
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst-free regioselective acetylation of primary hydroxy groups in partially protected and unprotected thioglycosides with acetic acid†
Polina I. Abronina *a,
Nelly N. Malyshevaa,
Alexander I. Zinina,
Natalya G. Kolotyrkinaa,
Elena V. Stepanovaab and
Leonid O. Kononov*a
*a,
Nelly N. Malyshevaa,
Alexander I. Zinina,
Natalya G. Kolotyrkinaa,
Elena V. Stepanovaab and
Leonid O. Kononov*a
aN. K. Kochetkov Laboratory of Carbohydrate Chemistry, N. D. Zelinsky Institute of Organic Chemistry, Leninsky prosp. 47, 119991 Moscow, Russian Federation. E-mail: Polina-Abronina@yandex.ru; leonid.kononov@gmail.com; kononov@ioc.ac.ru; Web: [https://carbohydrates.ru]
bResearch School of Chemistry and Applied Biomedical Sciences, Tomsk Polytechnic University, Lenin avenue 30, Tomsk 634050, Russian Federation
First published on 6th October 2020
Abstract
Highly regioselective acetylation of primary hydroxy groups in thioglycoside derivatives with gluco- and galacto-configurations was achieved by treatment with aqueous or anhydrous acetic acid (60–100% AcOH) at elevated temperatures (80–118 °C), avoiding complex, costly and time-consuming manipulations with protective groups. Acetylation of both 4,6-O-benzylidene acetals and the corresponding diols as well as the unprotected tetraol with AcOH was shown to lead selectively to formation of 6-O-acetyl derivatives. For example, the treatment of phenyl 1-thio-β-D-glucopyranoside with anhydrous AcOH at 80 °C for 24 h gave the corresponding 6-O-acetylated derivative in 47% yield (71% based on the reacted starting material) and unreacted starting tetraol in 34% yield, which can easily be recovered by silica gel chromatography and reused in further acetylation.
Introduction
Regioselective synthesis is a prominent challenge in organic chemistry and especially in carbohydrate chemistry since carbohydrates contain several hydroxyl groups of similar reactivity.1–5 Although primary hydroxy groups are generally considered more reactive in esterification reaction,6,7 regioselective acetylation of primary hydroxy groups in carbohydrates is surprisingly complicated4 and is usually conducted using specific catalysts and conditions.8,9 Disadvantages of many known acetylation methods are related to the use of environmentally toxic and expensive reagents.Typical examples of selective 6-O-acetylation in monosaccharides include the use of EtOAc in combination with acidic10,11 or basic12,13 catalysis, vinyl acetate and germanium compounds as catalysts,14 acetic acid (AcOH) and uronium-based coupling agents,15 1-acetyloxybenzotriazole16,17 as well as acetyl chloride or acetic anhydride in the presence of bases,18–22 iodine,23 Al2O3 (ref. 24) or 4 Å molecular sieves.25 Reactions employing organotin4,26 and organosilicon27 derivatives were used for regioselective acetylation of carbohydrates. It was noted that the selectivity of acetylation is dramatically influenced by the nature of counter-ion of acetylating agent and reaction conditions.28 Based on these findings, regioselective acetylation of diols and polyols using catalytic amounts of acetate ion has been developed.29 Lewis acid catalysed removal of 6-O-protective group in per-O-trimethylsilylated30,31 or benzylated32,33 compounds with subsequent one-pot acetylation of monosaccharides affording 6-O-acetylated fully protected sugars has been described. A method involving N-acetylthiazolidine-2-thione34 as acetylation agent has also been developed. 1-Acetylimidazole was used for selective acetylation of the primary hydroxy groups in derivatives of methyl glycosides in 1,2-dichloroethane at 80 °C (ref. 35) or, more recently, in water in the presence of tetramethylammonium hydroxide (TMAH).36 Highly selective but less accessible chiral organocatalysts37,38 for direct acetylation have also been proposed. Recently, 6-O-acetylation by migration of acetyl group from O-2 has been developed.39 Enzymatic acetylation40–42 has also been applied for the selective introduction of acetyl groups at primary hydroxy groups of sugars.
Acetylation at O-6 is common for carbohydrate low-molecular weight metabolites in plants.43–47 Bacterial polysaccharides often contain 6-O-acetyl groups, which play a complex role in the functional immune response.48–51 Hence, the development of ways for selective acetylation of unprotected and partially protected carbohydrates would contribute greatly to the study of naturally occurring carbohydrate molecules.
The non-catalysed (auto-catalysed) acetylation of simple alcohols is well-known.52–59 It has been extensively studied for industrial purposes like reactive distillation,60,61 which is used for esterification or for recovery of dilute acetic acid from wastewater.62–65 However, only few examples of the use of this reaction for acetylation of carbohydrates are known. The primary hydroxy group of several carbohydrates was selectively esterified by treatment with 50% AcOH at 100 °C for 24 h. For example, after removal of excess of AcOH in vacuo the syrup containing unchanged D-glucose, 6-O-acetyl-D-glucose, and a di-O-acetyl-D-glucose was obtained.66 The preparation of 6-O-acetyl-D-glucose by esterification of D-glucose was also performed by treatment with 75% AcOH at 100 °C for 8 h.67 Preparative yields of 6-O-acetylated products were moderate (10–30%).
Herein, we describe a method for regioselective acetylation of primary hydroxyl group in glycosides using acetic acid as single reagent in the absence of added catalyst. We focused on regioselective acetylation of thioglycoside diols and tetraol with 60–100% (v/v) AcOH using Methods A–I (Table 1).
| Method | Concentration of AcOH (%, v/v) | Temperatureb (°C) | Time (h) |
|---|---|---|---|
| a Aqueous or anhydrous AcOH, heating at indicated temperature.b Bath temperature.c Reflux.d Bp 118 °C.e Bp 102 °C.f Bp 105 °C. | |||
| A | 60 | 80 | 24 |
| B | 80 | 80 | 24 |
| C | 100 | 80 | 24 |
| D | 100 | 100 | 24 |
| E | 100 | 130c,d | 24 |
| F | 100 | 130c,d | 2.5 |
| G | 100 | 130c,d | 2 |
| H | 60 | 117c,e | 24 |
| I | 80 | 117c,f | 24 |
Results and discussion
Cleavage of 4,6-O-benzylidene group by aqueous AcOH with concomitant acetylation
Selective cleavage of 4,6-O-benzylidene group68,69 in various carbohydrate derivatives is commonly achieved by acidic hydrolysis70 (heating with aq AcOH,71 treatment with trifluoroacetic acid (TFA) in CH2Cl2–ethylene glycol72 or aq TFA in CH2Cl2 (ref. 73 and 74)), which leads to formation of the corresponding 4,6-diol. This procedure is considered to be robust and efficient and is widely used in protective group strategies.1,75We have recently reported that the cleavage of 4,6-O-benzylidene group in derivatives of β-configured ethyl 1-thio-β-D-galactopyranoside with silyl (TIPS or TBDPS) groups at O-2 and O-3 with 80% aq AcOH (80 °C, 1 h) gave the expected 4,6-diols in 68–75% yields, while treatment with aq TFA in CH2Cl2 led to pyranose ring contraction.76 To study the effect of anomeric configuration on the result of treatment of benzylidene acetals with acids, we synthesised ethyl 4,6-O-benzylidene-1-thio-α-D-galactopyranoside (1) and the corresponding 2,3-O-TIPS (2) and TBDPS (3) derivatives, starting from the known77 ethyl 1-thio-α-D-galactopyranoside.
Surprisingly, the reaction of α-configured ethyl 4,6-O-benzylidene-2,3-bis-O-triisopropylsilyl-1-thio-α-D-galactopyranoside (2) with 80% aq AcOH under similar conditions was not complete in 1–2 h. After 24 h of treatment with 80% aq AcOH at 80 °C (method B, Table 1, Fig. 1) the formation of the expected diol 4 (19%) was accompanied by considerable amounts (22%) of 6-O-acetylated product 5. Besides, the minor 4-O-acetylated product 7 and 6-O-acetylated thioglycoside 6 with a single TIPS group at O-2 were also obtained.
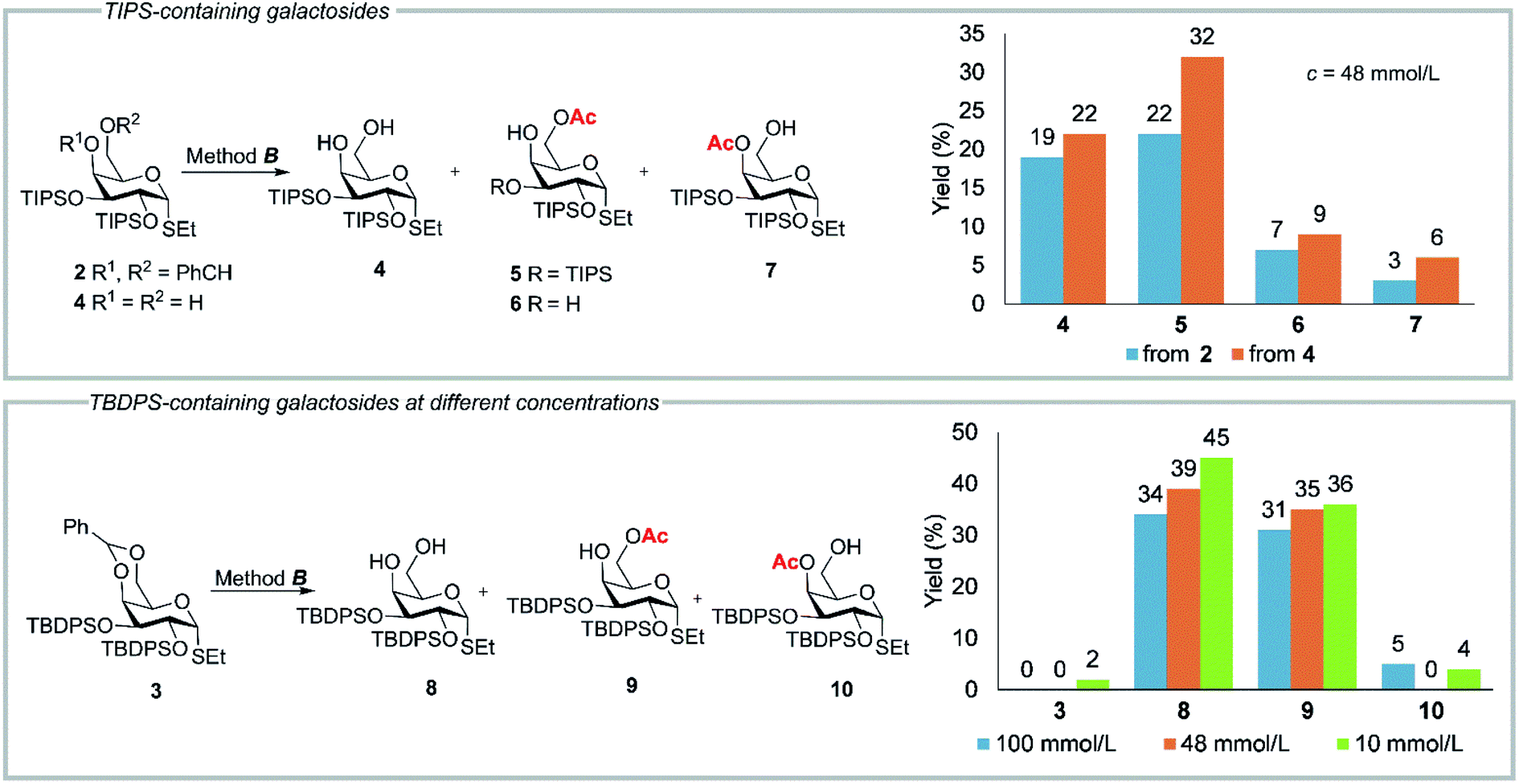 | ||
| Fig. 1 Results of acetylation of TIPS-containing compounds 2, 4 and TBDPS-containing compound 3 according to method B. TIPS = (i-Pr)3Si. TBDPS = t-BuPh2Si. See also Tables 1S and 2S in ESI.† | ||
Unlike the cleavage of 4,6-O-benzylidene group in TIPS-containing derivative 2, the treatment of thiogalactopyranoside 3 with TBDPS groups at O-2 and O-3 under identical conditions (according to method B) (Fig. 1) at the same concentration (c = 48 mmol L−1) gave diol 8 and 6-O-acetylated product 9 in 39 and 35% yields, respectively (Fig. 1). Apparently, the TBDPS groups are more stable under acidic conditions than the TIPS groups. A study of influence of the concentration on the outcome of acetylation revealed an additional formation of a minor amount of 4-O-acetylated product 10 at higher and lower concentrations (Fig. 1).
To check if the preferential formation of the product of O-acetylation of primary hydroxy group is related to limited solubility of extremely hydrophobic silyl-containing ethyl thio-α-D-galactopyranosides 2–4 we tested the known ethyl 2,3-di-O-benzyl-4,6-O-benzylidene-1-thio-β-D-galactopyranoside (11)71 with less hydrophobic benzyl groups at O-2 and O-3 in the reaction with acetic acid according to method B (Table 1) at two similar yet different concentrations (c = 58 and 68 mmol L−1) (Fig. 2). The yield of 6-O-acetylated thiogalactoside 14 was 41 and 43%, respectively. Diol 12 (ref. 71) was isolated in 36 and 30% yields, when the reaction was performed at 58 and 68 mmol L−1 of 11, respectively (Fig. 2). Besides, 4-O-acetylated thiogalactoside 13 was also obtained.
 | ||
| Fig. 2 The yields of products 11–14 after acetylation of compound 11 according to method B at different concentrations of substrate 11. See also Table 3S in ESI.† | ||
Similar results were obtained in gluco-series. Thus, the cleavage of benzylidene group in phenyl 2,3-di-O-benzyl-4,6-O-benzylidene-1-thio-β-D-glucopyranoside (15) with 80% aq AcOH at 80 °C under conditions of method B (Table 1) gave diol 16 and 6-O-acetylated product 17 in 43 and 45% yields, respectively (Fig. 3). 4-O-Acetylated thiogalactoside 18 was also isolated in 6% yield. A decrease in AcOH concentration to 60% led to the expected decrease in the proportion of 6-O-acetylated product 17, which was obtained in 27% yield (Fig. 3) under conditions of method A (Table 1).
 | ||
| Fig. 3 The yields of products 16–19 obtained from compounds 15–17 under acetylation conditions according to methods A–C. Concentration of substrate (15, 16 or 17) c = 20 mmol L−1. See also Table 4S in ESI.† | ||
Acetylation of 4,6-diols with aqueous AcOH
Recently, the transformation of mono- and oligosaccharide 4,6-O-benzylidene acetals to the corresponding 6-O-acetyl derivatives with free 4-hydroxy group by treatment with 60% aq AcOH at 80 °C has been reported.78 The authors emphasise that the presence of 4,6-O-benzylidene acetal is critical for the success of the transformation; no acetylation occurs with the corresponding 4,6-diols under identical conditions. In particular, according to the authors,78 when diol 16 was treated with 60% aq AcOH “no primary O-acetylation product could be identified even after 24 h” at 80 °C (these conditions mirror conditions of method A, Table 1). Our data do not support these findings. In our hands, after the treatment of diol 16 with 60% aq AcOH at 80 °C according to method A (Fig. 3) 6-O-acetylated product 17 was isolated in 28% yield. Unreacted diol 16 was recovered in 51% yield. Besides, the minor amount of 4-O-acetylated thioglucoside 18 was isolated in 2% yield.Note that the increase in concentration of AcOH to 80% while keeping the temperature at 80 °C (method B) for acetylation of diol 16 with gluco-configuration led to comparable yield (30%) of 6-O-acetylated product 17 (Fig. 3). Similar results were obtained in acetylation of diol 4 with galacto-configuration under identical conditions (80% aq AcOH 80 °C, method B), which gave 6-O-acetylated thioglycoside 5 (32%) as well as the product with acetyl group at O-4 (7, 6%) and 6-O-acetylated thioglycoside 6 (9%) with a single TIPS group at O-2 (Fig. 1).
Esterification of alcohols with AcOH is known to be a reversible process that occurs under thermodynamic control.52–59 Indeed, when 6-O-acetylated product 17 was treated under the reaction conditions (80% aq AcOH, 80 °C, 24 h; method B) the parent diol 16 (24%) and 4-acetate 18 (4%) were isolated from the reaction mixture in addition to the starting 6-acetate 17 (53%) (Fig. 3).
Acetylation of 4,6-diols with anhydrous AcOH
In accordance with this view on the mechanism of esterification of alcohols with aqueous AcOH,52–59 the use of anhydrous AcOH shifted the esterification equilibrium towards the formation of acetylated products. Thus, the treatment of gluco-diol 16 with anhydrous AcOH at 80 °C according to method C (Fig. 3) increased the yield of 6-O-acetylated product 17 (42%). Small amounts of 4-O-acetylated (18, 4%) and 4,6-di-O-acetylated (19 (ref. 79), 3%) derivatives were also isolated. It should be noted that unreacted diol 16 (35%) can be easily recovered by silica gel chromatography and reused in further acetylation.Thus, we demonstrated that not only 4,6-O-benzylidene acetals (as claimed in ref. 78) but also the corresponding 4,6-diols can form 6-O-acetylated thioglycosides by treatment with aqueous or anhydrous AcOH. Importantly, the regioselectivity of acetylation does not depend on configuration of monosaccharide: thioglycosides with both gluco- and galacto-configurations (with equatorial and axial 4-OH, respectively) perform equally well in the acetylation reactions.
Acetylation of tetraol with aqueous or anhydrous AcOH
The next step was to study if the found conditions for acetylation of carbohydrate diols with AcOH would be applicable for carbohydrate derivatives with larger number of hydroxyl groups.Treatment of unprotected phenyl 1-thio-β-D-glucopyranoside (20)80 with anhydrous AcOH at 80 °C for 24 h according to method C (Fig. 4, 5) gave the corresponding 6-O-acetylated derivative 21 in 47% yield and unreacted tetraol 20 in 34% yield along with minor amounts of mono- (26) and diacetylated derivatives 27 as well as 3,6-di-O-acetylated thioglycoside 22 (Fig. 4 and 5). This result is remarkable considering that the unreacted starting tetraol 20 can easily be recovered by silica gel chromatography and reused in further acetylation (thus making the yield of 21 fairly practical – 71% based on the reacted starting material). The only, although minor, drawback of this method for selective acetylation is the incomplete conversion of the starting tetraol 20 even after the long reaction time.
 | ||
| Fig. 4 The results of acetylation of compound 20 according to methods C–I. Concentration of 20 c = 20 mmol L−1. See also Table 5S in ESI.† | ||
 | ||
| Fig. 5 The effect of temperature on selectivity of acetylation. The yields of acetylated derivatives 20–28 isolated after acetylation of compound 20 with anhydrous AcOH at 80, 100 and 118 °C according to methods C, D, E, respectively. Concentration of 20 c = 20 mmol L−1. See also Table 5S in ESI.† | ||
Increasing the reaction temperature to 100 °C (24 h, method D) resulted in almost complete conversion of 20 (95%) and a decrease in the yield of 6-O-acetylated derivative 21 to 34%. Substantial amounts of di- and triacetylated products 27 and 28 in addition to minor amounts of monoacetylated derivative 26 were formed after 24 h at 100 °C (Fig. 4 and 5).
Reflux of tetraol 20 in more dilute AcOH solutions at ca. 100 °C for 24 h was not very effective either (Fig. 4). The conversion of tetraol 20 dropped to 50% in 60% AcOH (bp 102 °C, method H) as did the yield of 6-O-acetylated derivative 21 (33%). Although the yield of 6-O-acetylated derivative 21 somewhat increased to 42% in 80% AcOH (bp 105 °C, method I), the amount of recovered tetraol 20 decreased to 19%.
Further increase in reaction temperature resulted in preferential formation of di-, tri- and tetraacetylated products 22–28 when tetraol 20 was treated with anhydrous AcOH at reflux (bp 118 °C) for 24 h according to method E (Fig. 4 and 5). Importantly, neither tetraol 20 nor monoacetate 21 were detected under these conditions.
Variation of the reaction time (methods F and G) revealed that the treatment of tetraol 20 with anhydrous AcOH at reflux according to method F allows the preparation of the 6-O-acetylated derivative 21 in 49% yield, virtually identical to that achieved (47%) under conditions of method C, but in much shorter time (2.5 h vs. 24 h), albeit accompanied by a decrease in the yield of unreacted starting tetraol 20 (20%).
Our data are in qualitative agreement with the known results for the preparation of 6-O-acetyl-D-glucose by esterification of D-glucose with 50–75% AcOH at 100 °C,66,67 although the yields achieved in our work are considerably higher.
Note that the highest yields of 6-O-acetylated derivatives achieved by esterification of tetraol (49%), 4,6-diols (42%) or 4,6-O-benzylidene derivatives (45%) by aqueous or anhydrous AcOH are similar.
A possibility of esterification of carbohydrate alcohols by aqueous or anhydrous AcOH should be considered seriously as this reaction may constitute an important side process considerably diminishing the yield of the target transformation performed in the presence of AcOH.
Conclusions
We have developed an efficient and green procedure for the highly regioselective acetylation of primary hydroxy groups in thioglycosides, which was achieved by treatment with aqueous or anhydrous AcOH, avoiding complex, costly and time-consuming manipulations with protective groups. To this end, acetylation of thioglycoside derivatives with gluco- and galacto-configurations at various temperatures (80–118 °C) and concentrations of AcOH (60–100%) was studied. Acetylation of both 4,6-O-benzylidene acetals and the corresponding diols as well as the unprotected tetraol with AcOH was shown to lead selectively to formation of 6-O-acetyl derivatives. Unreacted starting polyol can easily be recovered by silica gel chromatography and reused in further acetylation, which is important from the preparative point of view.Experimental
General procedure for deprotection of benzylidene acetals 2, 3, 11, 15 and acetylation of alcohols 4, 16, 17
To thioglycosides 2, 3, 4, 11, 15–17 (0.1–0.5 mmol) 60–100% (v/v) AcOH was added and the reaction mixture (c = 10–100 mmol L−1) was stirred at T °C according to methods A–C (Table 1). Then the reaction mixture was diluted with CH2Cl2 (50 mL) and washed with satd aq NaHCO3 (50 mL). The aqueous layer was extracted with CH2Cl2 (2 × 5 mL) and the combined organic extracts were filtered through a cotton wool plug, concentrated, and dried in vacuo. The products were purified by silica gel column chromatography in gradient of EtOAc in light petroleum (1 → 20%) for isolating of silyl (TBDPS or TIPS)-containing thioglycosides 4–10 and in gradient of EtOAc in light petroleum (5 → 60%) for isolating of benzyl-containing thioglycosides 16–19. The yields of purified products are shown in Fig. 1–3 (see also Tables 1S–4S in ESI†). See ESI† for the characterisation data and copies of NMR spectra.General procedure for acetylation of thioglycoside 20
To thioglycoside 20 (150 mg, 0.55 mmol) 60–100% (v/v) AcOH (27.5 mL) was added and the reaction mixture (c = 20 mmol L−1) was stirred at T °C according to methods C–I (Table 1). Then the reaction mixture was concentrated under reduced pressure and dried in vacuo. The products were purified by silica gel column chromatography in gradient of EtOAc in light petroleum (5 → 60%) to afford acetylated products 21–28. The yields of purified products are shown in Fig. 4 and 5 (see also Table 5S in ESI†). See ESI† for the characterisation data and copies of NMR spectra.Phenyl 6-O-acetyl-1-thio-β-D-glucopyranoside (21)
Method C. Yield 47% (71% based on the reacted starting material). Rf = 0.30 (light petroleum–EtOAc 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9). [α]D23 −61.7 (c 2.0 in CHCl3). 1H NMR (300 MHz; CD3OD): δ = 2.04 (s, 3H, CH3CO), 3.22 (dd, J2,3 = 8.5 Hz, J2,1 = 9.7 Hz, 1H, H-2), 3.24–3.33 (m, 1H, H-4), 3.40 (dd ∼ t, Japp = 8.8 Hz, 1H, H-3), 3.50 (ddd, J5,6b = 2.2 Hz, J5,6a = 6.6 Hz, J5,4 = 9.8 Hz, 1H, H-5), 4.19 (dd, J6a,5 = 6.6 Hz, J6a,6b = 11.9 Hz, 1H, H-6a), 4.40 (dd, J6b,5 = 2.2 Hz, J6b,6a = 11.9 Hz, 1H, H-6b), 4.59 (d, J1,2 = 9.7 Hz, 1H, H-1), 7.19–7.38 (m, 3H, PhS (H-3, H-4, H-5)), 7.45–7.61 (m, 2H, PhS (H-2, H-6)); 13C NMR (75 MHz, MeOD): δ = 20.8 (
:9). [α]D23 −61.7 (c 2.0 in CHCl3). 1H NMR (300 MHz; CD3OD): δ = 2.04 (s, 3H, CH3CO), 3.22 (dd, J2,3 = 8.5 Hz, J2,1 = 9.7 Hz, 1H, H-2), 3.24–3.33 (m, 1H, H-4), 3.40 (dd ∼ t, Japp = 8.8 Hz, 1H, H-3), 3.50 (ddd, J5,6b = 2.2 Hz, J5,6a = 6.6 Hz, J5,4 = 9.8 Hz, 1H, H-5), 4.19 (dd, J6a,5 = 6.6 Hz, J6a,6b = 11.9 Hz, 1H, H-6a), 4.40 (dd, J6b,5 = 2.2 Hz, J6b,6a = 11.9 Hz, 1H, H-6b), 4.59 (d, J1,2 = 9.7 Hz, 1H, H-1), 7.19–7.38 (m, 3H, PhS (H-3, H-4, H-5)), 7.45–7.61 (m, 2H, PhS (H-2, H-6)); 13C NMR (75 MHz, MeOD): δ = 20.8 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3CO), 64.9 (C-6), 71.4 (C-4), 73.7 (C-2), 79.0 (C-5), 79.4 (C-3), 89.0 (C-1), 128.5 (PhS (C-4)), 129.8 (PhS (C-3, C-5)), 133.0 (PhS (C-2, C-6)), 134.8 (PhS (C-1)), 172.7 (CO). HRMS (ESI): m/z calcd for C14H18O6S + Na+: 337.0716 [M + Na]+; found: 337.0715.
H3CO), 64.9 (C-6), 71.4 (C-4), 73.7 (C-2), 79.0 (C-5), 79.4 (C-3), 89.0 (C-1), 128.5 (PhS (C-4)), 129.8 (PhS (C-3, C-5)), 133.0 (PhS (C-2, C-6)), 134.8 (PhS (C-1)), 172.7 (CO). HRMS (ESI): m/z calcd for C14H18O6S + Na+: 337.0716 [M + Na]+; found: 337.0715.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Foundation for Basic Research (Project No. 20-03-00465-a).Notes and references
- S. Oscarson, in The Organic Chemistry of Sugars, ed. D. E. Levy and P. Fügedi, CRC Press, Taylor & Francis Group, Boca Raton, FL, 2005, pp. 73–107 Search PubMed.
- V. Dimakos and M. S. Taylor, Chem. Rev., 2018, 118, 11457–11517 CrossRef CAS.
- B. Ren, L. Zhang and M. Zhang, Asian J. Org. Chem., 2019, 8, 1813–1823 CrossRef CAS.
- T. Wang and A. V. Demchenko, Org. Biomol. Chem., 2019, 17, 4934–4950 RSC.
- Protecting Groups: Strategies and Applications in Carbohydrate Chemistry, ed. S. Vidal, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2019 Search PubMed.
- M. Nahmany and A. Melman, Org. Biomol. Chem., 2004, 2, 1563–1572 RSC.
- J. Otera and J. Nishikido, Esterification: Methods, Reactions, and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010 Search PubMed.
- A. H. Haines, Adv. Carbohydr. Chem. Biochem., 1976, 33, 11–109 CrossRef CAS.
- D. Lee and M. S. Taylor, Synthesis, 2012, 44, 3421–3431 CrossRef CAS.
- S. S. Rana, J. J. Barlow and K. L. Matta, Tetrahedron Lett., 1981, 22, 5007–5010 CrossRef CAS.
- P. H. Liang, Y. J. Lu and T. H. Tang, Tetrahedron Lett., 2010, 51, 6928–6931 CrossRef CAS.
- N. Mallesha, S. P. Rao, R. Suhas and D. C. Gowda, J. Chem. Res., 2011, 35, 536–539 CrossRef CAS.
- X. Liu, B. Becker and M. A. Cooper, Aust. J. Chem., 2014, 67, 679–683 CrossRef CAS.
- K. Sugahara, N. Satake, K. Kamata, T. Nakajima and N. Mizuno, Angew. Chem., Int. Ed., 2014, 53, 13248–13252 CrossRef CAS.
- J.-d. A. K. Twibanire and T. B. Grindley, Org. Lett., 2011, 13, 2988–2991 CrossRef CAS.
- L. Greffe, M. T. Jensen, F. Chang-Pi-Hin, S. Fruchard, M. J. O'Donohue, B. Svensson and H. Driguez, Chem.–Eur. J., 2002, 8, 5447–5455 CrossRef CAS.
- H. M. Chen and S. G. Withers, Carbohydr. Res., 2018, 467, 33–44 CrossRef CAS.
- N. K. Kochetkov, N. E. Nifant'ev and L. V. Backinowsky, Tetrahedron, 1987, 43, 3109–3121 CrossRef CAS.
- K. Ishihara, H. Kurihara and H. Yamamoto, J. Org. Chem., 1993, 58, 3791–3793 CrossRef CAS.
- Y. P. Hu, Y. Q. Zhong, Z. G. Chen, C. Y. Chen, Z. H. Shi, M. M. L. Zulueta, C. C. Ku, P. Y. Lee, C. C. Wang and S. C. Hung, J. Am. Chem. Soc., 2012, 134, 20722–20727 CrossRef CAS.
- Q. W. Liu, H. C. Bin and J. S. Yang, Org. Lett., 2013, 15, 3974–3977 CrossRef CAS.
- S. Singh, Z. Su, M. Grossutti and F. I. Auzanneau, Carbohydr. Res., 2014, 390, 50–58 CrossRef CAS.
- K. P. R. Kartha and R. A. Field, Tetrahedron, 1997, 53, 11753–11766 CrossRef CAS.
- P. Tiwari and A. K. Misra, Carbohydr. Res., 2006, 341, 339–350 CrossRef CAS.
- M. Adinolfi, G. Barone, A. Iadonisi and M. Schiattarella, Tetrahedron Lett., 2003, 44, 4661–4663 CrossRef CAS.
- H. Dong, Z. Pei, S. Byström and O. Ramström, J. Org. Chem., 2007, 72, 1499–1502 CrossRef CAS.
- Y. Zhou, O. Ramström and H. Dong, Chem. Commun., 2012, 48, 5370–5372 RSC.
- E. Kattnig and M. Albert, Org. Lett., 2004, 6, 945–948 CrossRef CAS.
- B. Ren, M. Rahm, X. Zhang, Y. Zhou and H. Dong, J. Org. Chem., 2014, 79, 8134–8142 CrossRef CAS.
- A. A. Joseph, V. P. Verma, X. Y. Liu, C. H. Wu, V. M. Dhurandhare and C. C. Wang, Eur. J. Org. Chem., 2012, 2012, 744–753 CrossRef CAS.
- M. A. Witschi and J. Gervay-Hague, Org. Lett., 2010, 12, 4312–4315 CrossRef CAS.
- G. B. Yang, X. L. Ding and F. Z. Kong, Tetrahedron Lett., 1997, 38, 6725–6728 CrossRef CAS.
- Y. Cao, Y. Okada and H. Yamada, Carbohydr. Res., 2006, 341, 2219–2223 CrossRef CAS.
- D. Plusquellec and K. Baczko, Tetrahedron Lett., 1987, 28, 3809–3812 CrossRef CAS.
- R. V. Stick, D. M. G. Tilbrook and S. J. Williams, Aust. J. Chem., 1999, 52, 885–894 CrossRef CAS.
- Y. Lu, P. Wei, Y. Pei, H. Xu, X. Xin and Z. Pei, Green Chem., 2014, 16, 4510–4514 RSC.
- T. Kawabata, W. Muramatsu, T. Nishio, T. Shibata and H. Schedel, J. Am. Chem. Soc., 2007, 129, 12890–12895 CrossRef CAS.
- Y. Ueda, K. Mishiro, K. Yoshida, T. Furuta and T. Kawabata, J. Org. Chem., 2012, 77, 7850–7857 CrossRef CAS.
- D. A. Romanova, D. L. Avetyan, M. L. Belyanin and E. V. Stepanova, J. Nat. Prod., 2020, 83, 888–893 CrossRef CAS.
- H. G. Park, J. H. Do and H. N. Chang, Biotechnol. Bioprocess Eng., 2003, 8, 1–8 CrossRef CAS.
- A. M. Iribarren and L. E. Iglesias, RSC Adv., 2016, 6, 16358–16386 RSC.
- M. C. Villalobos, A. G. Gonçalves, M. D. Noseda, D. A. Mitchell and N. Krieger, Process Biochem., 2018, 73, 86–93 CrossRef CAS.
- C. S. Kim, L. Subedi, K. J. Park, S. Y. Kim, S. U. Choi, K. H. Kim and K. R. Lee, Fitoterapia, 2015, 106, 147–152 CrossRef CAS.
- B. Y. G. Mountessou, J. Tchamgoue, J. Paul Dzoyem, R. T. Tchuenguem, F. Surup, M. I. Choudhary, I. R. Green and S. F. Kouam, Tetrahedron Lett., 2018, 59, 4545–4550 CrossRef CAS.
- X.-J. Li, K.-W. Kim, D.-C. Kim, H. Oh, X.-Q. Liu and Y.-C. Kim, Nat. Prod. Res., 2019, 1–8 Search PubMed.
- A. Benmerache, A. Alabdul Magid, A. Kabouche, D. Harakat, L. Voutquenne-Nazabadioko and Z. Kabouche, Nat. Prod. Res., 2019, 1–7 CrossRef.
- M.-O. Sim, H. J. Lee, D. E. Jeong, J.-H. Jang, H.-K. Jung and H.-W. Cho, Chem.-Biol. Interact., 2019, 311, 108755 CrossRef CAS.
- Bacterial lipopolysaccharides: structure, chemical synthesis, biogenesis and interaction with host cells, ed. Y. A. Knirel and M. A. Valvano, Springer Vienna, Vienna, 2011 Search PubMed.
- J. J. Calix, J. S. Saad, A. M. Brady and M. H. Nahm, J. Biol. Chem., 2012, 287, 13996–14003 CrossRef CAS.
- F. Berti, R. De Ricco and R. Rappuoli, Molecules, 2018, 23, 1340 CrossRef.
- L. A. Mulard, in Carbohydrate Chemistry: Chemical and Biological Approaches, ed. A. P. Rauter, T. K. Lindhorst and Y. Queneau, The Royal Society of Chemistry, 2018, vol. 43, pp. 71–103 Search PubMed.
- A. T. Williamson and C. N. Hinshelwood, Trans. Faraday Soc., 1934, 30, 1145–1149 RSC.
- H. A. Smith, J. Am. Chem. Soc., 1939, 61, 254–260 CrossRef CAS.
- T. Pöpken, L. Götze and J. Gmehling, Ind. Eng. Chem. Res., 2000, 39, 2601–2611 CrossRef.
- N. Calvar, B. González and A. Dominguez, Chem. Eng. Process., 2007, 46, 1317–1323 CrossRef CAS.
- A. M. Toikka, M. A. Trofimova and M. A. Toikka, Russ. Chem. Bull., 2012, 61, 662–664 CrossRef CAS.
- M. Toikka, A. Samarov, M. Trofimova, A. Golikova, N. Tsvetov and A. Toikka, Fluid Phase Equilib., 2014, 373, 72–79 CrossRef CAS.
- A. M. Toikka, A. A. Samarov and M. A. Toikka, Russ. Chem. Rev., 2015, 84, 378–392 CrossRef CAS.
- A. Golikova, N. Tsvetov, A. Samarov, M. Toikka, I. Zvereva, M. Trofimova and A. Toikka, J. Therm. Anal. Calorim., 2020, 139, 1301–1307 CrossRef CAS.
- Reactive Distillation: Status and Future Directions, ed. K. Sundmacher and A. Kienle, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003 Search PubMed.
- A. A. Kiss, M. Jobson and X. Gao, Ind. Eng. Chem. Res., 2019, 58, 5909–5918 CrossRef CAS.
- S. Steinigeweg and J. Gmehling, Ind. Eng. Chem. Res., 2002, 41, 5483–5490 CrossRef CAS.
- R. S. Huss, F. R. Chen, M. F. Malone and M. F. Doherty, Comput. Chem. Eng., 2003, 27, 1855–1866 CrossRef CAS.
- Y. Liu, E. Lotero and J. G. Goodwin, J. Mol. Catal. A: Chem., 2006, 245, 132–140 CrossRef CAS.
- M. Schmitt and H. Hasse, Ind. Eng. Chem. Res., 2006, 45, 4123–4132 CrossRef CAS.
- R. B. Duff, J. Chem. Soc., 1957, 4730–4734 RSC.
- V. Bílik, Chem. Pap., 1972, 26, 82–83 Search PubMed.
- P. G. M. Wuts, Greene's protective groups in organic synthesis, John Wiley & Sons, Inc., Hoboken, NJ, 2014, pp. 414–428 Search PubMed.
- P. J. Kocienski, Protecting Groups, Georg Thieme Verlag, Stuttgart, 2004, pp. 137–150 Search PubMed.
- A. H. Haines, Adv. Carbohydr. Chem. Biochem., 1981, 39, 13–70 CrossRef CAS.
- P. J. Garegg, I. Kvarnström, A. Niklasson, G. Niklasson and S. C. T. Svensson, J. Carbohydr. Chem., 1993, 12, 933–953 CrossRef CAS.
- J. Lindberg, S. C. T. Svensson, P. Påhlsson and P. Konradsson, Tetrahedron, 2002, 58, 5109–5117 CrossRef CAS.
- H. Gold, R. G. Boot, J. Aerts, H. S. Overkleeft, J. D. C. Codée and G. A. van der Marel, Eur. J. Org. Chem., 2011, 1652–1663 CrossRef CAS.
- P. I. Abronina, A. I. Zinin, N. N. Malysheva, E. V. Stepanova, A. O. Chizhov, V. I. Torgov and L. O. Kononov, Synlett, 2017, 28, 1608–1613 CrossRef CAS.
- S.-C. Hung and C. C. Wang, in Glycochemical Synthesis: Strategies and Applications, ed. S.-C. Hung and M. M. L. Zulueta, John Wiley & Sons, Inc., Hoboken, NJ, 2016, ch. 2, pp. 35–68 Search PubMed.
- P. I. Abronina, N. N. Malysheva, V. V. Litvinenko, A. I. Zinin, N. G. Kolotyrkina and L. O. Kononov, Org. Lett., 2018, 20, 6051–6054 CrossRef CAS.
- L. Käsbeck and H. Kessler, Liebigs Ann./Recl., 1997, 169–173 CrossRef.
- M. M. Mukherjee, N. Basu, S. Nandi and R. Ghosh, Carbohydr. Res., 2019, 476, 36–43 CrossRef CAS.
- M. C. Galan, A. T. Tran and S. Whitaker, Chem. Commun., 2010, 46, 2106–2108 RSC.
- M. S. Motawia, C. E. Olsen, K. Enevoldsen, J. Marcussen and B. L. Møller, Carbohydr. Res., 1995, 277, 109–123 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental procedures, characterisation data and copies of NMR spectra. See DOI: 10.1039/d0ra07360a |
| This journal is © The Royal Society of Chemistry 2020 |