
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAggregation induced emission in one easy step: pyridinium AIEgens and counter ion effect†
Kaspars Leduskrasts and
Edgars Suna*
and
Edgars Suna*
Latvian Institute of Organic Synthesis, Aizkraukles 21, LV-1006, Riga, Latvia. E-mail: edgars@osi.lv
First published on 15th October 2020
Abstract
Protonation of pyridines with a strong acid is a general and straightforward approach to achieve efficient aggregation induced emission (AIE) in structurally remarkably simple organic molecules that lack any of the conventional luminophores. The relationship between the nature of counter ion and the AIE efficiency is demonstrated. The superiority of the perchlorate counter ion is attributed to efficient stabilization of the key intermolecular π+–π interactions between neighboring luminophore molecules in the crystal lattice.
A large variety of optoelectronic appliances such as photovoltaic devices,1 organic light emitting diodes,2 and organic field-effect transistors3 as well as a number of light harvesting applications including artificial photosynthesis4 and photon refining,5 rely on solid state luminophores. In 2001, Tang introduced a general method to achieve highly efficient solid state luminescence (SSL) in purely organic molecules, generally known as aggregation induced emission (AIE).6 The vast majority of AIE luminogens (AIEgens) are based on sterically hindered, bulky propeller-like molecular structures to reduce the planarity of luminophore,7 because the planar architecture is known to be detrimental to the AIE efficiency.8,7a In the meantime, the majority of luminophores with high emission intensity in solution feature a planar structure, so an approach that would allow for the design of the solid state luminogens based on the planar architecture of the known solution state emitters is highly desirable.
We have recently demonstrated that the formation of intermolecular interactions between quaternary nitrogen-containing heteroaromatic cations and aromatic π-system in the solid state is a convenient and general approach to achieve AIE and to turn-on high SSL in planar organic molecules.9 The high SSL has been attributed to intermolecular charge transfer (ICT) between quaternary heteroaromatic subunits such as N-methyl pyridinium and N-methyl imidazolium cations and aromatic π-systems.9b Herein we report on a further development of the conceptual approach to achieve AIE. Specifically, we demonstrate that efficient AIE can be achieved in planar organic luminophores by simple protonation of pyridine with a suitable acid (Fig. 1). Furthermore, our study provides an important insight into the relationship between the nature of counter ion and the AIE efficiency of pyridinium salts.
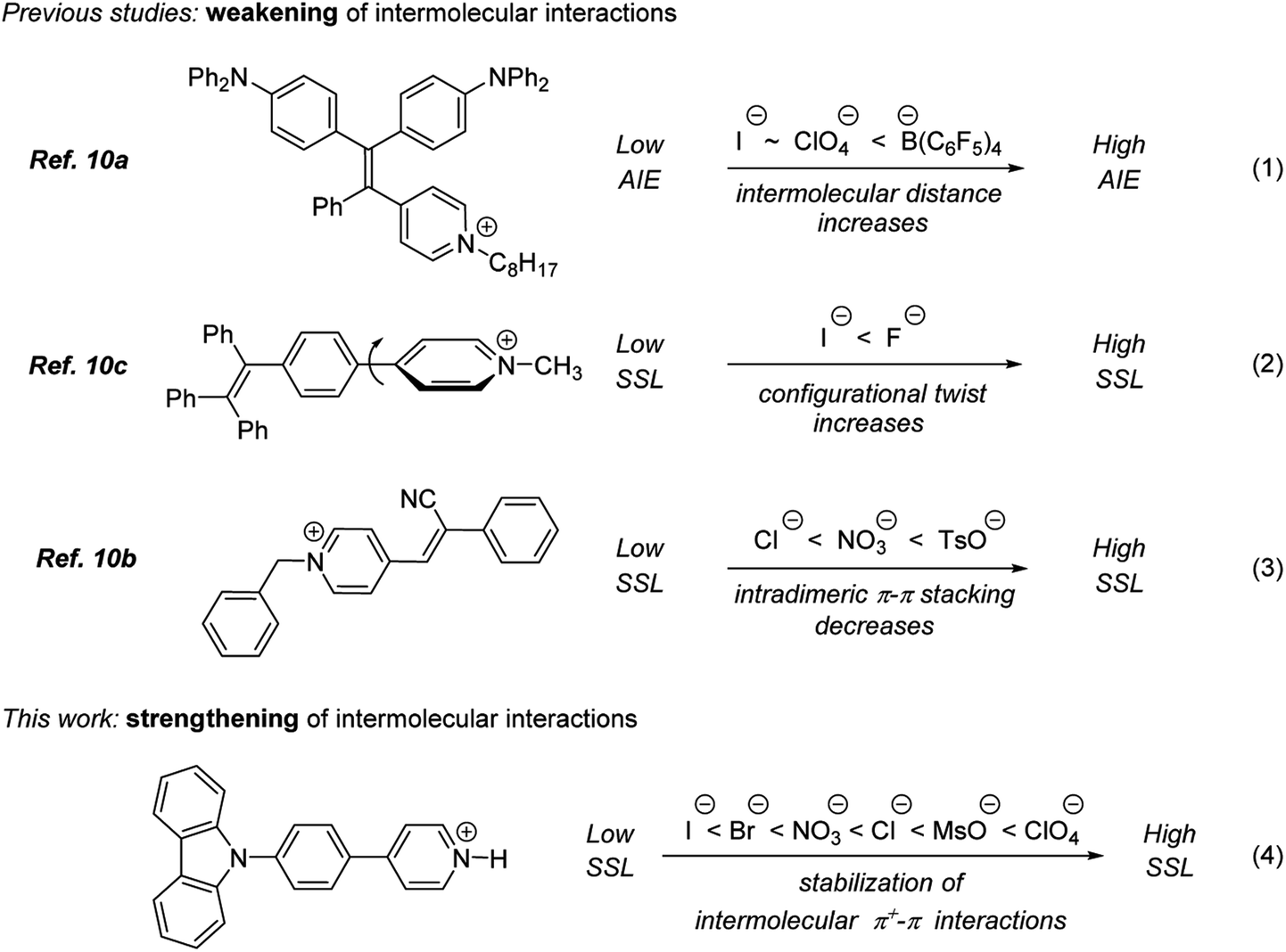 | ||
| Fig. 1 Effect of counter ions on AIE and SSL. | ||
