
The benzimidazole compounds are among the bioactive heterocyclic compounds, which exhibit a wide range of biological activities due to their imidazole nuclei.17 In particular, functional benzimidazole derivatives have been synthesized as anticancer drugs in medicinal chemistry and their efficacy has been demonstrated by patents.18 Benzimidazole compounds are fusion molecules, composed of the imidazole ring with a positive charge and a benzene ring with hydrophobic properties and a partial negative charge. Therefore, they are capable of interacting with both positive and negative regions of biological molecules. As is known, the DNA molecules have a partial negative charge due to the phosphate groups they contain, so the positive portions of the benzimidazole compounds tend to bind to DNA. There are a lot of studies on this topic and as a result, effective and remarkable data have been obtained and reported.17
In the literature, DNA binding modes include electrostatic interactions, intercalation interactions, binding to major and small grooves, and single-stranded DNA (ssDNA) base binding. Electrostatic interaction is the deterioration of the DNA charge balance due to interactions between the negatively charged phosphate backbone of DNA and the positively charged ends of the molecules.18 Intercalation occurs when an aromatic planar substituent is inserted between the DNA base pairs causing the unwinding and lengthening of the DNA helix.19 Groove binding is caused by groove binders connected to the major or minor grooves of the DNA. This involves hydrogen bonding or van der Waals interactions of the molecule with the nucleic acid without substantially disrupting the duplex structure, or bases, as opposed to intercalation.20 ssDNA base binding results from binding to the single-stranded DNA of some proteins.21
In this study, we report the synthesis and characterization of new p-tert-butylcalix[4]arenes with benzimidazole derivatives and a benzothiazole derivative. The DNA cleavage effects of the synthesized compounds were investigated using pBR322 plasmid DNA by the agarose gel electrophoresis method. The determination of the site of DNA cleavage with BamHI and HindIII restriction enzymes was carried out for all compounds. In another study, the effects of synthesized calixarene compounds against CT-DNA were investigated using fluorescence and UV-vis. spectrometry. Binding constants (Kb) and quenching constants (Ksv) were calculated for some compounds that were effective in fluorescence spectrometry. Finally, competitive binding experiments with ethidium bromide were performed and quenching constants (Ksv) were determined. To support the experimental results with theoretical studies, molecular docking studies of the synthesized compounds with the DNA were also performed.
Results and discussion
Synthesis of p-benzazole derivatives of calix[4]arenes
The starting compound, p-tert-butylcalix[4]arene (1) and derivative compounds 2 and 3 were synthesized according to the literature procedures (Scheme 1).22–24 | ||
| Scheme 1 Schematic route for the synthesis of p-benzimidazole derivatives of calix[4]arenes. | ||
To obtain benzimidazole compounds that have different structures, the dialdehyde-derived calix[4]arene compound 3 was reacted with o-phenylenediamine and its 4-substituted derivatives separately in the presence of KI in DMF (Scheme 1). Before the mixtures were boiled in a domestic microwave oven for a few minutes, they were heated at 80 °C under a reflux condenser. The reactions were monitored by thin-layer chromatography (TLC). After purification, the FTIR spectrum showed that the peak at 1680 cm−1 of the aldehyde carbonyl was completely gone and the peaks belonging to the benzimidazole group (approximately 1660 cm−1 and 1620 cm−1) were formed. Also, –NH protons belonging to the benzimidazole group were determined by 1H NMR at 12.54 ppm for compound 4, 12.76 ppm for compound 5, 12.80 ppm for compound 6, and 13.30 ppm for compound 7; NH protons were in resonance. The confirmations of the cone conformations of the compounds obtained were carried out by 1H NMR to give the following results for ArCH2Ar protons: compound 4 had a doublet pair at 3.57 ppm and 4.24 ppm J = 13.1 Hz; compound 5 had a doublet pair at 3.61 ppm and 4.27 ppm, J = 12.8 Hz; compound 6 had a doublet pair at 3.60 ppm and 4.28 ppm, J = 12.8 Hz; compound 7 had a doublet pair at 3.60 ppm and 4.26 ppm, J = 13.1 Hz. The characterizations of the synthesized compounds were supported by 13C NMR and MALDI TOF MS analyses (Fig. S1 and S2†).
In the second part of the synthesis, to obtain N-substituted benzimidazole derivatives, o-phenylenediamine was reacted with benzyl bromide and p-nitrobenzyl bromide and converted into N-substituted o-phenylenediamine derivatives (8 and 9) (Scheme 2) as in the literature,25 and the structural analyses were performed using FTIR and 1H NMR. The obtained compounds were reacted with dialdehyde-derived calixarene (3) and compounds 10 and 11 were synthesized. In the FTIR analysis of these compounds, the peak of the aldehyde group disappeared and the benzimidazole imine peak was formed; 1H NMR spectra were also determined. The confirmations of the cone conformations of the compounds were again provided by 1H NMR analysis. For the synthesis of the benzothiazole derivative of calixarene (12), the synthetic pathway is given in Scheme 2. Compound 3 and 2-aminothiophenol were heated in the presence of KI in DMF by using the synthesis-type microwave oven for 3 hours and compound 12 was obtained. Structure analysis was performed by FTIR and 1H NMR. In the FTIR spectrum of this compound, the carbonyl band of the aldehyde group disappeared while the imine band appeared at 1658 cm−1. In the 1H NMR spectrum, ArCH2Ar protons were exposed as 3.57 ppm and 4.36 ppm as a doublet pair (J = 13.3 Hz). 13C NMR and MALDI-TOF MS analyses were performed for all compounds and their structures were illuminated (Fig. S3 and S4†).
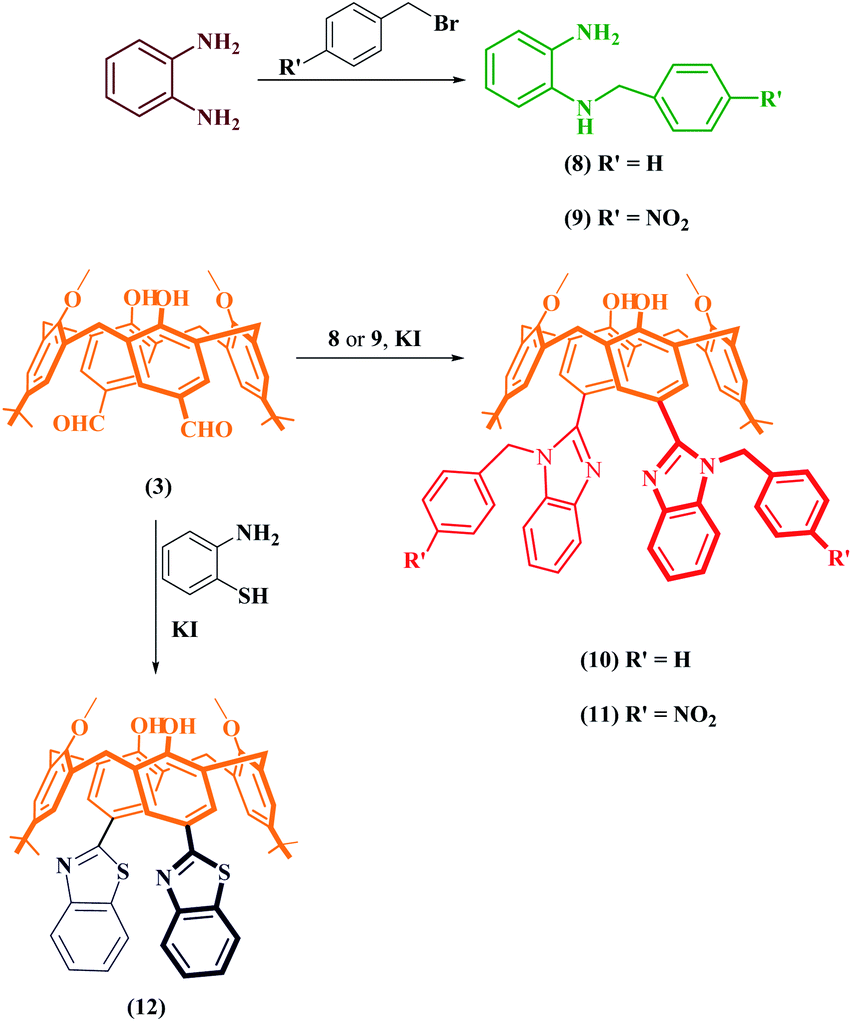 | ||
| Scheme 2 Schematic route for the synthesis of functional p-benzimidazole derivatives and a benzothiazole derivative of calix[4]arenes. | ||
DNA cleavage studies with pBR322 plasmid DNA
Plasmid DNAs are fragments of protective DNA that are contained within the bacterial cell that can copy itself, apart from the chromosomal DNA of the bacterium. When bacteria are exposed to any external agent (various chemicals etc.), they use their DNA to protect themselves. Therefore, molecules that can damage or break down the plasmid DNA cause the bacteria to die by preventing their growth. For all these reasons, we investigated the activity of the synthesized molecules on this DNA using the pBR322 plasmid DNA. These studies were carried out with the aim of pioneering DNA studies, presenting the first ideas about the interaction of compounds with DNA.The interactions of synthesized calixarene compounds with supercoiled pBR322 DNA were investigated using agarose gel electrophoresis. When circular plasmid DNA is subjected to electrophoresis, the fastest migrating supercoiled Form I, the slower-moving open circular Form II, and the linear Form III will all be generated, with migration in between.26
Firstly, in order to determine whether dimethylformamide (DMF) as a solvent in the various concentrations of the compound samples affected pBR322 plasmid DNA, incubation with plasmid DNA for the purpose of control in the same proportions was performed using the same conditions. According to the obtained results, the solvent had little effect on the plasmid DNA (Fig. 1a). According to the electrophoresis image seen in Fig. 1a, compound 4 is not effective on the plasmid DNA at all the studied concentrations. In the embodiment of Fig. 1b, compound 5 destroyed the Form II (open circular form) structure that was present in the plasmid DNA at the first three concentrations. However, the Form I (supercoiled form) structure remained. In the same image, for compound 6 on the right side of the gel, all bands of plasmid DNA (Form I and Form II) at the maximum concentration of 10000 μM disappeared, while the Form I structure was formed in more dilute concentrations, and the Form II structure was observed as very slim at the low concentration. We can say that this compound showed the cleavage effect on plasmid DNA at this concentration.7b
