
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of trisubstituted buta-1,3-dienes and α,β-unsaturated ketones via the reaction of functionalized vinyl phosphates and vinyl phosphordiamidates with organometallic reagents†
Petr Oeser‡
a,
Jakub Koudelka‡a,
Hana Dvořákováb and
Tomáš Tobrman *a
*a
aDepartment of Organic Chemistry, University of Chemistry and Technology, Prague, Technická 5, 166 28 Prague 6, Czech Republic. E-mail: tomas.tobrman@vscht.cz
bLaboratory of NMR Spectroscopy, University of Chemistry and Technology, Prague, Technická 5, 166 28 Prague 6, Czech Republic
First published on 22nd September 2020
Abstract
We studied the reactions of vinyl phosphates and vinyl phosphordiamidates containing an ester functional group with organometallic reagents. We found that the functionalized vinyl phosphates were smoothly converted into tri- and tetrasubstituted buta-1,3-dienes via the reaction with aryllithium reagents. Moreover, the vinyl phosphordiamidates were converted into α,β-unsaturated ketones using Grignard reagents. Based on the performed experiments, we proposed a reaction mechanism, which was confirmed by means of the isolation of key intermediates.
Introduction
A Kumada–Tamao–Corriu (KTC) reaction is a representative example of the traditional cross-coupling reaction of an electrophilic template with an organometallic reagent, as represented by Grignard reagents. This reaction is frequently used for the formation of a C–C bond.1 Among the various electrophilic templates that are suitable for use in a KTC reaction, substrates containing an activated C–O bond are considered particularly attractive. The reason behind the popularity of such substrates is their availability, which is associated with their preparation. Recent examples of the cross-coupling reactions of ethers,2 tosylates,3 and triflates4 with Grignard reagents all illustrate the KTC reaction potential of compounds with an activated C–O bond. Using this approach, a wide variety of substances can be prepared, including substituted alkenes.The stereoselective synthesis of di-, tri-,5 and tetrasubstituted6 double bonds has been the subject of significant research attention in recent decades. Vinyl phosphates also play a key role in the synthesis of substituted alkenes. Unsurprisingly, a number of KTC reactions have been described in which the reaction of the vinyl phosphates has been catalyzed by iron,7 nickel,8 or palladium9 catalysts in order to prepare the substituted double bond.
Despite considerable progress having been made in relation to the preparation and application of functionalized Grignard reagents,10 the preparation of alkenes with functional groups that react with Grignard reagents using the KTC reaction remains difficult (Scheme 1). The vinyl phosphates 1 with a carbonyl group attached to the vinyl unit have been used in the stoichiometric synthesis of organoselenium11 and organotellurium compounds.12 A methodology for the stoichiometric cross-coupling reaction of the functionalized vinyl phosphates with dialkylcuprates has also been described.13 This methodology has been successfully used in the field of organic synthesis14 as well as in the total synthesis of naturally occurring compounds.15 The catalytic reactions of the functionalized vinyl phosphates represented by the general structure 1 include reactions with organoaluminum compounds,16 iron-catalyzed reactions with methylmagnesium halides,17 and nickel-catalyzed cross-coupling reactions with trimethylsilylmethylmagnesium chloride.18 These prior results contrast with a recent report of the KTC reaction of functionalized vinyl tosylates being performed in dry acetonitrile at 45 °C.3b The absence of a general methodology for the KTC reaction of the functionalized vinyl phosphates 1 with Grignard reagents may indicate that this type of vinyl phosphate exhibits low stability in the presence of Grignard reagents. Our review of the literature indicated that a systematic study of the reactivity of the functionalized vinyl phosphates 1 with organometallic reagents has not previously been reported. Thus, based on our earlier studies of the cross-coupling reactions of vinyl phosphates,19 we decided to investigate the stability of the functionalized vinyl phosphates 2a–2c in the presence of Grignard reagents.
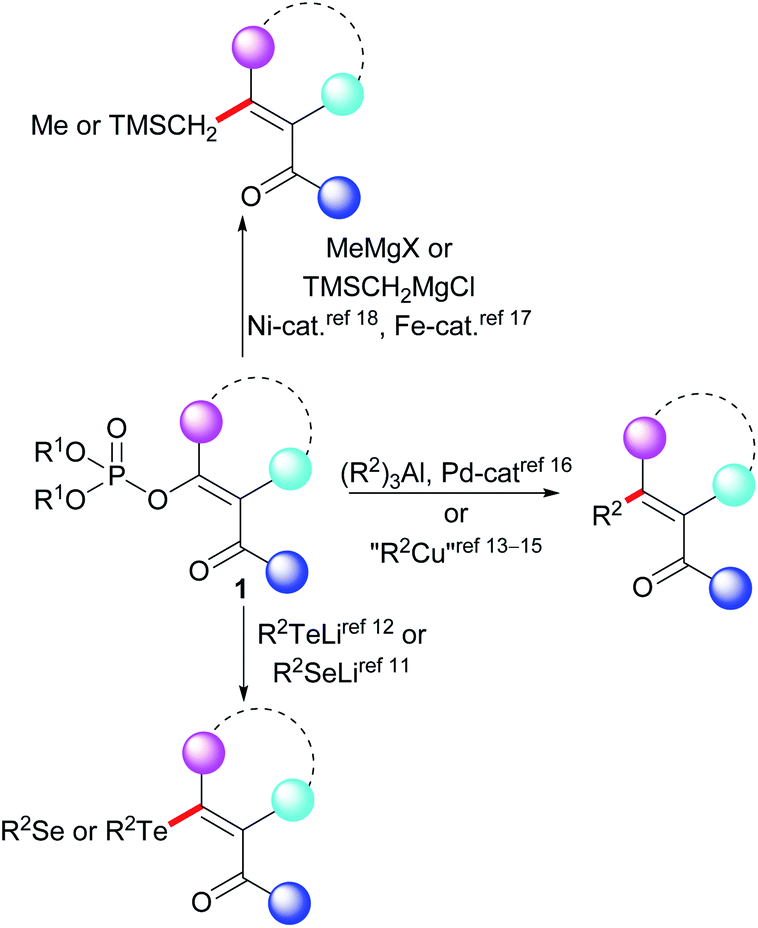 | ||
| Scheme 1 General scheme representing the replacement of the phosphate group of the functionalized vinyl phosphates 1. | ||
Results and discussion
We began our study by synthesizing the vinyl phosphate 2a by means of the enolization of ethyl acetoacetate, followed by the reaction of the formed enolate with diethyl chlorophosphate. The prepared phosphate 2a was mixed with phenylmagnesium chloride, and then the reaction mixture was stirred for 2 hours at 23 °C. After the reaction mixture was quenched with aqueous ammonium chloride, the formation of a complex mixture of products was observed based on analysis of the 1H nuclear magnetic resonance (1H NMR) spectrum of the mixture. However, we isolated the trisubstituted diene 3a as a pure compound in an isolated yield of 27%. This finding was particularly interesting, as the synthesis of trisubstituted dienes is generally limited. The palladium-catalyzed coupling of tosylhydrazones with aryl halides,20 the selective arylation of vinylarenes,21 and diethyl-phosphite-promoted carbonyl olefination22 are recently reported methods for the synthesis of substituted buta-1,3-dienes. We observed that the isolated yield of the diene 3a strongly depended on the reaction workup, with comparatively higher yields of the alkene 3a being achieved with hydrochloric acid, sulfuric acid, and phosphoric acid (Table 1, entries 3–5). The isolated yield of the alkene 3a did not affect the increased equivalents of phenylmagnesium chloride or the use of the diphenylphosphate 2b (Table 1, entries 6 and 7). The best isolated yield of the diene 3a was obtained when phenyllithium was used in combination with the phenyl ester 2c (Table 1, entry 9).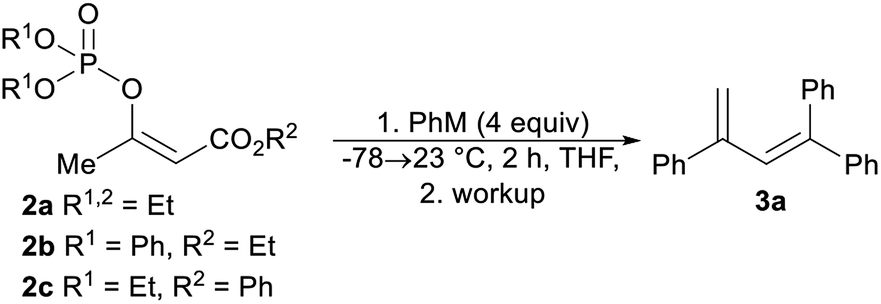
Optimized reaction conditions were used to evaluate the scope of the reaction (Scheme 2). The diethyl phosphate 2c reacted with the aryllithium reagents, thereby affording the corresponding dienes 3a–3d in a single step and in good isolated yields. 2-Methoxyphenyllithium, as an example of an ortho-substituted phenyl, gave the product 3e in a high isolated yield. A similar reactivity pattern was observed with regard to the diethyl phosphate with an extended alkyl chain 2d. In this case, the dienes 3f–3i were obtained in similar isolated yields to the dienes 3a–3c. The phosphate 2d was also reacted with 2-methoxyphenyllithium and 2-thienyllithium, as an example of a heteroaryl reagent, in order to give the dienes 3j and 3k. The dienes 3f–3k were obtained as an inseparable mixture of the (E)- and (Z)-stereoisomers and the double-bond geometry was determined by means of nuclear Overhauser effect (NOE) experiments. The use of an alkyllithium reagent, for example, n-butyllithium, failed to give the expected product. Instead, a complex reaction mixture was formed, as observed via the 1H NMR spectroscopy of the crude reaction mixture. It is worth noting that all the dienes 3a–3e were stored in a freezer, as significant decomposition was observed when they were stored at room temperature. However, the tetrasubstituted dienes 3f–3k showed excellent long-term storage stability.
