
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivatable superparamagnetic iron oxide nanoparticles scavenge reactive oxygen species in macrophages and endothelial cells†
Chukwuazam Nwasike a,
Eunsoo Yooa,
Erin Purra and
Amber L. Doiron*b
a,
Eunsoo Yooa,
Erin Purra and
Amber L. Doiron*b
aDepartment of Biomedical Engineering, Binghamton University (SUNY), Binghamton, NY 13902, USA
bDepartment of Electrical and Biomedical Engineering, University of Vermont, Burlington, VT 05405, USA. E-mail: amber.doiron@uvm.edu; Tel: +1-802-656-3382
First published on 12th November 2020
Abstract
Reactive oxygen species (ROS) are key markers of inflammation, with varying levels of superoxide indicating the degree of inflammation. Inflammatory diseases remain the leading cause of death in the developed world. Previously, we showed that interpolymer complexed superparamagnetic iron oxide nanoparticles (IPC-SPIOs) are capable of decomplexing and activating T2 magnetic resonance (MR) contrast in superoxide-rich environments. Here, we investigate the ability of IPC-SPIOs to scavenge ROS in immune and endothelial cells which should activate the superparamagnetic core. In exogenously generated superoxide, ROS scavenging by the nanoparticles was concentration dependent and ranged from 5% to over 50% of available ROS. A statistically significant reduction in ROS was observed in the presence of IPCSPIOs compared to poly(ethylene glycol)-coated SPIOs (PEG-SPIOs). During in vitro cellular assays, a reduction in ROS was observed in macrophages, monocytes, and human endothelial cells. Macrophages and endothelial cells experienced significantly higher ROS reduction compared to monocytes. ROS scavenging peaked 12 hours post-exposure to IPC-SPIOs in most studies, with some cell samples experiencing extended scavenging with increasing IPC-SPIO concentration. At the tested concentrations, particles were not cytotoxic, and confocal imaging showed localization of particles within cells. These findings demonstrate the potential of IPC-SPIOs as activatable MR contrast agents capable of activating under inflammation-induced cellular redox conditions as reporters of inflammatory disease severity or staging.
Introduction
Reactive oxygen species (ROS) generation is critical to the initiation and progression of several inflammatory diseases or conditions such as atherosclerosis, cancer, neurotoxicity, and pulmonary inflammation.1–11 ROS are produced as part of normal homeostatic processes in response to metabolic activities as well as apoptosis and cellular injuries,11–14 yet excessive ROS production can result in tissue injury and endothelial dysfunction.15 Recently, studies have linked high levels of ROS production to a wide array of proinflammatory disorders; superoxide anions (O2˙−) and hydrogen peroxide (H2O2) are the most biologically relevant ROS in inflammatory diseases.16–18 These chemical species are generated in vascular cells by different oxidases including Nicotinamide adenine dinucleotide phosphate oxidase, lipoxygenase, xanthine oxidase, cytochrome p450, uncoupling of endothelial nitric oxide synthase (eNOS), and uncoupling of the mitochondrial respiratory chain.19–23 Several antioxidants such as superoxide dismutases, catalase, glutathione peroxidase, and peroxiredoxins counterbalance ROS production in the cell.Reactive oxygen species are produced by a wide variety of cells in response to different conditions and stimuli. Superoxide, which are the most abundant form of ROS in the body, are mostly produced in the mitochondria during Krebs cycle at complexes I and III.24 Superoxide are excreted by phagocytic cells like macrophages as well as non-phagocytic cells, such as endothelial cells.25,26 Macrophages play a key role in inflammation and inflammatory disorders by initiating, maintaining, and resolving inflammation.27 When M2 macrophages are triggered to differentiate from monocytes, superoxide is produced and plays a key role for the biphasic extracellular-signal regulated kinase (ERK) pathway, which is necessary for macrophage differentiation.28 On the other hand, endothelial cells generate ROS after cellular damage, which triggers a key inflammatory process involving macrophages. Enhanced ROS secretion results in vascular permeability, allowing movement of substances across the endothelial barrier. ROS-induced interendothelial junction opening allows movement of inflammatory cells such as macrophages across the endothelial barrier to resolve the inflammation; however, persistent and ineffective immune responses result in additional tissue injury, which leads to chronic inflammatory disorders.29,30
Significant resources have been invested into deciphering the role of the immune system in disease pathophysiology and understanding the detrimental effects of excessive inflammation.31–34 Findings have identified several causative agents and symptoms of inflammatory diseases; however, detection and diagnosis of these inflammatory conditions remains difficult.35–38 In terms of imaging, most inflammatory diseases are detected by targeting the upregulation and trafficking of immune cells as they respond to stimuli. In recent decades, magnetic resonance imaging (MRI)-based nanoprobes have gained popularity with the introduction of different magnetic nanoparticles that are functionalized to target specific biomolecules and other cell constituents.39–41 These magnetic nanoparticles accumulate on target sites due to the prevalence of biomolecules, resulting in shortening of T1 and T2 relaxation times of surrounding tissues and causing signal reduction in the resulting MR images.42 This strategy enhances contrast and improves image quality significantly.
As the field of molecular imaging pushes towards the ability to measure and stage disease severity, functional MR scans have become imperative to improve diagnosis and monitor therapeutic outcomes. Recently, we showed that interpolymer complexed superparamagnetic iron oxide nanoparticles (IPC-SPIOs) are capable of decomplexing and activating T2 MR signal under oxidative conditions.43 IPC-SPIOs are comprised of superparamagnetic iron oxide nanoparticles (SPIOs) coated in poly(ethylene glycol) (PEG), which are complexed through hydrogen bonding with the pseudotannin poly(gallol). The complexed polymers exclude water from the radius immediately surrounding the SPIO core, which results in a very low T2 relaxtivity of the complexed MR contrast agent, similar to signal arising from water alone in the absence of a contrast agent. However, in the presence of oxidative species, poly(gallol) is oxidized, the polymers decomplex, and the particle coating swells resulting in water interacting with the SPIOs and the MR relaxivity increasing over 10-fold. Here we investigate the uptake and ROS scavenging activities of complexed IPC-SPIOs under in vitro conditions with monocytes, macrophages, and endothelial cells to extend our understanding of the potential of IPC-SPIOs as activatable MR contrast agents.
Materials and methods
Synthesis of uncoated SPIOs, PEGylated SPIOs, and IPC-SPIOs
Using previously optimized protocols,43 uncoated SPIOs were synthesize via coprecipitation method, SPIOs were PEGylated with 300 kDa PEG, and IPC-SPIOs were complexed using PEG 300 kDa and poly(gallol) made from 200 kDa dextran. Briefly, uncoated SPIOs were synthesized via the coprecipitation method under N2 atmosphere and vigorous magnetic stirring from a mixture of FeCl2 (Sigma) and FeCl3 (Sigma) in DI water.43,44 The mixture was heated to 80 °C, and ammonium hydroxide (NH4OH) (BDH; Poole, Dorset, UK) was added dropwise and allowed to stir for 5 hours to complete the reaction. Uncoated SPIOs were purified and collected via magnetic decantation and centrifugation. SPIOs were PEGylated by addition of PEG 300 kDa (Sigma-Aldrich; St. Louis, MO, USA; 40 mg mL−1) into the suspension of uncoated SPIOs (3 mg mL−1) in water resulting in 20 mg mL−1 PEG SPIOs concentration. The mixture was stirred using a magnetic stir bar for 24 hours at 1000 rpm. PEGylated SPIOs were purified by magnetic decantation resulting in a final PEG-SPIOs concentration of 2 mg mL−1. For nanoparticles conjugated with FITC to enable in vitro tracking, 0.2 mg of FITC-PEG-OH 5000 MW (NANOCS; NY, USA) was added to 2 mg mL−1 PEG-SPIOs solution and stirred for 24 hours at 1000 rpm before proceeding to complexation. Complexation was accomplished by mixing 0.25 mg mL−1 aqueous poly(gallol) solution with 2 mg mL−1 PEGSPIOs or FITC-PEG-SPIOs for one hour before adding 1 M of sodium phosphate buffer saline in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9:2 volume ratio of poly(gallol) solution to (FITC)-PEG-SPIOs to sodium phosphate buffer saline. The reaction was left stirring overnight. On the next day, unreacted reagents were removed, and the supernatant was collected for experiment.
