
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, and investigation of a visible light-driven photo-switching macromolecule
Juan Pang,
Xincheng Mao,
Jialin Xu,
Xiang Zhao,
Jingyang Kong and
Xiaohong Hu *
*
School of Material Engineering, Jinling Institute of Technology, Nanjing, 211169, China. E-mail: huxiaohong07@163.com; Fax: +86 25 86188617; Tel: +86 25 86188617
First published on 23rd September 2020
Abstract
The application of azobenzene (AZO) as a kind of photo-switch is restricted by its excitation source, i.e., UV light. Hence, visible light-driven azobenzene-based photo-switching is needed and has been designed in the work. In order to forecast the optimal triggered wavelength, the electrostatic potential, theoretical UV-vis spectra, as well as the energy gap for focused structures was calculated to describe the energy and orbit status of the molecules by DFT. According to the theoretical optimization results, m-Methyl Red (m-MR) containing copolymer was successfully synthesized as a visible light-driven photo-switch. Further, for performance evaluation, the efficiency and effectiveness of different excitation wavelengths was firstly evaluated for the copolymer using m-MR and m-Methyl Red acrylic anhydride (m-MRAA) as the controls. Compared with m-MR and m-MRAA, the copolymer exhibited outstanding characteristics as a photo-switch according to its response–recovery behavior. At the same time, blue light proved to be the most efficient excitation light source. Moreover, the equilibrium response time and recovery time showed some dependence on the excitation wavelength. Secondly, the influence of the light intensity on the isomerization transition was investigated. A relatively low light density could lead to a relatively low degree of the final cis form and needed more equilibrium time for trans to cis transformation but showed little effect on the recovery process. Thirdly, repeatable on/off irradiation was used to evaluate the fatigue resistance of the copolymer. Good fatigue resistance without photobleaching was verified from the results. Fourthly, the influence of the solvent on visible light-driven isomerization was also evaluated. Finally, the synthesized copolymer still had the characteristic of a pH indicator with a critical point at pH 5.0 and exhibited an obvious fluorescent characteristic.
Introduction
Azobenzene (AZO), as a kind of photo-switch, has attracted increasing interest due to the reversible isomerization in the field of smart materials using light as an excitation source.1–6 Generally, on excitation by UV light, the AZO structure isomerizes from the trans form to the cis form. From the aspect of energy, the trans isomer has a lower intrinsic energy than the cis isomer.7 Thus, the AZO structure recovers spontaneously from the cis form to the trans form after removing the excitation source.7 Simultaneously, the cis form has worse thermostability and shorter lifetime due to its higher energy status.7 Moreover, the photobleaching phenomenon was also found in the small AZO molecule derivatives.8 These drawbacks restrict their application as photo-switches. Numerous attempts have been made to increase the thermostability of the cis form, including the immobilization of AZO onto material surfaces, grafting of AZO to a macromolecule, copolymerization, and cyclization.2,4,6,8,9 Similar to these studies, we designed a polysaccharide derivative and AZO copolymers as photo-switches, and found that the thermostability of the cis form could be improved and controlled by environmental parameters as well as structural parameters such as temperature, light density, solvent, and chain characteristics.9Although these progresses have realized the application of AZOs as photo-switches, ultraviolet band excitation source has some disadvantages in extending the applications of smart molecules. For example, UV light has weaker penetration than longer wavelength light. Further, it could harm the cells and tissues of biological systems. From the aspect of energy sources, solar energy is an economical and easily available green energy, which contains γ rays, X rays, UV light, visible light, infrared light, microwaves, and radio waves.10–15 Among these waves, the energy of UV light only makes up 7% of the total solar energy, while the energy of visible light makes up 50% of the total solar energy. Besides these advantages, visible light has slow depletion in the long range and is not harmful to the human tissue. Therefore, visible light is a better choice as an excitation source. Based on the above discussion, the design of visible light-driven azobenzene-based photo-switching molecules has become a research hot topic in the field.15–18 It had been found in theoretical research that the maximum absorption peak of the UV-vis spectrum obviously shifted towards the visible band when AZO was para-substituted by a couple of electron-donating and electron-withdrawing groups.16,17 However, its thermal transformation from cis to trans was so quick that its cis isomer could not be detected. Recently, researchers have found that when the four ortho-position of the para-substituted AZO was substituted by electron-donating groups such as –OCH3, –Cl, and –F, its thermal transformation (cis → trans) became slow, as observed from the theoretical calculation and simulation.15–18
Although these designs have provided feasible structures for visible light-triggered azobenzene-based photo-switches, there is still a long way to go from molecular synthesis to their actual application because the controllability and processability for small molecules is not good enough for practical application. Moreover, organic synthesis is also a complex process. Actually, it is inferred from previous research that the four ortho-position substitution increases the steric hindrance towards isomerization, thus slowing the speed of the cis → trans transformation.9 Generally, the polymer could play a similar role in the steric hindrance so as to control the thermal transition on account of its time dependence of the chain or domain motion, which was confirmed by the previous research on UV light-driven AZO copolymers.9 Moreover, polymers have good processing properties and relatively low toxicity, which is suitable for making a carrier or bridge from molecular design to practical applications. Therefore, visible light-driven macromolecular photo-switches have been designed in the research. In order to avoid complex organic synthesis, a commercial AZO derivative containing electron-donating and electron-withdrawing groups was chosen for theoretical calculations and design of the copolymer. In the copolymer design, hydrophilic monomers for chain adjustment and functional monomers for function extension were simultaneously considered for its future application. Finally, the synthesized copolymer was investigated in detail to evaluate its performance as a photo-switch.
Although some visible light-driven azobenzene-based photo-switches have been designed and confirmed, research has provided another effective and efficient approach to obtain a functional and adjustable switch for its potential applications. Another merit of the research was that it focused on the commercial organic AZO derivative, which could be directly synthesized form the monomer and then copolymerized with other monomers. This would be helpful for its promotion and application. Moreover, the research could provide basic data for the clarification of the isomerization mechanism and the amendment of the theoretical model for AZO-based photo-switches.
Experimental
Materials
N-Hydroxysuccinimide (NHS), acryloyl chloride, m-Methyl Red (m-MR), p-aminoazobenzene (m-AZO) hydroxyethyl methylacrylate (HEMA), and N-vinyl-2-pyrrolidone (NVP) were purchased from Aladdin. N-Acrylate succinimide (NAS) was obtained by the synthesis in our lab. Dichloromethane (DCM), diethyl ether, ethyl alcohol, dioxane, sodium chloride (NaCl), tetrahydrofuran (THF), benzoyl peroxide (BPO), triethylamine (TEA), ethyl alcohol, and dimethylsulfone (DMSO) were obtained from Sinopharm Chemical Reagent Co., Ltd, China. All other reagents and solvents were of analytical grade and used as received.DFT calculations for azobenzene-based molecules
The calculation model was constructed using azobenzene-based structural units, including several nearby atoms, after the chosen azobenzene-based molecules (m-MR and m-AZO) would have been polymerized with other monomers. All calculations were carried out with the Gaussian 09 programs at the CAM-B3LYP/6-31G (d, p) level.19–21 The electrostatic potentials of the molecules were exhibited, in which the positive and negative regions appeared to be red and blue, respectively. The UV-vis spectra and the vertical excitation potential for the first singlet excited state (S1) were calculated by time-dependent DFT (TDDFT) calculations at the optimized S0 geometry at the same level. Solvent effects were taken into account within the polarizing continuum model (PCM) framework by using DMSO in all the geometry optimization and excited state calculations.Synthesis of azobenzene-based photo-switch
The functional monomer (NAS) was previously synthesized by the acylchlorination of NHS, which was confirmed by 1H nuclear magnetic resonance spectroscopy (1H NMR, Bruker, AV300).9 The AZO-containing monomer based on m-MR was synthesized by a similar method. Briefly, 5–10% excess acryloyl chloride was reacted with m-MR using TEA as the acid absorbent in sealed anhydrous DCM with a concentration of about 1 M. Before the beginning of the reaction, acryloyl chloride was added dropwise to the system within 1 h in an ice-water bath. The reaction would have lasted for 4–5 h at room temperature. The synthesized monomer (m-Methyl Red acrylic anhydride, m-MRAA) was obtained by the following procedure including filtration to remove the salt, washing with saturated NaCl solution, and separation several times to remove the unreacted reactants, followed by distillation under reduced pressure. The final products were characterized by NMR (AV300) using D6-DMSO as the solvent.The visible light-driven azobenzene-based photo-switching macromolecule was obtained by radical copolymerization with HEMA, NVP, and NAS. Briefly, the monomers were dissolved in dioxane with a total concentration of 0.3 M, into which BPO was added as an initiator in the form of dioxane solution with the concentration of 15 mM. The monomer feeding ratio was fixed as 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:3:1 (m-MRAA:HEMA:NVP:NAS) and the initiator molar ratio was fixed to 5% of the total monomer. Before polymerization, oxygen in the system was removed by purging with nitrogen. The reaction would have lasted for 24 h at 70 °C in a sealed anaerobic environment. The copolymer was obtained after purification (precipitated in diethyl ether and dissolved in THF several times) and freeze-drying (−50 °C, 7–8 Pa). The final product was characterized by 1H NMR (AV300) using D6-DMSO as the solvent.
