
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparative study of gold and silver interactions with amino acids and nucleobases†
Andrey A.
Buglak
 * and
Alexei I.
Kononov
* and
Alexei I.
Kononov
St. Petersburg State University, 199034 Saint-Petersburg, Russia. E-mail: andreybuglak@gmail.com
First published on 15th September 2020
Abstract
Metal nanoclusters (NCs) have gained much attention in the last decade. In solution, metal nanoclusters can be stabilized by proteins, and, thus, exhibit many advantages in biocatalysis, biosensing, and bioimaging. In spite of much progress in the synthesis of polypeptide-stabilized gold (Au) clusters, their structure, as well as amino acid-cluster and amino acid–Au+ interactions, remain poorly understood. It is not entirely clear which amino acid (AA) residues and sites in the protein are preferred for binding. The understanding of NC-protein interactions and how they evolve in the polypeptide templates is the key to designing Au NCs. In this work, binding of gold ion Au+ and diatomic neutral gold nanocluster Au2 with a full set of α-proteinogenic amino acids is studied using Density Functional Theory (DFT) and the ab initio RI-MP2 method in order to find the preferred sites of gold interaction in proteins. We demonstrated that the interaction of gold cations and clusters with protonated and deprotonated amino acid residues do not differ greatly. The binding affinity of AAs to the Au2 cluster increases in the following order: Cys(−H+) > Asp(−H+) > Tyr(−H+) > Glu(−H+) > Arg > Gln, His, Met ≫ Asn, Pro, Trp > Lys, Tyr, Phe > His(+H+) > Asp > Lys(+H+) > Glu, Leu > Arg(+H+) > Ile, Val, Ala > Thr, Ser > Gly, Cys, which agrees with the available experimental data that gold cluster synthesis occurs in a wide range of pH – amino acid residues with different protonation states are involved in this process. The significant difference in the binding energy of metal atoms with nucleobases and amino acids apparently means that unlike on DNA templates, neutral metal atoms are strongly bound to amino acid residues and can't freely diffuse in a polypeptide globula. This fact allows one to conclude that formation of metal NCs in proteins occurs through the nucleation of reduced Au atoms bound to the neighboring amino acid residues, and the flexibility of the amino acid residue side-chains and protein chain as a whole plays a significant role in this process.
1. Introduction
Metal nanoclusters (NCs) have gained much attention in the last decade in biocatalysis, biosensing, and bioimaging due to their high biocompatibility, small size, and high sensitivity to molecular environment.1–3 NCs have been used for the detection of metal ions, simple organic compounds, thiols, polypeptides, proteins, and nucleic acids.4–7 In solution, NCs can be stabilized by various polymer templates. In particular, DNA-stabilized8,9 and protein-protected6,7 NCs exhibit many advantages for biosensing and bioimaging: ultrasmall size, photostability, biocompatibility, and brightness. Noble metal NCs, in particular, silver (Ag) and gold (Au) clusters, in comparison with other NCs, exhibit excellent stability, facile synthesis, and low toxicity.10 Metal clusters emitting in the visible range have been synthesized using DNA,8,9 amino acids,11,12 peptides,13,14 and proteins such as bovine serum albumin,15–19 human serum albumin, egg albumin,20,21 lysozyme,22 and immunoglobulin.5In spite of much progress in the synthesis of a wide variety of NCs, their structure, as well as ligand–cluster interactions, remains poorly understood. For protein-stabilized NCs, it is not entirely clear which amino acid residues and sites in the polypeptide are preferred for binding. The understanding of NCs–polypeptide interactions and how they evolve in the polypeptide matrices is the key to design the functional fluorescent biolabels. We have investigated earlier the interactions of amino acids with silver ions and clusters.23 We showed that deprotonated amino acid residues are preferable for binding with silver clusters, which is in line with experimental data: the formation of silver clusters on protein templates occurs predominantly at alkaline pH. On the contrary, gold clusters are synthesized in a wide range of pH.24 Several factors may influence this process: the ability of different amino acid functional groups to reduce gold, charge, and binding energy with gold.
This paper focuses on the Au2 cluster/amino acid (AA) interactions and on the Au+/AA interactions as a precursor of the cluster. In a typical synthesis, hydrogen tetrachloroaurate is usually taken as a source of gold Au3+ ions. At the first stage, proteins reduce most of the Au3+ to Au1+.25 It is generally believed that at the first step of Au NPs synthesis Au+–thiolate complexes are formed.26–28 In proteins, Au ions may bind to various AAs able to interact with gold. Many AAs can reduce the ions.29–33
It is known that Au can interact with O, N and S atoms. At the same time, it is believed that gold in protein templates interacts most effectively with sulfur, especially with cysteine. Indeed, cysteine is considered as the preferential binding site for gold in proteins, which is confirmed by theoretical calculations.34
The interaction of gold cation (Au+) and gold nanoparticles with individual amino acids and proteins has been investigated earlier.29–33,35–37 It was shown experimentally that low temperature and acidic pH favors the growth of gold nanoparticles on protein template.35 On contrary, we showed that deprotonated amino acid residues are preferable for binding with silver clusters,23 which is in line with experimental data: the formation of silver clusters on protein templates occurs predominantly at alkaline pH.5,18,38–40 Gold clusters are synthesized in a wide range of pH.24,41–44 The synthesis of gold nanostructures through the photo-reduction of amino acids in water is also possible: the most stable structures are produced by arginine, cysteine, threonine, methionine, tryptophan, and phenylalanine.36 The interaction of gold cation (Au+), gold clusters, and nanoparticles with individual amino acids and proteins was investigated theoretically earlier.37,45–47 Using molecular dynamics, it was shown that negatively charged atoms play a significant role in adsorption of amino acids on the gold nanoparticles.37 It was shown that the interaction energy with neutral Au3 cluster is higher for the glycine bearing a negative charge than for glycine with charge 0 or +1, the same is true for cysteine.45 In the case of neutral and negatively charged glycine, the interaction occurs with the nitrogen atom, while in the case of protonated glycine interaction with Au3 cluster proceeds through the carboxyl. In the case of cysteine, neutral amino acid interacts with the cluster through the nitrogen, negatively charged amino acid forms a bond between Au and sulfur, while for positively charged Gly interaction with Au occurs through both sulfur and hydroxyl of the carboxyl group. They also showed that two major bonding factors are: (1) Au–N, Au–O, and Au–S anchoring bonding; and (2) nonconventional OH⋯Au and NH⋯Au hydrogen bonding.45 Rai and co-authors performed theoretical calculations for proline and Au3 cluster. The interaction of gold cluster with proline occurred predominantly through amide terminal.46 Investigation of the interactions of Au8 and Au20 clusters with alanine and tryptophan showed that these clusters prefer single-site interactions through the amino-group for the amino acids.47 In these works, the authors regarded only alanine, cysteine, glycine, proline, and tryptophan interaction with gold nanoclusters and how the interplay of gold with the remaining 15 amino acids occurs was still unknown.
In our quantum-chemical investigation we focused on the interactions of the neutral Au2 cluster and Au+ cation with a full set of proteinogenic α-amino acids. We tried to identify favorable sites of clusters formation on protein templates. Also, we took into account the effect of amino acid side chain protonation/deprotonation on the effectiveness of these interactions. The Au2 cluster was chosen as a model object since it is a minimal singlet cluster with a neutral charge. Au2 diatomic cluster protected by organic ligands is a classical object of nanocluster research.48,49 We calculated the binding Gibbs free energies between Au2 and amino acids in the neutral, protonated, and deprotonated forms of the side chain. We established the amino acids, which are more preferable for the interaction with Au2 and Au+. Next, we compared gold–amino acid binding energies with silver binding energies. Also, we examined the interactions in the complexes using Bader's quantum theory “Atoms-in-molecules” approach and natural bond orbital (NBO) analysis.
There is a certain interest to compare the binding energies of gold and silver clusters with protein and DNA matrices. It was shown experimentally that both Au and Ag NCs on protein matrices consist mostly of neutral metal atoms.15–17,22 In contrast, DNA-stabilized NCs are positively charged clusters.50–52 We performed the calculations for gold and silver ions and clusters with cytosine and adenine, the preferred binding residues for Ag and Au clusters in DNA,53,54 and compared them with the literature data on Au and Ag interactions with DNA.
2. Methods
Equilibrium geometry optimizations and corresponding hessian calculations of complexes were done with usage of density functional theory (DFT) at the PBE55 level, and resolution-of-the-identity second order Moller–Plesset perturbation theory (RI-MP2) realized in Orca 3.0 program package.56 Since DFT does not include dispersion forces, the atom-pair wise dispersion correction with Becke–Johnson damping was used.57 Karlsruhe basis set def2-TZVP was used in all the calculations, gold atoms were treated with def2-TZVP effective core potential (ECP).58 The initial geometries of amino acid–Au2 complexes for optimization were constructed by placing gold atoms near the active sites of amino acids. The active sites of the amino acids are the amino group, the carboxyl group, and sulfur. These groups possess electron-rich nitrogen, oxygen, and sulfur, which may donate electron density to Au from their lone electron pairs. Binding Gibbs free energies (ΔG) were calculated for the reactions:| Au+ + AA → AA–Au+ |
| Au2 + AA → AA–Au2 |
The NBO analysis59 was performed for the amino acid–Au2 complexes using NBO.5 program at the RI-MP2/def2-TZVP level of theory. The NBO analysis was done in order to obtain natural charges and Wiberg bond indices. The NBO orbitals of several complexes were plotted using the Chemcraft program. The atoms-in-molecules (AIM) analysis was performed with the Multiwfn program package60 to calculate the properties of bond critical points (BCPs).
