DOI:
10.1039/D0RA07078E
(Paper)
RSC Adv., 2020,
10, 36930-36940
Synergistic effect of Ni–Ag–rutile TiO2 ternary nanocomposite for efficient visible-light-driven photocatalytic activity†
Received
17th August 2020
, Accepted 24th September 2020
First published on 7th October 2020
Abstract
P25 comprising of mixed anatase and rutile phases is known to be highly photocatalytically active compared to the individual phases. Using a facile wet chemical method, we demonstrate a ternary nanocomposite consisting of Ni and Ag nanoparticles, decorated on the surface of XTiO2 (X: P25, rutile (R)) as an efficient visible-light-driven photocatalyst. Contrary to the current perspective, RTiO2-based Ni–Ag–RTiO2 shows the highest activity with the H2 evolution rate of ∼86 μmol g−1 W−1 h−1@535 nm. Together with quantitative assessment of active Ni, Ag and XTiO2 in these ternary systems using high energy synchrotron X-ray diffraction, transmission electron microscopy coupled energy dispersive spectroscopy mapping evidences the metal to semiconductor contact via Ag. The robust photocatalytic activity is attributed to the improved visible light absorption, as noted by the observed band edge of ∼2.67 eV corroborating well with the occurrence of Ti3+ in Ti 2p XPS. The effective charge separation due to intimate contact between Ni and RTiO2 via Ag is further evidenced by the plasmon loss peak in Ag 3d XPS. Moreover, density functional theory calculations revealed enhanced adsorption of H2 on Ti8O16 clusters when both Ag and Ni are simultaneously present, owing to the hybridization of the metal atoms with d orbitals of Ti and p orbitals of O leading to enhanced bonding characteristics, as substantiated by the density of states. Additionally, the variation in the electronegativity in Bader charge analysis indicates the possibility of hydrogen evolution at the Ni sites, in agreement with the experimental observations.
1. Introduction
The lack of clean energy resources is one of the severe problems being presently faced globally, which necessitates the development of next-generation environmentally benign and sustainable energy-efficient technologies. The use of photocatalytic systems, harvesting inexhaustible, and clean solar energy to split water and produce molecular oxygen/hydrogen has been considered as one of the best approaches so far.1,2 However, even after decades of active global research, the photocatalytic reaction system remains highly challenging.
Bulk and/or nanostructured semiconductors with superior physicochemical and optical properties are employed for photocatalysis. The semiconductor acts as an active photocatalyst for the light-induced photochemical reactions because of its unique electronic structure characterized by a filled valence band and an empty conduction band. Among various semiconductors, bulk and nanostructured TiO2 have stood as the central facet because of their high photocatalytic efficiencies, photochemical stability, nontoxicity, and high reactivity.3,4 However, the large bandgap of TiO2 (∼3.2 eV for anatase and brookite, ∼2.9 eV for rutile) requires an excitation wavelength that falls in the UV region, which consists of less than ∼5% of the total sunlight (solar light consists of ∼5% UV, ∼45% visible, and ∼50% infrared) incident at the earth's surface. Thus, the utilization of solar light for a photocatalytic or photoelectrochemical process can be enhanced by tuning the bandgap of TiO2 to the visible or infrared region.5 Apart from that, conventional TiO2-based homogeneous photocatalysts have inherent drawbacks, such as the fast recombination of photo-induced electron–hole pairs. Whereas, the heterogeneous photocatalysis has proven to be more appealing than its homogeneous counterpart due to the manipulation of the bandgap and enhancement of charge carrier separation.6–9 Thus, tailoring the physicochemical and optical properties of TiO2 is indispensable in utilizing a large fraction of the solar spectrum to realize the photocatalyst's indoor applications.
Heterostructures of nano-sized TiO2 with 0 dimensional (0D) noble nanoparticles,6,10,11 and 2D materials12 have shown remarkable photocatalytic properties. Among 0D particles deposited TiO2, Pt and Pd co-catalyst have shown exceptional photocatalytic properties. However, both these materials cannot be used for practical applications; whereas, the usage of metallic Ag is cost-effective.13 It is well known that Ag exhibits poor photocatalytic activity because of the weaker binding energy of Ag–H (37 kcal mol−1). Few of the recent report shows that present of Ni along with Ag is expected to enhance the H2 generation rate due to stronger binding energy of Ni–H (52 kcal mol−1)13 and reduced contact resistance14 in ternary nanocomposite. Further, it is to be noted that, among the various available form of TiO2, the photocatalytic activity of rutile TiO2 (RTiO2) is rarely reported as compared with the anatase TiO2 and P25 (a mixture of 80%wt anatase and 20%wt rutile) because of its lower photocatalytic activity.15,16 The lower activity of RTiO2 has been attributed to its low specific surface area, fast recombination, and positive location of its conduction band minimum.17,18 However, a recent report has shown that RTiO2 can also act as an active photocatalyst under the appropriate experimental conditions; although, the photocatalytic activity is noted in the UV region.19
In this work, we report a thorough and systematic experimental study on Ni, Ag, and XTiO2 (X: P25 and rutile)-based ternary nanocomposite synthesized using a wet chemical method to study the synergistic effects of multiple nano-junctions on the visible-light-driven H2 generation rate (HER). Synthesized nanocomposites are denoted as Ni–Ag–P25 (PAN), and Ni–Ag–RTiO2 (RAN) throughout the manuscript. Observed results are further supported with the density functional theory (DFT) simulations, which brings out the significance of both Ni and Ag in a ternary nanocomposite for H2 evolution.
2. Experimental methods
2.1. Synthesis of Ni–Ag–XTiO2 composites samples
Ni–Ag–XTiO2 (X: P25, and rutile (R)) ternary nanocomposite were synthesized by utilizing sonication based wet chemical synthesis following the method reported earlier.20 Materials for composites include Ni nanopowder with an average diameter of 200 nm (Ningxia Orient Tantalum Industry, Co. Ltd.), rutile TiO2 (99.5% assay, Alfa Aesar), P25 (>99.5%, Aeroxide, Evonik), AgNO3 (0.01 mol L−1 purchased from Sigma-Aldrich). De-ionized (DI) water from the laboratory water supply was used for work. Based on the previous report,20 Ni (6%wt)–Ag (10%wt)–XTiO2 (84%wt) has been prepared. We have prepared a whole batch of 100 mg of each Ni–Ag–XTiO2 ternary composite as follows. Synthesis of the batch was carried out as two 50 mg doses in two 100 mL Erlenmeyer flasks with 75 mL DI water. After the initial mixing of the Ni, TiO2, and DI water, water dissolved AgNO3 was added. The material synthesis was carried out by sonication using a Skymen JTS-1018 water bath ultrasonic cleaner. The cleaner was pre-heated to 70 °C, and the materials were then sonicated @39 kHz using maximum power (∼3 A current) for a duration of 4 h. After the continuous sonication, the samples were kept still at room temperature overnight until separation through sedimentation of particles had cleared the water. After sedimentation, water was carefully rinsed without losing synthesized particles, followed by decanting the synthesized particles. Drying the remaining water off from the as-prepared samples was carried out by evaporation of the water in an open beaker by utilizing a hot plate to heat samples to approximately 100 °C in the ambient air. Dried samples were then scraped off the beaker and stored in sample bottles as a dry powder. In a similar fashion, controlled samples of Ag–XTiO2 binary nanocomposites were also prepared. During the growth process, it was noted that Ni–XTiO2 (P25, R) could not be achieved, which further support the idea of required noble metal glue (Ag) to contact Ni with TiO2.
