
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDonor–acceptor duality of the transition-metal-like B2 core in core–shell-like metallo-borospherenes La3&[B2@B17]− and La3&[B2@B18]−†
Xiao-Yun
Zhao
ab,
Miao
Yan
a,
Zhihong
Wei
*a and
Si-Dian
Li
 *a
*a
aKey Laboratory of Materials for Energy Conversion and Storage of Shanxi Province, Institute of Molecular Science, Shanxi University, Taiyuan 030006, China. E-mail: weizhihong@sxu.edu.cn; lisidian@sxu.edu.cn
bDepartment of Applied Chemistry, Yuncheng University, Yuncheng 044000, China
First published on 15th September 2020
Abstract
Transition-metal doping induces dramatic structural changes and leads to earlier planar → tubular → spherical → core–shell-like structural transitions in boron clusters. Inspired by the newly discovered spherical trihedral metallo-borospherene D3h La3&B18− (1) (Chen, et al., Nat. Commun., 2020, 11, 2766) and based on extensive first-principles theory calculations, we predict herein the first and smallest core–shell-like metallo-borospherenes C2v La3&[B2@B17]− (2) and D3h La3&[B2@B18]− (3) which contain a transition-metal-like B2 core at the cage center with unique donor–acceptor duality in La3&Bn− spherical trihedral shells (n = 17, 18). Detailed energy decomposition and bonding analyses indicate that the B2 core in these novel complexes serves as a π-donor in the equatorial direction mainly to coordinate three La atoms on the waist and a π/σ-acceptor in the axial direction mainly coordinated by two B6 triangles on the top and bottom. These highly stable core–shell complexes appear to be spherically aromatic in nature in bonding patterns. The IR, Raman, and photoelectron spectra of 2 and 3 are computationally simulated to facilitate their spectroscopic characterizations.
1. Introduction
As a prototypical electron-deficient element, boron exhibits diverse geometrical structures and bonding patterns in both bulk allotropes and polyhedral molecules.1,2 Persistent joint photoelectron spectroscopy (PES) and first-principles theory investigations in the past two decades have unveiled a rich landscape for size-selected boron nano-clusters (Bn−/0) from planar or quasi-planar (2D) structures (n = 3–38, 41–42)3–8 to cage-like borospherenes (C3/C2 B39− and D2d B40−/0) which are all characterized with delocalized multi-centre bonding.9,10 Seashell-like borospherenes C2 B28− and Cs B29− were later confirmed in PES measurements to be minor isomers of the monoanions.11,12 The borospherene family has been systematically expanded at first-principles theory level to the cage-like Bnq series (n = 36–42, q = n − 40) in different charge states.13–16 More complicated structural competitions exist in medium-sized boron clusters, with B46 being theoretically predicted to be the smallest core–shell-like boron cluster reported to date, while B48, B54, B60, and B62 are proposed to possess bilayer structures.17,18 Joint ion-mobility experiments and density functional theory (DFT) investigations, on the other hand, indicated that bare Bn+ boron cluster monocations possess double-ring tubular structures in the size range between n = 16–25.19Transition-metal-doping induces dramatic structural changes and leads to earlier planar → tubular → spherical → core–shell-like structural transitions in boron clusters. Perfect transition-metal-centred 2D boron wheels D8h Co@B8−, D9h Ru@B9−, and D10h Ta@B10−, half-sandwich SmB6−, PrB7−, CoB12−, and RhB12−,20–23 double-ring tubular CoB16− (ref. 24) and RhB18−,25 and perfect inverse sandwich D7h La2B7−,26D8h La2B8−,27 and D9h La2B9− (ref. 26) have been successively characterized in PES experiments. The first experimentally observed tri-lanthanide-doped inverse triple-decker C2v La3B14− (ref. 28) contains two conjoined B8 rings which share a B2 unit on the waist. With four more B atoms added in, the first perfect spherical trihedral metallo-borospherene D3h La3&B18− (1) with three equivalent deca-coordinate La atoms as integral parts of the cage surface were discovered very recently in a joint PES experimental and theoretical investigation.29 Exohedral metallo-borospherenes M&B40 (M = Be, Mg)30 and Nin&B40 (n = 1–4)31 were previously proposed in theory which contain hepta-coordinate metal centres in η7-B7 rings on the cage surface of B40. Our group proposed very recently at first-principles theory level the smallest perfect spherical trihedral metallo-borospherenes D3h Ta3&B12− which contains three equivalent octa-coordinate Ta centres as integral parts of the cage surface.32 However, to the best of our knowledge, there have been no core–shell-like metallo-borospherenes reported to date in either theory or experiments.