 | ||
| Fig. 1 Electrophoresis images of pBR322 plasmid DNA interacting with compounds. P: only plasmid DNA, M: DNA marker. (a) Compound 4, 1→4: 10000 μM, 5000 μM, 2500 μM, 1250 μM; 5→8: only solvent (DMF) 100%, 50%, 25% and 12.5%. (b) Compound 5, 1→4: 10000 μM, 5000 μM, 2500 μM, 1250 μM; compound 6, 5→8: 10000 μM, 5000 μM, 2500 μM, 1250 μM. (c) Standard DNA marker (1 Kb DNA ladder). | ||
The image on the left (column 2–5) of Fig. 2a shows the interaction of the pBR322 plasmid DNA with compound 7. Accordingly, at all concentrations of this compound, a higher band was observed showing that the molecular weight of the Form I structure (supercoiled) was increased. A similar situation was observed in the 100% DMF sample used for the control, but a band closer to Form I was observed below. From this, we can say that compound 7 decreases the mobility and density of the Form I structure. Similarly, results were obtained in the literature where functional calixarene molecules reduced the mobility of the Form I structure of plasmid DNA.7b,15 Calixarene molecules, which are large in structure, can bind to the supercoiled structure of DNA and limit its mobility. The same image (Fig. 2a) on the right side (column 6–9) shows the interaction of compound 11 with pBR322 plasmid DNA. According to this, at the highest concentration of this compound (11), the supercoiled DNA fragment (Form I) and the open circular form (Form II) are broken and are, therefore, converted into smaller DNA fragments. At the lower concentrations, the Form I band was observed, albeit higher. In the electrophoresis image shown in Fig. 2b (column 2–5) is the interaction of compound 10 with plasmid DNA. Accordingly, all bands of the plasmid DNA (Form I and Form II) at the highest concentration (10000 μM) disappeared. A cleavage effect was observed at this concentration.7b Form I structure was observed at lower concentrations. On the other hand, the Form II structure was not observed. Fig. 2c shows the interaction of plasmid DNA with compound 12 on the left side (column 1–4) of the electrophoresis image. At the highest concentration of this compound, both the density and the mobility of the supercoiled Form I structure of the plasmid DNA decreased. On the other hand, the density of the open circular structure, Form II, increased. At lower concentrations, the bands appeared as in the control plasmid.
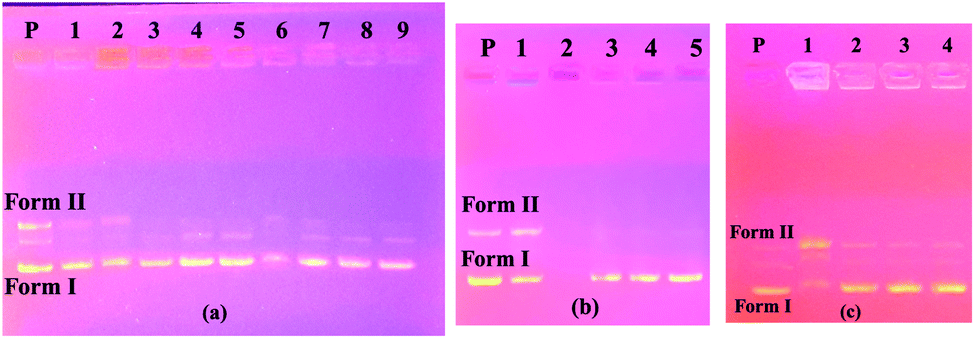 | ||
| Fig. 2 Electrophoresis images of pBR322 plasmid DNA interacting with compounds. P: only plasmid DNA. (a) 1: DMF control (100%), compound 7, 2→5: 10000 μM, 5000 μM, 2500 μM, 1250 μM; compound 11, 6→9: 10000 μM, 5000 μM, 2500 μM, 1250 μM. (b) Compound 10, 1: DMF control (100%), 2→5: 10000 μM, 5000 μM, 2500 μM, 1250 μM. (c) Compound 12, 1→4: 10000 μM, 5000 μM, 2500 μM, 1250 μM. | ||
According to the obtained results, the two most effective compounds were compounds 6 and 10, which have methyl and benzyl groups, respectively. The benzyl group has a rigid and planar structure due to the benzene ring it contains. Therefore, it was expected that compound 10 would approach the aromatic core bases of DNA. Unlike other benzimidazole compounds, the methyl group in compound 6 may bind hydrophobically to DNA due to its hydrophobic properties, as reported studies in the literature.37,38
In the second part of the study, to determine the ability of the synthesized heterocyclic p-tert-butylcalix[4]arene derivatives to cleave DNA molecules at the positions at which particular short sequences of bases are present, we used BamHI and HindIII enzymes. Restriction endonuclease analyses of compound-pBR322 plasmid DNA inserts were performed using these enzymes. BamHI and HindIII enzymes cut DNA at specific sequences as follows: 5′-G/GATCC-3′ and 5′-A/AGCTT-3′. As a result of the cutting process, Form I (supercoiled form) DNA and Form II (nicked form) DNA constructs were converted into Form III DNA in linear form.
Fig. 3a shows the results of the experiment with the BamHI enzyme. On the right side of the figure, it was found that the BamHI enzyme cut the plasmid DNA at all concentrations of compound 4. The electropherograms on the left side of the same figure were formed by the control samples made as a control.
 | ||
| Fig. 3 Electrophoresis images of compound-pBR322 plasmid DNA mixture after interactions with the enzymes BamHI (a) and HindIII (b). P: plasmid DNA, CP: cut plasmid DNA, 1→4: only solvent (DMF) 100%, 50%, 25%, 12.5%; compound 4, 5→8: 10000 μM, 5000 μM, 2500 μM, 1250 μM. | ||
Accordingly, the enzyme made the cuts in the highest DMF ratios. In Fig. 3b, a gel image of the HindIII segment is shown. On the right side of this figure, the enzyme cuts the plasmid DNA at all concentrations of compound 4. On the left side of the same figure, the enzyme again cuts in the samples containing only the solvent.
At the highest concentration of compound 5 on the left side of Fig. 4a, it was observed that plasmid DNA was almost absent and the linear Form III structure had very low density and BamHI enzyme was cut at lower concentrations of this compound. On the right side of the same figure, plasmid DNA was not seen at the first high concentration of compound 5 and the BamHI enzyme was cut at lower concentrations. In Fig. 4b, the gel image of the HindIII segment is given. From the figure, it is seen that at the highest concentrations of compounds 5 and 6 do not have visible plasmid DNA bands (columns 1 and 5), whereas, at lower concentrations of these compounds, the enzyme cut the plasmid DNA.
 | ||
| Fig. 4 Electrophoresis images of compound-pBR322 plasmid DNA mixture after interactions with the enzymes BamHI (a) and HindIII (b). P: plasmid DNA, CP: cut plasmid DNA, compound 5, 1→4: 10000 μM, 5000 μM, 2500 μM, 1250 μM; compound 6, 5→8: 10000 μM, 5000 μM, 2500 μM, 1250 μM. | ||
In Fig. 5a, it was found that plasmid DNA did not appear in both compounds 7 and 11 at the first high concentrations, whereas in lower concentrations, the BamHI enzyme cut the plasmid DNA. The HindIII enzyme cut image in Fig. 5b shows that there are no plasmid DNA bands at the highest concentrations of both compounds 7 and 11, while the enzyme cut the plasmid DNA partially at the lower concentrations of these compounds.
 | ||
| Fig. 5 Electrophoresis images of compound-pBR322 plasmid DNA mixture after interactions with the enzymes BamHI (a) and HindIII (b). P: plasmid DNA, CP: cut plasmid DNA, compound 7, 1→4: 10000 μM, 5000 μM, 2500 μM, 1250 μM; compound 11, 5→8: 10000 μM, 5000 μM, 2500 μM, 1250 μM. | ||
Fig. 6a and c show the results of the experiments with compound 10 and compound 12 of the BamHI enzyme, respectively. At the first high concentration of both compounds, all the bands of plasmid DNA were not found whereas, at lower concentrations of these compounds, the BamHI enzyme cut the plasmid DNA. Similar results were observed in the experimental results with the HindIII enzyme (Fig. 6b and d).
 | ||
| Fig. 6 Electrophoresis images of the compound-pBR322 plasmid DNA mixture after interaction with the enzymes BamHI (a and c) and HindIII (b and d), (a and b). P: plasmid DNA, CP: cut plasmid DNA, compound 10, 1→4: 10000–1250 μM; (c and d) compound 12, 1→4: 10000–1250 μM. | ||
The obtained results showed that the compounds had no interest in the specific binding regions of the enzymes.