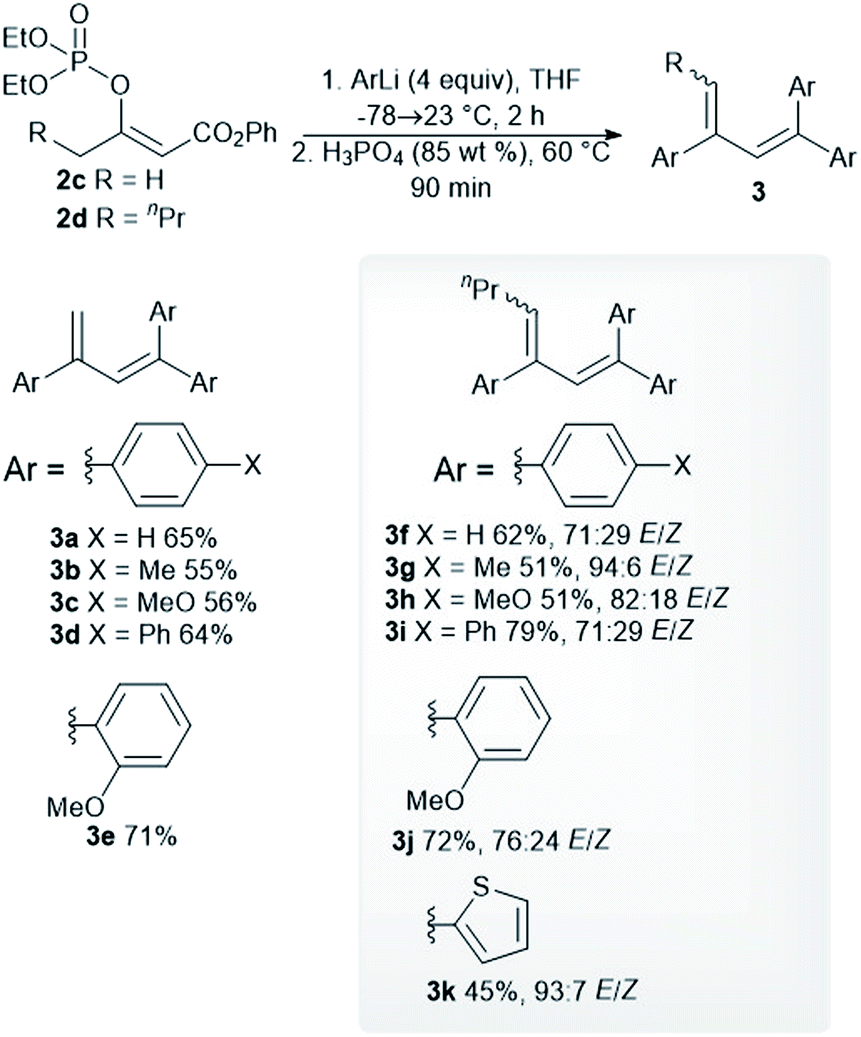 | ||
| Scheme 2 Reaction of the phosphates 2c and 2d with aryllithium reagents. | ||
We synthesized the vinyl phosphordiamidates 2e–2g so as to test their behavior during the developed one-pot synthesis of the substituted buta-1,3-dienes 3. Surprisingly, the vinyl phosphate 2e reacted smoothly with phenylmagnesium chloride to give the α,β-unsaturated ketone 4a in a high isolated yield (Scheme 3). α,β-Unsaturated ketones are known for their medicinal applications,23 and they are also valuable building blocks in relation to organic synthesis.24 Therefore, significant research efforts have been dedicated to identifying a novel approach to conjugated enones.25 Nickel-catalyzed 1,2-acyl migration,26 nucleophilic addition to enones,27 the cross-coupling reaction between acid fluorides and vinyl triflates,28 and carbonyl group olefinations29 are examples of such reactions. The conversion of the vinyl phosphordiamidate 2e into the ketone 4a observed in our study represents a hitherto undescribed reaction of phosphordiamidate en route to becoming α,β-unsaturated ketones. Further investigation in this regard showed that a significantly lower isolated yield of the ketone 4a was obtained with phenyllithium. Uniformly high yields of the corresponding ketones were obtained with the other para-, meta-, and ortho-substituted phenylmagnesium halides 4b–4f. Almost the same isolated yields of the ketones 4h–4m were obtained for the vinyl phosphordiamidate 2f. Additionally, the 2-thienylmagnesium bromide 4n, benzylmagnesium chloride 4o, and octylmagnesium chloride 4p reacted well with the phosphordiamidate 2f under the tested reaction conditions. The examination of the reactivity of the variously substituted vinyl phosphordiamidates was also extended to a phosphordiamidate with a phenyl substituent rather than an alkyl group, and the alkenes 4q and 4r were obtained in quantitative isolated yields.
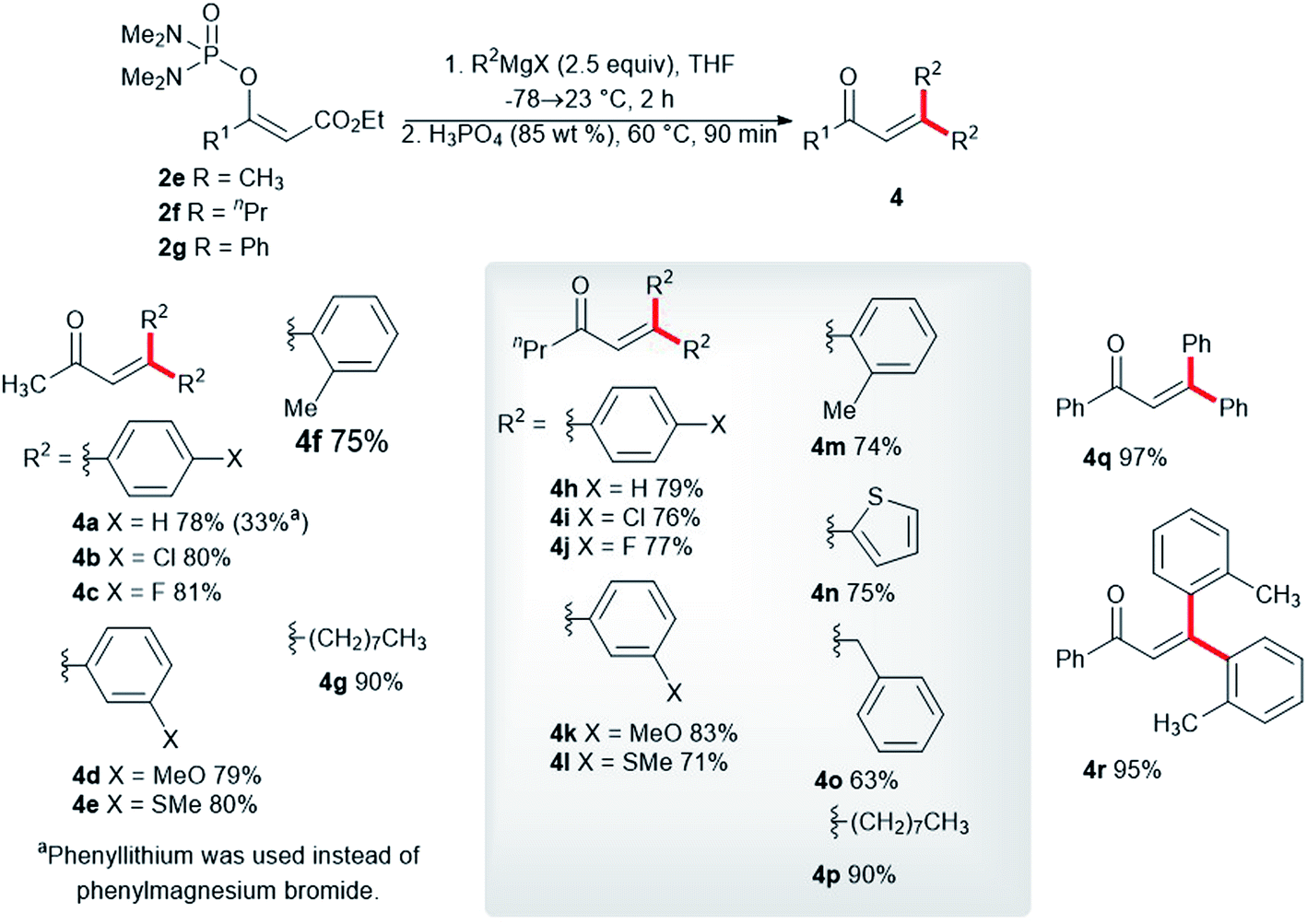 | ||
| Scheme 3 Synthesis of trisubstituted α,β-unsaturated ketones via the reaction of the phosphordiamidates 2e–2g with Grignard reagents. | ||
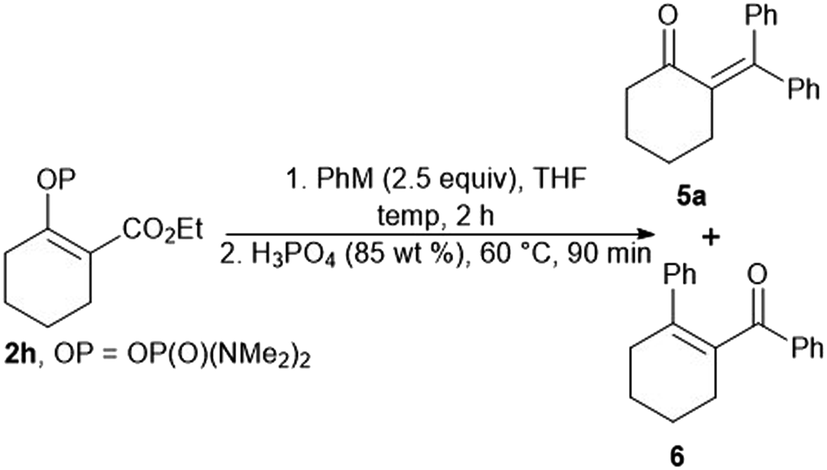
Our attempts to prepare enones with a tetrasubstituted double bond by means of the reaction of the vinyl phosphordiamidate 2h with phenylmagnesium chloride proved to be less effective, with the formation of the ketones 5a and 6 being observed (Table 2, entry 1). Therefore, we tried to optimize the studied reaction in order to obtain only the ketone 5a. However, the use of phenylmagnesium chloride always gave a mixture of the ketones 5a and 6, regardless of the reaction conditions (Table 2, entries 2 and 3). The desired outcome was ultimately achieved through the reaction between phenyllithium and the phosphordiamidate 2h, and the ketone 5a was obtained in an isolated yield of 67% (Table 2, entry 4).
The extension of the studied reaction to the cyclic vinyl phosphordiamidates 2i and 2j was successful in terms of the preparation of substituted cyclopentanones and cycloheptanones. In both cases, the expected ketones 5b, 5c, 5d, and 5e were obtained in very good isolated yields. Similar results were obtained for the acyclic phosphordiamidate 2k. Only the use of 2-thienyllithium proved less effective, and the reaction products 5f and 5i were obtained in low isolated yields (Scheme 4).
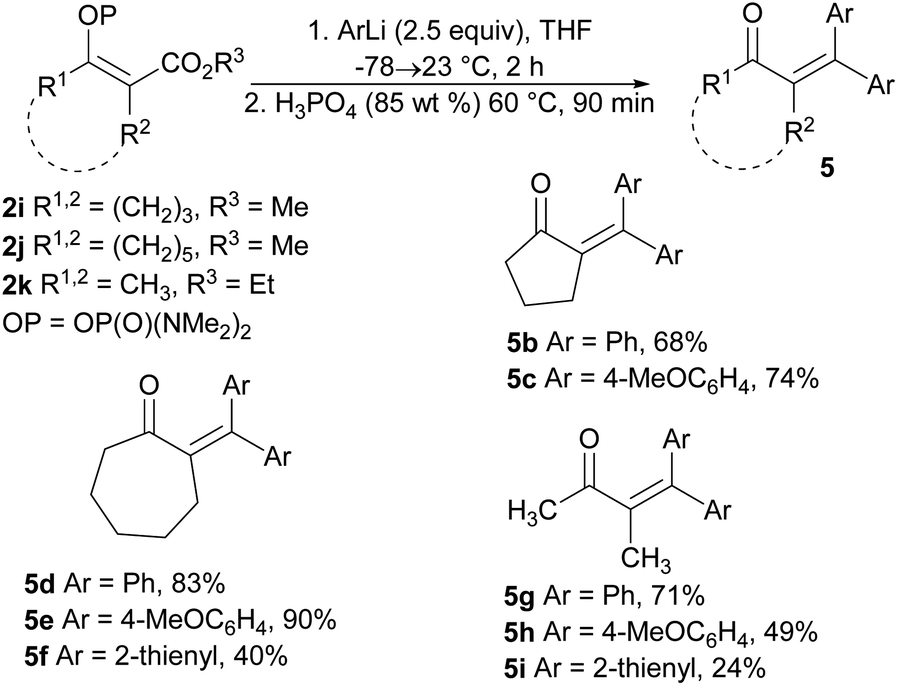 | ||
| Scheme 4 Synthesis of tetrasubstituted α,β-unsaturated ketones via the reaction of the phosphates 2i–2k with organolithium reagents. | ||
As a result of the performed experiments, we were able to propose a mechanism for the observed transformations. We assumed that during the first step, an organometallic reagent addition to the ester functional group occurred and formed a common intermediate 7. The intermediate 7 with a phosphate group subsequently underwent trans-phosphorylation, with the resultant elimination forming an unsaturated ketone 4a, which was obtained in a low isolated yield of 35% when the crude reaction mixture was quenched at −78 °C. The resulting ketone 4a reacted with an organolithium reagent to form the corresponding tertiary alcohol, which was then dehydrated by phosphoric acid to give the diene 3a. This hypothesis was confirmed by the reaction of the ketone 4a with phenyllithium, with the subsequent dehydration giving the diene 3a in a 93% isolated yield. We also verified that ethyl acetoacetate did not provide the ketone 4a or the diene 3a under the studied conditions. In the case of the intermediate 7 with phosphordiamidate moiety, the trans-phosphorylation reaction is sluggish and the intermediate 7 was converted into the tertiary alcohol 8. The formation of this intermediate was confirmed by means of the hydrolysis of the crude reaction mixture with water, and the corresponding alcohol 8 was obtained in a 79% isolated yield. The formation of the ketone 4a was completed by the alcohol 8 dehydration. This was again confirmed by the dehydration of the independently prepared alcohol 8 into the product 4a in a quantitative isolated yield. Alternatively, the dehydration of the alcohol 8 could be performed using phosphoryl chloride in the presence of triethylamine in a similar isolated yield (Scheme 5).
