
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectronic and optical properties of [Au(CH3CSS)]4 cluster. A quantum chemistry study†
Fernando Mendizabal *a and
Sebastián Miranda-Rojas*b
*a and
Sebastián Miranda-Rojas*b
aDepartamento de Química, Facultad de Ciencias, Universidad de Chile, P.O. Box 653, Las Palmeras 3425, Ñuñoa, Santiago, Chile. E-mail: hagua@uchile.cl
bDepartamento de Ciencias Químicas, Facultad de Ciencias Exactas, Universidad Andrés Bello, Avenida República 275, Santiago, Chile. E-mail: sebastian.miranda@unab.cl
First published on 10th September 2020
Abstract
The uses of the sulfur–gold bond in the design of new molecular clusters have gained increasing attention in recent years. Their size and shape are diverse providing a wide variety of optical and electronic properties. Here we present a computational study of the absorption and emission properties of a small [Au(dithioacetate)]4 cluster as a model for these systems. The electronic structure of the Au4S8 core of this cluster permits rationalization of the source of the optical properties and how these are connected with that specific structural scaffold. Due to the complex nature of the aurophilic intramolecular interactions taking place in this system, several methods were used, such as the MP2, SCS-MP2, PBE-D3, and TPSS-D3 levels; both in gas and solvent phases. The absorption spectra of the cluster were calculated by the single excitation time-dependent-DFT (TD-DFT) method, CC2, SCS-CC2, and ADC(2) levels. The ab initio correlated calculations and previously reported experimental data have been used to assess the performance of our calculations. Moreover, the emission T1–So transition was calculated, where the SCS-CC2 level showed an excellent agreement with the experimental results. The core Au4S8 was identified as mainly responsible for the absorption and emission transitions according to the theoretical model.
Introduction
The inorganic molecular clusters formed by transition metals with d8 and d10 electronic configuration represent a particular class. These chemical systems have geometries obtained from various types of noncovalent interactions.1–4 The versatility of the heavy transition metal coordination modes has enabled wide structure and topology variations of the complexes. The chemical systems involve inter- and intramolecular interactions that lead to the formation of dimers, oligomers, chains, sheets, clusters, and nanoparticles; generating systems of high complexity.5–18 In addition to the aforementioned noncovalent interactions, metal–metal interactions in the complexes have also been associated with the direct formation of self-assemblies in the solid-state.19The closed-shell metal–metal contacts have been named as metallophilic interaction, and in the particular case of gold as aurophilic interaction.20–28 These closed-shell interactions are estimated to be energetically between 20 and 50 kJ mol−1 in the case of gold(I) and to be weaker for other metals such as silver(I), copper(I), thallium(I), mercury(II), and platinum(II).1–4,29–31 Such interaction has been evidenced experimentally by solid-state X-ray diffraction32–38 and Nuclear Magnetic Resonance (NMR) measurements.39,40 From a theoretical point of view, the nature of this weak metallic contact in complexes and clusters has been understood as composed by two principal contributions: dispersion and ionic.21 Moreover, the relativistic effects are present in the gold–gold interaction, contributing approximately 20% to the interaction energies;22 while the dispersion interaction contribution is recovered in the electronic correlation. Thus, the theoretical calculations used to describe such interactions are post-Hartree–Fock methods such as second-order Møller–Plesset perturbation theory (MP2), spin-component-scaled Møller–Plesset perturbation theory (SCS-MP2), Coupled-Cluster Single, Double and Triple (CCSD(T)). In addition to that, Density Functional Theory (DFT) with dispersion (Grimme approximation) correction has been used for the same purpose.41–45 In the particular case of the DFT methods, it is often used for chemical systems of higher scale like large molecular clusters or for solid-state with band theory.
Accompanying the increase in the variability of structures with metallophilic interactions, it has emerged a rich chemistry in terms of the availability of a new diversity of excited states with unusual electronic and luminescence properties.46–48 In systems involving gold and others transition metals, the resulting compounds have shown a similar behavior to that observed in organic-light emitting diodes (OLEDs),49,50 with a wide range of emission colors. This relationship is an essential feature as it allows understanding the structural properties of these complexes through the analysis of the luminescent properties of the materials. This will contribute to the design and optimization of new materials with optical properties.46,51,52 Many complexes and clusters involving aurophilic interactions reported to this date have shown luminescence properties, indirectly evidencing the relevance of this interaction in the optical response.2,44
Within this broad group of systems, gold(I) chalcogenides represent an interesting subclass of absorption and luminescent gold(I) clusters.2,44 We have focused on a small cluster of gold thiolate: [Au(dta)]4 (dta = dithioacetate, S2CCH3), in which the four gold atoms are arranged to form a rhombic structure.53,54 The average Au(I)–Au(I) distance is 301.3 pm. The optical properties of this cluster are interesting and can allow describing larger systems. The absorption spectrum of this cluster in carbon disulfide (CS2) phase shows a band at the 407 nm peak maximum and a long-wavelength shoulder at 430 nm.55 The cluster also shows a moderate intense emission band at 743 nm in ethanol glass at 77 K, where emission could be assigned to gold ions in the excited state.
This work aims to understand the process of absorption and emission of energy in the gold cluster of the type d10–d10 [Au(dithioacetate)]4. The absorption and emission processes in the cluster will be studied using the approximate Second-Order Coupled Cluster (CC2 and SCS-CC2), algebraic diagrammatic construction through second-order level (ADC(2)) and time-dependent density functional theory (TDDFT) calculations.