DNA interaction studies with calf thymus DNA
In this study, we also followed the absorbances of the compounds (4–7 and 10–12). Upon the addition of increasing amounts of CT-DNA, a significant hyperchromism was observed for all compounds except compound 7 (Fig. 7).26 In the absorption spectrum of compound 7, a partial increase in electronic absorption (at 396 nm) was observed with the increase in the DNA concentration as in other compounds; however, the graph was not included here because the increase remained low as compared to other compounds.
 | ||
| Fig. 7 The obtained UV-vis absorption spectra of the compounds on adding CT-DNA in increasing concentrations (from bottom to top (0–50 μM)) in increments of 2.5 μM of CT-DNA, additionally 75 μM and 100 μM CT-DNA. (a) Compound 4, (b) compound 5, (c) compound 6, (d) Compound 10, (e) compound 11, (f) compound 12. The arrows show the changes upon increasing the amounts of CT-DNA. | ||
As shown in Fig. 7, there was a regular increase in the absorbance of compound 4 at 318 and 333 nm, compound 5 at 322 and 338 nm, and compound 6 at 317 and 320 nm on increasing the CT-DNA concentration. Likewise, a steady increase in CT-DNA concentration was observed with the absorbances of compound 10 at 316 and 330 nm, compound 11 at 304 nm and compound 12 at 330 nm. When the all spectra were examined, it was observed that there was no shift in wavelengths and only the intensity of absorption increased.
Although there were no appreciable changes in the absorption spectra of the compounds, the positions of the intra-compound bands were observed, and a strong chromic effect was observed with increased CT-DNA concentration. This observed change shows that the compounds most likely interacted with DNA in the groove binding mode.27a–c This observed hyperchromism may have occurred as a result of external contact, such as surface bonding with the DNA double-strand.27a This surface contact may have occurred as a result of hydrogen bonding, electrostatic interactions, or π-interactions between aromatic groups. The compounds that contain imidazole groups, other functional groups (such as Cl, NO2, or methyl) and aromatic rings can cause these conditions.
Based on these results, we can say or suggest that the compounds most likely bind to CT-DNA. However, extra studies were needed to obtain accurate and reliable results and to determine the mode of binding of the compounds with CT-DNA; hence, fluorimetry and competitive binding studies with EtBr, known as an intercalator, were performed to clarify the DNA-binding mode.
The quantitative estimation of extinguishing experiments in terms of the binding constant was conducted using the following Stern–Volmer equation:28
| F0/F = 1 + Ksv[Q] |
Another modified Stern–Volmer equation was used to calculate the DNA binding constants of the compounds from both the increase and decrease in emission intensities:28
| log(F0 − F)/F = logKb + nlog[DNA] |
An increase in the emission intensity of compound 4 at 401 nm was observed with increasing DNA concentration according to the emission spectrum obtained using 330 nm excitation (Fig. 8a). The binding constant Kb was determined from the plot of log(F0 − F)/F vs. log[DNA] and was found to be 8.6 × 102 M−1 (Fig. 8b). A regular increase in the emission intensity of compound 5 at 431 nm was observed with increasing the DNA concentration; the emission maximum wavelength of this compound shifted slightly (1 nm) (Fig. 9a). The Kb constant calculated from the linear graph obtained was 9.3 × 102 M−1 (Fig. 9b).
 | ||
| Fig. 8 Fluorescence spectra obtained by adding CT-DNA in increasing concentrations (from bottom to top (0–30 μM)) with increments of 5 μM of CT-DNA of compound 4 (a); the linear graph from which the Kb constant was calculated (b). The arrow shows the changes upon increasing the amounts of CT-DNA. | ||
 | ||
| Fig. 9 Fluorescence spectra obtained by adding CT-DNA in increasing concentrations (from bottom to top (0–30 μM) with increments of 5 μM of CT-DNA, additionally 75 μM and 100 μM CT-DNA) of compound 5 (a); the linear graph from which the Kb constant was calculated (b). The arrow shows the changes upon increasing the amounts of CT-DNA. | ||
The fluorescence spectra obtained using the excitation wavelength at 300 nm for compound 10 are shown in Fig. 10a. When we look at the fluorescence spectrum of compound 10, it can be said that a different state occurred. A slight decrease in fluorescence the intensity was observed with the addition of DNA. The quenching constant Ksv 9.8 × 103 M−1 was calculated from the fluorescence quenching measurement at 381 nm (Fig. 10b). The calculated Kb constant from the linear graph obtained was 7.1 × 103 M−1 (Fig. 10c).
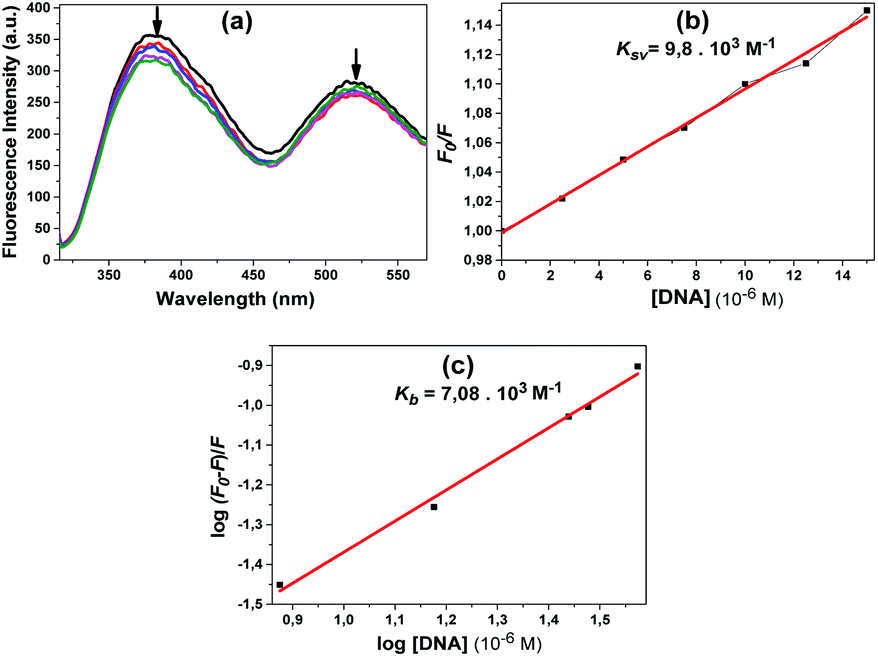 | ||
| Fig. 10 Fluorescence spectra obtained by adding CT-DNA in increasing concentrations [from bottom to top (0–50 μM) with increments of 2.5 μM of CT-DNA and additionally, 75 μM and 100 μM CT-DNA] of compound 10 (a); the linear graph from which the Kb constant was calculated (b); the linear graph from which the Kb constant was calculated (c). The arrows show the changes upon increasing the amounts of CT-DNA. | ||
Compound 11 also showed a result similar to compound 10; the fluorescence spectra obtained using the excitation wavelength at 300 nm are shown in Fig. 11a. A slight decrease in fluorescence intensity was observed with the addition of DNA. The quenching constant Ksv 7.8 × 103 M−1 was calculated from the fluorescence quenching measurement at 386 nm (Fig. 11b).
 | ||
| Fig. 11 Fluorescence spectra obtained by adding CT-DNA in increasing concentrations (from bottom to top (0–50 μM)) with increments of 5 μM of CT-DNA of compound 11 (a); the linear graph from which the Ksv constant was calculated (b); the linear graph from which the Kb constant was calculated (c). The arrow shows the changes upon increasing the amounts of CT-DNA. | ||
Fig. 12a shows the fluorescence spectra obtained at the excitation wavelength of 300 nm for 12. With increasing DNA concentration, the emission band of the compound at 384 nm showed a steady decrease (Fig. 12a). The quenching constant Ksv, 3.3 × 103 M−1, was calculated using the Stern–Volmer equation (Fig. 12b).
 | ||
| Fig. 12 Fluorescence spectra obtained by adding CT-DNA in increasing concentrations (from bottom to top (0–50 μM) with increments of 2.5 μM of CT-DNA and additionally, 75 μM and 100 μM CT-DNA) of compound 12 (a); the linear graph from which the Ksv constant was calculated (b); the linear graph from which the Kb constant was calculated (c). The arrow shows the changes upon increasing the amounts of CT-DNA. | ||
According to the obtained results, the fluorescence intensities of some of the compounds increased with the addition of CT-DNA while others decreased. The compounds that had decreased fluorescence intensities were compounds 10, 11 and 12. The compounds that had increased emission intensities were compounds 4 and 5. The calculated mathematical data are given in Table 1. According to these results, the compound that had the highest binding constant was 10. DNA binding strength ranking was 5 > 4 > 11 > 12. The quenching constants were also calculated from here, as the emission intensity of the compounds decreased with increasing DNA concentration for compounds 4, 5 and 12. The found Ksv constants are compatible with the results in the Kb binding constants. The numbers n found for all compounds are approximately equal to one, indicating that the compounds bind 1:1 with DNA. Both results showed that the functional calixarene compounds interacted by binding to CT-DNA.