The relationship between the structure of counter ion and luminescence efficiency has been studied in several pyridinium-containing AIE luminogens.10 The highest luminescence efficiency was observed for pyridinium salts possessing a counter ion that helped to avoid the detrimental aggregation-induced quenching (ACQ) effect by minimizing intermolecular π–π interactions between planar luminophore molecules (eqn (1) and (3), Fig. 1)10a,b or by stabilizing twisted conformation of luminogens (eqn (2), Fig. 2).10c In sharp contrast, a completely opposite counter ion effect has been observed in this study. Specifically, counter ions that contributed to strengthening the intermolecular interactions between planar pyridinium luminophores have helped to achieve the highest SSL efficiency (eqn (4), Fig. 1). Perchlorate was found to be superior as the counter-ion, whereas the corresponding mesylate, nitrate as well as halides were inferior. The latter showed apparent negative correlation between the polarizability of the counter ion and SSL emission intensity (Cl > Br > I; see eqn (4), Fig. 1.). The superiority of perchlorate has been rationalized based on X-ray crystallographic analysis as demonstrated below. The striking difference of counter-ion effects in this work (eqn (4)) and in the earlier studies (eqn (1)–(3)) points to an apparent difference in mechanisms that are responsible for the AIE (Fig. 1).
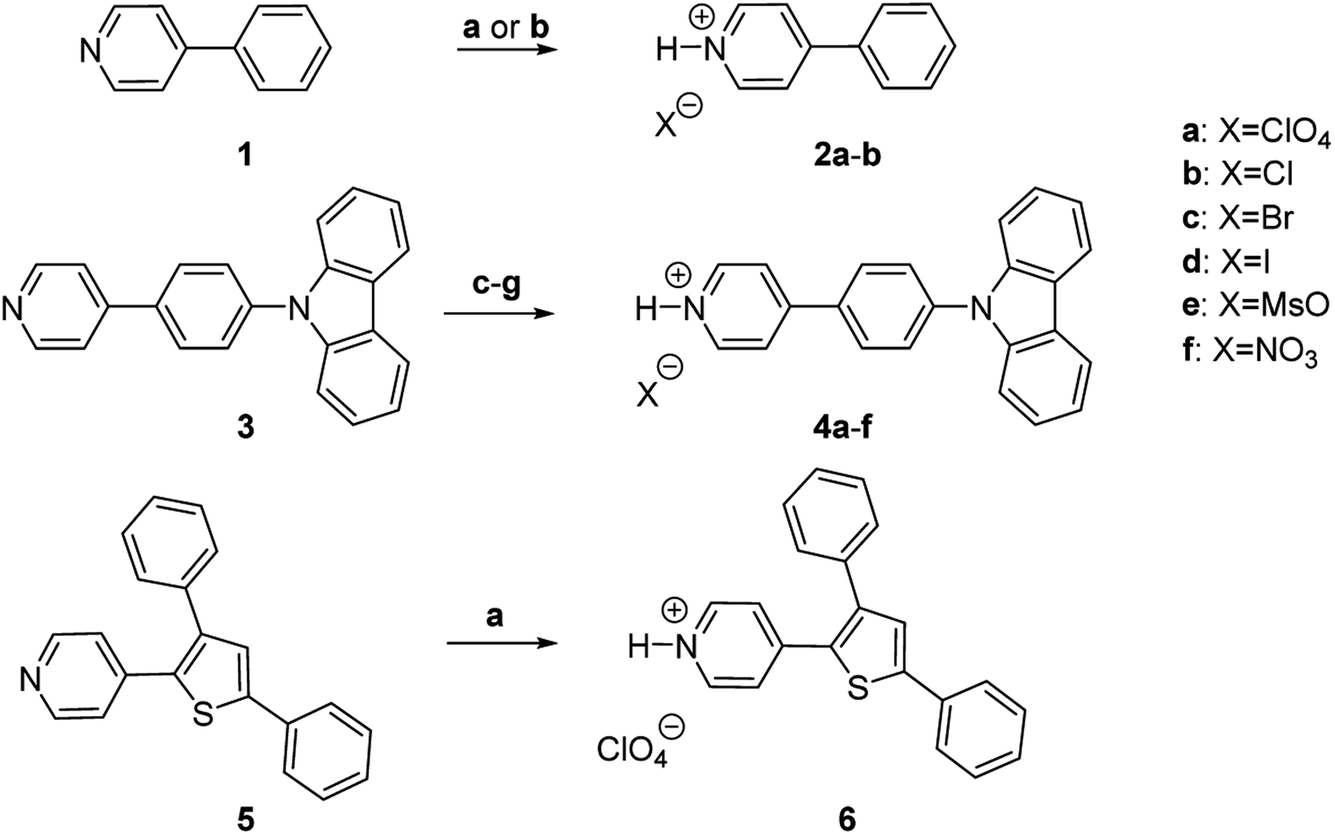 | ||
Fig. 2 Synthesis of pyridinium salts 2a,b and 4a–f. Reagents and conditions: (a) aq. HClO4, MeOH or MeCN, rt, 10 min, 83% (2a); 99% (4a); 32% (6). (b) 4 M HCl in dioxane, EtOAc, rt, 10 min, 97% (2b). (c) 4 M HCl in dioxane, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 CH2Cl2:hexane, rt, 10 min, 93% (4b). (d) aq. HBr, MeOH, rt, 10 min, 91% (4c). (e) aq. HI, EtOH, rt, 1 h, 61% (4d). (f) MeSO3H, MeCN, rt, 10 min, 94% (4e). (g) HNO3, MeCN, rt, 30 min, 99% (4f). :2 CH2Cl2:hexane, rt, 10 min, 93% (4b). (d) aq. HBr, MeOH, rt, 10 min, 91% (4c). (e) aq. HI, EtOH, rt, 1 h, 61% (4d). (f) MeSO3H, MeCN, rt, 10 min, 94% (4e). (g) HNO3, MeCN, rt, 30 min, 99% (4f). | ||
Pyridinium salts 2a,b were obtained by protonation of commercially available 4-phenylpyridine 1 with aqueous HClO4 and hydrochloric acid, respectively. The protonation of previously reported 39b with HClO4, HCl, HBr, HI, MsOH and HNO3 resulted in the formation of pyridinium salts 4a–f (Fig. 2). All pyridinium salts 2a,b and 4a–f were crystalline materials.
UV-vis spectra of all pyridinium salts 2a,b and 4a–f and parent heteroaromatic compounds 1 and 3 were measured in MeCN solutions at room temperature under ambient atmosphere at ca. 10−5 M concentration (see ESI, pages S11–S28†). Pyridine 1 displayed one absorption band at 251 nm (Table 1, entry 1). This single absorption band was red-shifted by 40 nm in the corresponding pyridinium salts 2a,b (Table 1, entries 2 and 3). Pyridine 3 displayed 3 absorption bands at 238, 292 and 322 nm (Table 1, entry 4). The absorption band of pyridine 3 at 238 nm did not change upon protonation, and remained in the range of 236–238 nm for salts 4a–f (Table 1 entries 7–14). The absorption band of free-base 3 at 292 nm experienced slight hypsochromic shift to 281–288 nm in protonated forms 4a–c,e,f. However, the largest difference was observed for the absorption band at 322 nm, which featured bathochromic shift of 52–56 nm in salts 4a–c,e,f. Pyridinium iodide 4d displayed two absorption bands at 314 and 338 nm (Table 1, entry 12).