Based on extensive global minimum (GM) searches and first-principles theory calculations, we predict herein the first and smallest core–shell-like (CSL) spherical trihedral metallo-borospherenes C2v La3&[B2@B17]− (2) and D3h La3&[B2@B18]− (3) which contain three equivalent La atoms as integral parts of cage surface and, more importantly, a B2 core with unusual donor–acceptor duality at the cage centre (Fig. 1 and TOC) which exhibits obvious transition-metal-like behaviour. With the formation of effective coordination interactions between B2 and La3@Bn− spherical shells (n = 17, 18), the B2 core plays an essential role in stabilizing these spherically aromatic CSL complexes. La3&[B2@B17]− (2) and La3&[B2@B18]− (3) prove to possess the optimum core–shell combinations to demonstrate the donor–acceptor duality of a B2 core in La3&Bn− spherical shells (n = 17, 18).
 | ||
| Fig. 1 Side (a) and top (b) views of the global minimum structures of D3h La3&B18− (1), C2v La3&[B2@B17]− (2), and D3h La3&[B2@B18]− (3) at PBE0/B6-311+G(d)/La/ECP28MWB level, with the B2 core highlighted in red and important bond lengths indicated in Å. | ||
2. Theoretical procedure
Extensive global minimum (GM) searches were performed on BnLa3− (n = 19 and 20) using the TGmin 2.0 algorithms,33,34 in conjunction with manual structural constructions. Approximately 2000 trial structures were probed on the potential energy surface for each species in both singlet and triplet states. The low-lying isomers were then fully re-optimized at both the hybrid PBE0 (ref. 35) and TPSSh36 DFT levels with the 6-311+G(d)37 basis set for B and Stuttgart relativistic small-core pseudopotential (ECP28MWB) for La38,39 using the Gaussian 09 program suite (with the self-consistent-field convergence criteria of scf = tight),40 with vibrational frequencies checked to make sure all the low-lying isomers obtained are true minima of the systems. Relative energies of the five lowest-lying isomers were further refined at the more accurate CCSD(T) level41–43 at PBE0 geometries using the MOLPRO program,44 with the same basis sets. Bonding analyses were performed using the adaptive natural density partitioning (AdNDP)45,46 approach. Born–Oppenheimer molecular dynamics (BOMD) simulations were performed on La3&[B2@B17]− (2) and La3&[B2@B18]− (3) for 30 ps at different temperatures using the CP2K software package47 with the time step of 10 fs at the PBE/TZVP level, starting from the equilibrium GM geometry with random velocities assigned to the atoms in a cubic box with 15 Å on each side. The initial conditions were chosen to correspond to a microcanonical ensemble with the energy cutoff of 300 eV. The iso-chemical shielding surfaces (ICSSs)48,49 were generated with the Multiwfn 3.7 code.50 A detailed energy decomposition analysis with natural orbitals for chemical valence (EDA-NOCV)51–53 was carried out using the ADF program package54 at the PBE0/TZP-ZORA level where scalar relativistic effects were considered for the metals using the zeroth-order regular approximation (ZORA).55–57 The frozen core approximation was not employed in these computations. In the EDA analysis, the interaction energy (ΔEint) between two fragments is decomposed into the electrostatic interaction energy (ΔEelstat), the Pauli repulsion (ΔEPauli), and the orbital interaction energy (ΔEorb) in eqn (1).| ΔEint = ΔEelstat + ΔEPauli + ΔEorb | (1) |
3. Results and discussions
We start from closed-shell D3h La3&[B2@B18]− (3, ), the smallest perfect CSL metallo-borospherene which can be obtained by adding a B2 core directly inside the experimentally observed La3&B18− (1) shell29 (Fig. 1). Extensive global searches indicate that La3&[B2@B18]− (3) is the well-defined GM of La3B20− in thermodynamics in both singlet and triplet states, with the lowest vibrational frequency of νmin = 113.2 cm−1 (e′) and large HOMO–LUMO gap of ΔEgap = 2.30 eV at PBE0/B6-311+G(d)/La/ECP28MWB level.37–39 It lies 0.51, 0.52, 0.29 eV lower than the second lowest-lying triple-decker Cs La3&B20− at PBE0,35 TPSSh,36 and CCSD(T)41–43 levels with the same basis sets, respectively (Fig. S1†). The D3h La3&B18− shell in La3&[B2@B18]− (3) contains three equivalent deca-coordinate La atoms at the centres of three conjoined η10-B10 rings which share two B6 triangles on the top and bottom interconnected by three B2 units on the waist. All the fifteen lowest-lying isomers within 1.1 eV possess 3D structures in singlet states, with the second, third, and fourth CSL La3&[Bn@B20−n]− isomers (n = 1–3) lying 0.65, 0.88, and 1.08 eV higher than the GM at PBE0 level, respectively. Triplet isomers are found to be much less stable than the GM, with the first triplet structure Cs La3&[B2@B18]− (14) possessing the relative energies +0.97 eV at PBE0 (Fig. S1†). Removing one B atom from a B2 unit on the waist in La3&[B2@B18]− (3) results in the doublet C2v La3&[B2@B17]− (2, 2A1) which, with one η10-B10 ring in the front and two equivalent η9-B9 rings on the back, is also the GM of the system (Fig. S2†).