3. Results and discussion
In our previous study, we showed that RI-MP2 method in combination with def2-TZVP basis set gives reasonable results predicting Gibbs free energy of interaction between simple organic molecules and silver.61 In our next study, we showed that PBE-D3 method along with RI-MP2 give fruitful results when calculating Gibbs free energy of interaction between silver and amino acid residues.23 Thus, in this study we used both PBE-D3 and RI-MP2 method with def2-TZVP basis set and def2-TZVP ECP for another noble metal–gold.3.1 Amino acid interaction with gold cation Au+
We started with the analysis of amino acid interactions with gold cation Au+. Among the neutral amino acids, arginine had the highest binding free energy (ΔG) with Au+: −130.7 kcal mol−1 and −140.7 kcal mol−1, according to RI-MP2/def2-TZVP and PBE-D3/def2-TZVP, respectively (Table 1). ΔG was the lowest one for glycine: −60.1 kcal mol−1 and −73.4 kcal mol−1, according to RI-MP2/def2-TZVP and PBE-D3/def2-TZVP, respectively. Thus, PBE-D3 tends to overestimate the binding free energy for approximately 10–15 kcal mol−1 as compared to RI-MP2, which is considered as a more precise method. For this reason, PBE-D3 method due to its low computational costs was used for the pretreatment of initial amino acid–Au+ geometries (attachment to different sites of amino acid and different conformations were compared) while RI-MP2 was used for the precise calculation of the final geometries and Gibbs free energy.| Amino acid | RI-MP2 | PBE-D3 |
|---|---|---|
| Amino acids with deprotonated side chain | ||
| Asp(−H+) | −169.8 | −183.3 |
| Cys(−H+) | −194.1 | −207.4 |
| Glu(−H+) | −150.0 | −166.9 |
| Tyr(−H+) | −177.2 | −184.3 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Neutral amino acids | ||
| Ala | −63.3 | −76.7 |
| Arg | −130.7 | −140.7 |
| Asn | −73.0 | −82.8 |
| Asp | −65.4 | −79.3 |
| Cys | −74.9 | −88.7 |
| Gln | −79.1 | −91.2 |
| Glu | −69.0 | −85.5 |
| Gly | −60.1 | −73.4 |
| His | −93.1 | −102.7 |
| Ile | −64.0 | −77.4 |
| Leu | −63.5 | −78.0 |
| Lys | −116.8 | −124.6 |
| Met | −94.8 | −107.9 |
| Phe | −82.3 | −91.2 |
| Pro | −68.5 | −82.3 |
| Ser | −65.1 | −75.3 |
| Thr | −65.2 | −77.4 |
| Trp | −95.9 | −105.3 |
| Tyr | −83.6 | −93.8 |
| Val | −63.8 | −77.7 |
Cys(−H+) had the highest ΔG among all amino acids (see Fig. 1 for complex geometry). Generally, the binding affinity of AAs increases in the following order: Cys(−H+) > Tyr(−H+) > Asp(−H+), Glu(−H+) > Arg, Lys > Trp > Met > His > Tyr > Phe > Gln > Cys > Asn > Glu > Pro > Asp > Thr, Ser > Ile, Val, Leu, Ala > Gly. Obviously, we may conclude that Au2 will preferably bind to the deprotonated residues of the amino acids rather than to the protonated ones in a peptide or protein. Aliphatic AAs are less preferable for Au+ binding. It is also evident that the binding order (preferred binding sites) depends strongly on the pH conditions. Neutral cysteine attaches Au+ to both the nitrogen of the amino-group and the sulphur atom while deprotonated through the side-chain anionic cysteine (pK = 8.3) forms a monodentate complex with Au+ and attaches gold atom solely to the sulphur. ΔG rises from −74.9 kcal mol−1 (−88.7 kcal mol−1) to −194.1 kcal mol−1 (−207.4 kcal mol−1) during the deprotonation of the cysteine side-chain. Neutral Asp and Glu attach Au+ solely to the nitrogen of the amino-group while deprotonated anionic variants of these amino acids attach Au+ to the nitrogen and one of the oxygens of the carboxylate group (Fig. 1 and S1†). ΔG rises from −65.4 kcal mol−1 (−79.3 kcal mol−1) to −169.8 kcal mol−1 (−183.3 kcal mol−1) during the deprotonation of the Asp side-chain (pK = 3.7). For Glu (pK = 4.3), ΔG rises from −69.0 kcal mol−1 (−85.5 kcal mol−1) to −150.0 kcal mol−1 (−166.9 kcal mol−1) upon deprotonation of the side-chain. Both neutral Tyr and Tyr(−H+) with deprotonated side-chain (pK = 10.5) attach Au+ to the amino-group and form a cation–pi interaction with the six-membered ring (Fig. S1†). ΔG rises from −83.6 kcal mol−1 (−93.8 kcal mol−1) to −177.2 kcal mol−1 (−184.3 kcal mol−1) upon deprotonation.
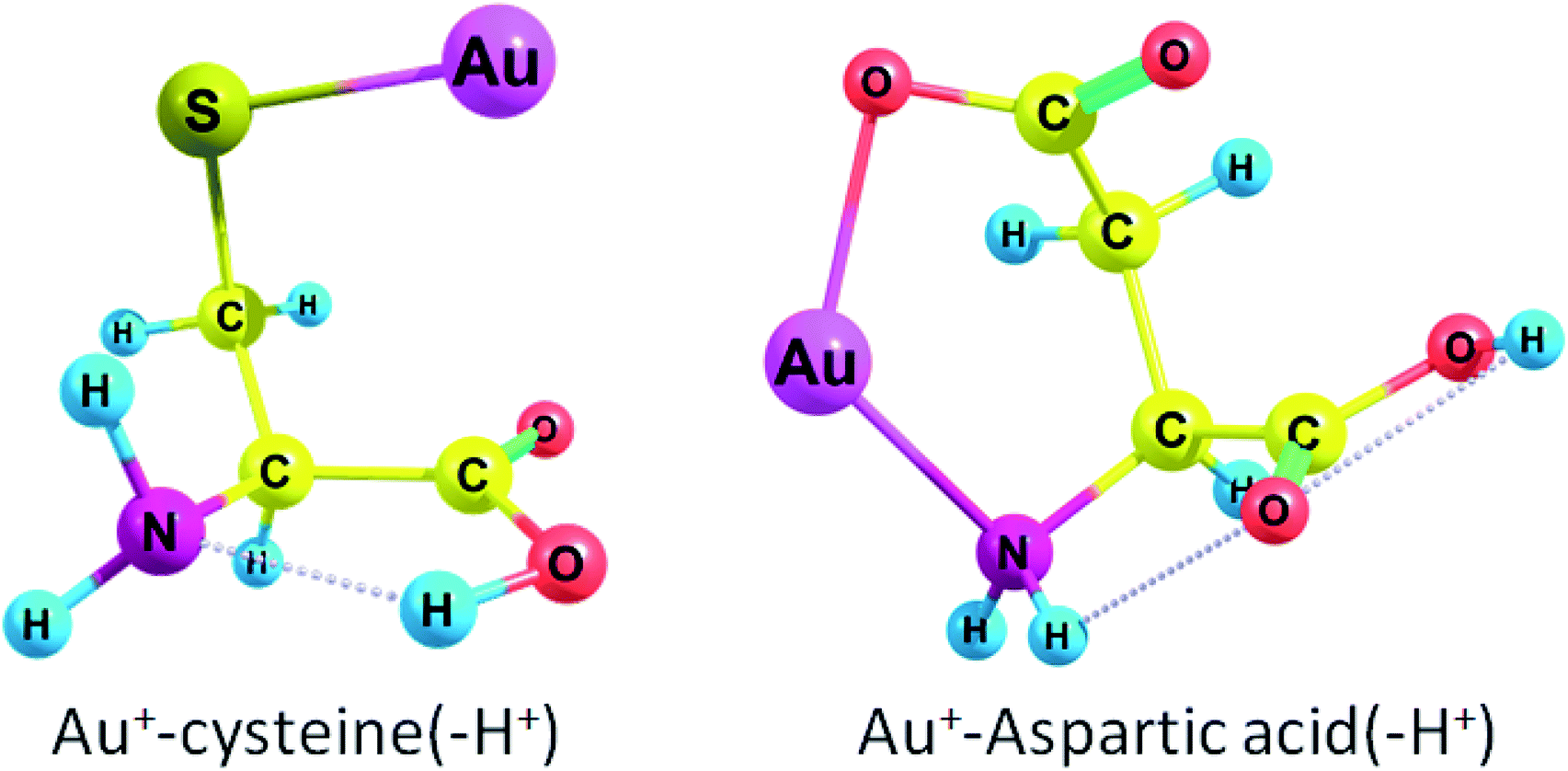 | ||
| Fig. 1 Geometries of deprotonated anionic amino acids bound to gold cation. | ||
Thereby, all amino acids form either monodentate or bidentate complexes with Au+ (see Fig. S1 in ESI†). Every amino acid except deprotonated cysteine forms a bond between Au+ and the nitrogen of the amino group. Aliphatic amino acids (except methionine), Asp, Glu, Pro, Ser, and Thr form a monodentate complex with Au+. Cys and Met form a bidentate complex while second bond occurs between sulphur and Au. For Arg, His, and Lys, the second site is the nitrogen of the radical side chain; this second site of Au+ attraction may become single in a polypeptide since the first site, amino-group, will participate in the formation of a peptide bond. Trp attaches Au+ to one of the carbon atoms of the six-membered ring. Phe, Tyr, and Tyr(−H+) form a cation–pi interaction between Au+ and a six-membered ring. Asn and Gln attach Au+ to the carbonyl of the side chain (Fig. S1†). Hence, the Gibbs free energies for the interaction of Au+ with Asp, Cys, Glu, Tyr bearing deprotonated side chain radicals appear to be higher than those for the neutral forms of these amino acids (Table 1).