:9:2 volume ratio of poly(gallol) solution to (FITC)-PEG-SPIOs to sodium phosphate buffer saline. The reaction was left stirring overnight. On the next day, unreacted reagents were removed, and the supernatant was collected for experiment.
Dynamic light scattering (DLS)
The hydrodynamic size (nm), zeta-potential (mV), and polydispersity index (PDI), of uncoated SPIOs, PEG-SPIOS, and IPC-SPIOs were measured by electrophoretic dynamic light scattering (Zetasizer, NanoZS Malvern, Worcestershire, UK) at room temperature. All samples were suspended in water and placed in a folded capillary cuvette cell for DLS readings.Cell culture
Human umbilical vein endothelial cells (HUVEC, Lonza, Walkersville, MD, USA) were seeded onto an 8 μg cm−2 collagen-coated 96-well plate and cultured in supplemented endothelial cell growth medium-2 (Lonza) under standard conditions at 37 °C with 5% CO2 and humidity. RAW264.7 murine cells (ATCC® TIB71™; Manassas, VA, USA) were cultured in RPMI-1640 with L-glutamine, 10% (v/v) fetal bovine serum (FBS) (Seradigm, Radnor, PA, USA), and 1% (v/v) penicillin streptomycin glutamine (Life Technologies, Burlington, ON, Canada). RAW264.7 cells were cultured in a 96 well plate under standard conditions at 37 °C with 5% CO2 and humidity. RAW264.7 monocytes were incubated with 100 ng mL−1 of lipopolysaccharides (LPS, Sigma) for 48 hours to induce macrophage differentiation.45,46Ex vivo superoxide scavenging assay
In the presence of superoxide, soluble nitroblue tetrazolium (NBT) salt is converted to insoluble NBT-diformazam, which is blue in color. Xanthine oxidase (0.015 units, XOD, Sigma) in 0.1 M sodium phosphate buffered saline (PBS) was added to 30 mM hypoxanthine (HX, Amresco, Solon, OH, USA) suspended in 50 mM potassium hydroxide (KOH, Research Products International, Mt. Prospect, IL, USA) to generate superoxide. The oxide-indicating NBT (3 mM, Sigma) in 70% N,N-dimethylformamide (DMF, Sigma) was prepared fresh just before use. The superoxide-containing solution was added to 150, 100, and 75 μg mL−1 concentrations of PEG-SPIOs or IPC-SPIOs. After 6 hours, the NBT solution was added to all nanocomplex suspensions to determine the remaining superoxide in solution. A volume ratio of 20 parts nanoparticle suspension: 2 parts HX solution: 0.7 parts XOD solution: 2 parts NBT solution was used, with superoxide mixed with NBT serving as the control.43,47 Absorbance was measured at 560 nm using a plate reader (Synergy H1, BioTek; Winooski, VT).Cell viability assay
Stock solutions of PEG-SPIOs and IPC-SPIOs were diluted to 150, 100, and 75 μg mL−1 in cell medium. Confluent cells were exposed to nanoparticles for 24 hours or controls of fresh cell media. After exposure, the cell media was aspirated, and the cells were rinsed three times with PBS supplemented with calcium and magnesium. A 100 μL aliquot of diluted cell counting kit-8 (CCK-8, Sigma) reagent (10% CCK-8 in cell media) was added into each well, wells were incubated for 1 hour, and absorbance was measured at 450 nm.In vitro ROS scavenging assay
HUVEC, LPS-stimulated RAW264.7 (macrophages), and not stimulated RAW264.7 (monocytes) were seeded into Corning® 96-well Flat Clear Bottom Black Polystyrene TC-treated Microplates (Corning, NY). HUVEC were seeded into 8 μg cm−2 collagen coated plates. Quadruplicate wells were exposed to 150, 100, or 75 μg mL−1 of PEG-SPIOs or IPCSPIOs in cell culture media for 0, 1, 2, 4, 8, 12, 16, 20, and 24 hours. Cell media was aspirated, and cells were rinsed three times with PBS supplemented with calcium and magnesium. ROS indicator 2′,7′-dichlorofluorescin diacetate (DCFDA; 100 μL of 20 μM; Sigma) diluted in PBS was added to each well for 30 minutes, the supernatant aspirated, and cells were rinsed with PBS three times. The fluorescence intensity of each well was measured with a plate reader at excitation and emission wavelengths of 495 nm and 529 nm, respectively. Controls without particles were used to obtain baseline ROS, and all other data points were normalized as percentage based on this cell-only sample.Fixation and staining cells
Cells were cultured on a LabTek 8 Chambered Coverglass slide (Thermo Fisher Scientific™, Waltham, MA, USA). FITC conjugated IPC-SPIOs (150 μg mL−1) were added to wells for a 3 hour or 24 hour incubation. After incubation, cells were rinsed and fixed with 4% paraformaldehyde at room temperature for 10 minutes. Cells were rinsed again and stained for 10 minutes at 37 °C with CellMask Deep Red (Life Technologies, diluted 1:1000 in PBS). The supernatant was aspirated, cells were rinsed three times with PBS, and the slide was mounted, and cover slipped with Prolong Diamond antifade reagent containing DAPI (Thermo Fisher Scientific™). Slides were stored at 4 °C until imaging.
Confocal microscopy for cell uptake
Cells were imaged using LSM 880 NOL 3-channel multiphoton confocal laser scanning microscope (Zeiss; Oberkochen, Germany) with the following parameters: ex 488 nm/em 493–563 nm for FITC signal, ex 633 nm/em 638–755 nm for CellMask deep red, and ex 405 nm/em 410–502 nm for DAPI. All images were obtained using a water-immersion 40× objective and Zeiss Efficient Navigation (ZEN) 2.3 software.Statistics
