DOI:
10.1039/D0RA06611G
(Paper)
RSC Adv., 2020,
10, 34869-34884
Design and formulation of polymeric nanosponge tablets with enhanced solubility for combination therapy
Received
30th July 2020
, Accepted 6th September 2020
First published on 21st September 2020
Abstract
Three drugs namely caffeine, paracetamol, and aceclofenac are commonly used for treating various acute and chronic pain related ailments. These 3 drugs have varied solubility profiles, and formulating them into a single tablet did not have the desired dissolution profile for drug absorption. The objective of the present research was to tailor the drug release profile by altering drug solubility. This was achieved by loading the drug into nanosponges. Here, three-dimensional colloidal nanosponges were prepared using β-cyclodextrin with dimethyl carbonate as a cross-linker using the hot-melt compression method. The prepared nanosponges were characterized by FTIR, 1H NMR spectroscopy, DSC, XRPD studies and SEM. The FTIR and DSC results obtained indicated polymer-drug compatibility. The 1H NMR spectroscopy results obtained indicated the drug entrapment within nanosponges with the formation of the inclusion complex. XRPD studies showed that the loaded drug had changed crystalline properties altering drug solubility. SEM photographs revealed the porous and spongy texture on the surface of the nanosponge. Box–Behnken experimental design was adopted for the optimization of nanosponge synthesis. Among the synthesized nanosponges containing paracetamol, aceclofenac and caffeine, batch F3–P31, F3–A31 and F3–C31 were considered optimized. Their particle size was 185, 181 and 199 nm with an entrapment efficiency of 81.53, 84.96, and 89.28% respectively. These optimized nanosponges were directly compressed into tablets and were studied for both pre and post-compression properties including in vitro drug release. The prepared tablet showed desired drug dissolution properties compared to the pure drug. The above outcomes indicated the applicability of nanosponges in modulating the drug release with varied solubility for combination therapy.
Introduction
Nanosponges are colloidal structures that can encapsulate a wide variety of substances such as antineoplastic drugs, proteins and peptides, volatile oil, DNA, etc. Nanosponges have advantages over microsponges, because the diameter of the former is less than 1 μm and that of the latter is approximately 10–25 μm with a void size of 5–300 μm. Nanosponges are strong and stable up to temperature of 300 °C, while microsponges are stable up to temperature of 130 °C and are fragile.1 They are lipophilic in nature, and spread in water as a moving medium helping mask unpleasant flavors and shifting the compound's physical state from liquid to solid. Biodegradable polyesters such as polyglycolic acid (PGA) and cyclodextrin-dependent items measure the beneficial charge of the medication.2
The nanosponge is about the size of a virus, and a naturally degradable polyester providing a backbone to a substance (a scaffolding structure). In solution, the long polyester threads are combined with small molecules, called cross-linkers, which are preferred for certain polyester parts.3 They cross-connect the pieces of polyester to create a spherical shape, that has many spaces or cavities. These nanosponges are a series of novel nanoparticles, that are typically extracted from natural derivatives. They are porous, non-toxic and insoluble in water and organic solvents, as compared to the other nanoparticles.4
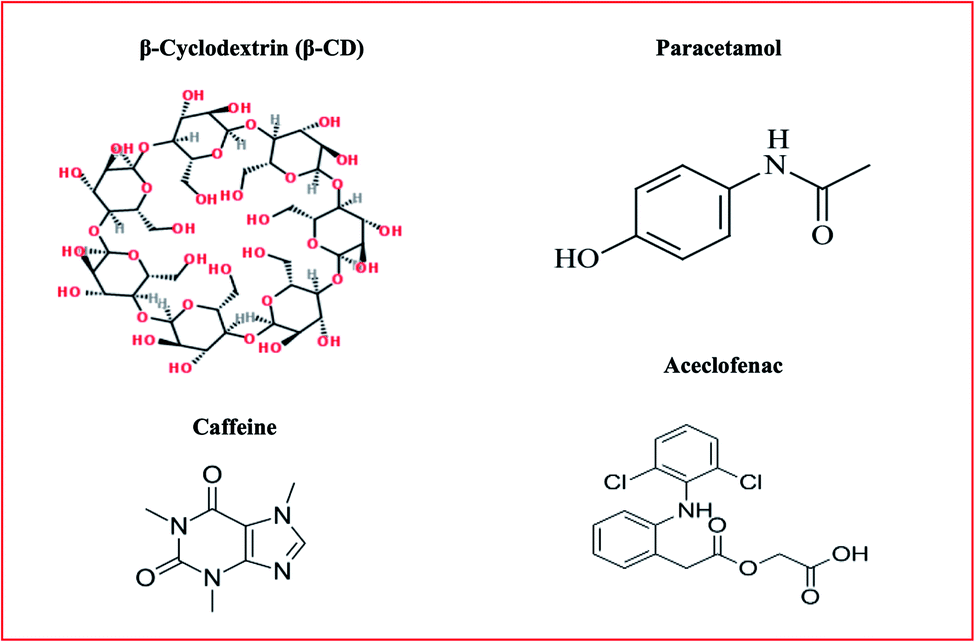
Nanosponges can be used as a tool for therapeutic ideas to improve the lipophilic aqueous solubility, maintain degradable molecules, and build pathways for the transmission of medicines for different routes of administration in addition to oral.5 Preparation is simple with slight manipulation, basic chemistry of polymers and appropriate use of cross-linkers. Hence, this approach is easy for commercial production scale-up. Nanosponges are soluble in water, but under sunlight, they are not biologically breakable. It can be used to cover up undesirable aromas, turning oils into solids. The chemical connections allow the nanosponges to attach favorably to the target site.6 Chemical structures of excipients used for nanosponge synthesis is depicted in Fig. 1.
 |
| | Fig. 1 Chemical structures of excipients used for nanosponge synthesis. | |
Cyclodextrin can produce molecules of size and polarity in line with its internal lipophilic cavity. Natural cyclodextrin cannot contain other hydrophilic compounds or large molecules. Therefore, many native cyclodextrin chemical modifications have been recorded to address those limitations.7 Among several solutions to the synthesis of cyclodextrin polymers using cross-linking agents, literature has suggested cyclodextrin polymers using cross-linking agents, or cyclodextrin-based nanosponges. Several cyclodextrin interlinks with the connectors throughout the cyclodextrin nanosponge (CD-NS). A central inner cavity serves as a pore of a natural sponge capable of drawing liquids into it, so-called “CD-NS”.8
Insoluble cyclodextrin polymers synthesized with parent cyclodextrin reactions have long been reported with cross-linkers such as epichlorohydrin, dialdehydes, diacyl chlorides, epoxides, etc., but the term CD-NS was first used by DeQuan Li and Min Ma in 1998.9 CD-NS shows very high efficiency in handling the poorly soluble molecules by incorporation and non-inclusion complexes. The existence of the lipophilic cavities of cyclodextrin monomers and the porous nature of hydrophilic CD-NS channels allows the introduction of a wide variety of compounds. Besides, the CD and cross-linker ratio needs to be changed to obtain a polymer with a specific product load capacity and an ideal release profile. Nanosponge has a unique property of forming nanosuspension, when vapor is dispersed.10
Paracetamol is world's most widely over-the-counter prescribed analgesic. It is the first step of medication in the WHO pain scale, and is widely prescribed through many international recommendations as first-line pharmacological treatment for acute and chronic painful conditions.11 On the other hand, aceclofenac is the diclofenac pro-drug, a non-steroidal anti-inflammatory drug (NSAID), widely used for treating both acute and chronic pain.12 There are no current systematic studies on its analgesic effects in acute post-operative pain. It functions by inhibiting the COX (cyclooxygenase) enzymes that further hinder prostaglandin's secretion. Furthermore, caffeine is a methylxanthine derivative, called IUPAC 3, 7-dihydro-1,3,7-trimethyl-1H-purine-2,6-dione. In many plants, it is a natural alkaloid that has the most important sources: guarana seeds, coffee seeds, tea leaves, cola nuts, and cacao beans. The regular use of caffeine are in tea, coffee, energy drinks, and other caffeine-based soft drinks. The caffeine is also used to enhance the solubility of the drug as aceclofenac and paracetamol is poorly soluble.13 Present study aims to develop a nanosponge tablet, that can increase the solubility of the class II drugs. There are no other combination of these triple drugs (paracetamol, aceclofenac and caffeine), which can be used for rapid onset of action against severe pain. Hence, these dosage form can be considered as the best anti-inflammatory drug.14
Experimental
Materials
Paracetamol, aceclofenac, caffeine (Merck® Specialties Private Limited, Mumbai, India), β-cyclodextrin (Mengzhou Hauxing, China), dimethyl carbonate (DMC) and cross povidone (Loba Chemie® Private Limited, Mumbai, India).
0.01 M phosphate buffer solution (PBS)
6.80 g of potassium dihydrogen phosphate was correctly weighed and dissolved in sufficient purified water to achieve an amount of 1000 ml. Glacial acetic acid was added to adjust the pH to 5.5.
Methods
Synthesis of β-cyclodextrin nanosponge. β-Cyclodextrin (β-CD) was cross-linked using cross-linking polymer namely DMC to prepare the nanosponge. Specific polymer-to-cross linker molar ratios were used for the nanosponge preparation. The polymer-to-cross linker mixture was taken as mentioned in Table 1 and allowed to react for 5–7 h at around 90 °C. To collect the resulting solid, the reaction mixture was kept aside to cool, and then filtered. Subsequently, the formed solid particles were broken down by gentle grinding, and Soxhlet installed extraction using ethanol for around 30 min to remove unreacted cross-linkers and other impurities. The reaction was conducted with an excess DMC, and the resulting nanosponge was deposited at 25 °C after purification until further use.8
Table 1 Ingredients used for preparing nanosponges with the different amount
| Ingredients |
F1 |
F2 |
F3 |
F4 |
| β-Cyclodextrin (mg) |
600 |
600 |
600 |
600 |
| Dimethyl carbonate (ml) |
1.2 |
2.4 |
4.8 |
6 |
After nanosponges synthesis, three varied formulations were prepared after drug loading into the nanosponge. Then, loaded nanosponge formulations were directly compressed into tablet formulation and finally characterized and evaluated.
Characterization of active ingredient
Fourier-transform infrared spectroscopy (FTIR). FTIR spectroscopic analysis was carried out for pure drugs, polymers used, and their physical mixture for the compatibility evaluation of drugs and polymers. FTIR spectrophotometer (8400S, Shimadzu®, Kyoto, Japan) was used to obtain the spectra.2
Differential scanning calorimetry (DSC). DSC studies was carried out for pure drugs, polymers used, and their physical mixture for the compatibility evaluation of drugs and polymers. It was conducted with Shimadzu®, DSC 60 thermal analyzer, Japan, using a liquid nitrogen cooling accessory.8
X-ray diffraction studies (XRD). Drug XRD patterns were recorded at ambient temperature, using an X-ray diffractometer (Rigaku®, Japan). Ni-filtered Cu Kα radiation at intervals 2 = 10–50°, 40 kV voltage, 20 mA current, and 2 s scanning speed of 0.01° s−1 were used to record diffraction patterns.15
Evaluation of nanosponge
Production yield (%). All formulations of the prepared nanosponge were correctly weighed and noted. The output yield for the nanosponge was then calculated using the following equation.16| | |
Production yield (%) = practical mass of nanosponge × 100/theoretical mass (polymer + drug)
| (1) |
Nanosponge drug loading. The aqueous nanosponge suspensions was taken, which is mentioned in the Table 2. Allowed to stir in a magnetic stirrer for 24 h.17 These suspensions were then centrifuged at 2000 rpm for 10 min to isolate the non-complexed content as a residual. Freeze-drying was applied to the colloidal supernatants to remove the drug-loaded NS.