3.2 AIM analysis of amino acid complexes with gold cation Au+
Next, we used AIM analysis62 to study the nature of bonding interactions: amino acid interactions with gold cation Au+ were analyzed in terms of electron density and its derivatives. We used several AIM parameters that are presented in Table 2: the density of all electrons (ρ(r)), the Laplacian of electron density (∇2ρ(r)), the Lagrangian kinetic energy term (G(r)), the potential energy density (V(r)), and the energy density (H(r)).| Complex | BCP | ρ, hartree | ∇2ρ, hartree | G(r), hartree | V(r), hartree | H(r), hartree | E bond, kcal mol−1 |
|---|---|---|---|---|---|---|---|
| Arg–Au+ | Au–NH2 | 0.12875 | 0.46379 | 0.16682 | −0.21769 | −0.05087 | 68.30 |
| Arg–Au+ | Au–NH | 0.14096 | 0.49987 | 0.18537 | −0.24578 | −0.06040 | 77.11 |
| Cys–Au+ | Au–SH | 0.10009 | 0.21356 | 0.08981 | −0.12623 | −0.03642 | 39.61 |
| Cys–Au+ | Au–NH2 | 0.08420 | 0.31613 | 0.09839 | −0.11776 | −0.01936 | 36.95 |
| Cys(−H+)–Au+ | Au–S | 0.13265 | 0.12066 | 0.09774 | −0.16531 | −0.06757 | 51.87 |
| Gly–Au+ | Au–NH2 | 0.11226 | 0.40289 | 0.13730 | −0.17388 | −0.03658 | 54.55 |
| Tyr–Au+ | Au–C | 0.09177 | 0.19938 | 0.08127 | −0.11269 | −0.31425 | 35.36 |
| Tyr–Au+ | Au–NH2 | 0.09620 | 0.35979 | 0.11642 | −0.14289 | −0.02647 | 44.83 |
| Tyr(−H+)–Au+ | Au–C | 0.10641 | 0.18719 | 0.09018 | −0.13357 | −0.04338 | 41.91 |
| Tyr(−H+)–Au+ | Au–NH2 | 0.08936 | 0.34254 | 0.10804 | −0.13044 | −0.02240 | 40.93 |
A positive value of ∇2ρ(r) indicates depletion of electronic charge along the bond, which is typical for electrostatic interaction, while a negative value of ∇2ρ(r) indicates that electronic charge is located between the nuclei, which is a feature of electron-sharing and covalent interaction. All bond critical points (BCPs) in Table 2 have a positive ∇2ρ(r) value, which means that all interactions are electrostatic.
The electronic energy density term H(r) is a sum of kinetic and potential components:
| H(r) = G(r) + V(r) | (1) |
The virial theorem states that G(r) and V(r) are related to Laplacian through the equation:
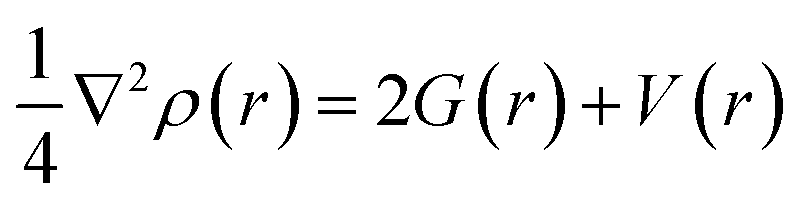 | (2) |
If H(r) is positive then accumulation of charge at this point is destabilizing. If H(r) is negative then accumulation of charge is stabilizing. The negative value of H(r) indicates the presence of a covalent bond. From Table 2 we may see that in all the cases the electrostatic interactions are stabilizing: all H(r) values are negative. The positive value of ∇2ρ(r) and negative value of H(r) mean that Au–X bonds are partially covalent and partially electrostatic. That is true for the complexes with both neutral and deprotonated amino acids. For example, AIM parameters calculated for the Au–N bond in the Gly–Au+ complex are following: ρ = 0.11226 hartree, ∇2ρ = 0.40289 hartree, and V(r) = −0.17388 hartree. The V(r) values allow to calculate the energy of Au–X bond as follows:63
| Ebond = −V(r) × 627.51/2 | (3) |
As expected, arginine had the highest Au–X bond energy value (77.1 kcal mol−1 for the Au–NH bond) among the regarded complexes. This in line with the Gibbs free energy calculations, which show that Au2–Arg complex possesses the highest binding energy among neutral AAs. In Au2–Arg complex Au interacts both with the nitrogen of the NH2 group (68.3 kcal mol−1) and with the NH group of the side chain (for geometry see Fig. S1†). Generally, the deprotonated amino acids had higher bond energy values than the neutral amino acids (this fact is supported by Gibbs free energy calculations): Au–S bond energy was equal to 51.9 kcal mol−1 for Au+–Cys(−H+) and 39.6 kcal mol−1 for Au+–Cys, Au–C bond energy was equal to 41.9 kcal mol−1 for Au+–Tyr(−H+) and 35.4 kcal mol−1 for Au+–Tyr.
3.3 Interaction of amino acids with Au2 cluster
Next, we studied the complexes of amino acids with a minimal neutral cluster Au2. In the equilibrium geometry of bare Au2, the bond length r(Au1–Au2) was equal to 2.465 Å, according to RI-MP2/def2-TZVP method. This bond length tends to slightly diminish in the complexes: for example, r(Au1–Au2) was equal to 2.461 Å in Au2–Gly.Conformations of amino acids were carefully analyzed both free and in complex with Au2 (see ESI† for cartesian coordinates). All unionized amino acids attach Au2 to the nitrogen of the amino group, except arginine and histidine, which attach Au2 to the nitrogen of the side-chain (these interactions between Au NCs and side-chains of amino acid residues may become even more important in a protein since amino group does participate in the formation of a peptide bond). Also, in the case of arginine gold plays the role of a proton acceptor and forms a nonconventional H-bond with the hydroxyl group (O–H⋯Au) of the carboxyl (Fig. 2). Amino acid complexes with gold are always monodentate, except for arginine, which forms a bidentate complex and involves hydrogen bonding. The structural parameters of the complex between arginine and Au2 are presented on Fig. 2.
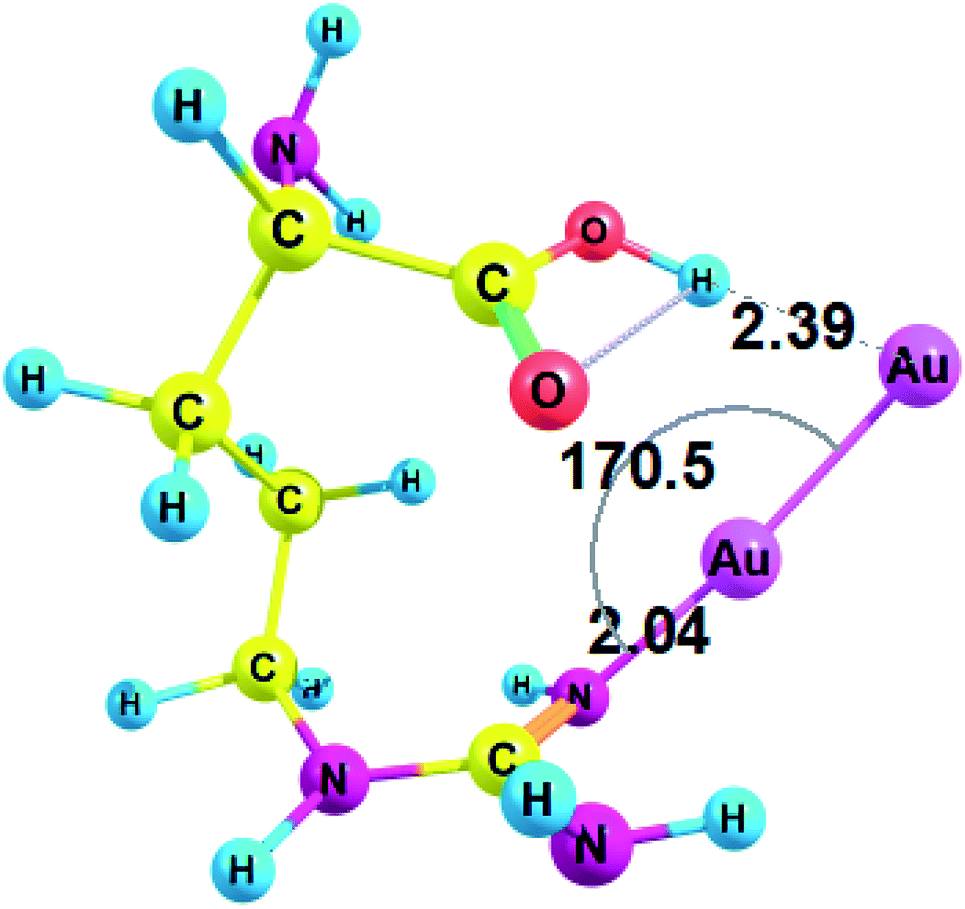 | ||
| Fig. 2 Complex of arginine and Au2 cluster optimized with RI-MP2/def2-TZVP method. | ||
Thus, arginine forms an Au–N coordination bond with a length of 2.040 Å and a valence angle N–Au–Au equal to 170.5° (Fig. 2). For Arg–Au2 complex the C–NH bond is increased by 0.028 Å after the interaction with Au2 cluster: the C–NH bond is equal to 1.308 for the Arg–Au2 complex while for free Arg C–NH bond is equal to 1.280 Å. In addition, the stretching mode of ν(C–N) undergoes a blue shift with respect to that of the uncoordinated C–N group: from 1730.5 cm−1 to 1635.5 cm−1.
Amino acids with deprotonated side chain surpass ΔG values of the neutral amino acids while Cys(−H+) possesses the highest ΔG among the deprotonated amino acids. Cys(−H+) attaches the cluster through the sulfur atom. Cys(−H+) forms the Au–S bond with a length of 2.260 Å and a valence angle C–S–Au equal to 102.6° (Fig. 3). For Cys(−H+)–Au2 complex, the C–S bond stays intact after the interaction with Au2 cluster: the C–S bond is equal to 1.821 Å for the Cys(−H+)–Au2 complex and the same C–S bond is equal to 1.821 Å for the free Cys(−H+).
