
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular synthesis and transition metal-free alkynylation/alkenylation of Castagnoli–Cushman-derived N,O- and N,S-heterocyclic vinyl chlorides†
Timothy K. Beng *,
Abdikani Omar Farah and
Victoria Shearer
*,
Abdikani Omar Farah and
Victoria Shearer
Department of Chemistry, Central Washington University, Ellensburg, WA 98926, USA. E-mail: Timothy.beng@cwu.edu
First published on 7th October 2020
Abstract
A modular and functional group-tolerant protocol for the transition metal-free coupling of novel N,O- and N,S-heterocyclic vinyl chlorides with terminal acetylenes and styrenes has been developed, leading to the epimerization-free synthesis of fully carbofunctionalized dihydro-1,4-oxazines/thiazines. Bicyclic morpholines have also been prepared through the interrogation of newly synthesized cross-conjugated dienes in Diels–Alder reactions. The use of environmentally benign reaction media endows the current strategy with a practical advantage.
Introduction
Partially and fully reduced 1,4-oxazines and thiazines (examples of which are depicted in Fig. 1) constitute the core of several alkaloid natural products, corrosion inhibitors, optical bleaching agents, textile dying agents, fruit preservation agents, ligands and pharmaceuticals.1 The biological relevance of these heterocycles as well as their architectural complexity continue to endear them to the medicinal and synthesis communities, thus, inspiring the development of increasingly more cost-effective and environmentally friendly strategies for their construction, peripheral functionalization, and evaluation of structure–activity relationships (i.e., S.A.R.). However, designing selective, efficient, cost-effective, and modular strategies for accessing N,O- and N,S-heterocycles can be quite daunting mainly due to conformational constraints and their insatiable appetite for ring opening. These challenges notwithstanding, several transition metal-catalyzed strategies for accessing C-substituted dihydro-1,4-oxazines and 1,4-thiazines have emerged, including those employed by Katukojvala2 (using diazoenals), Saa3 (using catalytic ruthenium carbenes derived from alkynals and alkynones), Bode4 (using SnAP and SLAP reagents), Tiecco5 (using vinyl selenones), and Carreira6 (using spirocyclic 3-oxetanones). | ||
| Fig. 1 Examples of bioactive 1,4-oxazine and thiazine derivatives. | ||
Alkynes are frequently used as building blocks in organic synthesis and as catalysts in asymmetric catalysis owing to their rich reactivity profiles.7 Since the seminal work of Sonogashira and co-workers,8 Sonogashira-type cross-couplings have become one of the most reliable methods for the construction of internal alkynes with differential substitution.9 Recognizing the merits of the Castagnoli–Cushman reaction (e.g., the use of feedstock chemicals such as amines, aldehydes and cyclic anhydrides to readily access nonplanar sp3-enriched core structures bearing vicinal stereocenters), our group10 as well as that of Krasavin11 previously disclosed that lactams of type 3 are obtainable from imines such as 1 and cyclic anhydrides of type 2 (Fig. 2, top). Desiring a modular and operationally simple approach to alkynylated dihydro-1,4-oxazines and thiazines, it was surmised that transition metal-free late-stage coupling of densely populated precursors such as 4 with terminal alkynes offered an excellent starting point (Fig. 2). The appendage of an alkynyl motif to the skeleton of a cyclic enamine (see 5) offers several advantages for the synthesis of alkaloids and nitrogen-containing pharmaceuticals. As has been previously demonstrated by Toste12 and Occhiato,13 alkyne-substituted enamines may be engaged in Conia-ene and carboauration-type transformations. Meanwhile, the synthesis of conjugated dienes of type 6 via a Heck-type coupling protocol could pave the way for the construction of polycyclic morpholines.14 We were not oblivious to the challenges that belie such transformations seeing as vinyl chlorides such as 4 seldom feature in mild cross-couplings, even under transition metal-catalyzed conditions. This is presumably due to challenges associated with an extremely slow oxidative addition step.15 The successful implementation of the planned strategy would also hinge on our ability to negate incessant ring-opening events that are typically associated with morpholines and thiomorpholines.16 We were also keen on accessing 4 upon recognizing that it retains latent functionality throughout the periphery of the heterocycles that could facilitate further manipulation. For example, the enal subunit may be engaged in conventional olefination/cyclization technologies to arrive at intermediates of type 7. Additionally, the C–C double bond resident in 4 could pave the way for harnessing several reactivity modes, including hydroarylation,17 oxoamination,18 dioxygenation19 trifluoromethylation,20 aziridination,21 pentannulation,22 and boration.23 Herein, detailed efforts toward the elicitation of our ideals are described.