Table 2 Different formulations used for preparing the drug-loaded nanosponges
| Ingredients |
FA1 |
FA2 |
FA3 |
| Paracetamol (mg) |
325 |
325 |
325 |
| Aceclofenac (mg) |
125 |
125 |
125 |
| Caffeine (mg) |
50 |
50 |
50 |
| Nanosponge (mg) |
263 |
525 |
1050 |
| Ethanol (ml) |
25 |
25 |
25 |
Design of experiment
Design Expert® (Version 12.0.2, Stat-Ease Inc., Minneapolis, MN, USA) software was used for the illustration of the response surface model by getting the combination of values. A three-factor, three-level Box–Behnken experimental design was adopted for the optimization of nanosponge synthesis. A design of three parts, each of two fully leveled factors, and a third factor set at zero level. The quadratic response surfaces were represented by the second-order polynomial model.18 The independent and dependent variables selected are presented in Table 3, and the results of the trial are generated by the quadratic equation:| | |
Y = b0 + b1X1 + b2X2 + b3X3 + b12X1X2 + b13X1X3 + b23X2X3 + b11X12 + b22X22 + b33X32
| (2) |
where,
Table 3 Independent and dependent variables for analysis
| Independent variables |
Dependent variables |
| Polymer concentration (mol) |
R1: Particle size (nm) |
| Crosslinker concentration (mol) |
R2: Entrapment efficiency (%) |
| Reaction time (h) |
|
Y is a determined result correlated with the combination each of the factor level; b0 is an intercept; b1 to b33 are coefficients of regression derived from Y's observed experimental values; X1, X2, and X3 are marked levels of independent variables.
The terms XiXj i = 1, 2 or 3 and j = 1, 2 or 3 and Xi2 (i = 1, 2 or 3) symbolize the relation and the quadratic terms, respectively. Three factors and just twelve runs, plus three replicates at a center point are required. Thereby, indicating the less time as well as energy consumption.19 Additionally, each factor is studied and coded at three basic levels. It does not concern the factors at extremely high or extremely low levels for avoiding the experiments in extreme conditions which may lead to undesirable results.
Checkpoint analysis and optimization of nanosponge
Provided that our reasoning was to maximize both nanosponge particle size and trap efficiency concurrently, multi-criteria decision analysis (MCDA) was used in the output data to merge the responses obtained of two responses into a single unified response system. It offers a systemic solution by requiring options to be chosen depending on several parameters, and thus overcoming the drawbacks of unstructured decision-making. The desired points were selected for each response based on importance, which may affect the characteristics of a set goal.20 Graphical optimization has also been followed post numerical optimization. Overlay plot, as well as desirability plot, were then constructed for investigating the relationship between factors and response. The optimum formula was selected based on the criteria of optimum particle size and maximum entrapment.
Evaluation of drug-loaded nanosponge
Drug content (%). Extracted from the prepared paracetamol, aceclofenac and caffeine formulation with 30 ml of ethanol containing 1 g of drug equivalent to 10 mg. This was taken in a volumetric flask and made upto 100 ml using pH 5.5 acetate buffer. Using UV spectrophotometer 1800, Shimadzu®, Japan, the absorption of the resulting solution was measured at different nm after appropriate dilutions.20 The formulation's drug content was calculated using the following equation:| | |
% Drug content = actual concentration of drug in the formulation × 100/theoretical concentration of drug
| (3) |
Entrapment efficiency. Accurately weighed drug-loaded nanosponges were crushed in a mortar and pestle. 5 ml of ethanol was added to the standard 100 ml flask and made up the volume using acetate buffer with pH 5.5. Mixture was kept aside for 1 h with regular shaking to remove if any undissolved particulate of nanosponge. It was then filtered and the filtrate absorbance was estimated at 243, 275, and 273 nm, after sufficient dilutions. Calibration curve was then plotted and the substance content was expressed as actual drug content in nanosponge. The efficiency percentage of nanosponge drug trapping was calculated using the following equation.21| | |
Drug entrapment efficiency (%) = experimental drug loading × 100/theoretical drug loading
| (4) |
Scanning electron microscope (SEM). SEM was used to report the surface morphology of nanosponges. Suitable tests were mounted on a double-sided adhesive tape stub made of aluminum. Firstly, the tape was firmly attached to the stub, and sample powder was carefully dispersed over its surface. To keep the specimens conductive, the stub with the substance was then sputter-coated with a thin layer of gold.22
1H NMR spectroscopy. To establish the formation of inclusion complex between drug and CD-NS, 1H NMR spectroscopy has been done. 1H NMR allows for a clear disparity between inclusion and any other possible external interaction with the large effects observed on protons inside the hydrophobic CD cavity (H-3 and H-5, with H-5, more pronouncedly affected), thus clearly proving the inclusion. Besides, the internal protons chemical environment varies, resulting in a change in the protons chemical shifts due to the effects of shielding or de-shielding.
Preparation of nanosponge loaded tablets. Nanosponge loaded with paracetamol, aceclofenac and caffeine is equivalent to 500 mg distributed equally to the weigh excipients. Cross povidone was taken in varied concentration for the better solubility (Table 4).23
Table 4 Tablets compression with the list of ingredients and excipient used
| Ingredients |
Quantity (mg) |
| Paracetamol, aceclofenac and caffeine loaded nanosponge |
525 |
| Cross povidone |
25 |
Characterization and evaluation of drug loaded nanosponge
Physical properties. The prepared formulations for nanosponge were inspected visually for their color and appearance (white colored nanosponge loaded with drug particles).
Particle size analysis. The mean particle size of the nanosponge was calculated using an optical microscope. A stage micrometer had been fitted to the microscope to calibrate the eye piece micrometer. One field estimated the diameter of 30 particles on average. The mean size of the particles was determined using the equation below.
Pre-formulation study. The pre-formulation study was carried out to show the good flow property and meeting the pharmacopeia specifications.24
Angle of repose. Friction forces were measured by the resting angle in a loose powder. That is an indication of the flow properties of the powder. When possible, it is defined as the maximum angle between the surface of the powder pile and the horizontal plane.| | |
Angle of repose = tan−1(2h/d)
| (6) |
Bulk density. The powder was poured into the weighing jar and the volume was noted. It is called sample weight, separated by untapped volume and transmitted as g cm−3.
Tapped density. The weighed powder amount was poured into the weighing jar, and the volume was measured. The cylinder tapped at a height of 10 cm on a hard surface for 100 times, until the gap volume was lowered and final reading was taken.
Carr's compressibility index. The Carr scale is an indication of a compressibility of the powder. It can be calculated using a formula that is mentioned below.| | |
C = 100 × (1 − bulk density/tap density)
| (7) |
Hausner's ratio. This displays the properties of the powder flow, and is expressed in the ratio of tapped and bulk mass. If the value is less than 1.25, this shows good flow.
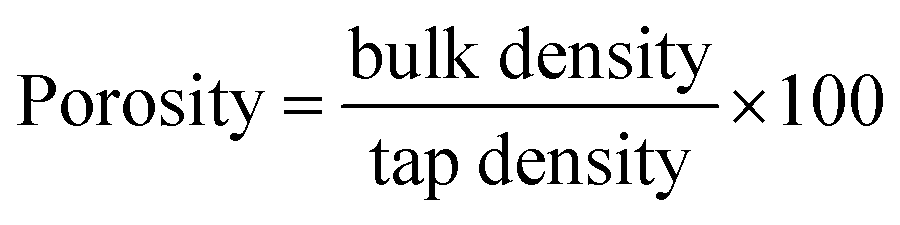
Porosity. The study of porosity was conducted to check the extent of the nanosponge. The tapped and untapped (bulk) densities were determined by marking a small cuvette with known volume, then inserting a small known powder mass into the cuvette (bulk density) and tapping it 50 times (tapped density) vertically against a padded benchtop. The mass was broken down by the initial and final quantities.19 True density has been calculated by dividing sample weight by volume of the sample. Nanosponge exhibits higher porosity compared to the parent polymer and co-polymers used to make the system, due to their porous nature. Percent porosity is given by the equation.| |
 | (8) |
Evaluation parameters of tablets dosage form
Friability test. Twenty tablets of each formulation were weighed and abrasive by employing for 4 min at 25 revolutions per min. The tablets were then measured and compared to their original weights and were obtained a percentage of friability.25| | |
% Friability = 1 − (final weight/initial weight) × 100
| (9) |
Tablet breaking force (hardness) test. The crushing strength was determined using Automatic Tablet Hardness Tester (Pfizer® Hardness Tester). From each formulation, 3 tablets were randomly selected, and the pressure at which each tablet crushed was registered.26
Average weight. Ten tablets were randomly selected, and individually measured. Average weight was then calculated, and equated of the actual tablet. Not more than two of the individual weights deviate by more than the limit. From the average weight number of tablet taken as 10 and none of the batch deviated by more than twice the percent.| | |
Average wt = total wt of 10 tablets in gram × 1000/10
| (10) |
In vitro disintegration time test. The time test for in vitro disintegration is a significant feature needed for tablets that are released immediately. The time of dosage forms to disintegrate must be within 25 min. The time for complete disintegration of the tablet was measured in min, with no observable mass remaining in the apparatus.27
In vitro dissolution. It was performed using the type II USP apparatus (ELECROLAB® TDT 06 T, Mumbai, India). At the pH 5.5 buffer, the dissolution medium consisting 900 ml of buffer held at 37 ± 0.5 °C and stirred at 50 RPM. Samples (10 ml) were removed at predetermined 5, 10, 15, 20, 25 and 30 min time intervals. Equal amount of the fresh dissolution medium, retained at the same temperature, was immediately replaced. The solution absorbance was measured against blank at approximately 243, 274 and 273 nm. It has been made clear that the test did not interfere with any of the ingredients used in the matrix formulation. The percentage of drug release from the prepared standard curve was measured.16
Release kinetics. Release kinetic studies was carried out using mathematical models for the cumulative drug release. The mathematical models used are zero, first, Higuchi, and Korsmeyer–Pepas model that was done by using BCP software.
Stability studies. Stability is characterized as the degree to which a commodity retains the same properties and characteristics it possessed at the time of its manufacture, within defined limits, and over its complete storage and usage duration.28 The optimized formulation was packed in a tightly closed container and held in an International Council of Harmonisation approved stability chamber at 40 ± 2 °C and 75 ± 5% RH for one month. The formulation was tested at periodic intervals before and after, for the improvement of appearance, pH, drug quality, and in vitro drug release.
Results and discussion
Compatibility study
Study on compatibility was performed to test for any specific interference between the drug and excipients used. Drug–excipient interaction was assessed by FTIR and DSC analysis.
