
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResearch on the deactivation mechanism of a denitration catalyst WO3–V2O5/TiO2 at a coal-fired power plant†
Xianghui Liu and
Qiaowen Yang *
*
School of Chemical & Environmental Engineering, China University of Mining &Technology, Beijing 100083, China. E-mail: zz2bb4ry@163.com
First published on 11th December 2020
Abstract
The spent and fresh V2O5–WO3/TiO2 monolith catalysts were collected from a coal-fired power plant. The de-NOx efficiency dropped by 20% after the fresh catalyst was used for 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h. Then, the catalysts and the fly ash attached to spent catalysts were collected and analyzed. It was found that the relative amount of Si and Al increased by 80.84% and 2.26 times, respectively, which indicated that a lot of sediments deposited on the surface of the catalyst. Moreover, the content of Na, K, Ca and Fe increased in different degrees. A few new elements, such as Cl, Zn and Pb, appeared on the surface of the deactivated catalyst, and all of these elements had bad effects on the activity. Some kinds of ammonium salts and sulfates emerged on the fly ash, which showed that the catalysts were poisoned by SO2. The special area decreased only by 4.39 m2 g−1. The V3+/(V4+ + V5+) ratio in the catalyst increased from 0.09 to 0.45 after deactivation, and V4+ decreased by about 39.54%, which caused the deactivation of the catalyst. The surface acidity of the deactivated catalyst decreased a lot, which might be the immediate cause of deactivation. The particle size of TiO2 increased due to sintering. The main causes for the deactivation may be described as active sites decreased, poisoned and covered.
000 h. Then, the catalysts and the fly ash attached to spent catalysts were collected and analyzed. It was found that the relative amount of Si and Al increased by 80.84% and 2.26 times, respectively, which indicated that a lot of sediments deposited on the surface of the catalyst. Moreover, the content of Na, K, Ca and Fe increased in different degrees. A few new elements, such as Cl, Zn and Pb, appeared on the surface of the deactivated catalyst, and all of these elements had bad effects on the activity. Some kinds of ammonium salts and sulfates emerged on the fly ash, which showed that the catalysts were poisoned by SO2. The special area decreased only by 4.39 m2 g−1. The V3+/(V4+ + V5+) ratio in the catalyst increased from 0.09 to 0.45 after deactivation, and V4+ decreased by about 39.54%, which caused the deactivation of the catalyst. The surface acidity of the deactivated catalyst decreased a lot, which might be the immediate cause of deactivation. The particle size of TiO2 increased due to sintering. The main causes for the deactivation may be described as active sites decreased, poisoned and covered.
1. Introduction
Nitrogen oxides (NOx) lead to a number of environmental problems, such as photochemical smog, acid rain, haze and greenhouse effect.1,2 Most of NOx come from coal-fired power plants in China. To eliminate the NOx pollution, the selective catalytic reduction (SCR) of NOx with NH3 is used for its high activity.3,4 Numerous types of catalytic systems have been extensively investigated.5 Moreover, two dimensional materials became an important platform to design single site catalysts for various reactions, such as CO2 reduction and CO oxidation.5–14 However, among these catalysts, V2O5–WO3/TiO2 is the most effective and popular catalyst used in most of coal-fired power plants for its high de-NOx efficiency and its tolerance towards SO2.15,16However, the V2O5–WO3/TiO2 catalyst, similar to most of catalysts, faces the problem of deactivation after being used for a long term.17 The lifespan of the V2O5–WO3/TiO2 catalyst is closely related to the type of coal, the combustion method, and working conditions in coal-fired power plants.18 Meanwhile, the cost of catalyst occupies most of SCR system. In general, the V2O5–WO3/TiO2 catalyst cannot play the role of de-NOx after being used for about 3 years. To extend the life-span of the catalyst by modification, it is necessary to identify the possible causes of its deactivation.
Otherwise, the spent catalyst faces the disposal problem.19 A few valuable metals still remain in the spent catalyst, such as W, V and Ti. On one hand, the deactivated catalyst may be used again after being regenerated by a proper way. On the other hand, recovering the valuable metals from the spent catalyst by suitable methods is also an effective way to reuse the resource. Different regeneration methods are based on the specific causes of deactivation.20,21 The recovery products are affected by the substances deposited on the surface of the catalyst. Therefore, it is essential to study the deactivation mechanism of SCR catalysts at coal-fired power plant.
The spent honeycomb monolith SCR catalyst was obtained from a coal fired power plant, which was used continuously for about 30000 h. The fresh catalyst was also collected for a comparison in the experiment. In this study, the catalytic activity of fresh and spent catalysts was studied in a laboratory, and the deactivation mechanism of the spent catalyst was discussed.
2. Material and methods
2.1 Commercial SCR catalysts
The specimens of fresh and deactivated catalysts were deposed in the middle part of the catalyst in the plant. The ash was collected from the superficies of the deactivated catalysts by an air blower. The pore diameter of the fresh and deactivated catalysts were 8.08 mm and 8.04 mm, respectively, while the wall thickness were 1.00 mm and 1.10 mm, respectively. There were a few gray and red-gray sediments on the surface of the pore. The color of the fly ash was gray, and a few granulated substances were found in the ash. The fly ash on the surface of the deactivated catalyst was also collected for the research. For further studying, we defined the fresh catalyst as FC, the deactivated catalyst as DC and the fly ash on the catalyst as FA.2.2 Catalytic activity tests
FC and AC were smashed to below 149 μm for the test. The SCR catalytic activity tests of FC and AC were carried out in a fixed-bed stainless steel reactor tube with 0.6 g of the samples. The test temperature was in the range from 240 °C to 420 °C. The test system consisted of four parts: simulated gases and mass flow meters, a gas mixing section, a reactor and an analyzer section. The experimental layout of the catalytic activity test is shown in Fig. 1. The components of the simulated gas stream were: c(NO) = 500 mg m−3, c(NH3) = 500 mg m−3, ϕ(O2) = 5% with N2 as the balance gas in the experiment. The de-NOx efficiency of FC and DC was calculated by eqn (1).