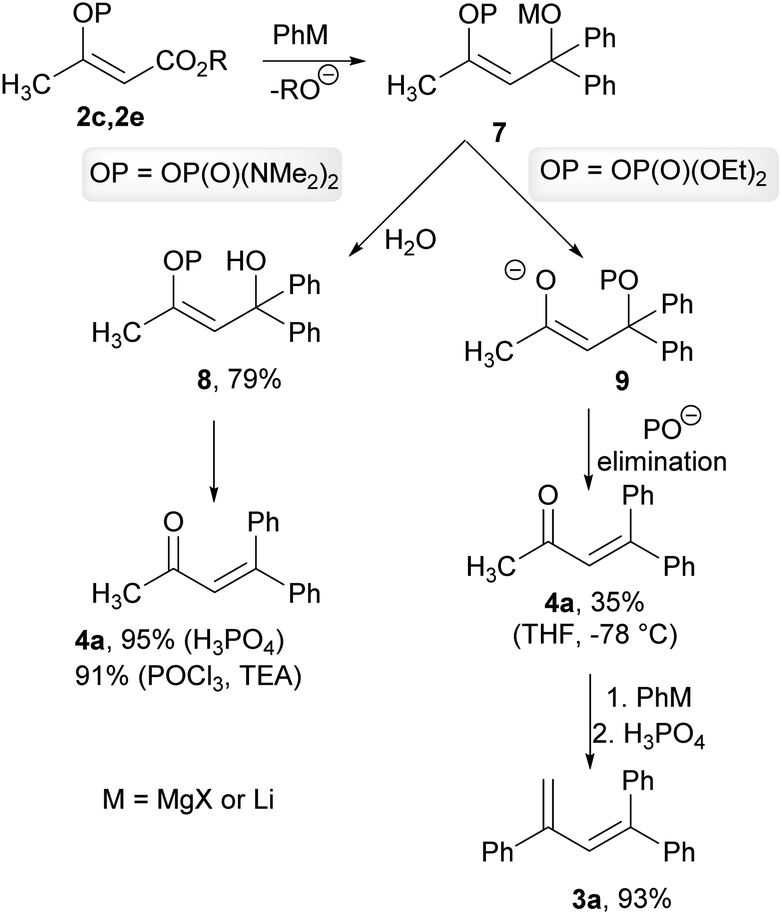 | ||
| Scheme 5 Proposed mechanism for the reaction of the vinyl phosphate 2c and the vinyl phosphordiamidate 2e with organometallic reagents. | ||
Conclusion
In conclusion, we studied the reactivity of both vinyl phosphates and vinyl phosphordiamidates. We found that vinyl phosphates can be converted into tri- and tetrasubstituted buta-1,3-dienes via the reaction with aryllithium reagents in dry THF at −78 °C to 23 °C. The tetrasubstituted dienes were isolated as a mixture of the (E)- and (Z)-stereoisomers. In the case of the vinyl phosphordiamidates, the reaction with Grignard reagents under the same reaction conditions resulted in the formation of unsaturated ketones, which were isolated in yields ranging from 24% to 97%. Based on the experimental results, we were able to propose a mechanism explaining the origins of both products.Experimental section
Materials and methods
All reactions were performed under argon atmosphere. NMR spectra were measured on Varian MercuryPlus 300 (1H, 300.13 MHz; 13C, 75.46 MHz), Agilent 400 MR DD2 (1H, 400.13 MHz; 13C, 100.61 MHz) or Bruker Avance III 500 (31P, 202.45 MHz) spectrometer at 298 K. Chemical shifts of 31P NMR spectra are referenced to the signal of 85% H3PO4 that was assigned the chemical shift of 0. Mass spectra were measured on ZAB-SEQ (VG Analytical). The dry and degassed THF was prepared by PureSolv MD7. Silica gel (Merck, Silica Gel 60, 40–63 μm or Merck Silica Gel 60, 63–200 μm) was used for column chromatography. A phosphate 2a was prepared according to a published procedure.30 n-BuLi (2.5 M solution in hexane), and other compounds were purchased from Sigma-Aldrich, FLuorochem and Acros Organics. Concentration of BuLi was determined by titration using menthol and 1,10-phenanthroline before use.Ethyl (Z)-3-((diphenoxyphosphoryl)oxy)but-2-enoate (2b). Prepared according to the GP1 from ethyl acetoacetate (0.260 g, 2.0 mmol), NaH (0.060 g, 2.50 mmol) and difenyl chlorophosphate (0.67 g, 2.5 mmol). Column chromatography (hexane/AcOEt 3
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, Rf = 0.45) gave 0.67 g (93%) of the title compound as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 7.39–7.16 (m, 10H), 5.43 (s, 1H), 4.11 (q, J = 7.1 Hz, 2H), 2.18–2.14 (m, 3H), 1.25 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 163.2 (d, J = 1.7 Hz), 156.6 (d, J = 7.2 Hz), 150.4 (d, J = 7.6 Hz), 129.7, 125.5 (d, J = 1.2 Hz), 120.1 (d, J = 4.9 Hz), 106.7 (d, J = 8.6 Hz), 60.0, 21.5 (d, J = 1.2 Hz), 14.0; HRMS (ESI) m/z: calcd for C18H20O6P [M + H]+ 363.0992; found 363.0992.
:1, Rf = 0.45) gave 0.67 g (93%) of the title compound as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 7.39–7.16 (m, 10H), 5.43 (s, 1H), 4.11 (q, J = 7.1 Hz, 2H), 2.18–2.14 (m, 3H), 1.25 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 163.2 (d, J = 1.7 Hz), 156.6 (d, J = 7.2 Hz), 150.4 (d, J = 7.6 Hz), 129.7, 125.5 (d, J = 1.2 Hz), 120.1 (d, J = 4.9 Hz), 106.7 (d, J = 8.6 Hz), 60.0, 21.5 (d, J = 1.2 Hz), 14.0; HRMS (ESI) m/z: calcd for C18H20O6P [M + H]+ 363.0992; found 363.0992.
Phenyl (Z)-3-((diethoxyphosphoryl)oxy)but-2-enoate (2c). Prepared according to the GP1 from phenyl acetoacetate (1.780 g, 10.0 mmol), NaH (0.480 g, 12.0 mmol) and diethyl chlorophosphate (2.070 g, 12.0 mmol). Column chromatography (hexane/AcOEt 2
:1, Rf = 0.21) gave 2.61 g (83%) of the title compound as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 7.41–7.38 (m, 2H), 7.26–7.23 (m, 1H), 7.14–7.12 (m, 2H), 5.57 (s, 1H), 4.31–4.25 (m, 4H), 2.30 (s, 3H), 1.38–1.35 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 161.7 (d, J = 1.7 Hz), 160.2 (d, J = 6.3 Hz), 150.4, 129.2, 125.5, 121.6, 104.3 (d, J = 8.5 Hz), 64.9 (d, J = 6.5 Hz), 21.7 (d, J = 1.2 Hz), 15.9 (d, J = 7.1 Hz); 31P NMR (202 MHz CDCl3) δ −8.38 (s); HRMS (ESI) m/z: calcd for C14H19O6P [M + Na]+ 337.0812; found 337.0815.
Phenyl (Z)-3-[(diethoxyphosphoryl)oxy]hept-2-enoate (2d). Prepared according to the GP1 from phenyl 3-oxoheptanoate (3.524 g, 16.0 mmol), NaH (0.768 g, 19.20 mmol) and diethyl chlorophosphate (2.899 g, 16.80 mmol). Column chromatography (hexane/AcOEt 3
:1, Rf = 0.26) gave 4.048 g (71%) of the title compound as a yellowish oil. 1H NMR (300 MHz, CDCl3): δ 7.38–7.35 (m, 2H), 7.23–7.19 (m, 1H), 7.12–7.09 (m, 2H), 5.57 (s, 1H), 4.29–4.17 (m, 4H), 2.54–2.50 (m, 2H), 1.64–1.58 (m, 2H), 1.44–1.38 (m, 2H), 1.32 (td, J = 7.1, 1.2 Hz, 6H), 0.95 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 164.1 (d, J = 6.9 Hz), 162.0 (d, J = 1.9 Hz), 150.4, 129.3, 125.6, 121.7, 104.1 (d, J = 7.8 Hz), 64.9 (d, J = 6.4 Hz), 35.0 (d, J = 1.0 Hz), 28.4, 22.0, 16.0 (d, J = 7.3 Hz), 13.7; HRMS (APCI) m/z: calcd for C17H25O6P [M + H]+ 357.1462; found 357.1463.
Ethyl (Z)-3-[(bis(N,N-dimethylamino)phosphoryl)oxy]but-2-enoate (2e). Prepared according to the GP2 from ethyl acetoacetate (13.01 g, 100.0 mmol), NaH (4.42 g, 110.0 mmol) and bis(N,N-dimethylamino)phosphoryl chloride (18.83 g, 110.0 mmol). Column chromatography (AcOEt, Rf = 0.33) gave 12.74 g (54%) of the title compound as a yellowish oil. 1H NMR (500 MHz, CDCl3) δ 5.16 (s, 1H), 4.07 (q, J = 7.1 Hz, 2H), 2.69 (d, J = 10.2 Hz, 12H), 2.11 (s, 3H), 1.20 (dd, J = 7.8, 6.5 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 164.0, 159.7 (d, J = 5.8 Hz), 103.6 (d, J = 7.5 Hz), 59.4, 36.4, 22.1 (d, J = 2.1 Hz), 14.3; 31P NMR (202 MHz, CDCl3) δ 15.02 (s); HRMS (ESI) m/z: calcd for C10H21N2O4P [M + Na]+ 287.1131; found 287.1135.
Ethyl (Z)-3-[(bis(N,N-dimethylamino)phosphoryl)oxy]hex-2-enoate (2f). Prepared according to the GP2 from ethyl 3-oxohexanoate (4.746 g, 30.0 mmol), NaH (1.441 g, 36.0 mmol) and bis(N,N-dimethylamino)phosphoryl chloride (5.373 g, 31.50 mmol). Column chromatography (AcOEt, Rf = 0.32) gave 4.648 g (53%) of the title compound as a yellowish oil. 1H NMR (500 MHz, CDCl3) δ 5.24–5.22 (m, 1H), 4.12–4.07 (m, 2H), 2.72 (d, J = 10.2 Hz, 12H), 2.44–2.38 (m, 2H), 1.61–1.54 (m, 2H), 1.26–1.21 (m, 3H), 0.97–0.92 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 164.1, 163.5 (d. J = 6.2 Hz), 103.1 (d, J = 7.2 Hz), 59.5, 37.3, 36.6 (d, J = 4.3 Hz), 20.0, 14.3, 13.5 (d, J = 1.4 Hz); HRMS (APCI) m/z: calcd for C12H25N2O4P [M + H]+ 293.1625; found 293.1628.