FTIR analysis
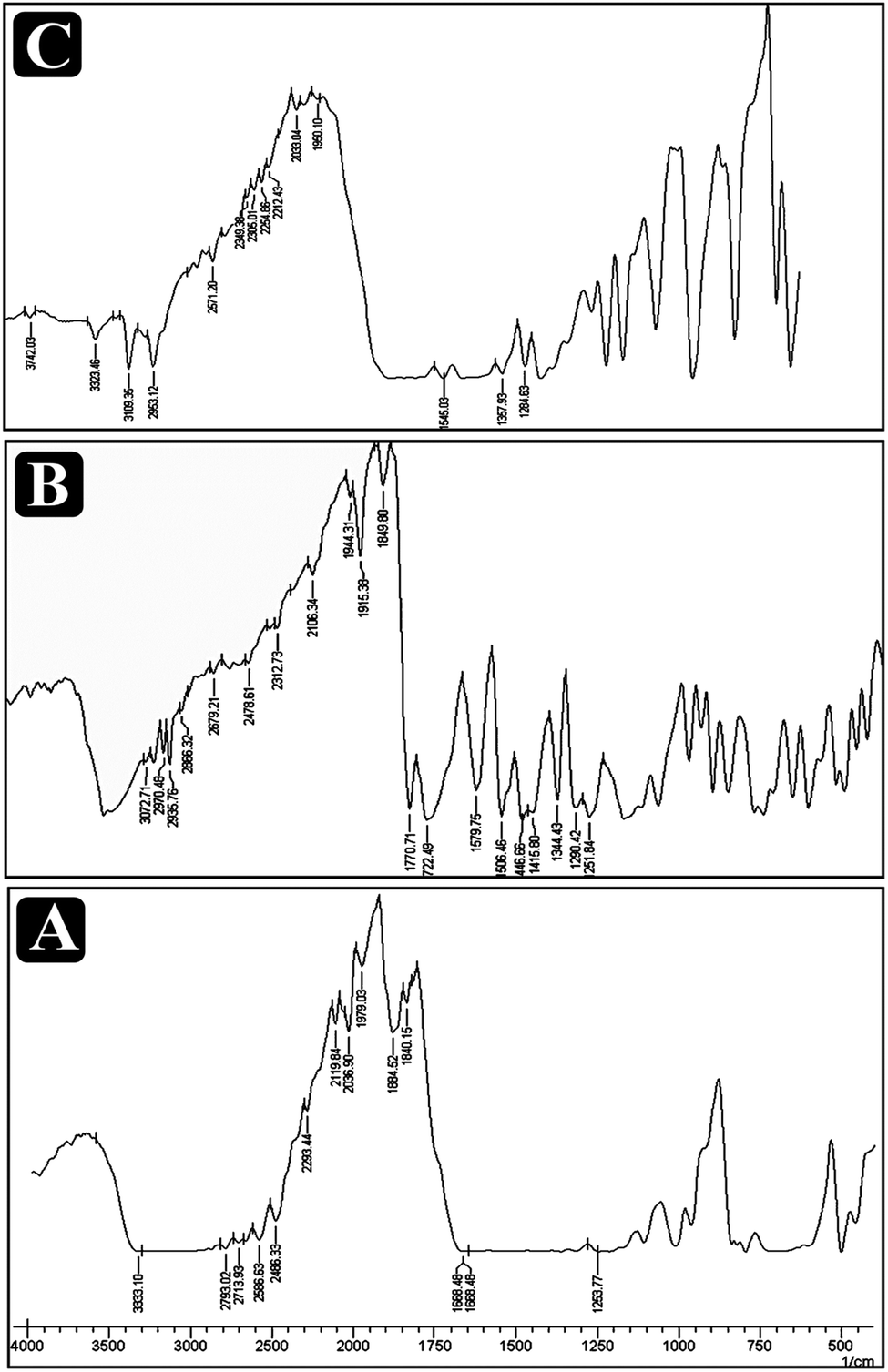
IR spectroscopic studies performed and results obtained are presented in Fig. 2. No newpeaks, major shift or loss of characteristic peaks was observed for drug–excipient physical mixture spectra, in comparision to pure drug spectra indicating drug–excipient compatibility with all the excipients. The drug and excipients interaction between the drug paracetamol, aceclofenac and caffeine was carried out with that of the excipients and the spectrum appeared found to be of paracetamol as vibrational peak for O–H and CH3 at 3326 and 3126 cm−1 and asymmetrical stretching C–H bond at 1507 cm−1 and C–C stretching takes place as 1435 cm−1. The mixture of paracetamol with the excipient's forms peak at 3437 cm−1, but there is no disappearing of the peaks. Aceclofenac has a characteristic stretching at 965.21 cm−1 O–H bond 3319.73 cm−1 at N–H stretching, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band at 1577.9 cm−1 with the addition of excipients there found to be no interaction with the peaks or disappearing but new peaks formed at 3496.63 cm−1. Caffeine has the functional group found for C–H and CH3 wavelength at 3134 cm−1 and 3850 cm−1, CO at 1690 cm−1, C–N at 1240 cm−1 with that of physical mixture found to form extra peaks at 3356 cm−1 but found to be no disappearing of the peak.
O band at 1577.9 cm−1 with the addition of excipients there found to be no interaction with the peaks or disappearing but new peaks formed at 3496.63 cm−1. Caffeine has the functional group found for C–H and CH3 wavelength at 3134 cm−1 and 3850 cm−1, CO at 1690 cm−1, C–N at 1240 cm−1 with that of physical mixture found to form extra peaks at 3356 cm−1 but found to be no disappearing of the peak.
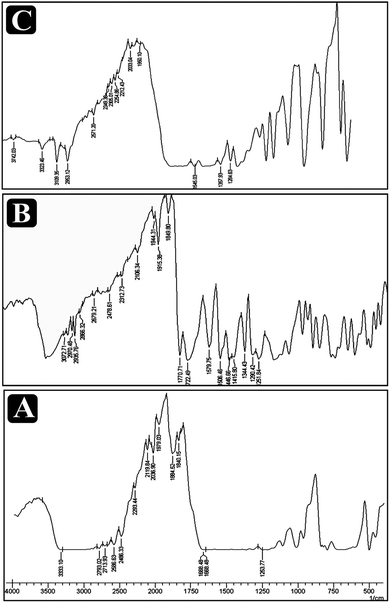 |
| | Fig. 2 Overlain FTIR spectra of paracetamol, aceclofenac and caffeine with the β-cyclodextrin and physical mixture. | |
DSC analysis
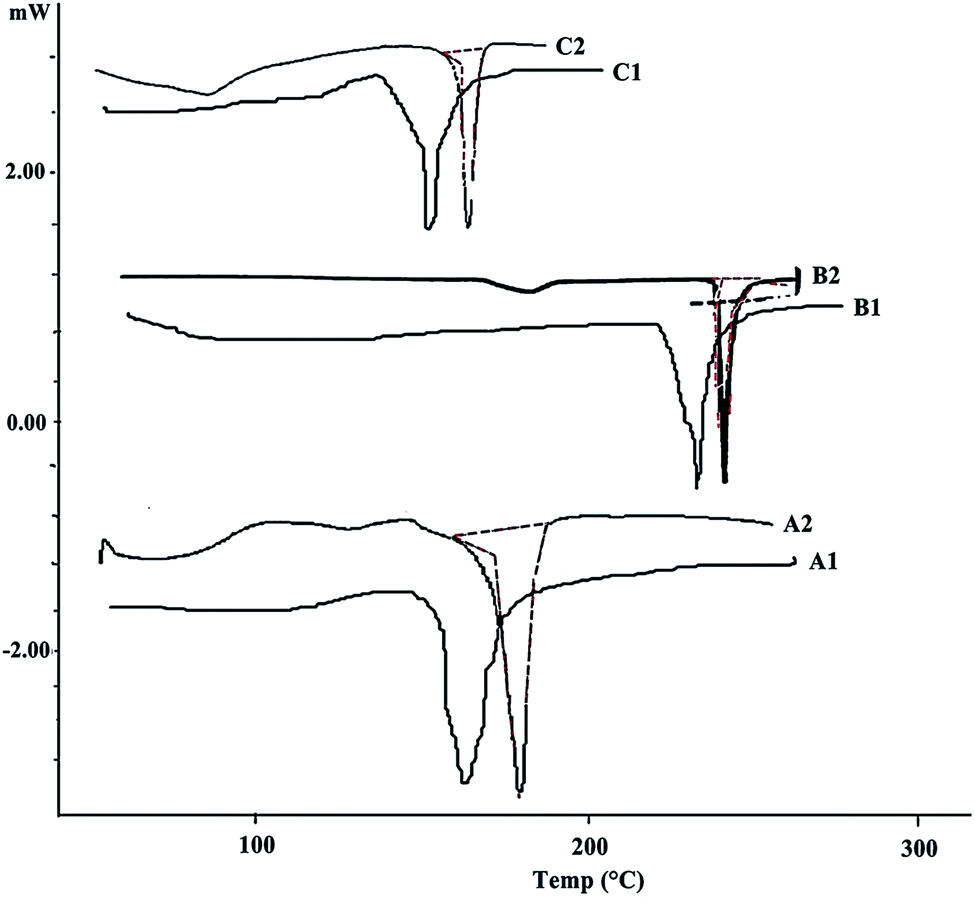
The overlaying DSC thermograms of paracetamol, aceclofenac, caffeine and β-CD, with their physical mixture are depicted in Fig. 3. Endotherm corresponding to melting of paracetamol, aceclofenac, caffeine and β-CD were observed in respective thermograms at 169 °C, 156.1 °C and 236 °C, respectively. Compatibility analysis via DSC revealed that paracetamol, aceclofenac, and caffeine dispersed in polymer, exhibited same thermal behavior as that of pure sample. As the corresponding endothermic peaks of the drug, as well as polymer were adequately retained in their physical mixture. It was inferred that drug did not interact while in contact with the polymer, suggesting good compatibility among them.
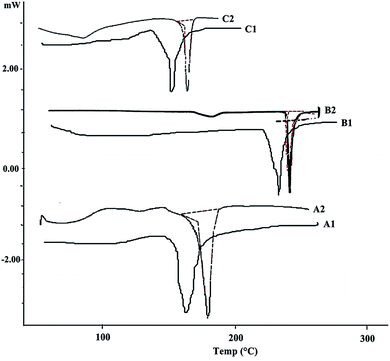 |
| | Fig. 3 Overlain DSC thermograms of (A1 and A2) paracetamol, physical mixture; (B1 and B2) aceclofenac, physical mixture; (C1 and C2) caffeine, physical mixture. | |
Synthesis of CD-NS
The synthesis of CD-NS involves three steps: (i) synthesis of CD-based nanosponge via optimum cross-linking of polymer using cross-linker, (ii) purification of nanosponge to remove unreacted cross-linker, and other chemical impurities, and (iii) drug loading into blank nanosponge. In this study, we have selected β-CD as polymer and DMC as a cross-linker. In the nanosponge synthesis, the ratio of polymer to cross-linker plays a crucial role. It should be such that, to lead the optimum cross-linking among CD molecule (neither to higher extent nor to lesser) and should also aid in the formation of rigid, nano-sized, stable nanosponge. The higher percent of polymer with respect to cross-linker results in incomplete cross-binding of CD cups. Consequently, resulting in the formation of drug-CD complex instead of nanosponge. Also, the inclusion and non-inclusion complexation of drugs in the resultant complex will be quite less. On the contrary, if the cross-linker concentration is excessively more to polymer; it leads to extremely high degree of cross-linking, that ultimately ends in a lack of binding sites on nanosponge for the drug molecule to bind. Hence, an optimum balance of the two is expected to attain nanosponge with superior drug entrapment and other best possible characteristics. Taking into consideration the underlying facts, trial and error experiments, and available literature, the ratios of polymer and cross-linker were selected. Further, for statistical validation and optimization of the same, the QbD approach was embraced by applying Box–Behnken Design (BBD). The nanosponge was prepared by the classical hot melt method as outlined in the Methodology section (Table 5).