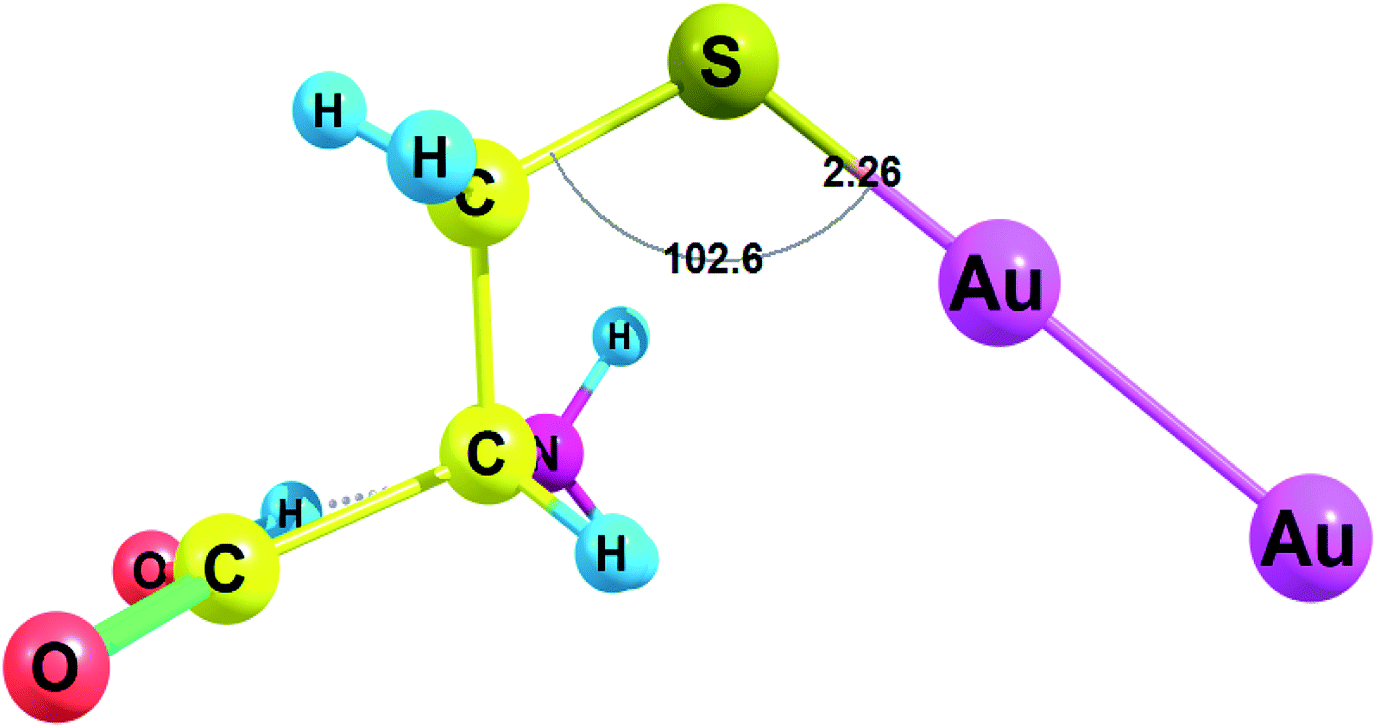 | ||
| Fig. 3 Complex of cysteine(−H+) with Au2 cluster optimized with RI-MP2/def2-TZVP. | ||
Cys(−H+) had the highest ΔG among all amino acids (see Fig. 3 for complex geometry). Generally, the binding affinity of AAs to Au2 cluster increases in the following order: Cys(−H+) > Asp(−H+) > Tyr(−H+) > Glu(−H+) > Arg > Gln, His, Met ≫ Asn, Pro, Trp > Lys, Tyr, Phe > His(+H+) > Asp > Lys(+H+) > Glu, Leu > Arg(+H+) > Ile, Val, Ala > Thr, Ser > Gly, Cys. Obviously, we may conclude that Au2 will preferably bind to the deprotonated residues of the amino acids rather than to the protonated ones in a peptide or protein. Surprisingly, neutral cysteine has the lowest binding energy among all the amino acids, which results in the highest ΔG difference between the neutral and deprotonated amino acid. Generally, the interaction of Au2 with protonated and deprotonated AAs do not differ greatly.
It is generally believed that noble metal atoms preferably interact with sulfur in peptides and amino acids.45,61 For cysteine ΔG was equal to −19.1 kcal mol−1 (−17.7 kcal mol−1), and for deprotonated cysteine it was found to be −53.5 kcal mol−1 (−50.8 kcal mol−1) (Table 3). Cysteine has a pKa of 8.3, which means that alkalization of the solution containing Cys and Au2 from neutral to alkaline pH would give the increase in interaction energy equal to 34.4 kcal mol−1 (33.1 kcal mol−1). The interaction energy of Au2 with methionine, which also occurs through the sulfur, was equal to −28.1 kcal mol−1 (−24.5 kcal mol−1), which was one of the highest ΔG among the neutral amino acids. The disulfide bond is less favorable for the formation of a complex with the gold cluster: the energy of the interaction between dimethyldisulfide and Au2 was equal to −23.4 kcal mol−1, according to RI-MP2/def2-TZVP method (for geometry of the complex see Fig. S2†); it was equal to −22.7 kcal mol−1, according to the PBE-D3/def2-TZVP calculation.
| Amino acid | RI-MP2 | PBE-D3 |
|---|---|---|
| Amino acids with deprotonated side chain | ||
| Asp(−H+) | −41.1 | −40.6 |
| Cys(−H+) | −53.5 | −50.8 |
| Glu(−H+) | −35.4 | −33.5 |
| Tyr(−H+) | −39.4 | −36.6 |
|
||
| Neutral amino acids | ||
| Ala | −20.6 | −17.9 |
| Arg | −34.3 | −30.7 |
| Asn | −26.9 | −21.5 |
| Asp | −22.7 | −21.1 |
| Cys | −19.1 | −17.7 |
| Gln | −28.5 | −23.1 |
| Glu | −22.1 | −18.2 |
| Gly | −19.2 | −17.2 |
| His | −28.3 | −26.7 |
| Ile | −21.0 | −17.9 |
| Leu | −22.1 | −20.7 |
| Lys | −23.3 | −19.7 |
| Met | −28.1 | −24.5 |
| Phe | −22.9 | −21.0 |
| Pro | −26.4 | −21.5 |
| Ser | −19.4 | −14.3 |
| Thr | −20.6 | −17.7 |
| Trp | −25.9 | −20.7 |
| Tyr | −23.1 | −21.2 |
| Val | −21.0 | −18.3 |
|
||
| Amino acids with protonated side chain | ||
| Arg(+H+) | −22.0 | −16.8 |
| His(+H+) | −22.9 | −17.1 |
| Lys(+H+) | −22.2 | −17.5 |
Deprotonated anionic amino acids Asp(−H+), Glu(−H+), and Tyr(−H+) attach Au2 to the side chain namely to the oxygen atom and have high ΔG values: −41.1 kcal mol−1 (−40.6 kcal mol−1), −35.4 kcal mol−1 (−33.5 kcal mol−1), and −39.4 kcal mol−1 (−36.8 kcal mol−1), respectively (Table 3). Tyrosine, which has a pKa of 10.5, has a much lower ΔG equal to −23.1 kcal mol−1 (−21.2 kcal mol−1) in the unionized form and that means that alkalization of the solution with Tyr and Au2 from neutral to alkaline pH would give an increase in the interaction energy equal to 16.3 kcal mol−1 (15.6 kcal mol−1). These data show why alkalization of the solution with the protein containing Tyr residues is advantageous when a gold cluster is formed.
Histidine (pKa 6.0) has ΔG equal to −28.3 kcal mol−1 (−26.7 kcal mol−1) in the unionized form and −22.9 kcal mol−1 (−17.1 kcal mol−1) with the protonated side chain. Alkalization of the solution with His and Au2 from acid to alkaline and neutral pH would give the increase of interaction energy equal to 5.4 kcal mol−1 (9.6 kcal mol−1). For lysine (pKa 10.5) ΔG rises from −22.2 kcal mol−1 (−17.5 kcal mol−1) for the cationic amino acid with protonated side chain to −23.3 kcal mol−1 (−19.7 kcal mol−1) for the neutral molecule. For arginine (pKa 12.5) ΔG changes from −22.0 kcal mol−1 (−16.8 kcal mol−1) for the cationic form with the protonated side-chain to −34.3 kcal mol−1 (−30.7 kcal mol−1) for the unionized molecule.
In general, our results are in agreement with experimental results showing that gold nanoparticles have the highest binding affinity with the peptides containing Cys, His, Met, and Tyr residues.33 In real experimental conditions, charge repulsion between negatively charged residues Asp(−H+), Glu(−H+) and chloroaurate anions frustrate the reduction reaction. However, some experimental protocols allow Asp and Glu residues to play significant role in the synthesis of gold nanoclusters.24 Moreover, the stability of Au2 complexes of all neutral and protonated amino acid residues is rather high (ΔG is more than −20 kcal mol−1 in absolute values), which indicates that all 20 proteinogenic AAs can stabilize gold NCs.
3.4 AIM and natural bond orbital (NBO) analysis of amino acid complexes with Au2 cluster
We used Bader's AIM analysis to study the nature of amino acid interactions with Au2 cluster in terms of electron density and its derivatives: the density of all electrons ρ(r), the Laplacian of electron density ∇2ρ(r), the Lagrangian kinetic energy term G(r), the potential energy density V(r), and the energy density H(r) are presented in Table 4.| Complex | BCP | ρ, hartree | ∇2ρ, hartree | G(r), hartree | V(r), hartree | H(r), hartree | E bond, kcal mol−1 |
|---|---|---|---|---|---|---|---|
| Arg–Au2 | Au1–NH | 0.12082 | 0.46238 | 0.15834 | −0.20108 | −0.04274 | 63.09 |
| Arg–Au2 | Au2⋯HO | 0.02065 | 0.04638 | 0.01193 | −0.01226 | −0.00033 | 3.85 |
| Cys–Au2 | Au–SH | 0.11312 | 0.25384 | 0.11014 | −0.15682 | −0.04668 | 49.20 |
| Cys(−H+)–Au2 | Au–S | 0.11773 | 0.18658 | 0.09891 | −0.15117 | −0.05226 | 47.43 |
| Gly–Au2 | Au–NH2 | 0.10571 | 0.42161 | 0.13766 | −0.16992 | −0.03226 | 53.31 |
| Tyr–Au2 | Au–NH2 | 0.10946 | 0.43010 | 0.14239 | −0.17726 | −0.03487 | 55.61 |
| Tyr(−H+)–Au2 | Au–O | 0.11164 | 0.52169 | 0.16384 | −0.19725 | −0.03342 | 61.89 |
One can see that in each case the electrostatic interaction is stabilizing since for each complex H(r) value is negative. The positive value of ∇2ρ(r) and negative value of H(r) in all cases means that Au–X bonds are partially covalent and partially electrostatic.