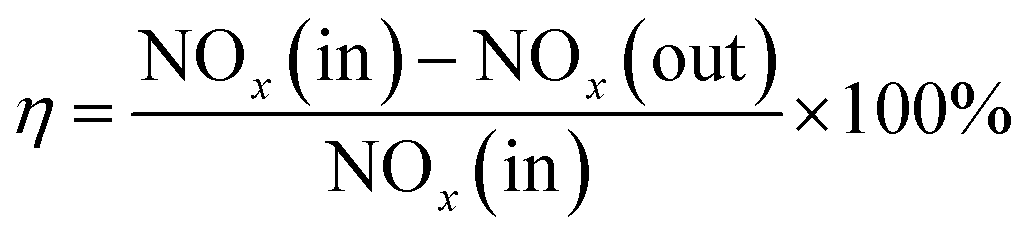 | (1) |
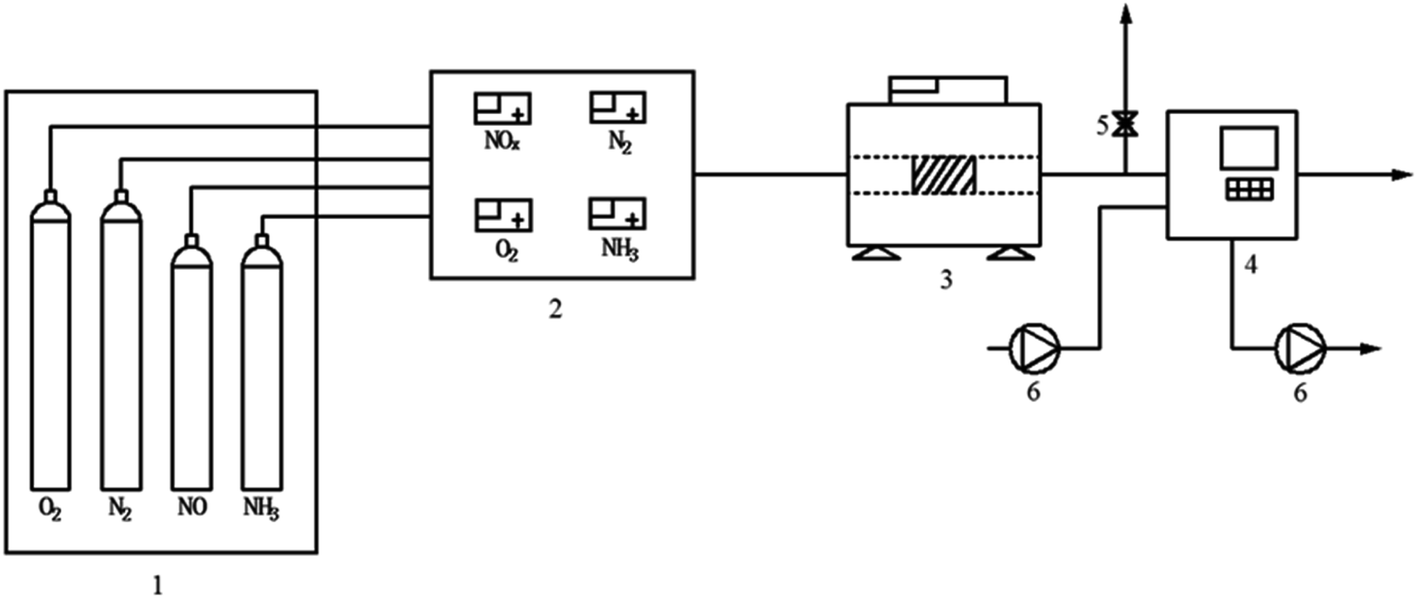 | ||
| Fig. 1 The experimental layout of the catalytic activity test equipment ((1) gas tanks and cylinders (2) mixing reactor (3) reactor (4) flue gas analyzer (5) air outlet (6) pump). | ||
2.3 Chemical characterization
The elemental content of DC, FC, and FA was evaluated on an X-ray fluorescence spectrometer (XRF, PFX-235, Thermo, USA). The valence of W, V, Ti and O was detected by X-ray photoelectron spectroscopy (XPS, QUANTUM 2000X, Physical Electronics Company, USA). The BET surface area of FC and DC was measured using a Micromeritics ASAP 2020 apparatus. The crystalline phase of FC, DC and FA was detected via X-ray diffraction (XRD, X'Pert Pro, PANalytical B.V., Holand). The particle morphology of the catalyst was examined by a scanning electron microscope (SEM, S-4800, Hitachi, Japan). The main chemical functional groups of the catalyst were identified by their Fourier transform infrared spectra (FT-IR, Nicolet iS10, Thermo, USA). NH3-TPD was detected by an AutoChem II 2920 (NH3-TPD, AutoChem II 2920).3. Results and discussion
3.1 De-NOx efficiency
The efficiency of FC and DC in the experiment is presented in Fig. 2. As shown in Fig. 2, the denitrification efficiency of FC was higher than 85% in a temperature range of 300–420 °C. However, the de-NOx efficiency of DC decreased significantly in the test temperature window. DC showed only 71.43% in the experiment at 420 °C, which cannot satisfy the requirement of the power plant. The value of the de-NOx efficiency of DC was below 80% in the test temperature window. The trend of the de-NOx efficiency of DC and FC was similar at the test temperature, which indicated that DC and FC showed a relatively high de-NOx efficiency at a high temperature. Moreover, the catalyst was a medium and high temperature denitration catalyst, which caused the de-NOx efficiency to increase with the test temperature in an almost linear relationship.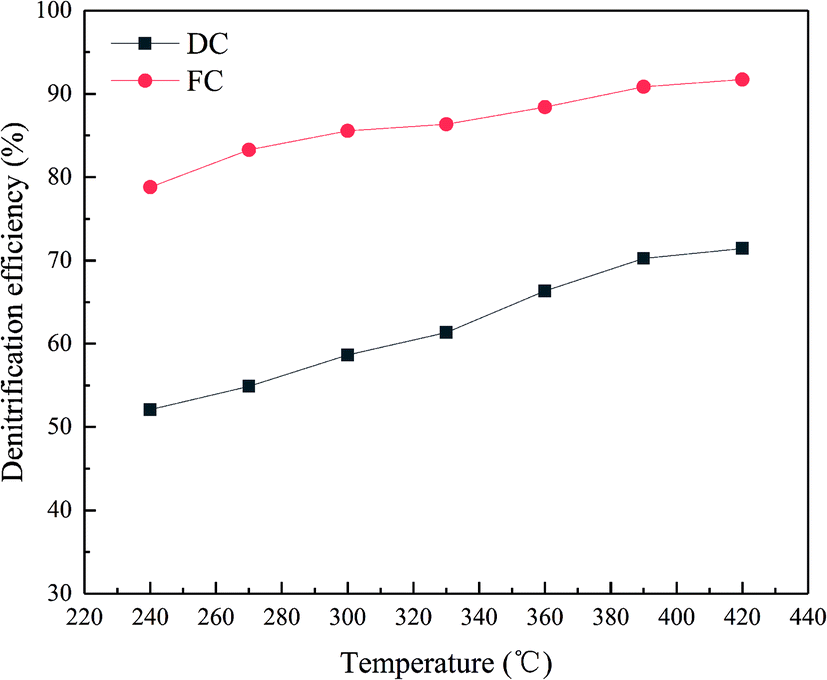 | ||
| Fig. 2 The de-NOx efficiency of FC and DC as a function of the test temperature. | ||
3.2 XRF
It was found that about 7 g of FA was collected when 100 g of DC gathered. Therefore, it can be inferred that about 7 g of FA deposited on the surface of 100 g of DC after being used for 30000 h. The composition of FC and DC was identified by XRF, and FA was also analyzed by XRF. The substances on the catalyst, appeared or vanished, may have a great influence on the NH3-SCR reaction. The content change of TiO2 could be ignored as compared with other ingredients. TiO2 was selected as the reference for its high content in the catalyst. So, the changing substances were considered in the research. The metal oxide ratio of MOx/TiO2 between FC and 100 g DC + 7 g FA was considered and presented in Fig. 3.
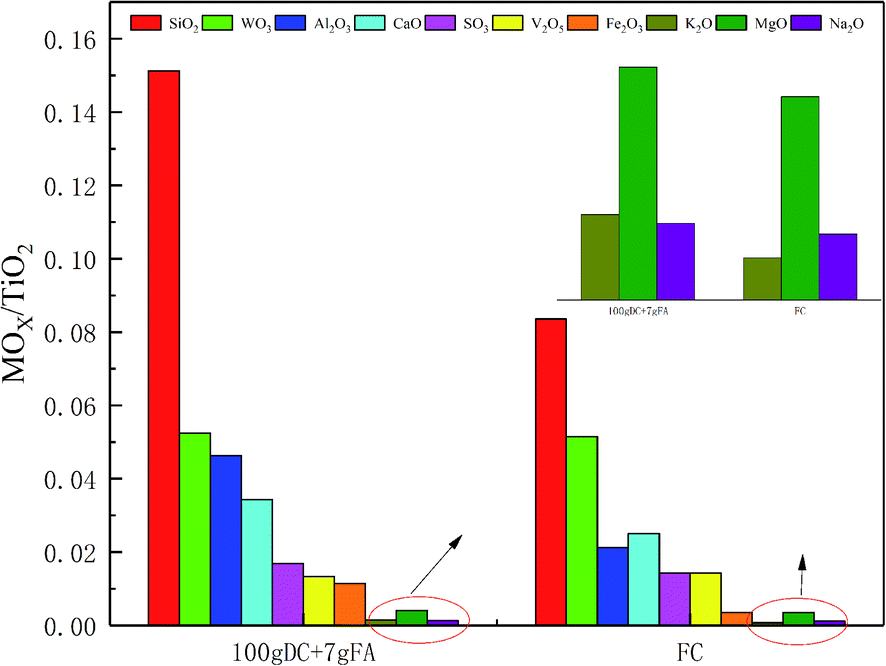 | ||
| Fig. 3 MOx/TiO2 between FC and DC + FA. | ||
As shown in Fig. 3, the oxide ratio of WO3/TiO2 remained constant, which indicated that WO3 was stable. The relative content of SiO2 increased by 80.84% in DC + FA. Additional SiO2 was from the fly ash in the flue gas. SiO2 may deposit on the surface or block the pore of the catalyst. The relative content of SO3 increased by 18.11%, which may have existed as CaSO4, (NH4)2SO4 or NH4HSO4. The sulfates had a negative effect on the NH3-SCR reaction,22 which could destroy V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.