| Aa | Bb | Kb | n | Ksv | R2 |
|---|---|---|---|---|---|
| a Compound.b Functional Group.c R2 values are given for Kb and Ksv constants, respectively. | |||||
| 4 | — | 8.6 × 102 | 0.37 | — | 0.991 |
| 5 | Chloro | 9.3 × 102 | 0.45 | — | 0.996 |
| 10 | Benzyl | 7.1 × 103 | 0.78 | 9.8 × 103 | 0.991/0.993c |
| 11 | p-Nitro benzyl | 7.2 × 102 | 0.39 | 7.8 × 103 | 0.99/0.993c |
| 12 | Sulfur (S) | 1.4 × 102 | 0.52 | 3.3 × 103 | 0.993/0.989c |
Competitive binding experiments in the presence of ethidium bromide
One of the useful experimental methods for understanding whether the compounds interact with CT-DNA is to monitor the fluorescence intensity of the EtBr–DNA complex using ethidium bromide (EtBr). EtBr is one of the most sensitive fluorescent probes due to its planar structure and is connected to the DNA by the intercalative mode.29 When the fluorescence spectrum in EtBr alone is taken up, an emission peak at about 600 nm is observed and its intensity is very low due to the solvent action.29 However, when CT-DNA is added to the environment, the emission intensity of EtBr increases due to the formed EtBr + DNA complex.29 If there is a substance that binds to DNA in the environment, the fluorescence intensity of the EtBr–DNA complex decreases29 because the substance inhibits EtBr interaction with DNA. From the resulting decrease, Stern–Volmer quenching constants28 can be calculated and the binding strength of molecules or substances to DNA can be compared.For these experiments, the CT-DNA + EtBr complex was formed by adding 2 μM EtBr to 20 μM CT-DNA. The prepared compound solutions were added to CT-DNA/EtBr solutions in concentrations from 2.5 μM to 35 μM increasing by 2.5 μM and incubated for 3 hours. Emission spectra ranking between 500–750 nm were recorded in each experiment using 480 nm as the excitation wavelength.
In the obtained emission spectra, the intensities at approximately 600 nm where maximum emission occurred were taken into consideration and the necessary calculations were made using the Stern–Volmer equation below:28
| F0/F = 1 + Ksv[Q] |
According to the results of the experiments with EtBr and compounds 4 and 5, the emission of the EtBr–DNA complex decreased with the increasing concentration of the compound (Fig. 13a and c). Here, we can say that the compounds bound to CT-DNA, preventing EtBr from binding. The Ksv constants calculated using the Stern–Volmer equation were found to be 4.3 × 103 M−1 and 5.9 × 103 M−1, respectively (Fig. 13b and d). Both compounds led to a gradual decrease in the emission spectrum and very close Ksv values appeared.
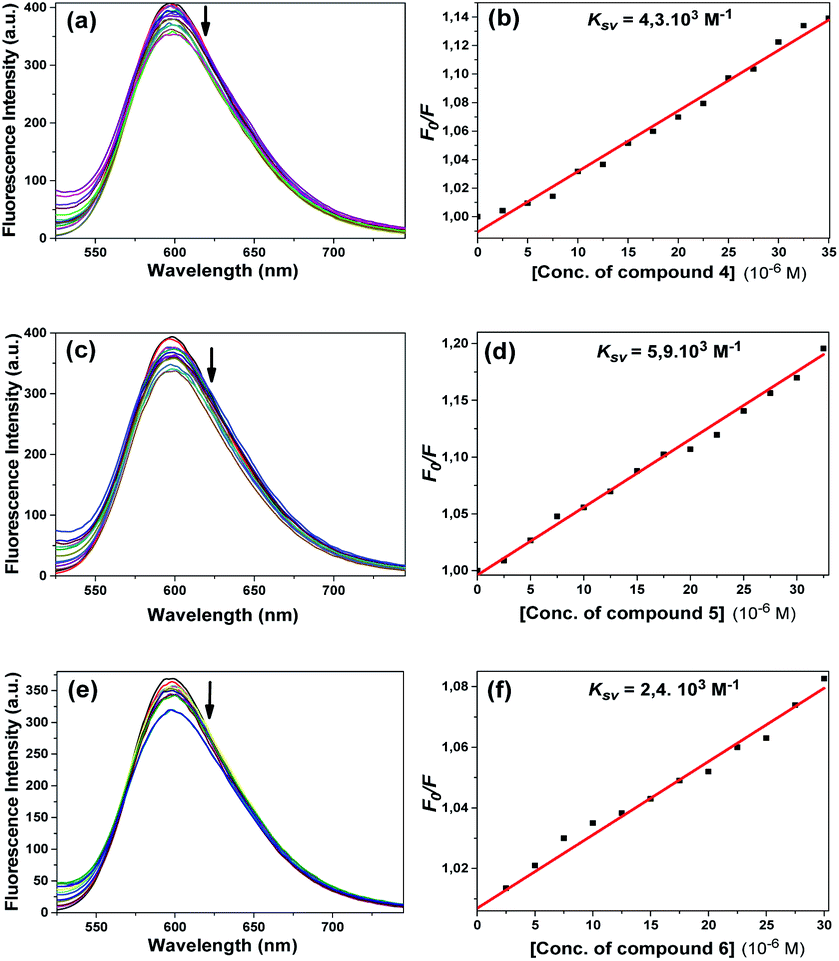 | ||
| Fig. 13 Fluorescence emission spectra of EtBr bound with CT-DNA ([DNA] = 2.0 × 10−5 M, [EtBr] = 2.0 × 10−6 M) in the absence (black) and presence (other colours) of compounds 4 (a), 5 (c), and 6 (e) in Tris–HCl/NaCl buffer upon addition of CT-DNA and EtBr [from top to bottom (0–35 μM)] with increments of 2.5 μM of the compounds. Arrows represent the changes in the fluorescence intensity upon increasing the compound concentration. For compounds 4 (b), 5 (d) and 6 (f), linear fitting graphs were used to calculate the quenching constant Ksv based on the Stern–Volmer equation. | ||
Fig. 13e shows that the emission intensity of the EtBr–DNA complex at about 600 nm decreased with the addition of compound 6 at increasing concentrations. The calculated Ksv constant was found to be 2.4 × 103 M−1 (Fig. 13f). As in compounds 4 and 5, it can be interpreted that this compound performed intercalative binding with DNA.
Compounds 10 and 12 had the same effect as the other compounds competitively substituted with EtBr, causing a slight reduction in the emission of the EtBr–DNA complex (Fig. 14a and c). Stern–Volmer quenching constants were also found to be 5.8 × 103 M−1 and 1.1 × 102 M−1, respectively (Fig. 14b and d). Unlike other compounds, compound 11 increased the EtBr emission intensity. The obtained emission spectrum is not included here because a different effect occurred. Some studies in the literature have shown that some molecules can bind to the cavity of the DNA by the groove binding mode through hydrophobic interactions, and in this case, the EtBr–DNA complex can cause an increase in the emission intensity.36 The same effect may apply for compound 11.
 | ||
| Fig. 14 Fluorescence emission spectra of EtBr bound with CT-DNA ([DNA] = 2.0 × 10−5 M, [EtBr] = 2.0 × 10−6 M) in the absence (black) and presence (other colours) of compounds 10 (a) and 12 (c) in Tris–HCl/NaCl buffer upon the addition of CT-DNA and EtBr [from top to bottom (0–35 μM)] with increments of 2.5 μM of the compounds. Arrows represent the changes in fluorescence intensity upon increasing the compound concentration. For compounds 10 (b) and 12 (d) linear fitting graphs were used to calculate the quenching constant Ksv based on the Stern–Volmer equation. | ||
The calculated quenching constants of the compounds using the reductions in the emission intensity of the CT-DNA + EtBr complex are given in Table 2. According to the calculated Ksv values, compounds 5 and 10 achieved the strongest binding with CT-DNA, while the compounds following them were 4 > 6 > 12, respectively. Compound 10 had the highest binding constant in fluorescence studies and it gave the same value as compound 5 in competitive displacement studies with EtBr.