2.2. Instrumentation
Room-temperature powder XRD patterns were recorded with Rigaku SmartLab 9 kW equipped with five-axis θ–θ goniometer and 1D solid-state detector and scintillator using Co-Kα. All three ternary nanocomposites were also measured at the Brockhouse High Energy Wiggler Beamline, CLS, Canada. All these powder samples were loaded in Kapton capillaries of 0.9 mm diameter. The data collection was performed with nominally 33 keV photons. The refined wavelength from a LaB6 calibrant was 0.35414 Å, and the sample to detector distance was 448.58 mm. An exposure time of 2–3 s was used, with 120 frames for each of these data collection using a PerkinElmer area detector, a flat panel X-ray detector, 200 μm pixel, 40 × 40 cm2. UV-vis spectroscopy was carried with Shimadzu UV-2600 Spectrometer. X-ray photoelectron spectroscopy (XPS) was performed with Al Kα using Thermo Fisher Scientific ESCALAB 250Xi XPS System. Field emission scanning electron microscope (FESEM)-energy dispersive spectroscopic (EDS) mapping was carried out with Zeiss Ultra plus FESEM. Transmission electron microscope (TEM)-EDS mapping was performed using JEOL JEM-2200FS EFTEM/STEM. For HER, samples were cycled in Perfect Light PCX50B photo reactor through eight monochromatic LED light sources covering near UV regime to visible light (in a range of 365 to 630 nm) equipped with magnetic stirring. Sampling and analysis of gaseous species were carried out using Agilent Micro 490 GC gas chromatograph (GC) equipped with an H2 sensitive column (10MS5A) after the end of each illumination round.
2.3. Photocatalytic reduction of water for H2 evolution
The catalytic activity of the pristine and synthesized nanocomposites was carried out in a photoreactor to measure any activity differences. Pristine TiO2, their binary and ternary nanocomposites were used to run experiments in the same conditions, such as identical cells, same volume, an equal amount of DI-water, a total weight of 5 mg of each sample, and fixed illumination time of 2 h. A magnetic stirring bar for stirring the suspension was carried out during the experiment. The 5 mg samples were added into a quartz bottle with a total volume of 68 mL. For each experiment, 3 s sonication was carried out to provide a consistent starting point in terms of the dispersion of the suspension. The gaseous content was flushed for a fixed repeatable starting point by using argon. In the first round of preparation, we applied careful Ar-flushing, including heating of the water, to minimize the water-dissolved air. The argon flush's primary function was to establish a reliable control method to monitor gas leaks that arose through effusion and, to a lesser extent, diffusion mechanisms, thus detecting potential abnormally high leak rates. By utilizing the available monochromatic wavelengths each at the time, eight experiments per sample were carried out. The repetitive nature of the experiments also provided initial results of the reusability of the samples.
2.4. Density functional theory
To corroborate the experimental findings of observed higher activity in the ternary nanocomposites, DFT simulations were carried out using the plane wave pseudo potential based, Vienna Ab initio Simulation Package (VASP) to corroborate with the experimental findings.21 We have modeled a Ti8O16 cluster using the super cell approach, such that the cluster is placed in the center of a cubic box with dimensions of 20 Å. Since a considerably large supercell is used to model the Ti8O16 clusters, a gamma centered k grid was used for the Brillouin zone integration. The plane waves included in the basis set were expanded using kinetic energy cut off of 400 eV, and Gaussian smearing was employed. The exchange and correlation were described using GGA in the PAW-PBE method.22,23 The atomic positions were relaxed with zero external stress and this is ensured by using energy and force tolerances of 10−6 eV and 0.001 eV Å−1, respectively. To account for the underestimation of band gaps caused by GGA, the Hubbard U parameter in the Dudarev's method is employed for Ti with the values, U = 4.5 eV and J = 0 eV.24
3. Results and discussions
3.1. X-ray diffraction
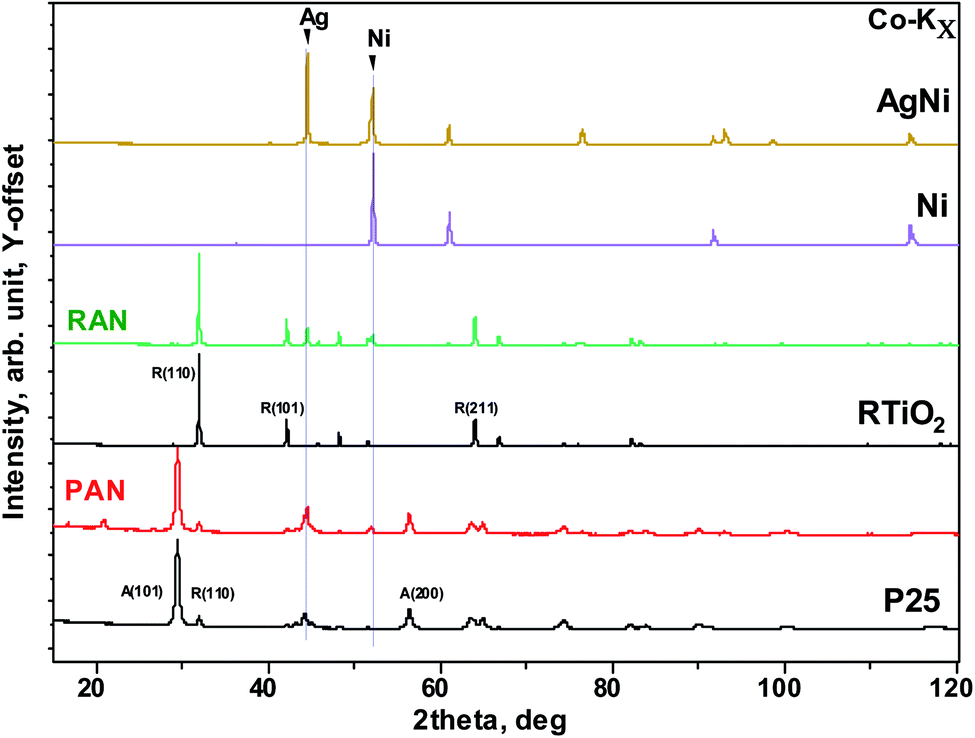
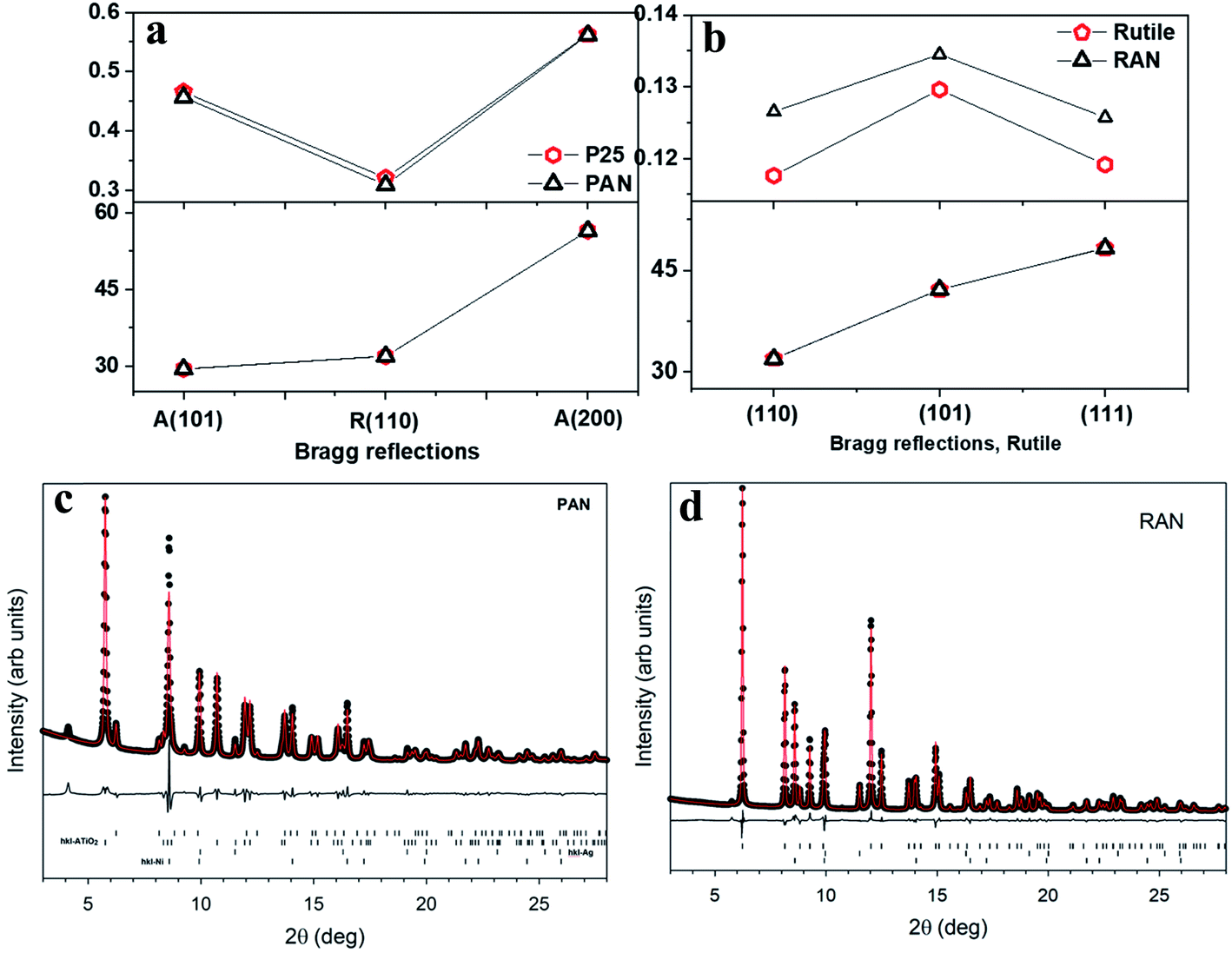