Ethyl (Z)-3-((bis(N,N-dimethylamino)phosphoryl)oxy)-3-phenylacrylate (2g). Prepared according to the GP2 from ethyl benzoylacetate (3.840 g, 20.0 mmol), NaH (0.960 g, 24.0 mmol) and bis(N,N-dimethylamino)phosphoryl chloride (4.120 g, 24.0 mmol). Column chromatography (AcOEt, Rf = 0.38) gave 3.86 g (55%) of the title compound as a yellowish oil. 1H NMR (500 MHz, CDCl3) δ 7.64–7.52 (m, 2H), 7.43–7.42 (m, 3H), 5.81 (s, 1H), 4.21 (q, J = 7.1 Hz, 2H), 2.61 (d, J = 10.1 Hz, 12H), 1.34 (t, J = 7.1 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 164.3 (d, J = 2.1 Hz), 158.6 (d, J = 6.7 Hz), 136.0 (d, J = 1.8 Hz), 130.2, 128.3, 127.4, 107.1 (d, J = 6.3 Hz), 60.1, 36.6 (d, J = 4.6 Hz), 14.3; 31P NMR (202 MHz, CDCl3) δ 15.23 (s). HRMS (ESI) m/z: calcd for C15H23N2O4P [M + Na]+ 349.1288; found 349.1294.
Ethyl 2-((bis(N,N-dimethylamino)phosphoryl)oxy)cyclohex-1-ene-1-carboxylate (2h). Prepared according to the GP2 from ethyl 2-oxocyclohexane-1-carboxylate (3.404 g, 20 mmol), NaH (0.960 g, 24 mmol) and bis(N,N-dimethylamino)phosphoryl chloride (3.582 g; 21.0 mmol). Column chromatography (AcOEt, Rf = 0.21) gave 4.578 g (75%) of the title compound as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 4.16 (qd, J = 7.2, 1.2 Hz, 2H), 2.68 (d, J = 10.1, 12H), 2.48–2.44 (m, 2H), 2.34–2.31 (m, 2H), 1.71–1.56 (m, 4H), 1.28–1.24 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 153.6 (d, J = 6.6 Hz), 113.9 (d, J = 7.4 Hz), 111.6 (d, J = 4.7 Hz), 60.0, 36.5 (d, J = 4.3 Hz), 29.3 (d, J = 1.7 Hz), 25.6, 22.3, 21.6, 14.2; 31P NMR (202 MHz, CDCl3): δ 14.88; HRMS (APCI) m/z: calcd for C13H25N2O4P [M + H]+ 305.1625; found 305.1628.
Methyl 2-[(bis(N,N-dimethylamino)phosphoryl)oxy]cyclopent-1-ene-1-carboxylate (2i). Prepared according to the GP2 from ethyl 2-oxocyclopentane-1-carboxylate (1.424 g, 10.0 mmol), NaH (0.480 g, 12.0 mmol) and bis(N,N-dimethylamino)phosphoryl chloride (1.791 g; 10.50 mmol). Column chromatography (AcOEt, Rf = 0.28) gave 2.127 g (77%) of the title compound as a yellowish oil. 1H NMR (400 MHz, CDCl3) δ 3.69 (s, 3H), 2.72–2.70 (m, 14H), 2.57–2.57 (m, 2H), 1.93–1.85 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 164.6, 159.6 (d, J = 5.3 Hz), 112.2 (d, J = 7.6 Hz), 50.9, 36.4 (d, J = 4.4 Hz), 33.5 (d, J = 1.9 Hz), 28.7, 19.3; 31P NMR (202 MHz, CDCl3): δ 15.63; HRMS (APCI) m/z: calcd for C11H21N2O4P [M + H]+ = 277.1312; found 277.1315.
Methyl 2-[(bis(N,N-dimethylamino)phosphoryl)oxy]cyclohept-1-ene-1-carboxylate (2j). Prepared according to the GP2 from ethyl 2-oxocycloheptane-1-carboxylate (2.558 g, 15.0 mmol), NaH (0.721 g, 18.0 mmol) and bis(N,N-dimethylamino)phosphoryl chloride (2.729 g, 16.0 mmol). Column chromatography (AcOEt, Rf = 0.25) gave 3.142 g (69%) of the title compound as a colorless liquid. 1H NMR (300 MHz, CDCl3) δ 3.55 (s, 3H), 2.58–2.43 (m, 14H), 2.26–2.26 (m, 2H), 1.59–1.37 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 167.6 (d, J = 1.7 Hz), 158.2 (d, J = 7.3 Hz), 118.7 (d, J = 7.4 Hz), 51.1, 36.1 (d, J = 4.2 Hz), 33.6 (d, J = 1.6 Hz), 31.2, 27.4, 25.8 (d, J = 1.4 Hz), 23.9 (d, J = 0.6 Hz); 31P NMR (202 MHz, CDCl3): δ 14.67; HRMS (APCI) m/z: calcd for C13H25N2O4P [M + H]+ 305.1625; found 305.1626.
Ethyl (Z)-3-[(bis(N,N-dimethylamino)phosphoryl)oxy]-2-methylbut-2-enoate (2k). Prepared according to the GP2 from ethyl 2-methylacetoacetate (2.88 g, 20 mmol), NaH (0.960 g, 24.0 mmol) and bis(N,N-dimethylamino)phosphoryl chloride (4.120 g, 24.0 mmol). Column chromatography (AcOEt, Rf = 0.33) gave 2.60 g (50%) of the title compound as a yellowish oil. 1H NMR (500 MHz, CDCl3) δ 4.17 (q, J = 7.1 Hz, 2H), 2.67 (d, J = 10.1 Hz, 12H), 2.11 (s, 3H), 1.83 (s, 3H), 1.27 (t, J = 7.1 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 167.3, 150.0 (d, J = 6.9 Hz), 112.5 (d, J = 7.8 Hz), 60.2, 36.5 (d, J = 3.8 Hz), 18.2 (d, J = 1.6 Hz), 14.7, 14.2; 31P NMR (202 MHz, CDCl3) δ 15.08 (s); HRMS (ESI) m/z: calcd for C11H23N2O4P [M + Na]+ 301.1288; found 301.1289.
1,1,3-Triphenylbuta-1,3-diene (3a). Prepared according to the GP3 from bromobenzene (0.320 g, 2.05 mmol), nBuLi (0.83 mL, 2.0 mmol, 2.4 M) and enol phosphate 2c (0.160 g, 0.5 mmol). Column chromatography (hexane/toluene 99
:1, Rf = 0.47) gave 0.092 g (65%) of the title compound as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.40–7.15 (m, 15H), 6.74 (d, J = 1.2 Hz, 1H), 5.39 (d, J = 1.4 Hz, 1H), 5.03 (t, J = 1.4 Hz, 1H), in accordance with literature.22
1,1,3-Tris(4-methylphenyl)buta-1,3-diene (3b). Prepared according to the GP3 from 4-bromotoluene (0.350 g, 2.05 mmol), nBuLi (0.83 mL, 2.0 mmol, 2.4 M) and enol phosphate 2c (0.160 g, 0.5 mmol). Column chromatography (hexane, Rf = 0.34) gave 0.089 g (55%) of the title compound as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.37–7.37 (m, 2H), 7.26–7.24 (m, 2H), 7.15–7.08 (m, 6H), 7.05–7.03 (m, 2H), 6.67–6.66 (m, 1H), 5.38–5.37 (m, 1H), 4.97–4.96 (m, 1H), 2.38 (s, 3H), 2.35 (s, 3H), 2.33 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 145.0, 144.4, 140.6, 138.1, 137.4, 137.3, 137.2, 136.6, 130.0, 128.9, 128.8, 128.7, 127.9, 127.5, 126.5, 115.9, 21.3, 21.2, 21.1; HRMS (APCI) m/z: calcd for C25H24 [M + H]+ 325.1951 found 325.1945.
1,1,3-Tris(4-methoxyphenyl)buta-1,3-diene (3c). Prepared according to the GP3 from 4-bromoanisole (0.38 g, 2.05 mmol), nBuLi (0.83 mL, 2.0 mmol, 2.4 M) and enol phosphate 2c (0.160 g, 0.5 mmol). Column chromatography (hexane/AcOEt 20
:1, Rf = 0.23) gave 0.104 g (56%) of the title compound as a yellowish oil. 1H NMR (400 MHz, CDCl3) δ 7.36–7.33 (m, 2H), 7.29–7.26 (m, 2H), 7.09–7.06 (m, 2H), 6.87–6.84 (m, 2H), 6.80–6.72 (m, 4H), 6.59–6.57 (m, 1H), 5.31–5.31 (m, 1H), 4.95–4.93 (m, 1H), 3.83 (s, 3H), 3.79 (s, 3H), 3.77 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 159.2, 159.1, 158.6, 144.9, 143.6, 136.1, 133.5, 132.7, 131.3, 129.2, 127.8, 126.6, 115.1, 113.5, 113.4, 113.3, 55.3, 55.3, 55.2. HRMS (APCI) m/z: calcd for C25H24O3 [M + H]+ 373.1798; found 373.1793.
1,1,3-Tris[(1,1′-biphenyl)-4-yl]buta-1,3-diene (3d). Prepared according to the GP3 from 4-bromobiphenyl (0.48 g, 2.05 mmol), nBuLi (0.83 mL, 2.0 mmol, 2.4 M) and enol phosphate 2c (0.160 g, 0.5 mmol). Column chromatography (hexane/AcOEt 20
:1, Rf = 0.57) gave 0.092 g (64%) of the title compound as a white solid, mp = 64.0–66.0 °C. 1H NMR (400 MHz, CDCl3) δ 7.65–7.25 (m, 17H), 6.89 (s 1H), 5.51 (s, 1H), 5.20 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 145.1, 143.9, 142.0, 140.82, 140.80, 140.7, 140.5, 140.3, 139.9, 139.6, 138.9, 130.7, 128.8, 128.71, 128.69, 128.53, 128.48, 127.4, 127.29, 127.22, 127.20, 127.1, 127.04, 127.02, 126.95, 126.8, 126.6, 118.0; HRMS (APCI) m/z: calcd for C40H30 [M + H]+ 511.2420; found 511.2397.
1,1,3-Tris(2-methoxyphenyl)buta-1,3-diene (3e). Prepared according to the GP3 from 2-bromoanisole (0.380 g, 2.05 mmol), nBuLi (0.83 mL, 2.0 mmol, 2.4 M) and enol phosphate 2c (0.160 g, 0.5 mmol). Column chromatography (hexane/AcOEt 20
:1, Rf = 0.22) gave 0.132 g (71%) of the title compound as a yellowish oil. 1H NMR (400 MHz, CDCl3) δ 7.30–7.19 (m, 2H), 7.12–7.10 (m, 1H), 7.03–6.87 (m, 4H), 6.84–6.81 (m, 2H), 6.76–6.72 (m, 1H), 6.70–6.66 (m, 1H), 6.51–6.45 (m, 2H), 5.36–5.35 (m, 1H), 5.21–5.20 (m, 1H), 3.71 (s, 3H), 3.61 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 157.3, 156.3, 156.0, 144.8, 136.0, 133.8, 132.2, 130.8, 130.4, 130.4, 130.1, 129.8, 128.1, 128.0, 127.7, 120.4, 120.3, 119.5, 119.1, 111.8, 109.5, 109.4, 55.8, 55.0, 54.9; HRMS (APCI) m/z: calcd for C25H24O3 [M + H]+ 373.1798; found 373.1789.
(E/Z)-1,1,3-Triphenylhepta-1,3-diene (3f). Prepared according to the GP3 from bromobenzene (0.320 g, 2.05 mmol), nBuLi (0.83 mL, 2.0 mmol, 2.4 M) and enol phosphate 2d (0.178 g, 0.5 mmol). Column chromatography (hexane, Rf = 0.35) gave 0.101 g (62%, E/Z = 79
:21) of the title compound as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 7.37–7.10 (m, 15H, E and Z), 6.69–6.68 (m, 1H, E and Z), 5.77–5.73 (m, 1H, Emajor), 5.64–5.61 (m, 1H, Zminor), 1.99–1.95 (m, 2H, E and Z), 1.24–1.15 (m, 2H, E and Z), 0.84 (t, J = 6.0 Hz, 3H, Emajor), 0.72 (t, J = 6.0 Hz, 3H, Zminor); 13C NMR (100 MHz, CDCl3) δ 145.1, 143.6minor, 143.3major, 141.8major, 141.6minor, 140.4minor, 140.3major, 139.7minor, 138.2minor, 137.3major, 135.6major, 131.6major, 131.3minor, 130.4, 129.9, 129.2, 128.2, 128.1, 128.0, 127.9, 127.7, 127.6, 127.5, 127.1, 127.0, 126.7, 126.50, 126.47, 126.4, 126.3, 31.9major, 31.2minor, 22.7minor, 22.2major, 14.0major, 13.7minor; HRMS (APCI) m/z: calcd for C25H24 [M + H]+ 325.1951; found 325.1953.