| Entry | Compound | Additive | λabs, nm | Solution λem, nm | Solid λem, nm | Solution, φ (%) | Solid, φ (%) |
|---|---|---|---|---|---|---|---|
| a Corresponds to wavelength of excitation.b Salt 4a was obtained by evaporating 10:1 MeCN:water solution of 4a under the reduced pressure at 20 °C.c Equilibrium concentration of free base 3 is responsible for the emission in the MeCN solution.d A batch of 4a that was recrystallized from MeCN.e Crystalline batch of 4a obtained by vapor diffusion from Et2O/MeOH. |
|||||||
| 1 | 1 | — | 251a | — | — | <0.1 | <0.1 |
| 2 | 2a | — | 291a | 378 | 417 | 30.0 | 59.6 |
| 3 | 2b | — | 291a | 378 | 434 | 24.6 | 28.5 |
| 4 | 3 | — | 238, 292, 322a | 442 | 371, 387, 407 | 73.1 | 5.7 |
| 5 | 3 | HClO4 (500 equiv.) | — | — | n/a | <0.1 | n/a |
| 6 | 3 | Bu4NClO4 (500 equiv.) | — | 442 | n/a | 62.1 | n/a |
| 7 | 4aAb | 237, 281, 377a | 442c | 496 | n/a | 5.5 | |
| 8 | 4aBd | — | 237, 281, 377a | 442c | 503 | n/a | 24.1 |
| 9 | 4aCe | 237, 281, 377a | 442c | 492 | n/a | 54.6 | |
| 10 | 4b | — | 238, 288, 374a | 442c | 496 | n/a | 42.4 |
| 11 | 4c | — | 236, 282, 378a | 442c | 514 | n/a | 18.2 |
| 12 | 4d | — | 238, 292, 314, 338a | 442c | — | n/a | <0.1 |
| 13 | 4e | — | 238, 287, 376a | 442c | 484 | n/a | 45.8 |
| 14 | 4f | — | 237, 288, 377a | 442c | 468 | n/a | 33.4 |
| 15 | 5 | — | 265, 329a | 416 | 414 | 2.1 | 2.0 |
| 16 | 6 | — | 254, 275, 388a | 489 | 524 | 2.8 | 17.2 |
Pyridine 1 was not emissive in solution, whereas the corresponding protonated species 2a,b displayed high emission in MeCN solution at 378 nm with 30.0% and 24.6% photoluminescence quantum yield (PLQY), respectively (Table 1, entries 2 and 3). Pyridine 1 did not display any observable emission also in the solid state. In sharp contrast, pyridinium salts 2a,b featured high SSL at 417 and 434 nm respectively. Notably, the solid state PLQY for salts 2a,b (59.6% and 28.5%, respectively) were higher than those in MeCN solution (Table 1, entries 2 and 3), and this observation highlights AIE enhancement properties of pyridinium salts 2a,b. Pyridine 3 and its salts 4a–f displayed identical emission wavelength at 442 nm in MeCN solution, suggesting that the same species might be responsible for the solution-state emission of both 3 and 4a–f. We hypothesized that the dissociation of salts 4a–f generates equilibrium concentration of free-base 3 that is responsible for the observed non-structured emission band at 442 nm. To verify this hypothesis, a large excess (500 equiv.) of HClO4 was added to the solution of 3 in MeCN to ensure that all pyridine 3 is protonated. Gratifyingly, a complete quench of the emission was observed for the acidified solution suggesting that protonated pyridines are non-emissive in MeCN solution (Table 1, entry 5). Excess of Bu4N–ClO4 (500 equiv.) was also added to a solution of 3 to verify whether perchlorate anion would affect the solution-state emission. Modest drop of the emission efficiency from 73.1 to 62.1% PLQY was observed when excess Bu4N–ClO4 (500 equiv.) was added to a solution of 3, however the emission spectra did not change (Table 1, entry 6). These experiments provide strong evidence that the equilibrium concentration of free base 3 is responsible for the observed solution state emission of pyridinium salts 4a–f.