Since we know V(r) values we can calculate bond energies. Arginine had the highest Au–X bond energy value equal to 63.1 kcal mol−1 (Table 4), and this fact is supported by Gibbs energy calculations, which state that arginine has the highest binding energy among neutral AAs. In the Arg–Au2 complex, gold interacts both with the nitrogen of the side chain and with hydrogen of the carboxyl group. The hydrogen bond energy is pretty small – 3.9 kcal mol−1, but it is a reasonable value for a hydrogen bond.36
Next, we performed the NBO analysis. Natural charges of gold atom and amino acid atom to which the cluster is attached are presented in Table 5 (qAu and qX, respectively). When Au and X both have positive charges (the case of Cys–Au2 complex), it obliquely indicates that Au–X bond has a covalent nature. When Au and X have opposite charges (all other cases), it indicates that Au–X bond has an electrostatic nature. In all the cases Au2 cluster had a negative charge (qcluster), which means that it oxidizes the coordinated amino acid. The bond orders were evaluated by using Wiberg's bond indices, which are presented in Table 5. The Wiberg bond indices were higher for the deprotonated negatively charged amino acid complexes with Au2 than for the neutral complexes: WAu1–X is equal to 0.366 for Cys–Au2 and is 0.580 for Cys(−H+)–Au2, WAu1–X is equal to 0.206 for Tyr–Au2 and is 0.247 for Tyr(−H+)–Au2. On contrary, the Au–Au bond order is lower for deprotonated amino acids.
| Complex | Bond type | W Au1–X | W Au1–Au2 | q X | q Au | q cluster |
|---|---|---|---|---|---|---|
| Au2 | — | — | 1.016 | — | 0 | 0 |
| Arg–Au2 | Au1–NH | 0.272 | 0.851 | −0.954 | 0.130 | −0.114 |
| Arg–Au2 | Au2⋯HO | 0.055 | 0.851 | 0.528 | −0.242 | −0.114 |
| Cys–Au2 | Au1–SH | 0.366 | 0.889 | 0.078 | 0.024 | −0.183 |
| Cys(−H+)–Au2 | Au1–S | 0.580 | 0.756 | −0.444 | 0.038 | −0.374 |
| Gly–Au2 | Au1–NH2 | 0.190 | 0.918 | −0.861 | 0.090 | −0.110 |
| Tyr–Au2 | Au1–NH2 | 0.206 | 0.922 | −0.862 | 0.073 | −0.124 |
| Tyr(−H+)–Au2 | Au1–O | 0.247 | 0.806 | −0.888 | 0.250 | −0.152 |
NBO analysis gives useful information when analyzing intramolecular bonding and interaction between bonds. The electron donor orbital and electron acceptor orbital occupancies, as well as the interacting stabilization energy E(2) obtained from the second-order perturbation theory analysis, are reported in Table 6. The larger the E(2) value, the more intensive the interaction between i electron donor orbital and j electron acceptor orbital, which means the more donating tendency from electron donor to electron acceptor. Delocalization of the electron density from occupied bonds or lone pair NBO orbitals to formally unoccupied antibond NBO orbitals indicates a stabilizing donor–acceptor interaction. The intramolecular interaction is formed by the orbital overlap between n, n*, σ and σ* bond orbitals, which leads to the intramolecular charge transfer (ICT) permitting the stabilization of the system. These interactions are observed as an increase in the electron density in anti–bonding orbital that weakens the respective bonds.
| Complex | Donor (i) | Type of orbital | Occupancy | Acceptor (j) | Type of orbital | Occupancy | E(2), kcal mol−1 | q CT |
|---|---|---|---|---|---|---|---|---|
| Arg–Au2 | N | n | 1.815 | Au | n* | 0.095 | 44.90 | 0.060 |
| Cys–Au2 | Au–Au | σ | 1.830 | Au–S | σ* | 0.176 | 113.84 | 0.220 |
| Cys(−H+)–Au2 | Au–Au | σ | 1.896 | Au–S | σ* | 0.120 | 65.05 | 0.121 |
| Gly–Au2 | N | n | 1.859 | Au1–Au2 | σ* | 0.086 | 39.32 | 0.058 |
| Tyr–Au2 | N | n | 1.849 | Au1–Au2 | σ* | 0.086 | 36.29 | 0.052 |
| Tyr(−H+)–Au2 | O | n | 1.826 | Au1–Au2 | σ* | 0.091 | 37.78 | 0.057 |
For each i donor orbital and j acceptor orbital, the stabilization energy E(2) of the i → j delocalization was calculated according to the following formula:
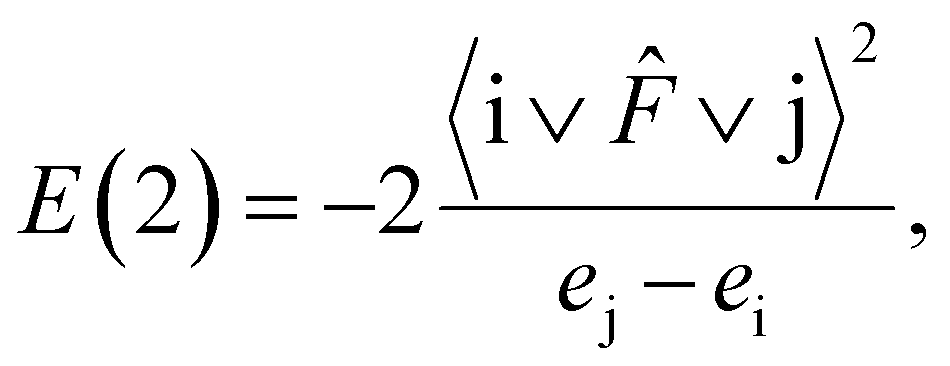 | (4) |
![[F with combining circumflex]](https://www.rsc.org/images/entities/i_char_0046_0302.gif) is the Fock operator.
is the Fock operator.
The amount of transferred charge from i donor orbital to j acceptor orbital was also calculated using the Fock operator and NBO orbital energies as follows:
 | (5) |
In Table 6, E(2) and qCT for the Au–X and Au–Au bonds of some complexes are presented. The charge is transferred from the lone pair of nitrogen and oxygen or σ Au–Au bond to n* orbital of Au, Au–S or Au–Au σ* anti-bond. In the case of Arg–Au2 complex, the charge is transferred from the nitrogen lone pair to the lone pair of Au. In the Gly–Au2 complex, the charge is transferred from the nitrogen lone pair to Au–Au σ* anti-bonding orbital (Fig. 4).
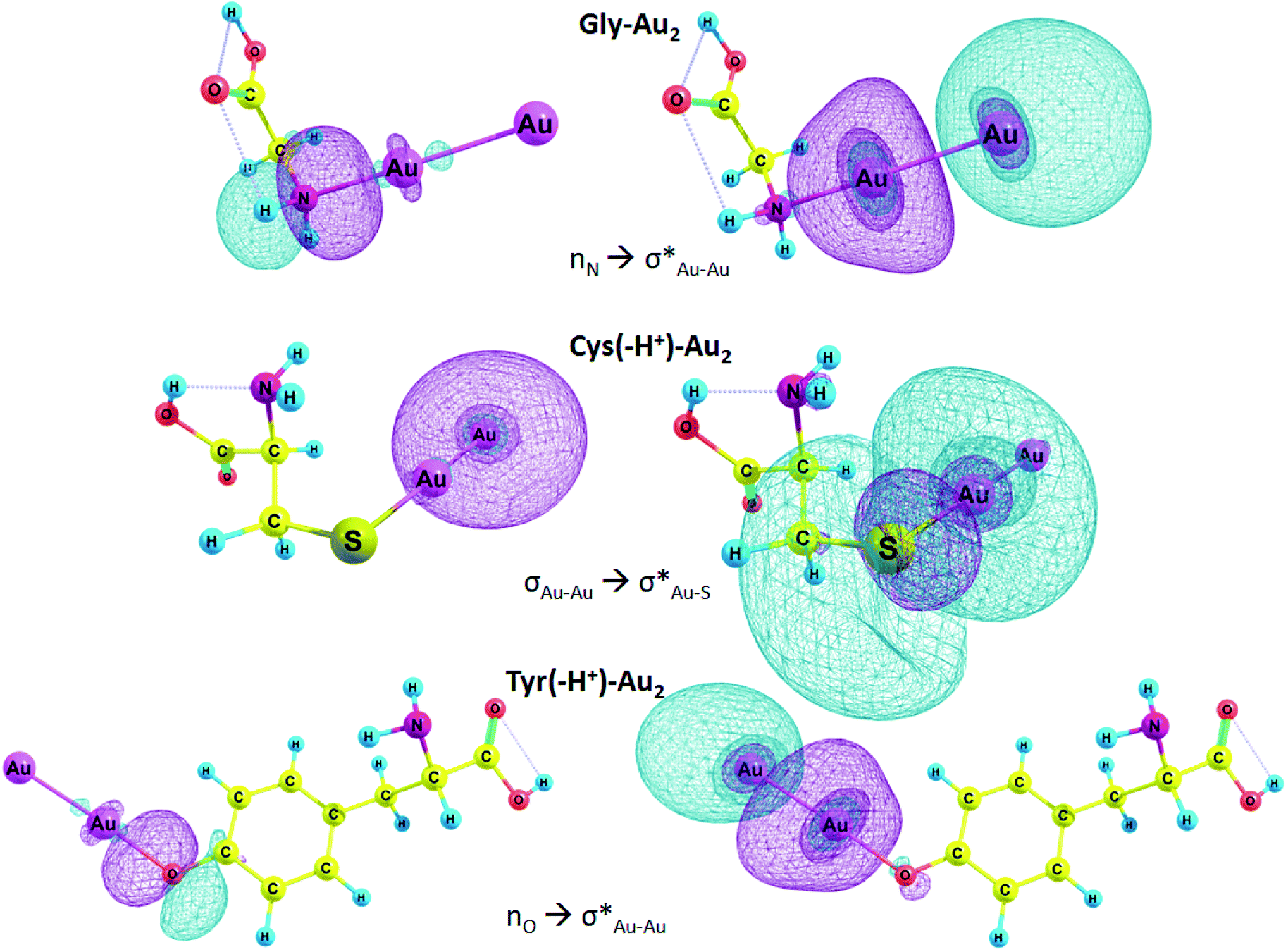 | ||
| Fig. 4 Selected NBO orbitals involved in charge transfer. | ||
We focused on the complexes of cysteine and tyrosine with the gold nanocluster since deprotonation of these amino acids gives the strongest energy gain when the AA–Au2 complex is formed (Table 3). Moreover, only these two amino acids exhibited capability to produce fluorescent complexes with silver clusters, as it was show experimentally.61 In the Cys–Au2 complex, the intramolecular charge transfer occurs from the σ Au–Au bond to Au–S σ* anti-bonding orbital and leads to delocalization of 113.8 kcal mol−1, which is the strongest stabilization energy among the regarded complexes. For the Cys(−H+)–Au2, the same ICT contributes to the stabilization energy of 65.1 kcal mol−1. In the Tyr–Au2 complex, ICT of the electron from the nitrogen lone pair to the σ* Au–Au anti-bond leads to delocalization of 36.3 kcal mol−1. In the Tyr(−H+)–Au2 complex, ICT of the n electron of oxygen lone pair to the σ* Au–Au anti-bond leads to delocalization equal to 37.8 kcal mol−1.