Data were collected in quadruplicates. All values were expressed as mean ± standard deviation. A two-way analysis of variance (ANOVA) was used, followed by Tukey's post hoc tests to compare means. p < 0.05 was considered statistically significant.Results
Nanoparticle synthesis and characterization
The synthesis and characterization of IPC-SPIOs was published previously and is summarized here.43,44 Briefly, transmission electron microscope (TEM) scans of uncoated SPIOs showed spherically shaped particles, and PEG SPIOs had gray halo around the much darker SPIO nanoparticles, demonstrating the polymer coating was present. The presence of iron and oxygen was validated using energy dispersive X-ray spectroscopy (EDS). The external morphology of uncoated SPIOs and PEG SPIOs was also investigated using scanning electron microscope (SEM). Nanoparticles had an external spherical morphology with an average diameter slightly larger than TEM scans.43,44 IPC-SPIOs are not suitable for TEM or SEM imaging as polymer coatings continued to degrade under the electron beam. Here, we characterized the hydrodynamic size, zeta potential, and polydispersity index of all nanoparticle formulations with DLS before use. Nanoparticle formulations showed a size increase as the iron oxide core was coated and complexed with polymers (Table 1). The size of IPC-SPIOs was 157 ± 2 nm, while PEG-SPIOs were smaller sized as expected at 112 ± 2 nm. Like previous studies, PEGylation and complexation of SPIOs shifted particle charge towards neutral.43 This is was attributed to charge shielding due to particle surface coating. The nanoparticles were very well dispersed with minimal aggregation as the polydispersity indexes were all around 0.1–0.15 (Table 1).| Nanoparticles | Hydrodynamic size (nm) | Zeta potential (mV) | Polydispersity index (PDI) |
|---|---|---|---|
| Uncoated SPIOs | 100 ± 0.4 | −47 ± 1 | 0.115 ± 0.014 |
| PEG-SPIOs | 112 ± 2 | −44 ± 1 | 0.104 ± 0.009 |
| IPC-SPIOs | 157 ± 2 | −39 ± 0.2 | 0.15 ± 0.007 |
IPC-SPIOs scavenge exogenous ROS
NBT was used to provide qualitative and quantitative evidence of the superoxide scavenging capabilities of IPC-SPIOs. Hypoxanthine and xanthine oxidase were used to produce excess superoxide in deionized water before exposing different concentrations of PEG-SPIOs and IPC-SPIOs to the superoxide-rich environment. After particles were allowed to react with the superoxide for a period of 6 hours, NBT was added to the solution to react with any remaining superoxide, which led to the development of blue color. IPC-SPIOs reduced the total amount of superoxide present in the solution in a concentration-dependent manner as shown by the less prominent blue observed in the NBT reactions (Fig. 1A). Quantification of blue NBT color showed that scavenging ranged from 5% in low concentration to over 50% in high concentration (Fig. 1B). Along with our previously published study, these results suggest that as IPC-SPIOs decomplex, they reduce the amount of superoxide species present in solution.43 | ||
| Fig. 1 (A) Sample image of NBT in a 96 well plate with PEG-SPIOs and IPC-SPIOs (nanoparticles are 300, 150, 100, and 75 μg mL−1 of SPIOs content from left to right). (B) Quantification of NBT samples relative to superoxide control in quadruplicate (n = 4) tests at various concentrations. Values shown are mean ± standard deviation. Data were analyzed by a one-way ANOVA and Tukey's post hoc test (P < 0.05). Data that do not share any letters are statistically significantly different. | ||
Establishing baseline cellular ROS over time
To establish baseline intracellular ROS, monocyte, macrophages, and HUVEC were exposed to DCFDA fluorescent dye in media. ROS levels were detected over 24 hours to determine levels of inflammation present in endothelial cells, LPS-stimulated macrophages, and monocytes. Interestingly, the highest change in ROS relative to time 0 hour was observed in HUVEC followed by monocytes and macrophages (Fig. 2). HUVEC ROS spiked within 1 hour of ROS measurement which was followed by rapidly decline and stabilization for 16 hours before another spike was detected. Macrophage ROS was relatively stable with minor changes detected over 24 hours. Changes in monocytic ROS followed an upwards trend with an initial minor spike in ROS observed at 3 hours before a gradual increase that plateaued at 20 hours. The different changes in ROS presented by each cell line is statistically different for all cell types at each time point except for HUVEC and monocytic ROS at 20 hours.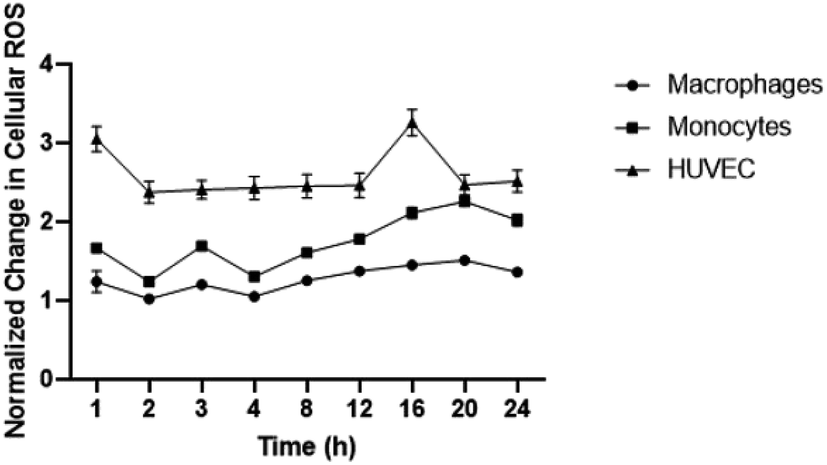 | ||
| Fig. 2 Normalized change in ROS for each cell type relative to time 0 hour. Values are shown as mean ± standard deviation. Data were analyzed by a one-way ANOVA and post hoc multiple t-test (p < 0.05). All three cell lines are statistically significantly different at each time point and compared to time zero; n = 4. | ||
IPC-SPIOs scavenge cellular ROS