Pyridine 3 displayed a structured vibronic emission in the solid state at 371, 387 and 407 nm. The considerable reduction of emission efficiency in the solid state (5.7% PLQY) as compared to that in the solution (73.1% PLQY) points to the aggregation caused quenching (ACQ) properties of luminophore 3 (Table 1, entry 4). The corresponding perchlorate 4a showed a broad non-structured charge transfer (CT) type11 solid state emission with maxima spanning the range from 492 to 503 nm (entries 7–9). Notably, efficiency of the SSL of perchlorate 4a highly depended on crystallinity of the solid material as well as on the properties of crystal lattice. Thus, a batch of perchlorate 4aA that was obtained by concentrating 10:1 MeCN:water solution of 4a under the reduced pressure at 20 °C showed a relatively low emission efficiency (5.5% PLQY; entry 7). X-Ray powder diffraction (XRPD) spectra of 4aA featured a distorted baseline, which is indicative of semi-crystalline character of the solid material (see ESI, page S30†). Notably, recrystallization of solid 4a from MeCN afforded crystalline 4aB that demonstrated considerably higher emission efficiency (24.1%, entry 8). Furthermore, when crystals of 4a were grown by vapour diffusion from Et2O/MeOH, the corresponding material 4aC featured the highest emission efficiency among all prepared salts of 3 (54.6% PLQY, entry 9). Importantly, XRPD spectra confirmed that 4aB and 4aC were different polymorphs (see ESI, page S30–S31†). These data show that the efficiency of SSL depends on the degree of crystallinity and on the properties of crystal lattice. It should be also noted that the solid state emission spectra of all batches 4aA–4aC were similar.
Pyridinium salts 4b,c,e,f displayed broad non-structured CT type emission11a in the solid state11b at 496, 514, 484 and 468 nm (entries 10, 11, 13 and 14, respectively). Pyridinium mesylate 4e showed the second highest emission efficiency after perchlorate 4aC (45.8% PLQY; entry 13). Pyridinium chloride 4b, bromide 4c and nitrate 4f were inferior with PLQY of 42.4, 18.2 and 33.4, respectively (entries 10, 11, 14), whereas pyridinium iodide 4d was completely non-emissive (<0.1% PLQY) in the solid state (entry 12). Notably, our data demonstrates an apparent negative correlation between solid state PLQY and the polarizability of the halide counter ion.12 It should be also noted, that the lack of solid-state emission for iodide-containing pyridinium salts has been observed previously.13,9b Importantly, the highest intensity of SSL has been observed for pyridinium salts 2a and 4aC possessing perchlorate counter ion (Table 1).