Table 5 Loading of the drug into nanosponge based on the different ratios
| Ingredients |
FA1 |
FA2 |
FA3 |
| Paracetamol (mg) |
325 |
325 |
325 |
| Aceclofenac (mg) |
125 |
125 |
125 |
| Caffeine (mg) |
50 |
50 |
50 |
| Nanosponge (mg) |
263 |
525 |
1050 |
| Ethanol (ml) |
25 |
25 |
25 |
Design of experiment (DoE)
The coded and real values of implemented Box–Behnken design based independent and dependent variables are shown in Table 6 along with their levels, such as high, medium, and low. The experimental nature based on this mixture of the component has resulted in 15 separate nanosponge formulation batches (counting 3 center points in construction). As indicated, numerous nanosponge lots were prepared and then assessed for each of the responses. The responses observed were fit to 15 runs, and it has been noted that the best fit model was the quadratic model for the two dependent variables. The significance of the model with that of comparing with the other model for the analysis by analysis of variance (ANOVA). In polynomial equations, positive sign before the factor shows the linear correlation between response and factor, while the negative sign shows the inverse relation between the same. All the responses recorded for 24 runs and the relation of independent and dependent variables are presented in Table 7.
Table 6 Box–Behnken design variables for drug-loaded nanosponge
| Used level, actual (coded) |
| Factors |
Low |
Medium |
High |
| Independent variables |
| Polymer concentration (mg) (A) |
200 |
400 |
500 |
| Cross-linker concentration (ml) (B) |
400 |
800 |
1200 |
| Reaction time (h) (C) |
2.5 |
5 |
7.5 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Dependent variables |
| R1 = particle size (nm) |
|
|
|
| R2 = entrapment efficiency (%) |
|
|
|
Table 7 Different formulations with dependent and independent variables used for the analysis
| Formulation code |
Polymer:cross-linker |
Drug:nanosponge |
Particle size (nm) |
% Drug content (%) |
Entrapment efficiency (%) |
| F1–P11 |
1:2 |
1:1 |
220 |
50 |
60 |
| F1–P12 |
1:2 |
264 |
60.8 |
65 |
| F1–A11 |
1:1 |
200 |
53 |
50 |
| F1–A12 |
1:2 |
214 |
68.9 |
40 |
| F1–C11 |
1:1 |
210 |
74 |
58 |
| F1–C12 |
1:2 |
218 |
70.1 |
60 |
| F2–P21 |
1:4 |
1:1 |
226 |
78 |
74 |
| F2–P22 |
1:2 |
244 |
70.6 |
56 |
| F2–A21 |
1:1 |
236 |
80 |
68 |
| F2–A22 |
1:2 |
224 |
76.4 |
52 |
| F2–C21 |
1:1 |
226 |
70.2 |
49 |
| F2–C22 |
1:2 |
212 |
75.3 |
59 |
| F3–P31 |
1:8 |
1:1 |
185 |
81.53 |
76 |
| F3–P32 |
1:2 |
200 |
79 |
60 |
| F3–A31 |
1:1 |
181 |
84.96 |
79 |
| F3–A32 |
1:2 |
220 |
80 |
74 |
| F3–C31 |
1:1 |
199 |
89.28 |
78 |
| F3–C32 |
1:2 |
280 |
78 |
60 |
| F4–P41 |
1:10 |
1:1 |
222 |
69.8 |
69 |
| F4–P42 |
1:2 |
242 |
68.1 |
62 |
| F4–A41 |
1:1 |
232 |
70 |
55 |
| F4–A42 |
1:2 |
202 |
78.9 |
58 |
| F4–C41 |
1:1 |
245 |
72 |
65 |
| F4–C42 |
1:2 |
218 |
63.2 |
58 |
Characterization-cum-optimization of paracetamol, aceclofenac and caffeine nanosponge
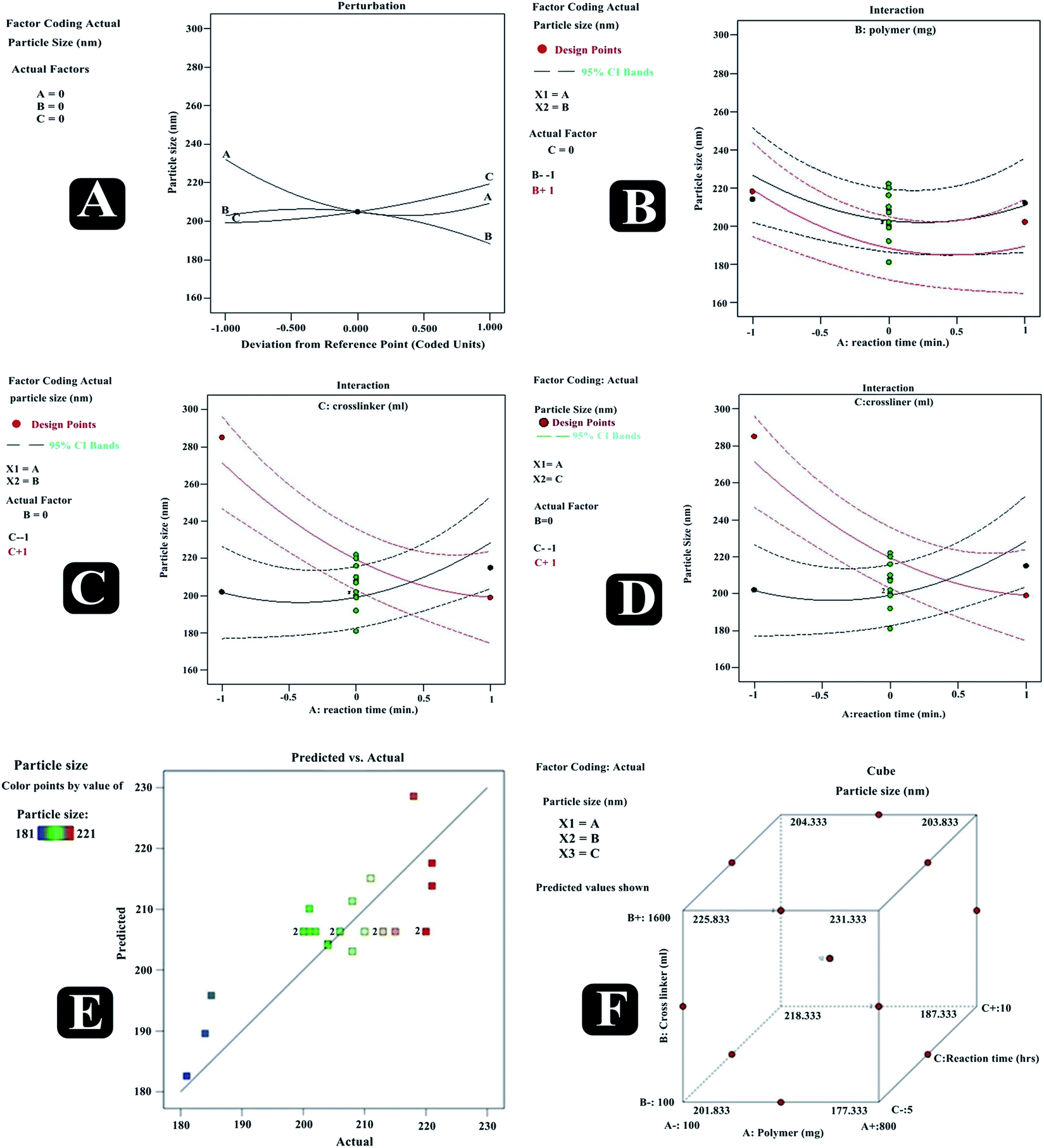
Particle size (R1) and zeta potential measurement. Particle size of nanosponge is a key factor for their performance, as it significantly impacts the rate and extent of drug release, and consequently its absorption. Smaller particle size improves the drug release as it offers a larger interfacial area for diffusion of the drug. Particle size of all suggested trial runs were assessed using size analyzer working on the basis of DLS technique, and obtained data were fed to DoE. Numerical and graphical analysis of the data was done via software. The derived equation from the best fit mathematical model that can be related to the response R1 and factors (A, B, and C) was R1 = +403.66 + 17.75A + 10.00B + 1.50C + 2.00AB + 1.00AC − 0.50BC − 15.83A2 + 21.66B2 − 16.83C2. Diverse graphs were also plotted for predicting the relative influence of factors on response R1. The 3D response surface (RE1, RE2, and RE3) and contour plots (CE1, CE2, CE3) of different factor combinations, affecting response particle size (R1) are shown in Fig. 4. It was evident from the polynomial equation as well as graphs, that polymer concentration had a much dominant positive or direct influence on particle size (R1), followed by cross-linker concentration, whereas, quite lesser influence existed between R1 and the reaction time.
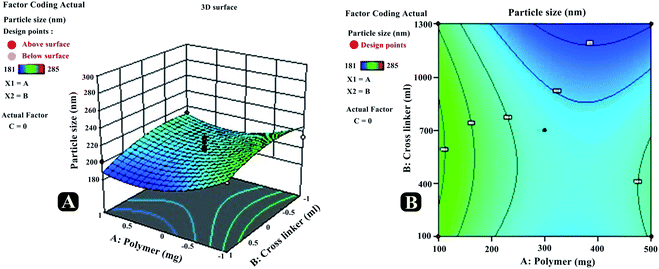 |
| | Fig. 4 Three-dimensional response surface plots (RE1, RE2, RE3) and contour plots (CE1, CE2, CE3) revealing relative effects of independent variables (A: polymer concentration; B: cross-linker concentration and C: reaction time) on dependent variable – particle size of paracetamol, aceclofenac and caffeine nanosponges. | |
With an increase in the number of RSM factors, response surface visualization becomes tedious with graphical tools. Under such circumstances, ‘perturbation’ and ‘interaction’ (special forms of response plots) are used for RSM data. Perturbation plot compares the effect of all factors at a particular point in RSM design space, and on the perturbation plot, a curvature or steep slope in an input variable shows a relatively higher sensitivity of response. Whereas, using interaction plot, the effect of a combination of independent variables can be traced independently by keeping other all variables constant. Fig. 5 presents predicted vs. actual, perturbation, factors interaction, and cube graphs for particle size (R1). A higher degree of linearity was reflected by predicted vs. actual plot for all the trial run formulations. Prominent positive impact on response R1 was noted in the case of factor A and factor B when deviated from −1 to +1 (Table 6). Also, a slight positive effect on response R1 had been noted by the factor C. Moreover, all interaction plots (revealing combined independent variables effect on a response) further established the impact of factor combinations on particle size and were in good agreement with outcomes of the polynomial equation.