Theoretical models and calculations
The [Au(dta)]4 (dta = dithioacetate, S2CCH3) cluster is modeled using the crystal state as reference. 53,54 We have used the experimental ligand. The theoretical structure is depicted in Fig. 1. The geometries are fully optimized at the scalar quasi-relativistic MP2, SCS-MP2,56 PBE,57 and TPSS58 levels. SCS-MP2 attenuates the overestimation given by the MP2 method. Also, Grimme's dispersion correction is used to incorporate a proper description of the weak interactions when using PBE and TPSS. Nowadays, this approach is known as the DFT-D3 level with Becke–Johnson (BJ) correction.59 The structure optimizations were performed with the gold cluster in gas and CS2 phases in the ground state. The solvent was modelled by using the conductor-like screening model (COSMO) with a dielectric constant (ε) of 2.6 for CS2.60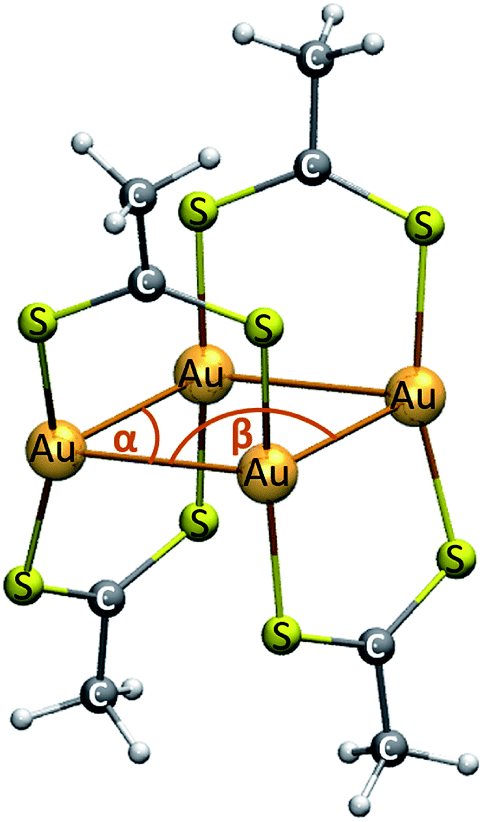 | ||
| Fig. 1 Model of the cluster of [Au(S2CCH3)]4. | ||
Single point calculations of the equilibrium geometries were used to study the excitation spectra by PBE and TPSS (DFT). The excitation energy was obtained using the time-dependent perturbation theory approach (TD).61 Also, the transition energies and oscillator strengths were calculated at the approximate second-order coupled cluster (CC2) and Spin-Component-Scaled (SCS)-CC2 levels of approximation.62 We have used the equilibrium distance (Re) estimated at the MP2 and SCS-MP2 levels to calculate the excitation spectrum at CC2 and SCS-CC2. Moreover, we have included the use of the ADC(2) method,62 which is an intermediate approach between the TDDFT and CC2 levels, for which we used the MP2 optimized geometry. The CC2 and ADC(2) methods involve the Laplace transformation (LT) algorithm and the reduced-virtual-space (RVS) approximation. The RVS cut-off threshold was 60 eV.62 We have used 11 electronic transitions. On the other hand, TDDFT is widely used in excited state studies of medium and large molecules, because of its excellent performance and its relatively low computational costs. However, since TDDFT calculations might suffer from charge transfer problems, the reliability of the obtained results should be assessed by comparing with the results calculated at higher levels of theory as CC2 and SCS-CC2 methods.63 Finally, we have computed the corresponding So–T1 vertical excitations using the optimized first triplet excited state structures in the methods mentioned above (in ethanol solvent with COSMO). Only energy values are given for electronic transitions. All UV/Vis spectra showed a Gaussian curve of full-width at half-maximum (FWHM) of 0.5 eV.
The calculations were carried out using the Turbomole 7.6 (ref. 64) program. For the 19 valence-electrons (VE) of Au quasi-relativistic (QR) pseudo-potential (PP) of Andrae65 were employed. We used three f-type and two g-type polarization functions on gold (αf = 1.41, 0.40, 0.15; αg = 1.20, 0.40). Also, the C and S atoms were treated through PPs, using double-zeta basis sets with the addition of two d-type polarization functions.66 For the H atom, a double-zeta basis set plus one p-type polarization function was used.67
Intending to have a complete representation of the interactions in the cluster, we calculated the non-covalent interaction index (NCI).68,69 This index allows to identify long range interactions based on the reduced density gradient (s) and the electronic density (ρ); which is possible through the definition of the low-density regions where s tends to zero. From this analysis, it is possible to obtain a representation in the real space of the non-covalent interactions taking place between the gold(I) atoms and others. Also, it allows to distinguish between attractive and repulsive interactions inside of the cluster. For interpretative purposes, the regions of the surface colored in blue denote strong stabilizing interactions; green indicates weak interactions usually associated with van der Waals interactions, and the colored in red are indicative of repulsive interactions. The analysis was performed from the output densities obtained at the TPSS-D3 level as no major differences were observed between this and PBE-D3 results.
Results and discussion
Geometrical structure and non-covalent interaction analysis (NCI)
Some geometric parameters of the optimized gold cluster at the different methods used in this study are shown in Table 1. We report the averaged values of the parameters depicted from Fig. 1. The resulting Au–Au distance and Au–Au–Au angles are in close agreement with the experimental values used to validate our models.53,54 In addition, the obtained optimized structures reproduce the rhombic geometry of the four gold centers. As evidenced from the data in Table 1, MP2 overestimates the correlation effects leading towards a shorter Au–Au distances and a lower Au–Au–Au angle; while SCS-MP2 and DFT-D3 methods were close to the crystal structure. According to this, the Grimme correction method allowed to obtain a good description of the Au–Au distance at the DFT level provided by the comparison with the experimental values. The coordinates of the optimized structures for each calculation method used in this study are included in the ESI (Table S1†).| Method | Au–Au | Au–S | S–C | C–C | α(Au–Au–Au) | β(Au–Au–Au) |
|---|---|---|---|---|---|---|
| MP2 | 282.4 | 229.5 | 168.1 | 150.3 | 61.6 | 118.4 |
| SCS-MP2 | 298.5 | 229.4 | 167.2 | 151.1 | 63.7 | 114.5 |
| PBE-D3 | 301.1 | 233.5 | 169.7 | 149.5 | 64.9 | 115.1 |
| TPSS-D3 | 296.0 | 234.1 | 169.8 | 149.9 | 64.2 | 115.8 |
| Exp.53,54 | 300.5 | 229.7 | 166.1 | 151.3 | 66.4 | 114.6 |
The geometry obtained at the MP2 level showed an Au–Au average distance of 282.4 pm. It is known that the MP2 approximation exaggerates such week attractive interactions; however, it gives a good indication of their existence.20,27,28 The results at the SCS-MP2 and DFT-D3 levels are very similar (298.5, 301.1, and 296.0 pm, respectively) and show a Au–Au distance slightly closer to that of the experimental structure. In all methods, the Au–Au contacts are within the range described for the classical aurophilic interaction.20 Regardless of the method used, the rest of the geometric parameters such as Au–S, S–C, C–C, and Au–Au–Au angles remained without significant changes.
Through the use of the NCI index, a representation in real space of the non-covalent interactions was obtained with the purpose of characterizing the inner forces that stabilize the complex, analysis shown in Fig. 2. The gold cluster is expected to be stabilized by the formation of aurophilic interactions. From this analysis, it is possible to clearly visualize the dispersive contribution to this interaction depicted by the regions of the surface colored in green at three different visualization angles of the cluster. These regions are localized between each pair of gold atoms. This feature is also present between the two gold atoms at opposite position within the cluster but at a shorter distance, as depicted by the perspective provided by Fig. 2b. The dispersion forces are also revealed at the interface between the dithioacetate ligands, which adopt a coplanar interaction conformation that favors the dispersive interaction between these ligands. Lastly, there is a ring-shaped surface moiety at the Au–S interaction axis that denotes a region of strong attractive interaction at the inside of the ring and a strong repulsive interaction at the outer region. According to our previous works on Au–chalcogen interaction,70–72 this interaction feature raises at the surrounding of a strong ionic interaction that takes part between gold and sulfur. In general, the results from the NCI index reveal the widespread relevance of the dispersion forces for the stability of the complex, which also suggests that the systems adopt a conformation that allows to maximize this contribution.