| Compound | Functional group | Ksv (M−1) | R2 |
|---|---|---|---|
| 4 | — | 4.3 × 103 | 0.989 |
| 5 | Chloro | 5.9 × 103 | 0.989 |
| 6 | Methyl | 2.4 × 103 | 0.987 |
| 10 | Benzyl | 5.8 × 103 | 0.997 |
| 12 | Sulfur(S) | 1.1 × 102 | 0.988 |
Molecular docking studies
To predict the noncovalent binding conformations and affinities of the newly synthesized compounds toward DNA, molecular docking was carried out. The predicted free energy values of binding are given in Table 3. Active compounds 10, 5, and inactive compound 12, were selected for comparison according to the experimental results of their binding strength to DNA. The most energetically profitable poses of 10, 5 and 12 in the DNA binding site are given in Fig. 15 with both the three-dimensional (3D) structures and the molecular surface representations (Fig. 15a–c) and two-dimensional (2D) interaction diagrams (Fig. 15d–f). Docking figures for the other compounds 4, 6, 7 and 11 are presented as ESI (Fig. S5†). As seen in Fig. 15, the interaction of the compounds with the DNA was dominated by noncovalent C–H⋯π, hydrogen bonding and water-mediated contacts. For the docking of the compounds 10, 5 and 12 to DNA, binding free energy values of −20.1, −15.2, and −10.0 kcal mol−1 were predicted, respectively. Lower binding energy values indicate more preferable bonding interactions between compound–DNA complexes. It was observed that compound 12 with the highest binding energy, interacts less with DNA than other compounds. Five- and six-membered rings of benzothiazole groups of 12 interacted with the DNA residues A17 and A18 via C–H⋯π interactions. In the case of compound 10, it was predicted that there were many more C–H⋯π interaction networks with DNA residues as compared to 12: two between the toluene ring bound to benzimidazole group and DNA residues C11 and A17, five- and six-membered rings of other benzimidazole groups and G12, the ring of the tert-butyl methoxybenzene group of 10 and the G16 residue. For the docking of compound 5, hydrogen bonding and O–H⋯π interactions as well as C–H⋯π interactions with G12, T19, A18 residues, and water molecules were observed.| Compound | Binding free energy (kcal mol−1) |
|---|---|
| 4 | −10.9 |
| 5 | −15.2 |
| 6 | −11.4 |
| 7 | −10.9 |
| 10 | −20.1 |
| 11 | −13.5 |
| 12 | −10.0 |
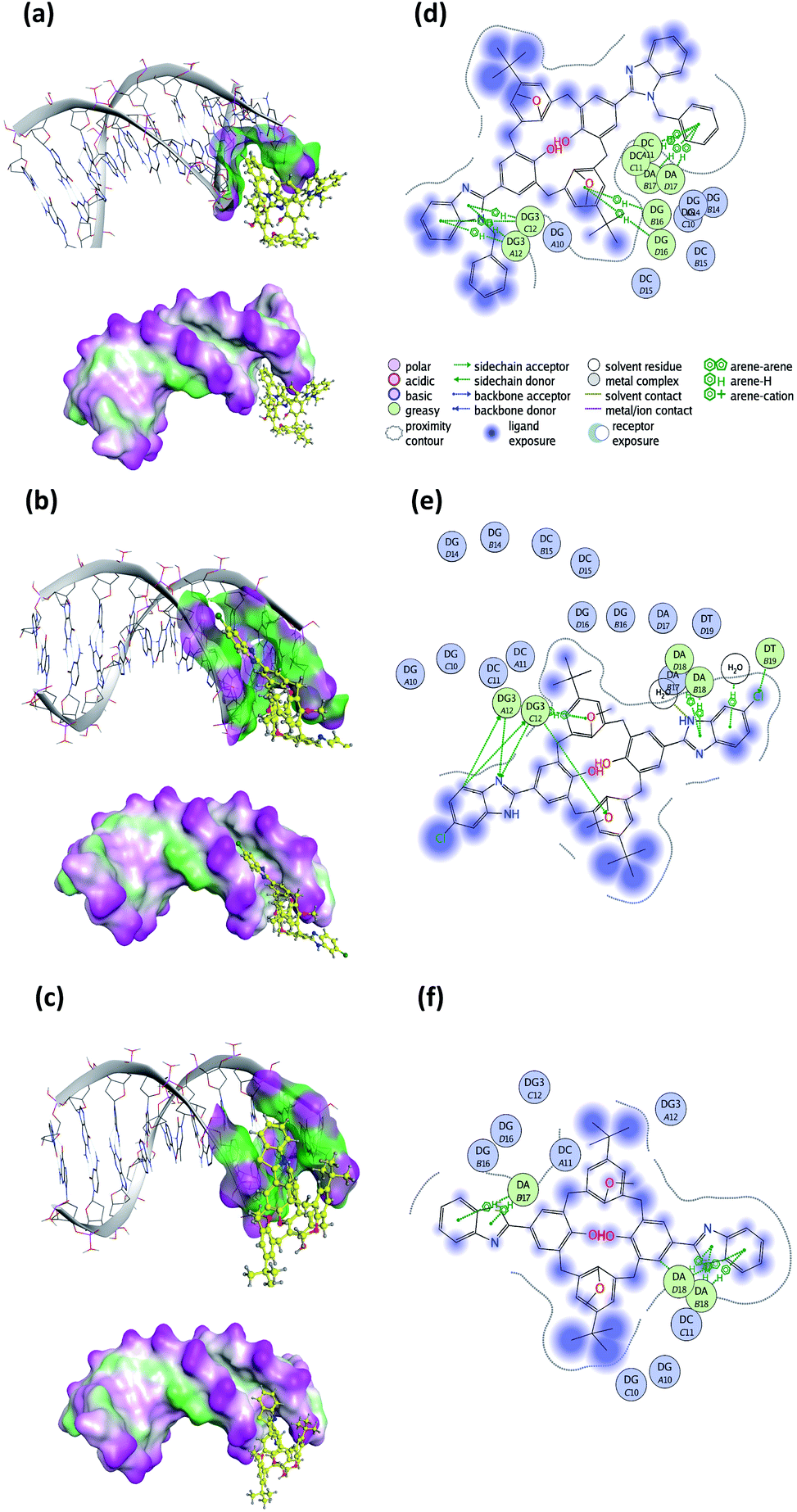 | ||
| Fig. 15 Three-dimensional (3D) docked structures with molecular surface representations (left column) and two-dimensional (2D) interaction plots (right column) obtained for the most energetically profitable poses of the compound–DNA complexes: (a) and (d) compound 10; (b) and (e) compound 5; (c) and (f) compound 12 (PDB ID: 1BNA). | ||
Experimental
Reagents and techniques
Chemicals and solvents were obtained from commercial sources (Merck and Sigma) and were used without further purification. DNA (supercoiled pBR322) and restriction enzymes (BamHI and HindIII) were purchased from Thermo-Fischer Scientific. Calf thymus DNA was supplied by Sigma. Measurements of 1H NMR and 13C NMR spectra were recorded in CDCl3 and DMSO-d6 on a Varian MR 400 MHz spectrometer using TMS as an internal standard. A Perkin Elmer 100 FTIR spectrophotometer was used to record the infrared spectra of all compounds (4000–400 cm−1). Mass spectra were obtained using a Bruker Microflex LT MALDI-TOF mass spectrometer. Elemental analyses (C, H, and N) were performed using a Leco 932 CHNS analyzer. The UV-vis. measurements were performed on a Shimadzu UV 1800 UV-vis spectrometer. A Perkin Elmer LS 55 Fluorimeter was used for fluorescence spectra. Microwave irradiated reactions were performed using a CEM MDS-2000, and a household microwave oven. The determination of the melting points was performed using a Büchi B-540 instrument. Analytical TLC was performed on precoated silica gel plates (SiO2, Merck PF254). All aqueous solutions were prepared with purified water or ultrapure water purified by a Millipore Milli-Q Plus water purification device.Synthetic procedures
The p-tert-butylcalix[4]arene 1 and its derivatives 2 and 3, and 8 and 9 were synthesized according to the literature procedures.22–25![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). 1H NMR (DMSO-d6): δ ppm 1.05 (s, 18H, But), 3.57 (d, J = 13.1 Hz, 4H, ArCH2Ar), 3.94 (s, 6H, OCH3), 4.24 (d, J = 13.1 Hz, 4H, ArCH2Ar), 7.12 (bs, 8H, Calix. ArH, Benzim. ArH), 7.54 (dd, J = 7.04 Hz, 4H, Benzim. ArH), 8.05 (s, 4H, Calix. ArH), 8.74 (s, 2H, OH), 12.54 (s, 2H, Benzim. NH). 13C NMR (DMSO-d6): δ ppm 155, 153.2, 152.2, 151.7, 150.5, 147.7, 132.4, 130.7, 128.8, 127.5, 126.4, 123.7, 121.9, 121.6, 117.7, 64.0, 34.5, 31.6, 31.5. MALDI-TOF MS: (CHNO = 796.4 m/z) 796.12 [M+] (Fig. S1†). Anal. cal. for C52H52N4O4: C, 78.36; H, 6.58; N, 7.03%. Found: C, 78.0; H, 6.8; N, 7.1%.