 | ||
| Fig. 2 Proposed plan for the transition metal-free functionalization of N,O- and N,S-heterocyclic vinyl chlorides. | ||
Results and discussion
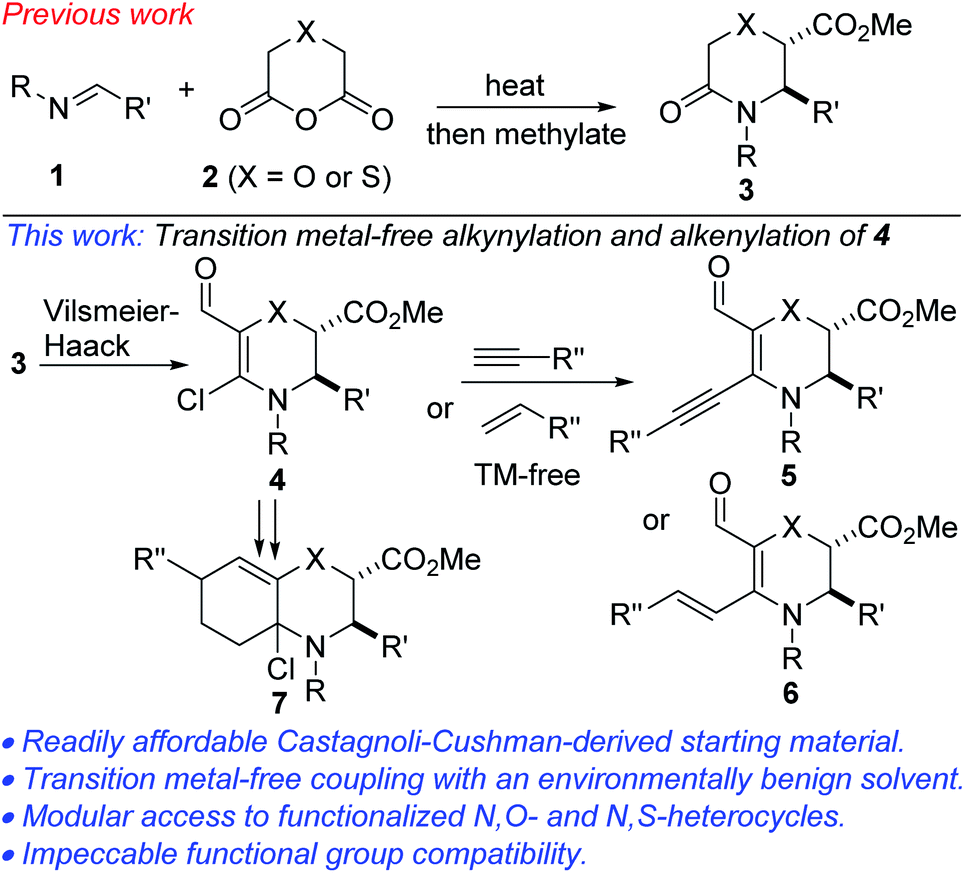
We initiated studies toward the transition metal-free coupling of N,O- and N,S-heterocyclic vinyl chlorides by attempting to find general Vilsmeier–Haack conditions for preparing precursor 4. In the event, we found that under reaction conditions analogous to those previously optimized on the more stable piperidine system,24 morpholinones bearing electron-rich-, electron-neutral- and electron-deficient N-aryl substituents as well as alkyl and benzyl substituents underwent productive lactam-selective Vilsmeier–Haack functionalization (Scheme 1). In all cases, satisfactory yields were obtained (see 4a1–4a18). As we have previously noted,24 the silica/Et3N system employed in these studies for mild hydrolysis of the intermediate chloromethylene iminium salts is desirable given that the use of conventional bases such as NaOH, KOH or NH4OAc led to poor yields, undesirable epimerization, and ring-opened side products. | ||
| Scheme 1 Synthesis of N,O-heterocyclic vinyl chlorides. | ||
The corresponding dihydro-1,4-thiazines have also been prepared (Scheme 2). However, low temperatures and shorter reaction times were required. The variable diastereomeric ratios reported in these cases reflect those of the lactam precursors rather than epimerization during the Vilsmeier–Haack reaction. The slight decrease in yields is a reflection of the somewhat fragile nature of the products.