O.
The relative content of Fe2O3 increased by 2.26 times, and the large amount of Fe was from the coal. The Fe might take place the V active sites.23 The relative content of MgO increased by 1.18 times. Although the poisoning effect of Mg on the catalyst was less than that of K, Na and Ca, it could also lead to a decrease in activity.24 The relative content of Al2O3 increased by 2.26 times, and the large amount of Al was from the coal. Al may deposit on the surface of the catalyst. A few researchers found Al2(SO4)3, which could decrease the specific surface area.25 The relative content of CaO increased by 36.71%, which could decrease the adsorbed ammonia amount.26 Moreover, Ca could work together with a few inorganic matters, such as NO, C, SO2 or P2O5.24,27
The oxide ratio of K2O/TiO2 and Na2O/TiO2 increased by 102.76% and 15.79%, respectively. Many researchers have carried out experiments on the effect of alkali metals on catalysts.28 The alkali metals can cause lots of changes in the catalyst, such as blocked catalyst channels, decreased reducibility and reduced surface acidity sites. The poisoning effect of a few alkali salts on the V2O5–WO3/TiO2 catalyst follows the sequence Na2CO3 < K2CO3 < Na2SO4 < K2SO4 < NaCl < KCl.29 K can coordinate to Brønsted acidic sites.30,31 Also, substances containing K, such as KCl can induce the catalysts sintered, decrease V atoms on the surface, and change the valence state of V.32
Besides, a few elements such as Pb, Cl, As and Zn appeared in DC and FAwere also proved to be poisonous and harmful to the catalyst. Pb could poison the active polytungstate species, inhibit NH3 adsorption, and decrease the Brønsted acidic sites.33,34 In addition, the collective effect of Pb and SO2 could not only decrease the surface acidic sites but also decrease the redox sites.35 This can promote to form N2O above 300 °C, and destroy the active V–OH sites.36 Both the Lewis and Brønsted acidic sites decreased when H2AsO4 or HAsO4− formed. The surface active oxygen increased and the oxidizability of catalysts enhanced, when the catalyst was poisoned by As, which resulted in increased NH3 oxidation and reduced SCR selectivity at high temperatures.37
Moreover, the effect of Zn poisoning for V2O5–WO3/TiO2 was rarely reported. The amount of Mn4+ and chemisorbed oxygen species in the Mn/TiO2 catalyst decreased after being poisoned by Zn. Zn also affected the reducibility, surface acidity and NO adsorption ability of the catalyst.38,39
In addition, a certain amount of V was found in FA, which indicated an outflow of some active components during the operational process.
3.3 XRD
Fig. 4a shows the XRD patterns of FC, DC and FA. Only the peaks of anatase TiO2 are displayed for FC and DC. There were no obvious peaks of SiO2, V2O5 and WO3 in FC and DC due to their low concentrations and high dispersion. It could be clearly seen that the crystal form of TiO2 did not change between FC and DC.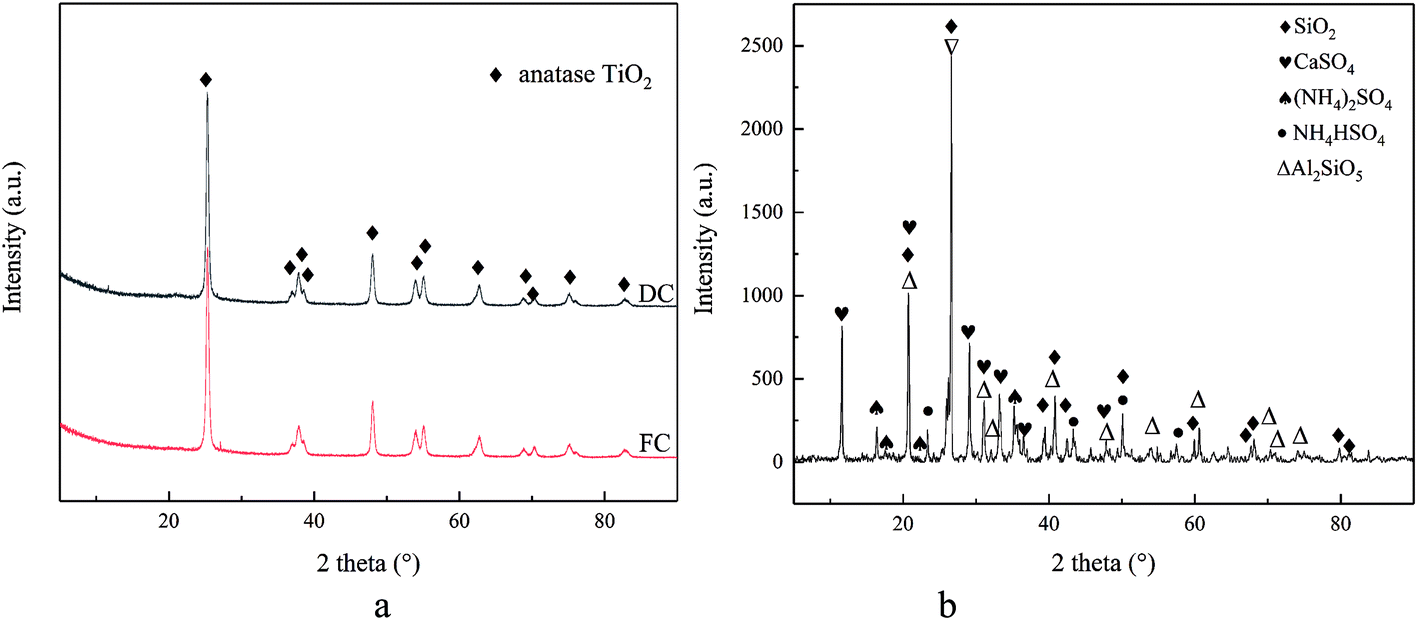 | ||
| Fig. 4 The XRD patterns of FC, DC (a) and FA (b). | ||
The XRD patterns of FA are also shown in Fig. 4b. The diffraction peak of SiO2 appeared in FA according to Fig. 4b. Some SiO2 were added as a skeleton material with other ingredients in the catalyst preparation stage. Most of SiO2 was from the coal of combustion, which covered the active sites on the catalyst surface. Too much SiO2 and other ingredients on the surface of the catalyst may lead to a decrease in its efficiency. Meanwhile, CaSO4 was found in FA. CaSO4 deposited on the surface of the catalyst could restrict the NH3-SCR reaction. Furthermore, (NH4)2SO4 and NH4HSO4 were also observed. (NH4)2SO4 and NH4HSO4 were formed from NH3 and SO2 during the operational stage, which were harmful for the active site V2O5 in the catalyst.
Former researches had pointed out that the main sediment sulfates of (NH4)2SO4 and NH4HSO4 were formed under the temperature of 300 °C, while CaSO4 was generated as the main sulfate at higher temperatures.40,41 In addition, Al2SiO5 emerged in FA, which may form from SiO2 and Al2O3 in the catalyst or in the flue gas, which may also cover the active sites.
On the basis of the Scherrer equation, the mean dimensions of TiO2 particles for FC and DC were 18.9 and 20.3 nm, respectively. Hence, it could be observed that DC was sintered, which led to an increase in the mean particle size of TiO2.