To demonstrate the scope and generality of the protonation approach as a means to achieve AIE and high SSL, perchlorate salt 6 of a known luminophore 5 (ref. 14) possessing a pyridine core with 3,5-diphenylthiophene substituent was prepared (Fig. 2). Pyridine 5 displayed two absorption bands at 265, 329 nm and perchlorate 6 showed 3 absorption bands at 254, 275 and 388 nm in MeCN solution. A broad non-structured emission at 416 nm was observed for pyridine 5 in the solution. Protonation of 5 resulted in a red-shift of the emission to 489 nm in perchlorate 6. Pyridine 5 showed low emission efficiency both in solution and in the solid state with PLQY of 2.1% (at 416 nm) and 2.0% (at 414 nm), respectively (Table 1, entries 15 and 16). In sharp contrast, pyridinium perchlorate 6 featured pronounced AIE properties, as evidenced by the increased SSL (17.2% PLQY) as compared to the emission efficiency in the solution (2.8% PLQY). Furthermore, noticeable red-shift of 35 nm was also observed for the solid state emission maxima (524 nm) vs. that in the solution (489 nm; entry 14). The observed AIE properties and increased SSL upon the protonation of 5 by perchloric acid has confirmed that the protonation is a general approach to achieve AIE and high SSL in pyridine-containing organic luminophores.
X-ray crystallography provided an important insight into key interactions underlying the observed AIE properties and high SSL for pyridinium luminophores (Table 2). Single crystals of 2a, 2b, 4aB, 4aC, 4b, 4f and 6 suitable for X-ray crystallography were obtained by Et2O vapour diffusion into the corresponding MeCN and MeOH solutions or by crystallization from MeCN. Importantly, all pyridinium salts 2a, 2b, 4aB, 4aC, 4b, 4f and 6 featured π+–π interactions between pyridinium cation and aromatic π-system in the crystal lattice (Table 2). For example, pyridinium salt 2a formed a strictly parallel head-to-tail packing with 3.751 Å distance for the π+–π interactions of pyridinium subunit with phenyl ring. Likewise, pyridinium salt 2b formed a near parallel head-to-tail packing with 3.776 and 3.900 Å distances between the pyridinium and phenyl ring centroids (Table 2). Similar head-to-tail packing was also observed for pyridinium salt 4aC, which feature nearly parallel off-center π+–π interactions between the charged pyridinium subunit and electron rich carbazole moiety. Furthermore, molecules of 4aC formed stacked dimers in the crystal lattice, where every molecule was engaged in two distinct π+–π interactions at 3.618 and 3.655 Å distances between respective carbazole and pyridinium moieties (Table 2). In contrast, the corresponding polymorph 4aB formed parallel displaced packing with only one intermolecular π+–π interactions at 4.215 Å distance between the pyridinium ring and the phenylene linker. Similar packing was also observed for pyridinium nitrate 4f with the corresponding π+–π interactions at 3.927 Å distance. Chloride 4b featured 3.535 Å distance for the off-center π+–π interactions between pyridinium and carbazole subunit, whereas non-parallel off-centered π+–π interactions between pyridinium heterocycle and phenyl moiety (3.714 Å distance) were observed for pyridinium perchlorate 6 (Table 2).
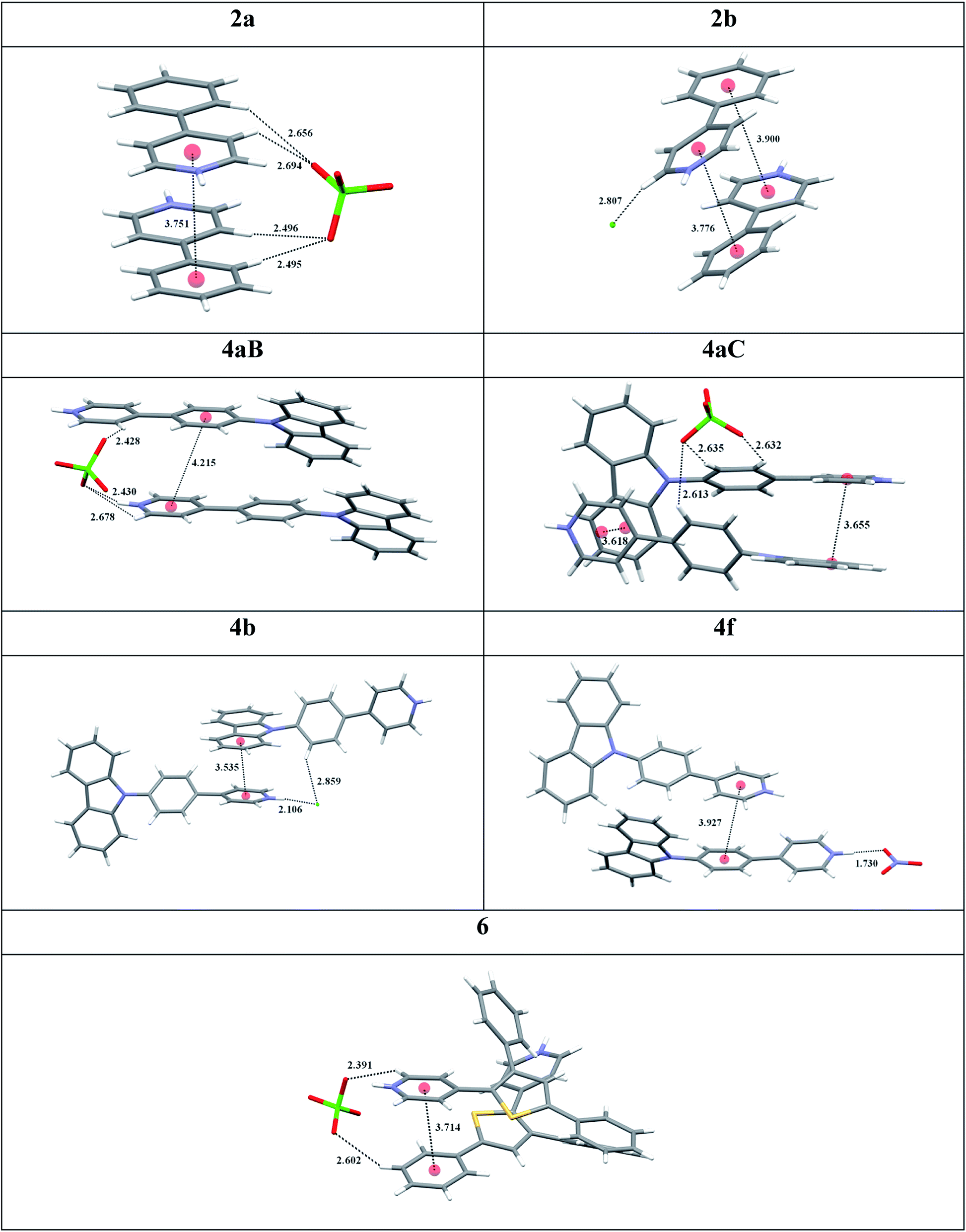 |