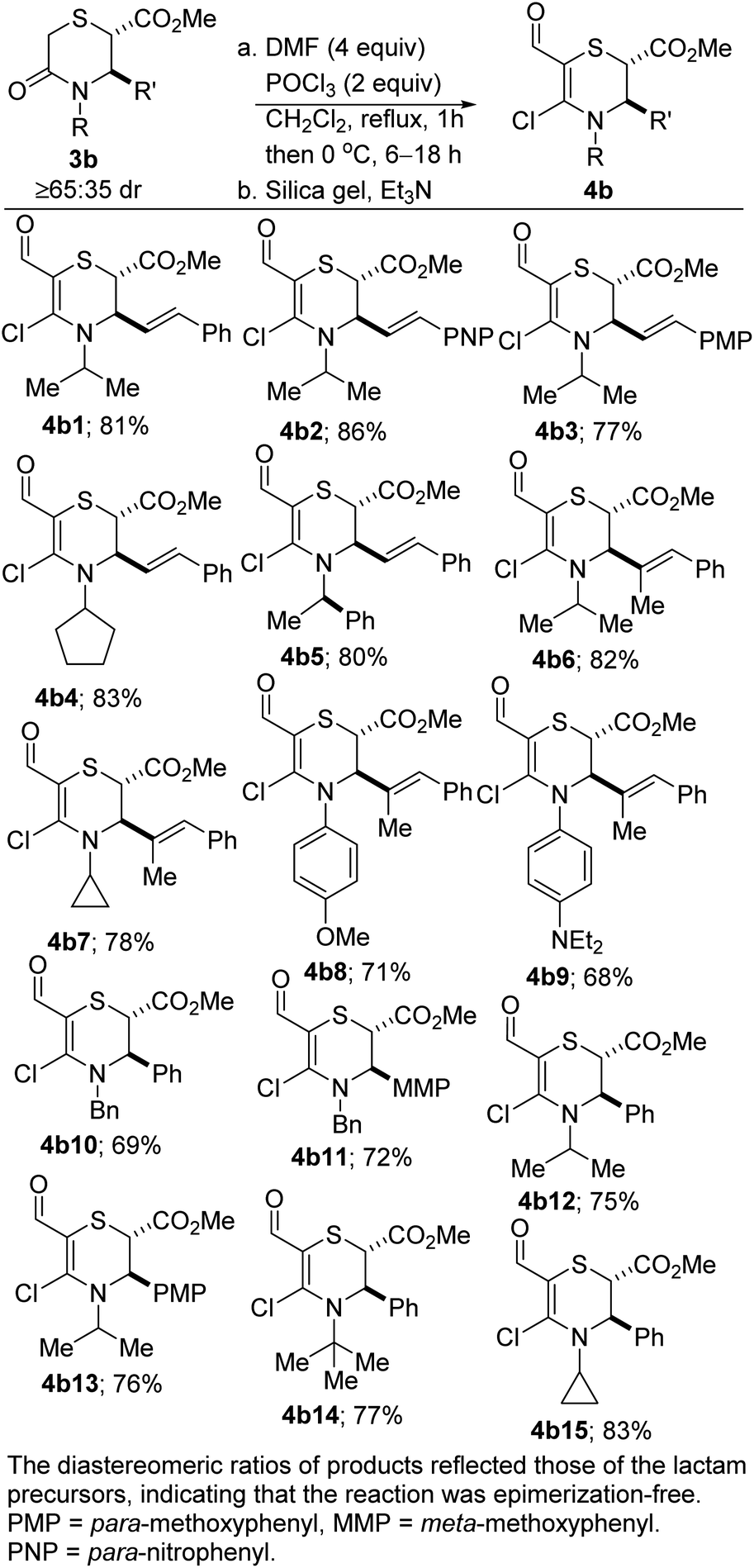 | ||
| Scheme 2 Synthesis of N,S-heterocyclic vinyl chlorides. | ||
With an assorted wardrobe of N,O- and N,S-heterocyclic vinyl chlorides in hand, we next sought efficient conditions for transition metal-free α-alkynylation. Despite the many merits of transition metal-catalyzed coupling of inert substrates of this class,24 one of our objectives was to avoid the drawbacks, including toxicity, cost, need for substrate-dependent designer ligands, and risk of product contamination. We also desired to utilize a sustainable solvent as the reaction medium for the alkynylation. After perusing the literature for some transition metal-free conditions,25 we were pleased to find that N,O-heterocyclic vinyl chlorides of type 4a undergo efficient alkynylation with triisopropylsilylacetylene to afford the products depicted in Scheme 3 (see 5a–g). These directing group-free conditions employ hindered and non-nucleophilic lithium tetramethylpiperidide (LiTMP) as the base and environmentally benign 2-MeTHF26 as the reaction medium. Differentially substituted N-aryl and N-alkyl vinyl chlorides have been surveyed and satisfactory outcomes were obtained in most cases. The ability to install a triisopropylsilyl motif bodes well for further diversification since its robust yet removable nature27 allows it to be employed as a terminal acetylene surrogate. The direct coupling of 4a with inherently inert and typically challenging alkyne coupling partners such as cycloalkenyl, chloroalkyl, and aryl acetylenes proceeds satisfactorily (see 5h–k). Congruent with 4a, the use of innately fragile N,S-heterocyclic vinyl chlorides of type 4b leads to productive alkynylation (see 5l–o).
 | ||
| Scheme 3 Transition metal-free alkynylation of N,O- and N,S-heterocyclic vinyl chlorides. | ||
Although the mechanistic underpinnings of the transformation are still under investigation, our working hypothesis is that homolytic cleavage of the C–Cl bond resident in 4a3 furnishes G, which reacts with the alkyne to generate vinyl radical H. H can then undergo termination with the free chlorine radical in solution to arrive at I. Finally, a base-promoted E2 reaction of newly formed vinyl chloride I affords the desired coupling product (Fig. 3).