), the smallest perfect CSL metallo-borospherene which can be obtained by adding a B2 core directly inside the experimentally observed La3&B18− (1) shell29 (Fig. 1). Extensive global searches indicate that La3&[B2@B18]− (3) is the well-defined GM of La3B20− in thermodynamics in both singlet and triplet states, with the lowest vibrational frequency of νmin = 113.2 cm−1 (e′) and large HOMO–LUMO gap of ΔEgap = 2.30 eV at PBE0/B6-311+G(d)/La/ECP28MWB level.37–39 It lies 0.51, 0.52, 0.29 eV lower than the second lowest-lying triple-decker Cs La3&B20− at PBE0,35 TPSSh,36 and CCSD(T)41–43 levels with the same basis sets, respectively (Fig. S1†). The D3h La3&B18− shell in La3&[B2@B18]− (3) contains three equivalent deca-coordinate La atoms at the centres of three conjoined η10-B10 rings which share two B6 triangles on the top and bottom interconnected by three B2 units on the waist. All the fifteen lowest-lying isomers within 1.1 eV possess 3D structures in singlet states, with the second, third, and fourth CSL La3&[Bn@B20−n]− isomers (n = 1–3) lying 0.65, 0.88, and 1.08 eV higher than the GM at PBE0 level, respectively. Triplet isomers are found to be much less stable than the GM, with the first triplet structure Cs La3&[B2@B18]− (14) possessing the relative energies +0.97 eV at PBE0 (Fig. S1†). Removing one B atom from a B2 unit on the waist in La3&[B2@B18]− (3) results in the doublet C2v La3&[B2@B17]− (2, 2A1) which, with one η10-B10 ring in the front and two equivalent η9-B9 rings on the back, is also the GM of the system (Fig. S2†).
Extensive BOMD simulations are performed on La3&[B2@B17]− (2) and La3&[B2@B18]− (3) at 300, 700 and 1000 K to check their dynamical stabilities in gas-phases, respectively (Fig. S3 and S4†). Both of these CSL species appear to be dynamically stable at 1000 K, with the small calculated average root-mean-square-deviations of RMSD = 0.13, 0.14 Å and maximum bond length deviations of MAXD = 0.40, 0.47 Å, respectively. No high-lying isomers were observed during the simulations.
Natural bonding orbital (NBO) analyses58 show that the three equivalent La centres in La3&[B2@B18]− (3) possess the electronic configuration of La [Xe]6s0.125d1.46, natural atomic charge of qLa = +1.28|e|, and total Wiberg bond index of WBILa = 3.11, respectively, indicating that each La centre in these complexes donates its 6s2 electrons almost completely to the B20 ligand, while, in return, accepts about half an electron in its partially filled 5d orbitals from the CSL B20 framework (Table S1†). As a commonly used indicator of bond order,13–16 the calculated B–B Wiberg bond index of WBIB–B = 0.73 in the B2 core indicates the formation of a B–B single bond, the B⋯B bond orders of WBIB–B = 0.27–0.43 between the two B6 triangles and B2 core suggest the existence of B6⋯B2 coordination interactions in axial direction, while the B⋯La bond order of WBILa⋯B = 0.26 between the B2 core and three La atoms evidences the formation of B2⋯La coordination interactions on the equator. The B2 core thus forms effective coordination interactions with the La3B18− shell around it, akin to a transition metal centre in traditional complexes.
Detailed EDA-NOCV analyses51–53 at PBE0/TZP-ZORA level35,55–57 – with various interacting fragments in different charge states59 considered (Table S2†) indicate that, with the smallest orbital interaction energy of ΔEorb = −717.9 kcal mol−1, the B2 and La3&B18− fragments interaction is best suited to describe the bonding scheme of La3&[B2@B18]− (3). Such a scheme is well in line with the experimental observation that a spherical trihedral La3B18− cage is highly stable in gas phase.29 As for the B2 core, previous theoretical and experimental investigations demonstrated that the isolation of B2 allotropes with a B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) B triple bond can be achieved in the formula of L→BB←L, where L is N-heterocyclic carbene (NHC),60,61 carbonyl (CO),62 or boronyl anion (BO−).63 The third excited state (1Σg+) of B2 with two electrons excited from 1σu to 1πu rather than the ground state (3Σg−) with the valence electron configuration of 1σ2g1σ2u1π2u was proven to have the most suitable electron configuration (1σ2g1π4u) to describe the bonding nature of (NHC)→BB←(NHC).64,65 For La3&[B2@B18]− (3), the B2 core in the third excited state [1Σg+] produces a smaller orbital interaction energy (ΔEorb) than that in the ground state [3Σg−] by 261.8 kcal mol−1. We thus choose to use B2 [1Σg+] and La3B18− [1A1] fragments as interacting species to demonstrate the bonding scheme of La3&[B2@B18]− (3) in Fig. 2.