:3:3:1 (m-MRAA:HEMA:NVP:NAS) and the initiator molar ratio was fixed to 5% of the total monomer. Before polymerization, oxygen in the system was removed by purging with nitrogen. The reaction would have lasted for 24 h at 70 °C in a sealed anaerobic environment. The copolymer was obtained after purification (precipitated in diethyl ether and dissolved in THF several times) and freeze-drying (−50 °C, 7–8 Pa). The final product was characterized by 1H NMR (AV300) using D6-DMSO as the solvent.
Effects of the excitation sources on the isomerization for the photo-switches
m-MR, m-MRAA, and the copolymer were dissolved respectively in DMSO to obtain two kinds of dilute solutions. Four different excitation sources, namely, UV lamp (240–365 nm, 3.6 W), blue light lamp (450–457 nm, 3.6 W), yellow light lamp (580–595 nm, 3.6 W), and red light lamp (620–625 nm, 3.6 W), were used to cause the isomerization for the three photo-switches including m-MR, m-MRAA, and synthesized copolymer with controllable light density and temperature. After light irradiation, the recovery process was realized in dark at different temperatures. Real-time UV-vis spectra were respective recorded in the irradiation process and the recovery process.Response–recovery performance of the synthesized copolymer
The photo-switching performance included the excitation process in the presence of a specific light source and the reversible recovery process in a dark environment. Besides the excitation sources, the effects of light density on the photo-switching performance of the synthesized copolymer were investigated. The excitation source was fixed at blue light. Similarly, the real-time UV-vis spectrum of the irradiation process under specific light density as well as that of the recovery process under a dark environment was recorded. In the next step, repeated irradiation and recovery method were applied to study the fatigue resistance of the copolymer. The light density was set at 1000 lux and the temperature was set at room temperature. Finally, the effects of the solvent on the response–recovery performance were also tracked and recorded. Besides DMSO, water and alcohol were used to adjust the property of the solvent.Additional properties of the photo-switches
The UV spectra of m-MR, m-MRAA, and the synthesized copolymer solutions with gradually changing pH values were also detected to evaluate their pH-dependent property. 10 M HCl or NaOH were used to adjust the pH value of the targeted solution.The fluorescence emission spectra of the m-MR, m-MRAA, and copolymer solutions with the same concentration was tracked by a spectrofluorometer (FS5). The excitation wavelength was set at 425 nm and the slit width was set at 1 nm.
Results and discussion
DFT calculations for azobenzene-based molecules
In order to find an effective molecule for further polymerization, DFT was used to calculate the electrostatic potential, UV-vis spectra, and the corresponding molecular orbital transitions from HOMO to LUMO. These results are shown in Fig. 1. It was found from Fig. 1a that the m-MR structure based on the commercial m-MR molecule had obvious electron-donating and electron-withdrawing characteristics across the main AZO structure, while the m-AZO structure based on the previously polymerized macromolecule showed different electronic effects with electron-donating and electron-withdrawing groups on one side of its main structure. Furthermore, compared with that of m-AZO, the theoretical UV-vis spectra of m-MR was obviously red-shifted by about 42 nm (Fig. 1b). Simultaneously, the energy gap between the HOMO and LUMO of m-MR were also lower than that of m-AZO (Fig. 1b). Generally, the electronic effects of the electron-donating and electron-withdrawing groups would increase the conjugative effect (electron motility) of the structural system, which made electron transition easier for m-MR. Thus, the excitation energy of m-MR from S0 (base state for trans isomer) to S1 (excited state) was lower than that of m-AZO, which also induced red-shifting of the maximum absorbance peak since the UV-vis absorbance is generally associated with electronic transitions. On the other hand, the trans → cis transition included two process, from S0 up to S1 and from S1 down to S2 (base state for cis isomer) according to the existing mechanism. Therefore, the effective excitation wavelength for the trans → cis transition was related to the maximum UV-vis absorbance wavelength.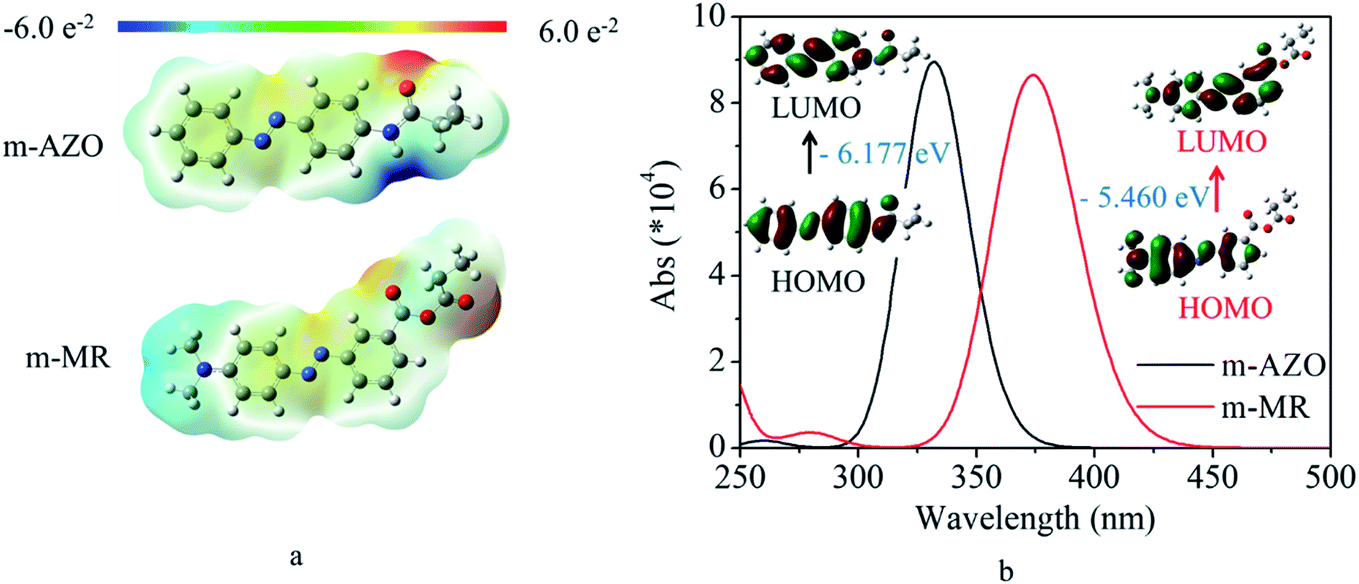 | ||
| Fig. 1 (a) Electrostatic potential of the trans isomers for the azobenzene-based structural unit; (b) Calculated UV-vis spectra and the corresponding molecular orbital transitions from HOMO to LUMO for the azobenzene-based structural unit. The energy gap is also illustrated in the figure. | ||
Actually, the maximum absorbance peak of the m-AZO-containing copolymer was 350 nm, which was red-shifted by about 18 nm away from the theoretical UV-vis spectrum.9 The phenomenon was reasonable as the theoretical calculation could not include all the effects of the practical experiment. However, the red-shifting tendency would not change, which is a generally acknowledged fact. From the above results and discussion, it was expected that the m-MR-containing copolymer would have visible light-responsive performance. Therefore, the m-MR molecule was used to design and synthesize a visible light-driven photo-switching macromolecule for further investigation in the next step.