 | ||
| Fig. 3 Tentative mechanism for transition metal-free alkynylation of activated vinyl chlorides. | ||
Our studies have revealed that N,O-heterocyclic vinyl chlorides of type 4a are amenable to transition metal-free cross-coupling with styrenes (Scheme 4). Electron-deficient, electron-rich, and electron-neutral styrenes couple satisfactorily to afford these Diels–Alder-suitable cross-conjugated dienes. The orthogonality of the coupling strategy is highlighted by observations that substrates bearing an aryl chloride motif undergo site-selective coupling (see 6b–e). The coupling presumably proceeds via a radical pathway.28 Although not surprising, it is disappointing that N,S-heterocyclic vinyl chlorides of type 4b are fragile under this tert-BuOK conditions and undergo undesirable ring-opening.
 | ||
| Scheme 4 Transition metal-free alkenylation of N,O-heterocyclic vinyl chlorides. | ||
As articulated previously, we were drawn toward vinyl chlorides of type 4 after recognizing that they harbor versatile functional handles that could facilitate fragment-based discovery. To elucidate this point, we have subjected aldehyde-bearing chloroenaminol ether 4a9 to the catalytic hydrogenation conditions described in Scheme 5. In the event, reduction of the styrenyl double bond was accompanied by hydrodeoxygenation of the aldehyde functionality and concomitant dehalogenation, affording methyl-bearing enamine 8 in high yield. This cascade reduction appears to be general given that alkyne 5c reacts in similar fashion to afford tris-alkylated dihydro-1,4-oxazine 9. The failure to reduce the endocyclic C–C double bond speaks to the challenges associated with reducing a tetrasubstituted enamine.
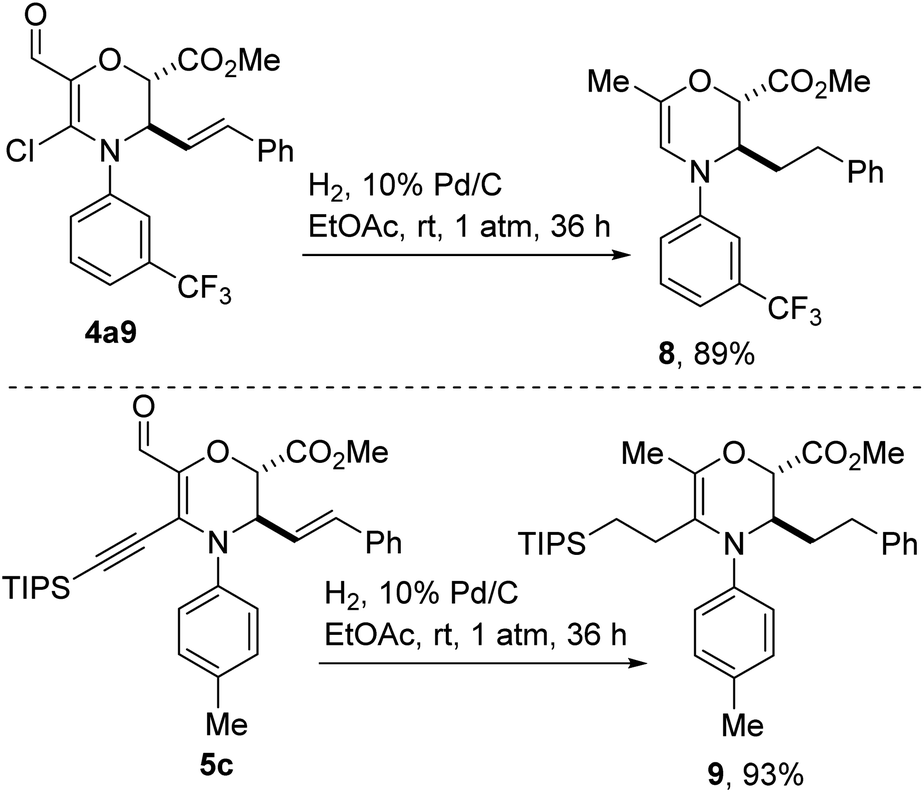 | ||
| Scheme 5 Cascade reduction of functionalized dihydro-1,4-oxazines. | ||
Medicinal chemists are becoming increasingly keen on escaping flatland in view of exploring 3D-structural space. Accordingly, bicyclic morpholines are viewed as valued targets for pharmaceutical companies. Pleasingly, Wittig olefination of enals of type 4a followed by a room temperature normal electron-demand Diels–Alder cycloaddition with tetracyanoethylene proceeds efficiently to afford oxazabicycles such as 7a and 7b, which bear three contiguous tetrasubstituted sp3 carbons (Scheme 6). These mild reaction conditions are noteworthy since they allow for the retention of the pharmaceutically pertinent α-chloroamino motif. Unambiguous establishment of the relative configuration of 7a/b has not been achieved at this point.
 | ||
| Scheme 6 Synthesis of bicyclic and tricyclic morpholines. | ||
Of note, when the Diels–Alder reaction is performed at slightly elevated temperatures, a cascade process ensues whereby the cycloaddition is accompanied by 1,4-elimination (see 10). A similar mode of reactivity is operative when diene 3a5 and bromomaleic anhydride are heated to 90 °C, leading to the formation of benzomorpholine 11 whose benzenoid portion is highly functionalized.