B triple bond can be achieved in the formula of L→BB←L, where L is N-heterocyclic carbene (NHC),60,61 carbonyl (CO),62 or boronyl anion (BO−).63 The third excited state (1Σg+) of B2 with two electrons excited from 1σu to 1πu rather than the ground state (3Σg−) with the valence electron configuration of 1σ2g1σ2u1π2u was proven to have the most suitable electron configuration (1σ2g1π4u) to describe the bonding nature of (NHC)→BB←(NHC).64,65 For La3&[B2@B18]− (3), the B2 core in the third excited state [1Σg+] produces a smaller orbital interaction energy (ΔEorb) than that in the ground state [3Σg−] by 261.8 kcal mol−1. We thus choose to use B2 [1Σg+] and La3B18− [1A1] fragments as interacting species to demonstrate the bonding scheme of La3&[B2@B18]− (3) in Fig. 2.
 | ||
| Fig. 2 Bonding scheme of D3h La3&[B2@B18]− (3) using the fragments B2 (1Σg+) and La3&B18− (1A1) as interacting species at PBE0/TZP-ZORA level. | ||
The bonding molecular orbitals (MOs) 29e′, 15e′′,  and
and  which represent coordination bonding between the B2 core and La3B18− shell are connected with the corresponding fragmental orbitals by red lines in Fig. 2, with the orbital compositions listed in Table S3.† The doubly degenerate MOs 29e′ are mainly composed of contributions from the occupied 1e′ of B2 core with π character and vacant 29e′ of La3B18− with π feature. The doubly degenerate 15e′′ are a linear combination of the occupied 15e′′ of La3B18− with π feature and vacant 1e′′ of B2 core with π* antibonding character. The non-degenerate
which represent coordination bonding between the B2 core and La3B18− shell are connected with the corresponding fragmental orbitals by red lines in Fig. 2, with the orbital compositions listed in Table S3.† The doubly degenerate MOs 29e′ are mainly composed of contributions from the occupied 1e′ of B2 core with π character and vacant 29e′ of La3B18− with π feature. The doubly degenerate 15e′′ are a linear combination of the occupied 15e′′ of La3B18− with π feature and vacant 1e′′ of B2 core with π* antibonding character. The non-degenerate  and
and  mainly originate from the occupied
mainly originate from the occupied  and
and  of La3B18− with π character and vacant
of La3B18− with π character and vacant 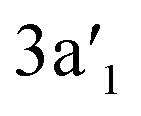 and
and  of B2 core with σ bonding and σ* antibonding characters, respectively. The non-degenerate
of B2 core with σ bonding and σ* antibonding characters, respectively. The non-degenerate  originates almost completely from the 2a′ of B2 which represents the B–B σ single bond in the B2 core. As detailed in Table 1, EDA analyses clearly demonstrate that the interaction energy ΔEint between the B2 core and La3&B18− shell in La3&[B2@B18]− (3) consists of Pauli repulsion ΔEPauli, coulombic attraction ΔEelstat, and orbital interaction ΔEorb, with slightly more covalent contribution (51.2%) than electrostatic contribution (48.8%). The decompositions of the orbital interactions ΔEorb into pairwise contributions between occupied and vacant MOs of the fragments provide quantitative insight into the charge flow. The strongest orbital interaction ΔEorb(1) (43.9%) arises mainly from [B2 (π)] → [La3B18− (πLa(s+d))] in which the B2 core serves as a π-donor in equatorial direction mainly to coordinate the three La atoms on the waist, while the relatively weaker orbital interaction ΔEorb(2) (17.8%) originates from [B2 (π*)] ← [La3B18− (πB(p))] where the B2 core is a π*-acceptor in axial direction mainly coordinated by two B6 triangles on the top and bottom. The orbital interactions ΔEorb(3) (19.4%) and ΔEorb(4) (11.0%) correspond to [B2 (σp)] ← [La3B18− (πB(p))] and [B2 (
originates almost completely from the 2a′ of B2 which represents the B–B σ single bond in the B2 core. As detailed in Table 1, EDA analyses clearly demonstrate that the interaction energy ΔEint between the B2 core and La3&B18− shell in La3&[B2@B18]− (3) consists of Pauli repulsion ΔEPauli, coulombic attraction ΔEelstat, and orbital interaction ΔEorb, with slightly more covalent contribution (51.2%) than electrostatic contribution (48.8%). The decompositions of the orbital interactions ΔEorb into pairwise contributions between occupied and vacant MOs of the fragments provide quantitative insight into the charge flow. The strongest orbital interaction ΔEorb(1) (43.9%) arises mainly from [B2 (π)] → [La3B18− (πLa(s+d))] in which the B2 core serves as a π-donor in equatorial direction mainly to coordinate the three La atoms on the waist, while the relatively weaker orbital interaction ΔEorb(2) (17.8%) originates from [B2 (π*)] ← [La3B18− (πB(p))] where the B2 core is a π*-acceptor in axial direction mainly coordinated by two B6 triangles on the top and bottom. The orbital interactions ΔEorb(3) (19.4%) and ΔEorb(4) (11.0%) correspond to [B2 (σp)] ← [La3B18− (πB(p))] and [B2 ( )] ← [La3B18− (πB(p))], respectively, in which the B2 core functions as both σ- and σ*-acceptors in axial direction coordinated mainly by two B6 triangles on the top and bottom. Fig. S5† further shows the deformation densities Δρ associated with the pairwise interactions ΔEorb(1)–(4) in La3&[B2@B18]− (3).