Fig. 1 shows the experimental X-ray diffraction (XRD) patterns for the pristine XTiO2 and ternary nanocomposites along with the XRD pattern of Ag and Ni. Both the ternary nanocomposites, that is, PAN, and RAN contain all the Bragg peaks associated with their respective three individual components, thus, suggesting the successful preparation of ternary nanocomposites. The intensity, peak position and full width at half maximum (FWHM) being the three crucial features of an XRD pattern, have been used to determine the structural effect of Ni and Ag on the unit cell size and shape of TiO2 (via peak position) and crystalline quality (via peak width). Thus, the insight into XRD analysis has been carried out using representative Bragg reflections of P25 vs. PAN, and RTiO2 vs. RAN (Fig. 2). No significant change in the relative intensities of XRD peaks suggests no observable changes in the atomic arrangements of TiO2. Fig. 2 indicates the possible changes in PAN, and RAN compared to their pristine samples. No significant changes in the peak positions indicate the unit cell's unaltered size in these ternary nanocomposites. Further, no significant changes in the FWHM are observed for PAN as compared to P25. However, RAN shows a systematic increase in the FWHM as compared to RTiO2. Such compensating change corresponds to the refinement of the local atomic arrangement in RAN with degraded crystallinity compared to the pristine RTiO2, possibly due to changes in the dielectric environment of RTiO2. Rietveld refinement on the high-energy synchrotron XRD data has been further carried out (Table 1) to elucidate the phase fraction of Ni, Ag, and XTiO2 in ternary nanocomposites. According to the previous report,20 Ni (6%wt)–Ag (10%wt)–XTiO2 (84%wt) was chosen for the synthesis process. However, Rietveld refinement results (Table 1) show the wt% contribution of TiO2 as 90% in both the ternary nanocomposites. However, the %wt of Ni and Ag is ∼5 in RAN, whereas, for PAN, it is ∼2 and 8, respectively, although the growth parameters used were kept the same.
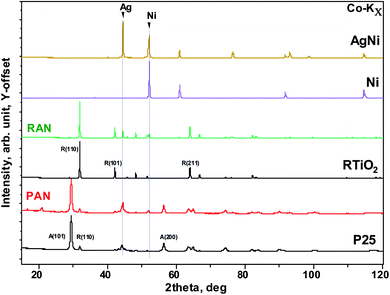 |
| | Fig. 1 Co-Kα source-based X-ray diffraction patterns of the selected pristine (such as Ni, rutile, and P25) as well as nanocomposite (PAN, and RAN). Two vertical lines correspond to Ag and Ni main Bragg peak. | |
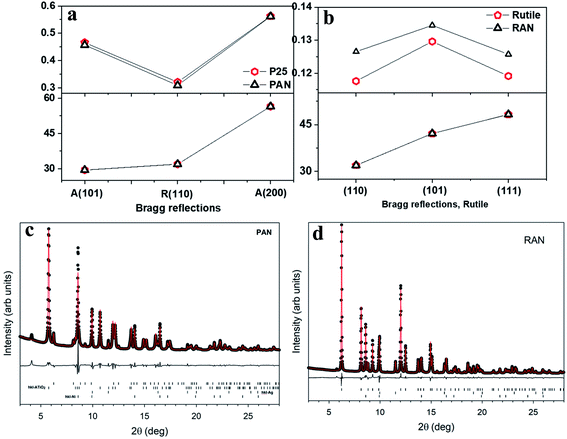 |
| | Fig. 2 Changes in the XRD parameters in PAN (a), and RAN (b) as compared to their pristine counterparts, taken from lab source. Rietveld refinement for PAN (c) and RAN (d) using three phase refinement. The black circles are the data points, red lines are the fits, and the difference curves are shown beneath. The tick marks show the allowed hkl reflections of rutile, anatase, Ni, Ag from top to bottom. Ticks are only shown for phases present in each sample. | |
Table 1 Results of the Rietveld refinements on the synchrotron powder diffraction data
| Wt% |
PAN |
RAN |
| Rutile |
9.7 (2) |
89.7 (2) |
| Anatase |
76.8 (4) |
|
| Ni |
2.96 (8) |
4.60 (3) |
| Ag |
10.50 (8) |
5.68 (2) |
| Fit Rw |
5.29 |
3.95 |
3.2. UV-vis spectroscopy
The UV-vis spectra of P25, PAN, RTiO2, and RAN ternary nanocomposites are shown in Fig. 3a. The bandgap energy of the prepared nanocomposites is further determined from (αhν)2 versus hν.25 For indirect transition, that is, (αhν)1/2 versus hν, the Eg estimated from the α = 0 extrapolation is ∼2.25 and 2.1 eV for P25 and RTiO2, respectively, which does not seem realistic. Thus, the direct transition is considered to be most probable in the current case, which is a better fit than the corresponding indirect bandgap fit, resulting in bandgap values of 3.1 eV (P25) and 2.85 eV (RTiO2). Fig. 3b shows that the optical bandgap of PAN, and RAN ternary nanocomposites is ∼2.85, and 2.67 eV, which is relatively lower than that of P25 (3.1 eV), and RTiO2 (2.85 eV). The absorption edge of the PAN, and RAN ternary nanocomposites extended an unambiguous red shift compared to the P25, and RTiO2, which reflects that the electronic structure and optical properties of ternary nanocomposites have been modified due to the presence of Ag and Ni nanoparticles. These results thus suggest that the incorporation of Ag and Ni with pristine TiO2 caused a redshift of the UV-vis absorption spectrum. Moreover, the PAN and RAN ternary nanocomposites exhibited higher absorption, suggesting that the enhanced electron transfer among the individual components. Benefiting from such visible light absorption and high absorption intensity, the ternary nanocomposite is expected to show improved utilization of solar energy and perhaps possesses an enhanced visible-light-driven photocatalytic performance. The observed redshift in the band edge can be understood due to band edge alignment when metal gets in contact with a semiconductor.26 Further, it is to be noted that no significant change in the band gap of Ag–P25, and Ag–RTiO2 binary nanocomposites (ESI Fig. S1†) has been found.