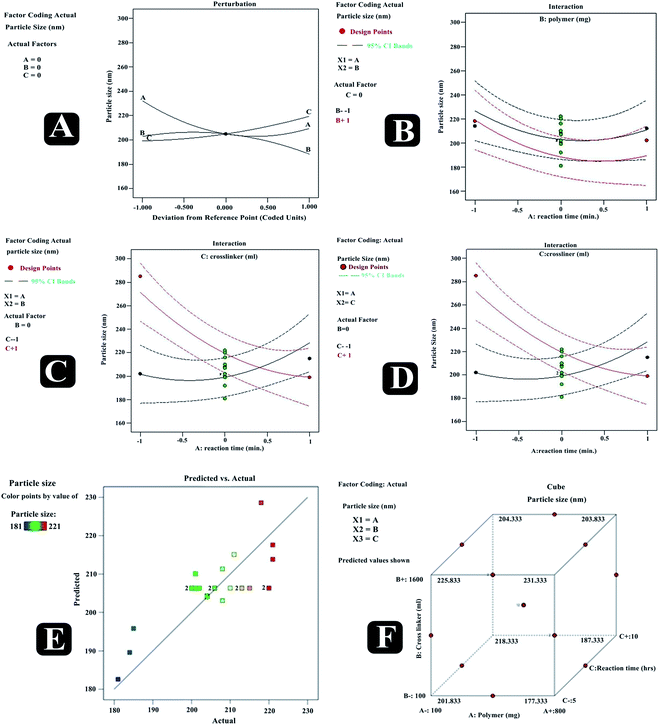 |
| | Fig. 5 Predicted vs. actual (A), perturbation (B), interaction (C–E) and cube (F) plots revealing relative effects of independent variables (A: polymer concentration; B: cross-linker concentration and C: reaction time) on dependent variable – particle size of paracetamol, aceclofenac and caffeine nanosponges. | |
The results have depicted that with an increase in polymer concentration from 1 to 3 (mol), the mean particle size increased from 353 to 440 nm. This could be attributed to the fact that at higher concentrations, the mixed polymeric matrix leads to the formation of cross-linked aggregates; which consequently fallout to increase nanosponge particle size. Also at extreme higher polymer concentration devoid of appropriate cross-linker amount, there may be the only formation of CD complexes with drug instead of nanosponge (having size in micro or macro range). As the cross-linker amount increases in the reaction mix, it facilitates cross-linking and accelerates the formation of larger size cross-linked polymeric agglomerates. The reaction time is equally crucial as it directly affects the extent or degree of cross-linking; hence affect particle size in a direct mode. ANOVA of equation has shown model F value 36.68 and P-value <0.0005; indicating the significance of the model. Furthermore, predicted R-squared 0.9618 was in a reasonable concurrence with adjusted R-squared 0.9582, concluding that the obtained polynomial equation signifies the good fit of response variables at diverse levels of factors (Table 6).
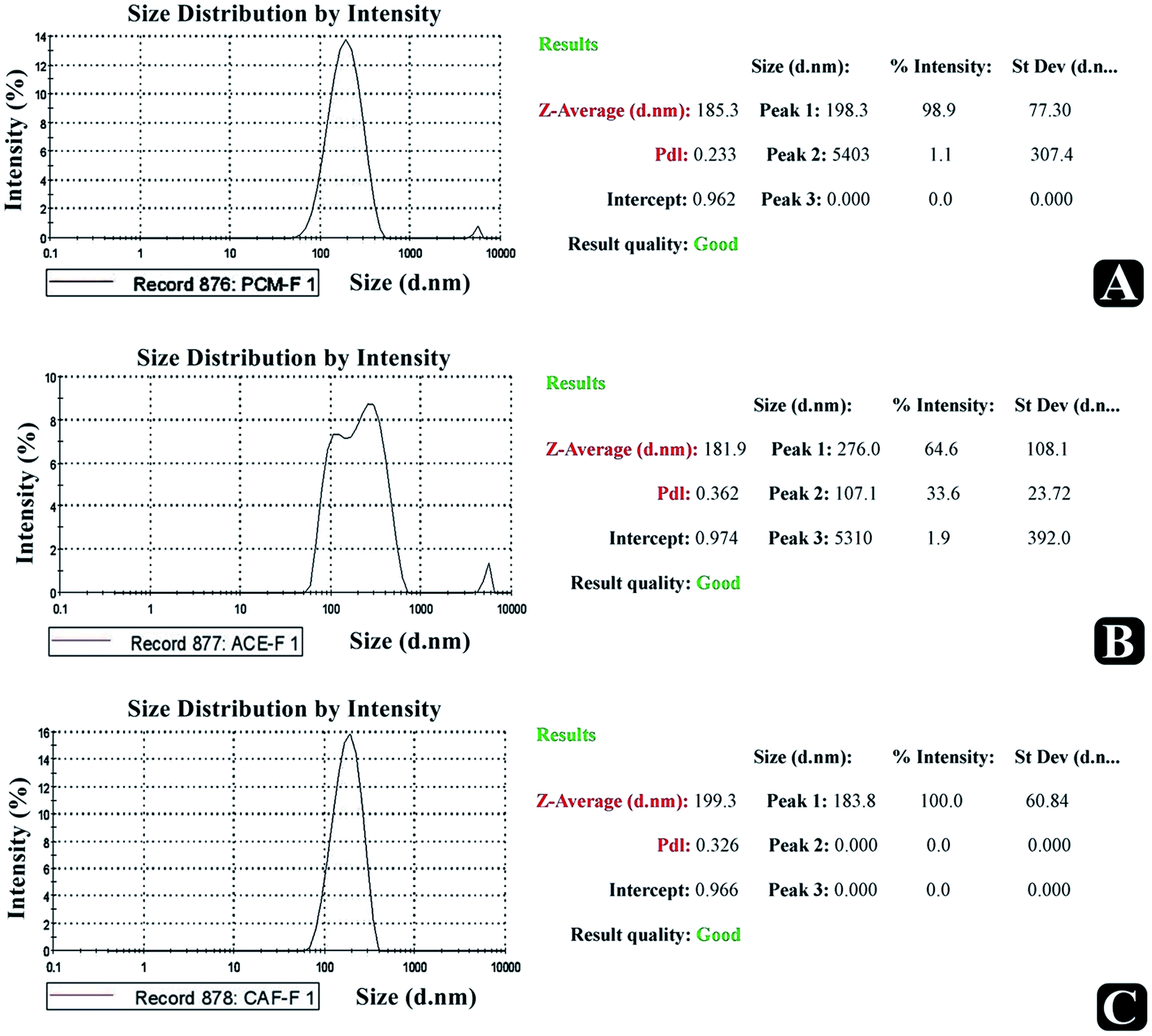
To find out the particle size and size distribution pattern, optimized paracetamol, aceclofenac, and caffeine loaded nanosponge were subjected to particle size analysis. The recorded size distribution pattern is presented in Fig. 6. The average diameter and PDI of the optimized paracetamol, aceclofenac and caffeine nanosponge were 185, 181 and 183 nm, 0.923 as PDI respectively.
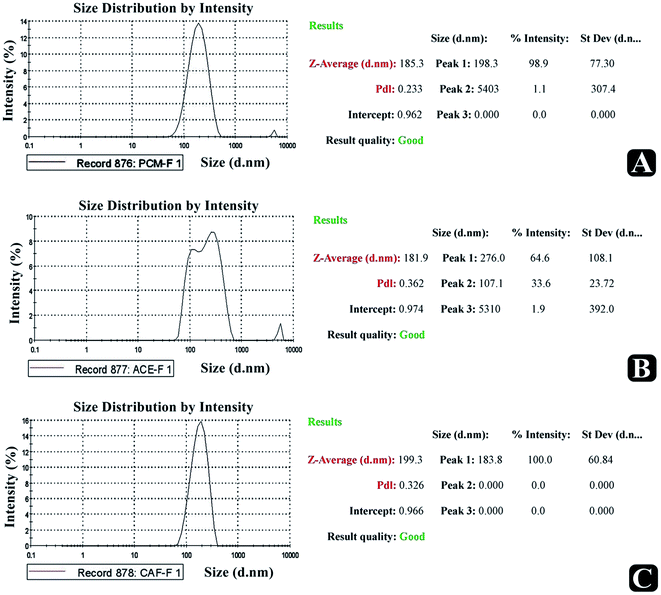 |
| | Fig. 6 Particle size analysis report. | |
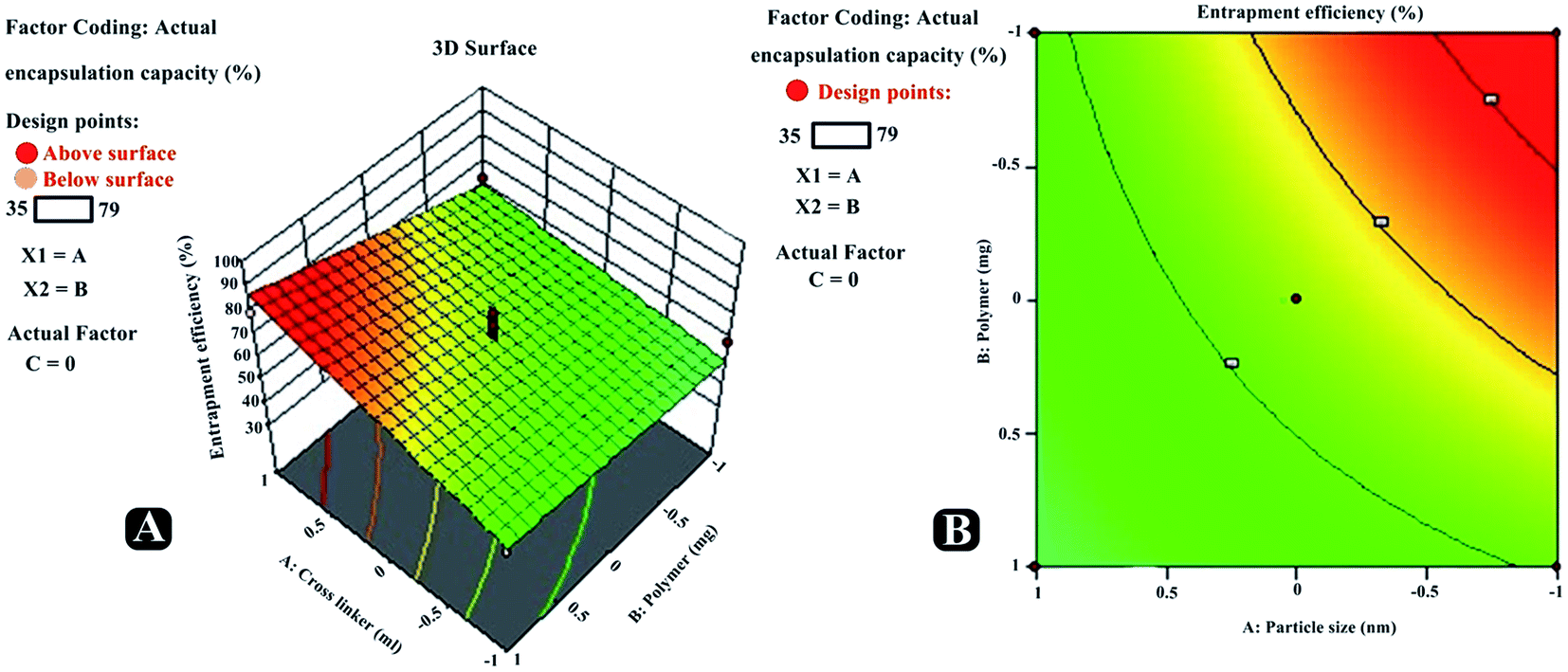
Entrapment efficiency. Various parameters were investigated to understand their effect on the entrapment efficacy for all the batches prepared using BBD, and results acquired are presented in Table 7. The independent variables showed a positive effect on the entrapment efficiency (R2). The entrapment efficiency for the software suggested batches were recorded in the range from 39.16 to 81.64%. Equation obtained from the best fit mathematical model was R2 = +57.33 + 8.04A + 7.26B + 5.12C + 12.65AB − 4.17AC + 2.12BC + 5.17A2 − 8.62B2 +2.43C2. It was experienced that with increasing concentration of polymer, entrapment efficiency significantly increases [Fig. 7 (RE1, CE1; RE2, CE2)].