N). 1H NMR (DMSO-d6): δ ppm 1.05 (s, 18H, But), 3.57 (d, J = 13.1 Hz, 4H, ArCH2Ar), 3.94 (s, 6H, OCH3), 4.24 (d, J = 13.1 Hz, 4H, ArCH2Ar), 7.12 (bs, 8H, Calix. ArH, Benzim. ArH), 7.54 (dd, J = 7.04 Hz, 4H, Benzim. ArH), 8.05 (s, 4H, Calix. ArH), 8.74 (s, 2H, OH), 12.54 (s, 2H, Benzim. NH). 13C NMR (DMSO-d6): δ ppm 155, 153.2, 152.2, 151.7, 150.5, 147.7, 132.4, 130.7, 128.8, 127.5, 126.4, 123.7, 121.9, 121.6, 117.7, 64.0, 34.5, 31.6, 31.5. MALDI-TOF MS: (CHNO = 796.4 m/z) 796.12 [M+] (Fig. S1†). Anal. cal. for C52H52N4O4: C, 78.36; H, 6.58; N, 7.03%. Found: C, 78.0; H, 6.8; N, 7.1%.General procedure for synthesis of compounds 5–7 and 10, 11
Here, 0.5 g (0.805 mmol) of the dialdehyde compound (3), 1.691 mmol of o-phenylenediamine derivatives [4-methyl-o-phenylenediamine (0.21 g), 4-nitro-o-phenylenediamine (0.26 g), and 4-chloro-o-phenylenediamine (0.24 g), N-benzyl-o-phenylenediamine (0.34 g), and N-p-nitrobenzyl-o-phenylenediamine (0.41 g), respectively] were weighed. Then, 0.28 g (1.691 mmol) of KI was added, followed by 50 mL of DMF, and kept in the household microwave device for approximately 1–2 min. After being removed from the device and cooled slightly, it was heated to 80 °C by connecting it back to the condenser for 24–96 hours (96 hours for 5, 10 and 11, 72 hours for 6 and 24–48 hours for 7). It was cooled when the reaction was determined to be over by TLC and FTIR. To the mixture was added 50 mL of ethyl acetate and water. The mixture was taken into the separating funnel, shaken and allowed to separate. The ethyl acetate phase was obtained, dried with CaCl2, then the solvent was removed and washed with ethanol.N). 1H NMR (DMSO-d6): δ ppm 1.07 (s, 18H, But), 3.61 (d, J = 12.8 Hz, 4H, ArCH2Ar), 3.97 (s, 6H, OCH3), 4.27 (d, J = 12.8 Hz, 4H, ArCH2Ar), 7.07–7.29 (overlapped, 6H, Calix. ArH), 7.43–7.72 (m, 4H, Calix. ArH, Benz. ArH), 8.05 (s, 4H, Benzyl. ArH), 8.81 (s, 2H, OH), 12.76 (s, 2H, Benzyl. NH). 13C NMR (DMSO-d6): δ ppm 155.3, 147.7, 145.4, 142.7, 135.1, 134.8, 132.3, 132.1, 128.8, 128, 127.7, 126.5, 125.3, 124.7, 118.1, 64.0, 34.5, 31.6, 31.5. MALDI-TOF MS: (CHClNO = 864.32 m/z) 864.57 [M+] (Fig. S1†). Anal. calc. for C52H50Cl2N4O4: C, 72.1; H, 5.82; N, 6.47%. Found: C, 71.6; H, 6.2; N, 5.9%.N). 1H NMR (DMSO-d6): δ ppm 1.08 (s, 18H, But), 2.46 (s, 6H, ArCH3), 3.60 (d, J = 12.8 Hz, 4H, ArCH2Ar), 3.97 (s, 6H, OCH3), 4.28 (d, J = 12.8 Hz, 4H, ArCH2Ar), 6.99 (d, J = 8.2 Hz, 2H, Benzyl. ArH), 7.15 (s, 4H, Calix. ArH), 7.35 (s, 2H, Benz. ArH), 7.44 (d, J = 8.2 Hz, 2H, Benzyl. ArH), 7.77–8.19 (m, 5H, Calix. ArH, OH), 8.77 (s, 1H, OH), 12.8 (bs, 2H, Benz) NH. 13C NMR (DMSO-d6): δ ppm 154.9, 151.8, 151.7, 147.7, 132.4, 132.1, 131.3, 130.8, 129.3, 128.8, 127.4, 126.5, 126.4, 123.5, 121.5, 64, 34.5, 31.5, 31.4, 21.8. MALDI-TOF MS: (CHNO = 824.43 m/z) 824.07 [M+] (Fig. S2†). Anal. Cal. for C54H56N4O4: C, 78.6; H, 6.84; N, 6.79%. Found: C, 77.6; H, 7.2; N, 6.5%.N), 1521 cm−1 (NO2). 1H NMR (DMSO-d6): δ ppm 1.09 (bs, 18H, But), 3.60 (d, J = 13.1 Hz, 4H, ArCH2Ar), 3.96 (bs, 6H, OCH3), 4.26 (d, J = 13.1 Hz, 4H, ArCH2Ar), 6.64–6.81 (m, 4H, Calix. ArH), 7.07–7.37 (m, 4H, Calix. ArH), 7.85–7.93 (m, 4H, Benzyl. ArH), 8.04–8.21 (bs, 2H, Benz. ArH), 8.59 (s, 1H, OH), 9.04 (s, 1H, OH), 13.32 (s, 2H, Benzyl. NH). 13C NMR (DMSO-d6): δ ppm 159.6, 156.6, 151.6, 151.3, 147.9, 147.7, 136.5, 135.0, 132.8, 130.5, 129.2, 128.3, 126.5, 124.2, 112.8, 64, 34, 31.5, 31.3. MALDI-TOF MS: (CHNO = 886.37 m/z) 886.60 [M+] (Fig. S2†). Anal. cal. for C52H50N4O4: C, 70.4; H, 5.68; N, 9.47%. Found: C, 70.6; H, 6.2; N, 9.5%.N). 1H NMR (DMSO-d6): δ ppm 1.08 (s, 18H, But), 3.61 (d, J = 13.5 Hz, 4H, ArCH2Ar), 3.95 (bs, 6H, OCH3), 4.10–4.45 (m, 4H, ArCH2Ar), 5.31 (s, 4H, NCH2), 6.73–7.37 (m, 12H, Calix. ArH, ArH), 7.37–7.78 (m, 6H, Calix. ArH, ArH), 7.79–8.17 (m, 5H, ArH) 8.18–8.45 (m, 2H, ArH), 8.55 (s, 1H, ArH), 8.82 (s, 2H, ArOH). MALDI-TOF MS: (CHNO = 976.49 m/z) 976.04 [M+] (Fig. S3†). Anal. cal. for C66H64N4O4: C, 81.1; H, 6.60; N, 5.73%. Found: C, 80.6; H, 6.8; N, 5.6%.N), 1519 cm−1 (NO2). 1H NMR (DMSO-d6): δ ppm 0.96 (s, 18H, But), 3.51 (d, J = 13.0 Hz, 4H, ArCH2Ar), 3.91 (bs, 6H, OCH3), 4.18 (d, J = 13.0 Hz, 4H, ArCH2Ar), 5.71 (s, 4H, NCH2), 6.89 (m, 2H, Calix. ArH), 7.03–7.50 (m, 11H, Calix. ArH, ArH), 7.50–7.89 (m, 6H, ArH), 7.97–8.06 (m, 1H, ArH), 8.16 (d, J = 8.61 Hz, 4H, ArH), 8.30–8.47 (m, 1H, ArOH), 8.55–8.77 (m, 1H, ArOH). 13C NMR (DMSO-d6): δ ppm 159.2, 154.9, 154.1, 151.6, 147.3, 145.3, 143.2, 136, 132.2, 131.3, 130.8, 129.9, 128.9, 127.8, 126.4, 124.4, 122.7, 119.5, 111.2, 63.9, 47.7, 34.3, 34.1, 31.3. MALDI-TOF MS: (CHNO = 1066.46 m/z) 1066.05 [M+] (Fig. S3†). Anal. cal. for C66H62N6O8: C, 74.3; H, 5.86; N, 7.87%. Found: C, 74.6; H, 6.0; N, 7.7%.Synthesis of compound 12
Here, 0.62 g (1 mmol) of compound 3 was weighed and 1.068 mL (10 mmol) of 2-aminothiophenol and 0.83 g (5 mmol) of KI were added. Next, 50 mL of DMF was added and refluxed for 2–3 hours in a laboratory microwave. To the cooled mixture was added 50 mL of ethylacetate and water. The mixture was taken into the separating funnel, shaken and allowed to separate. The ethyl acetate phase was separated off, dried with MgSO4, and the solvent removed and purified from hot ethanol. Compound 12 was obtained in 55% yield. Mp 258–262 °C FTIR: 1658 cm−1 (CN), 1373 cm−1 (C–S). 1H NMR (CDCl3): δ ppm 0.99 (s, 18H, But), 3.57 (d, J = 13.3 Hz, 4H, ArCH2Ar), 4.03 (s, 6H, OCH3), 4.36 (d, J = 13.3 Hz, 4H, ArCH2Ar), 6.60 (bs, 1H, Calix. ArH), 6.72 (bs, 1H, Calix. ArH), 6.84 (s, 1H, Calix. ArH), 6.94 (s, 2H, Calix. ArH), 7.18 (d, J = 6.5 Hz, 2H, Thia. ArH), 7.35 (t, J = 6.5 Hz, 2H, Thia. ArH), 7.48 (t, J = 6.5 Hz, 2H, Thia. ArH), 7.69 (s, 1H, Calix. ArH), 7.82–7.90 (m, 4H, Calix. ArH, ArOH), 8.04–8.08 (m, 2H, Thia. ArH). 13C NMR (CDCl3): δ ppm 156.2, 154.3, 151.3, 148.6, 148, 137, 136.8, 134.8, 131.6, 129.1, 127.8, 125.9, 122.5, 121.4, 118.3, 63.6, 34.1, 31.2, 31.1. MALDI-TOF MS: (CHNOS = 830.32 m/z) 830.70 [M+] (Fig. S4†). Anal. cal. for C52H50N2O4S2: C, 75.15; H, 6.06; N, 3.37; S, 7.72%. Found: C, 74.8; H, 6.2; N, 3.4; S, 7.80%.
DNA studies
000 MM. The buffer solution prepared for both DMF and DNA was used in half-dilution processes (5000, 2500, 1250, 625 μM). The prepared stock DNA solution contained 0.5 μg mL−1 plasmid DNA for each substance (30 μL samples) and was incubated in the dark in a water bath at 37 °C for 24 hours. At the end of this period, 10 μL samples were taken from each sample and mixed with 3 μL of dye and loaded onto 1% agarose gel [prepared with 1XTAE (Tris + acetic acid + EDTA)] and electrophoresed in 1XTAE buffer at 60 V for 2 or 3 hours. At the end of this time, the gel was removed and 10 μL of 0.5 mg mL−1 (100 μM) EtBr solution was shaken for 30 minutes, after which the images of the gels were recorded under a UV lamp. In experiments with restriction enzymes, 8 μL samples were taken from samples incubated overnight for both BamHI and HindIII. Then, 1 unit of enzyme (0.1 μL) and buffer (1 μL) was added to these samples and incubated for one hour at 37 °C in a hot water bath. At the end of this period, the samples were removed from the hot water bath and immediately transferred to an ice bath. The same operations described above were performed and the images were recorded.Molecular docking computations
Before the molecular docking, the geometries of the initial structures of the ligands considered were optimized by using the Gaussian 09 software33 with the semi-empirical parametric method 6 (PM6).34 The crystal structure of the DNA dodecamer was retrieved from the RCSB Protein Data Bank (http://www.rcsb.org/pdb/), under the accession code 1BNA.28b Molecular Operating Environment (MOE) software35 was used for molecular docking studies. The co-crystallized water molecules farther than 4.5 Å from ligand or DNA were deleted from the crystal structure. DNA–ligand complexes were energy-minimized to a gradient of 0.01 kcal (mol−1 Å−1), and protonated by the force field AMBER99. Charges on the DNA and ligands were assigned using the force fields AMBER99 and MMF94X, correspondingly. Possible ligand-binding sites were identified by the “Site Finder” module of the MOE. The Triangle Matcher Algorithm and scoring functions of London dG and GBVI/WSA dG were used to produce 30 poses of each ligand. All poses generated with docking were analyzed and the best-scored pose with the lowest binding energy and root mean square value for each compound was selected for further investigation of interactions with the DNA.Conclusions
In this study, the synthesis and characterization of six different p-benzimidazole-derived and a benzothiazole-derived p-tert-butylcalix[4]arene compounds were performed and the interactions of the compounds obtained with pBR322 plasmid DNA and CT-DNA were evaluated by various methods. Compounds showing cleavage effects on pBR322 plasmid DNA were 6 and 10 (at the highest concentration, 10000 μM), while other compounds 5, 7, 11 and 12 caused different changes in the structure of the plasmid DNA. CT-DNA interaction experiments were evaluated using UV-vis and molecular fluorescence spectral studies. The results obtained from UV-vis studies showed that there was a hyperchromic effect, proving that all of the compounds, except compound 7, interact with DNA. The stoichiometries of the binding modes of the compounds with DNA and the evaluation of the interaction strengths of the complexes formed were performed by fluorescence spectroscopy. The first of these studies showed that the emission intensities of compounds 4 and 5 were increased by the addition of DNA, whereas those of compounds 10–12 decreased. DNA binding constants (Kb) and the number of molecules that bind to DNA were calculated for the compounds giving both conditions. Compounds 7 and 10 had the highest Kb constants (9.3 × 102 M−1 and 7.1 × 103 M−1, respectively). In the displacement experiments performed with EtBr, results in line with previous studies were obtained. Compounds 5 and 10 had the highest Ksv constants (5.9 × 103 M−1 and 5.8 × 103 M−1, respectively). Consequently, when the obtained results were combined, it was evaluated that the compounds showed binding strengths toward CT-DNA of the order of 10 > 5 > 4 > 11 > 6 > 12.
Docking of the synthesized compounds into the DNA binding site was carried out. Docking results complemented well the experimental results on binding strength of the compounds to DNA. Lower binding free energies and much more interaction networks were observed for compounds that more strongly interacted with the DNA than weaker ones. For the docking of compounds 10, 5 and 12, the DNA residues C11, G12, G16, A17, A18 and T19 played a major role in the binding site.
Experimental results have shown that the benzyl ring of compound 10 is more potent between the nucleotides of DNA than other compounds because of its aromatic and planar structure. In a similar study, it was determined that the benzimidazole compounds with substituted benzyl groups are effective on CT-DNA.36 Compound 11, unlike 10, has a nitro group attached to the benzyl ring. However, the fact that compound 7 with the nitro group had no activity toward the CT-DNA led to the idea that it was due to the structure of the nitro group. We, therefore, thought that compound 11 did not show activity as compound 10. Secondly, compound 5, which strongly binds to DNA, has the chloro-group; chlorine, an electronegative atom, increased its ability to bind to the DNA. It has been reported that compounds containing halogens show activity toward DNA.30,31 Compound 4 is a calixarene compound without a substituent attached to the ring and its binding to DNA was stronger than the remaining compounds. Compounds 10 and 5 took first place in the ranking according to the calculated Kb and Ksv constants. Finally, when comparing studies with benzothiazole compound (12), it was concluded that benzimidazole compounds bind more strongly with CT-DNA than a benzothiazole compound.
Some of the compounds synthesized in this study showed activity against bacterial plasmid DNA, some against CT-DNA and some against both DNA types. While compounds 4–6, and 10 were found to bind to DNA in the intercalative mode, compound 11 was also bound to DNA, possibly with the groove binding mode.
The development of new and effective drugs has become essential and important due to antibiotic resistance, which is a major problem in our age that has become more and more scary day by day. For this reason, the design and synthesis of new compounds with antibacterial properties have been among the subjects that scientists have worked hard on. For this purpose, it was seen in this study that two of the synthesized compounds could break down the plasmid DNA, which is the protective DNA of bacteria.
On the other hand, the absence of the drug(s) that completely cure cancer, one of the biggest health problems of today, requires the synthesis of new and effective molecules. For this purpose, the studies carried out with CT-DNA are preliminary studies and the active compounds can be used in future experimental in vitro anticancer studies.12,16,30,32,37–39
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Research Foundation of Selcuk University, Konya Turkey [grant number BAP 2017/17201115] for its financial support of this work produced from a part of Ş. Ç. Özkan's PhD thesis. In addition, the author would like to thank the Department of Chemistry of Gebze Technical University for its laboratory and device facilities.Notes and references
- (a) A. Villiers, C. R. Acad. Sci., 1891, 536–538 Search PubMed; (b) F. Schardinger and Z. Unters, Nahr. Genussm., 1903, 6, 865–880 CAS.
- (a) C. J. Pedersen, J. Am. Chem. Soc., 1967, 89(26), 7017–7036 CrossRef CAS; (b) C. J. Pedersen, J. Am. Chem. Soc., 1967, 89(10), 2495–2496 CrossRef CAS.
- C. D. Gutsche, Calixarenes; Monographs in Supramolecular Chemistry, ed. J. F. Stoddart, The Royal Society of Chemistry, Cambridge, 1989, p. 1 Search PubMed.
- (a) D. Baurer, M. Blumberg, M. Köckerling and C. Mamat, RSC Adv., 2019, 9, 32357–32366 RSC; (b) J. Rodríguez-Lavado, A. Lorente, E. Flores, A. Ochoa, F. Godoy, P. Jaque and C. Saitz, RSC Adv., 2020, 10, 21963–21973 RSC; (c) M. Phichi, A. Imyim, T. Tuntulani and W. Aeungmaitrepirom, Anal. Chim. Acta, 2020, 1104, 147–155 CrossRef CAS.
- (a) B. Mokhtari and K. Pourabdollah, Asian J. Chem., 2013, 25(1), 1–12 CrossRef CAS; (b) F. K.-W. Hau, H.-S. Lo and V. W.-W. Yam, Chem.–Eur. J., 2016, 22, 3738–3749 CrossRef CAS; (c) S. Erdemir, B. Tabakci and M. Tabakci, Sens. Actuators, B, 2016, 228, 109–116 CrossRef CAS.
- (a) M. Kandpal, A. K. Bandela, V. K. Hinge, V. R. Rao and C. P. Rao, ACS Appl. Mater. Interfaces, 2013, 5, 13448–13456 CrossRef CAS; (b) Z. Zheng, W.-C. Geng, J. Gao, Y.-J. Mu and D.-S. Guo, Org. Chem. Front., 2018, 5, 2685–2691 RSC; (c) H. Kaur, N. Singh, N. Kaur and D. O. Jang, Sensor. Actuator. B Chem., 2019, 284, 193–201 CrossRef CAS.
- (a) M. Mourer, R. E. Duval, P. Constant, M. Daffé and J.-B. Regnouf-de-Vains, ChemBioChem, 2019, 20, 911–921 CrossRef CAS; (b) Ş. Ç. Özkan, A. Yılmaz, E. Arslan, L. Açık, Ü. Sayın and E. Gülbahçe Mutlu, Supramol. Chem., 2015, 27, 255–267 CrossRef.
- (a) M. N. Soares Jr, T. M. Gáscon, F. L. A. Fonseca, K. S. Ferreira and I. A. Bagatin, Mater. Sci. Eng. C, 2014, 40, 260–266 CrossRef; (b) Z. D. Gezelbash and K. A. Dilmaghani, J. Chin. Chem. Soc., 2020, 67, 1446–1452 CrossRef.
- Y. Tauran, J. P. Cerón-Carrasco, M. Rhimi, F. Perret, B. Kim, D. Collard, A. W. Coleman and H. Pérez-Sánchez, Antibiotics, 2019, 8(73), 1–15 Search PubMed.
- (a) M. Mourer, N. Psychogios, G. Laumond, A.-M. Aubertin and J.-B. Regnouf-de-Vains, Bioorg. Med. Chem., 2010, 18, 36–45 CrossRef CAS; (b) F. N. Pur, Mol. Divers., 2020 DOI:10.1007/s11030-020-10042-0.
- G. Granata, I. Paterniti, C. Geraci, F. Cunsolo, E. Esposito, M. Cordaro, A. R. Blanco, S. Cuzzocrea and G. M. L. Consoli, Mol. Pharm., 2017, 14(5), 1610–1622 CrossRef CAS.
- (a) R. P. M. Dings, J. I. Levine, S. G. Brown, L. Astorgues-Xerri, J. R. MacDonald, T. R. Hoye, E. Raymond and K. H. Mayo, Invest. New Drugs, 2013, 31, 1142–1150 CrossRef CAS; (b) B. Yilmaz, A. T. Bayac and M. Bayrakci, Appl. Biochem. Biotechnol., 2020, 190, 1484–1497 CrossRef CAS; (c) L. An, C. Wang, L. Han, J. Liu, T. Huang, Y. Zheng, C. Yan and J. Sun, Front. Chem., 2019, 7, 856 CrossRef CAS; (d) M. Oguz, A. Gul, S. Karakurt and M. Yilmaz, Bioorg. Chem., 2020, 94(1–8), 103207 CrossRef CAS; (e) E. B. Noruzi, B. Shaabani, S. Geremia, N. Hickey, P. Nitti and H. S. Kafil, Molecules, 2020, 25(1–19), 370 CrossRef CAS.
- (a) S. B. Nimse and T. Kim, Chem. Soc. Rev., 2013, 42, 366–386 RSC; (b) Y. Tauran, M. Kumemura, M. C. Tarhan, G. Perret, F. Perret, L. Jalabert, D. Collard, H. Fujita and A. W. Coleman, Sci. Rep., 2019, 9(1–13), 5816 CrossRef; (c) M. Oguz, E. Kalay, S. Akocak, A. Nocentini, N. Lolak, M. Boga, M. Yilmaz and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2020, 35(1), 1215–1223 CrossRef.
- (a) F. J. Ostos, J. A. Lebrón, M. L. Moyá, M. Deasy and P. López-Cornejo, Colloids Surf., B, 2015, 127, 65–72 CrossRef CAS; (b) R. V. Rodik, A.-S. Anthony, V. I. Kalchenko, Y. Mély and A. S. Klymchenko, New J. Chem., 2015, 39, 1654–1664 RSC.
- (a) Ş. Ç. Özkan, A. Yılmaz and İ. Özmen, Supramol. Chem., 2014, 26, 25–31 CrossRef; (b) K. Samanta, D. S. Ranade, A. Upadhyay, P. P. Kulkarni and C. P. Rao, ACS Appl. Mater. Interfaces, 2017, 9(6), 5109–5117 CrossRef CAS; (c) R. Salvio, S. Volpi, T. Folcarelli, A. Casnati and R. Cacciapaglia, Org. Biomol. Chem., 2019, 17, 7482–7492 RSC.
- (a) M. Mirza-Aghayan, M. Yarmohammadi, R. Zadmard and R. Boukherroub, Supramol. Chem., 2014, 26(5–6), 442–449 CrossRef CAS; (b) F. J. Ostos, J. A. Lebrón, P. López-Cornejo, M. López-López, M. García-Calderón, C. B. García-Calderón, I. V. Rosado, V. I. Kalchenko, R. V. Rodik and M. L. Moyá, J. Mol. Liq., 2020, 304(1–14), 112724 CrossRef CAS.
- Salahuddin, M. Shaharyar and A. Mazumder, Arabian J. Chem., 2017, 10, 157–173 CrossRef.
- H. M. Refaat, Eur. J. Med. Chem., 2010, 45, 2949–2956 CrossRef CAS.
- (a) S. U. Rehman, T. Sarwar, M. A. Husain, H. M. Ishqi and M. Tabish, Arch. Biochem., 2015, 576, 49–60 CrossRef; (b) X. L. Li, Y. J. Hu, H. Wang, B. Q. Yu and H. L. Yue, Biomacromolecules, 2012, 13, 873–880 CrossRef CAS; (c) W. D. Sasikala and A. Mukherjee, J. Phys. Chem. B, 2012, 116, 12208–12212 CrossRef CAS.
- (a) A. Mukherjee, R. Lavery, B. Bagchi and J. T. Hynes, J. Am. Chem. Soc., 2008, 130, 9747–9755 CrossRef CAS; (b) B. A. D. Neto and A. A. M. Lapis, Molecules, 2009, 14, 1725–1746 CrossRef CAS.
- S. Kunzelmann, C. Morris, A. P. Chavda, J. F. Eccleston and M. R. Webb, Biochemistry, 2010, 49(5), 843–852 CrossRef CAS.
- C. D. Gutsche, M. Iqbal and D. Stewart, J. Org. Chem., 1986, 51, 742–745 CrossRef CAS.
- A. Casnati, A. Arduini, E. Ghidini, A. Pochini and R. Ungaro, Tetrahedron, 1991, 47, 2221–2228 CrossRef CAS.
- A. Sap, B. Tabakci and A. Yilmaz, Tetrahedron, 2012, 68, 8739–8745 CrossRef CAS.
- (a) The commercial product; (b) A. E. Sparke, C. M. Fisher, R. E. Mewis and S. J. Archibald, Tetrahedron Lett., 2010, 51, 4723–4726 CrossRef CAS.
- A. Zianna, G. Psomas, A. Hatzidimitriou and M. Lalia-Kantouri, J. Inorg. Biochem., 2016, 163, 131–142 CrossRef CAS.
- (a) A. Haleel, D. Mahendiran, V. Veena, N. Sakthivel and A. Kalilur Rahiman, Mater. Sci. Eng. C, 2016, 68, 366–382 CrossRef CAS; (b) P. Gurumoorthy, D. Mahendiran and A. Kalilur Rahiman, Chem. Biol. Interact., 2016, 248, 21–35 CrossRef CAS; (c) F. Arjmand, S. Parveen, M. Afzal and M. Shahid, J. Photochem. Photobiol., B, 2012, 114, 15–26 CrossRef CAS.
- (a) J. R. Albani, Principles and Applications of Fluorescence Spectroscopy, Wiley-Blackwell, Hoboken, 2007 CrossRef; (b) M. M. Silva, F. C. Savariz, E. F. Silva-Júnior, T. M. de Aquino, M. H. Sarragiotto, J. C. C. Santos and I. M. Figueiredo, J. Braz. Chem. Soc., 2016, 27(9), 1558–1568 CAS.
- (a) J. R. Lakowicz and G. Weber, Biochemistry, 1973, 12, 4161–4170 CrossRef CAS; (b) G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed; (c) S. Scheiner, Hydrogen Bonding, A Theoretical Perspective, Oxford University Press, Oxford, 1997 Search PubMed.
- M. Sirajuddin, S. Ali and A. Badshah, J. Photochem. Photobiol., B, 2013, 124, 1–19 CrossRef CAS.
- M. Ariyaeifar, H. A. Rudbari, M. Sahihi, Z. Kazemi, A. A. Kajani, H. Zali-Boeini, N. Kordestani, G. Bruno and S. Gharaghani, J. Mol. Struct., 2018, 1161, 497–511 CrossRef CAS.
- W. Hu, C. Blecking, M. Kralj, L. Šuman, I. Piantanida and T. Schrader, Chem.–Eur. J., 2012, 18(12), 3589–3597 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian Inc., Wallingford, CT, USA, 2009 Search PubMed.
- J. J. P. Stewart, J. Mol. Model., 2007, 13(12), 1173–1213 CrossRef CAS.
- Molecular Operating Environment (MOE), C.C.G.I., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2014.
- P. Singla, V. Luxami and K. Paul, J. Photochem. Photobiol., B, 2017, 168, 156–164 CrossRef CAS.
- M. M. Silva, E. O. O. Nascimento, E. F. Silva-Júnior, J. X. de Araújo-Júnior, C. C. Santana, L. A. M. Grillo, R. S. de Oliveira, P. R. R. Costa, C. D. Buarque, J. C. C. Santos and I. M. Figueiredo, Int. J. Biol. Macromol., 2017, 96, 223–233 CrossRef CAS.
- W. Zhou, W. Zhang, Y. Peng, Z.-H. Jiang, L. Zhang and Z. Du, Molecules, 2020, 25, 3180–3197 CrossRef CAS.
- Y.-Y. Qi, Q. Gan, Y.-X. Liu, Y.-H. Xiong, Z.-W. Mao and X.-Y. Le, Eur. J. Med. Chem., 2018, 154, 220–232 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07486a |
| This journal is © The Royal Society of Chemistry 2020 |