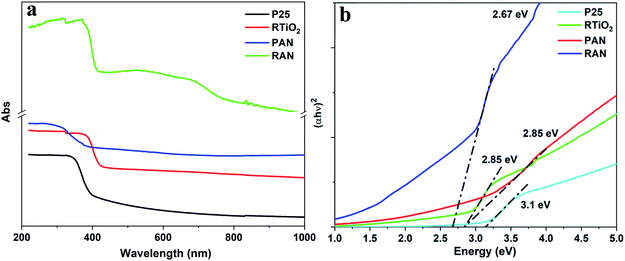 |
| | Fig. 3 (a) UV-vis spectra and corresponding (b) (αhν)2 vs. hν for P25, RTiO2, PAN, and RAN. | |
Apart from the above observations, RAN shows a clear absorption peak in the range of 500–700 nm (Fig. 3a), which could be assigned to Ag NPs (absorption peak ∼ 539 nm) and Ni NPs (∼700 nm).
3.3. X-ray photoelectron spectroscopy
X-ray photoelectron spectroscopy (XPS) analysis is carried out to determine the composition and identify the chemical states of grown nanocomposites (Table 2). The high-resolution XPS spectra of Ti 2p (Fig. 4(a) and (c)) for pristine P25 and RTiO2 show the two strong peaks corresponding to Ti 2p3/2 and Ti 2p1/2. The observed peak position (Table 2) and peak separation of 5.8 eV of the Ti 2p doublet agree well with that reported for TiO2. Fig. 4(b) and (d) present the O 1s XPS spectra of the above two samples. The peaks located at ca. 530.5 eV corresponds to the O 1s electron binding energy (B.E.) for TiO2. Further, a weak shoulder observed at a higher binding energy of 531 eV can be attributed to the O–H from the absorbed H2O.
Table 2 Comparison of XPS analysis
| Sample |
Peak position/FWHM (eV) |
| Ti4+ |
Ti3+ |
O 1s |
| 2P3/2 |
2P1/2 |
2P3/2 |
2P1/2 |
TiO2 |
O–H |
Ti–OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
| P25 |
458.50/0.96 |
464.20/1.86 |
|
|
530.44/1.07 |
531/1.93 |
|
| PAN |
459.00/1.32 |
464.70/2.26 |
|
|
530.24/1.25 |
531/2.55 |
|
| RTiO2 |
459.00/1.04 |
464.76/2.03 |
|
|
530.34/1.01 |
531/2.14 |
|
| RAN |
459.33/1.15 |
465.41/1.98 |
458.97/1.05 |
464.56/2.04 |
530.40/1.25 |
531.50/1.75 |
532.96/3.50 |
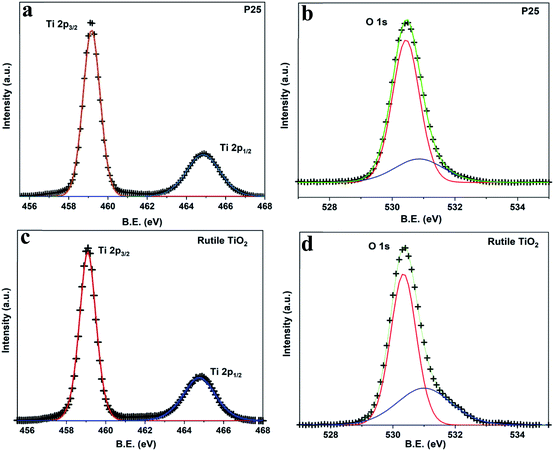 |
| | Fig. 4 High-resolution XPS spectra of (a) Ti 2p, (b) O 1s from P25 and (c) Ti 2p, (d) O 1s from RTiO2. | |
XPS spectra of PAN and RAN are shown in Fig. 5. Fig. 5(a–c) shows XPS spectra for Ti 2p, O 1s, and Ag 3d for PAN. Apart from an increase in the B.E. by ∼0.5 eV and an increase in FWHM (Table 2), no other changes are observed for the Ti 2p spectrum. Observed changes in B.E. and FWHM could be due to change in the chemical environment due to the presence of Ag and Ni. Further, no changes are observed in the O 1s XPS spectrum. The Ag 3d spectrum for the PAN consists of two individual peaks located at ca. 368 eV and ca. 374 eV, which can be assigned to Ag 3d5/2 and Ag 3d3/2 of metallic Ag, respectively. For Ag 3d, a broad peak at ca. 368.70 eV is also detected in addition to the metallic peak at 368 eV. This peak has been attributed to the presence of metallic sub-nanoparticles that are smaller than 4 nm.27 This peak has been shown to change its position depending on the size and distribution of sub-nanoparticle. The observed large FWHM (∼1.36 eV) of this peak points to a large distribution of sizes. Thus, no significant changes are observed for PAN as compared to P25.
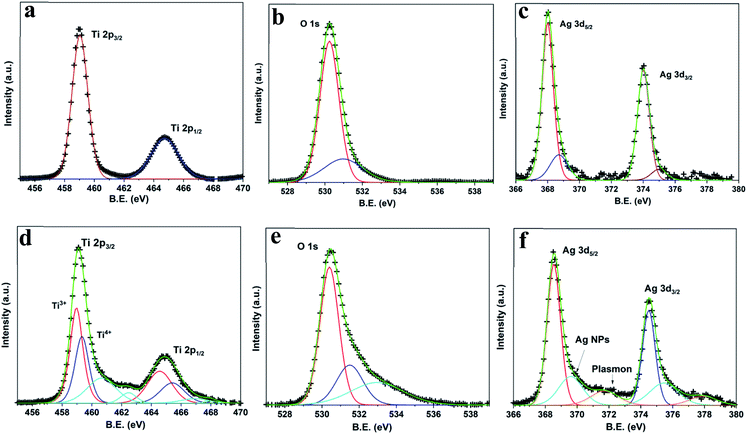 |
| | Fig. 5 High-resolution XPS spectra of from PAN ((a) Ti 2p, (b) O 1s, (c) Ag 3d) and RAN ((d) Ti 2p, (e) O 1s, (f) Ag 3d). | |
Fig. 5(c–e) shows XPS spectra for Ti 2p, O 1s, and Ag 3d for RAN. Changes in the XPS spectra of RAN have been observed as compared to RTiO2 (Table 2). High-resolution XPS spectrum for Ti 2p can be divided into two different peaks, one centered at ca. 458.97 eV, ascribed to Ti3+ species, and the other centered at ca. 459.33 eV, corresponding to Ti4+. Observance of Ti4+ with increased B.E. and Ti3+ peak as compared to bare RTiO2 suggests a lower electron density of the TiO2, possibly due to intimate contact of RTiO2 with Ag and/or Ni nanoparticles. The presence of Ti3+ oxide with a narrow band gap28 (well corroborates with UV-vis spectrum) can be advantageous to the higher photocatalytic activity of the RAN driven by visible light. The presence of satellite peak suggests changes in the coulombic potential of RTiO2 as the photo-ejected electron passes through the valence band, which is possibly due to the presence of Ag/Ni. Further, the wide and asymmetric O 1s spectra indicate that there would be more than two components, which can be further fitted into three peaks including crystal lattice oxygen (OTi–O at ca. 529.4 eV), surface hydroxyl groups (OO–H at ca. 531.7 eV), and adsorbed O2 (at ca. 533.5 eV). In Fig. 5e, the Ag 3d spectrum for the RAN consists of two individual peaks corresponding to Ag 3d5/2 and Ag 3d3/2, a broad peak corresponding to metallic sub-nanoparticles as also observed for PAN. Further, the peak related to plasmon loss in Ag has also been detected for RAN, as evidenced by the separation of ∼4 eV between the main peak and plasmon peak.29,30 The presence of a plasmon loss peak suggests the charge transfer from RTiO2 to Ag, indicating the intimate interaction between Ag and TiO2 via O in RAN, which is further supported via DFT simulation discussed later.
Furthermore, the absence of Ti3+ oxide peak in the XPS spectrum for Ti 2p (ESI Fig. S2†) in Ag–RTiO2 nanocomposite corroborates well with no change in the band gap in the same.