3.4 Surface morphology
The SEM images of FC and DC are shown in Fig. 5. As shown in Fig. 5a, FC showed the relatively regular smooth morphology without an impurity particle distribution. In contrast, DC illustrates a coarse surface and a particle agglomeration or hardness to some extent. As shown in Fig. 5b, c, e and f, a few substances with a trod-like structure, a block structure or a globular structure appeared on the surface of DC. There were a certain amount of SiO2, Al2O3 and CaO as skeletal structures in FC. The skeletal structures did not appear on the surface of FC for low activity. After eroding for 30000 h continuously, a few Si–Ca–Al skeletal structures bared as trod-like structures on the surface of DC. The block structure matters were sintered with the catalyst and other ingredients in the flue gas. A lot of substances generated in the flue gas might deposit on the surface of the catalyst, and a few of them reacted with the flue gas and ingredients of the catalyst. These matters exhibited all kinds of morphologies after deposited or reacted for a certain period of time. A certain amount of S emerged on the surface of DC might be sulfate. SO2 existed in the flue gas, and V2O5 could be the catalyst for SO2 to be converted to SO3. SO3 was transformed to ammonium sulfate and calcium sulfate via reacting with other ingredients, such as NH3 or CaO. The substances deposited on the catalyst surface could be considered as contaminants. They could not only cover the active sites and destroy the pores, but also change the structure of the active sites.
 | ||
| Fig. 5 SEM photos of samples (FC (a–d); DC (b–e), (c–f)). | ||
3.5 Specific surface area
The specific surface area, pore structure and pore volume were also under the attention for their influence on the catalytic reaction. In the research, the value of specific surface area of FC was 64.30 m2 g−1, while the corresponding value for DC was 59.91 m2 g−1. A decrease in the specific surface area was limited, which indicated that the specific surface area did not play a main role in the deactivation.The curves about adsorption–desorption isotherms and pore distribution of FC and DC are also shown in Fig. 6a and b, respectively. Both FC and DC exhibited type IV adsorption–desorption hysteresis loops, which indicated that FC and DC both were porous materials. The pore size distribution of FC and DC was calculated by the DFT approach according to adsorption–desorption hysteresis loops. The major pores of the FC and DC were mesoporous. As shown in Fig. 6a, the meso-pore peak centers of FC were at 9, 10 and 13 nm, while the peaks of DC were at 13, 15 and 20 nm. The major pores did not show obvious changes between FC and DC. Above all, the specific surface area, pore structure and pore volume did not exhibit an important influence on the deactivation.
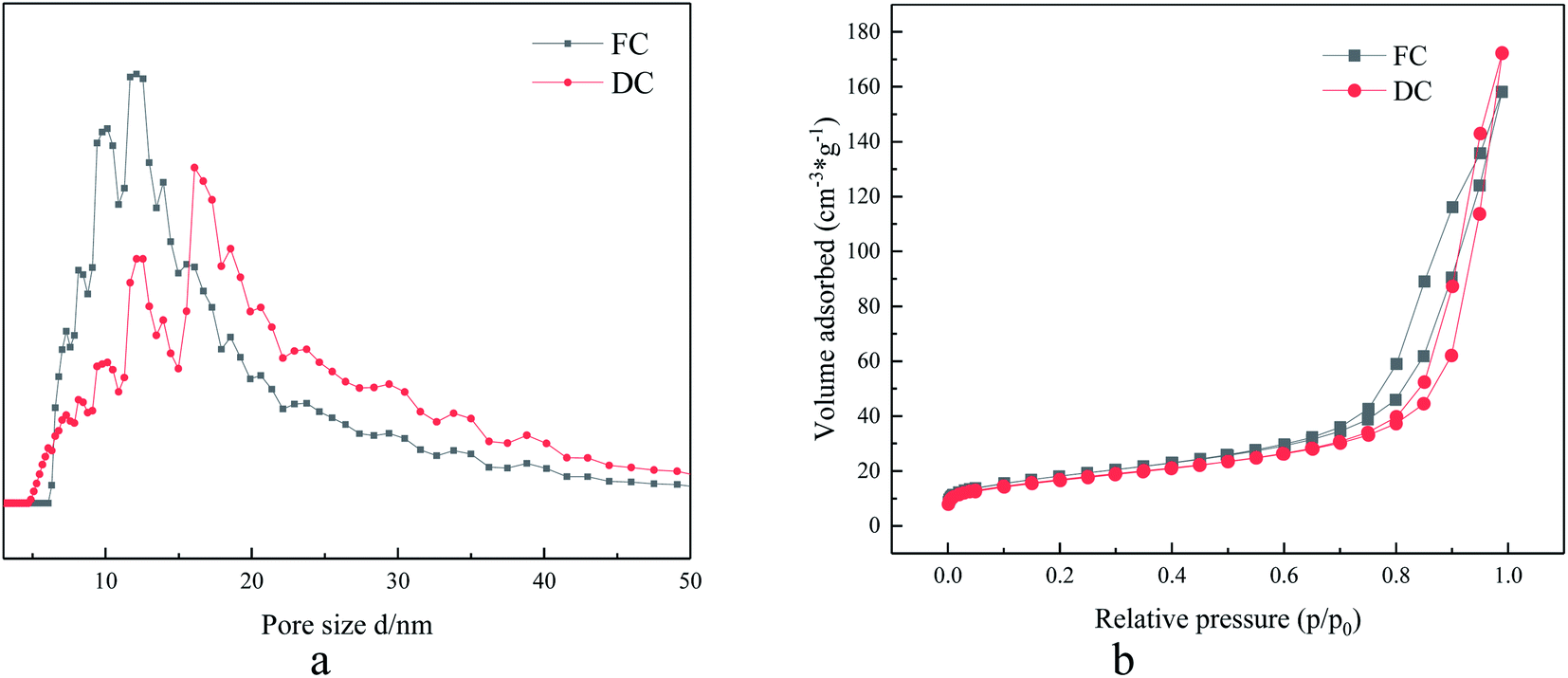 | ||
| Fig. 6 Pore size distribution (a) and adsorption–desorption curve of FC and DC (b). | ||
3.6 XPS
The XPS analysis was also conducted to understand the chemical states on the surfaces of FC and DC. A special attention was paid to elements of Ti, W, V and O, which are displayed in Fig. 7.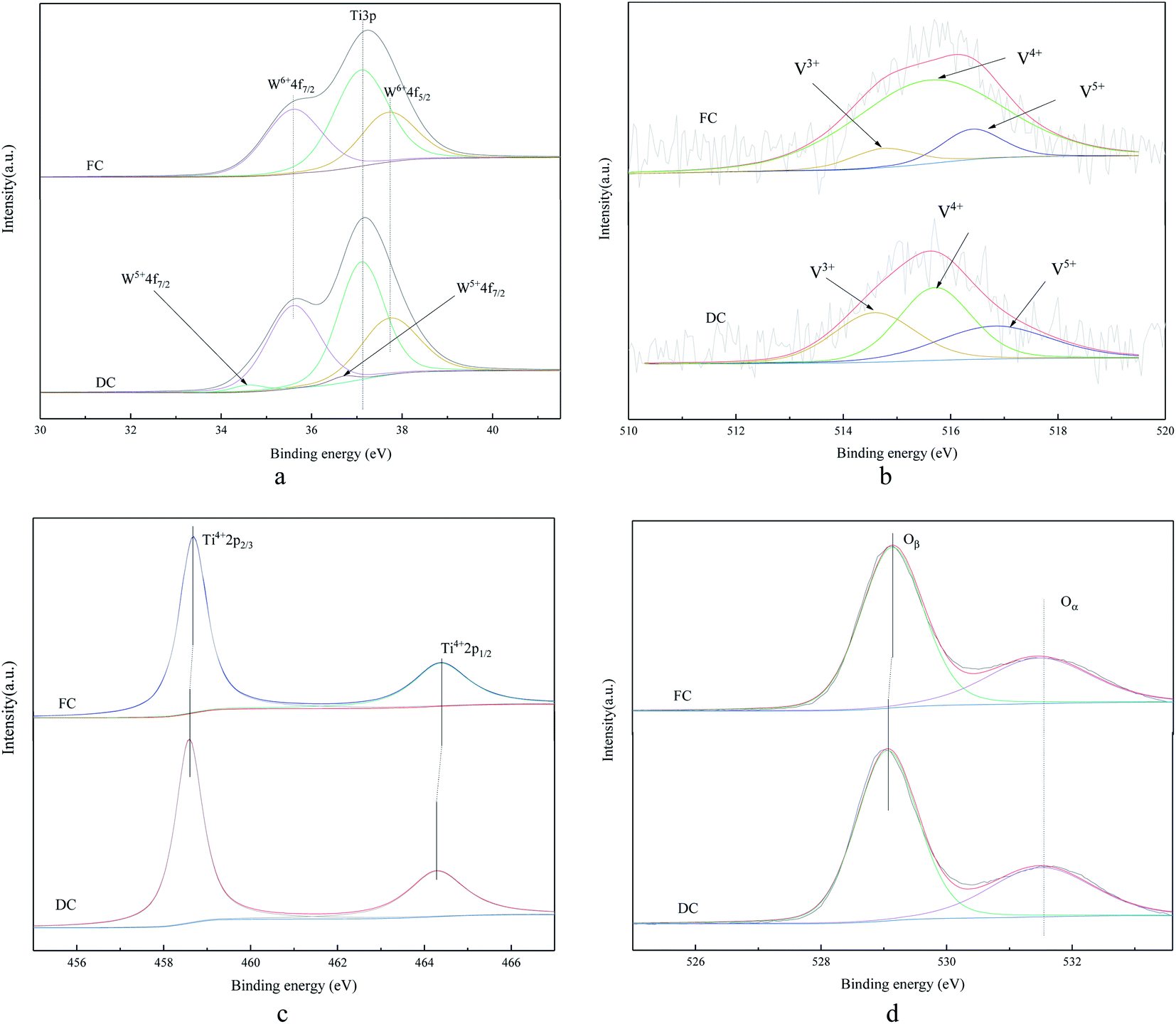 | ||
| Fig. 7 The XPS spectrum of FC and DC ((a) W; (b) V; (c) Ti; (d) O). | ||