3.5 Binding energies between nucleobases and metals
We calculated the interaction energies of metal cations (Ag+ and Au+), neutral metal atoms (Ag0 and Au0), and diatomic metal clusters (Ag2 and Au2) with nucleobases, namely cytosine (Cyt) and adenine (Ade). Later, we compared the interaction energies between silver and gold, between different types of metal particles, between nucleobases and selected amino acids (Table 7). Our results are consistent with previously reported data for the nucleobases. Thus, for example, the interaction energy of silver atom Ag0 and cytosine was equal to −3.5 kcal mol−1, according to Gwinn with co-authors.53 The same complex interaction energy was equal to −7.0 kcal mol−1, according to Volkov et al.64 In our case the result was −8.3 kcal mol−1. The advantage of our work is that the vibrational spectra were calculated and Gibbs free energies of complex formation were determined along with ΔEtotal. Thus, for Ag0 complex with cytosine ΔG is equal to −1.3 kcal mol−1. We have also investigated the complexes of Au2 with adenine and protonated cytosine; for the latter it was done for the first time.| Complex | Bond | ΔG | ΔE |
|---|---|---|---|
| Nucleobases | |||
| Ag+_Cyt | Ag–N1, Ag–O | −50.1 (ref. 64) | |
| Ag–N1, Ag–O | −55.2 | −64.0 | |
| Ag0_Cyt | Ag–N1 | −3.5 (ref. 53) | |
| Ag–O | −7.0 (ref. 64) | ||
| Ag–N1 | −1.3 | −8.3 | |
| Ag–O | 2.2 | −5.4 | |
| Ag2_Cyt | Ag–N1 | −11.5 (ref. 53) | |
| Ag–O | −22.7 (ref. 64) | ||
| Ag–N1 | −9.3 | −21.3 | |
| Ag–O | −4.9 | −16.6 | |
| Au+_Cyt | Au–N1 | −74.3 | −83.6 |
| Au–O | −67.9 | −77.0 | |
| Au0_Cyt | Au–N1 | −5.7 | −13.4 |
| Au–O | 0.3 | −6.9 | |
| Au2_Cyt | Au–N1 | −24.2 (ref. 65) | |
| Au–N1 | −26.2 | −38.2 | |
| Au–O | −14.9 | −27.4 | |
| Ag0_Cyt(+H+) | Ag⋯HN1 | 1.9 | −5.9 |
| Ag2_Cyt(+H+) | Ag–O, Ag⋯HN1 | −6.1 | −15.9 |
| Au0_Cyt(+H+) | Au⋯HN1 | 1.1 | −5.7 |
| Au2_Cyt(+H+) | Au–O, Au⋯HN1 | −5.0 | −16.9 |
| Ag+_Ade | Ag–N7, Ag–NH2 | −43.2 | −52.9 |
| Ag0_Ade | Ag–N7 | 0.1 | −6.9 |
| Ag2_Ade | Ag–N7 | −7.8 | −16.9 |
| Au+_Ade | Au–N7 | −62.6 | −71.4 |
| Au0_Ade | Au–N7 | −3.9 | −13.0 |
| Au2_Ade | Au–N7 | −25.3 | −38.3 |
|
|||
| Amino acids | |||
| Ag+_Cys(−H+) | Ag–S | −151.8 (ref. 23) | −162.0 |
| Ag0_Cys(−H+) | Ag–S | −16.7 (ref. 23) | −26.1 |
| Ag2_Cys(−H+) | Ag–S | −30.1 (ref. 23) | −43.1 |
| Au+_Cys(−H+) | Au–S | −194.1 | −204.3 |
| Au0_Cys(−H+) | Au–S | −27.6 | −36.9 |
| Au2_Cys(−H+) | Au–S | −53.5 | −68.0 |
| Ag+_Asp(−H+) | Ag–O | −152.8 (ref. 23) | −163.6 |
| Ag0_Asp(−H+) | Ag–O | −15.3 (ref. 23) | −24.2 |
| Ag2_Asp(−H+) | Ag–O | −30.7 (ref. 23) | −42.3 |
| Au+_Asp(−H+) | Au–O | −169.8 | −180.9 |
| Au0_Asp(−H+) | Au–O | −16.9 | −26.2 |
| Au2_Asp(−H+) | Au–O | −41.1 | −54.5 |
Yet, we didn't limit our study to only Me–N bonding, we also analyzed the interactions between metal atoms and the carbonyl oxygen of the cytosine. However, Me–O binding energies concede to Me–N interactions. Thus, for example, in Ag0_Cyt complex ΔG for the Ag–N binding is equal to −1.3 kcal mol−1, while for Ag–O binding ΔG is only 2.2 kcal mol−1 (Table 7), so it's highly likely that Ag–O bond doesn't occur. The same is true for Au0_Cyt complex: ΔG for Au–O interaction is also positive: 0.3 kcal mol−1.
Geometry of the complexes between gold and silver ions and nucleobases, namely cytosine and adenine, are presented on Fig. 5. Ag+_Cyt and Au+_Cyt complexes have certain differences: the silver ion forms bonds with both nitrogen and oxygen while gold ion attaches only to N1. The same is true for Ag+_Ade and Au+_Ade complexes: Ag+ forms bonds with N7 of the imidazole ring and nitrogen of the amino-group, Au+ forms a bond only with N7. ΔG binding energy is higher for Au+_Cyt than for Ag+_Cyt complex: −74.3 kcal mol−1 and −55.2 kcal mol−1, respectively. Au+_Cyt complex with Au–O bonding is less stable than the complex with Au–N bond: −67.9 kcal mol−1 and −74.3 kcal mol−1, respectively (Table 7). Once again it shows that Me–N bonding is more stable than Me–O bonding.
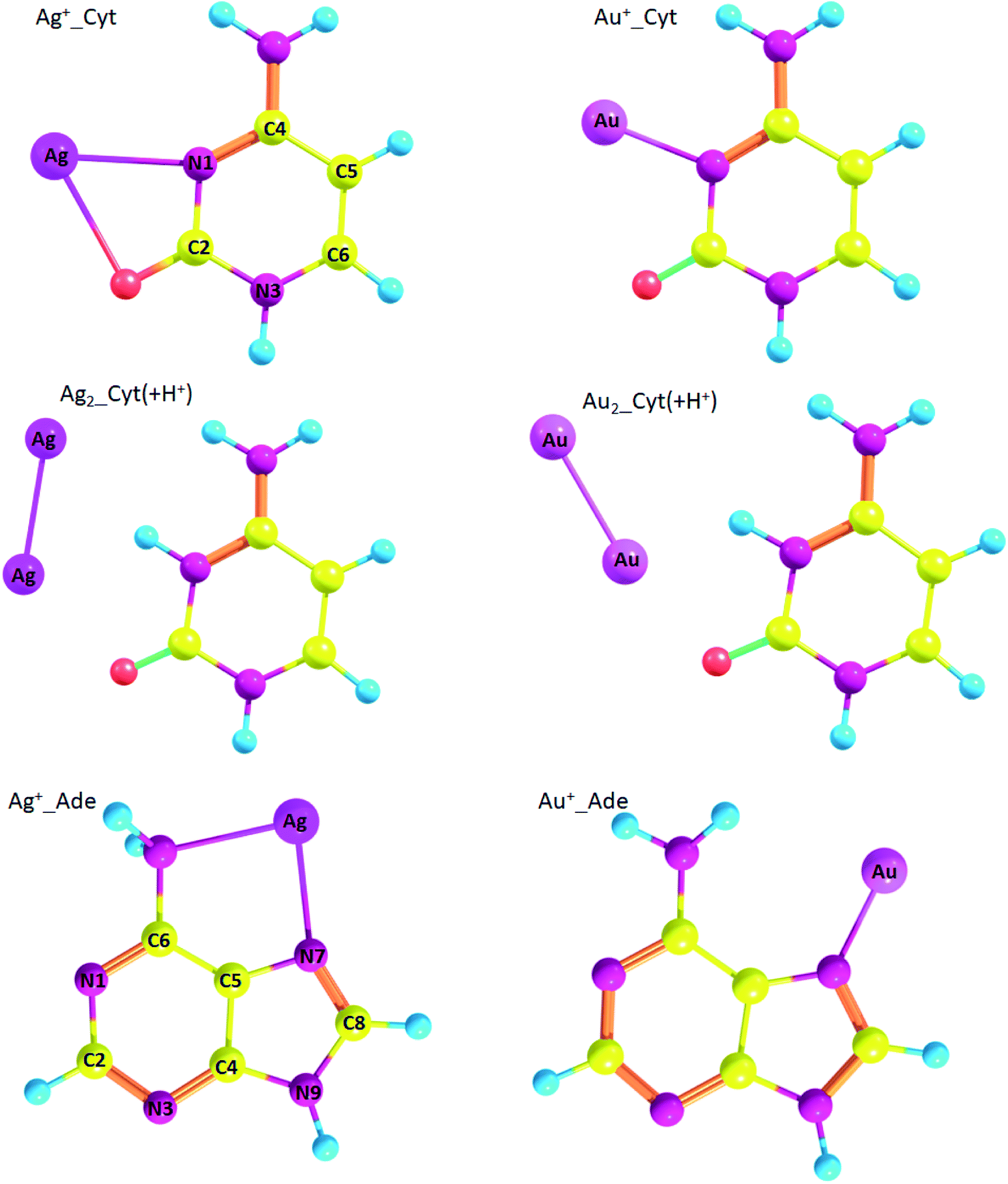 | ||
| Fig. 5 Geometry of metal/nucleobase complexes (optimized at RI-MP2/def2-TZVP level of theory) with atom numbering. | ||
Speaking about adenine, complexes with gold are more stable than silver complexes. Thus, for Ag+_Ade, Ag0_Ade, and Ag2_Ade ΔG is equal to −43.2 kcal mol−1, 0.1 kcal mol−1, and −7.8 kcal mol−1, respectively, while for Au+_Ade, Au0_Ade, and Au2_Ade ΔG is equal to −62.6 kcal mol−1, −3.9 kcal mol−1, and −25.3 kcal mol−1 (Table 7), respectively.