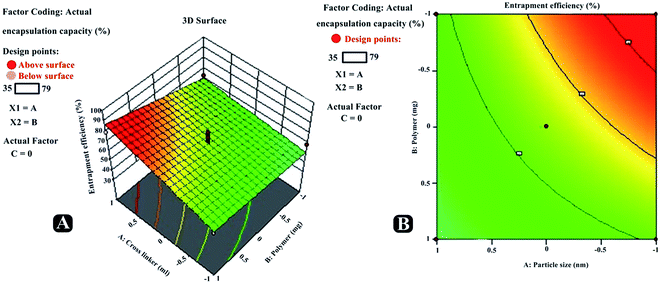 |
| | Fig. 7 Three-dimensional response surface plots (RE1, RE2, RE3) and contour plots (CE1, CE2, CE3) revealing relative effects of independent variables (A: polymer concentration; B: cross-linker concentration and C: reaction time) on dependent variable – entrapment efficiency paracetamol, aceclofenac and caffeine of nanosponge. | |
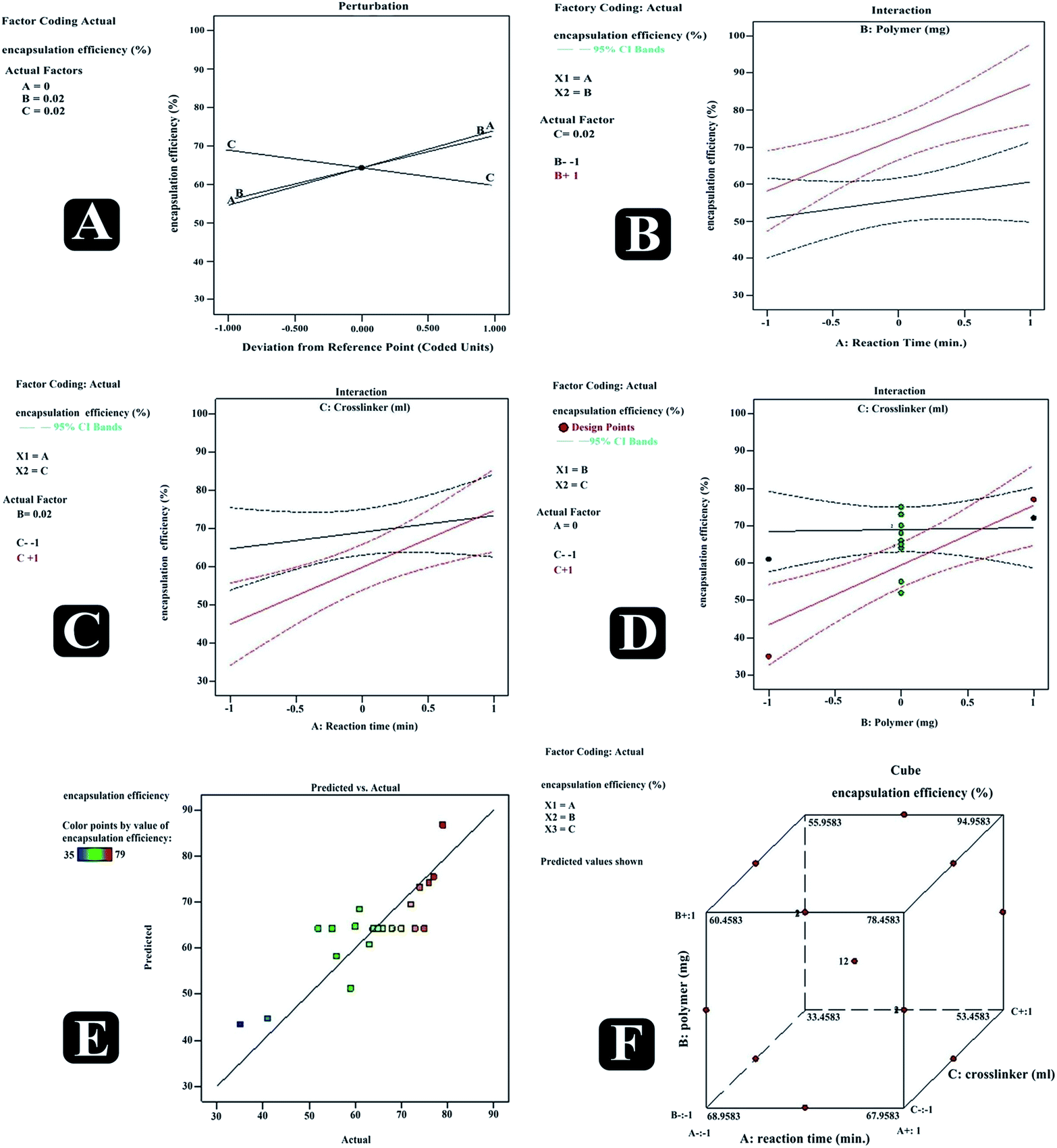
Also, increasing the cross-linker content decreases the escaping tendency of drug into external phase, and thus it was noticed that increasing the cross-linker concentration had increased drugs entrapment efficiency in nanosponges. Furthermore, increase in the reaction time had also led to the positive effect on the entrapment efficiency. Predicted vs. actual perturbation, factors interaction and cube graphs for entrapment efficiency (R2) [Fig. 8 (RE2, CE2; RE3, CE3)]. A higher degree of linearity was reflected by predicted vs. actual plot for all the trial run formulations. Prominent positive impact on response R2 can be traced in case of all the three factors (A, B, C); when deviated from −1 to +1. Moreover, all interaction plots further established the impact of factor combinations on entrapment efficiency and were in good agreement with outcomes of polynomial equation.
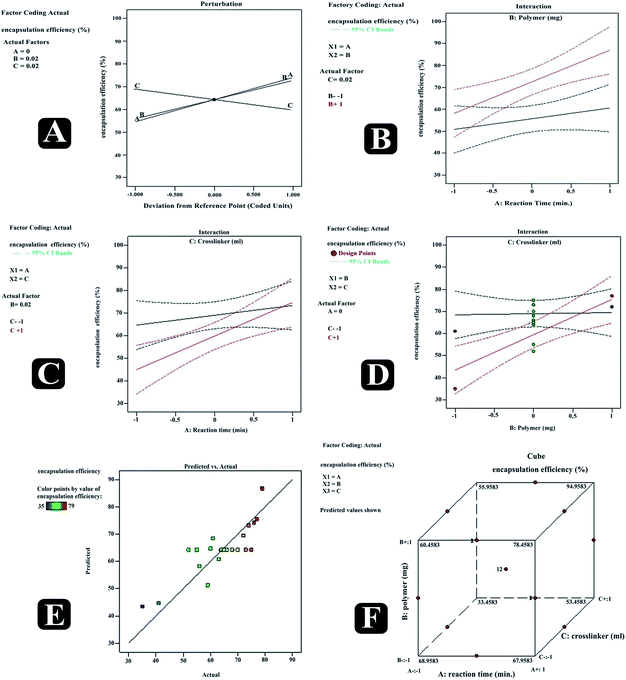 |
| | Fig. 8 Predicted vs. actual (A), perturbation (B), interaction (C–E) and cube (F) plots revealing relative effects of independent variables (A: polymer concentration; B: cross-linker concentration and C: reaction time) on dependent variable – entrapment efficiency of paracetamol, aceclofenac and caffeine nanosponges. | |
ANOVA represented the model F value 98.62 and P value < 0.0001 indicating the significant model. Moreover, predicted R-squared 0.9150 was in a reasonable concurrence with adjusted R-squared 0.9843, concluding that the obtained polynomial equation signifies the good fit of response variables at diverse levels of factors.
Optimized paracetamol, aceclofenac and caffeine loaded nanosponge batch F3–P31, F3–A31 and F3–C31 has shown the entrapment efficiency of 81.53, 84.96 and 89.28%. The encapsulation of the drug with the inner structure of nanosponges takes place by the degree of cross-linker between the β-CD and DMC. The difference that takes place in the entrapment of paracetamol, aceclofenac and caffeine among the diverse batches of experiment has already shown the effect of amount of cross-linking on capacity of nanosponges to form the complexes with drug. According to the finding, a smaller volume of cross-linker established the network with insufficient CD cross-linking with a decreased number of locations for complexing paracetamol, aceclofenac and caffeine; thus, large quantities of paracetamol, aceclofenac and caffeine were not integrated. However, the existence of further cross-linkers resulted in a greater degree of β-CD cross-linking, and thus further associations between the cavities of paracetamol, aceclofenac and caffeine and β-CD, resulting in improved trapping.
Check point analysis and optimization of paracetamol, aceclofenac and caffeine nanosponges. BBD design, a rotatable or nearly rotatable second-order design, is mainly based on the three-level incomplete factorial designs. To investigate the combined effect of polymer concentration (mg), cross-linker concentration (ml), and reaction time on mean particle size (nm) and entrapment efficiency (%) of paracetamol, aceclofenac and caffeine nanosponges over three levels, a three variable BBD response surface methodology was applied.BBD design is helpful for studying the quadratic response surfaces and is a second degree polynomial model, which eventually used for optimizing the various process variables with minimum experiment number. While, an overlay plot and desirability plot were used for optimizing the design space. CN16 was found to be the optimized formulation by applying constraints which were common for all the formulations, like optimum particle size (341–440 nm range) and maximum entrapment efficiency. By applying the DoE, recommended concentrations of the factors were calculated from the overlay plot of solution with highest desirability i.e. near to 1.0. With the help of overlay plot, which is a type of contour plots, the design space can be visually examined. Overlay plot shows the highlighted area, where the chances to get an optimized formulation with desired responses are higher. In the overlay plot, the final optimized formulation with obtained desirability flag was plotted. The most favourable values of selected variables indicated by DoE were 3.000 of A, 8.11631 of B and 5.000 of C.
Swaminathan S. et al., (2007) have prepared itraconazole nanosponges to enhance the solubility using nanosponges of β-CD and also studied the effect of a ternary component copolyvidonum on the solubility. Because itraconazole in nanosponges was effectively solubilised, it was immediately available, and the phase-to-phase transition which limits bioavailability was eliminated. Solid dispersion technique had been used to incorporate drug into nanosponges. It was found that the solubility of itraconazole was enhanced more than 50 folds with a ternary solid dispersion system. Using copolyvidonum in conjunction with nanosponges helped to increase the solubilisation efficiency of nanosponges as evident from the results of phase solubility studies.29
Characterization and evaluation of optimized nanosponges
Production yield. The production yield for optimized paracetamol, aceclofenac and caffeine nanosponges formulation (F3–P31, F3–A31 and F3–C31) was 48.63 ± 1.3%, 46.34 ± 1.3% and 47.73.±1.3%, which was calculated based on theoretical weight and practical weight of product that had been obtained. Srinivas et al., 2015 have formulated and evaluated voriconazole loaded nanosponges for oral and topical delivery using three different polymers i.e. ethyl cellulose, poly(methyl methacrylate) and Pluronic F-68 using polyvinyl alcohol (PVA) as surfactant by emulsion solvent evaporation method.24 The effect of drug: polymer ratios, surfactant concentration, stirring speeds and time, sonication time on physical characteristics of the nanosponges as well as the drug entrapment efficiency of the nanosponges were investigated. The particle size of the optimized formulation was in the range of 200–400 nm and the drug entrapment efficiency was found to be in the range of 69.8–72.5%. These nanosponge formulations were prepared as gel using carbopol 971P and studied for pH, viscosity, in vitro drug release and antimicrobial activity.