Conclusions
In summary, chemoselective Vilsmeier–Haack functionalization of morpholinonates and thiomorpholinonates has been successfully implemented, leading to the modular synthesis of N,O- and N,S-heterocyclic vinyl chlorides. The versatility of these densely populated organic chlorides has been amply demonstrated through transition metal-free late-stage alkynylation and alkenylation protocols, under environmentally benign conditions. Bicyclic and tricyclic morpholines have also been assembled through the interrogation of cross-conjugated 1,3-dienes in Diels–Alder reactions. The current work stands as an advance over existing methodologies given that the construction of these versatile intermediates can be achieved in modular step-economical, directing/protecting group-free, high-yielding, and transition metal-free fashions. The amenability of these highly functionalized intermediates to C–C bond forming transformations bodes well for future late-stage assembly of complex morpholine/thiomorpholine-bearing bioactive molecules.Experimental
All experiments involving air and moisture sensitive reagents were carried out under an inert atmosphere of nitrogen and using freshly distilled solvents. Column chromatography was performed on silica gel (230–400 mesh). Thin-layer chromatography (TLC) was performed using Silicycle Siliaplate™ glass backed plates (250 μm thickness, 60 Å porosity, F-254 indicator) and visualized using UV (254 nm) or KMnO4 stain. Unless otherwise indicated, 1H, 13C, and DEPT-135 NMR, and NOESY spectra were acquired using CDCl3 solvent at room temperature. Chemical shifts are quoted in parts per million (ppm). HRMS-EI+ data were obtained using either electronspray ionization (ESI) or electron impact (EI) techniques. High-resolution ESI was obtained on an LTQ-FT (ion trap; analyzed using Excalibur). High resolution EI was obtained on an Autospec (magnetic sector; analyzed using MassLynx).General procedure A
General procedure B
General procedure C
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20). Yield = 9.416 g, 74%. 1H NMR (400 MHz, chloroform-d) δ 9.70 (s, 1H), 7.67 (d, J = 8.1 Hz, 2H), 7.40–7.22 (m, 2H), 6.88 (d, J = 8.2 Hz, 2H), 6.63 (d, J = 15.8 Hz, 1H), 6.21 (dd, J = 15.9, 6.5 Hz, 1H), 5.01 (s, 1H), 4.89 (d, J = 6.5 Hz, 1H), 3.62 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 180.4, 167.7, 142.2, 138.5, 135.2, 134.1, 132.8, 131.5, 130.3, 128.8, 127.2, 122.4, 119.6, 91.8, 74.6, 62.6, 52.7. HRMS calc. for C21H17ClINO4, 508.9891, found 509.9899.
:20). Yield = 9.416 g, 74%. 1H NMR (400 MHz, chloroform-d) δ 9.70 (s, 1H), 7.67 (d, J = 8.1 Hz, 2H), 7.40–7.22 (m, 2H), 6.88 (d, J = 8.2 Hz, 2H), 6.63 (d, J = 15.8 Hz, 1H), 6.21 (dd, J = 15.9, 6.5 Hz, 1H), 5.01 (s, 1H), 4.89 (d, J = 6.5 Hz, 1H), 3.62 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 180.4, 167.7, 142.2, 138.5, 135.2, 134.1, 132.8, 131.5, 130.3, 128.8, 127.2, 122.4, 119.6, 91.8, 74.6, 62.6, 52.7. HRMS calc. for C21H17ClINO4, 508.9891, found 509.9899.Note: all other N,O-heterocyclic vinyl chlorides depicted in Scheme 1 were prepared as described above. Spectroscopic data can be found in the ESI.†