Concerning the question of the chemical state of Ni, it was clearly seen by XAS-photoemission electron microscopy14 (a highly surface sensitive local probe down to 10 nm spatial resolution) that the Ni–Ag–TiO2 ternary composites do not show any higher states of Ni, for example, Ni2+.
Thus, XPS analysis indicates that Ni and TiO2 are connected to each other via Ag. The observed interaction between Ni and TiO2 via Ag in RAN is further confirmed using field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM)-based energy dispersive spectroscopy (EDS) mapping.
3.4. Energy dispersive spectroscopy (EDS) mapping
A morphological examination of RAN is carried out with FESEM-EDS (Fig. 6). The FESEM imaging, along with EDS mapping, suggests the homogenous distribution of RTiO2, Ag, and Ni nanoparticles in RAN. Further, TEM-EDS imaging (Fig. 7) suggests the successful formation of RAN ternary nanocomposite, wherein Ni nanoparticle of size ∼200 nm is attached to RTiO2 via Ag nanoparticle of size ∼20 nm. However, the same could not be observed for PAN. For instance, in the case of PAN, Ag decorated onto P25 is mainly observed, that is, the formation of the binary nanocomposite. This observation suggests RTiO2 as the primary phase to provide the base for the formation of the targeted nanocomposite.
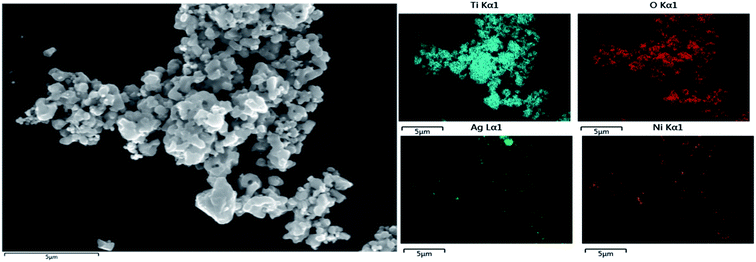 |
| | Fig. 6 FESEM-EDS mapping showing homogenous distribution of RTiO2, Ag, and Ni nanoparticles in RAN. | |
 |
| | Fig. 7 TEM and corresponding EDS mapping showing formation of heterojunction between Ni (red)–RTiO2 (blue) via Ag (green) in RAN. | |
Based on these observations, the HER efficiency is accordingly expected to be highest in RAN followed by PAN, wherein, our primary goal was to study the potential activation and increment of HER in the visible light range in these ternary nanocomposites as discussed in the next sub-section.
3.5. HER experiment
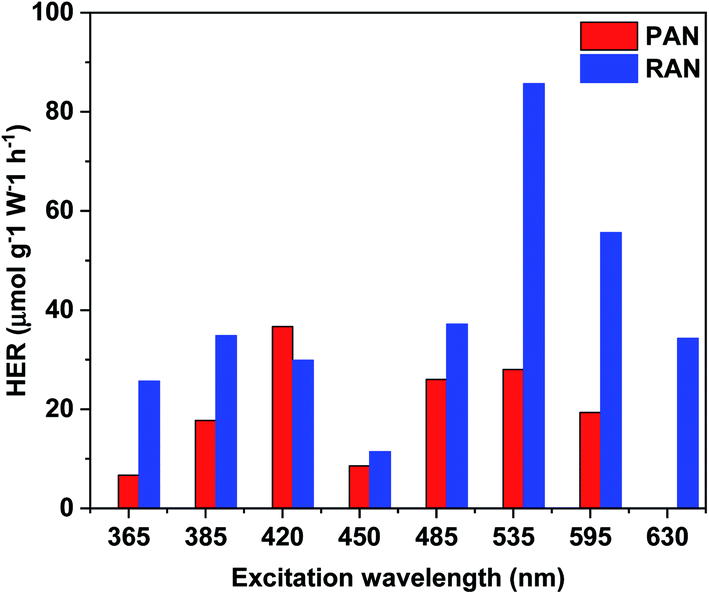
TiO2 based ternary nanocomposites (PAN and RAN) dispersed in DI water were used for photocatalytic H2 generation in the light range of 365 to 630 nm, thus covering a significant part of sunlight. In a control experiment, samples of bare XTiO2 and binary Ag–XTiO2 were also tested for comparison. The most significant and distinctive difference being that hydrogen evolution can also be observed in the visible light range. Fig. 8 shows the H2 production rate of PAN and RAN. It is to be noted that the HER of RAN (86 μmol g−1 W−1 h−1) is highest at the excitation wavelength of 535 nm, which is 3 folds higher than PAN (28 μmol g−1 W−1 h−1) at the same irradiation. Further, the observed HER of RAN ternary nanocomposites is found to be much higher than the pristine XTiO2, which showed negligible H2 yield in the above-noted excitation range and Ag–RTiO2 binary nanocomposite (3.23 μmol g−1 W−1 h−1@535 nm). The observation of hydrogen formation in the experiments is systematic and repeatable. The observed higher hydrogen yield of RAN compared to PAN over the whole excitation range corroborates well with the formation of intimate contact between Ni and RTiO2 via Ag, as evidenced by XPS and EDS mapping. The observed superior photocatalytic activity of rutile TiO2-based RAN as compared to P25-based PAN is intriguing and contrast with the previous reports, which shows higher catalytic efficiency of P25 as compared to rutile TiO2.16 The observed intriguing results of RTiO2 outperforming anatase-rich P25 can be attributed to the formation of intimate contact between Ni and RTiO2 via Ag, the %wt of Ni and Ag, absorption edge in the visible region and electron transfer among the individual components in RAN, as evidenced by the EDS mapping and Ti3+ in the XPS. This observation is further supported with the observed lower H2 yield in RA nanocomposite, which is attributed to the absence of Ti3+ oxide peak in the XPS spectrum for Ti 2p and no change in band gap.
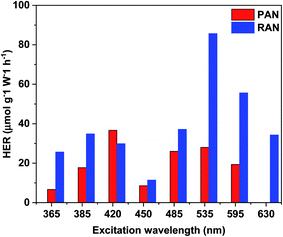 |
| | Fig. 8 Plots of the photocatalytic H2-evolution rate vs. various excitation wavelength for PAN, and RAN. | |
3.6. Density functional theory
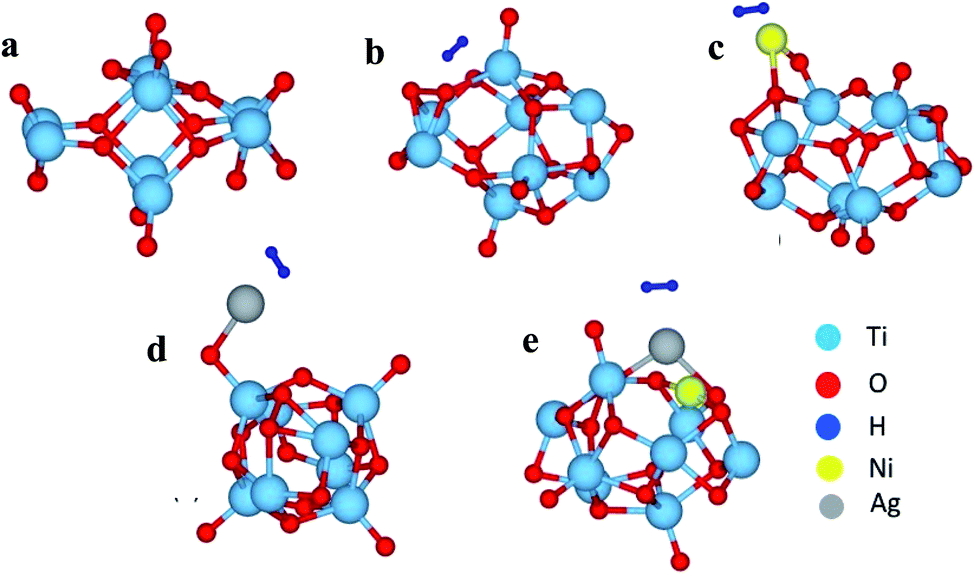
To get further insight into the observed properties, we have modeled a Ti8O16 cluster using the supercell approach, and total energy optimization was carried out to find the most stable geometry. Using this optimized geometry, H2 adsorption was studied for pristine as well as Ag and/or Ni adsorbed clusters. The optimized geometries are shown in Fig. 9. The adsorption energies calculated to understand the energetics are shown in Tables 3 and 4.