Scavenging of cellular ROS was investigated in macrophages, monocytes, and HUVEC for a 24 hour time period using DCFDA. All cell types showed concentration-dependent ROS scavenging with the highest scavenging detected in macrophages and HUVEC. In macrophages, maximum ROS scavenging was detected from 4–12 hours before a rapid restoration to initial cellular ROS (Fig. 3A). Return to initial cellular ROS was detected 16 hours post-nanoparticle exposure. For HUVEC, maximum ROS scavenging was achieved faster compared to the other two cells. Peak ROS scavenging was detected from 2–12 hours, which was the longest IPC-SPIOs scavenging activity detected (Fig. 3B). Like macrophages, ROS recovery was detected after 16 hours exposure to IPC-SPIOs. However, ROS recovery was significantly slower in HUVEC. Finally, ROS scavenging was also investigated in monocytes. Monocytes experienced the least ROS scavenging activity, which peaked from 4–12 hours post IPC-SPIOs treatment (Fig. 3C). Like macrophages, return to normal cellular ROS was rapid and occurred 16 hours post treatment. In order to understand the effect of IPC-SPIOs on all three cell lines together, ROS scavenging was further analyzed 8 hours post IPC-SPIOs treatment. It was observed that 150 μg mL−1 of IPC-SPIOs scavenged the same level (no statistical difference) of ROS in macrophages, monocytes, and HUVEC (Fig. 3D). Therefore, 150 μg mL−1 of IPC-SPIOs hold the most potential for scavenging ROS in either immune or endothelial cells during inflammation (full statistical comparisons between all time points are found in Fig. S3.† Additionally, raw data for ROS scavenging are also found in Fig. S6†). Further, 150 μg mL−1 of IPC-SPIOs was investigated in macrophages using CellROX assays and T2 – weighted MRI scans (Fig. S1 and S2†). ROS decline was detected in the extracellular and intracellular membranes. ROS reduction was also detected in the mitochondria but relative to time 0 hour and not baseline cellular ROS. T2 – weighted MR scans showed cellular ROS mediated IPC-SPIOs activation and contrasting of MRI signals. IPC-SPIOs exhibited pre-to post-cellular ROS exposure T2 values of 179 ms to 149 ms.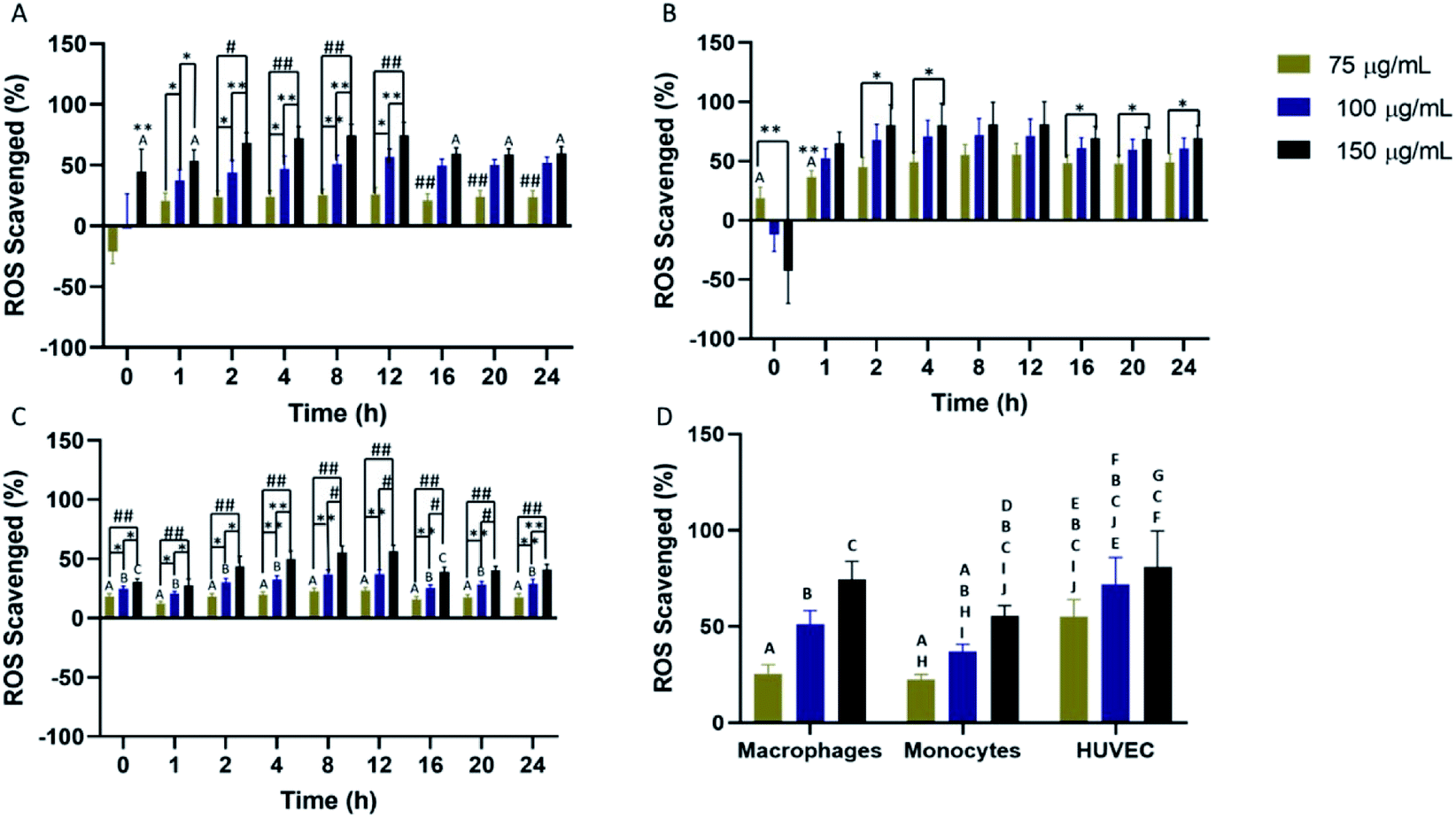 | ||
| Fig. 3 (A) Macrophages, (B) HUVEC, and (C) Monocytes exposed to different concentrations of IPC-SPIOs. ROS scavenging was detected over 24 hours using the DCFDA assay. (D) Analysis of ROS scavenging in all cell types after 8 hours of particle exposure. Values are shown as mean ± standard deviation. Data were analyzed by a two-way ANOVA and Tukey post hoc (*P < 0.05, **P < 0.01, #P < 0.001, ##P < 0.0001). Data that do not share any letters are statistically significantly different. n = 4. | ||
Relative percent change in scavenging differs based on cell type
Percent change in scavenging relative to initial time of exposure (0 hour) was investigated to understand the kinetics of reduction in cellular ROS. Firstly, macrophages, monocytes, and HUVEC were exposed to different concentrations of IPC-SPIOs. Cellular ROS post exposure was obtained from time 0–24 hours. Time 0 hour was used as a baseline to detected subsequent changes in ROS at each timepoint. Relative percent change in scavenging was dependent on cell type. Macrophages showed the highest percent change (over 5000%) in ROS relative to time 0 hour compared to other cells. Even though scavenging was stable over the 24 hour period, the highest percent change was detected at 12 hours for macrophages treated with 100 μg mL−1 of IPC-SPIOs (Fig. 4A), while the highest rate of ROS scavenging was detected at 1 hour (Fig. S5A†). Monocytes experienced the lowest percent change in ROS relative to initial time of exposure. The different IPC-SPIOs concentrations followed the same scavenging trend with a concentration dependent percent change in scavenging (Fig. 4B). The highest percent change in monocytes were at 12 hours for cells treated with 150 μg mL−1 of IPC-SPIOs, which experienced over 220% change in ROS compared to initial exposure time. In contrast, IPC-SPIOs exhibited strong relative change in ROS across all concentrations in HUVEC. Like macrophages, HUVEC treated with 100 μg mL−1 of IPCSPIOs induced the greatest percent change in ROS compared to initial time of exposure (Fig. 4C).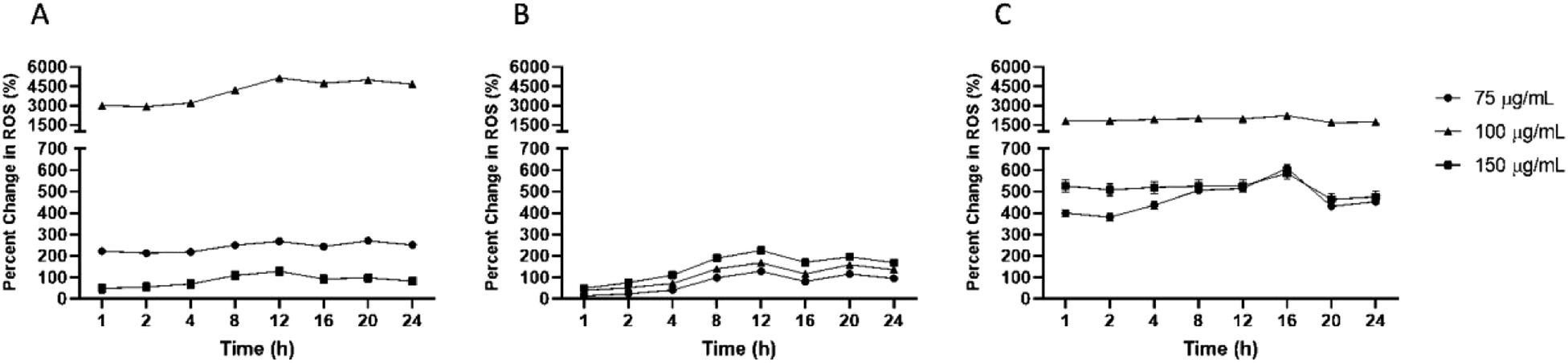 | ||
| Fig. 4 Percent change in cellular ROS reduction in (A) macrophages, (B) monocytes, and (C) HUVEC exposed to different concentrations of IPC-SPIOs. Data obtained from time 1–24 hours were normalized as percent change relative to time 0 hour. Values are shown as mean ± standard deviation. Data were analyzed by a two-way ANOVA and Tukey post hoc. All data points are statistical significantly different except for HUVEC from 8–24 for 75 μg mL−1 and 150 μg mL−1 as well as monocytes at all time points. n = 4. | ||
This highest percent change occurred 16 hours post exposure which is different to macrophages and monocytes, of while the rate of ROS scavenging peaked 1 hour after nanoparticle exposure (Fig. S5B†). Taken together, 12 hours post IPC-SPIOs treatment seems to be the most important time to detect maximum percent change in scavenging in monocytes and macrophages, and 16 hours appears to be the optimal time for HUVEC.