:20). Yield = 296.3 mg, 81%, oily substance.1H NMR (400 MHz, chloroform-d) δ 9.81 (s, 1H), 7.44–7.28 (m, 5H), 6.49 (dd, J = 32.9, 15.7 Hz, 1H), 6.16 (dd, J = 15.8, 6.8 Hz, 1H), 4.94 (ddd, J = 6.9, 2.9, 1.1 Hz, 1H), 4.85 (p, J = 6.7 Hz, 1H), 3.88 (dd, J = 2.8, 1.0 Hz, 1H), 3.71 (s, 3H), 1.42–1.16 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 185.5, 185.0, 169.4, 142.4, 135.6, 133.8, 132.7, 128.8, 128.7, 128.6, 128.5, 128.4, 126.9, 126.8, 126.4, 122.5, 102.5, 56.8, 56.4, 53.1, 52.9, 52.6, 47.9, 44.6, 22.3, 21.3, 20.7, 19.6. HRMS calc. for C18H20ClNO3S 365.0852, found 365.0855.Note: all other N,S-heterocyclic vinyl chlorides depicted in Scheme 2 were prepared as described above. Spectroscopic data can be found in the ESI.†
:5). Yield = 221 mg, 79%, 95:5 dr. 1H NMR (400 MHz, chloroform-d) δ 9.68 (s, 1H), 7.41–7.22 (m, 6H), 7.15–7.06 (m, 2H), 6.89–6.78 (m, 2H), 6.56 (d, J = 15.8 Hz, 1H), 6.27 (dd, J = 15.8, 7.7 Hz, 1H), 4.99 (d, J = 1.7 Hz, 1H), 4.73 (dt, J = 7.6, 1.4 Hz, 1H), 3.78 (t, J = 3.3 Hz, 7H), 0.91 (d, J = 4.3 Hz, 21H). 13C NMR (101 MHz, CDCl3) δ 180.60, 168.30, 158.60, 138.54, 136.50, 135.62, 133.9, 128.7, 128.5, 127.8, 126.9, 123.5, 114.3, 104.5, 95.2, 74.8, 62.4, 55.6, 52.8, 18.4, 11.0. HRMS calc. for C33H41NO5Si, 559.2754, found 559.2759.Note: all other enynes depicted in Scheme 3 were prepared as described above. Spectroscopic data can be found in the ESI.†
:20). Yield = 103.8 mg, 83%, 95:5 dr. 1H NMR (400 MHz, CDCl3) δ 9.78 (s, 1H), 7.79 (d, J = 14.2 Hz, 1H), 7.53–7.02 (m, 13H), 6.99 (d, J = 14.2 Hz, 1H), 6.73 (d, J = 12.5 Hz, 1H), 6.49 (dd, J = 12.6, 1.3 Hz, 1H), 5.08 (dd, J = 5.8, 1.3 Hz, 1H), 4.78 (d, J = 1.3 Hz, 1H), 3.75 (s, 3H), 2.22 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 180.6, 180.5, 167.9, 138.6, 134.2, 129.6, 128.9, 128.8, 127.3, 127.2, 126.9, 126.6, 122.7, 122.4, 74.7, 74.7, 64.2, 63.9, 52.8, 52.8, 21.4. HRMS calc. for C30H26ClNO4, 499.1550, found 499.1555.Note: all other dienes depicted in Scheme 4 were prepared as described above. Spectroscopic data can be found in the ESI.†
:15). Yield = 90.2 mg, 89%, 95:5 dr. 1H NMR (400 MHz, chloroform-d) δ 7.42–7.29 (m, 6H), 7.14–6.71 (m, 3H), 5.63 (s, 1H), 4.82 (d, J = 1.7 Hz, 1H), 4.46–4.35 (m, 1H), 3.45 (s, 3H), 2.97–2.76 (m, 2H), 2.15–2.00 (m, 5H). 13C NMR (101 MHz, CDCl3) δ 169.1, 146.5, 140.7, 136.4, 132.1, 131.8, 131.5, 129.8, 128.7, 128.5, 128.4, 126.4, 125.6, 122.9, 117.4, 115.4, 110.7, 110.7, 102.0, 77.5, 77.2, 76.8, 75.3, 54.1, 51.9, 32.5, 31.0, 17.6. HRMS calc. for C22H22F3NO3, 405.1552, found 405.1557.Note: enamine 9 was prepared as described above. Spectroscopic data can be found in the ESI.†
:15). Yield = 264.0 mg, 88%, 90:10 dr. 1H NMR (400 MHz, CDCl3) δ 7.56–7.16 (m, 14H), 6.75 (d, J = 8.3 Hz, 1H), 6.29 (ddd, J = 57.5, 15.9, 6.7 Hz, 3H), 5.83–5.76 (m, 1H), 5.13 (d, J = 1.5 Hz, 1H), 4.72 (d, J = 1.5 Hz, 1H), 3.91 (s, 3H), 3.87 (d, J = 10.6 Hz, 1H), 2.23 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 168.3, 166.9, 153.5, 153.1, 141.3, 137.1, 136.6, 134.6, 134.5, 132.2, 129.9, 129.4, 129.2, 128.9, 128.6, 127.2, 126.8, 125.3, 118.8, 116.2, 114.8, 113.1, 112.7, 110.7, 86.6, 85.8, 67.2, 67.0, 54.0, 21.4. HRMS calc. for C35H26ClN5O3, 599.1724, found 599.1729.Note: bicyclic morpholine 7b was prepared as described above. Spectroscopic data can be found in the ESI.†