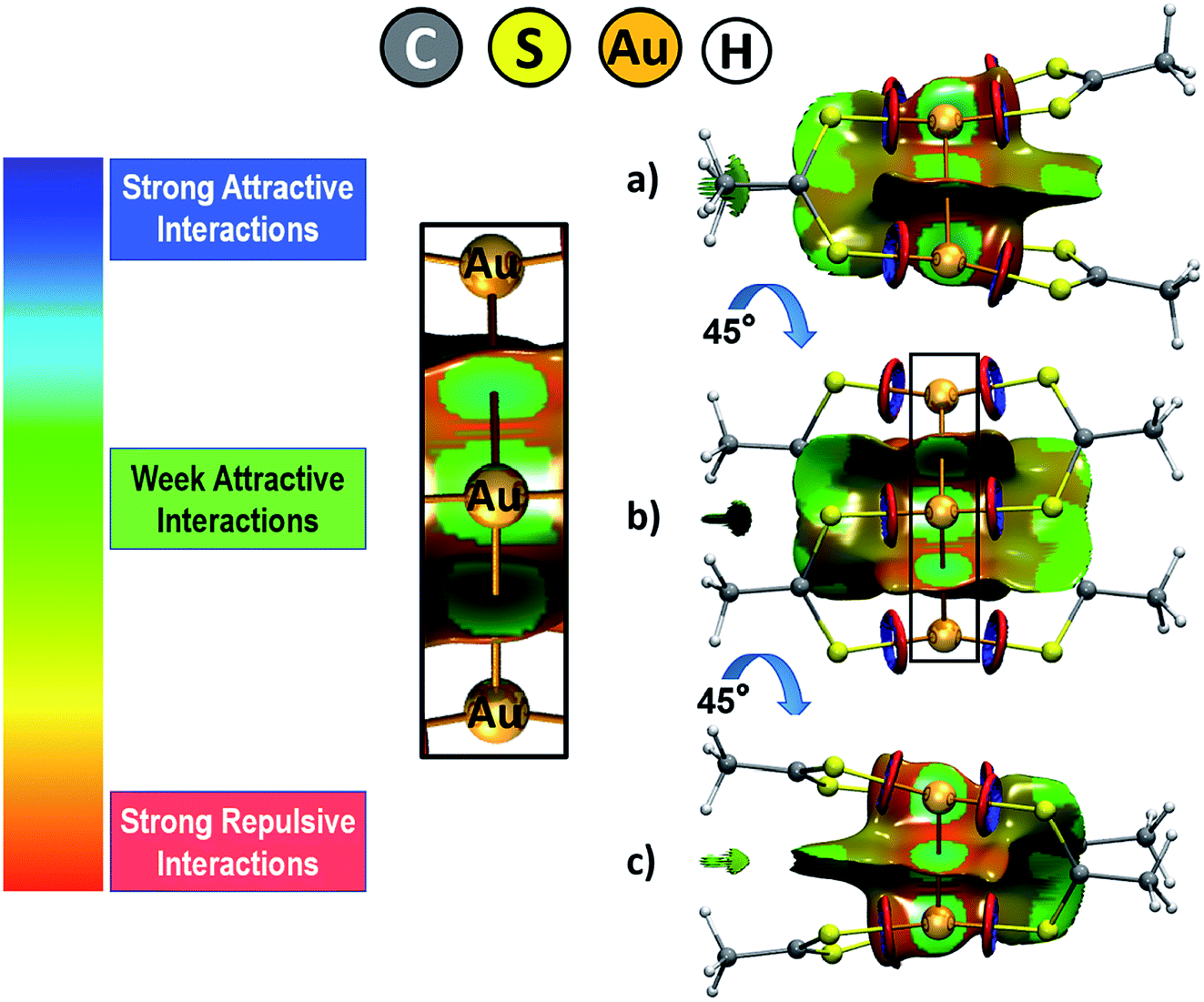 | ||
| Fig. 2 Graphical representation of the non-covalent interactions involved in the gold cluster at three different angle perspectives (a), (b) and (c) (the isovalue used as cutoff was set to 0.5). | ||
Excitation energy calculations
The absorption spectra have been calculated at the CC2, SCS-CC2, and ADC(2) levels. Also, the excitation energies of the model were analyzed at the TDDFT level using PBE and TPSS functionals. We calculated the allowed spin-singlet transition for these systems, based on the ground-state structures of [Au(S2CCH3)]4 cluster, in gas and CS2 phases. The principal vertical excitation energies calculated for the gold cluster are reported in Table 2 and all transitions are included in the ESI (Tables S2 and S3†).| Method | Principal transition |
|---|---|
| CC2 | 414 |
| CC2 (solv) | 410 |
| SCS-CC2 | 415 |
| SCS-CC2 (solv) | 409 |
| ADC(2) | 429 |
| ADC(2) (solv) | 431 |
| PBE | 436 |
| PBE (solv) | 430 |
| TPSS | 424 |
| TPSS (solv) | 419 |
| Exp.55 | 407 |
The experimental absorption spectra of [Au(S2CCH3)]4 cluster shows a band maximum at 407 nm with a shoulder at 430 nm in CS2 solvent.55 Very close excitation energies are obtained at the CC2 (410 nm) and SCS-CC2 (409 nm) levels in the same solvent phase. However, the band transition calculated at the ADC(2) and DFT levels is red shifted compared to the CC2 and SCS-CC2 values, with values of 431, 430, and 419 nm, respectively. All methods show a small red shift in the gas phase. The shape of the bands obtained at the CC2 and SCS-CC2 levels of theory are shown in Fig. 3 and 4. The experimental band has the same shape.55 For the other methods, this band pattern is similar, and it is shown in the ESI (Fig. F1–F3†). In all the methods is possible to observe three important transitions that we have designated with the letters (A), (B) and (C).