Synthesis of the photo-switching macromolecule
Since the effectiveness and efficiency of acylchlorination has been proven by previous research for double carbon modification of molecules containing reactive hydrogen, m-MRAA could be synthesized by this method. The 1H NMR spectrum of m-MRAA is shown in Fig. 2a, which was analyzed as follows: chemical shifts from 6.5 to 7.0 ppm were attributed to the three protons on the vinyl group at 9–10 positions with the integration of 0.38, chemical shifts from 7.4 to 8.5 ppm were attributed to the protons of the benzene ring with the integration of 1.00. The existence protons at 9–10 positions indicated the existence of the vinyl group and the consistency of the peak area ratio (1:0.38) to proton number ratio (8:3) confirmed the correct m-MRAA structure. Moreover, the chemical shifts were suitably assigned to the respective carbon atoms in the 13C NMR spectrum one by one, as shown in Fig. 2b, which was also consistent with the m-MRAA structure. Therefore, the results proved the successful synthesis of m-MRAA.
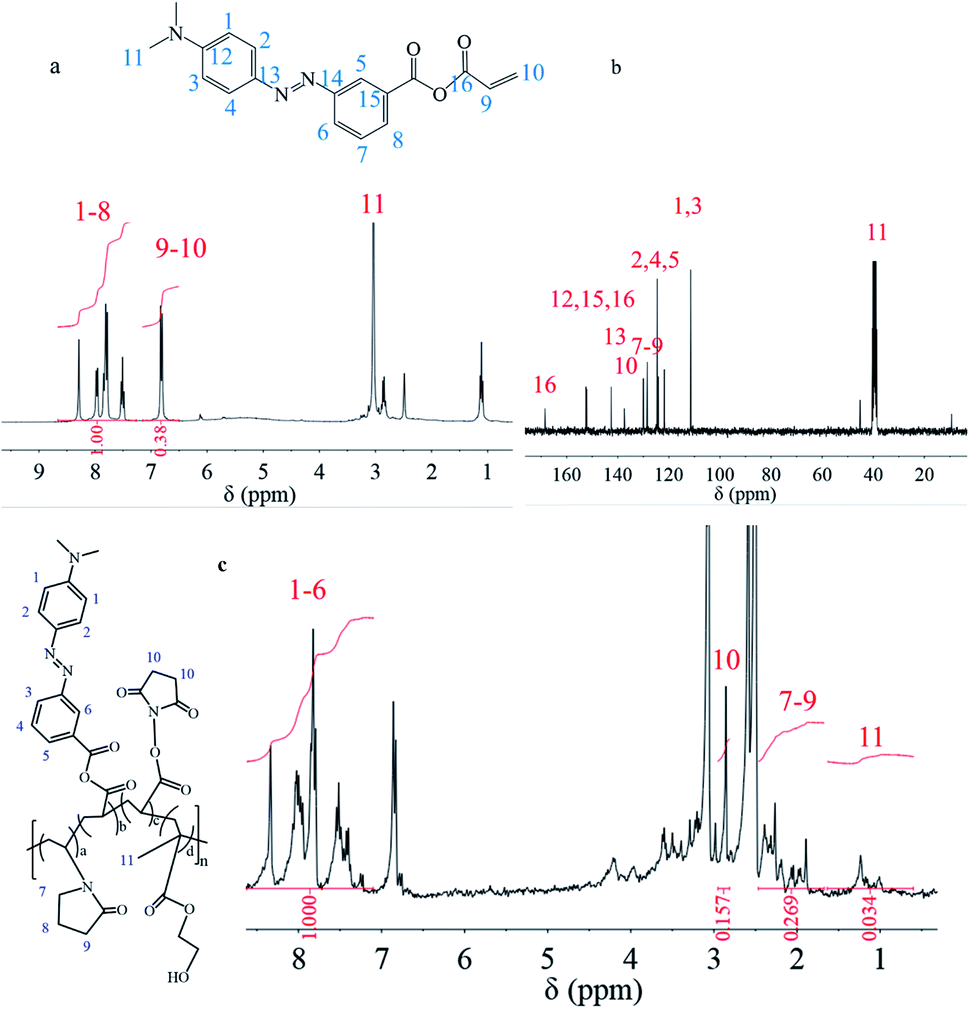 | ||
| Fig. 2 (a) 1H NMR spectrum of m-MRAA; (b) 13C NMR spectrum of m-MRAA; (c) 1H NMR spectrum of the copolymer. | ||
Similarly, the 1H NMR spectrum was also used to confirm the structure of the synthesized copolymer in Fig. 2c, which was analyzed as follows: chemical shifts from 7.1 to 8.6 ppm were attributed to the protons of the benzene ring on the m-MRAA domain at 1–6 positions with the integration of 1.000, the chemical shift at 2.7 ppm was attributed to the protons of the five-membered ring on the NAS domain at the 10 position with the integration of 0.157, chemical shifts from 1.7 to 2.5 ppm were attributed to the protons of the five-membered ring on the NVP domain at the 7–9 positions with the integration of 0.296, chemical shifts from 0.5 to 1.4 ppm were attributed to the protons of the methyl group on the HEMA domain at the 11 position with the integration of 0.034. These results indicated that four different monomers could be successfully copolymerized to form the copolymer with four different structural units. Besides the qualitative analysis, the relative content of each monomer could be calculated by the peak area since the areas of the resonance peaks were proportional to the number of protons. The contents of the structural units were calculated to be m-MRAA of 51%, NAS of 16%, NVP of 18%, and HEMA of 15%. Compared with the feeding ratio, more m-MRAA and NAS entered the copolymer chain but less HEMA and NVP entered the copolymer chain. The result indicated that m-MRAA and NAS might have more polymerization activity due to their conjugated structure. Moreover, the physical transition of the MR-containing copolymer was characterized by DSC. However, no physical transitions including Tg transition or phase transition were found in the DSC curve.