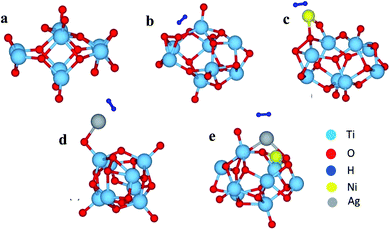 |
| | Fig. 9 The optimized cluster geometries of (a) Ti8O16, (b) Ti8O16H2, (c) Ti8O16NiH2, (d) Ti8O16AgH2 and (e) Ti8O16AgNiH2. | |
Table 3 The calculated adsorption energies of Ag and Ni on Ti8O16 cluster
| Cluster |
Adsorption energy (eV) |
| Ni-on-O (Ti8O16Ni) |
−5.4 |
| Ni-on-Ti (Ti8O16Ni) |
−4.99 |
| Ag-on-O (Ti8O16Ag) |
−3.34 |
| Ag-on-Ti (Ti8O16Ag) |
−1.78 |
| Ti8O16AgNi |
−6.44 |
Table 4 The calculated adsorption energies of hydrogen on Ti8O16 cluster
| Cluster |
Adsorption energy (eV) |
| Ti8O16AgNi |
−6.44 |
| Ti8O16H2 |
−1.32 |
| Ti8O16AgH2 |
−4.96 |
| Ti8O16NiH2 |
−7.11 |
| Ti8O16NiAgH2 |
−8.29 |
The calculated adsorption energies indicate that Ni and Ag atoms prefer to attach to the O atom rather than the Ti atom (Table 3). Hence, for further calculations involving H2 adsorption, these geometries were considered and compared with H2 adsorption on the pristine cluster. Comparing Ag and Ni, Ni lowers the adsorption energy for H2, and hence, when both Ag and Ni are present in the cluster, the adsorption happens more easily (Table 4). The bond lengths for the optimized clusters are presented in Table 5. For the pristine cluster, the Ti–O bond length in the cage is 2.13 Å, while for outer Ti–O, it is 1.69 Å. Interestingly, the H–H bond distances are larger when Ni and/or Ag are present in the cluster, and this indicates that the H2 molecule dissociates to H atoms, and that gets attached to the metal atoms in the ternary cluster. On the other hand, in the case of pristine Ti8O16, it stays as H2, during adsorption, retaining its molecular bond distance of 0.75 Å. It is also seen that there is a significant increase in bond distances when Ag and/or Ni are present in the cluster, and these changes in crystal environment will aid in the dissociative adsorption of hydrogen. Further, to understand the charge transfer mechanism, the charge density difference (CHD) between Ti8O16H2 and Ti8O16AgNiH2 is plotted in Fig. 10, and the Bader charge analysis is shown in Table 5.
Table 5 The bond distances and Bader charges for the clusters after H2 adsorption. The Bader charges are shown for the atoms either directly bonded or in the near vicinity of H
| Cluster |
Distances (Å) |
Charge |
| Ti |
O |
Ag |
Ni |
H |
| Ti8O16H2 |
H–H: 0.75 (Ti–O)surface: 1.68, (Ti–O)cage: 2.01 |
2.508 |
−1.105 |
|
|
−0.014, 0.013 |
| Ti8O16AgH2 |
H–H: 0.81, Ag–O: 2.05, H–Ag: 1.82, Ag–Ti: 3.0 (Ti–O)surface: 1.77, (Ti–O)cage: 1.89 |
2.543 |
−1.158 |
0.619 |
|
0.006, 0.080 |
| Ti8O16NiH2 |
H–H: 0.87, Ni–H: 1.57, Ni–O: 2.03 (Ti–O)surface: 1.77, (Ti–O)cage: 2.01 |
2.532 |
−0.768 |
|
0.810 |
0.002, −0.064 |
| Ti8O16NiAgH2 |
H–H: 0.80, Ag–H: 1.94, Ag–O: 2.38 (Ti–O)surface: 1.74, (Ti–O)cage: 2.07, Ni–O: 1.79, Ti–Ni: 2.66, Ag–Ti: 2.94 |
2.488 |
−0.675 |
0.665 |
0.270 |
0.024, 0.046 |
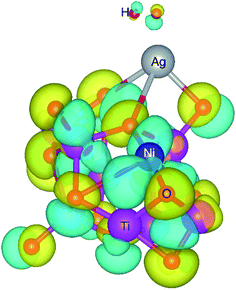 |
| | Fig. 10 The difference in charge density of Ti8O16H2 cluster with and without Ag–Ni adsorbed on it. The yellow and blue regions represent negative and positive charges respectively. The iso-surface level is 0.05. | |
The charge density difference shows that the O atoms gain charge, while the other atoms bonded to them lose charge. The more or less inert nature of Ag, as observed from the charge density difference plot, indicates that the electronegativity of Ag remains the same regardless of the presence of Ni. However, the significant charge isosurface surrounding Ni atom indicates that changes in the chemical environment in the near vicinity affects the electronegativity of Ni. This is evident from the Bader charge analysis, as Ni atom loses less in the presence of Ag, while for Ag, the Bader charges stay consistent irrespective of Ni atom's presence. It can be seen from Tables 3 and 4 that for all cases, O atoms being more electronegative, gain charge, whereas Ti, Ag, and Ni lose charge. The H–H bond distances increase in the presence of both Ag and Ni (0.80 to 0.87 Å) compared to that of the H2 bond length in the pristine Ti8O16 cluster (0.75 Å). This has also been evident from the increase in H–H bond distances and the shortened Ag/Ni–H bonds, compared to Ag/Ni–Ti and Ag/Ni–O bonds. This indicates that the presence of Ag and Ni helps the splitting up of hydrogen into H* atoms, which later recombines to evolve as H2 molecule. The change in electronegativity of Ni, as seen from the Bader charge analysis, indicates the possibility of hydrogen evolution happening at the Ni sites.