:20). Yield = 203.7 mg, 79%. 1H NMR (400 MHz, chloroform-d) δ 7.70–7.50 (m, 6H), 7.54–7.44 (m, 2H), 7.43–7.34 (m, 2H), 6.78 (s, 1H), 5.27 (d, J = 1.5 Hz, 1H), 5.16 (s, 1H), 4.09 (p, J = 8.3 Hz, 1H), 3.92 (s, 3H), 2.02–1.94 (m, 1H), 1.72–1.57 (m, 3H), 1.46–1.25 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 167.73, 166.69, 154.91, 153.84, 134.49, 133.84, 132.36, 129.75, 129.44, 129.32, 128.86, 126.42, 115.29, 114.51, 113.84, 113.16, 112.52, 78.65, 77.42, 77.11, 76.79, 62.68, 60.95, 53.91, 31.00, 30.11, 22.93, 22.73. HRMS calc. for C31H25N5O3, 515.1957, found 515.1954.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to Central Washington University for financial support through startup funds to T. K. B. The School of Graduate Studies and Research is thanked for partial support of this work through a Faculty Research Award to T. K. B. and the graduate fellowship to A. O. F. V. S. thanks the McNair Scholars Program for a research fellowship.Notes and references
- (a) F. J. R. Rombouts, G. Tresadern, O. Delgado, C. Martinez-Lamenca, M. Van Gool, A. Garcia-Molina, S. A. Alonso de Diego, D. Oehlrich, H. Prokopcova, J. M. Alonso, N. Austin, H. Borghys, S. Van Brandt, M. Surkyn, M. De Cleyn, A. Vos, R. Alexander, G. Macdonald, D. Moechars, H. Gijsen and A. A. Trabanco, J. Med. Chem., 2015, 58, 8216–8235 CrossRef CAS; (b) T. J. Sindhu, D. Paul, M. Chandran, A. R. Bhat and K. Krishnakumar, World J. Pharm. Pharmaceut. Sci., 2014, 3, 1655–1662 CAS; (c) V. A. Pal'chikov, Russ. J. Org. Chem., 2013, 49, 787–814 CrossRef; (d) J. W. Corbett, S. S. Ko, J. D. Rodgers, L. A. Gearhart, N. A. Magnus, L. T. Bacheler, S. Diamond, S. Jeffrey, R. M. Klabe, B. C. Cordova, S. Garber, K. Logue, G. L. Trainor, P. S. Anderson and S. K. Erickson-Viitanen, J. Med. Chem., 2000, 43, 2019–2030 CrossRef CAS; (e) J. Nozulak, J. M. Vigouret, A. L. Jaton, A. Hofmann, A. R. Dravid, H. P. Weber, H. O. Kalkman and M. D. Walkinshaw, J. Med. Chem., 1992, 35, 480–489 CrossRef CAS.
- J. Kalepu and S. Katukojvala, Angew. Chem., Int. Ed., 2016, 55, 7831–7835 CrossRef CAS.
- J. A. Varela and C. Saa, Synthesis, 2016, 48, 3470–3478 CrossRef CAS.
- (a) S.-Y. Hsieh and J. W. Bode, ACS Cent. Sci., 2017, 3, 66–72 CrossRef CAS; (b) M. U. Luescher and J. W. Bode, Angew. Chem., Int. Ed., 2015, 54, 10884–10888 CrossRef CAS; (c) M. U. Luescher, C.-V. T. Vo and J. W. Bode, Org. Lett., 2014, 16, 1236–1239 CrossRef CAS; (d) W.-Y. Siau and J. W. Bode, J. Am. Chem. Soc., 2014, 136, 17726–17729 CrossRef CAS; (e) C.-V. T. Vo and J. W. Bode, J. Org. Chem., 2014, 79, 2809–2815 CrossRef CAS; (f) C.-V. T. Vo, G. Mikutis and J. W. Bode, Angew. Chem., Int. Ed., 2013, 52, 1705–1708 CrossRef CAS.
- L. Bagnoli, C. Scarponi, M. G. Rossi, L. Testaferri and M. Tiecco, Chem.–Eur. J., 2011, 17, 993–999 CrossRef CAS.
- (a) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257–8322 CrossRef CAS; (b) J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Mueller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9052–9067 CrossRef CAS.
- (a) H. Ehrhorn and M. Tamm, Chem.–Eur. J., 2019, 25, 3190 CAS; (b) U. H. F. Bunz, Chem. Rev., 2000, 100, 1605 CrossRef CAS; (c) U. H. F. Bunz, K. Seehafer, M. Bender and M. Porz, Chem. Soc. Rev., 2015, 44, 4322 RSC; (d) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783 CrossRef CAS; (e) X. Ren, G. Li, S. Wei and H. Du, Org. Lett., 2015, 17, 990 CrossRef CAS; (f) A. Ahlers, T. de Haro, B. Gabor and A. Furstner, Angew. Chem., Int. Ed., 2016, 55, 1406 CrossRef CAS; (g) P. M. Cromm, S. Schaubach, J. Spiegel, A. Furstner, T. N. Grossmann and H. Waldmann, Nat. Commun., 2016, 7, 11300 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467 CrossRef.