X-ray crystallography data has also provided an evidence that perchlorate counter ion provides an important stabilization of the key π+–π interactions between pyridinium subunit and aromatic π system. Thus, the perchlorate forms a hydrogen–bonded bridge between two molecules of 2a in the crystal lattice as evidenced by the short H-bond contacts spanning a range from 2.495 to 2.694 Å (Table 2). The symmetric nature of these interactions not only helps to stabilize the intermolecular π+–π interactions, but also ensures the coplanar conformation around the biaryl bond in 2a. In contrast, the chloride in 2b is involved in hydrogen bonding with only one pyridinium subunit (2.807 Å bond length), which leads to 22.7° biaryl dihedral angle in 2b. The bridging effect of perchlorate apparently contributes to the higher crystal lattice energy for 2a vs. 2b, as evidenced by the considerably higher melting point for 2a (145 °C) as compared to that for 2b (74 °C). Notably, perchlorate 2a featured remarkably high SSL efficiency (59.6% PLQY), whereas chloride 2a was less efficient (28.5% PLQY; see entry 3 vs. 2, Table 1). The propensity of perchlorate ion to form the hydrogen–bonded bridge between two molecules in the crystal lattice has been also observed for luminogens 4aB (range of contacts from 2.428 to 2.678 Å), 4aC (2.613–2.635 Å) and 6 (2.391 and 2.602 Å). Among them, the perchlorate-bridged dimer 4aC possessed the highest SSL efficiency (54.6% PLQY, entry 9, Table 1). The corresponding chloride 4b, bromide 4c and mesylate 4e were less efficient (entries 10, 11, 13 vs. 9). Surprisingly, the hydrogen–bonded bridging interactions were not observed for nitrate 4f (Table 2). The lack of intermolecular stabilization in the crystal lattice has obviously resulted in inferior SSL for 4f as compared to perchlorate 4aC (entry 14 vs. 9, Table 1). Hence, among all single crystals of pyridinium salts examined, only the perchlorate counter ion contributes to stabilization of the key π+–π interactions, and this effect results in the higher SSL efficiency for perchlorates as compared to chlorides and bromides. The apparent relationship between the intermolecular π+–π interactions and SSL has been also demonstrated in previous study for structurally closely related N-methyl pyridinium perchlorate,9b and TDDFT calculations provided strong evidence that the π+–π interactions generated through-space CT bands in the crystal state, resulting in SSL.
Additional support for the involvement of through-space CT in the SSL of 2a, 2b, 4aB, 4aC, 4b, 4f and 6 was obtained by a series of control experiments. Thus, the emission of 3 + HCl15 in MeCN solution was measured at 298 K, 190 K and 77 K. At room temperature MeCN solution of 3 + HCl featured lack of emission (see also entry 5, Table 1), whereas in a frozen MeCN matrix at 190 K intense emission was observed for 3+HCl with maxima at 545 nm. Notably, a remarkable blue shift of emission maxima to 452 nm was measured upon further cooling to 77 K (Fig. 3A). The observed pronounced rigidochromic effect is characteristic of CT-type emission due to the polarization flip between ground and excited states.16 Furthermore, the observed hypsochromic shift also speaks against the possible involvement of excimers in the SSL of the protonated pyridine 3 + HCl, because eximer-driven emission typically features a batochromic shift of the emission maxima.17 An additional evidence against eximer formation was obtained by measuring the emission spectra of protonated pyridine 2a in MeCN at various concentrations. Pyridinium perchlorate 2a was chosen due to the relatively high solubility in organic solvents and observable emission in the monomeric state (entry 2, Table 1). The emission maxima for 2a did not change at concentrations in the range from 0.001 mM to 10 mM indicating that excimer formation is not responsible for the luminescence of 2a (Fig. 3B).