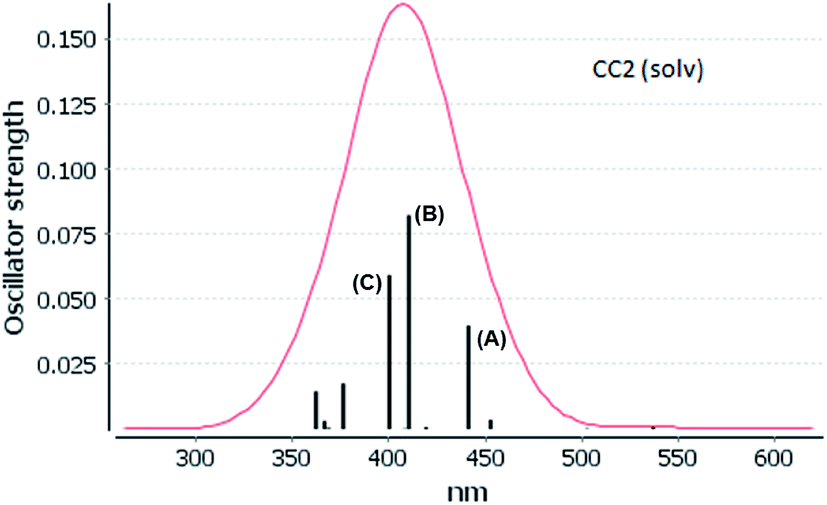 | ||
| Fig. 3 Electronic spectra at the CC2 level calculated by [Au(S2CCH3)]4 in CS2. | ||
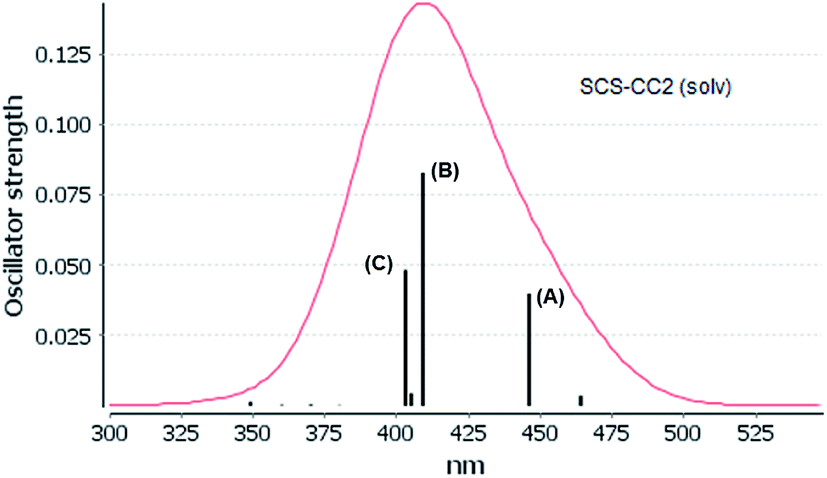 | ||
| Fig. 4 Electronic spectra at the SCS-CC2 level calculated by [Au(S2CCH3)]4 in CS2. | ||
To identify the nature of the electronic excitations, the expansion coefficients obtained by the different methods and the composition of the most important molecular orbitals that participate in the absorption process were used. The electronic excitations of the [Au(S2CCH3)]4 cluster are assigned to transitions from the ground state to: (i) metal–ligand-to-metal–ligand (MLML), (ii) metal–ligand-to-metal (MLM), and (iii) metal–ligand-to-ligand (MLL) charge transfers states. The excitation characters of the strong bands are listed in Tables 3 and 4 in CS2. We have used the wave function analysis to study the composition of each molecular orbital involved in electronic sates in the process of absorption.
| Method | λcalc/nm | fa | Contributionb | Transition type |
|---|---|---|---|---|
| a Oscillator strength.b Values are |coeff.|2 × 100. | ||||
| CC2 | 441 (A) | 0.0398 | 83a → 85a (70) | MLMLCT (sd + pz(S) → spz + pz(C)) |
| 410 (B) | 0.0824 | 81a → 87a (29) | LMMLCT (dyz + pz(S) → spz + pz(C)) | |
| 80a → 85a (24) | MLMLCT (dyz + pz(S) → spz + pz(C)) | |||
| 82a → 86a (21) | MLMCT (dyz + pz(S) → spz + pz(S) + pz(C)) | |||
| 400 (C) | 0.0593 | 82a → 87a (62) | MLMLCT (dyz + pz(S) → spz + pz(C)) | |
| SCS-CC2 | 446 (A) | 0.0399 | 83a → 85a (60) | MLMLCT (sd + pz(S) → spz + pz(C)) |
| 409 (B) | 0.0829 | 84a → 87a (31) | MMLCT (sdx2–y2 + pz(S) → spz + pz(C)) | |
| 82a → 85a (15) | MLMCT (dyz + pz(S) → spz + pz(C)) | |||
| 83a → 87a (15) | MLMLCT (sd + pz(S) → spz + pz(C)) | |||
| 403 (C) | 0.0483 | 82a → 87a (57) | MLMCT (dyz + pz(S) → spz + pz(C)) | |
| Method | λcalc/nm | fa | Contributionb | Transition type |
|---|---|---|---|---|
| a Oscillator strength.b Values are |coeff.|2 × 100. | ||||
| ADC(2) | 472 (A) | 0.0292 | 83a → 85a (62) | MLMCT (sd + pz(S) → spz + pz(C)) |
| 431 (B) | 0.1164 | 82a → 85a (37) | MLMCT (dyz + pz(S) → spz) | |
| 83a → 87a (28) | MLMCT (sd + pz(S) → spz + pz(S)) | |||
| 84a → 91a (14) | MLMCT (sd + pz(S) → spz) | |||
| 422 (C) | 0.0660 | 82a→ 87a (46) | MLMCT (dyz + pz(S) → spz + pz(S)) | |
| PBE | 432 (A) | 0.0240 | 80a → 87a (70) | MLMLCT(dxzdyz + pz(S) → s + pz(S) + pz(C)) |
| 430 (B) | 0.0296 | 79a → 86a (67) | MLMLCT (sd + pz(S) → pzdyz + pz(C)) | |
| 81a → 87a (12) | MLMLCT (dx2–y2dyz + pz(S) → s + pz(C)) | |||
| 427 (C) | 0.0143 | 83a → 88a (95) | MLLCT (sd + pz(S) → dyz + pz(S) + pz(C)) | |
| TPSS | 449 (A) | 0.0245 | 81a → 86a (46) | MLMLCT(dx2–y2dyz + pz(S) → pz + pz(S) + pz(C)) |
| 80a → 85a (45) | MLMLCT (dx2–y2dyz + pz(S) → pzdyz + pz(S) + pz(C)) | |||
| 420 (B) | 0.0253 | 80a → 87a (46) | MLMLCT (dx2–y2dyz + pz(S) → s + pz(S) + pz(C)) | |
| 79a → 86a (24) | MLMLCT (sd + pz(S) → pz + pz(S) + pz(C)) | |||
| 419 (C) | 0.0265 | 79a → 86a (44) | MLMLCT (sd + pz(S) → pz + pz(S) + pz(C)) | |
| 80a → 87a (24) | MLMLCT (dx2–y2dyz + pz(S) → s + pz(S) + pz(C)) | |||
The spectra calculated at the CC2 and SCS-CC2 levels showed transitions of the same type at similar energies. The composition of orbitals centered on the gold and sulfur (Au4S8 core) atoms predominate, as shown in Fig. 5 and 6. From the results obtained by the CC2 level, the main transitions were located at 441 nm (A), 410 nm (B), and 400 nm (C), which were assigned to 83a (HOMO−1) → 85a (LUMO) (A), 81a (HOMO−3) → 87a (LUMO+2); 80a (HOMO−4) → 85a (LUMO) (B), and 82a (HOMO−2) → 87a (LUMO+2) (C). These bands correspond to a MLML charge transfer. Here, the principal transition is (B), which is made up of the HOMO−3 → LUMO+2 (dyz + pz(S) → spz + pz(C)) and HOMO−3 → LUMO (dyz + pz(S) → spz + pz(C)) that can be defined as an MLMLCT excitation from antibonding to bond character. The starting orbitals have contributions from S (π*) orbitals and in a lower degree from gold orbitals. Meanwhile, the arrival orbitals have a strong character of gold orbitals. The orbitals are shown in Fig. 4.