Fig. 7a shows the W 4f XPS spectrum of FC and DC. The binding energy of 37.1 eV was attributed to Ti 3p,42 and the spectrum around 35.6 and 37.7 eV corresponded to W6+ 2p7/2 and W6+ 2p5/2.43 It can be seen from Fig. 7a that only spectra of Ti 3p and W6+ were detected over FC, revealing that tungsten oxide existed in a hexavalent state when FC was not used in the SCR system. The peaks located at 34.6 and 36.7 eV were attributed to W5+ 2p7/2 and W5+ 2p5/2, respectively.44 WO3 can store electrons45,46 and WO3–TiO2 can provide Lewis acidic sites for ammonia adsorbtion.47,48 As shown in Fig. 7a, W5+ 2p appeared in DC, which indicated that a certain amount pentavalent tungsten was also generated. The content of W5+ occupied 5.14% in DC, and W5+ might be poisoned by heavy metals, such as Pb, which could restrain the active polytungstate species to be formed again.34 There were a few clear evidences about the valence change of W in V2O5–WO3/TiO2, the decrease in W6+ might have some influence on the deactivation.
It is proposed that the peaks around 516.4–517.4 eV and 515.6–516.1 eV were attributed to V5+ and V4+, respectively, while the peak at 513.1–514.7 was assigned to V3+.49–51 As compared with FC, the valence states of V in DC showed a few changes. V3+/(V4+ + V5+) in the catalyst increased from to 0.09 to 0.45. As known, V3+ had no catalytic activity in the NH3-SCR reaction, while the higher concentration of V4+ could contribute to a high SCR activity.52–54 V4+ decreased from 80.14% in FC to 40.60% in DC, which contributed to the deactivation.
It can be seen from Fig. 7c that only Ti4+ was detected over FC and DC. TiO2 was stable when used for about 30000 h. As shown, a slight decrease about the binding energies of DC was observed with Ti4+ 2p3/2 shifted from 458.7 to 458.6 eV, and Ti4+ 2p1/2 shifted from 464.4 to 464.3 eV. The titanium species in DC had a higher density electron cloud than titanium species in FC according to the binding energies of Ti4+ 2p3/2 between FC and DC owing to the electrons around other species, such as tungsten, transferred to the titanium species. Moreover, the inductive effect of V was also related to the electron density of Ti4+.55
The binding energies of 529.0–530 eV and 531.3–531.9 eV were assigned to the lattice oxygen (Oβ) and chemisorbed oxygen (Oα), respectively.56–58 As shown in Fig. 7d, both Oα and Oβ were observed over FC and DC, and O 1s of DC shifted to the lower binding energy as compared to FC. Some higher electronegativity substances might reduce the binding energy of O. Surface chemisorbed oxygen was proven to be the most active oxygen during the SCR process. The Oα amount in DC decreased from 67.22% to 60.75%, which had a significant effect in the oxidation reaction.59
3.7 FT-IR
FT-IR was used to characterize the catalyst functional groups of FC and DC, and the results are shown in Fig. 8. The FT-IR spectra had bending vibrations of OH (3440) and HOH stretching (around 1636). Otherwise, the FT-IR spectra of FC and DC showed different shapes. The weak peak around 1020 cm−1 of FC may be assigned to VO.60 The bands at 1102 and 1190 cm−1 for DC could be attributed to the characteristic peaks of SO42−.61 It could be implied that a certain amount of sulfate was generated on the surface of DC, which was similar to the results of the elemental study. Due to SO2 in the flue gas, different sulfates, such as (NH4)2SO4, NH4HSO4 and CaSO4, formed on the used catalyst.61
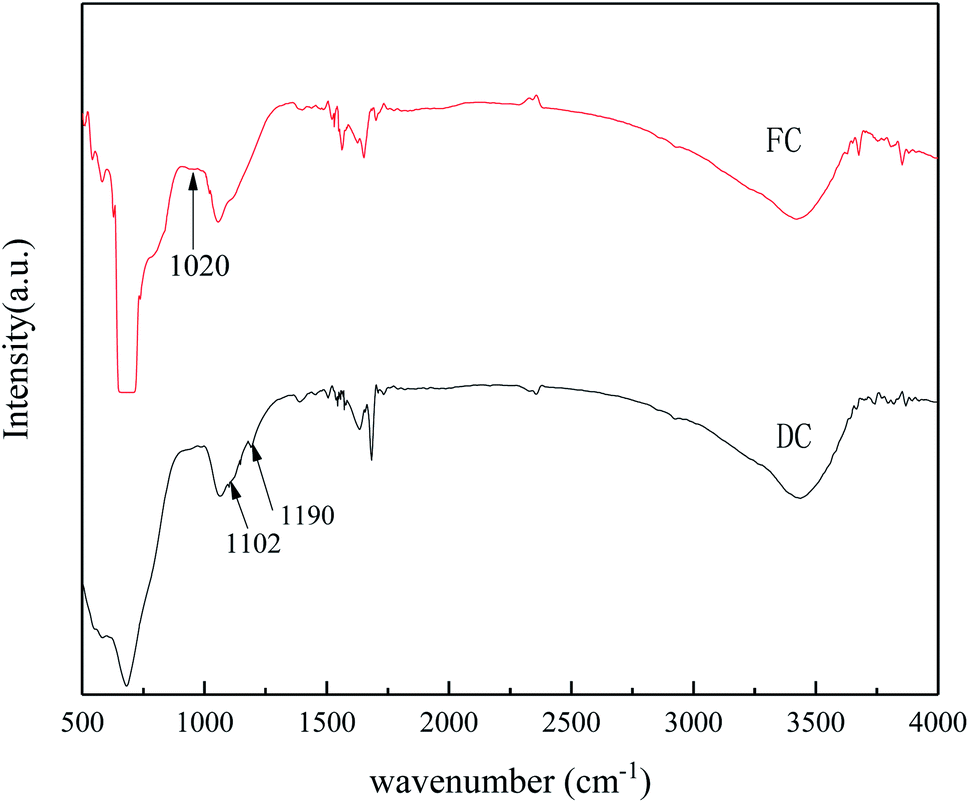 | ||
| Fig. 8 FT-IR spectra of FC and DC. | ||
3.8 NH3-TPD measurements