Also, we analyzed silver and gold complexes of protonated cytosine (Cyt(+H+)). Interestingly, Cyt(+H+) tends to form nonconventional hydrogen bonds with metal atoms Me0 and diatomic clusters (Fig. 5). However, Ag0_Cyt(+H+) and Au0_Cyt(+H+) complexes have positive ΔG values and highly likely are not formed: 1.9 kcal mol−1 and 1.1 kcal mol−1, respectively. ΔG values are negative for Ag2_Cyt(+H+) and Au2_Cyt(+H+) complexes: −6.1 kcal mol−1 and −5.0 kcal mol−1, respectively. The latter complexes are stabilized by Me–O bonds, as well as by nonconventional Me⋯H hydrogen bonding.
We compared metal complexes of nucleobases and amino acids. In all the cases amino acid complexes are more stable than analogous nucleobase complexes. Thus, for example, the most stable complex of Au+ is with cytosine (−74.3 kcal mol−1) while the binding energy of deprotonated cysteine with Au+ is equal to −194.1 kcal mol−1. Probably, the reason is that negative charge of the amino acid residue is favorable for high ΔG. For the neutral Cys, ΔG is comparable with Cyt: −74.9 kcal mol−1 (see Table 1).
The interaction of neutral metal atoms and diatomic clusters with AAs is high for both silver and gold. It is known that aspartic acid residues may play a significant role in the growth of protein-templated gold nanoclusters.24 For this reason the binding energy between gold and Asp are also high: for Au+, Au0, and Au2 ΔG is equal to −169.8 kcal mol−1, −16.9 kcal mol−1, and −41.1 kcal mol−1.
3.6 AIM analysis of metal complexes with cytosine
We used Bader's AIM analysis to study the nature of nucleobase interactions with metal ions, atoms, and nanoclusters in terms of electron density and its derivatives: ρ(r), ∇2ρ(r), G(r), V(r), and H(r) are presented in Table 8. Metal/nucleobase complex energy density H(r) value is negative for most of the complexes, which means that the electrostatic interaction between metal atoms and cytosine is stabilizing. The positive value of the Laplacian of electron density ∇2ρ(r) and negative value of H(r) in most of the cases means that Me–X bonds are partially covalent and partially electrostatic. In the case of Ag0_Cyt(+H+) with Ag⋯HN1 interaction and Ag2_Cyt(+H+) with Ag–O interaction the local kinetic energy G(r) outweighs V(r): internuclear charge concentration is destabilizing, which is typical for a nonbonded situation, which in the case of Ag0_Cyt(+H+) is also confirmed by positive ΔG value (1.9 kcal mol−1).| Complex | BCP | ρ, hartree | ∇2ρ, hartree | G(r), hartree | V(r), hartree | H(r), hartree | E bond, kcal mol−1 |
|---|---|---|---|---|---|---|---|
| Ag+_Cyt | Ag–N1 | 0.05707 | 0.25983 | 0.07118 | −0.07741 | −0.00622 | 24.29 |
| Ag+_Cyt | Ag–O | 0.04822 | 0.24258 | 0.06458 | −0.06852 | −0.00394 | 21.50 |
| Au+_Cyt | Ag–N1 | 0.13522 | 0.46462 | 0.17029 | −0.22442 | −0.05413 | 70.41 |
| Ag0_Cyt | Ag–N1 | 0.05308 | 0.24042 | 0.06496 | −0.06981 | −0.00485 | 21.90 |
| Au0_Cyt | Au–N1 | 0.08761 | 0.35086 | 0.10845 | −0.12919 | −0.02074 | 40.53 |
| Ag2_Cyt | Ag–N1 | 0.07269 | 0.35332 | 0.09808 | −0.10784 | −0.00975 | 33.84 |
| Au2_Cyt | Au–N1 | 0.11349 | 0.46240 | 0.15277 | −0.18995 | −0.03718 | 59.60 |
| Ag0_Cyt(+H+) | Ag⋯HN1 | 0.00866 | 0.01478 | 0.00355 | −0.00339 | 0.00015 | 1.06 |
| Au0_Cyt(+H+) | Au⋯HN1 | 0.02276 | 0.05869 | 0.01501 | −0.01535 | −0.00034 | 4.82 |
| Ag2_Cyt(+H+) | Ag⋯HN1 | 0.01995 | 0.03969 | 0.01041 | −0.01090 | −0.00049 | 3.42 |
| Ag2_Cyt(+H+) | Ag–O | 0.02725 | 0.13028 | 0.03252 | −0.03246 | 0.00005 | 10.18 |
| Au2_Cyt(+H+) | Au⋯HN1 | 0.03923 | 0.09874 | 0.03033 | −0.03598 | −0.00565 | 11.29 |
| Au2_Cyt(+H+) | Au–O | 0.05692 | 0.28276 | 0.07701 | −0.08333 | −0.00632 | 26.15 |
Ag+_Cyt complex attracted our interest since silver cation forms bonds with both nitrogen and oxygen atoms while Au+ forms a bond only with N1 atom of cytosine. The higher energy of potential energy density for Au–N BCP results in the higher energy of the bond equal to 70.4 kcal mol−1 as compared with Ag–N (24.3 kcal mol−1) and Ag–O (21.5 kcal mol−1), which is supported by Gibbs energy calculations (see Table 7). In the case of neutral metal atoms the situation is similar: for Ag0_Cyt complex Ag–N bond energy is equal to 21.6 kcal mol−1 while Au–N bond energy in Au0_Cyt complex is much higher: 40.53 kcal mol−1. The same is true for Ag2_Cyt and Au2_Cyt complexes: bond energies are equal to 33.8 kcal mol−1 and 59.6 kcal mol−1, respectively. These data are in agreement with Gibbs free energies for metal binding with nucleobases (Table 7): the binding energies are higher for gold atoms than for silver.
4. Conclusion
Binding energies between 19 amino acids and gold nanoparticles have been studied previously using molecular dynamics.66 In this study, Gibbs free energies of interaction of gold cation Au+ and diatomic neutral Au2 cluster with the full set of proteinogenic α-amino acids have been calculated by two methods: RI-MP2 and PBE-D3. The complexes of Au+ and Au2 with deprotonated side chains of all 20 amino acids exhibit the higher values of ΔG than that with the neutral and protonated amino acids, which is in agreement with the data obtained earlier for triatomic gold nanocluster Au3 complexes with glycine and cysteine.45 However, the stability of Au2 complexes with neutral and protonated amino acid residues is also rather high (ΔG is more than −20 kcal mol−1 in absolute values), which indicates that practically all 20 amino acids can stabilize gold NCs and nanoparticles. The binding affinity of AAs to the Au2 cluster increases in the following order: Cys(−H+) > Asp(−H+) > Tyr(−H+) > Glu(−H+) > Arg > Gln, His, Met > Asn, Pro, Trp > Lys, Tyr, Phe > His(+H+) > Asp > Lys(+H+) > Glu, Leu > Arg(+H+) > Ile, Val, Ala > Thr, Ser > Gly, Cys. Generally, the interaction of gold atoms with protonated and deprotonated amino acid residues do not differ greatly, which is in agreement with the experimental evidences that gold cluster synthesis occurs in a wide range of pH.24,67 The fact that deprotonated cysteine has the highest binding energy with both Au2 and Au+ among all the amino acids explains the fine synthesis of gold nanoclusters on thiolates.68,69Our results suggest that binding energy between neutral silver clusters and DNA is rather weak (ΔG is equal to −1 to −9 kcal mol−1) as compared to charged particles (−55 to −63 kcal mol−1). This is in line with experimental fact that DNA-templated metal clusters are positively charged.50–52 The interaction between Au0 atoms and nucleobases is also rather weak (−4 to −6 kcal mol−1); however, the binding energy between diatomic cluster and DNA is higher (−25 to −26 kcal mol−1). This obstacle explains the existence of neutral gold nanoclusters stabilized by poly-cytosine and poly-adenine.54
Speaking about protein templates, the interaction of neutral metal atoms and diatomic clusters with amino acid residues is high for both silver and gold. This is in agreement with the fact that a large part of protein-templated nanoclusters consists of mostly reduced metal atoms.15–17,22
Moreover, the significant difference in the binding energy of neutral gold and silver atoms with nucleobases and amino acids apparently means that unlike DNA template neutral metal atoms are strongly bound to amino acid residues and can't freely diffuse in a polypeptide globula. This fact allows to make a conclusion that formation of metal nanoclusters in proteins occurs through the nucleation of Au atoms located on the neighboring amino acid residues, and the flexibility of the amino acid residue side chains and protein chain as a whole plays a significant role in this process.