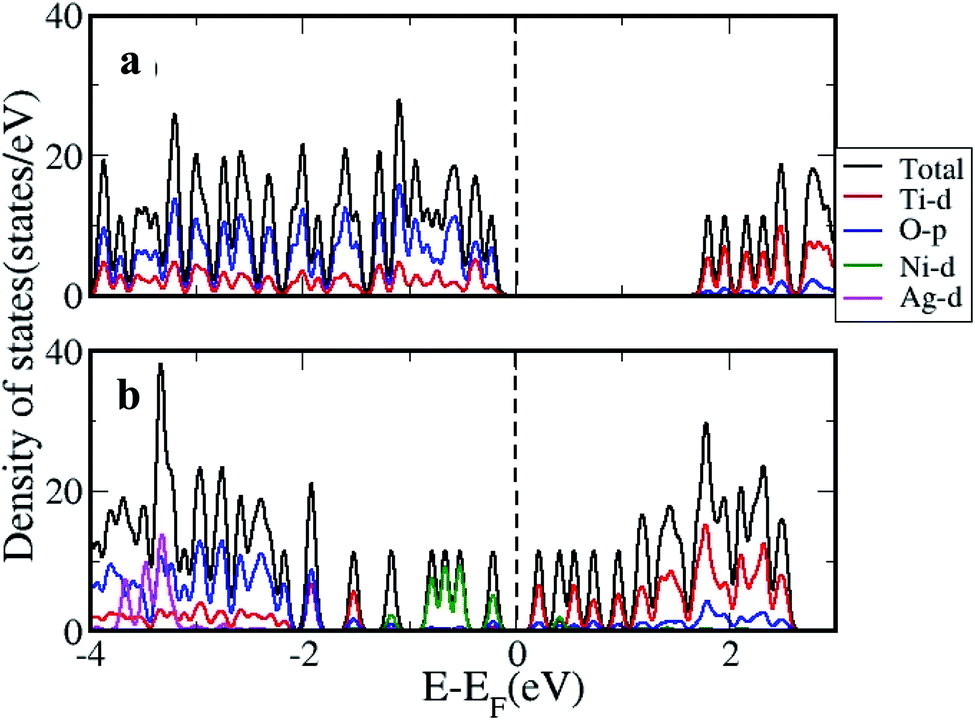
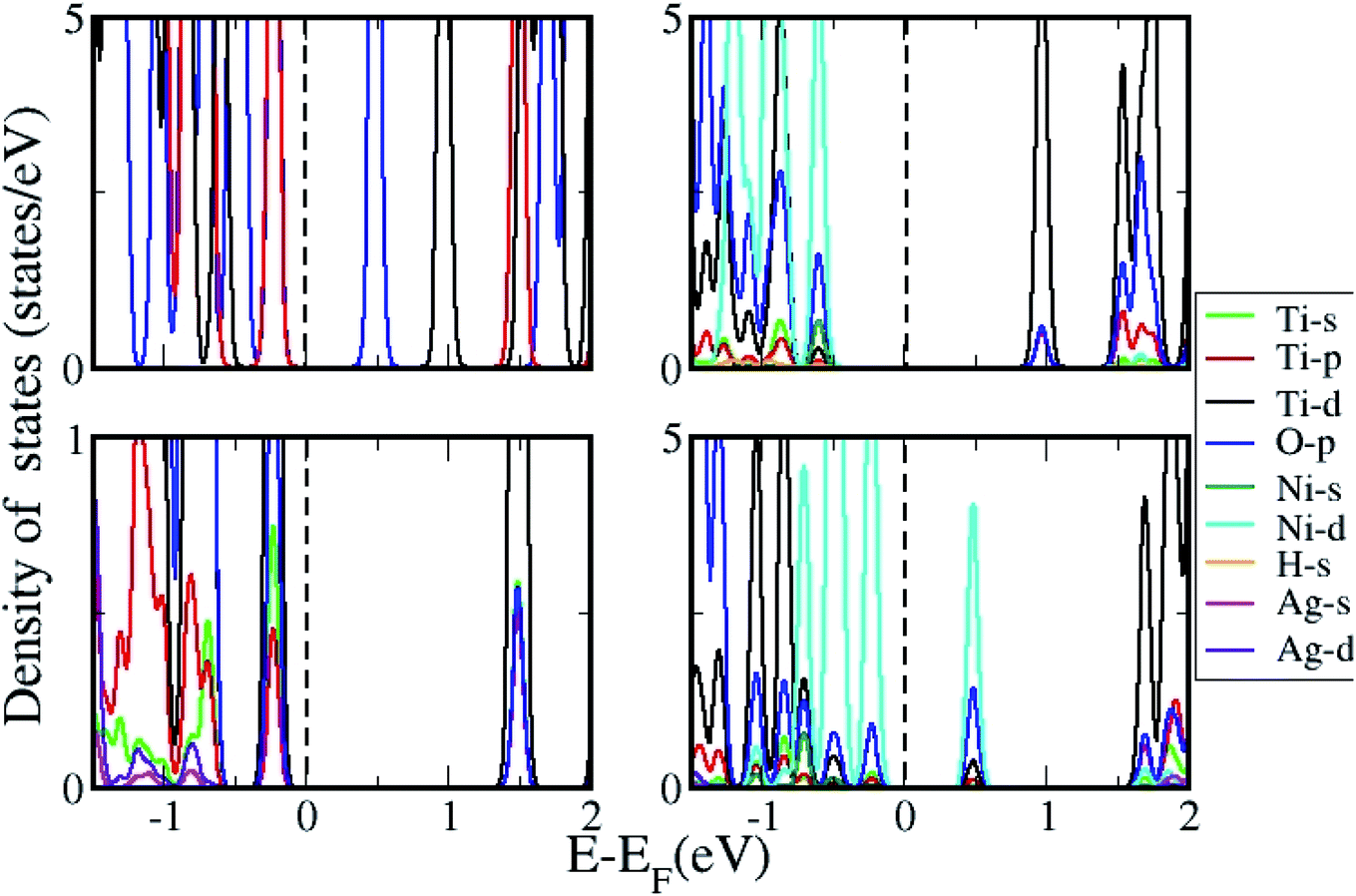
The density of states (DOS) was further calculated for the clusters with and without H2 adsorption using the generalized gradient approximation (GGA)+U approximation. The DOS plotted for pristine, and AgNi adsorbed clusters are shown in Fig. 11, while that with H2 adsorption is shown in Fig. 12. It can be seen from Fig. 12 that pristine Ti8O16 is an insulator with a gap of 1.58 eV. Whereas in the presence of Ag + Ni adsorbed on the Ti8O16 cluster, the bandgap is considerably reduced to 0.04 eV. This indicates that in the presence of metallic atoms, the bandgap of Ti8O16 clusters can be tuned for application purposes. Further, DOS shows that in the presence of Ni and Ag, the CB minimum (CBM) moves towards VB maximum (VBM), as new states are introduced in the energy gap. The shift in CBM towards the lower energy region is more pronounced with Ni addition than that of Ag, while with Ag addition, the VBM moves to a higher energy region. The net effect in both cases is the reduced bandgap and the introduction of additional energy levels in the gap. Nevertheless, when both Ag and Ni are together adsorbed on the cluster, the VBM moves to a higher energy region, combined with additional band gap levels. The atom and orbital resolved DOS presented in Fig. 13 shows Ni alone in the cluster pushes the VBM to lower energy levels and, on the other hand, moves the VBM to higher energy levels. The possibility of hybridization of the transition metals with d orbital of Ti and p orbitals of O is evident from the DOS, leading to enhanced bonding characteristics. When both Ag and Ni are present, this hybridization is more strengthened due to the introduction of multiple levels in the energy gap, further corroborating the experimental findings.
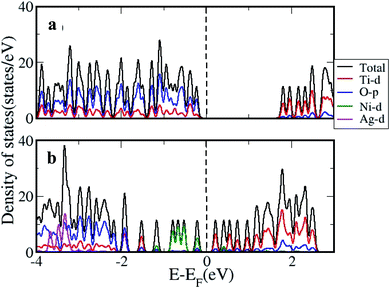 |
| | Fig. 11 The DOS calculated using GGA+U approximation for (a) pristine and (b) Ti8O16AgNi. | |
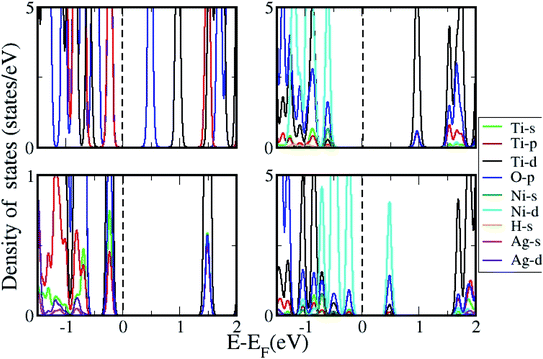 |
| | Fig. 12 DOS for Ti8O16 with H2 adsorption in presence of Ni, Ag and NiAg. | |
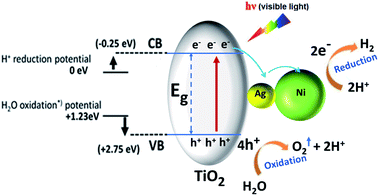 |
| | Fig. 13 Schematic illustration of photocatalytic H2 generation by Ni–Ag–RTiO2 ternary nanocomposite under simulated visible-light irradiation. | |
3.7. The photocatalytic mechanism for water splitting
Based on the above experimental and theoretical findings, the remarkable enhancement in the photocatalytic activity of ternary nanocomposites can be attributed to the unique design of Ni–Ag–RTiO2 nanocomposite that allows for efficient charge separation in the visible region. A tentative mechanism proposed for the high H2 production is illustrated in schematic in Fig. 13. Upon light excitation, the VB electrons are excited to the CB of TiO2, leaving holes in the VB. Previous studies have shown that the CB electrons of TiO2 can be rapidly injected into Ag nanoparticle due to the formation of heterojunction between semiconductor and metal nanoparticle.31 The rapidly injected electrons can either reduce H+ to H2, or they can further move to Ni nanoparticle, which can also act as an active site for H2 generation. Thus, the mediator Ag nanoparticle has a double role, (i) rapidly capture photogenerated electrons to reduce H+ and (ii) transfers these captured electrons to Ni nanoparticle to carry out the H2 evolution reaction step. Hence, the observed HER difference might arise from the lower bandgap allowing activity due to lower energy photon excitation by visible light and possibly delayed recombination of the electron–hole pair due to intimate contact between various components in ternary heterostructure to allow more time for redox chemistry occurrence. We would like to emphasize here that, despite the possibility of oxidation of Ni, which is in fact a natural phenomenon for transition metal nanoparticle, we believe that the proposed synergetic effect primarily appears due to the Ni metallic nature only based on the observation of Ni0 in the X-ray absorption spectroscopy-photoemission electron microscopy14.