PEG SPIOs exhibits antioxidant properties in cells
The effect of PEG-SPIOs on cellular ROS was also investigated over 24 hours. All cell types experienced a decrease in cellular ROS. In macrophages, reduction in ROS was more pronounced in cells treated with 150 μg mL−1 of PEG SPIOs at 2–24 hours (Fig. 5A). Return to initial cellular ROS level was not detected as antioxidant activities remained high. HUVEC also experienced similarly high ROS reduction after exposure to PEG SPIOs. The lowest concentration of PEG SPIOs did not statistically alter HUVEC ROS compared to initial time of treatment, but higher concentrations induced significant ROS reduction without returning the cells to initial ROS level (Fig. 5B). Monocytes experienced the least reduction in ROS compared to other cells. In fact, none of the PEG SPIOs concentration induced statistically significant reduction in ROS compared to initial ROS detected immediately after PEG SPIOs exposure (Fig. 5C). Unlike macrophages and HUVEC which experienced peak ROS reduction 8–12 hours post exposure at higher concentrations, monocytes remained stable. Full statistical comparisons between all time points are found in Fig. S4.† Additionally, raw data for PEG interaction with cellular ROS are also found in Fig. S6.†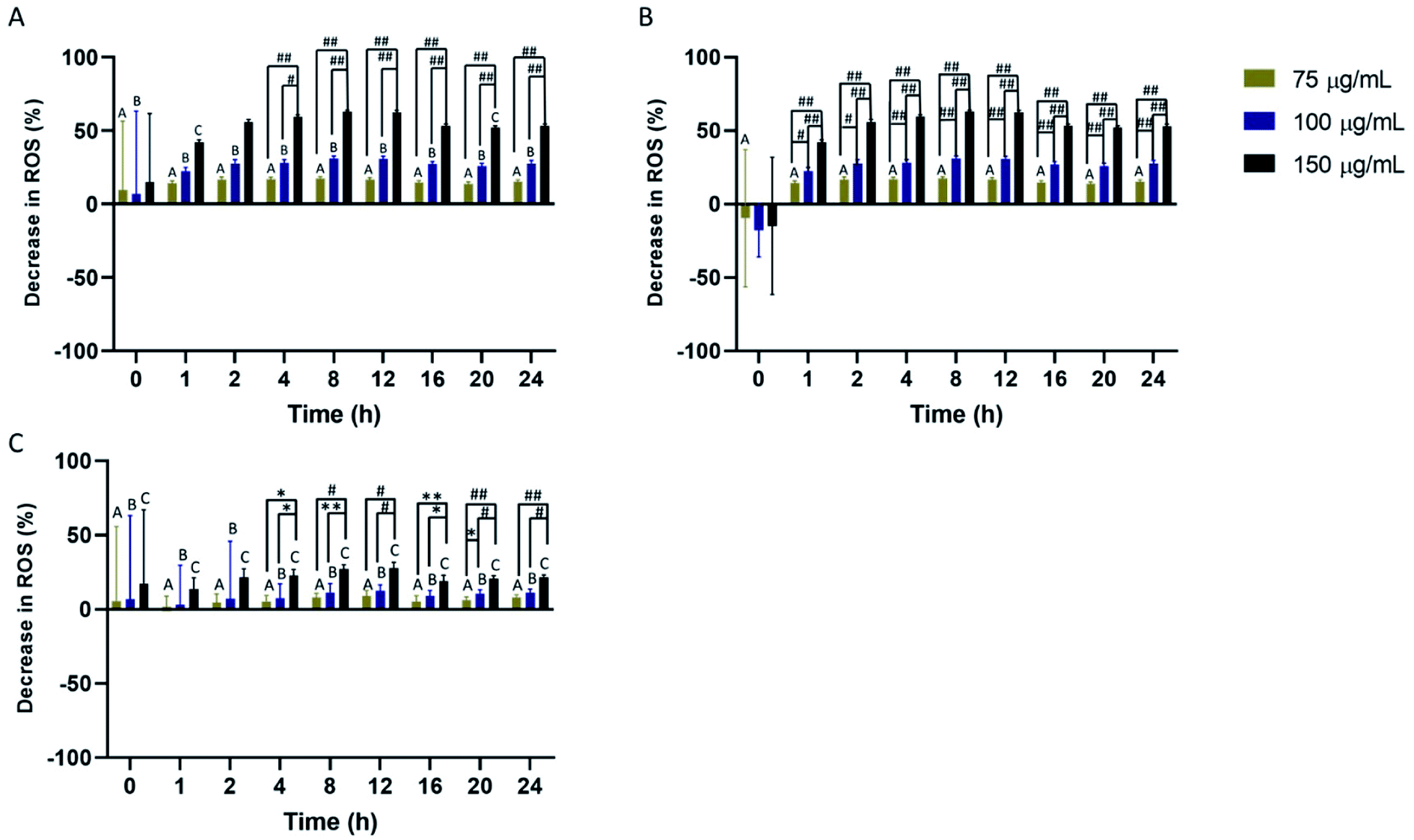 | ||
| Fig. 5 (A) Macrophages, (B) HUVEC, and (C) monocytes exposed to different concentrations of PEG SPIOs. ROS scavenging was detected over 24 hours using the DCFDA assay. Values are shown as mean ± standard deviation. Data were analyzed by a two-way ANOVA and Tukey post hoc (*P < 0.05, **P < 0.01, #P < 0.001, ##P < 0.0001). Data that do not share any letters are statistically significantly different n = 4. | ||
Uptake of IPC-SPIOs by cells
Macrophages, monocytes, and HUVEC were exposed to 150 μg mL−1 of FITC conjugated IPC-SPIOs (green) for 3 or 24 hours. Cells were stained with DAPI (blue) for nucleus and CellMask (red) for cell membrane and were imaged to determine uptake of nanoparticles. The control contained no particles and media was replaced at time 0 hours. After 3 hours of exposure, macrophages showed lower nanoparticle uptake as weaker green signals were detected. However, after 24 hour exposure, green signal became visibly stronger as a greater number of FITC IPCSPIOs accumulated inside of the cell (Fig. 6). As expected, due to a lower phagocytic capacity, weaker FITC signals were detected at 3 and 24 hours for monocytes (Fig. 6). This weaker uptake validates the poor ROS scavenging detected in monocytes. On the other hand, HUVEC showed high FITC IPC-SPIOs uptake, which was comparable to macrophages. In fact, after 3 hours exposure, HUVEC visibly took up more FITC IPC-SPIOs compared to macrophages. However, after 24 hours, nanoparticle uptake appeared similar in both cell lines. The higher FITC signals were observed 24 hours post exposure in all cell lines suggest time-dependent uptake of nanoparticles by cells. These observations follow a time-dependent uptake trend reported by different studies for similar size particles.48,49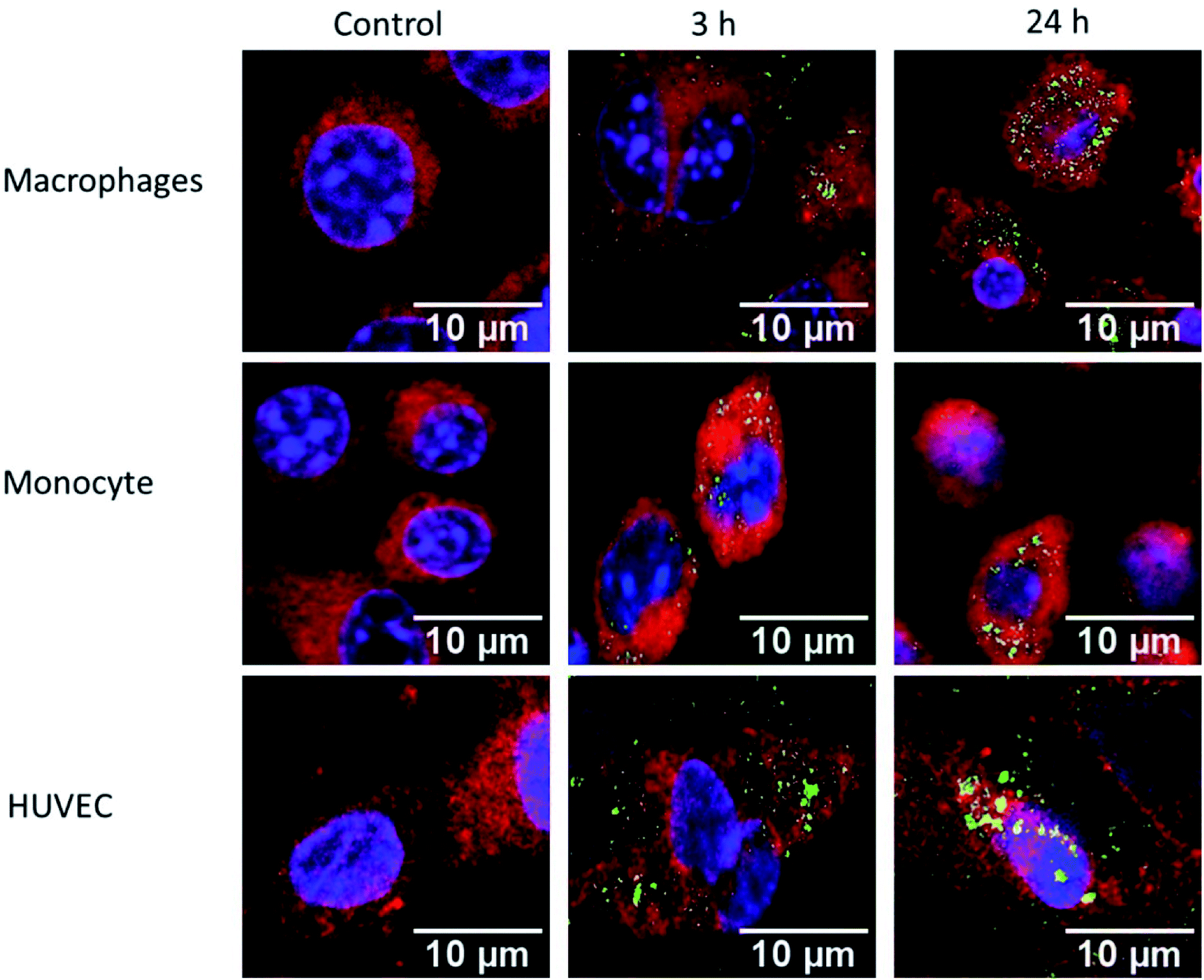 | ||
| Fig. 6 Confocal images of FITC IPC-SPIOs (green) cellular uptake. Macrophages, monocytes and HUVEC exposed to 150 μg mL−1 of FITC-IPC-SPIOs for 3 hours and 24 hours. Control cells were not exposed to particles. Cells were stained with CellMask (red) for cell membrane and DAPI (blue) for nuclei. | ||
Cellular viability after exposure to IPC-SPIOs
To determine the cytotoxicity of IPC-SPIOs, HUVEC and macrophages were exposed to different concentrations of PEG-SPIOs and IPC-SPIOs for 24 hours. The colorimetric CCK-8 assay was used to investigate the effects of nanoparticles on cells. Cells only, which serves as control, are cells treated with fresh media without particle exposure. Compared to controls, both HUVEC and macrophages showed no statistically significant reduction in viability as a result of exposure to any of the particles tested at 150 μg mL−1, 100 μg mL−1, or 75 μg mL−1. For HUVEC, 100 μg mL−1 particles induced significant effects on viability compared to other concentrations (Fig. 7A). At that concentration, PEG-SPIOs positively improved viability, while IPC-SPIOs were near 100% viable (Fig. 7A). On the other hand, macrophages showed a significant increase in viability for most particle concentrations compared to control (Fig. 7B). The greatest difference was observed between cells treated with 150 μg mL−1 PEG-SPIOs vs. IPC-SPIOs. Viability significantly increased for cells treated with PEG-SPIOs but statistically remained the same as the control for cells treated with IPC-SPIOs. Taken together, IPC-SPIOs were not observed to reduce cell viability and in some conditions improved viability.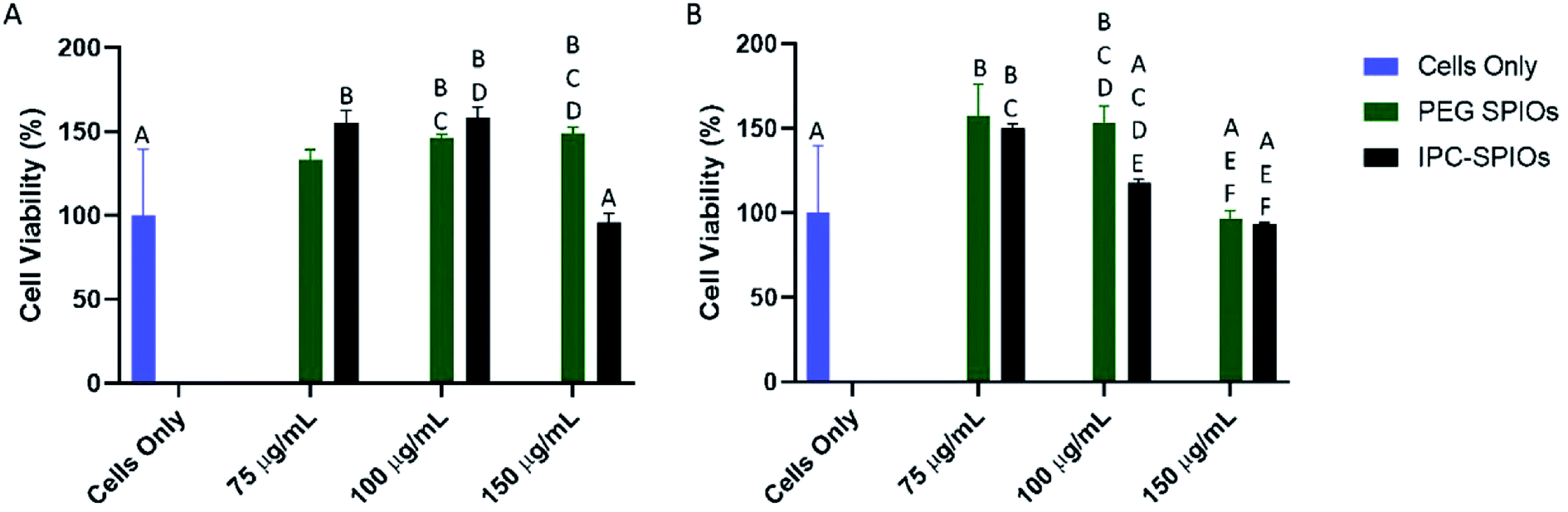 | ||