Effects of the excitation sources on the isomerization
For photo-switches, reversible response and recovery performance are indispensable properties that need to be investigated in detail. The photo-switching of the copolymer was evaluated by the transformation between the trans and cis isomers, which was characterized by UV-vis spectroscopy. Firstly, four excitation sources with different wavelengths were used to evaluate the feasibility and controllability of the isomerization transformation of the synthesized copolymer. In contrast, m-MR and m-MRAA were evaluated together. It was found that m-MR and m-MRAA gave nearly similar UV-vis spectra with the maximum absorbance at 425 nm, which was red-shifted by about 51 nm from the calculated value (Fig. 3). Nevertheless, the UV-vis spectra showed no change after the m-MR solution was excited by any of the light sources within the observable time. Similarly, UV light, yellow light, and red light could not induce visible absorbance changes in the UV-vis spectrum of the m-MRAA solution (Fig. 3a, b, and d). On the other hand, the absorbance value at 425 nm in the UV-vis spectrum decreased to 79% of the original value after m-MRAA solution irradiation by blue light for 2 min (Fig. 3c). Just as that discussed in the theoretical part, the absorbance peak at 425 nm of the UV-vis spectrum was related to the trans isomer in the system. These results indicated that no visible isomerization transformation happened for m-MR excited by any of the light sources and for m-MRAA excited by light sources except blue light. In addition, blue light might induce some visible isomerization transformation in m-MRAA.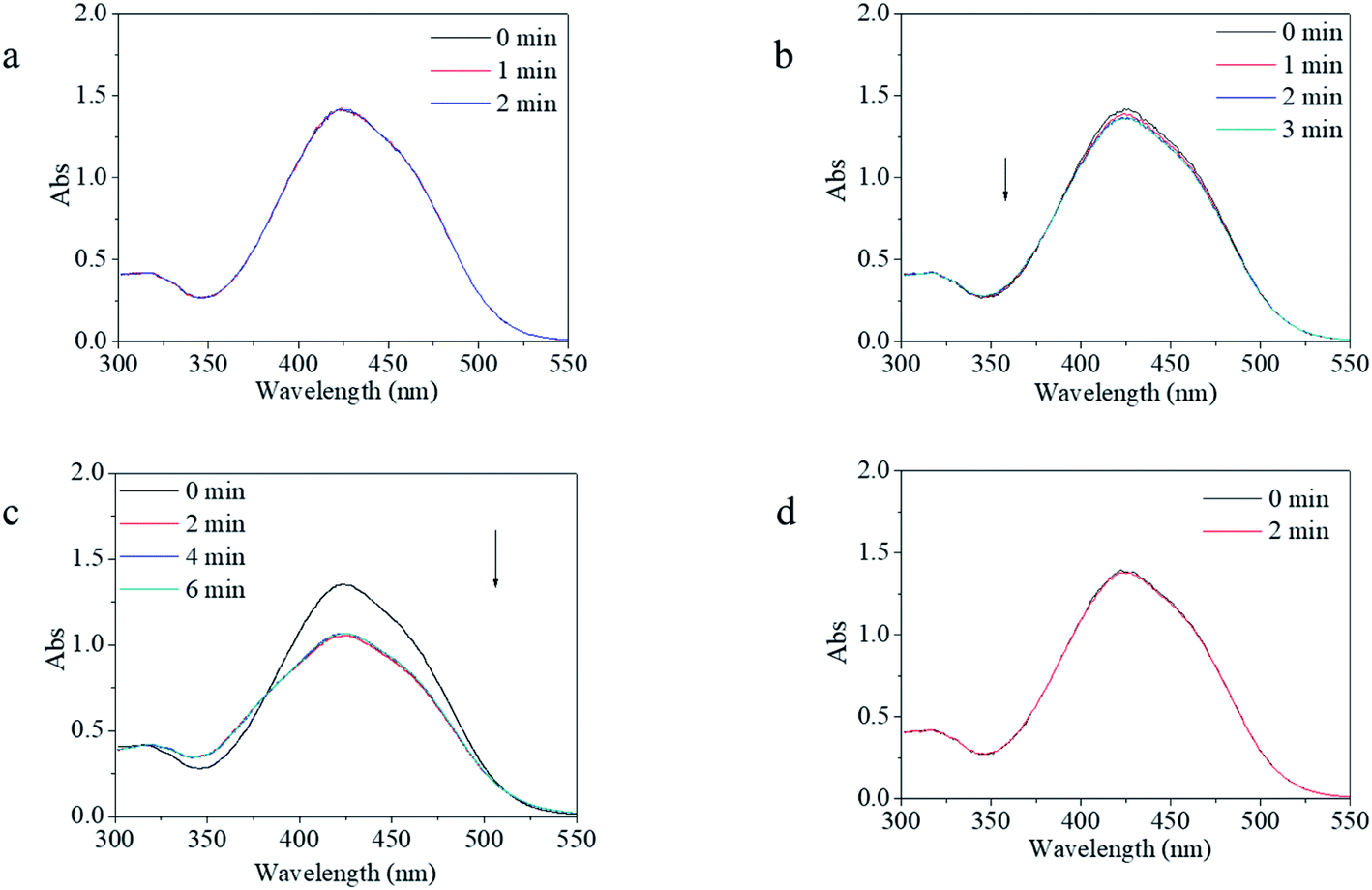 | ||
| Fig. 3 UV-vis spectra of the m-MRAA solution as a function of irradiation time by (a) red light, (b) yellow light, (c) blue light, and (d) purple light. The light density was set at 1000 lux at the room temperature. | ||
On the whole, undetectable or unclear isomerization transformation for m-MR or m-MRAA might be caused due to two reasons. One is because the isomerization transformation of m-MR or m-MRAA could not be excited by the excitation source. The other was that the speed of isomerization transformation for m-MR or m-MRAA was too fast to be detected. According to the proposed energy theory of instability of the cis form, we preferred the latter. A little difference in the blue light-excited response process between m-MR and m-MRAA might come from several atoms' difference in the molecular structure between m-MR and m-MRAA. Therefore, increasing stability and controllability of the cis form was the focus of the following investigation based on the theory that chain movement of the polymer had time dependence and was restricted by the free volume.
Although the synthesized copolymer possessed a similar UV-vis absorbance peak as that of m-MR or m-MRAA, its response–recovery performance was completely different (Fig. 4). Every excitation source could induce a decrease in the absorbance value but to a different final value (Fig. 4a, c, e, and g). However, the efficiency of excitation by different excitation sources was different according to the lowest absorbance value at 425 nm in the UV-vis spectrum since it is related to the trans form of the AZO structure. On excitation by red light, the absorbance value at 425 nm decreased gradually with time and finally stabilized at 76% of its original value with an equilibrium time of 10 min (Fig. 4a). On excitation by yellow light, the decreased absorbance value finally stabilized at 70% of its original value with an equilibrium time of 8 min (Fig. 4c). On excitation by blue light, the decreased absorbance value finally stabilized at 52% of its original value with an equilibrium time of 5 min (Fig. 4e). On excitation by UV light, the decreased absorbance value finally stabilized at 85% of its original value with an equilibrium time of 30 s (Fig. 4g). Obviously, the most efficient excitation source was blue light, next was yellow light, and the less efficient one was red light or UV light. In other words, the excitation efficiency decreased with increasing difference between the excitation wavelength and the maximum UV-vis absorbance wavelength (25 nm for blue light, 155 nm for yellow light, 195 nm for red light, 165 nm for UV light), which was consistent with the theoretical analysis in the beginning. Moreover, the equilibrium irradiation time was shortened along with the decrease in the excitation wavelength for the reason that light with a shorter excitation wavelength has a larger energy, which accelerated the speed of the trans → cis transition. In short, blue light was considered to be the most efficient excitation source with a proper response time for further investigations.