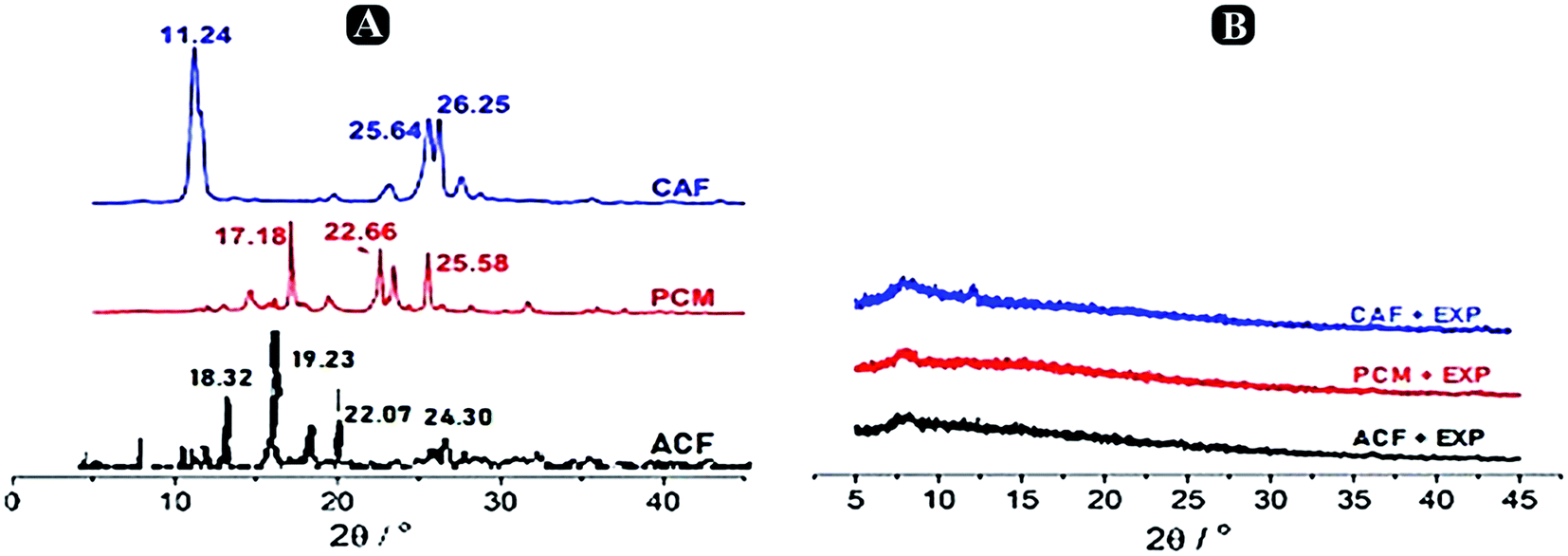
X-ray powdered diffraction analysis. To describe the crystalline structure of paracetamol, aceclofenac and caffeine inside the nanosponges to determine the mode of interaction between them, medication details (paracetamol, aceclofenac and caffeine), β-CD, product-nanosponge physical mixture, and the sample of paracetamol, aceclofenac and caffeine loaded β-CD nanosponges were collected. The drug's encapsulation crystalline structure of the β-CD nanosponges is changed by allowing to enter into the amorphous, therefore losing the crystallinity, as Shende et al., 2013 stated earlier.30 XRPD pattern of pure drug is shown in Fig. 8A, indicating its crystalline nature at 2θ and the values were as 17.18°, 22.66° and 25.58° followed by aceclofenac peaks found at 18.32°, 19.23°, 22.07° and 24.30° and caffeine peaks at 11.24°, 25.64° and 26.25° as reported earlier. There were no strong diffraction peaks in the XRPD pattern of β-CD, but rather a large peak was traced in 2 ranges 20–30°; confirming its amorphous form. In comparison, strong peaks relating to paracetamol, aceclofenac and caffeine showed that its crystalline form was preserved in nanosponge physical mixture in Fig. 9. In the other side, some of the peaks of paracetamol, aceclofenac and caffeine were less extreme here, which may be attributed to the β-CD dilution impact. Crystallinity of paracetamol, aceclofenac and caffeine had decreased when it was encapsulated by the nanosponge, which subsequently contributed to its amorphous existence and hence the lack of crystalline peaks in the XRPD form of the paracetamol, aceclofenac and caffeine loaded β-CD nanosponge sample (Fig. 9). Strober et al., 2009 furnished identical results earlier.31 Such observations from the XRPD study revealed that the complexes produced by the compound are not just due to the mechanical mixing of components, along with the results of the FTIR and DSC.
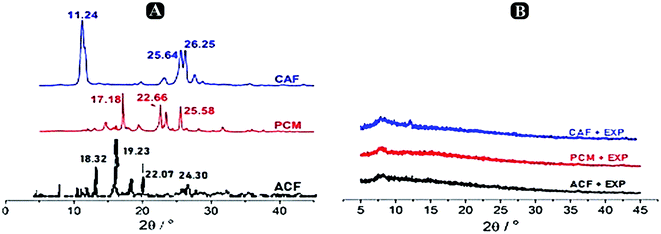 |
| | Fig. 9 X-ray diffractograms of (A) paracetamol, aceclofenac and caffeine. (B) Physical mixture of paracetamol, aceclofenac and caffeine loaded optimized nanosponges and β-CD. | |
1H NMR spectroscopy. NMR spectroscopy has been implied in our present work to establish the formation of inclusion complexation among drug and CD nanosponge. NMR allows for a strong difference between inclusion and some other potential external contact with the broad effects found on protons within the hydrophobic cavity of the CD (H-3 and H-5, with H-5 more pronouncedly affected), thereby strongly illustrating the inclusion. In addition, the internal protons chemical atmosphere varies, contributing to a difference in the protons chemical shifts due to the shielding or deshielding effects. Conversely, the protons on the outside experience so little or no effect. The acquired 1H NMR spectrum (Fig. 10A and B) of paracetamol (a), aceclofenac (b) and caffeine (c) exhibited all characteristic peaks. Fig. 10A shows the NMR of paracetamol, a singlet peak at 1.96 ppm indicates the presence of three protons of –COCH3, a singlet at 9.11 and 9.62 ppm shows the presence of hydroxyl group present at 4th position of the aromatic ring and one proton of the amine group of –NHCOCH3. Fig. 10B shows the NMR of aceclofenac, a singlet peak at 12.9 ppm indicates the presence of one proton of a hydroxyl group of –COOH and remaining proton reflect the presence of aceclofenac. Fig. 10C depicts the NMR of caffeine, a singlet peak at 3.32 ppm indicating the presence of 9 protons of three methyl groups and a singlet peak at 7.98 ppm indicating the presence of –CH2 of the purine ring. The acquired 1H NMR spectra of β-CD and paracetamol, aceclofenac and caffeine loaded optimized nanosponges are presented in Fig. 10. In case of β-CD NMR spectrum, major changes in the chemical shift values were noted, with a great effect on protons inside the hydrophobic cavity (i.e. H-3 and H-5 with δ values of 3.5 and 3.6). This inferred to the inclusion of guest molecules in CD hydrophobic cavity. The H-3 and H-5 protons of β-CD displayed an up-field shift in paracetamol, aceclofenac and caffeine loaded nanosponge spectra, notifying that these protons were situated near p-electron cloud of the aromatic nucleus (of paracetamol, aceclofenac and caffeine), resulted in an up-field shift due to its magnetic anisotropy. Furthermore, as formerly reported in literature an up-field shift displacement is most likely because of the local polarity difference as these protons are within the CD cavity and imply a weaker contact with the hydrogen atoms (a shielding effect attributed to the van der Waals interactions between the product and the carbohydrate chains). From all these NMR results, paracetamol, aceclofenac and caffeine embedding in the cavity of β-CD nanosponges can be concluded; also these findings were in great agreement with the outcomes of FTIR analysis. Moreover, the shift observed for an internal proton H-5, suggested that the paracetamol, aceclofenac and caffeine has entered the CD cavity for the formation of an inclusion complex. Also, there were no visible proton shifts suggesting that the matrix formed could be described as a complex with inclusions.
 |
| | Fig. 10 Overlain NMR of physical mixtures of (A) paracetamol, (B) aceclofenac and (C) caffeine. | |
Scanning electron microscopy. Using SEM, the configured nanosponges were checked for the surface morphology. Nanosponges with spongy and spherical nature was observed. SEM images represented in Fig. 11 reveals that, the formed nanosponges were having number of voids with fine surface and formed due to solvent diffusion. The findings were in good alignment with reports from the study of particle size. Also, no residual intact crystals of paracetamol, aceclofenac and caffeine were observed on the nanosponge surface, establishing the matrix on the nanosponge fact that was entirely formed by paracetamol, aceclofenac and caffeine and β-CD.
 |
| | Fig. 11 Microscopic analysis of the nanosponges in 500× and 2000×. | |
Characterization and evaluation of optimized formulation
Comparison of pre-formulation parameters results. The pre-formulation studies conducted for the tablet formulation with the three trials that have been complied together and T2 values has been found to be within the Indian pharmacopeia standards (Table 8).
Table 8 Pre-formulation characters
| Formulation code |
Angle of repose (θ) |
Bulk density (Bd) (g ml−1) |
Tapped density (Td) (g ml−1) |
Carr's index (%) |
Hausner's ratio |
| T1 |
17 |
0.806 |
1.31 |
38.47 |
1.62 |
| T2 |
26.3 |
1.33 |
1.35 |
1.503 |
1.01 |
| T3 |
33.6 |
2.35 |
2.81 |
19.57 |
1.19 |
| T4 |
40.6 |
2.91 |
3.36 |
13.39 |
1.15 |
Evaluation studies of tablet. The prepared tablets were evaluated for post compression properties such as percentage drug content, hardness, friability, average weight, and disintegration time as per the Indian pharmacopeia. The results obtained are presented in the Table 9. The formulation T2 passes the test as per the pharmacopeial specification for the post compression evaluations.
Table 9 Post-compression evaluation parameters for tablets
| Batch code |
Drug content (%) |
Hardness (kg cm−2) |
Friability (%) |
Average weight (mg) |
Disintegration time (min) |
| Mean ± SDa |
Mean ± SDa |
Mean ± SDa |
Mean ± SDa |
Mean ± SDa |
| Standard deviation, mean N = 4. |
| T1 |
73 ± 1.3 |
2.5 ± 0.5 |
0.3 ± 0.6 |
0.508 ± 0.002 |
20 ± 5.57 |
| T2 |
87 ± 1.3 |
4.2 ± 0.3 |
0.6 ± 0.3 |
0.519 ± 0.001 |
25 ± 0.57 |
| T3 |
35 ± 1.3 |
4.9 ± 0.4 |
0.2 ± 0.7 |
0.522 ± 0.003 |
32 ± 6.43 |
| T4 |
25 ± 1.3 |
3.2 ± 0.3 |
0.15 ± 0.75 |
0.531 ± 0.004 |
38 ± 12.43 |
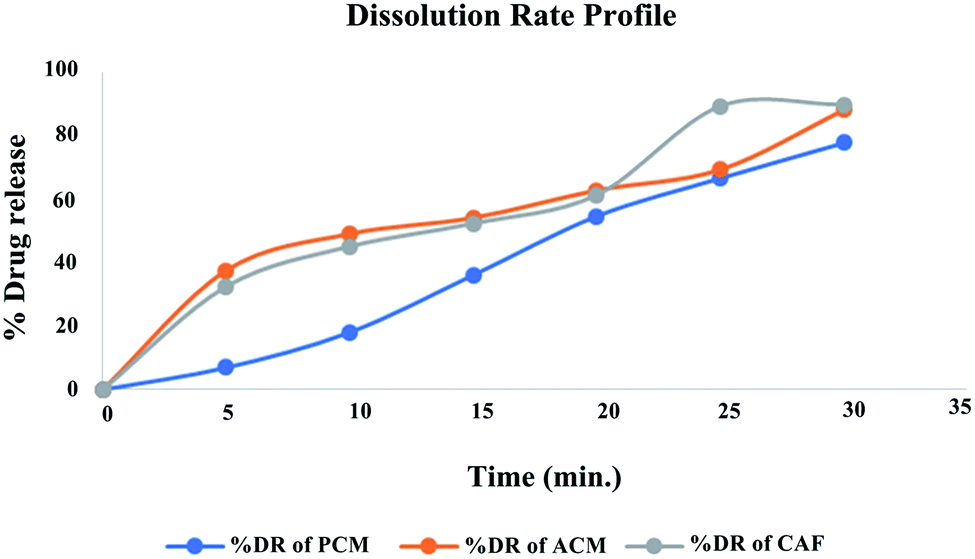
Dissolution studies. The prepared nanosponge compressed into tablet (formulation T2) was evaluated for % cumulative drug release. It indicated rapid dissolution in pH 5.5 phosphate buffer with 32.4% of drug released at 5th min and 89.6% of the drug by the end of 30th min meeting the excepted outcome. The dissolution rate profile for the formulation T2 is presented in the Fig. 12. The release data was fitted into various equations to understand the order of release and mechanism. The data obtained with regression value (R2) is presented in the Table 10. The drug release follows first order kinetics and is concentration dependent as the regression value was found to be 0.9891. The release fitted well in Higuchi equation indicating the drug release was by diffusion mechanism and follows Fickian kinetics as evident from the regression value of Korsmeyer–Pepas equation. The chemistry behind the drug loading, followed by its release is explained as follows; the fabricated nanocarriers were nanosponges that comprised of complex networks of cross-linked cyclodextrins with a roughly spherical structure, about the size of a protein, having channels, pores and numerous interconnected voids inside.32,33 The nanosponges are made up of a ‘backbone’ (a scaffold structure) of naturally degradable polyester. The polyester strands are mixed in solution with small molecules called cross-linkers that have an affinity for certain portions of the polyester. They ‘cross link’ segments of the polyester to form a porous structure that has many pockets (or cavities), where drugs gets entrapped. The drugs get inclusively complexed and/or molecularly dispersed into these porous cages.34 Post administration, over the passing time, the biodegradable cyclodextrin polymer gets slowly degraded to release the entrapped drugs.