The surface acidity of the NH3-SCR catalyst plays a crucial role in the reaction process. The NH3 adsorption step on the surface is important to covert NOx to N2. To explore the changes of the surface acidity between FC and DC, NH3-TPD was conducted in the research (Fig. 9). The TPD curve for FC showed three peaks around 213 °C, 385 °C and 530 °C. As shown in Fig. 9, the intensities of the desorption peaks of DC decreased a lot, which indicated that the acidic sites of the catalyst were seriously poisoned.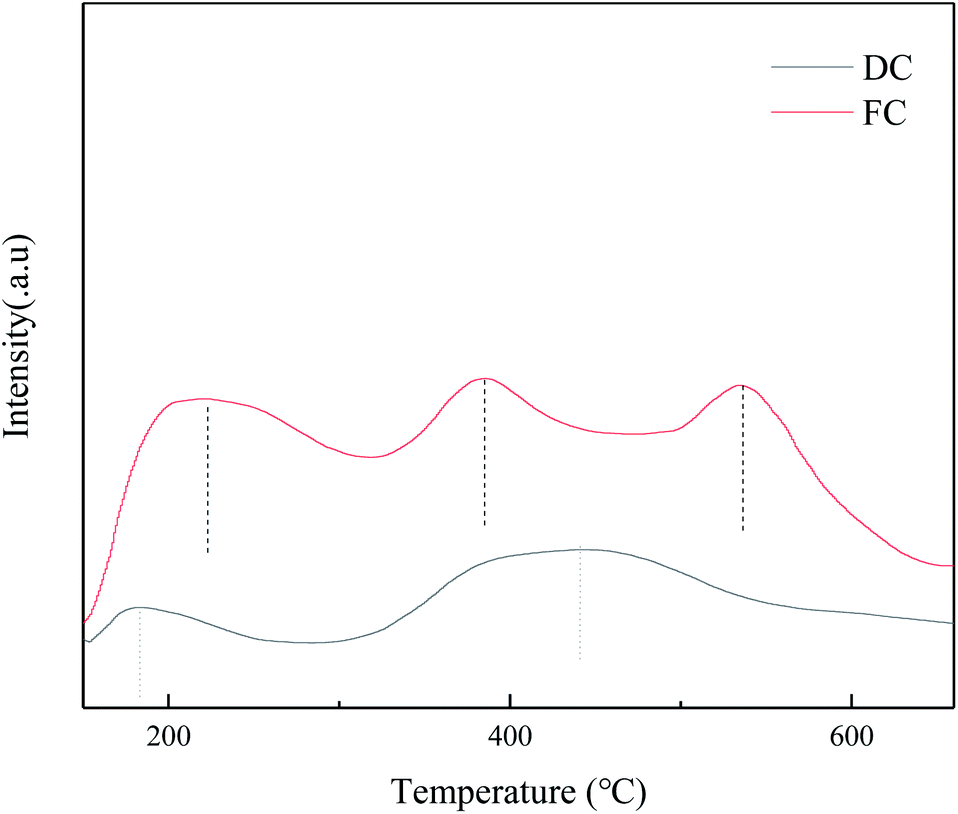 | ||
| Fig. 9 The NH3 adsorption–desorption spectrum of FC and DC. | ||
4. Conclusions
It is not easy to conclude the specific factors about the impacts on the SCR catalyst activity due to the complexity of the substances and the variable conditions of the flue gas. According to the experimental results of this research, the relative amount of Si and Al increased by 80.84% and by 2.26 times, respectively, which severely blocked and covered the active sites. The amount of alkali metals, such as K and Na increased a lot, which were harmful to the acidic sites. Other elements (Fe, Mg, Ca, etc.) could cover the active sites, which could lead to the deactivation of the catalyst. A few substances, particularly As, could terribly destroy the acidic sites and V–OH. The new substances played a great role on the catalyst deactivation, such as the sulfates formed on the catalyst. The special area decreased by 4.39 m2 g−1, which was not a main factor for deactivation. V3+/(V4+ + V5+) in the catalyst increased from to 0.09 to 0.45 after deactivation, and the V4+ amount decreased by about 39.54%. The surface acidity decreased a lot after being used for a long time. The valence change of V atoms might affect the reducibility of the catalyst surface, and lead to the deactivation of catalyst. The substances deposited on the surface could lead to a change in the special surface area and cover the active sites, causing the catalyst deactivated. Sulfates, such as CaSO4, (NH4)2SO4, and NH4HSO4, could cover the active sites and reduce the NH3 adsorption amount. In short, the deactivation pathways can be explained as three ways (Fig. 10): (1) the active sites were reduced, and some V2O5 was washed away. (2) The active sites were poisoned by various elements (K, Na, S, As, Pb, Zn, Ca, etc.), which caused V3+/(V4+ + V5+) to increase and NH3 adsorption amount to decrease. (3) The active sites were covered by substances, such as SiO2, CaSO4, (NH4)2SO4 and NH4HSO4, which restricted the catalytic reaction.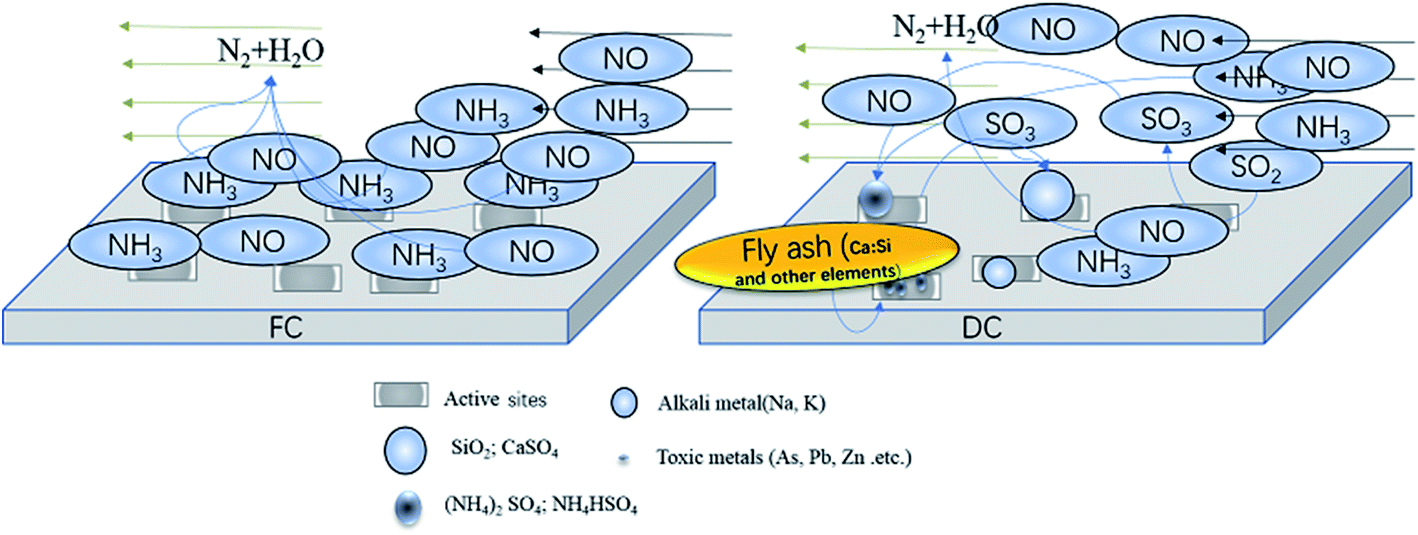 | ||
| Fig. 10 The deactivation pathways. | ||
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the Fundamental Research Funds for the Central Universities (2010YH14).References
- C. Paolucci, I. Khurana, A. A. Parekh, S. Li, A. J. Shih, H. Li, J. R. Di Iorio, J. D. Albarracin-Caballero, A. Yezerets, J. T. Miller, W. N. Delgass, F. H. Ribeiro, W. F. Schneider and R. Gounder, Dynamic multinuclear sites formed by mobilized copper ions in NOx selective catalytic reduction, Science, 2017, 357(6354), 898 CrossRef CAS.
- H. He, Y. Wang, Q. Ma, J. Ma, B. Chu, D. Ji, G. Tang, C. Liu, H. Zhang and J. Hao, Mineral dust and NOx promote the conversion of SO2 to sulfate in heavy pollution days, Sci. Rep., 2014, 4, 6092 CrossRef CAS.
- R. Khodayari and C. U. Ingemar Odenbrand, Regeneration of commercial SCR catalysts by washing and sulphation: effect of sulphate groups on the activity, Appl. Catal., B, 2001, 33(4), 277–291 CrossRef CAS.