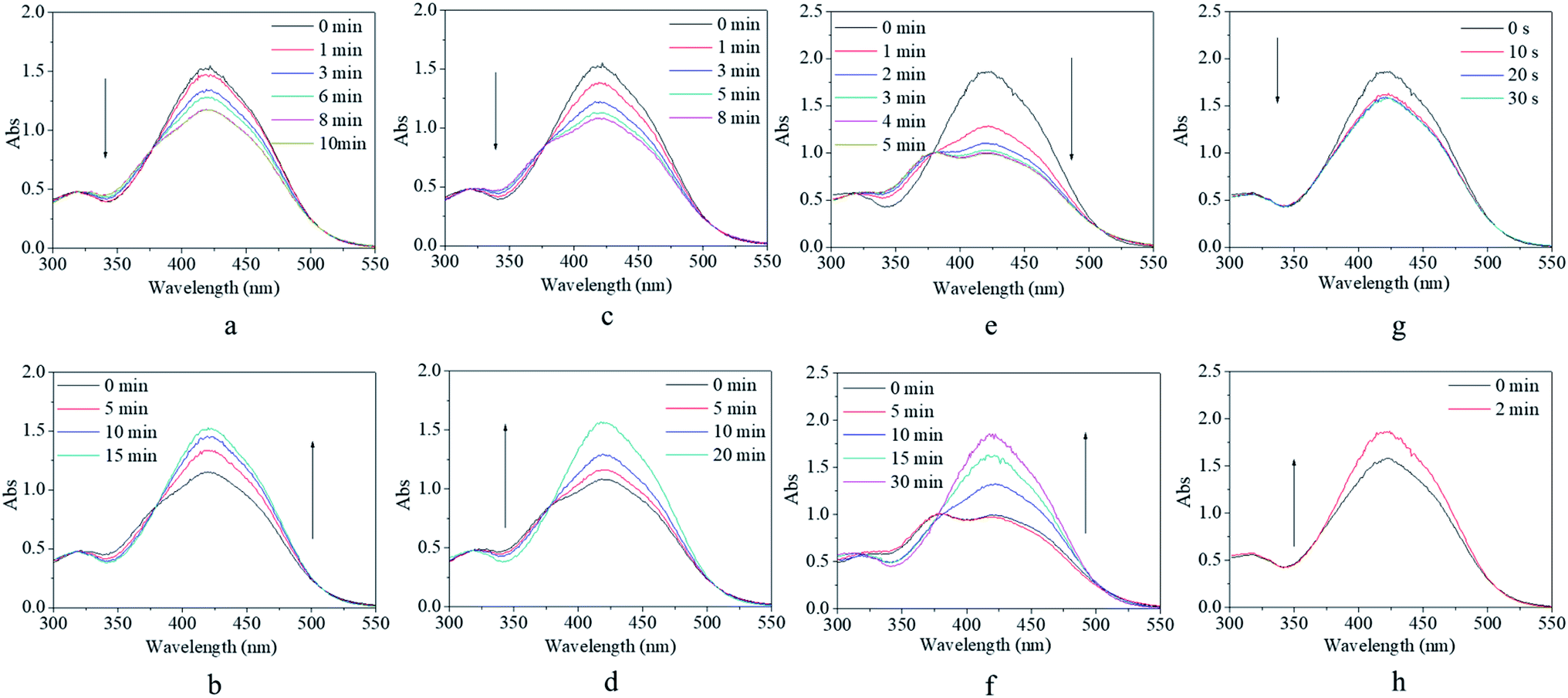 | ||
| Fig. 4 UV-vis spectra of the copolymer solution as a function of irradiation time (a, c, e, and g) and recovery time (b, d, f, and h) by (a andb) red light, (c and d) yellow light, (e and f) blue light, and (g and h) purple light. The light density was set at 1000 lux at room temperature. | ||
After irradiation, all the decreased absorbance values could gradually recover to their original value in a dark environment regardless of their original excitation source (Fig. 4b, d, f, and h). However, the time consumed for the recovery process varied from 2 min after UV irradiation (Fig. 4h) to 30 min after blue light irradiation (Fig. 4h), including two median values of 15 min after red light irradiation (Fig. 4b) and 20 min after yellow light irradiation (Fig. 4d). Since the content of the cis form in the system was related to the extent of decrease in the absorbance, as discussed above, more cis isomers naturally needed a longer time to realize their cis → trans recovery.
On comparing the results of Fig. 3 and 4, it was inferred that copolymerization could not change the energy characteristic brought by the electronic effects for the m-MR structure but could increase the steric hindrance effects in isomerization by the free volume of the copolymer. In other words, this research attempted to resolve the problem of stability and controllability for m-MR structure application as a photo-switch from the dynamic aspect of molecule movement.
Response–recovery performance of the synthesized copolymer
In general, light density is also a factor that influences isomerization, which was researched next. The effects of the light density on the response–recovery performance are shown in Fig. 5. It was found that blue light with a light density ranging from 150 lux to 1000 lux could excite the absorbance at 425 nm, decreasing though the end point of decreasing absorbance and the response time showed some dependence on the light density (Fig. 5a, c, e, and g). Elaborately, the absorbance at 425 nm decreased to 66% of the original value within 30 min of excitation by light with 150 lux (Fig. 5a), 64% of the original value within 12 min of excitation by light with 250 lux (Fig. 5c), 59% of the original value within 10 min of excitation by light with 400 lux (Fig. 5e), and 55% of the original value within 5 min of excitation by light with 700 lux of light density (Fig. 5g). Generally, the light intensity is a secondary factor that influences the excitation energy, which slightly influences the efficiency of isomerization transformation. Consequently, relatively low excitation energy led to relatively low degree of the final cis form and needed more equilibrium time for isomerization transformation. Combined with the results of Fig. 4e, the light density of 1000 lux was assumed to be a relative equilibrium value for excitation and was used for further investigation, according to the discussed variation tendency in Fig. 5. After removing the excitation source, the decreased absorbance peak could gradually recover to its original shape within 17–25 min under a dark environment (Fig. 5b, d, f, and h). The light density in the irradiation process showed no significant effects on the recovery process including the recovery time because the difference in the cis form content after irradiation was small.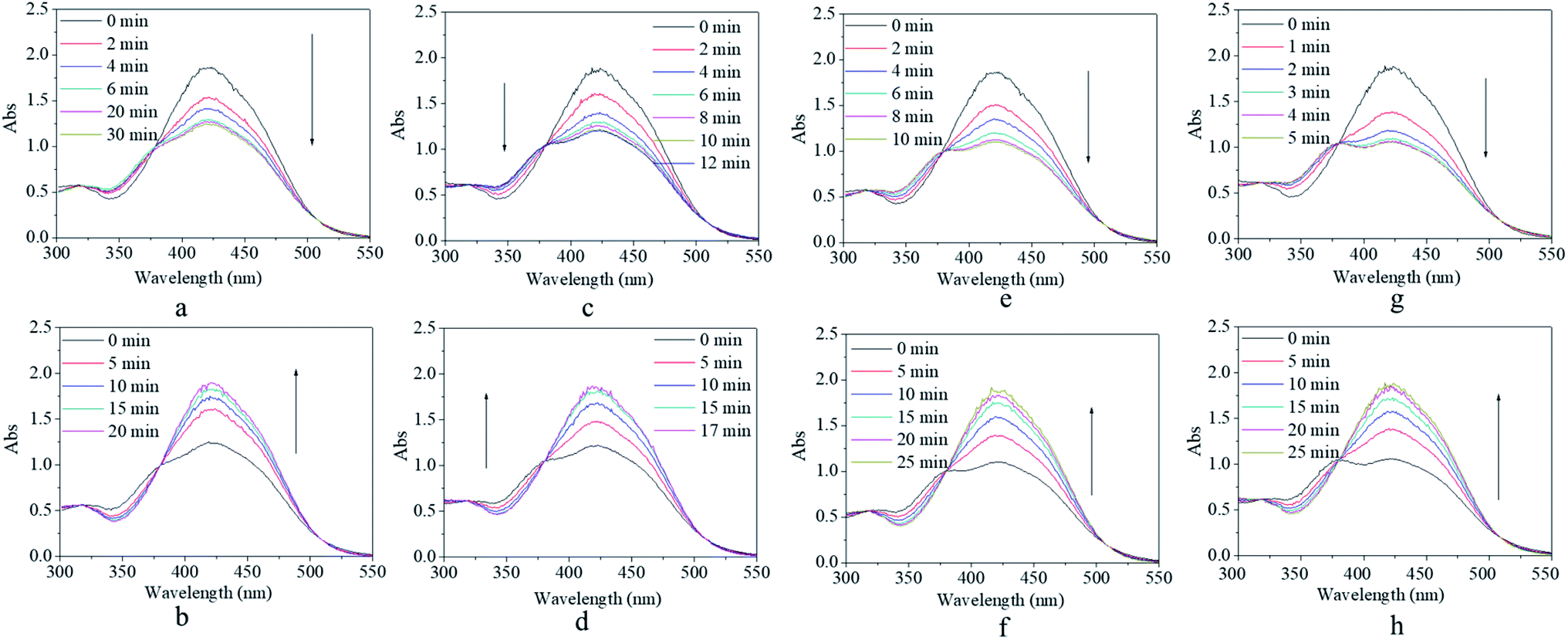 | ||
| Fig. 5 UV-vis spectra of the copolymer solution as a function of irradiation time by blue light (a, c, e, and g) and recovery time (b, d, f, and h) at room temperature. The blue light density was set at (a and b) 150 lux; (c and d) 250 lux; (e and f) 400 lux; (g and h) 700 lux. | ||
Besides the stable and controllable response–recovery process, fatigue resistance is also an essential property for the practical application of photo-switches. Hence, the fatigue resistance of the copolymer was evaluated by repeated on and off irradiation (Fig. 6). In order to simplify and emphasize the crucial results in the response–recovery process, only the maximum and minimum absorbance at 425 nm of the copolymer solution as a function of the cycle number were recorded, which are shown in Fig. 6a. It was found that the maximum absorbance was either the initial or after the recovery was stabilized at about 1.65–1.80 at the early stage and at about 1.85–1.95 at a later period; simultaneously, minimum absorbance after irradiation was stabilized at about 0.90 at an early stage and at about 1.05 at a later period of circle time (Fig. 6a). The slight difference in the maximum/minimum absorbance between the early stage and the later period might be ascribed to the increase in the polymer concentration by uncontrollable solvent volatilization during the long duration experiment. Besides the absorbance, the irradiation response time was fixed at 5 min and the recovery response time was fixed at 30 min regardless of the circle time (Fig. 6b). These results verified that the trans → cis transition and cis → trans recovery process could be stably repeated without any sign of fatigue or photobleaching occurring in the AZO small molecule derivative. Since effective structural change, stable and controllable response–recovery performance, and fatigue resistance are all desirable properties for an ideal photo-switch, the synthesized copolymer exhibited typical characteristics of a photo-switch from these results.