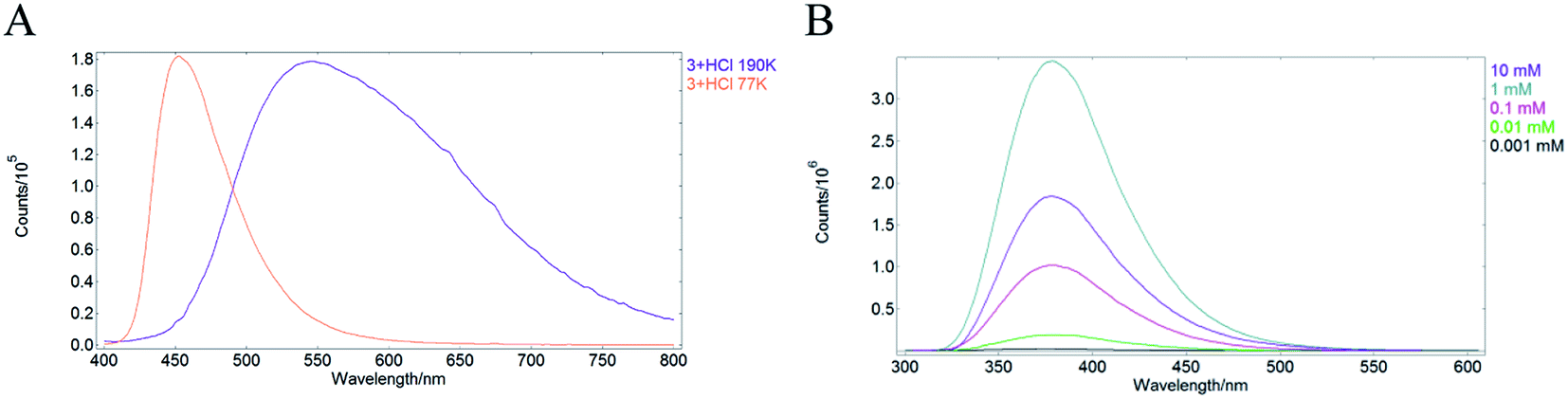 | ||
| Fig. 3 (A) Solid state emission of 3 + HCl in MeCN at 190 K (purple) and 77 K (pale red); (B) emission spectra of 2a in MeCN solution at various concentrations. | ||
In summary, this study demonstrates a straightforward and versatile approach to achieve efficient AIE in structurally diverse purely organic luminogens. The developed method relies on a simple protonation of pyridine derivatives by a strong acid to generate pyridinium cation for the key non-covalent π+–π interactions with aromatic π-system that leads to high SSL through the intermolecular charge transfer (ICT) mechanism.9b Hence, the protonation of pyridines is a highly straightforward and complementary approach to the previously developed N-alkylation/ion exchange sequence as a means of achieving the SSL. High solid state emission efficiency (up to 60% PLQY) has been attained by the protonation of structurally remarkably simple organic molecules such as 4-phenyl pyridine that lacks any of the conventional luminophores. The versatility of the protonation approach is demonstrated in three structurally distinct series of pyridine-containing luminogens with both planar and twisted architectures. The solid state emission efficiency depends strongly on the degree of crystallinity of the solid luminogens as well as on the structure of the pyridinium counter ion. Perchlorate as the counter ion was superior in terms of SSL efficiency as compared to chloride, bromide, mesylate and nitrate whereas iodide affected complete luminescence quench. X-Ray crystallographic analysis provided evidence that perchlorate counter-ion can increase AIE effect by (a) forming a H-bonded bridge between the neighbouring luminophore molecules in the crystal lattice, thus providing an additional stabilization of the key intermolecular π+–π interactions, (b) enabling H-bond assisted planarization of the luminophore molecule. Hence, these insights together with control experiments provide an evidence against the RIM AIE mechanism as well as excimer formation in protonated pyridine luminophores. We believe that the remarkable operational simplicity of the protonation as a general approach to achieve high SSL and the elucidation of counter-ion effect will provide ample opportunities for the rational design and development of solid state emitters.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by ERDF (project no. 1.1.1.1/18/A/063 “Next Generation Aggregation Induced Emission Luminogens for Artificial Lighting Sources”). We thank Dr S. Belyakov for X-ray crystallographic and XRPD analysis.References
- (a) B. Mi, Y. Dong, Z. Li, J. W. Y. Lam, M. Haussler, H. H. Sung, H. S. Kwok, Y. Dong, I. D. Williams, Y. Liu, Y. Luo, Z. Shuai, D. Zhu and B. Z. Tang, Chem. Commun., 2005, 3583–3585 RSC; (b) Y. Li, Z. Li, Y. Wang, A. Compaan, T. Ren and W.-J. Dong, Energy Environ. Sci., 2013, 6, 2907–2911 RSC.