- P. Forzatti, Environmental catalysis for stationary applications, Catal. Today, 2000, 62(1), 51–65 CrossRef CAS.
- L. Han, S. Cai, M. Gao, J. Hasegawa, P. Wang, J. Zhang, L. Shi and D. Zhang, Selective catalytic reduction of NOx with NH3 by using novel catalysts: state of the art and future prospects, Chem. Rev., 2019, 119(19), 10916–10976 CrossRef CAS.
- L. Yang, V. Bacic, I. A. Popov, A. I. Boldyrev, T. Heine, T. Frauenheim and E. Ganz, Two-dimensional Cu2Si monolayer with planar hexacoordinate copper and silicon bonding, J. Am. Chem. Soc., 2015, 137(7), 2757–2762 CrossRef CAS.
- J. Liu, L. Yang and E. Ganz, Efficient and selective electroreduction of CO2 by single-atom catalyst two-dimensional TM-Pc monolayers, ACS Sustainable Chem. Eng., 2018, 6(11), 15494–15502 CrossRef CAS.
- L. Xu, L. Yang and E. Ganz, Mn-graphene single-atom catalyst evaluated for CO oxidation by computational screening, Theor. Chem. Acc., 2018, 137(7), 98 Search PubMed.
- J. H. Liu, L. M. Yang and E. Ganz, Two-dimensional organometallic TM3-C12S12 monolayers for electrocatalytic reduction of CO2, Energy Environ. Mater., 2019, 2(3), 193–200 Search PubMed.
- B. Song, Y. Zhou, H. Yang, J. Liao, L. Yang, X. Yang and E. Ganz, Two-dimensional anti-van't Hoff/Le Bel array AlB6 with high stability, unique motif, triple dirac cones, and superconductivity, J. Am. Chem. Soc., 2019, 141(8), 3630–3640 CrossRef CAS.
- J. Liu, L. Yang and E. Ganz, Electrochemical reduction of CO2 by single atom catalyst TM-TCNQ monolayers, J. Mater. Chem. A, 2019, 7(8), 3805–3814 RSC.
- J. Liu, L. Yang and E. Ganz, Electrocatalytic reduction of CO2 by two-dimensional transition metal porphyrin sheets, J. Mater. Chem. A, 2019, 7(19), 11944–11952 RSC.
- J. Liu, L. Yang and E. Ganz, Efficient electrocatalytic reduction of carbon dioxide by metal-doped beta(12)-borophene monolayers, RSC Adv., 2019, 9(47), 27710–27719 RSC.
- J. Yang, X. Wang, Y. Qu, X. Wang, H. Huo, Q. Fan, J. Wang, L. Yang and Y. Wu, Bi-based metal-organic framework derived leafy bismuth nanosheets for carbon dioxide electroreduction, Adv. Energy Mater., 2020, 10(36), 2001709 CrossRef CAS.
- S. Chansai, R. Burch, C. Hardacre, J. Breen and F. Meunier, Investigating the mechanism of the H2-assisted selective catalytic reduction (SCR) of NOx with octane using fast cycling transient in situ DRIFTS-MS analysis, J. Catal., 2010, 276(1), 49–55 CrossRef CAS.
- P. Forzatti, Present status and perspectives in de-NOx SCR catalysis, Appl. Catal., A, 2001, 222(1), 221–236 CrossRef CAS.
- L. Lisi, G. Lasorella, S. Malloggi and G. Russo, Single and combined deactivating effect of alkali metals and HCl on commercial SCR catalysts, Appl. Catal., B, 2004, 50(4), 251–258 CrossRef CAS.
- K. Qiu, J. Song, H. Song, X. Gao, Z. Luo and K. Cen, A novel method of microwave heating mixed liquid-assisted regeneration of V2O5-WO3/TiO2 commercial SCR catalysts, Environ. Geochem. Health, 2015, 37(5), 905–914 CrossRef CAS.
- Y. Huo, Z. Chang, W. Li, S. Liu and B. Dong, Reuse and valorization of vanadium and tungsten from waste V2O5-WO3/TiO2 SCR Catalyst, Waste Biomass Valorization, 2015, 6(2), 159–165 CrossRef CAS.
- R. Khodayari and C. U. I. Odenbrand, Regeneration of commercial TiO2-V2O5-WO3 SCR catalysts used in bio fuel plants, Appl. Catal., B, 2001, 30(1), 87–99 CrossRef CAS.
- Y. Peng, J. Li, W. Si, J. Luo, Y. Wang, J. Fu, X. Li, J. Crittenden and J. Hao, Deactivation and regeneration of a commercial SCR catalyst: comparison with alkali metals and arsenic, Appl. Catal., B, 2015, 168–169, 195–202 CrossRef CAS.
- R. Khodayari, C. U. Ingemar Odenbrand, O. C. E. Department, U. Lund, F. K. Institutionen and U. Lunds, Regeneration of commercial SCR catalysts by washing and sulphation: effect of sulphate groups on the activity, Appl. Catal., B, 2001, 33(4), 277–291 CrossRef CAS.
- G. Wu, J. Li, Z. Fang, L. Lan, R. Wang, T. Lin, M. Gong and Y. Chen, Effectively enhance catalytic performance by adjusting pH during the synthesis of active components over FeVO4/TiO2-WO3-SiO2 monolith catalysts, Chem. Eng. J., 2015, 271, 1–13 CrossRef CAS.
- M. Klimczak, P. Kern, T. Heinzelmann, M. Lucas and P. Claus, High-throughput study of the effects of inorganic additives and poisons on NH3-SCR catalysts—part I: V2O5-WO3/TiO2 catalysts, Appl. Catal., B, 2010, 95(1–2), 39–47 CrossRef CAS.
- Y. Yu, C. He, J. Chen, L. Yin, T. Qiu and X. Meng, Regeneration of deactivated commercial SCR catalyst by alkali washing, Catal. Commun., 2013, 39, 78–81 CrossRef CAS.
- D. Nicosia, I. Czekaj and O. Kröcher, Chemical deactivation of V2O5-WO3/TiO2 SCR catalysts by additives and impurities from fuels, lubrication oils and urea solution, Appl. Catal., B, 2008, 77(3–4), 228–236 CrossRef CAS.
- O. Kröcher and M. Elsener, Chemical deactivation of V2O5-WO3/TiO2 SCR catalysts by additives and impurities from fuels, lubrication oils, and urea solution, Appl. Catal., B, 2008, 77(3–4), 215–227 CrossRef.
- Y. Yu, C. He, J. Chen, L. Yin, T. Qiu and X. Meng, Regeneration of deactivated commercial SCR catalyst by alkali washing, Catal. Commun., 2013, 39, 78–81 CrossRef CAS.
- L. Deng, X. Liu, P. Cao, Y. Zhao, Y. Du, C. Wang and D. Che, A study on deactivation of V2O5-WO3/TiO2 SCR catalyst by alkali metals during entrained-flow combustion, J. Energy Inst., 2017, 90(5), 743–751 CrossRef CAS.
- H. Kamata, K. Takahashi and C. U. I. Odenbrand, The role of K2O in the selective reduction of NO with NH3 over a V2O5(WO3)/TiO2 commercial selective catalytic reduction catalyst, J. Mol. Catal. A: Chem., 1999, 139(2), 189–198 CrossRef CAS.
- Y. Zheng, A. D. Jensen and J. E. Johnsson, Deactivation of V2O5-WO3/TiO2 SCR catalyst at a biomass-fired combined heat and power plant, Appl. Catal., B, 2005, 60(3–4), 253–264 CrossRef CAS.
- Y. Jiang, X. Gao, Y. Zhang, W. Wu, Z. Luo and K. Cen, Effect of KCl on the selective catalytic reduction of NO with NH3 over vanadia-based catalysts for biomass combustion, Environ. Prog. Sustainable Energy, 2014, 33(2), 390–395 CrossRef CAS.
- S. Wang, R. Guo, W. Pan, M. Li, P. Sun, S. Liu, S. Liu, X. Sun and J. Liu, The deactivation mechanism of Pb on the Ce/TiO2 catalyst for the selective catalytic reduction of NOx with NH3: TPD and DRIFT studies, Phys. Chem. Chem. Phys., 2017, 19(7), 5333–5342 RSC.