4. Conclusion
The Ni–Ag–RTiO2 ternary nanocomposite was successfully constructed through the decoration of Ni nanoparticles onto Ag–RTiO2. The results of XRD, FESEM/TEM-EDS mapping, and XPS spectroscopy demonstrated that the successful formation of rutile TiO2-based ternary nanocomposite. This ternary design builds up a unique transfer path for the photoexcited electrons of RTiO2 to the Ni nanoparticles via Ag nanoparticle bridge and thus effectively suppresses the electron–hole recombination in the RTiO2 photocatalyst. This ternary nanocomposite exhibits a remarkably high photocatalytic H2-generation rate under visible-light irradiation compare to that of pristine RTiO2, Ag–RTiO2 binary nanocomposite, and Ni–Ag–P25TiO2 ternary nanocomposite. We believe that this work demonstrates a promising way for the rational design of metal–semiconductor hybrid photocatalysts that can achieve a high photocatalytic efficiency for use in solar fuel production. Observed higher activity of ternary nanocomposites is further supported with DFT simulation carried out on Ti8O16 clusters.
The results on these ternary systems show ∼3 times higher photocatalytic performances in RTiO2-based ternary nanocomposite over P25-based ternary system and ∼28 times higher than Ag–RTiO2 binary nanocomposite. Intriguingly, the highest activity of the rutile TiO2-based system is in contrast with the reported literature, which suggests that P25 has the highest activity among different forms of TiO2. Thus, this work opens a new preposition in clean energy technology and production.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the European Regional Development Funding and the Oulu Council. The authors thank the Center of Microscopy and Nanotechnology of the University of Oulu. Computing resources were provided by CSC – IT Center for Science, Finland. Academy of Finland grant # 311934 is acknowledged. Part of the research described in this paper was performed at the Canadian Light Source, a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation (CFI), the Natural Sciences and Engineering Research Council (NSERC), the National Research Council (NRC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan.
References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- G. K. Mor, H. E. Prakasam, O. K. Varghese, K. Shankar and C. A. Grimes, Nano Lett., 2007, 7, 2356–2364 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS.
- S. G. Kumar and L. G. Devi, J. Phys. Chem. A, 2011, 115, 13211–13241 CrossRef CAS.
- J. Fang, L. Xu, Z. Zhang, Y. Yuan, S. Cao, Z. Wang, L. Yin, Y. Liao and C. Xue, ACS Appl. Mater. Interfaces, 2013, 5, 8088–8092 CrossRef CAS.
- F. J. Sheu and C. P. Cho, ChemistrySelect, 2018, 3, 354–362 CrossRef CAS.
- X. Yang, J. Qin, Y. Jiang, K. Chen, X. Yan, D. Zhang, R. Li and H. Tang, Appl. Catal., B, 2015, 166–167, 231–240 CrossRef CAS.
- Z. Zhang, Y. Ma, X. Bu, Q. Wu, Z. Hang, Z. Dong and X. Wu, Sci. Rep., 2018, 8, 1–11 CrossRef.
- A. Zielińska-Jurek, J. Nanomater., 2014, 2014, 208920 Search PubMed.
- R. Kavitha and S. G. Kumar, Review on bimetallic-deposited TiO2: preparation methods, charge carrier transfer pathways and photocatalytic applications, Springer International Publishing, 2020, vol. 74 Search PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS.
- D. Gao, W. Liu, Y. Xu, P. Wang, J. Fan and H. Yu, Appl. Catal., B, 2020, 260, 118190 CrossRef CAS.
- X. Shi, S. Posysaev, M. Huttula, V. Pankratov, J. Hoszowska, J. C. Dousse, F. Zeeshan, Y. Niu, A. Zakharov, T. Li, O. Miroshnichenko, M. Zhang, X. Wang, Z. Huang, S. Saukko, D. L. González, S. van Dijken, M. Alatalo and W. Cao, Small, 2018, 14, 1–10 Search PubMed.
- Y. K. Kho, A. Iwase, W. Y. Teoh, L. Mädler, A. Kudo and R. Amal, J. Phys. Chem. C, 2010, 114, 2821–2829 CrossRef CAS.
- Z. Rui, S. Wu, C. Peng and H. Ji, Chem. Eng. J., 2014, 243, 254–264 CrossRef CAS.
- J. Shi, J. Chen, Z. Feng, T. Chen, Y. Lian, X. Wang and C. Li, J. Phys. Chem. C, 2007, 111, 693–699 CrossRef CAS.
- N. G. Park, J. Van De Lagemaat and A. J. Frank, J. Phys. Chem. B, 2000, 104, 8989–8994 CrossRef CAS.
- F. Amano, M. Nakata, A. Yamamoto and T. Tanaka, Catal. Sci. Technol., 2016, 6, 5693–5699 RSC.
- X. Shi, S. Posysaev, M. Huttula, V. Pankratov, J. Hoszowska, J. C. Dousse, F. Zeeshan, Y. Niu, A. Zakharov, T. Li, O. Miroshnichenko, M. Zhang, X. Wang, Z. Huang, S. Saukko, D. L. González, S. van Dijken, M. Alatalo and W. Cao, Small, 2018, 14, 1–10 Search PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- V. Çelik, H. Ünal, E. Mete and Ş. Ellialtioǧlu, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 1–12 CrossRef.
- K. Madhusudan Reddy, S. V. Manorama and A. Ramachandra Reddy, Mater. Chem. Phys., 2003, 78, 239–245 CrossRef.
- I. Tunc, M. Bruns, H. Gliemann, M. Grunze and P. Koelsch, Surf. Interface Anal., 2010, 42, 835–841 CrossRef CAS.
- V. S. Calderon, et al., J. Phys. D: Appl. Phys., 2013, 46, 325303 CrossRef.
- C. Su, L. Liu, M. Zhang, Y. Zhang and C. Shao, CrystEngComm, 2012, 14, 3989–3999 RSC.
- R. A. Pollak, L. Ley, F. R. McFeely, S. P. Kowalczyk and D. A. Shirley, J. Electron Spectrosc. Relat. Phenom., 1974, 3, 381–398 CrossRef CAS.
- P. M. T. M. Van Attekum and J. M. Trooster, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 20, 2335–2340 CrossRef CAS.
- H. Liang, Z. Jia, H. Zhang, X. Wang and J. Wang, Appl. Surf. Sci., 2017, 422, 1–10 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07078e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Assa Aravindh Sasikala Devia,
Harishchandra Singha,
Graham King
*a,
Assa Aravindh Sasikala Devia,
Harishchandra Singha,
Graham King