)] ← [La3B18− (πB(p))], respectively, in which the B2 core functions as both σ- and σ*-acceptors in axial direction coordinated mainly by two B6 triangles on the top and bottom. Fig. S5† further shows the deformation densities Δρ associated with the pairwise interactions ΔEorb(1)–(4) in La3&[B2@B18]− (3).
| Energy terms | Orbital interaction | B2 [1Σg+] + B18La3− [1A1] |
|---|---|---|
| a The value in parentheses gives the percentage contribution to the total attractive interactions (ΔEelstat + ΔEorb). b The value in parentheses gives the percentage contribution to the total orbital interactions. | ||
| ΔEint | −349.9 | |
| ΔEPauli | 1053.5 | |
| ΔEelstata | −685.5 (48.8%) | |
| ΔEorba | −717.9 (51.2%) | |
| ΔEorb(1)b | [B2 (π)] → [La3B18−(πLa(s+d))] | −315.4 (43.9%) |
| ΔEorb(2)b | [B2 (π*)] ← [La3B18− (πB(p))] | −127.6 (17.8%) |
| ΔEorb(3)b | [B2 (σp)] ← [La3B18− (πB(p))] | −139.3 (19.4%) |
| ΔEorb(4)b | [B2 (σs*)] ← [La3B18− (πB(p))] | −79.1 (11.0%) |
| ΔEorb(rest)b | −57.2 (7.9%) | |
Our EDA-NOCV results quantitatively indicate that the B2 core in the spherically trihedral La3&[B2@B18]− (3) is a considerably strong π-donor to three equivalent La centres in equatorial directions rather than mainly a σ-acceptor in axial direction in the previously reported linear L→BB←L complexes.60–63 The π-donation of the B2 core effectively weakens the B–B π interactions in the B2 core and results in a B–B single bond in La3&[B2@B18]− (3) with the B–B bond length of rB–B = 1.63 Å (rather than a BB triple bond with the B–B distances of rBB = 1.45–1.48 Å in L→BB←L complexes).60–63 The π-donation of B2 core in La3&[B2@B18]− (3) to three equivalent La centers evenly distributed in three equatorial directions is also different from the activation of CO by transition-metal-like NHC→BB←NHC in one direction in which an HOMO–LUMO swap occurs in B2(NHC)2 to form effective electron back-donations with the π* MO of CO.65 The B2 unit at the centre serves as both π-donor and σ-acceptor in B2(NHC)2.60,61,65
Detailed adaptive natural density partitioning (AdNDP) bonding analyses45,46 recover both the localized and delocalized bonds of the concerned complexes. As shown in Fig. 3 in the first row, La3&[B2@B18]− (3) possesses 9 localized 2c–2e σ bonds on three edges on the waist, 8 3c–2e σ bonds on two B6 triangles on the top and bottom, 1 2c–2e B–B σ bond inside the B2 core, and 2 7c–2e B6(π) → B(σ) coordination bonds between the B2 core and two B6 triangles on the top and bottom (in which the B2 core serves as a σ-acceptor). The 3 5c–2e B4(π)–La(dσ) bonds in axial direction in the second row mainly represent B2 → La coordination interactions between the B2 core and three La atoms (in which the B2 core functions as a π-donor). There exist 3 13c–2e B12(π)–La(dπ) bonds in axis direction in the third row (in which the B2 core serves as a π*-acceptor), 3 13c–2e B12(π)–La(dπ) bonds in equatorial direction in the fourth row, and 3 13c–2e B12(π)–La(dσ) bonds in equatorial direction in the fifth row. The nine delocalized (p–d) π interactions evenly distributed on the cage surface form a local 6π-aromatic system over each La&B10 pyramidal subunit (similar to the 6π-aromatic system in benzene) and render overall spherical aromaticity to La3&[B2@B18]− (3). The 3 13c–2e (p–d) δ bonds in the sixth row help to further stabilize the CSL complex. As shown in Fig. S6,† a closed-shell C2v La3&[B2@B17]2− (1A1) possesses a similar bonding pattern with La3&[B2@B18]−(3). The open-shell C2v La3&[B2@B17]− (2) has the same bonding pattern as La3&[B2@B17]2−, with the 5c–2e B4(π)–La(dσ) bond at the centre in the second row in Fig. S6† singly occupied (which corresponds to its singly occupied HOMO (a1)).
 | ||
| Fig. 3 AdNDP bonding patterns of D3h La3&[B2@B18]− (3) with the occupation numbers (ON) indicated. | ||
The spherical aromaticity of La3&[B2@B17]− (2) and La3&[B2@B18]− (3) is further evidenced by their calculated negative nucleus-independent chemical shift (NICS) values66 of NICS = −80.05 and −80.18 ppm at the cage centres, respectively. Based on the calculated NICS-ZZ components, Fig. S7† plots their iso-chemical-shielding surfaces (ICSSs)48,49 with Z-axis parallel to the designated C2 molecular axes of the systems to illuminate the chemical shielding around the La&B10 pyramids in these complexes. Obviously, the space inside the spherical trihedron and within about 1.0 Å above the La centres in vertical direction belongs to the chemical shielding region with negative NICS-ZZ values, while the chemical de-shielding areas with positive NICS values are located outside the B10 ring in the horizontal direction. The ICSSs of these complexes in C2 axial directions appear to be similar to that of the prototypical aromatic benzene C6H6 in C6 axial direction (Fig. S7†).
Finally, we simulate the IR, Raman, and PES spectra of La3&[B2@B18]− (3) in Fig. 4 to facilitate its future spectroscopic characterizations in gas-phases. Its major IR bands occur at 365(e′), 517(e′), 728(e′), 824(e′), 1169(e′) and 1421(e′) cm−1, with the corresponding Raman active vibrations located at 158(e′′), 366(e′), 521( ), 824(e′) and 1006(
), 824(e′) and 1006( ) cm−1, respectively. Detailed vibrational analyses indicate that the symmetrical vibrations at 521(
) cm−1, respectively. Detailed vibrational analyses indicate that the symmetrical vibrations at 521( ) cm−1 represent typical radial breathing modes (RBMs) of the boron skeleton which can be used to characterize single-walled hollow boron nanostructures.67 The calculated PES spectrum of La3&[B2@B18]− in Fig. 4c exhibits major spectral features at 2.46, 2.51, 2.98, 3.73–4.94, 5.81 and 6.02 eV, respectively, which correspond to vertical electronic transitions from the ground state of the anion to the ground and excited states of the neutral at the ground-state geometry of the anion. The simulated IR, Raman, and photoelectron spectra of La3&[B2@B17]− (2) are depicted in Fig. S8.†
) cm−1 represent typical radial breathing modes (RBMs) of the boron skeleton which can be used to characterize single-walled hollow boron nanostructures.67 The calculated PES spectrum of La3&[B2@B18]− in Fig. 4c exhibits major spectral features at 2.46, 2.51, 2.98, 3.73–4.94, 5.81 and 6.02 eV, respectively, which correspond to vertical electronic transitions from the ground state of the anion to the ground and excited states of the neutral at the ground-state geometry of the anion. The simulated IR, Raman, and photoelectron spectra of La3&[B2@B17]− (2) are depicted in Fig. S8.†
 | ||
| Fig. 4 Simulated (a) IR, (b) Raman and (c) photoelectron spectra of La3&[B2@B18]− (3) at PBE0/B6-311+G(d)/La/ECP28MWB level. | ||
4. Summary
In summary, we have presented in this work a comprehensive first-principles theory investigation on the first and smallest spherically aromatic CSL metallo-borospherenes La3&[B2@B17]− and La3&[B2@B18]− in which the B2 core and La3&Bn− spherical trihedral shells (n = 17 and 18) match both geometrically and electronically. The donor–acceptor duality of the transition-metal-like B2 core plays an essential role in stabilizing these novel complexes, in particular, its strong π-donation to the three equivalent La atoms in equatorial directions makes major contributions to the overall orbital interaction. Initial results indicate that the experimentally observed spherical trihedral D3h Tb3&B18− (ref. 29) can also be used as a cage-like shell to host a B2 core with donor–acceptor duality. A similar B2 core in a tubular molecular rotor La2&[B2@B18]− has been reported recently.68 It is anticipated that more Bn units with donor–acceptor dualities could exist in 3D complexes or low-dimensional nanostructures which may have important applications in materials science, molecular devices, and chemical catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21720102006 and 21973057 to S.-D. Li). We thank the helpful discussions on EDA-NOCV analysis with Prof. Lili Zhao from Nanjing Tech University and Dr Jun Liu from FermiTech (Beijing) Co. Ltd.Notes and references
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley-Interscience, New York, 6th edn, 1999, p. 1355, ISBN 0-471-19957-5 Search PubMed.
- L. S. Wang, Int. Rev. Phys. Chem., 2016, 35, 69 Search PubMed.
- H. J. Zhai, A. N. Alexandrova, K. A. Birch, A. I. Boldyrev and L. S. Wang, Angew. Chem., Int. Ed., 2003, 42, 6004–6008 CrossRef CAS.
- A. P. Sergeeva, D. Y. Zubarev, H. J. Zhai, A. I. Boldyrev and L. S. Wang, J. Am. Chem. Soc., 2008, 130, 7244–7246 CrossRef CAS.
- W. Huang, A. P. Sergeeva, H. J. Zhai, B. B. Averkiev, L. S. Wang and A. I. Boldyrev, Nat. Chem., 2010, 2, 202–206 CrossRef CAS PubMed.
- Q. Chen, T. T. Chen, H. R. Li, X. Y. Zhao, W. J. Chen, H. J. Zhai, S. D. Li and L. S. Wang, Nanoscale, 2019, 11, 9698 RSC.
- T. Jian, X. N. Chen, S. D. Li, A. I. Boldyrev, J. Li and L. S. Wang, Chem. Soc. Rev., 2019, 48, 3550 RSC.
- H. Bai, T. T. Chen, Q. Chen, X. Y. Zhao, Y. Y. Zhang, W. J. Chen, W. L. Li, L. F. Cheung, B. Bai, J. Cavanagh, W. Huang, S. D. Li, J. Li and L. S. Wang, Nanoscale, 2019, 11, 23286–23295 RSC.
- H. J. Zhai, Y. F. Zhao, W. L. Li, Q. Chen, H. Bai, H. S. Hu, Z. A. Piazza, W. J. Tian, H. G. Lu, Y. B. Wu, Y. W. Mu, G. F. Wei, Z. P. Liu, J. Li, S. D. Li and L. S. Wang, Nat. Chem., 2014, 6, 727 CrossRef CAS.
- Q. Chen, W. L. Li, Y. F. Zhao, S. Y. Zhang, H. S. Hu, H. Bai, H. R. Li, W. J. Tian, H. G. Lu, H. J. Zhai, S. D. Li, J. Li and L. S. Wang, ACS Nano, 2015, 9, 754 CrossRef CAS.
- Y. J. Wang, Y. F. Zhao, W. L. Li, T. Jian, Q. Chen, X. R. You, T. Ou, X. Y. Zhao, H. J. Zhai, S. D. Li, J. Li and L. S. Wang, J. Chem. Phys., 2016, 144, 064307 CrossRef.
- H. R. Li, T. Jian, W. L. Li, C. Q. Miao, Y. J. Wang, Q. Chen, X. M. Luo, K. Wang, H. J. Zhai, S. D. Li and L. S. Wang, Phys. Chem. Chem. Phys., 2016, 18, 29147 RSC.
- Q. Chen, S. Y. Zhang, H. Bai, W. J. Tian, T. Gao, H. R. Li, C. Q. Miao, Y. W. Mu, H. G. Lu, H. J. Zhai and S. D. Li, Angew. Chem., Int. Ed., 2015, 54, 8160 CrossRef CAS PubMed.
- Q. Chen, H. R. Li, C. Q. Miao, Y. J. Wang, H. G. Lu, Y. W. Mu, G. M. Ren, H. J. Zhai and S. D. Li, Phys. Chem. Chem. Phys., 2016, 18, 11610 RSC.
- W. J. Tian, Q. Chen, H. R. Li, M. Yan, Y. W. Mu, H. G. Lu, H. J. Zhai and S. D. Li, Phys. Chem. Chem. Phys., 2016, 18, 9922 RSC.
- Q. Chen, H. R. Li, W. J. Tian, H. G. Lu, H. J. Zhai and S. D. Li, Phys. Chem. Chem. Phys., 2016, 18, 14186 RSC.
- L. W. Sai, X. Wu, N. Gao, J. J. Zhao and R. B. King, Nanoscale, 2017, 9, 13905–13909 RSC.
- L. Pei, Y.-Y. Ma, M. Yan, M. Zhang, R.-N. Yuan, Q. Chen, W.-Y. Zan, Y.-W. Mu and S. D. Li, Eur. J. Inorg. Chem., 2020, 3296–3301 CrossRef CAS.
- E. Oger, N. R. M. Crawford, R. Kelting, P. Weis and M. M. K. R. Ahlrichs, Angew. Chem., Int. Ed., 2007, 46, 8503 CrossRef CAS PubMed.
- T. R. Galeev, C. Romanescu, W. L. Li, L. S. Wang and A. I. Boldyrev, Angew. Chem., Int. Ed., 2012, 51, 2101–2105 CrossRef CAS.
- C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, J. Chem. Phys., 2013, 138, 134315 CrossRef.
- C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, Acc. Chem. Res., 2013, 46, 350–358 CrossRef CAS.
- I. A. Popov, W. L. Li, Z. A. Piazza, A. I. Boldyrev and L. S. Wang, J. Phys. Chem. A, 2014, 118, 8098–8105 CrossRef CAS PubMed.
- I. A. Popov, T. Jian, G. V. Lopez, A. I. Boldyrev and L. S. Wang, Nat. Commun., 2015, 6, 8654 CrossRef CAS.
- T. Jian, W. L. Li, X. Chen, T. T. Chen, G. V. Lopez, J. Li and L. S. Wang, Chem. Sci., 2016, 7, 7020–7027 RSC.
- T. T. Chen, W. L. Li, J. Li and L. S. Wang, Chem. Sci., 2019, 10, 2534–2542 RSC.
- W. L. Li, T. T. Chen, D. H. Xing, X. Chen, J. Li and L. S. Wang, PNAS, 2018, 115, E6972–E6977 CAS.
- T. T. Chen, W. L. Li, W. J. Chen, J. Li and L. S. Wang, Chem. Commun., 2019, 55, 7864–7867 RSC.
- T. T. Chen, W. L. Li, W. J. Chen, X. H. Yu, X. R. Dong, J. Li and L. S. Wang, Nat. Commun., 2020, 11, 2766 CrossRef CAS PubMed.
- H. Bai, Q. Chen, H. J. Zhai and S. D. Li, Angew. Chem., Int. Ed., 2015, 54, 941 CrossRef CAS PubMed.
- H. R. Li, X. X. Tian, X. M. Luo, M. Yan, Y. W. Mu, H. G. Lu and S. D. Li, Sci. Rep., 2017, 7, 5701 CrossRef.
- Y. Zhang, X. Y. Zhao, M. Yan and S. D. Li, RSC Adv., 2020, 10, 29320 RSC.
- Y. F. Zhao, X. Chen and J. Li, Nano Res., 2017, 10, 3407–3420 CrossRef CAS.
- X. Chen, Y. F. Zhao, L. S. Wang and J. Li, Comput. Theor. Chem., 2017, 1107, 57–65 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- D. Feller, J. Comput. Chem., 1996, 17, 1571 CrossRef CAS.
- L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045 CrossRef.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- J. Čížek, Adv. Chem. Phys., 1969, 14, 35 CrossRef.
- G. D. Purvis III and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910 CrossRef.
- K. Raghavachari, G. W. Trucks, J. A. People and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479 CrossRef CAS.
- H. J. Werner, et al., Molpro, version 2012.1 Search PubMed.
- N. V. Tkachenko and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2019, 21, 9590 RSC.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103 CrossRef CAS.
- S. Klod and E. Kleinpeter, J. Chem. Soc., Perkin Trans. 2, 2001, 1893 CAS.
- E. Kleinpeter, S. Klod and A. Koch, J. Mol. Struct., 2007, 811, 45 CrossRef CAS.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS.
- T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef CAS.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS.
- M. Mitoraj and A. Michalak, Organometallics, 2007, 26, 6576–6580 CrossRef CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. Chang, M. Pelissier and P. Durand, Phys. Scr., 1986, 34, 394–404 CrossRef CAS.
- J. L. Heully, I. Lindgren, E. Lindroth, S. Lundqvist and A. M. Martensson-Pendrill, J. Phys. B: At. Mol. Phys., 1986, 19, 2799–2815 CrossRef CAS.
- E. v. Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- L. Zhao, S. Pan, M. Zhou and G. Frenking, Science, 2019, 365, eaay5021 CrossRef CAS PubMed.
- G. Frenking and N. Holzmann, Science, 2012, 336, 1394–1395 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420–1422 CrossRef CAS.
- M. Zhou, N. Tsumori, Z. Li, K. Fan, L. Andrews and Q. Xu, J. Am. Chem. Soc., 2002, 124, 12936–12937 CrossRef CAS PubMed.
- S. D. Li, H. J. Zhai and L. S. Wang, J. Am. Chem. Soc., 2008, 130, 2573–2579 CrossRef CAS.
- N. Holzmann, M. Hermann and G. Frenking, Chem. Sci., 2015, 6, 4089–4094 RSC.
- H. Zhang, Z. Cao, W. Wu and Y. Mo, Angew. Chem., Int. Ed., 2018, 57, 13076–13081 CrossRef CAS PubMed.
- P. v. R. Schleyer and C. Maerker, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- D. Ciuparu, R. F. Klie, Y. M. Zhu and L. Pfefferle, J. Phys. Chem. B, 2004, 108, 3967–3699 CrossRef CAS.
- X. Q. Lu, Q. Chen, X. X. Tian, Y. W. Mu, H. G. Lu and S. D. Li, Nanoscale, 2019, 11, 21311–21316 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06769e |
| This journal is © The Royal Society of Chemistry 2020 |