- Y. Peng, W. Si, X. Li, J. Chen, J. Li, J. Crittenden and J. Hao, Investigation of the poisoning mechanism of lead on the CeO2-WO3 Catalyst for the NH3-SCR reaction via in situ IR and Raman spectroscopy measurement, Environ. Sci. Technol., 2016, 50(17), 9576–9582 CrossRef CAS.
- Y. Peng, D. Wang, B. Li, C. Wang, J. Li, J. Crittenden and J. Hao, Impacts of Pb and SO2 poisoning on CeO2-WO3/TiO2-SiO2 SCR catalyst, Environ. Sci. Technol., 2017, 51(20), 11943–11949 CrossRef CAS.
- X. Li, J. Li, Y. Peng, H. Chang, T. Zhang, S. Zhao, W. Si and J. Hao, Mechanism of arsenic poisoning on SCR catalyst of CeW/Ti and its novel efficient regeneration method with hydrogen, Appl. Catal., B, 2016, 184, 246–257 CrossRef CAS.
- Y. Peng, W. Si, X. Li, J. Luo, J. Li, J. Crittenden and J. Hao, Comparison of MoO3 and WO3 on arsenic poisoning V2O5/TiO2 catalyst: DRIFTS and DFT study, Appl. Catal., B, 2016, 181, 692–698 CrossRef CAS.
- R. Guo, Q. Wang, W. Pan, Q. Chen, H. Ding, X. Yin, N. Yang, C. Lu, S. Wang and Y. Yuan, The poisoning effect of heavy metals doping on Mn/TiO2 catalyst for selective catalytic reduction of NO with NH3, J. Mol. Catal. A: Chem., 2015, 407, 1–7 CrossRef CAS.
- M. D. Amiridis, R. V. Duevel and I. E. Wachs, The effect of metal oxide additives on the activity of V2O5/TiO2 catalysts for the selective catalytic reduction of nitric oxide by ammonia, Appl. Catal., B, 1999, 20(2), 111–122 CrossRef CAS.
- J. R. Strege, C. J. Zygarlicke, B. C. Folkedahl and D. P. McCollor, SCR deactivation in a full-scale cofired utility boiler, Fuel, 2008, 87(7), 1341–1347 CrossRef CAS.
- C. R. Crocker, S. A. Benson and J. D. Laumb, SCR catalyst blinding due to sodium and calcium sulfate formation, J. Am. Chem. Soc., 2004, 227(1), U1080 Search PubMed.
- P. Cheng, C. Deng, X. Dai, B. Li, D. Liu and J. Xu, Enhanced energy conversion efficiency of TiO2 electrode modified with WO3 in dye-sensitized solar cells, J. Photochem. Photobiol., A, 2008, 195(1), 144–150 CrossRef CAS.
- H. Al-Kandari, F. Al-Kharafi, N. Al-Awadi, O. M. El-Dusouqui and A. Katrib, Surface electronic structure-catalytic activity relationship of partially reduced WO3 bulk or deposited on TiO2, J. Electron Spectrosc. Relat. Phenom., 2006, 151(2), 128–134 CrossRef CAS.
- S. Zhang, H. Li and Q. Zhong, Promotional effect of F-doped V2O5-WO3/TiO2 catalyst for NH3-SCR of NO at low-temperature, Appl. Catal., A, 2012, 435–436, 156–162 CrossRef CAS.
- T. Tatsuma, S. Takeda, S. Saitoh, Y. Ohko and A. Fujishima, Bactericidal effect of an energy storage TiO2-WO3 photocatalyst in dark, Electrochem. Commun., 2003, 5(9), 793–796 CrossRef CAS.
- T. Tatsuma, S. Saitoh, P. Ngaotrakanwiwat, Y. Ohko and A. Fujishima, Energy storage of TiO2-WO3 photocatalysis systems in the gas phase, Langmuir, 2002, 18(21), 7777–7779 CrossRef CAS.
- L. Lietti, J. Svachula, P. Forzatti, G. Busca, G. Ramis and P. Bregani, Surface and catalytic properties of vanadia-titania and tungsta-titania systems in the selective catalytic reduction of nitrogen oxides, Catal. Today, 1993, 17(1), 131–139 CrossRef CAS.
- L. J. Alemany, F. Berti, G. Busca, G. Ramis, D. Robba, G. P. Toledo and M. Trombetta, Characterization and composition of commercial V2O5/WO3-TiO2 SCR catalysts, Appl. Catal., B, 1996, 10(4), 299–311 CrossRef CAS.
- M. Shen, C. Li, J. Wang, L. Xu, W. Wang and J. Wang, New insight into the promotion effect of Cu doped V2O5/WO3-TiO2 for low temperature NH3-SCR performance, RSC Adv., 2015, 5(44), 35155–35165 RSC.
- B. M. Reddy, P. M. Sreekanth, E. P. Reddy, Y. Yamada, Q. A. Xu, H. Sakurai and T. Kobayashi, Surface characterization of La2O3-TiO2 and V2O5/La2O3-TiO2 catalysts, J. Phys. Chem. B, 2002, 106(22), 5695–5700 CrossRef CAS.
- D. W. Kwon, K. H. Park and S. C. Hong, The influence on SCR activity of the atomic structure of V2O5/TiO2 catalysts prepared by a mechanochemical method, Appl. Catal., A, 2013, 451, 227–235 CrossRef CAS.
- F. Castellino, S. B. Rasmussen, A. D. Jensen, J. E. Johnsson and R. Fehrmann, Deactivation of vanadia-based commercial SCR catalysts by polyphosphoric acids, Appl. Catal., B, 2008, 83(1), 110–122 CrossRef CAS.
- M. C. Paganini, L. Dall'Acqua, E. Giamello, L. Lietti, P. Forzatti and G. Busca, An EPR study of the surface chemistry of the V2O5-WO3/TiO2 catalyst: redox behaviour and state of V(IV), J. Catal., 1997, 166(2), 195–205 CrossRef CAS.
- N. Y. Topsoe, H. Topsoe and J. A. Dumesic, Vanadia/titania catalysts for selective catalytic reduction (SCR) of nitric-oxide by ammonia: I. Combined temperature-programmed in situ FTIR and on-line mass-spectroscopy studies, J. Catal., 1995, 151(1), 226–240 CrossRef CAS.
- Y. Qiu, R. Liu, B. Liu, Z. Liu, J. Du, Q. Tang and C. Tao, The monolithic cordierite supported V2O5-MoO3/TiO2 catalyst for NH3-SCR, Chem. Eng. J., 2016, 294, 264–272 CrossRef CAS.
- J. Fang, X. Bi, D. Si, Z. Jiang and W. Huang, Spectroscopic studies of interfacial structures of CeO2–TiO2 mixed oxides, Appl. Surf. Sci., 2007, 253(22), 8952–8961 CrossRef CAS.
- M. Kang, E. D. Park, J. M. Kim and J. E. Yie, Manganese oxide catalysts for NOx reduction with NH3 at low temperatures, Appl. Catal., A, 2007, 327(2), 261–269 CrossRef CAS.
- Z. Wu, R. Jin, Y. Liu and H. Wang, Ceria modified MnOx/TiO2 as a superior catalyst for NO reduction with NH3 at low-temperature, Catal. Commun., 2008, 9(13), 2217–2220 CrossRef CAS.
- D. Yu, Y. Liu and Z. Wu, Low-temperature catalytic oxidation of toluene over mesoporous MnOx-CeO2/TiO2 prepared by sol-gel method, Catal. Commun., 2010, 11(8), 788–791 CrossRef CAS.
- Z. Huang, Z. Zhu, Z. Liu and Q. Liu, Formation and reaction of ammonium sulfate salts on V2O5/AC catalyst during selective catalytic reduction of nitric oxide by ammonia at low temperatures, J. Catal., 2003, 214(2), 213–219 CrossRef CAS.
- J. R. Strege, C. J. Zygarlicke, B. C. Folkedahl and D. P. McCollor, SCR deactivation in a full-scale cofired utility boiler, Fuel, 2008, 87(7), 1341–1347 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06812h |
| This journal is © The Royal Society of Chemistry 2020 |