1,1,3-Tris(4-methylphenyl)hepta-1,3-diene (3g). Prepared according to the GP3 from 4-bromotoluene (0.212 g, 1.23 mmol), nBuLi (0.49 mL, 2.40 M, 1.18 mmol), and enol phosphate 2d (0.107 g, 0.30 mmol). Column chromatography (hexane, Rf = 0.28) gave the title compound 0.056 g (51%, E/Z = 83
:17) as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 7.31–7.24 (m, 3H), 7.14–6.95 (m, 9H), 6.56–6.55 (m, 1H, Zminor), 6.53–6.52 (s, 1H, Emajor), 5.68 (t, J = 6.0 Hz, 1H, Emajor), 5.53 (t, J = 6.0 Hz, 1H, Zminor), 2.37 (s, 3H, Emajor), 2.32 (s, 3H, Zminor), 2.30 (s, 3H, Emajor), 2.29 (s, 3H, Zminor), 2.27 (s, 3H, Emajor), 1.96–1.84 (m, 2H, E and Z), 1.19–1.09 (m, 2H, E and Z), 0.80 (t, J = 6.0 Hz, 3H, Emajor), 0.69 (t, J = 6.0 Hz, 3H, Zminor); 13C NMR (75 MHz, CDCl3, only major peaks are reported) δ 144.7, 140.7, 138.9, 137.5, 137.2, 136.9, 136.5, 136.1, 130.4, 130.2, 129.7, 129.6, 129.0, 128.9, 128.8, 128.7, 128.7, 128.3, 128.1, 127.5, 126.4, 125.4, 31.9, 22.2, 21.2, 21.1, 21.0, 14.0; HRMS (APCI) m/z: calcd for C28H30 [M + H]+ 367.2420; found 367.2419.
1,1,3-Tris(4-methoxyphenyl)hepta-1,3-diene (3h). Prepared according to the GP3 from 4-bromoanisole (0.230 g, 1.23 mmol) a nBuLi (0.49 mL, 2.40 M, 1.17 mmol) and enol phosphate 2d (0.107 g, 0.30 mmol). Column chromatography (hexane/EtOAc 20
:1, Rf = 0.35) gave 0.063 g (51%, E/Z = 82:18) of the title compound as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 7.31–7.26 (m, 4H), 7.04–6.65 (m, 8H), 6.54–6.53 (m, 1H, Zminor), 6.48–6.47 (m, 1H, Emajor), 5.63 (t, J = 6.0 Hz, 1H, Emajor), 5.58 (t, J = 6.0 Hz, 1H, Zminor), 3.83 (s, 3H, Emajor), 3.79 (s, 3H, Zminor), 3.77 (s, 3H, Emajor), 3.76 (s, 3H, Zminor), 3.75 (s, 3H, Emajor), 1.98–1.89 (m, 2H, E and Z), 1.26–1.15 (m, 2H, E and Z), 0.83 (t, J = 6.0 Hz, 3H, Emajor), 0.74 (t, J = 6.0 Hz, 3H, Zminor); 13C NMR (100 MHz, CDCl3) δ 159.1, 158.5, 158.4, 143.9, 136.8, 136.2, 134.6, 133.0, 131.0, 129.3, 129.2, 127.6, 124.4, 113.4, 113.3, 113.1, 113.0, 55.3, 55.2, 55.1, 31.8major, 31.3minor, 22.9minor, 22.3major, 14.0major, 13.8minor; HRMS (APCI) m/z: calcd for C28H30O3 [M + H]+ 415.2268; found 415.2264.
1,1,3-Tris[(1,1′-biphenyl)-4-yl]hepta-1,3-diene (3i). Prepared according to the GP3 from 4-bromobiphenyl (0.287 g, 1.23 mmol), nBuLi (0.49 mL, 2.40 M, 1.17 mmol) and enol phosphate 2d (0.107 g, 0.30 mmol). Column chromatography (hexane → hexane/EtOAc 30
:1, Rf = 0.1) gave 0.119 g (72%, E/Z = 86:14) of the title compound as a yellowish solid, mp = 49.0–50.0 °C. 1H NMR (300 MHz, CDCl3) δ 7.65–7.06 (m, 27H, E and Z), 6.85 (s, 1H, Zminor), 6.82 (s, 1H, Emajor), 5.86–5.78 (m, 1H, E and Z), 2.10–2.01 (m, 2H, E and Z), 1.30–1.23 (m, 2H, E and Z), 0.88 (t, J = 7.3 Hz, 3H, Emajor), 0.76 (s, J = 7.3 Hz, 3H, Zminor); 13C NMR (75 MHz, CDCl3) δ 144.3, 142.4, 142.0, 141.0, 140.9, 140.8, 140.8, 140.7, 140.4, 139.8, 139.3, 139.3, 139.2, 139.0, 138.5, 138.4, 137.0, 136.6, 132.0, 131.6, 130.8, 130.4, 129.8, 128.8, 128.7, 128.6, 128.6, 128.0, 127.3, 127.2, 127.1, 127.0, 126.9, 126.9, 126.8, 126.7, 126.4, 126.2, 32.0major, 31.4minor, 22.8minor, 22.3major, 14.1major, 13.8minor; HRMS (APCI) m/z: calcd for C43H36 [M + H]+ 553.2890; found 553.2894.
1,1,3-Tris(2-methoxyphenyl)hepta-1,3-diene (3j). Prepared according to the GP3 from 2-bromoanisole (0.230 g, 1.23 mmol), nBuLi (0.49 mL, 2.40 M, 1.17 mmol) and enol phosphate 2d (0.107 g, 0.30 mmol). Column chromatography (hexane/EtOAc 50
:1, Rf = 0.14) gave 0.090 g (72%, E/Z = 71:29) of the title compound as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 7.27–7.12 (m, 3H, E and Z), 6.96–6.36 (m, 10H, E and Z), 5.80 (t, J = 6.0 Hz, 1H, Zminor), 5.49 (t, J = 6.0 Hz, 1H), 3.71 (s, 3H, Emajor), 3.68 (s, 3H, Zminor), 3.62 (s, 3H, Zminor), 3.60 (s, 3H, Emajor), 3.59 (s, 3H, Zminor), 3.44 (s, 3H, Emajor), 2.24 (q, J = 7.4 Hz, 2H, Emajor), 1.78 (q, J = 7.4 Hz, 2H, Zminor), 1.46–1.40 (m, 2H, Emajor), 1.25–1.22 (m, 2H, Zminor), 0.95 (t, J = 7.3 Hz, 3H, Emajor), 0.95 (t, J = 7.3 Hz, 3H, Zminor); 13C NMR (100 MHz, CDCl3) δ 157.4, 156.3, 156.1, 136.4, 136.1, 134.8, 134.0, 133.9, 132.3, 130.9, 130.8, 130.3, 129.7, 127.7, 127.4, 127.0, 120.3, 119.5, 119.0, 112.1, 109.6, 109.4, 55.9, 55.1, 54.7, 31.3minor, 31.1major, 22.7major, 22.6minor, 14.0major, 13.9minor; HRMS (APCI) m/z: calcd for C28H30O3 [M + H]+ 415.2268; found 415.2277.
1,1,3-Tris(2-thienyl)hepta-1,3-diene (3k). Prepared according to the GP3 from thiophene (0.108 g, 1.28 mmol), nBuLi (0.51 mL, 2.40 M, 1.22 mmol), dry THF (3 mL). The reaction mixture was stirred for 30 min at 0 °C followed by addition of enol phosphate 2d (0.110 g, 0.31 mmol). Column chromatography (hexane, Rf = 0.14) gave 0.048 g (45%, E/Z = 93
:7) of the title compound as a greenish oil. 1H NMR (300 MHz, CDCl3) δ 7.28–7.26 (m, 1H), 7.23–7.21 (m, 1H), 7.13–7.12 (m, 1H), 7.09–7.02 (m, 3H), 6.97–6.87 (m, 3H), 6.69 (d, J = 1.2 Hz, 1H, Emajor), 5.92 (td, J = 7.4, 1.3 Hz, 1H, Emajor), 5.92 (td, J = 7.4, 1.3 Hz, 1H, Zminor), 2.24–2.18 (m, 2H, Zminor), 2.07–2.01 (m, 2H, Emajor), 1.33–1.23 (m, 2H), 0.87 (t, J = 7.4 Hz, 3H, Emajor), 0.79 (t, J = 7.4 Hz, 3H, Zminor); 13C NMR (100 MHz, CDCl3) δ 146.9minor, 145.9, 145.3, 141.2minor, 140.2minor, 140.1, 136.2, 131.3, 130.6, 128.5, 127.3, 127.1, 126.5, 126.4, 126.2, 125.1, 125.1, 123.7, 123.3, 31.7major, 31.6minor, 22.6minor, 22.2major, 14.0major, 13.8minor; HRMS (APCI) m/z: calcd for C19H18S3 [M + H]+ 343.0643; found 343.0649.
4,4-Diphenylbut-3-en-2-one (4a). Prepared according to the GP4 from phosphordiamidate 2e (0.130 g, 0.5 mmol) and phenylmagnesium chloride (0.70 mL, 1.3 mmol). Column chromatography (hexane/AcOEt 9
:1, Rf = 0.42) gave 0.087 g (78%) of the title compound as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 7.43–7.39 (m, 3H), 7.36–7.28 (m, 5H), 7.25–7.20 (m, 2H), 6.58 (s, 1H), 1.88 (s, 3H), in accordance with literature.31
4,4-Bis(4-chlorophenyl)but-3-en-2-one (4b). Prepared according to the GP4 from phosphordiamidate 2e (0.130 g, 0.5 mmol) and 4-chlorophenylmagnesium chloride (1.30 mL, 1.3 mmol). Column chromatography (hexane/AcOEt 9
:1, Rf = 0.24) gave 0.116 g (80%) of the title compound as a yellowish oil. 1H NMR (400 MHz, CDCl3) δ 7.40–7.38 (m, 2H), 7.32–7.30 (m, 2H), 7.21–7.19 (m, 2H), 7.15–7.12 (m, 2H), 6.56 (s, 1H), 1.99 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 199.3, 151.5, 139.1, 137.0, 136.0, 135.2, 131.1, 129.8, 129.0, 128.9, 127.7, 31.0; HRMS (APCI) m/z: calcd for C16H12Cl2O1 [M + H]+ 291.0338; found 291.0329.
4,4-Bis(4-fluorophenyl)but-3-en-2-one (4c). Prepared according to the GP4 from phosphordiamidate 2e (0.130 g, 0.5 mmol) and 4-fluorophenylmagnesium chloride (1.30 mL, 1.3 mmol). Column chromatography (hexane/AcOEt 9
:1, Rf = 0.33) gave 0.105 g (81%) of the title compound as a yellowish oil. 1H NMR (400 MHz, CDCl3) δ 7.28–7.24 (m, 2H), 7.20–7.17 (m, 2H), 7.13–7.09 (m, 2H), 7.05–7.01 (m, 2H), 6.53 (s, 1H), 1.96 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 199.5, 163.6 (d, J = 251.5 Hz), 163.1 (d, J = 250.5 Hz), 151.8, 136.8 (d, J = 3.1 Hz), 134.6 (d, J = 3.5 Hz), 131.5 (d, J = 8.1 Hz), 130.4 (d, J = 9.1 Hz), 127.2 (d, J = 6.1 Hz), 115.7 (d, J = 5.1 Hz), 115.5 (d, J = 6.1 Hz), 30.6; HRMS (APCI) m/z: calcd for C16H12F2O1 [M + H]+ 259.0929; found 259.0929.