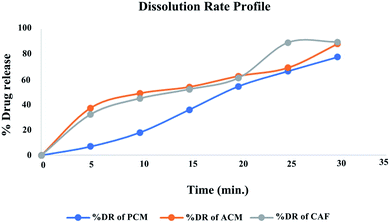 |
| | Fig. 12 Dissolution release profile of optimized formulation T2. | |
Table 10 Kinetic study of T2 formulation
| Formulation T2 |
| Kinetic model |
First order kinetics |
Zero order kinetics |
Higuchi model |
Korsmeyer–Pepas model |
| R2 |
0.9891 |
0.8436 |
0.8931 |
0.9081 |
Torne S. J. et al., (2010), have prepared and evaluated paclitaxel-loaded nanosponges for pharmacokinetic parameters in rats and the intrinsic effect of the dosage form on improvement of paclitaxel oral bioavailability. Paclitaxel-loaded nanosponges were prepared and characterized in terms of size distribution, drug solubilization and the kinetics of paclitaxel sedimentation. Taxol and paclitaxel loaded nanosponges were administered orally to rats. The relative oral bioavailability of paclitaxel-loaded nanosponges was found. After oral administration of loaded nanosponges, the area under the plasma concentration time curve was significantly increased by 3 fold in comparison to the control group. This indicated paclitaxel-loaded nanosponges as promising new formulation to enhance the oral bioavailability of paclitaxel. It also showed promising role in the solubility of the drug. Hence, nanosponges can be considered as a better choice of dosage form to increase the solubility and bioavailability.35
Stability studies. The formulation T2 was chosen for stability studies, as it was considered as the optimized formulation. The stability studies were conducted at temp 40 °C ± 2 °C and 75 ± 2% RH for the formula selected up to 45 days. The results obtained indicated no significant changes compared for parameters before and after study period, indicating that the formulation F3 was stable and passed the stability test (Table 11).
Table 11 Stability tests conducted for optimized formulation
| Formulation code |
Tested after time (in days) |
Hardness (kg cm−2) |
Disintegration time (min) |
Drug release (%) |
Drug content (%) |
| F3–P31 |
45 |
3.30 ± 0.04 |
25 ± 0.57 |
77.5 |
84.96 |
| F3–A31 |
87.84 |
84.96 |
| F3–C31 |
89.6 |
89.28 |
Conclusion
The drugs paracetamol, aceclofenac and caffeine had varied solubility profiles, and formulating them into single tablet did not have the desired dissolution profile for drug absorption. Therefore, they were successfully loaded into the nanosponge that was prepared by the hot-melt method. The nanosponge characterization studies confirmed the entrapment of the drug within the colloidal three-dimensional structure of β-CD with the formation of an inclusion complex. The entrapped drugs properties have been modified from crystalline to amorphous nature, which enhances drug solubility. The optimized nanosponge loaded formulations obtained by computer-based optimization technique was directly compressed into tablets with suitable diluents. The results of in vitro dissolution studies of nanosponge tablets indicated rapid dissolution due to changed solubility properties of the drug, compared to pure drug meeting the set objective enhanced absorption. The stability studies indicated the optimized formulation was stable at temp 40 °C ± 2 °C and 75 ± 2% RH for the formula selected up to 45 days. Thus, it can be concluded that drug with different solubility profile can be loaded into nanosponge that have optimized dissolution properties for combination therapy.
Conflicts of interest
The authors have no conflict of interest to declare.
References
- I. Krabicova, S. L. Appleton, M. Tannous, G. Hoti, F. Caldera, P. A. Rubin, C. Cecone, R. Cavalli and F. Trotta, Polymers, 2020, 12(5), 1122 CrossRef CAS.
- S. Pawar, P. Shende and F. Trotta, Int. J. Pharm., 2019, 565, 333–350 CrossRef CAS.
- O. M. Amin, A. Ammar and S. A. Eladawy, J. Pharm. Invest., 2020, 50, 399–411 CrossRef CAS.
- R. Cavalli, A. K. Akhter, A. Bisazza, P. Giustetto, F. Trotta and P. Vavia, Int. J. Pharm., 2010, 402, 254–257 CrossRef CAS.
- A. Singireddy, S. R. Pedireddi and S. Subramanian, J. Polym. Res., 2019, 26(4), 93 CrossRef.
- A. P. Sherje, B. R. Dravyakar, D. Kadam and M. Jadhav, Carbohydr. Polym., 2017, 173, 37–49 CrossRef CAS.
- S. Sapino, M. E. Carlotti, R. Cavalli, E. Ugazio, G. Berlier, L. Gastaldi and S. Morel, J. Inclusion Phenom. Macrocyclic Chem., 2013, 75, 69–76 CrossRef CAS.
- H. V. Gangadharappa, S. M. Prasad and R. P. Singh, J. Drug Delivery Sci. Technol., 2017, 41, 488–501 CrossRef CAS.
- D. Li and M. Ma, Cyclodextrin polymer separation materials, WO 9822197, 1998 May 28.
- M. F. Zidan, H. F. Ibrahim, M. I. Afouna and E. A. Ibrahim, Drug Dev. Ind. Pharm., 2018, 44, 1243–1253 CrossRef CAS.
- E. Roberts, V. Delgado Nunes, S. Buckner, S. Latchem, M. Constanti, P. Miller, M. Doherty, W. Zhang, F. Birrell, M. Porcheret, K. Dziedzic, I. Bernstein, E. Wise and P. G. Conaghan, Ann. Rheum. Dis., 2016, 75, 552–559 CrossRef CAS.
- J. Guo, Y. Xiao, Y. Lin, J. Crommen and Z. Jiang, J. Chromatogr. A, 2016, 1467, 288–296 CrossRef CAS.
- H. A. Hodali, R. S. Rawajfeh and N. A. Allababdeh, J. Dispersion Sci. Technol., 2017, 38, 1342–1347 CrossRef CAS.
- A. Vishwakarma, P. Nikam, R. Mogal and S. Talele, Int. J. Pharm. Tech. Res., 2014, 6, 11–20 CAS.
- N. M. P. Habeeba, K. Gladisa, Y. Anithaa and S. Mohammedc, Int. J. Biopharm., 2013, 4(1), 10–17 Search PubMed.
- S. M. Omar, F. Ibrahim and A. Ismail, Saudi Pharm. J., 2020, 28, 349–361 CrossRef CAS.
- C.-L. Lee, C.-C. Wu, H.-P. Chiou, C.-M. Syu, C.-H. Huang and C.-C. Yang, Int. J. Hydrogen Energy, 2011, 36, 6433–6440 CrossRef CAS.
- S. Honary, P. Ebrahimi and R. Hadianamrei, Pharm. Dev. Technol., 2014, 19, 987–998 CrossRef CAS.
- V. Singh, J. Xu, L. Wu, B. Liu, T. Guo, Z. Guo, P. York, R. Gref and J. Zhang, RSC Adv., 2017, 7, 23759–23764 RSC.
- A. E.-D. Omara, T. Elsakhawy, T. Alshaal, H. El-Ramady, Z. Kovács and M. Fári, Environment, Biodiversity and Soil Security, 2019, 3, 29–62 CrossRef.
- D. J. E. Francis and F. S. Yusuf, Univers. J. Pharm. Res., 2019, 4(1), 24–28 CAS.
- P. Jyoti, B. Tulsi, K. Popin and B. Chetna, Indo Global J. Pharm. Sci., 2016, 6, 59–64 CAS.
- S. C. B. Penjuri, N. Ravouru, S. Damineni, S. BNS and S. R. Poreddy, Turk. J. Pharm. Sci., 2016, 13, 304–310 CrossRef CAS.
- P. Srinivas and K. Sreeja, Int. J. Drug Dev. Res., 2013, 5, 55–69 CAS.
- B. B. Karad and A. D. Shinde, Indo Am. J. Pharm. Res., 2017, 7(08), 471–479 CAS.
- P. N. Remya, T. S. Saraswathi, S. Sangeetha, N. Damodharan and R. Kavitha, J. Pharm. Sci. Res., 2016, 8, 1258–1261 CAS.
- A. O. Abioye and A. Kola-Mustapha, Drug Dev. Ind. Pharm., 2016, 42, 39–59 CrossRef CAS.
- A. Vyas, S. Saraf and S. Saraf, J. Inclusion Phenom. Macrocyclic Chem., 2008, 62, 23–42 CrossRef CAS.
- S. Swaminathan, P. R. Vavia, F. Trotta and S. Torne, J. Inclusion Phenom. Macrocyclic Chem., 2007, 57, 89–94 CrossRef CAS.
- P. Shende, K. Deshmukh, F. Trotta and F. Caldera, Int. J. Pharm., 2013, 456, 95–100 CrossRef CAS.
- L. Strober, J. Englert, F. Munschauer, B. Weinstock-Guttman, S. Rao and R. Benedict, Mult. Scler. J., 2009, 15, 1077–1084 CrossRef CAS.
- S. D. Mhlanga, B. B. Mamba, R. W. Krause and T. J. Malefetse, J. Chem. Technol. Biotechnol., 2007, 82, 382–388 CrossRef CAS.
- F. Trotta and R. Cavalli, Compos. Interfaces, 2009, 16, 39–48 CrossRef CAS.
- R. K. Sharma and A. E. Yassin, Indian J. Ophthalmol., 2014, 62, 768 CrossRef.
- S. J. Torne, K. A. Ansari, P. R. Vavia, F. Trotta and R. Cavalli, Drug Delivery, 2010, 17, 419–425 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
N. K. Famna Roohib,
Syed Mohd Danish Rizvia,
Syed Amir Ashrafc,
Arif Jamal Siddiqui
a,
N. K. Famna Roohib,
Syed Mohd Danish Rizvia,
Syed Amir Ashrafc,
Arif Jamal Siddiqui