Finally, based on the AIM analysis, we have found that for all complexes amino acid–Au2 bonds are partially electrostatic and partially covalent, the same is true for amino acid complexes with Au+.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by the Russian Science Foundation grant 16-13-10090.References
- Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC.
- M. Liu, F. Tang, Z. Yang, J. Xu and X. Yang, J. Anal. Methods Chem., 2019, 2019, 1095148 Search PubMed.
- X. Du and R. Jin, ACS Nano, 2019, 13, 7383–7387 CrossRef CAS PubMed.
- L. Shang and G. U. Nienhaus, Mater. Today, 2013, 16, 58–66 CrossRef CAS.
- T. S. Sych, A. M. Polyanichko, L. V. Plotnikova and A. I. Kononov, Anal. Methods, 2019, 11, 6153–6158 RSC.
- R. Jin, Nanoscale, 2015, 7, 1549–1565 RSC.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- Z. Yuan, Y.-C. Chen, H.-W. Li and H.-T. Chang, Chem. Commun., 2014, 50, 9800–9815 RSC.
- E. Gwinn, D. Schultz, S. Copp and S. Swasey, Nanomaterials, 2015, 5, 180–207 CrossRef.
- Q. You and Y. Chen, J. Mater. Chem. C, 2018, 6, 9703–9712 RSC.
- Y.-S. Borghei, M. Hosseini and M. R. Ganjali, Curr. Nanosci., 2017, 13, 610–615 CAS.
- S. Roy and A. Benerjee, Soft Matter, 2011, 7, 5300–5308 RSC.
- J. Yu, S. A. Patel and R. M. Dickson, Angew. Chem., Int. Ed., 2007, 46, 2028–2030 CrossRef CAS.
- Y. Y. Cui, Y. L. Wang, R. Liu, Z. P. Sun, Y. T. Wei, Y. L. Zhao and X. Y. Gao, ACS Nano, 2011, 5, 8684–8689 CrossRef CAS PubMed.
- A. Mathew, P. R. Sajanlal and T. Pradeep, J. Mater. Chem., 2011, 21, 11205–11212 RSC.
- X. Le Guével, B. Hötzer, G. Jung, K. Hollemeyer, V. Trouillet and M. Schneider, J. Phys. Chem. C, 2011, 115, 10955–10963 CrossRef.
- L. Shang, R. M. Dörlich, V. Trouillet, M. Bruns and G. U. Nienhaus, Nano Res., 2012, 5, 531–542 CrossRef CAS.
- W. Zhang, J. Ye, Y. Zhang, Q. Li, X. Dong, H. Jiang and X. Wang, RSC Adv., 2015, 5, 63821–63826 RSC.
- T. Zhao, Z.-Q. Xuan, A. Wan and R. Gui, Mater. Technol., 2016, 2, 342–347 Search PubMed.
- T. Sych, A. Polyanichko and A. Kononov, J. Mol. Struct., 2017, 1140, 19–21 CrossRef CAS.
- D. Joseph and K. E. Geckeler, Colloids Surf., B, 2014, 115, 46–50 CrossRef CAS.
- T. Zhou, Y. Huang, W. Li, Z. Cai, F. Luo, C. J. Yang and X. Chen, Nanoscale, 2012, 4, 5312–5315 RSC.
- A. A. Buglak, R. R. Ramazanov and A. I. Kononov, Amino Acids, 2019, 51, 855–864 CrossRef CAS.
- H. Kawasaki, K. Hamaguchi, I. Osaka and R. Arakawa, Adv. Funct. Mater., 2011, 21, 3508–3515 CrossRef CAS.
- K. Chaudhari, P. L. Xavier and T. Pradeep, ACS Nano, 2011, 5, 8816–8827 CrossRef CAS.
- A. C. Templeton, W. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27–36 CrossRef CAS PubMed.
- C. J. Ackerson, P. D. Jadzinsky and R. D. Kornberg, J. Am. Chem. Soc., 2005, 127, 6550–6551 CrossRef CAS.
- C. A. Simpson, C. L. Farrow, P. Tian, S. J. L. Billinge, B. J. Huffman, K. M. Harkness and D. E. Cliffel, Inorg. Chem., 2010, 49, 10858–10866 CrossRef CAS.
- Z. Zhong, S. Patskovskyy, P. Bouvrette, J. H. T. Luong and A. Gedanken, J. Phys. Chem. B, 2004, 108, 4046–4052 CrossRef CAS.
- R. L. Willett, K. W. Baldwin, K. W. West and L. N. Pfeiffer, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7817–7822 CrossRef CAS.
- R. R. Naik, S. E. Jones, C. J. Murray, J. C. McAuliffe, R. A. Vaia and M. O. Stone, Adv. Funct. Mater., 2004, 14, 25–30 CrossRef CAS.
- S. Brown, M. Sarikaya and E. Johnson, J. Mol. Biol., 2000, 299, 725–735 CrossRef CAS.
- Y. N. Tan, J. Y. Lee and D. I. C. Wang, J. Am. Chem. Soc., 2010, 132, 5677–5686 CrossRef CAS PubMed.
- I. Tolbatov, C. Coletti, A. Marrone and N. Re, Int. J. Mol. Sci., 2019, 20, 820 CrossRef.
- J. Xie, J. Y. Lee and D. I. C. Wang, J. Phys. Chem. C, 2007, 111, 10226–10232 CrossRef CAS.
- N. K. Mishra, Catalytic Application of Nano-Gold Catalysts, IntechOpen, London, 2016 Search PubMed.
- F. Ramezani, M. Amanlou and H. Rafii-Tabar, Amino Acids, 2014, 46, 911–920 CrossRef CAS.
- M. Singh, I. Sinha and R. K. Mandal, Mater. Lett., 2009, 63, 425–427 CrossRef CAS.
- B. A. Lakkappa, S. C. Jasmith and M. G. Prabhuodeyara, Org. Med. Chem. Int. J., 2017, 3, 555622 Search PubMed.
- O. A. Baranova, S. D. Khizhnyak and P. M. Pakhomov, J. Struct. Chem., 2016, 57, 1203–1208 CrossRef CAS.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS.
- X. Le Guével, B. Hötzer, G. Jung, K. Hollemeyer, V. Trouillet and M. Schneider, J. Phys. Chem. C, 2011, 115, 10955–10963 CrossRef.
- W. Ding, Y. Liu, Y. Li, Q. Shi, H. Li, H. Xia, D. Wang and X. Tao, RSC Adv., 2014, 4, 22651–22659 RSC.
- B. A. Russell, B. Jachimska, P. Komorek, P. A. Mulheran and Y. Chen, Phys. Chem. Chem. Phys., 2017, 19, 7228–7235 RSC.
- H. Pakiari and Z. Jamshidi, J. Phys. Chem. A, 2007, 111, 4391–4396 CrossRef PubMed.
- S. Rai, N. V. Suresh Kumar and H. Singh, Bull. Mater. Sci., 2012, 35, 291–295 CrossRef CAS.
- M. H. Abdalmoneam, K. Waters, N. Saikia and R. Pandey, J. Phys. Chem. C, 2017, 121, 25585–25593 CrossRef CAS.
- Y. L. Wang, H. J. Zhai, L. Xu, J. Li and L. S. Wang, J. Phys. Chem. A, 2010, 114, 1247–1254 CrossRef CAS PubMed.
- S. Goel, K. A. Velizhanin, A. Piryatinski, S. A. Ivanov and S. Tretiak, J. Phys. Chem. C, 2012, 116, 3242–3249 CrossRef CAS.
- D. Schultz, K. Gardner, S. S. R. Oemrawsingh, N. Markešević, K. Olsson, M. Debord, D. Bouwmeester and E. G. Gwinn, Adv. Mater., 2013, 25, 2797–2803 CrossRef CAS.
- R. R. Ramazanov, T. S. Sych, Z. V. Reveguk, D. A. Maksimov, A. A. Vdovichev and A. I. Kononov, J. Phys. Chem. Lett., 2016, 7, 3560–3566 CrossRef CAS PubMed.
- J. T. Petty, M. Ganguly, I. J. Rankine, E. J. Baucum, M. J. Gillan, L. E. Eddy, J. C. Léon and J. Müller, J. Phys. Chem. C, 2018, 122, 4670–4680 CrossRef CAS.
- V. Soto-Verdugo, H. Metiu and E. Gwinn, J. Chem. Phys., 2010, 132, 195102 CrossRef.
- T. A. Kennedy, J. L. MacLean and J. Liu, Chem. Commun., 2012, 48, 6845–6847 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weinhold and C. R. Landis, Chem. Educ. Res. Pract., 2001, 2, 91–104 RSC.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. S. Sych, A. A. Buglak, Z. V. Reveguk, V. A. Pomogaev, R. R. Ramazanov and A. I. Kononov, J. Phys. Chem. C, 2018, 122, 26275–26280 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- I. L. Volkov, R. R. Ramazanov, E. V. Ubyivovk, V. I. Rolich, A. I. Kononov and N. A. Kasyanenko, ChemPhysChem, 2013, 14, 3543–3550 CrossRef CAS.
- M. K. Shukla, M. Dubey, E. Zakar and J. Leszczynski, J. Phys. Chem. C, 2009, 113, 3960–3966 CrossRef CAS.
- Q. Shao and C. K. Hall, Langmuir, 2016, 32, 7888–7896 CrossRef CAS.
- A. Tofanello, É. G. A. Miranda, I. W. R. Dias, A. J. C. Lanfredi, J. T. Arantes, M. A. Juliano and I. L. Nantes, ACS Omega, 2016, 1, 424–434 CrossRef CAS PubMed.
- O. Lopez-Acevedo, J. Akola, R. L. Whetten, H. Gronbeck and H. Häkkinen, J. Phys. Chem. C, 2009, 113, 5035–5038 CrossRef CAS.
- O. Lopez-Acevedo, H. Tsunoyama, T. Tsukuda, H. Hakkinen and C. M. Aikens, J. Am. Chem. Soc., 2010, 132, 8210–8218 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06486f |
| This journal is © The Royal Society of Chemistry 2020 |