- (a) Z. Ning, Z. Chen, Q. Zhang, Y. Yan, S. Qian, Y. Cao and H. Tian, Adv. Funct. Mater., 2007, 17, 3799–3807 CrossRef CAS; (b) W. Qin, Z. Yang, Y. Jiang, J. W. Y. Lam, G. Liang, H. S. Kwok and B. Z. Tang, Chem. Mater., 2015, 27, 3892–3901 CrossRef CAS; (c) J. Huang, N. Sun, Y. Dong, R. Tang, P. Lu, P. Cai, Q. Li, D. Ma, J. Qin and Z. Li, Adv. Funct. Mater., 2013, 23, 2329–2337 CrossRef CAS; (d) J. N. Moorthy, P. Venkatakrishnan, P. Natarajan, Z. H. Lin and T. J. J. Chow, J. Org. Chem., 2010, 75, 2599–2609 CrossRef CAS; (e) J. Huang, N. Sun, J. Yang, R. Tang, Q. Li, D. Ma and Z. Li, Adv. Funct. Mater., 2014, 24, 7645–7654 CrossRef CAS.
- (a) S. G. Surya, H. N. Raval, R. Ahmad, P. Sonar, K. N. Salama and V. R. Rao, TrAC, Trends Anal. Chem., 2019, 111, 27–36 CrossRef CAS; (b) Z. Zhao, Z. Li, J. W. Y. Lam, J.-L. Maldonado, G. Ramos-Ortiz, Y. Liu, W. Yuan, J. Xu, Q. Miao and B. Z. Tang, Chem. Commun., 2011, 47, 6924–6926 RSC; (c) M. P. Aldred, G.-F. Zhang, C. Li, G. Chen, T. Chen and M.-Q. Zhu, J. Mater. Chem. C, 2013, 1, 6709–6718 RSC.
- (a) M. Zhang, X. Yin, T. Tian, Y. Liang, W. Li, Y. Lan, J. Li, M. Zhou, Y. Ju and G. Li, Chem. Commun., 2015, 51, 10210–10213 RSC; (b) Y. Zeng, P. Li, X. Liu, T. Yu, J. Chen, G. Yang and Y. Li, Polym. Chem., 2014, 5, 5978–5984 RSC; (c) F. Qiao, L. Zhang, Z. Lian, Z. Yuan, C.-Y. Yan, S. Zhuo, Z.-Y. Zhou and L.-B. Xing, J. Photochem. Photobiol., A, 2018, 355, 419–424 CrossRef CAS.
- (a) P. Duan, D. Asthana, T. Nakashima, T. Kawai, N. Yanai and N. Kimizuka, Faraday Discuss., 2017, 196, 305–316 RSC; (b) L. Li, Y. Zeng, T. Yu, J. Chen, G. Yang and Y. Li, ChemSusChem, 2017, 10, 4610–4615 CrossRef CAS.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- (a) Y. Tang and B. Z. Tang, Principles and Applications of Aggregation-Induced Emission, Springer International Publishing, New York, 2019 CrossRef; (b) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS.
- (a) T. Forster and K. Z. Kasper, J. Phys. Chem., 1954, 1, 275 Search PubMed; (b) J. B. Birks, Photophysics of aromatic molecules, Wiley-Interscience, London, 1970 Search PubMed.
- (a) K. Leduskrasts and E. Suna, RSC Adv., 2019, 9, 460–465 RSC; (b) K. Leduskrasts, A. Kinens and E. Suna, Chem. Commun., 2019, 55, 12663–12666 RSC.
- (a) N. Adarsh and A. S. Klymchenko, Nanoscale, 2019, 11, 13977–13987 RSC; (b) G. Zhang, X. Zhang, L. Kong, S. Wang, Y. Tian, X. Tao and J. Yang, Sci. Rep., 2016, 6, 37609 CrossRef CAS; (c) M. Bineci, M. Bağlan and S. Atılgan, Sens. Actuators, B, 2016, 222, 315–319 CrossRef CAS.
- (a) X. Wang, S. Wang, J. Lv, S. Shao, L. Wang, X. Jing and F. Wang, Chem. Sci., 2019, 10, 2915–2923 RSC; (b) X.-H. Jin, C. Chen, C.-X. Ren, L.-X. Cai and J. Zhang, Chem. Commun., 2014, 15878 RSC.
- E. V. Anslyn and D. A. Dougherty, Modern physical chemistry, University Science, Sausalito, CA, 2004 Search PubMed.
- I. Richter, M. R. Warren, J. Minari, S. A. Elfeky, W. Chen, M. F. Mahon, P. R. Raithby, T. D. James, K. Sakurai, S. J. Teat, S. D. Bull and J. S. Fossey, Chem.–Asian J., 2009, 4, 194–198 CrossRef CAS.
- I. Karpaviciene, M. Jonusis, K. Leduskrasts, I. Misiunaite, E. Suna and I. Cikotiene, Dyes Pigm., 2019, 170, 107646 CrossRef CAS.
- Anhydrous HCl in dioxane (20 equiv.) was added to a solution of 3 in MeCN (ca. 10−5 mol L−1).
- A. J. Lees, Comments Inorg. Chem., 1995, 17, 319–346 CrossRef CAS.
- The formation of eximers was proposed to be responsible for the observed AIE effect in certain planar protonated pyridinium salts based on the large batochromic shift (50 nm) of the emission in thin films as compared to that in solution: X. Cui, Y. Hao, W. Guan, L. Liu, W. Shi and C. Lu, Adv. Opt. Mater., 2020, 2000125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1988911–1988916. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra07137d |
| This journal is © The Royal Society of Chemistry 2020 |