- For recent and representative examples, see: (a) E. Fernandez, M. Rivero Crespo, A. I. Dominguez, P. Rubio-Marques, J. Oliver-Meseguer, L. Liu, M. Cabrero-Antonino, R. Gavara, J. C. Hernandez-Garrido, M. Boronat, A. Leyva-Perez and A. Corma, J. Am. Chem. Soc., 2019, 141, 1928 CrossRef CAS; (b) T. Wang, J. Huang, H. Lv, Q. Fan, L. Feng, Z. Tao, H. Ju, X. Wu, S. L. Tait and J. Zhu, J. Am. Chem. Soc., 2018, 140, 13421 CrossRef CAS; (c) W. Liu, L. Li and C.-J. Li, Nat. Commun., 2015, 6, 6526 CrossRef CAS.
- H. Braunstein, S. Langevin, M. Khim, J. Adamson, K. Hovenkotter, L. Kotlarz, B. Mansker and T. K. Beng, Org. Biomol. Chem., 2016, 14, 8864–8872 RSC.
- D. Dar'in, O. Bakulina, M. Chizhova and M. Krasavin, Org. Lett., 2015, 17, 3930–3933 CrossRef.
- B. K. Corkey, S. T. Heller, Y.-M. Wang and F. D. Toste, Tetrahedron, 2013, 69, 5640–5646 CrossRef CAS.
- A. Oppedisano, C. Prandi, P. Venturello, A. Deagostino, G. Goti, D. Scarpi and E. G. Occhiato, J. Org. Chem., 2013, 78, 11007–11016 CrossRef CAS.
- D. P. Bassler, L. Spence, A. Alwali, O. Beale and T. K. Beng, Org. Biomol. Chem., 2015, 13, 2285–2292 RSC.
- C. Amatore, E. Carre, A. Jutand, H. Tanaka, Q. Ren and S. Torii, Chem.–Eur. J., 1996, 2, 957 CrossRef CAS.
- For evidence of ring-opening of this class of compounds, see: A. O. Farah, M. Rabah and T. K. Beng, RSC Adv., 2020, 10, 22454–22459 RSC.
- B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef CAS.
- P. K. Prasad, R. N. Reddi and A. Sudalai, Org. Lett., 2016, 18, 500–503 CrossRef CAS.
- T.-S. Zhang, Y.-J. Xiong, W.-J. Hao, X.-T. Zhu, S.-L. Wang, G. Li, S.-J. Tu and B. Jiang, J. Org. Chem., 2016, 81, 9350–9355 CrossRef CAS.
- A. Deb, S. Manna, A. Modak, T. Patra, S. Maity and D. Maiti, Angew. Chem., Int. Ed., 2013, 52, 9747–9750 CrossRef CAS.
- D. T. Smith and J. T. Njardarson, Angew. Chem., Int. Ed., 2014, 53, 4278–4280 CrossRef CAS.
- K. Hovenkotter, H. Braunstein, S. Langevin and T. K. Beng, Org. Biomol. Chem., 2017, 15, 1217–1221 RSC.
- W. Su, T.-J. Gong, X. Lu, M.-Y. Xu, C.-G. Yu, Z.-Y. Xu, H.-Z. Yu, B. Xiao and Y. Fu, Angew. Chem., Int. Ed., 2015, 54, 12957–12961 CrossRef CAS.
- A. O. Farah, R. Archibald, M. J. Rodriguez, A. Moreno, B. Dondji and T. K. Beng, Tetrahedron Lett., 2018, 59, 3495–3498 CrossRef CAS.
- (a) V. P. Mehta and B. Punji, RSC Adv., 2013, 3, 11957–11986 RSC; (b) R. A. D. Arancon, C. S. K. Lin, C. Vargas and R. Luque, Org. Biomol. Chem., 2014, 12, 10–35 RSC; (c) J. F. Gustavino, M. E. Buden and R. A. Rossi, J. Org. Chem., 2014, 79, 9104 CrossRef.
- A. R. Alcantara and P. D. de Maria, Curr. Green Chem., 2018, 5, 88 Search PubMed.
- T. W. Greene Protective Groups in Organic Synthesis, Wiley Interscience, New York, 1981 Search PubMed.
- (a) C.-L. Sun, Y.-F. Gu, B. Wang and Z.-J. Shi, Chem.–Eur. J., 2011, 17, 10844–10847 CrossRef CAS; (b) M. Rueping, M. Leiendecker, A. Das, T. Poisson and L. Bui, Chem. Commun., 2011, 47, 10629–10631 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data. See DOI: 10.1039/d0ra06619b |
| This journal is © The Royal Society of Chemistry 2020 |