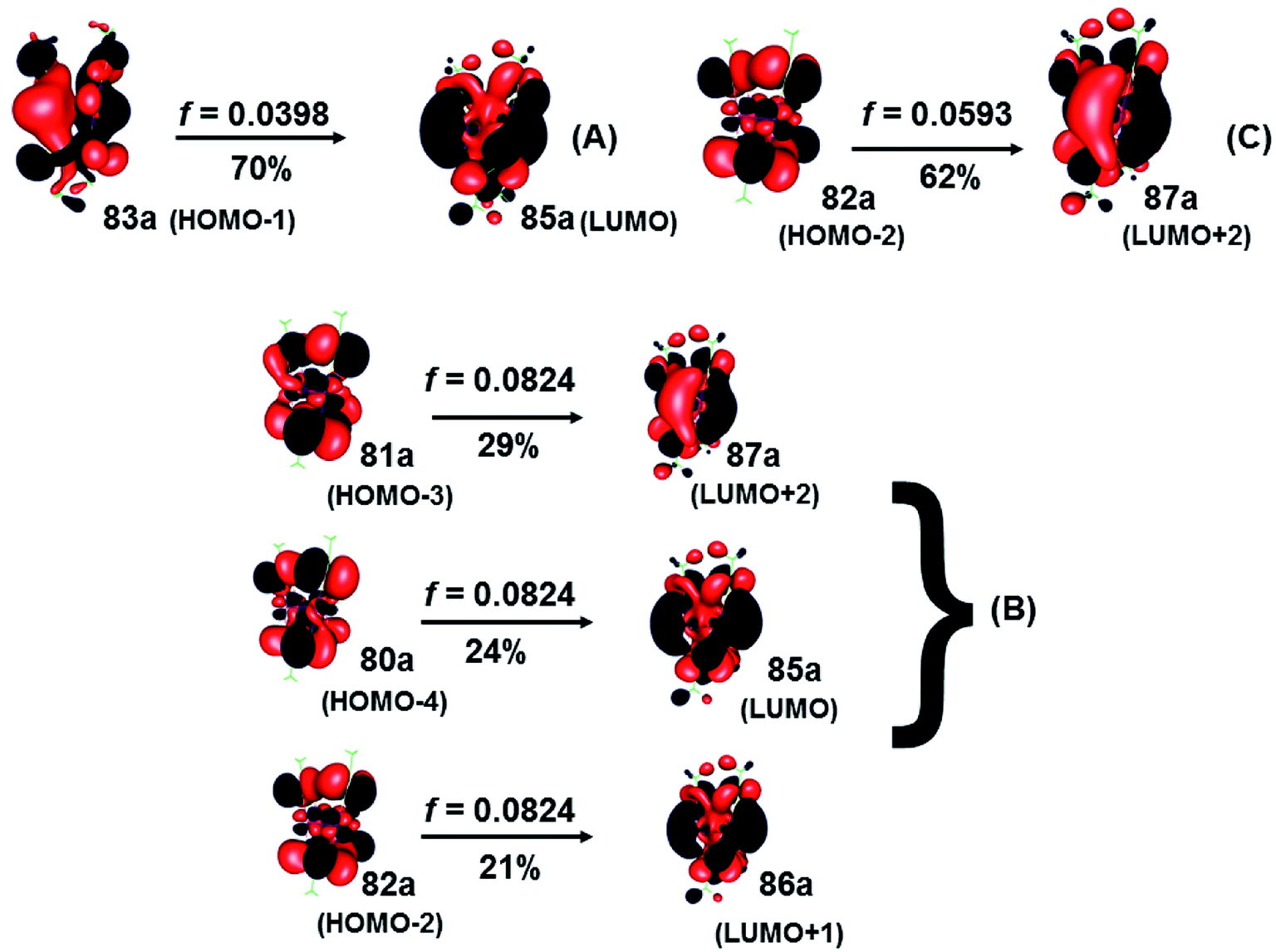 | ||
| Fig. 5 Most important active molecular orbitals in the electronic transitions of the [Au(S2CCH3)]4 at the CC2 level in CS2. | ||
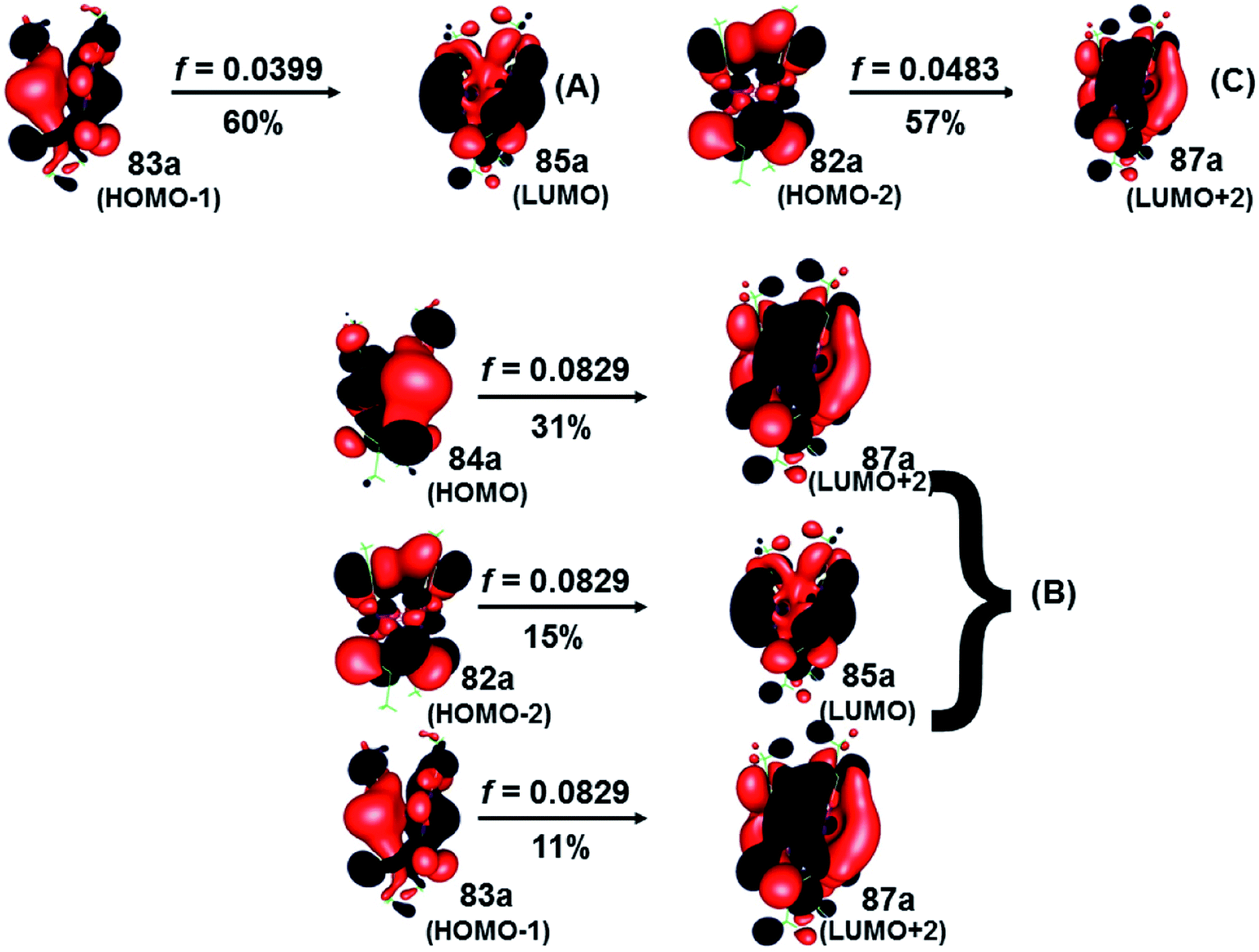 | ||
| Fig. 6 Most important active molecular orbitals in the electronic transitions of the [Au(S2CCH3)]4 at the SCS-CC2 level in CS2. | ||
On the other hand, the results of the SCS-CC2 level are very similar to that given above by CC2 calculated. The theoretical transitions were located at 446 nm (A), 409 nm (B), and 403 nm (C), which were assigned to 83a (HOMO−1) → 85a (LUMO) (A), 84a (HOMO) → 87a (LUMO+2), 82a (HOMO−2) → 85a (LUMO) (B), and 82a (HOMO−2) → 87a (LUMO+2) (C). Also, the bands described are associated with a MLML charge transfer. The principal transition is B built by the HOMO → LUMO+2 (sdx2–y2 + pz(S) → spz + pz(C)) and HOMO−2 → LUMO (dyz + pz(S) → spz + pz(C)) assigned as an MLMLCT excitation from antibonding to bond character. Once again, we can see that the starting orbitals have contributions from S (π*) orbitals and in a lower percentage to gold orbitals (spz); while the arrival orbitals have a strong character of gold. Fig. 5 shows the orbitals involved in these transitions. This description coincides with the one proposed by Vogler and Kunkely in their experimental work55 when trying to qualitatively explain the origin of the transition in the absorption spectrum. They were tentatively assigned to metal-to-ligand charge transfer with d/s mixing that leads to stabilization of the occupied orbitals. The principal transitions are derived from the 5d atomic orbitals. The description is in agreement with the calculations here presented.
The results from the ADC(2), PBE, and TPSS methods, showed a similar trend where the absorption bands from the gold cluster were slightly red shifted when compared to the corresponding absorption bands of the CC2 and SCS-CC2 levels described above. It is observed that TPSS transitions are closer to the two most precise methods, results listed in Table 4. The absorption bands and molecular orbitals are given in the ESI (Fig. F4–F6†). Furthermore, we can qualitatively appreciate that the composition of the transitions are of the same type as in those found from the CC2 and SCS-CC2 calculations. Once again, the principal transitions show that the starting orbitals have contributions from S (π*) orbitals and in a lower proportion from gold orbitals (spz); while the arrival orbitals have a strong character of gold orbitals. All transitions are centered on the Au4S8 core.
All methods used above show the same main electronic transition, from a S (π*) orbitals to the arrival orbitals of gold atoms (d), which can be explained by the short gold–gold distances described in the first section. The presence of aurophilic interactions conditions the location of the spectrum band. This has also been described in complexes with intermolecular gold–gold interactions such as in [Au(NH3)2]n(NO3)n (n = 2, 4, 8) and ([Au(NCH)2][AuCl4])n (n = 1, 2).28 In general, although the aurophilic interaction is not relevant to estimate the electronic properties directly, it is mainly the equilibrium distance in the ground state with which these optical properties are estimated. This is the starting point for describing the excited state that will be described in the next section.
Emission energies
The emission properties of the gold cluster were also studied. The first triplet excited state (T1) structures using ethanol as solvent were optimized at the same level of theory used for the ground state (MP2, SCS-MP2, PBE, TPSS). The coordinates of the optimized structures are presented in the Table S4 from the ESI.† The differences between the So ground state and excited T1 provide information about the role of the Au(I) and S in the cluster when the excitation process is produced.The most essential bond lengths and bond angles of the excited T1 state are summarized in Table 5. These data are compared with the So ground state given in Table 1. The optimized structure of the cluster at the excited T1 state show that with all the methods there are mainly two important changes: a shortening of Au–Au bonds, and a decrease in Au–Au–Au angles. The other parameters, such as Au–S, S–C, and C–C bonds, remain relatively invariant. We note that the MP2 and SCS-MP2 methods produce the lowest Au–Au distance, just as in the ground state. In general, the decreases in both geometric parameters manifest a binding state in T1. It was appreciated in the previous section when the main transition in the absorption spectrum had such character.
| Method | Au–Au | Au–S | S–C | C–C | α(Au–Au–Au) | β(Au–Au–Au) |
|---|---|---|---|---|---|---|
| MP2 | 270.6 | 229.9 | 168.7 | 149.7 | 65.1 | 114.9 |
| SCS-MP2 | 272.9 | 230.2 | 169.2 | 150.4 | 63.6 | 116.4 |
| PBE-D3 | 290.4 | 232.2 | 171.9 | 149.3 | 76.1 | 103.2 |
| TPSS-D3 | 285.9 | 233.0 | 171.9 | 149.7 | 68.4 | 110.5 |
We have computed the corresponding So–T1 vertical excitations using the optimized first triplet excited state structures in the methods above (in ethanol solvent) from the total energy for each state. The calculated emission wavelengths are shown in Table 6. The result for the SCS-CC2 level is 769 nm, which agreed well with the experimental data of 743 nm.55 This is the only methodology that comes close to experimental value. It is be due to the inclusion of the SCS correction. On the other hand, the CC2 method yields a longer transition of 932 nm. TDDFT calculations generate very long transitions due to further stabilization of the T1 state. The latter is expected since the DFT methods overestimate the energy of the excited state T1. The analysis described above is in agreement with that carried out experimentally by Vogler and Kunkely.55 They propose that the emission is yielded from a spin-forbidden transition of the type 3A1g → 1A2g. The large shift from absorption to emission is due to the reduction of the gold–gold bond in the cluster, which takes place in the excited state. This shift is experimentally located at 9800 cm−1.54 The excited state contraction of the gold–gold distance is verified by the calculations proposed in this work for all the methods used.
| Method | λemission [nm] |
|---|---|
| T1 → So | |
| CC2 | 932 |
| SCS-CC2 | 769 |
| PBE | 1088 |
| TPSS | 1285 |
| Exp.55 | 743 |
Conclusion
Theoretical calculations at the MP2, SCS-MP2, ADC(2), and DFT-D3 levels were suitable to describe the aurophilic interaction present in the proposed model of the cluster with configurations d10–d10. The weak gold interactions are described in the NCI analysis. Through CC2 and SCS-CC2 calculations, we were able to reproduce the experimental excitation spectra and rationalize the relationship between the gold–gold distance and the shift in the main absorption bands centered on the Au4S8 core. Finally, gold cluster's emission properties were computed the corresponding So–T1 vertical excitations given the SCS-CC2 level very good agreement with the experimental result.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Financial support of this work under Fondecyt projects 1180158 and 1181082 are gratefully appreciated.References
- K. M.-C. Wong, V. K.-M. Au and V. W.-W. Yam, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Oxford, UK, 2nd edn, 2013, pp. 59–130 Search PubMed.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS.
- M.-C. Tang, A. K.-W. Chan, M.-Y. Chan and V. W.-W. Yam, Top. Curr. Chem., 2016, 46, 374–385 Search PubMed.
- J. M. López-de-Luzuriaga, M. Monge and M. E. Olmos, Dalton Trans., 2017, 46, 2046–2067 RSC.
- S.-L. Zheng, C. L. Nygren, M. Messerschmidt and P. Coppens, Chem. Commun., 2006, 42, 3711–3713 RSC.
- R. L. White-Morris, M. M. Olmstead and A. L. Balch, Inorg. Chem., 2003, 42, 6741–6748 CrossRef CAS.
- M. Böge and J. Heck, Chem.–Eur. J., 2016, 22, 6787–6792 CrossRef.
- W. Lu, K. T. Chan, S.-X. Wu, Y. Chena and C.-M. Che, Chem. Sci., 2012, 3, 752–755 RSC.
- C. Yu, K. M.-C. Wong, K. H.-Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2005, 117, 801–804 CrossRef.
- R. Echeverría, J. M. López-de-Luzuriaga, M. Monge, S. Moreno and M. E. Olmos, Inorg. Chem., 2016, 55, 10523–10534 CrossRef.
- E. J. Fernández, A. Laguna, J. M. López-de- Luzuriaga, M. Monge, M. Nema, M. E. Olmos, J. Pérez and C. Silvestru, Chem. Commun., 2007, 43, 571–573 RSC.
- K. M.-C. Wong and V. W.-W. Yam, Acc. Chem. Res., 2011, 44, 424–434 CrossRef CAS.
- C.-K. Li, X.-X. Lu, K. M. Wong, C.-L. Chan, N. Zhu and V. W.-W. Yam, Inorg. Chem., 2004, 43, 7421–7430 CrossRef CAS.
- R. Donamaría, M. C. Gimeno, V. Lippolis, J. M. López-de-Luzuriaga, M. Monge and M. E. Olmos, Inorg. Chem., 2016, 55, 11299–11310 CrossRef.
- Q. -F. Sun, T. K.-M. Lee, T. Cheng, Y. -Z. Li and V. W. -W. Yam, Angew. Chem., Int. Ed., 2008, 120, 4627–4630 CrossRef.
- J. F. Parker, K. A. Kacprzak, O. Lopez-Acevedo, H. Haäkkinen and R. W. Murray, J. Phys. Chem. C, 2010, 114, 8276–8281 CrossRef CAS.
- J. Akola, K. A. Kacprzak, O. Lopez-Acevedo, M. Walter, H. Grönbeck and H. Häkkinen, J. Phys. Chem. C, 2010, 114, 15986–15994 CrossRef CAS.
- O. Bumbu, C. Ceamanos, O. Crespo, M. C. Gimeno, A. Laguna, C. Silvestru and M. D. Villacampa, Inorg. Chem., 2007, 46, 11457–11460 CrossRef CAS.
- C.-L. Margaret Yeung and V. W.-W. Yam, Chem. Soc. Rev., 2015, 44, 4192–4202 RSC.
- P. Pyykkö, N. Runeberg and F. Mendizabal, Chem.–Eur. J., 1997, 3, 1451–1457 CrossRef.
- N. Runeberg, M. Schütz and H.-J. Werner, J. Chem. Phys., 1999, 110, 7210–7215 CrossRef CAS.
- P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef.
- F. Mendizabal and P. Pyykkö, Phys. Chem. Chem. Phys., 2004, 4, 900–905 RSC.
- R.-F. Liu, C. A. Franzese, R. Malek, P. S. Zuchowski, J. G. Angyan, M. M. Szczesniak and G. Chalasinski, J. Chem. Theory Comput., 2011, 7, 2399–2407 CrossRef CAS.
- M. Andrejic and R. A. Mata, Phys. Chem. Chem. Phys., 2013, 15, 18115–18122 RSC.
- F. Mendizabal, S. Miranda-Rojas and L. Barrientos, Comput. Theor. Chem., 2015, 1057, 74–79 CrossRef CAS.
- L. Barrientos, S. Miranda-Rojas and F. Mendizabal, Int. J. Quantum Chem., 2019, 119, e25675 CrossRef.
- F. Mendizabal, S. Miranda-Rojas and P. Castro-Latorre, Mol. Simul., 2020, 46(7), 521–529 CrossRef CAS.
- E. J. Fernández, J. M. López-de-Luzuriaga, M. Monge, M. Montiel, M. E. Olmos, J. Pérez, A. Laguna, F. Mendizabal, A. A. Mohamed and J. P. Fackler, Inorg. Chem., 2004, 43, 3573–3581 CrossRef.
- R. Donamaría, M. C. Gimeno, V. Lippolis, J. M. López-de-Luzuriaga, M. Monge and M. E. Olmos, Inorg. Chem., 2016, 55, 11299–11310 CrossRef.
- D. J. Liptropt and P. P. Power, Nat. Rev. Chem., 2017, 1, 004 CrossRef.
- H. Schmidbaur, in Gold-Progress in Chemistry, Biochemistry and Technology, John Wiley & Sons, New York, 1999 Search PubMed.
- M. C. Gimeno and A. Laguna in Comprehensive Coordination Chemistry 1, ed. C. McCleverty and T. J. Meyer, Elsevier, Amsterdam, 2004, vol. 6, pp. 911–1145 Search PubMed.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC.
- Modern Supramolecular Gold Chemistry, ed. A. Laguna, Wiley-VCH, Weinheim, 2008 Search PubMed.
- X. Liu, J. Zhang, X. Guo, S. Wu and S. Wang, Nanotechnology, 2010, 21, 095501 CrossRef.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- J. P. Fackler, Inorg. Chim. Acta, 2015, 424, 83–90 CrossRef CAS.
- G. Tárkányi, P. Király, G. Pálinkás and A. Deák, Magn. Reson. Chem., 2007, 45, 917–924 CrossRef.
- A. Deák, T. Megyes, G. Tárkányi, P. Király, L. Biczók, G. Pálinkás and P. J. Stang, J. Am. Chem. Soc., 2006, 128, 12668–12670 CrossRef.
- F. Mendizabal, D. Reyes and C. Olea-Azar, Int. J. Quantum Chem., 2006, 106, 906–912 CrossRef CAS.
- L. Magnko, M. Schweizer, G. Rauhut, M. Schütz, H. Stoll and H.-J. Werner, Phys. Chem. Chem. Phys., 2002, 4, 1006–1013 RSC.
- P. Pyykkö, Chem. Soc. Rev., 2008, 37, 1967–1997 RSC.
- A. Fernando, K. L. D. Weerawardene, N. V. Karimova and C. M. Aikens, Chem. Rev., 2015, 115, 6112–6212 CrossRef CAS.
- J. R. Reimers, M. J. Ford, S. M. Marcuccio, J. Ulstrup and N. S. Hush, Nat. Rev. Chem., 2017, 1, 0017 CrossRef CAS.
- K. M.-C. Wong, V. K.-M. Au and V. W.-W. Yam, Comprehensive inorganic chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Oxford, UK, 2nd edn, 2013, pp. 59–130 Search PubMed.
- J. M. Lopez-de-Luzuriaga, M. Monge, M. E. Olmos, J. Quintana and M. Rodriguez-Castillo, Inorg. Chem., 2019, 58, 1501–1512 CrossRef CAS.
- C. L. M. Yeung and V. W.-W. Yam, Chem. Soc. Rev., 2015, 44, 4192–4202 RSC.
- Y. Chi and P. T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC.
- W. P. Li, S. J. Liu, T. Gong, Q. Zhao and W. Huang, Adv. Mater., 2014, 26, 570–606 CrossRef.
- J. M. Lopez-de-Luzuriaga, M. Monge, M. E. Olmos, J. Quintana and M. Rodriguez-Castillo, Inorg. Chem., 2019, 58, 1501–1512 CrossRef CAS.
- C. L. M. Yeung and V. W.-W. Yam, Chem. Soc. Rev., 2015, 44, 4192–4202 RSC.
- O. Piovesana and P. F. Zanazzi, Angew. Chem., Int. Ed. Engl., 1980, 19, 561–562 CrossRef.
- B. Chiari, O. Piovesana, T. Tarantelli and P. F. Zanazzi, Inorg. Chem., 1985, 24, 366–371 CrossRef CAS.
- A. Vogler and H. Kunkely, Chem. Phys. Lett., 1988, 150, 135–137 CrossRef CAS.
- S. Grimme, J. Chem. Phys., 2003, 118, 9095–9102 CrossRef CAS.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3869 CrossRef CAS.
- Y. Kanai, X. Wang and A. Selloni, J. Chem. Phys., 2006, 125, 234104–234110 CrossRef.
- S. Ehrlich, J. Moellmann, W. Reckien, T. Bredow and S. Grimme, ChemPhysChem, 2011, 12, 3414–3420 CrossRef CAS.
- A. Klamt and G. Schüürman, J. Chem. Soc., Perkin Trans. 2, 1993, 25, 799–805 RSC.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- N. O. Winter and C. Hättig, J. Chem. Phys., 2011, 134, 184101–184115 CrossRef.
- M. Gerenkamp and S. Grimme, Chem. Phys. Lett., 2004, 392, 229–235 CrossRef CAS.
- Turbomole: R. Ahlrichs, M. Bär, M. Häser, H. Horn and M. C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–1969 CrossRef CAS.
- D. Andrae, M. Häusserman, H. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- A. Bergner, M. Dolg, M. W. Küchle, H. Stoll and H. Preuss, Mol. Phys., 1993, 80, 1431–1443 CrossRef CAS.
- T. Dunning and P. Hay, in Modern Theoretical Chemistry, ed. H. Schaefer, Plenum Press, 1997, vol. 3, pp. 1–28 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef.
- S. Miranda-Rojas, A. Muñoz-Castro, R. Arratia-Pérez and F. Mendizabal, Phys. Chem. Chem. Phys., 2013, 15, 20363–20370 RSC.
- S. Miranda-Rojas, R. Salazar-Molina, J. Kästner, R. Arratia-Pérez and F. Mendizabal, RSC Adv., 2016, 6, 4458–4468 RSC.
- S. Miranda-Rojas and F. Mendizabal, Nanomaterials, 2020, 10(1237), 1–16 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 contains Cartesian coordinates (in Angstroms) for the optimized geometries of the systems studied in this work. Ground state (So). Tables S2 and S3 contain wavelengths (λ) and oscillator strengths (f) for the UV/Vis spectra of the [Au(S2CCH3)]4 CC2, SCS-CC2, ADC(2) and TDDFT (PBE and TPSS). All spectra were calculated in the gas phase and with the COSMO solvation model (epsilon = 2.6, CS2). Wavelengths (λ) are given in nm. Table S4 contains Cartesian coordinates (in Angstroms) for the optimized geometries of the systems studied in this work at first triplet excited state (T1) in the ethanol phase. Fig. F1, F2 and F3 shown calculated ADC(2), PBE, and TPSS electronic spectra of [Au(S2CCH3)]4 in CS2 phase, respectively. Fig. F4, F5, and F6 shown the most important active molecular orbitals in the electronic transitions of [Au(S2CCH3)]4 in the CS2 phase at the ADC(2), PBE, and TPSS levels, respectively. See DOI: 10.1039/d0ra06982e |
| This journal is © The Royal Society of Chemistry 2020 |