4,4-Bis(3-methoxyphenyl)but-3-en-2-one (4d). Prepared according to the GP4 from phosphordiamidate 2e (0.130 g, 0.5 mmol) and 3-methoxyphenylmagnesium bromide (1.30 mL, 1.3 mmol). Column chromatography (hexane/AcOEt 6
:1, Rf = 0.32) gave 0.112 g (79%) of the title compound as a yellowish oil. 1H NMR (400 MHz, CDCl3) δ 7.34–7.30 (m, 1H), 7.26–7.22 (m, 1H), 6.97–6.80 (m, 5H), 6.75–6.74 (m, 1H), 6.56 (s, 1H), 3.80 (s, 3H), 3.78 (s, 3H), 1.89 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 200.4, 159.6, 159.5, 153.5, 142.0, 140.2, 129.5, 129.4, 128.0, 122.1, 121.0, 115.0, 114.9, 114.4, 114.0, 55.3, 30.2; HRMS (APCI) m/z: calcd for C18H18O3 [M + H]+ 283.1329; found 283.1324.
4,4-Bis(3-(methylthio)phenyl)but-3-en-2-one (4e). Prepared according to the GP4 from phosphordiamidate 2e (0.130 g, 0.5 mmol) and (3-(methylthio)phenyl)magnesium bromide (2.60 mL, 1.3 mmol). Column chromatography (hexane/AcOEt 6
:1, Rf = 0.34) gave 0.126 g (80%) of the title compound as a yellowish oil. 1H NMR (400 MHz, CDCl3) δ 7.35–7.24 (m, 3H), 7.19–7.18 (m, 1H), 7.07–7.06 (m, 1H), 7.03–6.96 (m, 3H), 6.55 (s, 1H), 2.47 (s, 3H), 2.45 (s, 3H), 1.92 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 200.1, 152.9, 141.1, 139.2, 139.2, 139.1, 128.9, 128.8, 128.2, 127.4, 127.1, 126.7, 126.2, 126.1, 125.3, 30.4, 15.7, 15.6; HRMS (APCI) m/z: calcd for C18H18OS2 [M + H]+ 315.0872; found 315.0879.
4,4-Di-o-tolylbut-3-en-2-one (4f). Prepared according to the GP4 from phosphordiamidate 2e (0.130 g, 0.5 mmol) and o-tolylmagnesium chloride (1.30 mL, 1.3 mmol). Column chromatography (hexane/AcOEt 6
:1, Rf = 0.49) gave 0.094 g (75%) of the title compound as a yellowish solid, mp = 62.0–65.0 °C. 1H NMR (400 MHz, CDCl3) δ 7.28–7.03 (m, 8H), 6.31 (s, 1H), 2.35 (s, 3H), 2.10 (s, 3H), 1.85 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 200.2, 153.8, 140.3, 139.4, 135.7, 135.6, 132.3, 131.3, 130.6, 130.1, 129.9, 128.7, 128.4, 125.8, 125.8, 29.9, 21.0, 19.9; HRMS (APCI) m/z: calcd for C18H18O [M + H]+ 251.1430; found 251.1432.
4-Octyldodec-3-en-2-one (4g). Prepared according to the GP4 from phosphordiamidate 2e (0.13 g, 0.5 mmol) and octylmagnesium chloride (0.6 mL, 1.3 mmol). Column chromatography (hexane/AcOEt 6
:1, Rf = 0.41) gave 0.124 g (84%) of the title compound as a yellowish oil. 1H NMR (400 MHz, CDCl3) δ 6.01 (s, 1H), 2.55–2.51 (m, 2H), 2.16 (s, 3H), 2.12–2.08 (m, 2H), 1.46–1.25 (m, 24H), 0.90–0.85 (m, 6H); 13C NMR (101 MHz, CDCl3) δ 198.4, 163.8, 122.9, 38.7, 32.4, 31.92, 31.88, 31.85, 30.0, 29.44, 29.42, 29.3, 29.2, 28.6, 27.8, 22.67, 22.66, 14.12, 14.11; HRMS (APCI) m/z: calcd for C20H38O [M + H]+ 295.2995; found 295.2984.
1,1-Diphenylhex-1-en-3-one (4h). Prepared according to the GP4 from phosphordiamidate 2f (0.149 g; 0.51 mmol) and phenylmagnesium chloride (0.65 mL, 1.28 mmol). Column chromatography (hexane/AcOEt 20
:1, Rf = 0.23) gave 0.101 g (79%) of the title compound as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 7.41–7.29 (m, 8H), 7.21–7.18 (m, 2H), 6.58 (s, 1H), 2.22 (t, J = 7.3 Hz, 2H), 1.59–1.46 (m, 2H), 0.79 (t, J = 7.4 Hz, 3H), in accordance with literature.32
1,1-Bis(4-chlorophenyl)hex-1-en-3-one (4i). Prepared according to the GP4 from phosphordiamidate 2f (0.146 g; 0.50 mmol) and 4-chlorophenylmagnesium chloride (1.30 mL, 1.25 mmol). Column chromatography (hexane/AcOEt 20
:1, Rf = 0.31) gave 0.121 g (76%) of the title compound as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 7.37–7.35 (m, 2H), 7.32–7.29 (m, 2H), 7.21–7.19 (m, 2H), 7.12–7.10 (m, 2H), 6.57 (s, 1H), 2.33 (t, J = 7.3 Hz, 2H), 1.61–1.51 (m, 2H), 0.85 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 201.2, 150.8, 139.1, 136.9, 135.6, 134.7, 130.8, 129.6, 128.7, 128.6, 126.4, 45.6, 17.6, 13.7; HRMS (APCI) m/z: calcd for C18H16Cl2O [M + H]+ 319.0651; found 319.0650.
1,1-Bis(4-fluorophenyl)hex-1-en-3-one (4j). Prepared according to the GP4 from phosphordiamidate 2f (0.146 g; 0.50 mmol) and 4-fluorophenylmagnesium bromide (1.30 mL, 1.25 mmol). Column chromatography (hexane/AcOEt 20
:1, Rf = 0.24) gave 0.110 g (77%) of the title compound as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 7.27–7.24 (m, 2H), 7.18–7.14 (m, 2H), 7.10–7.00 (m, 4H), 6.53 (s, 1H), 2.30 (t, J = 7.3 Hz, 2H), 1.60–1.51 (m, 2H), 0.84 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 201.5, 163.5 (d, J = 250.4 Hz), 162.9 (d, J = 248.6 Hz) 151.2, 137.1 (d, J = 3.5 Hz), 134.6 (d, J = 3.5 Hz), 131.3 (d, J = 8.3 Hz), 130.2 (d, J = 8.4 Hz), 126.1, 115.5 (d, J = 10.9 Hz), 115.3 (d, J = 10.8 Hz), 45.5, 17.6, 13.7; HRMS (APCI) m/z: calcd for C18H16F2O [M + H]+ 287.1242; found 287.1243.
1,1-Bis(3-methoxyphenyl)hex-1-en-3-one (4k). Prepared according to the GP4 from phosphordiamidate 2f (0.146 g; 0.50 mmol) and 3-methoxyphenylmagnesium bromide (1.30 mL, 1.25 mmol). Column chromatography (hexane/AcOEt 20
:1, Rf = 0.18) gave 0.129 g (83%) of the title compound as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 7.32–7.22 (m, 2H), 6.95–6.71 (m, 6H), 6.55 (s, 1H), 3.78 (s, 3H), 3.77 (s, 3H), 2.21 (t, J = 7.3 Hz, 2H), 1.56–1.47 (m, 2H), 0.79 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 202.6, 159.4, 159.4, 152.4, 142.2, 140.2, 129.3, 127.0, 121.9, 120.9, 114.9, 114.5, 114.1, 114.0, 55.3, 55.2, 44.9, 17.8, 13.7; HRMS (APCI) m/z: calcd for C20H22O3 [M + H]+ 311.1642; found 311.1645.
1,1-Bis(3-(methylthio)phenyl)hex-1-en-3-one (4l). Prepared according to the GP4 from phosphordiamidate 2f (0.146 g; 0.50 mmol) and (3-(methylthio)phenyl)magnesium bromide (1.40 mL, 1.25 mmol). Column chromatography (hexane/AcOEt 30
:1, Rf = 0.11) gave 0.122 g (71%) of the title compound as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 7.30–7.18 (m, 5H), 7.05–6.94 (m, 3H), 6.54 (s, 1H), 2.46 (s, 3H), 2.46 (s, 3H), 2.45 (s, 3H), 2.24 (t, J = 7.3 Hz, 2H), 1.57–1.48 (m, 2H), 0.81 (t, J = 7.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 202.2, 151.9, 141.4, 139.3, 138.9, 138.8, 128.8, 128.6, 127.2, 127.16, 127.13, 126.5, 126.2, 126.1, 125.2, 45.1, 17.7, 15.7, 15.7, 13.7; HRMS (APCI) m/z: calcd for C20H22OS2 [M + H]+ 343.1185; found 343.1188.
1,1-Di-o-tolylhex-1-en-3-one (4m). Prepared according to the GP4 from phosphordiamidate 2f (0.146 g; 0.50 mmol) and o-tolylmagnesium chloride (1.30 mL, 1.25 mmol). Column chromatography (hexane/AcOEt 30
:1, Rf = 0.28) gave 0.103 g (74%) of the title compound as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 7.26–7.06 (m, 8H), 6.33 (s, 1H), 2.33 (s, 3H), 2.19 (t, J = 7.3 Hz, 2H), 2.10 (s, 3H), 1.54–1.49 (m, 2H), 0.79 (t, J = 7.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 202.3, 152.7, 140.7, 139.5, 135.7, 135.6, 131.2, 131.1, 130.4, 129.8, 129.8, 128.3, 128.2, 125.7, 125.6, 44.8, 20.9, 19.9, 17.7, 13.7; HRMS (APCI) m/z: calcd for C20H22O [M + H]+ 279.1743; found 279.1746.
1,1-Di(thiophen-2-yl)hex-1-en-3-one (4n). Prepared according to the GP4 from phosphordiamidate 2f (0.146 g; 0.50 mmol) and 2-thienylmagnesium bromide (1.30 mL, 1.26 mmol). Column chromatography (hexane/AcOEt 20
:1, Rf = 0.38) gave 0.098 g (75%) of the title compound as a brownish oil. 1H NMR (300 MHz, CDCl3) δ 7.48–7.46 (m, 1H), 7.38–7.36 (m, 1H), 7.15–7.12 (m, 1H), 7.10–7.08 (m, 2H), 7.03–7.00 (m, 1H), 6.61 (d, J = 1.7 Hz, 1H), 2.28 (td, J = 7.3, 1.8 Hz, 2H), 1.62–1.49 (m, 2H), 0.83 (td, J = 7.4, 1.8 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 201.2, 145.0, 138.6, 138.2, 129.9, 129.6, 128.1, 127.9, 127.7, 126.9, 125.5, 44.9, 17.9, 13.7; HRMS (APCI) m/z: calcd for C14H14OS2 [M + H]+ 263.0559; found 263.0551.
2-Benzyl-1-phenylhept-2-en-4-one (4o). Prepared according to the GP4 from phosphordiamidate 2f (0.146 g; 0.50 mmol) and benzylmagnesium chloride (1.30 mL, 1.26 mmol). Column chromatography (hexane/AcOEt 50
:1, Rf = 0.28) gave 0.088 g (63%) of the title compound as a yellowish oil. 1H NMR (300 MHz, CDCl3) δ 7.33–7.22 (m, 8H), 7.12–7.09 (m, 2H), 6.09 (s, 1H), 3.94 (s, 2H), 3.33 (s, 2H), 2.46 (t, J = 7.3 Hz, 2H), 1.68–1.62 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 201.3, 157.6, 138.9, 137.8, 129.3, 129.1, 128.5, 128.4, 126.6, 126.2, 125.6 46.5, 43.5, 36.8, 17.5, 13.8; HRMS (APCI) m/z: calcd for C20H22O [M + H]+ 279.1743; found 279.1738.