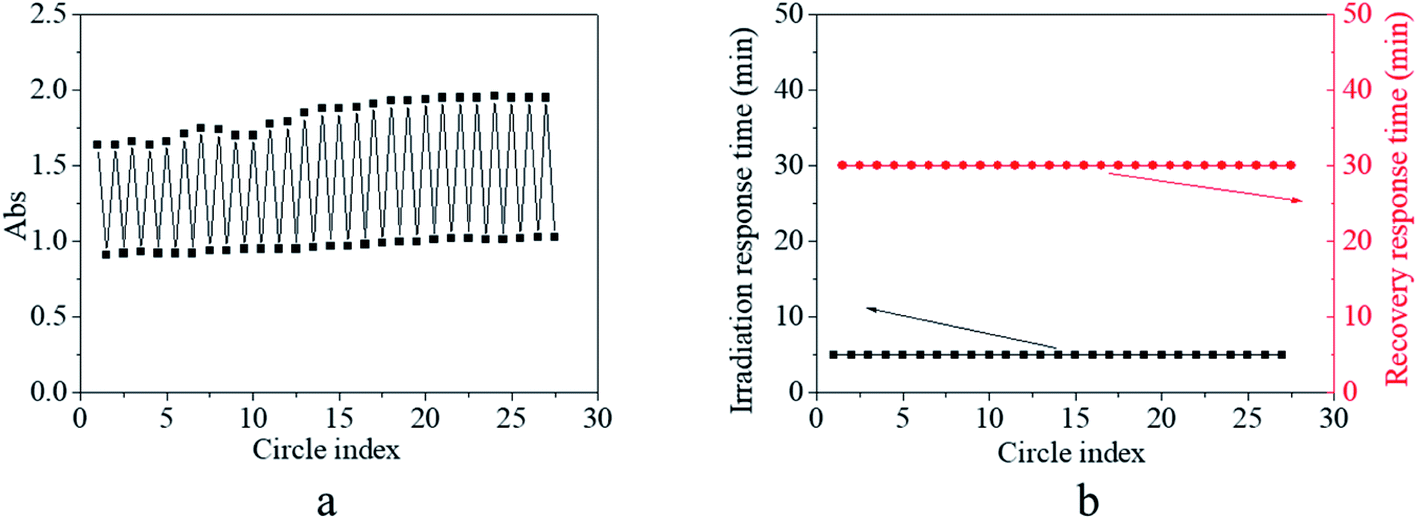 | ||
| Fig. 6 (a) Absorbance at 420 nm of the copolymer solution as a function of the circle index. (b) Irradiation response time by blue light and recovery response time in a dark environment as a function of the circle index. The light density was set at 1000 lux. | ||
In addition, the solvent effects on the trans → cis excitation process are investigated in Fig. 7 and 8. Alcohol and water were used to adjust the property of the solvent in consideration of the dissolvability and compatibility. With the addition of alcohol, the detectable isomerization process became unclear and even disappeared (Fig. 7). In detail, the absorbance at 425 nm decreased to 67% of the original value for the solution with 20% alcohol and 80% DMSO (Fig. 7a), 85% of the original value for the solution with 50% alcohol and 50% DMSO (Fig. 7b), and absorbance showed no variation for the solution with 70% alcohol and 30% DMSO (Fig. 7c). The effect of water on the trans → cis excitation process had a similar tendency but the extent of the water effect was acuter than that of the alcohol effect (Fig. 8). Also, the absorbance at 425 nm decreased to a relatively high value, 69% of the original value, when water occupied only 5% of the whole solvent (Fig. 8a). Also, the value reached 92% when water occupied only 10% of the whole solvent (Fig. 8b). Finally, the trans → cis transition could not be detected when water occupied only 15% of the whole solvent (Fig. 8c). Actually, the stable and controllable isomerization process of the previously synthesized AZO-HEMA-NVP-NAS copolymer in water solution could be realized by UV excitation according to previous research.9 Compared with the two copolymers, it was found that their dissolvability was different in water. The AZO-HEMA-NVP-NAS copolymer could be dissolved in water with the assistance of DMSO but the copolymer could not dissolve completely in water. In addition, this copolymer also had limited dissolvability in alcohol. Based on the fact that a poor solvent would induce domain aggregation, it was inferred that the aggregated m-MRAA domain increased the energy of isomerization transformation. Consequently, the trans → cis transition exhibited strong solvent dependence.