| Fig. 7 Viability of (A) HUVEC and (B) macrophages cell lines treated with different concentrations PEG-SPIOs, and complexed IPC-SPIOs for 24 hours. Values are normalized as percentage based on cells only and shown are mean ± standard deviation. Data were analyzed by a one-way ANOVA and Tukey's post hoc test. Data that do not share any letters are statistically significantly different n = 4. | ||
Discussion
Using ROS as an indicator of inflammatory diseases is an appealing strategy since they play an important role in most chronic and non-chronic inflammatory diseases. The use of a nanoprobe specifically designed to detect ROS levels in vivo could provide useful insight to severity staging of different inflammatory conditions and a metric for evaluating therapeutic efficacy. Therefore, we have demonstrated here the ROS-scavenging activity of activatable IPC-SPIOs.Most diagnostic investigations explore the use of functionalized nanoparticles to target specific epitopes or biomarkers.50–55 This study demonstrates for the first time interpolymer complexed superparamagnetic iron oxide nanoparticles scavenging free radicals in vitro. Tannins are naturally occurring macromolecular polyphenols present in the human diet.47,56,57 They are useful in biological systems as they act as antioxidants and have anti-inflammatory and antimicrobial properties.47 Given the difficulty with purifying plant extracts, pseudotannins were artificially synthesized to mimic the chemical structure of natural polyphenols and properties of tannins.47 Many researchers investigating activatable MRI detection of inflammatory diseases have taken different approaches to target inflammation including pH and enzyme specific targeting. One of the most interesting approaches is by Ta et al., who developed a thrombin activatable T1/T2 MRI nanosensor that selectively targets fibrin for non-invasive detection and characterization of inflammatory disease progression.58 The nanosensors are made up of iron oxide nanoclusters coated with a detachable layer of gadolinium and functionalized with fibrin binding peptide (Fb-Gd (IONC)). In the presence of thrombus, the nanosensors exhibit T1 contrast on MR signals, while in the absence of thrombus T2 contrast dominates the MR signal. In vitro activation of T1 and T2 signals using human fresh frozen plasma (FFP) resulted in bright T1 and negative T2 signals for FFP treated with Fb-Gd (IONC). The study showed the potential of detecting early stage blood clots and disease progression using activatable technologies. In an example of pH sensitive nanoparticles, Wang et al. developed a pH sensitive gadolinium metallofullerene (GMF), and doxorubicin nanoparticle encapsulated in a PEG-based polymer.59 In vitro sensitivity to pH was investigated using HeLa cells to determine drug release and enhancement of T1 MRI signals. Results show that pH sensitive gadolinium metallofullerene (GMF) nanoparticles did not activate T1 MRI signals in neutral pH but signal increased as pH became acidic. MR scans of HeLa tumor bearing mice which were treated with nanoparticle formulation showed 1.8-fold improvement signal to noise compared to non-pH sensitive particles. In contrast to pH and protein mediated nanoparticle activation, our study focuses on oxidation driven nanoparticle activation. Focusing on oxidative stress-based activation eliminates several issues with targeted nanoparticle activation such as nonspecific binding, degradation of proteins, and low nanoparticle activation due to poor sensitivity to target site. Therefore, for this study, poly(gallol) was the pseudotannin of choice used as the ROS scavenger that granted activatable properties to the IPC-SPIO nanoparticles.
ROS scavenging by IPC-SPIOs relies on the interaction between poly(gallol) and ROS. The mechanism involves oxidization of the phenolic groups on poly(gallol) by free radicals, which is detailed in previously published work.43 When IPC-SPIOs interact with cellular ROS, the ROS oxidizes the phenolic side of poly(gallol) resulting in the loss of hydroxyl by the polymer side. As this sequence occurs, the poly(gallol) becomes oxidized and it can no longer maintain its hydrogen-bonding-based interaction with PEG. The disruption to hydrogen bonding allows more water to interact with the SPIO core and T2 contrast is regained. It should also be noted that decomplexation does not result in complete severing of PEG-poly(gallol) attachment. Rather, decomplexation results in swelling of IPC-SPIOs, which grants the SPIO core more access to water. Some IPC-SPIOs are expected to decomplex in the extracellular space before uptake by cells while others will interact with cell membrane ROS and some nanoparticles localize within the cytosol.
Given the important roles of immune and endothelial cells during inflammation, macrophages and HUVEC were prioritized for investigating in vitro ROS scavenging by IPC-SPIOs. Initially, an Ex vivo NBT assay was successfully used to measure exogenous ROS scavenging providing evidence for non-physiological activation of IPC-SPIOs in a concentration dependent manner. In vitro DCFDA ROS studies showed promising cell-based activation of IPCSPIOs. ROS scavenging was very effective in HUVEC and macrophages with those cells presenting over 70% reduction in ROS for several hours. This is particularly interesting as ROS scavenging in HUVEC was not expected to keep up with macrophages. Macrophages are often referred to as “big eater” cells with high capacity for phagocytosis, so similar scavenging abilities detected in macrophages and HUVEC were unexpected. High scavenging activity detected in HUVEC could be attributed several factors including the level of inflammation present in the cell. It is well established that inflammation in the endothelium leads to endothelial gaps and leakages to facilitate recruitment of inflammatory mediators.60 It is possible that HUVEC were experiencing high inflammatory activity which would result in the appearance of endothelial gaps and leakages leading to easy flow of nanoparticles into the cell. Besides, this study has shown that HUVEC produced the highest change in ROS over 24 hours compared to other cells investigated. Therefore, nanoparticles could easily enter the cell and decomplex because of the abundance of free radicals thereby reducing ROS detected at each time point.
In our data, it does not appear that ROS scavenging is affected by the Fenton reaction. It is well established that iron oxide triggers increased reactive oxygen species production in cells through the Fenton reaction.61–63 Yarjanli et al. reported that iron oxide contributes to the Fenton reaction by a catalytic process that converts mitochondrial hydrogen peroxide to hydroxyl free radicals, thereby increasing intracellular ROS.64 The Fenton reaction occurs when ferric iron (Fe3+) reacts with cellular hydrogen peroxide to produce ferrous iron (Fe2+), hydroperoxyl free radical (HO2), and hydrogen ion (H+).65,66 The relationship between the Fenton reaction, IPC-SPIO uptake, and particle decomplexation will need to be investigated further. In this work, the polymer coating likely inhibited the reaction; however, the relationship between decomplexation and SPIO core induced Fenton reaction is currently not well understood.
It is important to mention that regardless of concentration, IPC-SPIOs did not cause any toxicity in cells. In fact, biocompatible PEG-SPIOs also induced a decline in cellular ROS. Previous studies have shown that PEG does not scavenge ROS nor suppress xanthine oxidase activity.67,68 Therefore, PEG-SPIOs mediated ROS decline was very interesting. Investigation by Luo et al. showed that PEG reduces ROS via PEG-mediated membrane repair, which inhibits ROS and lipid peroxidation.67 Hence, ROS decline detected is most likely due to membrane repair and inhibition of lipid peroxidation. Taken together, PEG-SPIOs and IPC-SPIOs does not present any toxicity threat to cells in fact PEG-SPIOs might be triggering cellular repair.
Conclusion
This study demonstrates the ROS scavenging activity of complexed IPC-SPIOs. IPCSPIOs were not toxic to HUVEC or macrophages at concentrations that scavenged ROS. Scavenging was concentration- and time-dependent with maximum scavenging ranging from 2–12 hours, depending on concentration. IPC-SPIOs hold strong potential as activatable contrast agents for detection of inflammatory conditions and diseases. Future work will include studying the intracellular trafficking of particles, the influence of targeting on biodistribution, and in vivo studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the NIH (1R21EB017504-01) and Clifford D Clark Diversity Fellowship at Binghamton University. The authors thank Yizhong Liu, PhD and Sebastian Freeman for support with cells studies and confocal imaging, respectively.References
- M. Mittal, M. R. Siddiqui, K. Tran, S. P. Reddy and A. B. Malik, Antioxid. Redox Signaling, 2014, 20, 1126–1167 CrossRef CAS.
- P. R. Kvietys and D. N. Granger, Free Radical Biol. Med., 2012, 52, 556–592 CrossRef CAS.
- P. J. Barnes, Free Radical Biol. Med., 1990, 9, 235–243 CrossRef CAS.
- H. Yokoyama, H. Kuroiwa, R. Yano and T. Araki, Neurol. Sci., 2008, 29, 293–301 CrossRef.
- P. C. Dedon and S. R. Tannenbaum, Arch. Biochem. Biophys., 2004, 423, 12–22 CrossRef CAS.
- J. Sund, H. Alenius, M. Vippola, K. Savolainen and A. Puustinen, ACS Nano, 2011, 5, 4300–4309 CrossRef CAS.
- F. Tao, B. Gonzalez-Flecha and L. Kobzik, Free Radical Biol. Med., 2003, 35, 327–340 CrossRef CAS.
- N. Azad, Y. Rojanasakul and V. Vallyathan, J. Toxicol. Environ. Health, Part B, 2008, 11, 1–15 CAS.
- H. U. Simon, A. Haj-Yehia and F. Levi-Schaffer, Apoptosis, 2000, 5, 415–418 CrossRef CAS.
- A. Harijith, D. L. Ebenezer and V. Natarajan, Front. Physiol., 2014 DOI:10.3389/fphys.2014.00352.
- E. Naik and V. M. Dixit, J. Exp. Med., 2011, 208, 417–420 CrossRef CAS.
- R. J. Mailloux and M.-E. Harper, Free Radical Biol. Med., 2011, 51, 1106–1115 CrossRef CAS.
- M. Sagi and R. Fluhr, Plant Physiol., 2006, 141, 336–340 CrossRef CAS.
- M. P. Murphy, Biochem. J., 2009, 417, 1–13 CrossRef CAS.
- D. C. Liemburg-Apers, P. H. G. M. Willems, W. J. H. Koopman and S. Grefte, Arch. Toxicol., 2015, 89, 1209–1226 CrossRef CAS.
- N. Neirynck, G. Glorieux, E. Schepers, A. Dhondt, F. Verbeke and R. Vanholder, Nephrol., Dial., Transplant., 2015, 30, 943–951 CrossRef CAS.
- Z. W. Zhang, X. C. Xu, T. Liu and S. Yuan, Oxid. Med. Cell. Longevity, 2016 DOI:10.1155/2016/6859523.
- J. A. Imlay, Annu. Rev. Biochem., 2008, 77, 755–776 CrossRef CAS.
- R. G. Alscher, N. Erturk and L. S. Heath, J. Exp. Bot., 2002, 53, 1331–1341 CrossRef CAS.
- I. M. Fearon and S. P. Faux, J. Mol. Cell. Cardiol., 2009, 47, 372–381 CrossRef CAS.
- K. Sugamura and J. F. Keaney, Free Radical Biol. Med., 2011, 51, 978–992 CrossRef CAS.
- J. C. Sullivan and J. S. Pollock, Circ. Res., 2006, 98, 717–719 CrossRef CAS.
- S. K. Wattanapitayakul and J. A. Bauer, Pharmacol. Ther., 2001, 89, 187–206 CrossRef CAS.
- D. B. Zorov, M. Juhaszova and S. J. Sollott, Physiol. Rev., 2014, 94, 909–950 CrossRef CAS.
- M. D. Kraaij, K. M. Koekkoek, S. W. Van der Kooij, K. A. Gelderman and C. Van Kooten, Cell. Immunol., 2013, 284, 1–8 CrossRef CAS.
- M. E. Widlansky and D. D. Gutterman, Antioxid. Redox Signaling, 2011, 15, 1517–1530 CrossRef CAS.
- H.-Y. Tan, N. Wang, S. Li, M. Hong, X. Wang and Y. Feng, Oxid. Med. Cell. Longevity, 2016 DOI:10.1155/2016/2795090.
- Y. Zhang, S. Choksi, K. Chen, Y. Pobezinskaya, I. Linnoila and Z.-G. Liu, Cell Res., 2013, 23, 898–914 CrossRef CAS.
- H. Guo, J. Zhang, M. Boudreau, J. Meng, J. Yin, J. Liu and H. Xu, Part. Fibre Toxicol., 2016, 13, 21 CrossRef.
- A. R. Fatkhullina, I. O. Peshkova and E. K. Koltsova, Biochemistry, 2016, 81, 1358–1370 CAS.
- N. Nanthakumar, D. Meng, A. M. Goldstein, W. Zhu, L. Lu, R. Uauy, A. Llanos, E. C. Claud and W. A. Walker, PLoS One, 2011, 6 DOI:10.1371/journal.pone.0017776.
- M. S. Yang, K. J. Min and E. Joe, J. Neurosci. Res., 2007, 85, 2298–2305 CrossRef CAS.
- D. Giugliano, A. Ceriello and K. Esposito, Part. Fibre Toxicol., 2006, 48, 677–685 CAS.
- E. K. Jo, D. M. Shin and A. M. K. Choi, Microbes Infect., 2012, 14, 119–125 CrossRef CAS.
- M. M. Moore, W. Chua, K. A. Charles and S. J. Clarke, Clin. Pharmacol. Ther., 2010, 87, 504–508 CrossRef CAS.
- K. Kalantar-Zadeh, T. A. Ikizler, G. Block, M. M. Avram and J. D. Kopple, Am. J. Kidney Dis., 2003, 42, 864–881 CrossRef.
- P. Libby, Nature, 2002, 420, 868–874 CrossRef CAS.
- T. Oka, S. Hikoso, O. Yamaguchi, M. Taneike, T. Takeda, T. Tamai, J. Oyabu, T. Murakawa, H. Nakayama, K. Nishida, S. Akira, A. Yamamoto, I. Komuro and K. Otsu, Nature, 2012, 485, 251–255 CrossRef CAS.
- C. Tarin, M. Carril, J. L. Martin-Ventura, I. Markuerkiaga, D. Padro, P. Llamas-Granda, J. A. Moreno, I. García, N. Genicio, S. Plaza-Garcia, L. M. Blanco-Colio, S. Penades and J. Egido, Sci. Rep., 2015, 5 DOI:10.1038/srep17135.
- K. C. Briley-Saebo, Y. S. Cho, P. X. Shaw, S. K. Ryu, V. Mani, S. Dickson, E. Izadmehr, S. Green, Z. A. Fayad and S. Tsimikas, Part. Fibre Toxicol., 2011, 57, 337–347 CAS.
- P. Caravan, B. Das, S. Dumas, F. H. Epstein, P. A. Helm, V. Jacques, S. Koerner, A. Kolodziej, L. Shen, W. C. Sun and Z. Zhang, Angew. Chem., Int. Ed., 2007, 46, 8171–8173 CrossRef CAS.
- H. Ittrich, K. Peldschus, N. Raabe, M. Kaul and G. Adam, Röfo, 2013, 185, 1149–1166 CAS.
- E. Yoo, H. A. Cheng, L. E. Nardacci, D. J. Beaman, C. T. Drinnan, C. Lee, K. W. Fishbein, R. G. Spencer, O. Z. Fisher and A. L. Doiron, Colloids Surf., B, 2017, 158, 578–588 CrossRef CAS.
- E. Yoo, Y. Liu, C. A. Nwasike, S. R. Freeman, B. C. DiPaolo, B. Cordovez and A. L. Doiron, Beilstein J. Nanotechnol., 2018, 9, 1228–1238 CrossRef CAS.
- M. Orecchioni, Y. Ghosheh, A. B. Pramod and K. Ley, Front. Immunol., 2019, 10, 1084 CrossRef CAS.
- R. E. Plevin, M. Knoll, M. McKay, S. Arbabi and J. Cuschieri, Shock, 2016, 45, 22–27 CrossRef CAS.
- H. A. Cheng, C. T. Drinnan, N. Pleshko and O. Z. Fisher, Soft Matter, 2015, 11, 7783–7791 RSC.
- R. Nicolete, D. F. Santos and L. H. Faccioli, Int. Immunopharmacol., 2011, 11, 1557–1563 CrossRef CAS.
- J. Blechinger, A. T. Bauer, A. A. Torrano, C. Gorzelanny, C. Bräuchle and S. W. Schneider, Small, 2013, 9, 3970–3980 CrossRef CAS.
- Y. Xiang, P. Wu, L. H. Tan and Y. Lu, Adv. Biochem. Eng./Biotechnol., 2014, 140, 93–120 CrossRef CAS.
- S. Jiang, K. Y. Win, S. Liu, C. P. Teng, Y. Zheng and M.-Y. Han, Nanoscale, 2013, 5, 3127 RSC.
- J. Li, F. Wang, J. Liu, Z. Xiong, G. Huang, H. Wan, Z. Liu, K. Cheng and H. Zou, Chem. Commun., 2015, 51, 4093–4096 RSC.
- Y. Yuan, J. Zhang, L. An, Q. Cao, Y. Deng and G. Liang, Biomaterials, 2014, 35, 7881–7886 CrossRef CAS.
- M. C. Smith, R. M. Crist, J. D. Clogston and S. E. McNeil, Anal. Bioanal. Chem., 2015, 407, 3705–3716 CrossRef CAS.
- C. Nwasike, K. Reeser, Y. Liu, J. S. Nagi, E. Purr, C. Han and A. L. Doiron, in Racing for the Surface, ed. B. Li, T. Moriarty, T. Webster and M. Xing, Springer, 1st edn, 2020, ch. 2, pp. 35–72 Search PubMed.
- O. Z. Fisher, B. L. Larson, P. S. Hill, D. Graupner, M. T. Nguyen-Kim, N. S. Kehr, L. De Cola, R. Langer and D. G. Anderson, Adv. Mater., 2012, 24, 3032–3036 CrossRef CAS.
- V. Barbosa, E. C. Ramires, I. A. T Razera and E. Frollini, Ind. Crops Prod., 2010, 32, 305–312 CrossRef CAS.
- H. A. Ta, N. Arndt, Y. Wu, H. J. Lim, S. Landeen, R. Zhang and Z. P. Xu, Nanoscale, 2018, 31, 15103–15115 RSC.
- S. Wang, Z. Zhou, Z. Wang, Y. Liu, O. Jacobson, Z. Shen and X. Chen, Small, 2019, 16, 1900691 CrossRef.
- D. Mcdonald, G. Thurston and P. Baluk, Microcirculation, 1999, 6, 7–22 CrossRef CAS.
- W. Zhang, S. Cao, S. Liang, C. H. Tan, B. Luo, X. Xu and P. E. Saw, Front. Bioeng. Biotechnol. DOI:10.3389/fbioe.2020.00537.
- B. Yu, Z. Wang, L. Almutairi, S. Huang and M.-H. Kim, Nanomedicine, 2020, 24, 102158 CrossRef CAS.
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman, M. Moseley, L. M. Coussens and H. E. Daldrup-Link, Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS.
- Z. Yarjanli, K. Ghaedi, A. Esmaeili, S. Rahgozar and A. Zarrabi, BMC Neurosci., 2018, 18, 51 CrossRef.
- G. Huang, H. Chen, Y. Dong, X. Luo, H. Yu, Z. Moore, E. A. Bey, D. A. Boothman and J. Gao, Theranostics, 2013, 3, 116–126 CrossRef CAS.
- M. C. Pereira, L. C. A. Oliveira, E. Murad and J. Adams, Clay Miner., 2012, 47, 285–302 CrossRef CAS.
- J. Luo, R. Borgens and R. Shi, J. Neurochem., 2002, 83, 471–480 CrossRef CAS.
- Y. Raveh, F. Ichinose, P. Orbach, K. Bloch and W. Zapol, Anesthesiology, 2002, 96, 926–933 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06683d |
| This journal is © The Royal Society of Chemistry 2020 |