6-Octyltetradec-5-en-4-one (4p). Prepared according to the GP4 from phosphordiamidate 2f (0.148 g; 0.51 mmol) and octylmagnesium chloride (0.63 mL, 1.26 mmol). Column chromatography (hexane/AcOEt 30
:1, Rf = 0.44) gave 0.147 g (90%) of the title compound as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 5.99 (s, 1H), 2.55–2.51 (m, 2H), 2.39 (t, J = 7.3 Hz, 2H), 2.12–2.08 (m, 2H), 1.64–1.58 (m, 2H), 1.44–1.25 (m, 24H), 0.94–0.86 (m, 9H); 13C NMR (75 MHz, CDCl3) δ 200.9, 163.3, 122.6, 46.4, 38.7, 32.5, 31.9, 31.8, 30.0, 29.5, 29.4, 29.4, 29.3, 29.2, 28.7, 27.8, 22.7, 22.6, 17.7, 14.1, 13.8; HRMS (APCI) m/z: calcd for C22H42O [M + H]+ 323.3308; found 323.3310.
1,3,3-Triphenylprop-2-en-1-one (4q). Prepared according to the GP4 from phosphordiamidate 2g (0.160 g; 0.50 mmol) and phenylmagnesium chloride (0.70 mL, 1.30 mmol). Column chromatography (hexane/AcOEt 9
:1, Rf = 0.32) gave 0.129 g (97%) of the title compound as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.92–7.90 (m, 2H), 7.48–7.46 (m, 1H), 7.41–7.36 (m, 7H), 7.28–7.26 (m, 3H), 7.19–7.12 (m, 2H), 7.12 (s, 1H), in accordance with literature.33
1-Phenyl-3,3-di-o-tolylprop-2-en-1-one (4r). Prepared according to the GP4 from phosphordiamidate 2g (0.160 g; 0.50 mmol) and 2-tolylmagnesium chloride (1.30 mL, 1.30 mmol). Column chromatography (hexane/AcOEt 9
:1, Rf = 0.35) gave 0.144 g (95%) of the title compound as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.90–7.88 (m, 2H), 7.50–7.46 (m, 1H), 7.40–7.36 (m, 2H), 7.25–7.06 (m, 8H), 6.98 (s, 1H), 2.36 (s, 3H), 2.09 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 192.0, 154.9, 141.1, 139.6, 138.3, 135.8, 135.7, 132.6, 131.2, 130.3, 129.9, 129.6, 128.5, 128.4, 128.3, 128.0, 128.0, 125.9, 125.5, 21.0, 20.1; HRMS (APCI) m/z: calcd for C23H20O [M + H]+ 313.1587; found 313.1591.
2-(Diphenylmethylene)cyclohexan-1-one (5a). Prepared according to the GP4 from bromobenzene (0.196 g, 1.25 mmol), nBuLi (0.50 mL, 1.20 mmol), phosphordiamidate 2h (0.152 g, 0.50 mmol). Column chromatography (hexane/AcOEt 20
:1, Rf = 0.17) gave 0.088 g, (67%) of the title compound as a white solid, mp = 130.0–131.0 °C. 1H NMR (400 MHz, CDCl3) δ 7.34–7.22 (m, 6H), 7.14–7.11 (m, 2H), 7.06–7.04 (m, 2H), 2.65–2.59 (m, 4H), 2.01–1.96 (m, 2H), 1.83–1.78 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 206.9, 144.2, 142.2, 140.8, 138.9, 129.9, 129.0, 128.0, 127.7, 127.3, 45.0, 34.1, 26.6, 26.3; HRMS (APCI) m/z: calcd for C19H18O [M + H]+ 263.1430; found 263.1434.
2-(Diphenylmethylene)cyclopentan-1-one (5b). Prepared according to the GP4 from bromobenzene (0.198 g, 1.26 mmol), nBuLi (0.50 mL, 1.20 mmol), phosphordiamidate 2i (0.138 g, 0.50 mmol). Column chromatography (hexane/AcOEt 20
:1, Rf = 0.08) gave 0.085 g (68%) of the title compound as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 7.34–7.31 (m, 6H), 7.21–7.17 (m, 2H), 7.14–7.10 (m,2H), 2.82 (t, J = 7.0 Hz, 2H), 2.38 (t, J = 7.7 Hz, 2H), 1.98–1.88 (m, 2H), in accordance with literature.34
2-(Bis(4-methoxyphenyl)methylene)cyclopentan-1-one (5c). Prepared according to the GP4 from 4-bromoanisole (0.237 g, 1.26 mmol), nBuLi (0.50 mL, 1.20 mmol), phosphordiamidate 2i (0.140 g, 0.51 mmol). Column chromatography (hexane/AcOEt 9
:1, Rf = 0.15) gave 0.115 g (74%) of the title compound as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.14–7.12 (m, 2H), 7.06–7.04 (m, 2H), 6.86–6.83 (m, 4H), 3.82 (s, 6H), 2.81 (t, J = 7.0 Hz, 2H), 2.37 (t, J = 7.7 Hz, 2H), 1.91 (p, J = 7.3 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 206.6, 159.7, 159.4, 148.2, 134.6, 132.6, 132.5, 131.6, 131.3, 113.2, 113.1, 55.3, 55.1, 40.0, 33.5, 20.7; HRMS (APCI) m/z: calcd for C20H20O3 [M + H]+ 309.1485; found 309.1489.
2-(Diphenylmethylene)cycloheptan-1-one (5d). Prepared according to the GP4 from bromobenzene (0.200 g, 1.28 mmol), nBuLi (0.51 mL, 1.22 mmol), phosphordiamidate 2j (0.160 g, 0.52 mmol). Column chromatography (hexane/AcOEt 20
:1, Rf = 0.15) gave 0.120 g (83%) of the title compound as a white solid, mp = 79.5–81.5 °C. 1H NMR (400 MHz, CDCl3) δ 7.37–7.19 (m, 8H), 7.11–7.08 (m, 2H), 2.50–2.47 (m, 2H), 2.39–2.36 (m, 2H), 1.92–1.88 (m, 2H), 1.72–1.61 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 211.2, 142.5, 141.8, 141.5, 140.9, 128.9, 128.6, 128.3, 128.1, 127.22, 127.20, 43.4, 30.3, 29.3, 29.2, 24.2; HRMS (APCI) m/z: calcd for C20H20O [M + H]+ 277.1587; found 277.1586.
2-(Bis(4-methoxyphenyl)methylene)cycloheptan-1-one (5e). Prepared according to the GP4 from 4-bromoanisole (0.240 g, 1.28 mmol), nBuLi (0.51 mL, 1.22 mmol), phosphordiamidate 2j (0.152 g, 0.50 mmol). Column chromatography (hexane/AcOEt 9
:1, Rf = 0.24) gave 0.152 g (90%) of the title compound as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.12–7.10 (m, 2H), 7.00–6.98 (m, 2H), 6.88–6.86 (m, 2H), 6.77–6.74 (m, 2H), 3.81 (s, 3H), 3.76 (s, 3H), 2.50–2.41 (m, 2H), 2.40–2.38 (m, 2H), 1.90–1.86 (m, 2H), 1.71–1.60 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 211.7, 158.7, 158.7, 141.4, 141.4, 134.4, 133.6, 130.4, 13.99, 113.0, 113.4, 55.19, 55.1, 43.4, 30.5, 29.4, 29.3, 24.3; HRMS (APCI) m/z: calcd for C22H24O3 [M + H]+ 337.1798; found 337.1802.
2-(Di(thiophen-2-yl)methylene)cycloheptan-1-one (5f). Prepared according to the GP4 from thiophene (0.104 g, 1.23 mmol), nBuLi (0.49 mL, 1.17 mmol), phosphordiamidate 2j (0.156 g, 0.51 mmol). Column chromatography (hexane/AcOEt 20
:1, Rf = 0.28) gave 0.059 g (40%) of the title compound as an orange oil. 1H NMR (400 MHz, CDCl3) δ 7.36–7.35 (m, 1H), 7.26–7.24 (m, 1H), 7.04–6.99 (m, 2H), 6.93–6.88 (m, 2H), 2.53–2.46 (m, 4H), 1.86–1.67 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 211.4, 145.1, 142.4, 141.6, 128.0, 127.8, 126.8, 126.7, 126.4, 126.3, 124.7, 42.7, 30.9, 28.4, 28.3, 23.7; HRMS (APCI) m/z: calcd for C16H16OS2 [M + H]+ 289.0715; found 289.0719.
3-Methyl-4,4-diphenylbut-3-en-2-one (5g). Prepared according to the GP4 from bromobenzene (0.20 g, 1.3 mmol), nBuLi (0.52 mL, 1.25 mmol), phosphordiamidate 2k (0.140 g, 0.50 mmol). Column chromatography (hexane/AcOEt 20
:1, Rf = 0.37) gave 0.084 g (71%) of the title compound as an yellowish oil. 1H NMR (400 MHz, CDCl3) δ 7.37–7.26 (m, 6H), 7.18–7.10 (m, 4H), 1.98 (s, 3H), 1.82 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 207.9, 145.1, 141.7, 141.1, 137.7, 129.8, 129.7, 128.4, 128.3, 128.1, 127.7, 30.2, 18.7; HRMS (APCI) m/z: calcd for C17H16O [M + H]+ 237.1274; found 237.1266.
4,4-Bis(4-methoxyphenyl)-3-methylbut-3-en-2-one (5h). Prepared according to the GP4 from 4-bromoanisole (0.240 g, 1.3 mmol), nBuLi (0.52 mL, 1.25 mmol), phosphordiamidate 2k (0.140 g, 0.50 mmol). Column chromatography (hexane/AcOEt 6
:1, Rf = 0.55) gave 0.072 g (49%) of the title compound as a white solid, mp = 58.0–61.0 °C. 1H NMR (400 MHz, CDCl3) δ 7.10–7.07 (m, 2H), 7.02–7.00 (m, 2H), 6.88–6.80 (m, 4H), 3.82 (s, 3H), 3.81 (s, 3H), 2.00 (s, 3H), 1.80 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 207.9, 160.0, 159.1, 145.6, 136.3, 134.6, 133.8, 131.4, 131.4, 113.7, 113.4, 55.3, 30.2, 19.0; HRMS (APCI) m/z: calcd for C19H20O3 [M + H]+ 297.1485; found 297.1489.
3-Methyl-4,4-di(thiophen-2-yl)but-3-en-2-one (5i). Prepared according to the GP4 from thiophene (0.110 g, 1.30 mmol), nBuLi (0.52 mL, 1.25 mmol), phosphordiamidate 2k (0.140 g, 0.50 mmol). Column chromatography (hexane/AcOEt 9
:1, Rf = 0.52) gave 0.029 g (24%) of the title compound as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.41–7.39 (m, 2H), 7.06–7.05 (m, 2H), 6.99–6.98 (m, 2H), 2.17 (s, 3H), 1.91 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 207.2, 143.2, 142.7, 139.8, 130.3, 129.9, 129.5, 128.5, 127.6, 127.2, 127.0, 29.5, 19.3; HRMS (APCI) m/z: calcd for C13H12OS2 [M + H]+ 249.0402; found 249.0406.
:1, Rf = 0.28) gave 0.062 g (46%) of the title compound as a white solid. 1H NMR (300 MHz, CDCl3) δ 7.69–7.66 (m, 2H), 7.32–7.31 (m, 1H), 7.23–7.19 (m, 2H), 7.09–6.99 (m, 5H), 2.53–2.45 (m, 4H), 1.91–1.82 (m, 4H), in accordance with literature.35:1, Rf = 0.53) gave 1.480 g (79%) of the title compound as a white solid, mp = 74.0–77.0 °C. 1H NMR (500 MHz, CDCl3) δ 7.59–7.57 (m, 4H), 7.30–7.27 (m, 4H), 7.18–7.15 (m, 2H), 6.22 (s, 1H), 5.74 (s, 1H), 2.42 (d, J = 10.1 Hz, 12H), 2.05 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 148.8, 145.5 (d, J = 7.6 Hz), 128.1, 126.4, 126.2, 122.7 (d, J = 6.6 Hz), 75.8, 36.2 (d, J = 4.7 Hz), 21.8; 31P NMR (202 MHz, CDCl3) δ 15.12 (s); HRMS (ESI) m/z: calcd for C20H27N2O3P [M + Na]+ 397.1652; found 397.1660.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported from the Grant Agency of the Czech Republic (Grant No 18-12150S).Notes and references
- (a) J. Adrio and J. C. Carretero, ChemCatChem, 2010, 2, 1384–1386 CrossRef CAS; (b) M. M. Heravi and P. Hajiabbasi, Monatsh. Chem., 2012, 143, 1575–1592 CrossRef CAS; (c) M. M. Heravi, V. Zadsirjan, P. Hajiabbasi and H. Hamidi, Monatsh. Chem., 2019, 150, 535–591 CrossRef CAS.
- (a) R. Ambre, H. Yang, W.-C. Chen, G. P. A. Yap, T. Jurca and T.-G. Ong, Eur. J. Inorg. Chem., 2019, 3511–3517 CrossRef CAS; (b) F. Fan, J. Tang, M. Luo and X. Zeng, J. Org. Chem., 2018, 83, 13549–13559 CrossRef CAS; (c) P.-P. Chen, E. L. Lucas, M. A. Greene, S.-Q. Zhang, E. J. Tollefson, L. W. Erickson, B. L. H. Taylor, E. R. Jarvo and X. Hong, J. Am. Chem. Soc., 2019, 141, 5835–5855 CrossRef CAS; (d) D. Ghorai, J. Loup, G. Zanoni and L. Ackermann, Synlett, 2019, 30, 429–432 CrossRef CAS.
- (a) A. Piontek, W. Ochędzan-Siodłak, E. Bisz and M. Szostak, Adv. Synth. Catal., 2019, 361, 2329–2336 CrossRef CAS; (b) Y. Sato, Y. Ashida, D. Yoshitake, M. Hoshino, T. Takemoto and Y. Tanabe, Synthesis, 2018, 50, 4659–4667 CrossRef CAS.
- C. A. Quesnelle and V. Snieckus, Synthesis, 2018, 50, 4395–4412 CrossRef CAS.
- (a) R. Manikandan and M. Jeganmohan, Org. Biomol. Chem., 2015, 13, 10420–10436 RSC; (b) E.-i. Negishi, G. Wang, H. Rao and Z. Xu, J. Org. Chem., 2010, 75, 3151–3182 CrossRef CAS; (c) W.-Y. Siau, Y. Zhang and Y. Zhao, in Stereoselective Alkene Synthesis, ed. J. Wang, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 33–58, DOI:10.1007/128_2012_315.
- (a) M. Drouin, J.-D. Hamel and J.-F. Paquin, Synthesis, 2018, 50, 881–955 CrossRef CAS; (b) M. Maraswami and T.-P. Loh, Synthesis, 2019, 51, 1049–1062 CrossRef CAS; (c) P. Polák, H. Váňová, D. Dvořák and T. Tobrman, Tetrahedron Lett., 2016, 57, 3684–3693 CrossRef.
- (a) G. Cahiez, V. Habiak and O. Gager, Org. Lett., 2008, 10, 2389–2392 CrossRef CAS; (b) G. Cahiez and H. Avedissian, Synthesis, 1998, 1199–1205 CrossRef CAS; (c) G. Cahiez, O. Gager and V. Habiak, Synthesis, 2008, 2636–2644 CrossRef CAS.
- (a) D. Fiorito, S. Folliet, Y. Liu and C. Mazet, ACS Catal., 2018, 8, 1392–1398 CrossRef CAS; (b) A. S. E. Karlström, K. Itami and J.-E. Bäckvall, J. Org. Chem., 1999, 64, 1745–1749 CrossRef; (c) C. Sahlberg, A. Quader and A. Claesson, Tetrahedron Lett., 1983, 24, 5137–5138 CrossRef CAS.
- (a) D. Gauthier, S. Beckendorf, T. M. Gøgsig, A. T. Lindhardt and T. Skrydstrup, J. Org. Chem., 2009, 74, 3536–3539 CrossRef CAS; (b) W. You, Y. Li and M. K. Brown, Org. Lett., 2013, 15, 1610–1613 CrossRef CAS; (c) J. A. Miller, Tetrahedron Lett., 2002, 43, 7111–7114 CrossRef CAS.
- (a) P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem., Int. Ed., 2003, 42, 4302–4320 CrossRef CAS; (b) H. Ila, O. Baron, A. J. Wagner and P. Knochel, Chem. Commun., 2006, 583–593 RSC; (c) R. Li-Yuan Bao, R. Zhao and L. Shi, Chem. Commun., 2015, 51, 6884–6900 RSC; (d) C. E. I. Knappke and A. Jacobi von Wangelin, Chem. Soc. Rev., 2011, 40, 4948–4962 RSC.
- C. C. Silveira, R. B. Guerra and J. V. Comasseto, Tetrahedron Lett., 2007, 48, 5121–5124 CrossRef CAS.
- R. E. Barrientos-Astigarraga, P. Castelani, C. Y. Sumida, J. Zukerman-Schpector and J. V. Comasseto, Tetrahedron, 2002, 58, 1051–1059 CrossRef CAS.
- F.-W. Sum and L. Weiler, Can. J. Chem., 1979, 57, 1431–1441 CrossRef CAS.
- (a) O. J. Yoshitaka, T. Tomofumi and M. Teruaki, Chem. Lett., 2003, 32, 994–995 CrossRef; (b) T. Bach and F. Höfer, J. Org. Chem., 2001, 66, 3427–3434 CrossRef CAS; (c) R. C. D. Brown, C. J. Bataille, R. M. Hughes, A. Kenney and T. J. Luker, J. Org. Chem., 2002, 67, 8079–8085 CrossRef CAS; (d) H. V. Thulasiram, R. M. Phan, S. B. Rivera and C. D. Poulter, J. Org. Chem., 2006, 71, 1739–1741 CrossRef CAS.
- Selected examples: (a) O. Piva, J. Org. Chem., 1995, 60, 7879–7883 CrossRef CAS; (b) K. P. Reber, J. Xu and C. A. Guerrero, J. Org. Chem., 2015, 80, 2397–2406 CrossRef CAS; (c) C. C. Nawrat and C. J. Moody, Org. Lett., 2012, 14, 1484–1487 CrossRef CAS; (d) K. Ishigami, S. Yamada and H. Watanabe, Tetrahedron Lett., 2015, 56, 5816–5819 CrossRef CAS.
- (a) S.-i. Hashimoto, M. Ban, Y. Yanagiya, S. Sakata and S. Ikegami, Tetrahedron Lett., 1991, 32, 4027–4030 CrossRef CAS; (b) D. Das, R. Kant and T. K. Chakraborty, Org. Lett., 2014, 16, 2618–2621 CrossRef CAS.
- (a) H. M. Cheng, W. Tian, P. A. Peixoto, B. Dhudshia and D. Y.-K. Chen, Angew. Chem., Int. Ed., 2011, 50, 4165–4168 CrossRef CAS; (b) N. Hayashi, K. Yamamoto, N. Minowa, M. Mitomi and M. Nakada, Org. Biomol. Chem., 2010, 8, 1821–1825 RSC; (c) N. Hayashi and M. Nakada, Tetrahedron Lett., 2009, 50, 232–235 CrossRef CAS; (d) C. M. Gampe, H. Tsukamoto, T.-S. A. Wang, S. Walker and D. Kahne, Tetrahedron, 2011, 67, 9771–9778 CrossRef CAS.
- (a) M. Polla and T. Frejd, Tetrahedron, 1993, 49, 2701–2710 CrossRef CAS; (b) L. Pettersson, G. Magnusson, T. Frejd, T. A. Pakkanen, J. C. Négrel, M. Chanon, C. Striley, J. Weidlein, A. Nasiri and Y. Okada, Acta Chem. Scand., 1993, 47, 196–207 CrossRef CAS; (c) V. K. Aggarwal, P. A. Bethel and R. Giles, Chem. Commun., 1999, 325–326 RSC.
- (a) P. Polák and T. Tobrman, Eur. J. Org. Chem., 2019, 957–968 CrossRef; (b) P. Polák and T. Tobrman, Org. Biomol. Chem., 2017, 15, 6233–6241 RSC; (c) V. Kotek, P. Polák, H. Dvořáková and T. Tobrman, Eur. J. Org. Chem., 2016, 5037–5044 CrossRef; (d) V. Kotek, H. Dvořáková and T. Tobrman, Org. Lett., 2015, 17, 608–611 CrossRef CAS.
- J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Adv. Synth. Catal., 2010, 352, 3235–3240 CrossRef CAS.
- Y. Zou, L. Qin, X. Ren, Y. Lu, Y. Li and J. Zhou, Chem.–Eur. J., 2013, 19, 3504–3511 CrossRef CAS.
- R. Wang and S. Zhang, RSC Adv., 2014, 4, 39497–39507 RSC.
- M. Hossain, U. Das and J. R. Dimmock, Eur. J. Med. Chem., 2019, 183, 111687 CrossRef CAS.
- (a) R. C. Boruah and G. S. Nongthombam, Heterocycles, 2019, 98, 19–62 CrossRef; (b) C. C. Meyer, E. Ortiz and M. J. Krische, Chem. Rev., 2020, 120, 3721–3748 CrossRef CAS; (c) J. Lam, K. M. Szkop, E. Mosaferi and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3592–3612 RSC.
- (a) A. Douchez, A. Geranurimi and W. D. Lubell, Acc. Chem. Res., 2018, 51, 2574–2588 CrossRef CAS; (b) S. Zhang, H. Neumann and M. Beller, Chem. Soc. Rev., 2020, 49, 3187–3210 RSC.
- C. Jiang, H. Lu, W.-H. Xu, J. Wu, T.-Y. Yu, P.-F. Xu and H. Wei, ACS Catal., 2020, 10, 1947–1953 CrossRef CAS.
- C. Nájera, L. K. Sydnes and M. Yus, Chem. Rev., 2019, 119, 11110–11244 CrossRef.
- F.-F. Pan, P. Guo, C.-L. Li, P. Su and X.-Z. Shu, Org. Lett., 2019, 21, 3701–3705 CrossRef CAS.
- (a) Y.-L. An, K. Li, Y. Shen, Z. Hong, L. Chen, Y. Hu, L. Zhou, D. Wang, X. Shi, S. Liu, W. Su, W. Cui, L. Kuai, H. Yang and X. Peng, Org. Lett., 2020, 22, 3931–3935 CrossRef CAS; (b) J. U. Rhee and M. J. Krische, Org. Lett., 2005, 7, 2493–2495 CrossRef CAS.
- N. Kreutzkamp, J. Pluhatsch and H. Schindler, Arch. Pharm., 1960, 293, 900–907 CrossRef CAS.
- X. Zeng, Z. Lu, S. Liu, G. B. Hammond and B. Xu, J. Org. Chem., 2017, 82, 13179–13187 CrossRef CAS.
- Y. Sugawara, W. Yamada, S. Yoshida, T. Ikeno and T. Yamada, J. Am. Chem. Soc., 2007, 129, 12902–12903 CrossRef CAS.
- L. Yang and Q. Zeng, Synthesis, 2017, 49, 3149–3156 CrossRef CAS.
- R. C. Larock and C. K. Reddy, Org. Lett., 2000, 2, 3325–3327 CrossRef CAS.
- Y. Miyahara and Y. N. Ito, J. Org. Chem., 2014, 79, 6801–6807 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07472a |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2020 |