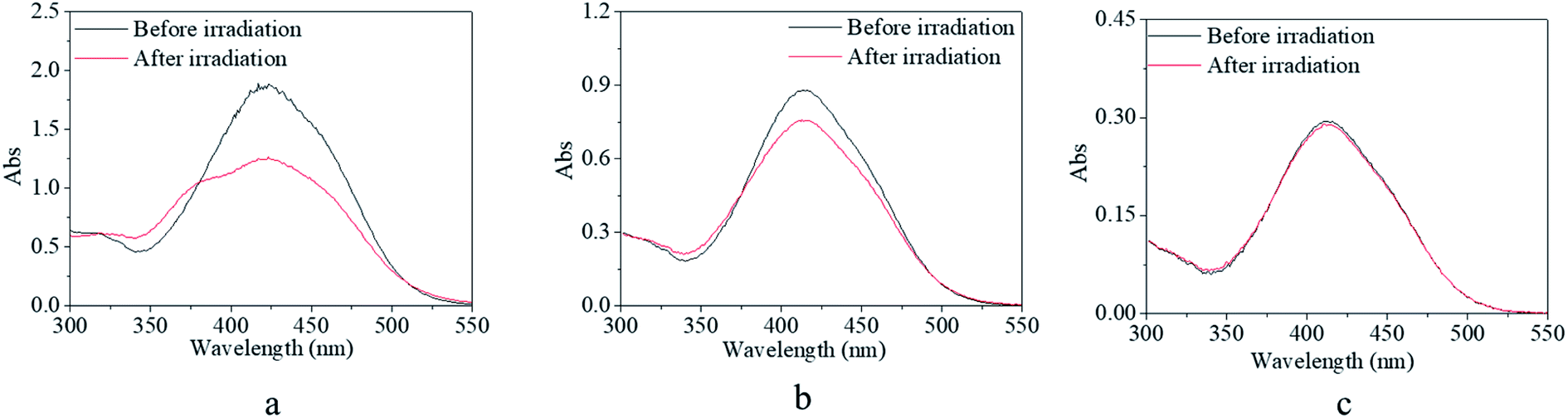 | ||
| Fig. 7 UV-vis spectra of the copolymer solution before and after irradiation by blue light with a light density of 1000 lux. (a) 80% DMSO and 20% ethyl alcohol; (b) 50% DMSO and 50% ethyl alcohol; (c) 30% DMSO and 70% ethyl alcohol. | ||
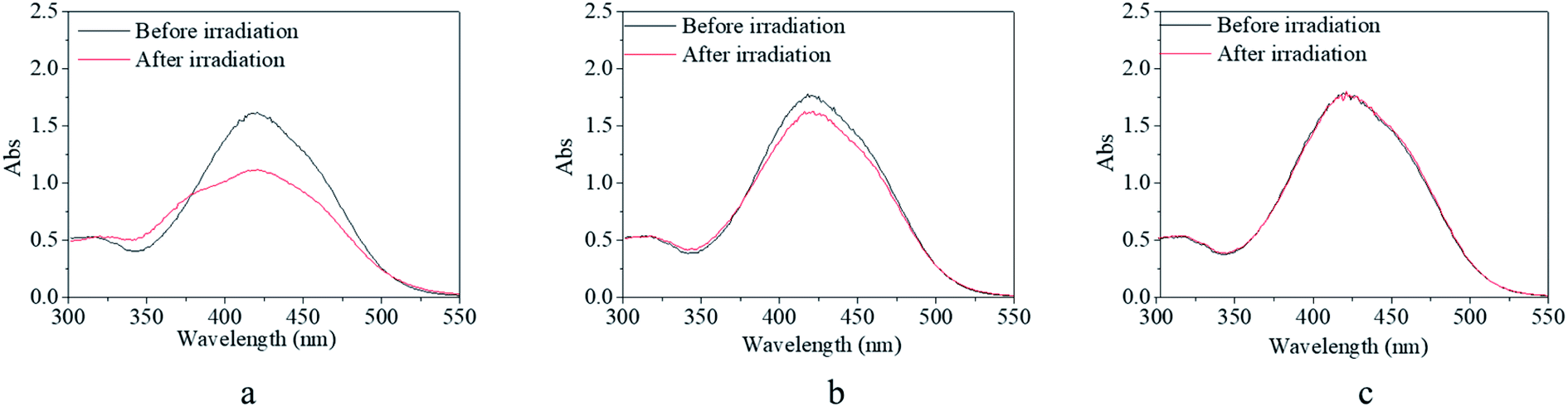 | ||
| Fig. 8 UV-vis spectra of the copolymer solution before and after irradiation by blue light with a light density of 1000 lux. (a) 95% DMSO and 5% water; (b) 90% DMSO and 10% water; (c) 85% DMSO and 15% water. | ||
Additional properties of the photo-switches
It is known that commercial m-MR is a commonly-used pH indicator. In order to clarify the pH-dependent property of its derivatives, UV-vis spectra and digital photos of their solution as a function of pH value were recorded, as shown in Fig. 9. For m-MR, the absorbance peak of the UV-vis spectra was blue shifted from 504 nm to 450 nm when the pH value was larger than 4, which was the reason for its discoloration, and also the mechanism as a pH indicator with a critical point of pH 4.0 (Fig. 9a). For m-MRAA, the absorbance peak of the UV-vis spectra was blue shifted with the increase in the pH value with a critical point of pH 5.0 (Fig. 9b). For the copolymer, the absorbance peak of the UV-vis spectra was identical to that for m-MRAA, with a critical point of pH 5.0 (Fig. 9c). The functional modification of m-MR might lead to the variation in the polarity and electron distribution, which resulted in the difference in the critical pH point between m-MR and the m-MRAA/copolymer. In brief, the synthesized copolymer still had the characteristics of a pH indicator with a critical point of pH 5.0. In addition, the pH critical point of m-MR, m-MRAA, or the copolymer was also confirmed by the color of the solutions with different pH values, as shown in Fig. 9d, e, and f.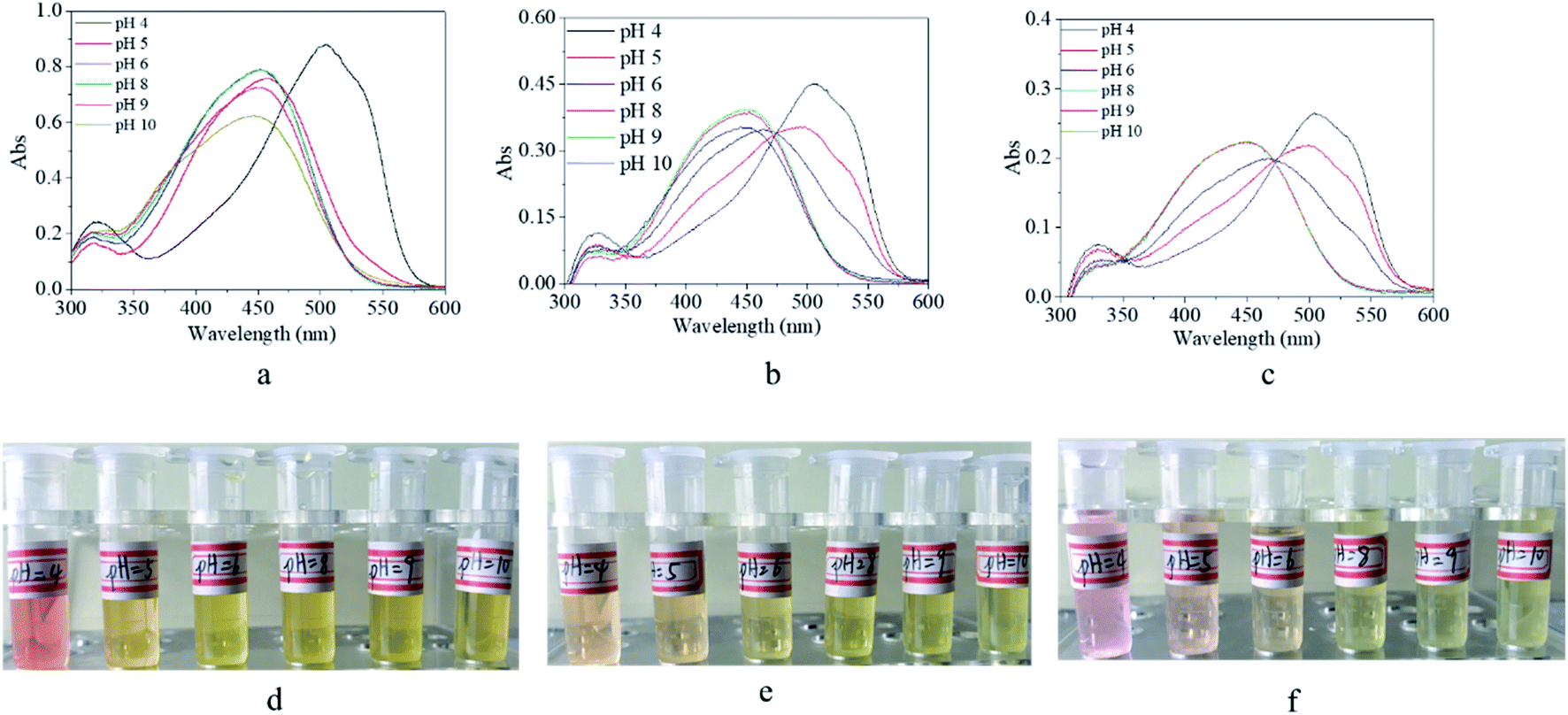 | ||
| Fig. 9 UV-vis spectra (a–c) and digital photos (d–f) of (a and d) m-Methyl Red solution, (b and e) m-MRAA solution, and (c and f) copolymer solution in different pH value environment. | ||
Finally, the fluorescent property of m-MR, m-MRAA, and the copolymer was checked to evaluate the overall performance, as shown in Fig. 10. It was found that m-MR and m-MRAA exhibited no fluorescent characteristics, while the copolymer exhibited an obvious fluorescent absorbance at 550 nm. Since the other structural units had no fluorescent characteristics, the m-MR structure contributed to the fluorescent characteristics of the copolymer. The conjugated azobenzene structure possessed a certain electron transition capacity, which was directly related to the fluorescent characteristics. It is natural that the structure possessed some fluorescent characteristics. However, the small molecule of m-MR and m-MRAA did not exhibit any fluorescence, which might be ascribed to interactions or aggregations between the two molecules, thus leading to the quenching of fluorescence. On the other hand, the polymer chain could reduce the interactions or aggregations between the two conjugated azobenzene structural units. Therefore, the copolymer could retain some fluorescent characteristics of the conjugated azobenzene structure.
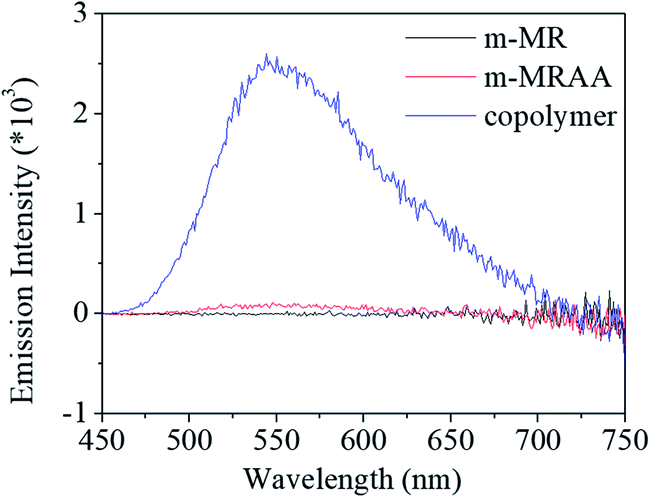 | ||
| Fig. 10 Fluorescence spectra of m-MR, m-MRAA, and the copolymer. | ||
Conclusion
The electrostatic potential described typical electron-donating and electron-withdrawing characteristics for the m-MR structure compared with the m-AZO structure. Further, the theoretical UV-vis spectra of m-MR exhibited an obvious red-shifting characteristic on the basis of that of m-AZO. Concurrently, the energy gap between the HOMO and LUMO for m-MR was calculated to be lower than that for m-AZO. From the theoretical calculation and analysis, the visible light-responsive performance of the m-MR-containing copolymer was expected. The synthesized monomer of m-MRAA was successfully synthesized with the functional monomers of HEMA, NVP, and NAS by the radical polymerization to form the copolymer, which contained the following domains: 51% m-MRAA, 16% NAS, 18% NVP, and 15% HEMA. Detectable isomerization transformation could be hardly be caused by the excitation sources for m-MR and m-MRAA, although slight trans → cis transition could be caused by blue light for m-MRAA. However, nearly all the used light sources, namely, red light, yellow light, blue light, and UV light, could cause detectable isomerization transformation for the synthesized copolymer although the excitation efficiency was different. It was confirmed that the excitation efficiency decreased with increasing difference between the excitation wavelength and the maximum UV-vis absorbance wavelength, while the equilibrium response time monotonically decreased with the decrease in the excitation wavelength. Therefore, the optimal excitation source was blue light, which was consistent with the theoretical analysis. In the dark environment, the cis form could recover to the original trans isomer. The recovery time ranged from 2 min to 30 min depending on the extent of the final decreasing absorbance value. As a secondary factor, the light intensity had a slight influence on the trans → cis transition. Relatively low light density led to relatively low degree of the final cis form and needed more equilibrium time for isomerization transformation due to its low energy but showed little effect on the recovery process. Good fatigue resistance of the copolymer without any sign of photobleaching was confirmed by the stable and repeatable trans → cis/cis → trans process. In addition, isomerization transformation exhibited strong solvent dependence. Finally, the synthesized copolymer still had a characteristic of the pH indicator with a critical point of pH 5.0 and exhibited obvious fluorescent characteristics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by Natural Science Foundation of China (21702082), Natural Science Foundation of Jiangsu Province (BK20171113), Six talent peaks project in Jiangsu Province (JY-071).References
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- Q. Bian, W. Wang, G. Han, Y. Chen, S. Wang and G. Wang, ChemPhysChem, 2016, 17, 2503–2508 CrossRef CAS.
- Q. Bian, W. Wang, S. Wang and G. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 27360–27367 CrossRef CAS.
- S. R. Deka, S. Yadav, M. Mahato and A. K. Sharma, Colloids Surf., B, 2015, 135, 150–157 CrossRef CAS.
- Z. Li, P. Wang, B. Liu, Y. Wang, J. Zhang, Y. Yan and Y. Ma, Soft Matter, 2014, 10, 8758–8764 RSC.
- L. R. Lin, X. Wang, G. N. Wei, H. H. Tang, H. Zhang and L. H. Ma, Dalton Trans., 2016, 45, 14954–14964 RSC.
- J. Henzl, M. Mehlhorn, H. Gawronski, K. H. Rieder and K. Morgenstern, Angew. Chem., 2006, 45, 603–606 CrossRef CAS.
- J. Pang, Z. Y. Gao, L. Zhang, H. M. Wang and X. H. Hu, Front. Chem., 2018, 6, 217 CrossRef.
- J. Pang, Z. Y. Gao, H. P. Tan, X. C. Mao, H. M. Wang and X. H. Hu, Front. Chem., 2019, 7, 86 CrossRef CAS.
- W. Ma, W. Li, R. Liu, M. Cao, X. Zhao and X. Gong, Chem. Commun., 2019, 55, 7486–7489 RSC.
- M. B. Zhu, Y. X. Li, S. Q. Tian, Y. Xie, X. J. Zhao and X. Gong, J. Colloid Interface Sci., 2019, 534, 509–517 CrossRef CAS.
- Z. L. Li, X. J. Zhao, C. B. Huang and X. Gong, J. Mater. Chem. C, 2019, 7, 12373–12387 RSC.
- Z. J. Wang, X. J. Zhao, Z. Z. Guo, P. Miao and X. Gong, Org. Electron., 2018, 62, 284–289 CrossRef CAS.
- J. Pang, Y. F. Ye, Z. Q. Tian, X. Y. Pang and C. Y. Wu, Comput. Theor. Chem., 2015, 1066, 28–33 CrossRef CAS.
- J. Pang, Z. Q. Tian and J. Ma, Chem. Phys. Lett., 2014, 613, 110–114 CrossRef CAS.
- S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, J. Am. Chem. Soc., 2013, 135, 9777–9784 CrossRef CAS.
- D. Bleger and S. Hecht, Angew. Chem., 2015, 54, 11338–11349 CrossRef CAS.
- M. Dong, A. Babalhavaeji, C. V. Collins, K. Jarrah, O. Sadovski, Q. Dai and G. A. Woolley, J. Am. Chem. Soc., 2017, 139, 13483–13486 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision A.02, Gaussian Inc, Wallingford, CT, 2009 Search PubMed.
- B. Ransil, J. Chem. Phys., 1961, 34, 2109 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |