
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiochemical assay for serum creatinine detection through a 1-methylhydantoin and cobalt complex
Pallavi Dasguptaa,
Vinay Kumarb,
Patnam R. Krishnaswamya and
Navakanta Bhat *a
*a
aCentre for Nanoscience and Engineering, Indian Institute of Science, Bengaluru, India. E-mail: navakant@iisc.ac.in
bPathShodh Healthcare Pvt. Ltd., Bengaluru, India
First published on 26th October 2020
Abstract
Creatinine is a reliable indicator of renal function and degradation of muscular metabolism. Current analytical techniques for its measurement are limited by their cost and requirement of sophisticated instruments. In this work, we report a highly sensitive amperometric biosensor for creatinine by utilizing the one-step selective conversion of creatinine by creatinine deiminase. The novelty of the proposed sensor relies on the measurement of N-methylhydantoin produced in the reaction. The sensing chemistry comprises of creatinine deiminase as the receptor for creatinine, cobalt chloride as the electrochemically active recognition element, and methylene blue as the redox mediator. The sensing chemistry is immobilized on the glassy carbon electrode surface by physisorption. We have been able to provide a standalone device that reliably quantifies creatinine in serum and even whole blood, without any sample pre-processing. It is possible to measure creatinine in the clinically relevant range from 0.8 to 4 mg dL−1 with this approach.
1 Introduction
The muscles of an individual contain creatine that serves as a reservoir of high energy phosphate that is released during muscular exertion, converting creatine to creatine phosphate. This reaction is reversible and both creatine (∼40%) and creatine phosphate (∼60%) are found in muscles and even in brain and blood to an extent.1 Depending on an individual's muscle mass, creatine phosphate is converted to creatinine by a non-enzymatic route at a constant rate. The creatinine released is not protein bound, physiologically inert, and is freely filtered by the kidneys into the urine. The urinary creatinine excretion is <100 mg/24 hours.1 Under physiological conditions, the baseline concentration of creatinine in serum is low, i.e., 0.5–1 mg dL−1 in females and 0.7–1.2 mg dL−1 in males owing to clearance of creatinine in urine.1 In case of alteration or decline in filtration by kidneys or muscle degradation due to muscular dystrophy, the creatinine levels in serum increase. An increase in serum creatinine concentration beyond 2 mg dL−1 indicates significant decline in renal function. Thus, measuring concentration of serum creatinine provides an easier and accurate reflection of renal efficiency.2,3Owing to the importance of serum creatinine as a vital clinical biomarker, investigation into techniques for its reliable estimation is still underway. In 1886, Max Jaffe identified a chromogen that formed due to interaction between creatinine and picric acid in an alkaline media. In 1904, Folin utilized this reaction to estimate creatinine in biological fluids. This technique remains the cornerstone for creatinine estimation in clinical laboratories, till date. However, it suffers from both, positive and negative interferences from non-creatinine chromogens leading to an overestimation of ∼20%.1 This necessitated the adoption of enzymatic assays for creatinine quantification. In 1976, Meyerhoff developed the first enzymatic sensor for creatinine in which creatinine was hydrolyzed by creatinine deiminase and liberated ammonia. It was a potentiometric sensor that relied on ammonia liberation.4 However, the major drawback of this sensor was interference from endogenous ammonia. This led to increased efforts by diverse groups to minimize influence from endogenous ammonia4 or even adopt different sensing techniques for reliable estimation of creatinine. Tsuchida and Yoda developed the first amperometric enzymatic sensor for creatinine using a cascaded sequence of enzymes—creatinine amidohydrolase, creatine amidinohydrolase, and sarcosine oxidase.5 Although majority of the existing sensors for creatinine estimation employ amperometric technique, increased complexity of the synergistic interaction among the multiple enzymes and a consequent decrease in its sensitivity limits its application as a point-of-care (POC) device. Hence, it is ideal to utilize a simple mono-enzymatic pathway for creatinine estimation,6–8 such as that involving creatinine deiminase, with no significant interferences.
In this regard, a recent work proposed a sensor that coupled creatinine deiminase with a specific quantum dot for estimating creatinine via fluorescence resonance energy transfer.9 The resulting output signal is proportional to the logarithm of creatinine concentration and is consequently insensitive to the lower concentration of serum creatinine. Another work developed an ISFET that involved immobilization of creatinine deiminase on a complex zeolite modified electrode with no investigation of response with real samples.10 Further, since the device still relies on change in ammonium ion concentration, it would observe interference from endogenous ammonia levels. In order to overcome the inherent disadvantages of potentiometric sensors, a conductometric sensor was also developed that immobilized creatinine deiminase on a composite film of PVA/PEI/AuNPs.11 Although the sensor exhibits an acceptable linear range, it highlights the need of a cumbersome fabrication for a viable sensor. Further, the behavior of the proposed device in a complex matrix of serum or blood has not been reported. Hence, there is a need for an amperometric device that assures a broad linear range of detection with a mono-enzymatic approach that provides simplicity and reliability of creatinine estimation.
Here, we report an amperometric sensor for creatinine estimation by one-step enzymatic hydrolysis of creatinine by creatinine deiminase. The mono-enzymatic route provides a simple yet reliable approach for creatinine quantification. Rather than measuring ammonia, we focus on the measurement of N-methylhydantoin, which is the other reaction product. The sensor couples the enzymatic conversion to an electrochemically active recognition element—cobalt chloride—in conjunction with an electron mediator—methylene blue. The sensor can estimate creatinine in serum and whole blood from 0.8 to 4 mg dL−1 within 1 minute without any sample pre-processing. The proposed sensor also contains the various components of the sensing chemistry immobilized on the electrode surface and hence, provides an independent device for creatinine estimation.
2 Principle of detection
The proposed sensor utilizes one step enzymatic conversion of creatinine to N-methylhydantoin (majority of which is 1-methylhydantoin) by creatinine deiminase (Scheme 1(a)). We focus on measuring the 1-methylhydantoin concentration to provide an indirect estimation of creatinine present in the physiological sample. The measurement of 1-methylhydantoin has been reported for the first time in this work, as a plausible route for quantification of creatinine. This is attained by developing an electrochemical biosensor for its quantification.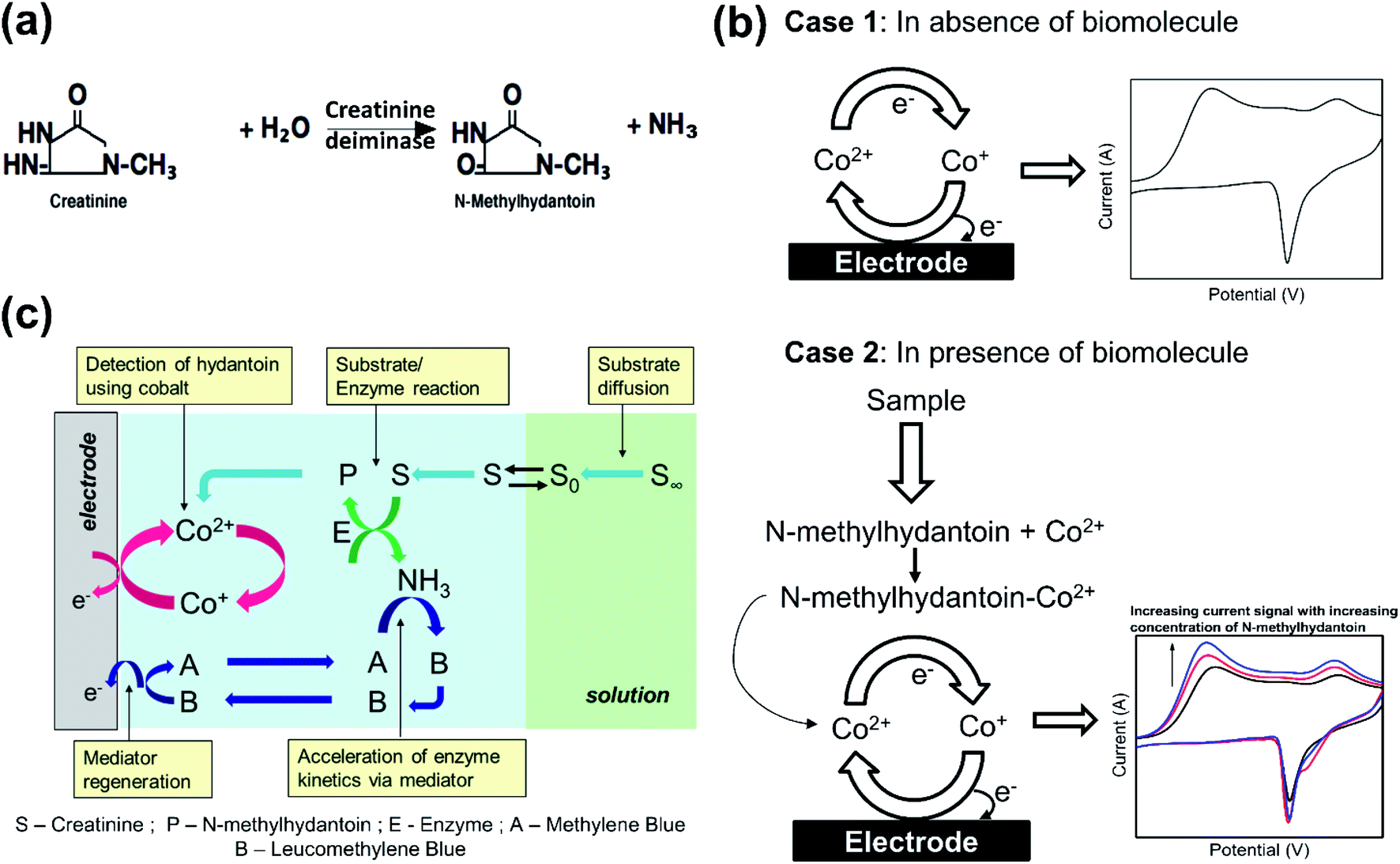 | ||
| Scheme 1 Principle of electrochemical estimation. (a) Mono-enzymatic conversion of creatinine by creatinine deiminase to form N-methylhydantoin and ammonia. (b) Cyclic voltammogram of cobalt is indicated in absence and presence of biomolecule. In absence of biomolecule, cobalt indicates a current signal owing to change in its oxidation state on application of potential. In presence of N-methylhydantoin, cobalt forms an electroactive complex with it and hence, current signal is associated with the redox transition of the newly formed complex. Here, the current signal increases with increasing concentration of N-methylhydantoin. (c) Illustration of simultaneous reactions in the electrode vicinity owing to interaction of sensing chemistry with the target analyte. Creatinine (S) from the bulk solution diffuses near the electrode surface, where it undergoes enzymatic conversion by creatinine deiminase (E) to form 1-methylhydantoin (P). The resulting N-methylhydantoin (P) then forms a complex with the cobalt ions that is subsequently detected by the electrode. Simultaneously, the enzyme kinetics is ensured by the presence of a redox mediator (A) that quenches the liberated ammonia and maintains optimal conditions for the enzyme activity. | ||
An electrochemical biosensor couples the recognition event of the target analyte to an electrochemical transducer that converts the recognition event to a measurable current signal. It requires a redox species, or a chemical that undergoes electron transfer to derive the current signal. The criteria are met inherently if the target analyte has a redox centre, otherwise it can be addressed by binding the target analyte with a redox chemical. In this case, 1-methylhydantoin does not undergo electron transfer and we utilize a transition metal such as cobalt to bind the analyte and derive a current signal. It is well reported that transition metals form a complex with hydantoin moieties by attaching at its N3 position.12–14 Since these metals can exist in multiple oxidation states, they readily undergo electron transfer. Although any transition metal such as nickel, copper, iron or cobalt12–16 may be utilized, the signal of cobalt ions was observed to be robust against interferences from insulating serum proteins, mainly albumin. Hence, on application of a potential sweep, cobalt exhibits an electrochemical current signal that reflects its concentration in accordance with the Faraday's law (Scheme 1(b)). However, in presence of a ligand, such as, 1-methylhydantoin that binds with cobalt, the intensity of the current signal will alter in proportion to its concentration, depending on the nature of the complex. In this case, the complex formed is electroactive and hence, the intensity of the current signal increases with increasing concentration of 1-methylhydantoin, due to increased formation of the cobalt–hydantoin complex (Scheme 1(b)). The electroactivity of cobalt–hydantoin complex is attributed to the ability of the conjugated cobalt ion to undergo electron transfer reaction. The cobalt ions when conjugated with hydantoin moiety undergo these electron transfer reactions more readily as compared to the solvated cobalt ions. This occurs due to the supply of delocalized electrons from the anionic hydantoin moiety conjugated with it. Thus, we can correlate the intensity of the current signal to the concentration of 1-methylhydantoin.
The electrochemical quantification of 1-methylhydantoin is then coupled with enzymatic conversion of creatinine to 1-methylhydantoin to yield an indirect estimation of creatinine. The kinetics of the enzymatic conversion can be accelerated by using a mediator, such as, methylene blue. The increase in the enzymatic kinetics is achieved by quenching of the liberated ammonia by methylene blue. As a result, methylene blue is converted to leucomethylene blue that is then regenerated via redox transition back to methylene blue. Depletion of ammonia by methylene blue creates a concentration gradient and thereby enhances the enzyme kinetics in the forward direction. This quenching also minimizes any local change in pH as a result of the enzymatic conversion. This ensures maintenance of optimal conditions for the enzyme. Although, the reduction of cobalt–hydantoin complex and regeneration of mediator occurs simultaneously, however, these two processes do not interfere. This is assured by employing a lower concentration of methylene blue in presence of a significantly higher concentration of cobalt chloride. Further, the reduction of cobalt–hydantoin complex occurs at ∼−0.9 V, while that of methylene blue occurs at ∼−0.4 V. This ensures that the electrochemical processes of each proceed un-influenced by the other.
Hence, the sensing chemistry comprises of cobalt chloride as the biorecognition element, creatinine deiminase enzyme as a receptor that hydrolyses creatinine to detectable 1-methylhydantoin, and methylene blue as a mediator for accelerated kinetics of enzymatic conversion, as indicated in Scheme 1(c).
The sensing chemistry is immobilized on the working electrode directly. The modified electrode is then utilized for electrochemical measurements to quantify 1-methylhydantoin in saline and also, from creatinine. Immobilized sensing chemistry ensures simultaneous enzymatic conversion accelerated by mediator and subsequent electrochemical detection. It also increases the accessibility of the proposed sensor for decentralized testing of creatinine. In addition, it minimizes intervention of users and requirement of trained personnel for performing the measurements. Further, it highlights the potential of further development of portable and disposable platforms with the immobilized sensing chemistry for routine and continuous measurements of creatinine.
3 Experimental section
3.1 Materials and methods
All the chemicals such as creatinine, 1-methylhydantoin, human serum albumin, sodium chloride, phosphate buffer (PBS), creatinine deiminase (21 units in 1 mg), and cobalt chloride were procured from Sigma Aldrich. All the solutions were prepared in deionized (DI) water. Cobalt chloride and methylene blue solutions were prepared in 0.085 M sodium chloride, while that of creatinine deiminase enzyme was prepared in 1 M PBS. The analyte, i.e., creatinine and 1-methylhydantoin along with albumin were prepared in physiological levels of saline (0.154 M sodium chloride).The experimental setup involves a three-electrode electro-chemical cell with glassy carbon as working electrode, platinum wire as reference electrode, and platinum foil as counter electrode. The total volume in the electrochemical cell was maintained at 10 mL. The electrodes were cleaned prior to electrochemical measurements. Glassy carbon electrode was cleaned by scrubbing with alumina powder on the micro-fibre cleaning pad and subsequent washing with DI water several times. The electrode was then dried under ambient conditions. The platinum electrodes were cleaned by dipping it in dilute hydrochloric acid followed by recurrent washing with DI water. Although a non-standard reference electrode was employed for the measurements, no drift of peak potentials or electrolysis of the supporting electrolyte was observed under the experimental conditions.
3.2 Preparation of modified electrode
The electrode surface was modified by drop-casting the sensing chemistry on the surface of glassy carbon electrode and thereafter dried in hot air oven at 45 °C for 20 min. The modified electrode was then used for electrochemical measurements. In case of estimation of different concentrations of 1-methylhydantoin from saline, only 4.5 mg cobalt chloride was dispensed on the electrode surface. However, for estimation from creatinine, the sensing chemistry solution was prepared by mixing 4.5 mg cobalt chloride, 0.5 mg methylene blue in 0.085 M NaCl, and 0.5 units of creatinine deiminase in 1 M PBS.The sensing chemistry has been affixed to the electrode surface by a simple process of physisorption that does not entail any irreversible chemical reaction. As the modified electrode is introduced to the electrochemical cell, the adsorbed sensing chemistry undergoes gradual dissolution or leaching. Although, immobilization by physisorption ensures easier accessibility of the ions of the sensing chemistry in the solvated form to the analyte in the electrochemical cell, it also involves loss of sensing chemistry from the electrode surface. However, this technique has been adopted to demonstrate the ability of immobilized sensing chemistry to respond to varying concentrations of the analyte. Furthermore, the minor losses of sensing chemistry by leaching is compensated by loading an excess amount of each component of the sensing chemistry.
4 Results and discussion
4.1 Electrochemical characterization of cobalt-modified electrode
Cyclic voltammograms of the cobalt modified electrode (Fig. 1(a)) was recorded in presence of 1-methylhydantoin in saline media as shown in Fig. 1(b). The immobilized cobalt gives rise to three reduction peaks at 0.1, −0.38, and −0.85 V, respectively for the stepwise change in oxidation state of cobalt. Two of these peaks are attributed to the conversion of immobilized Co(II) to Co(I) and finally to Co(0). The presence of reduction peak at −0.38 V indicates existence of an intermediate complex oxidation state of cobalt that gradually disappears in presence of increasing concentration of 1-methylhydantoin. The requirement of higher energy for conversion of Co(II) to lower oxidation states is reflected by the increased shift of the reduction potential in the negative direction.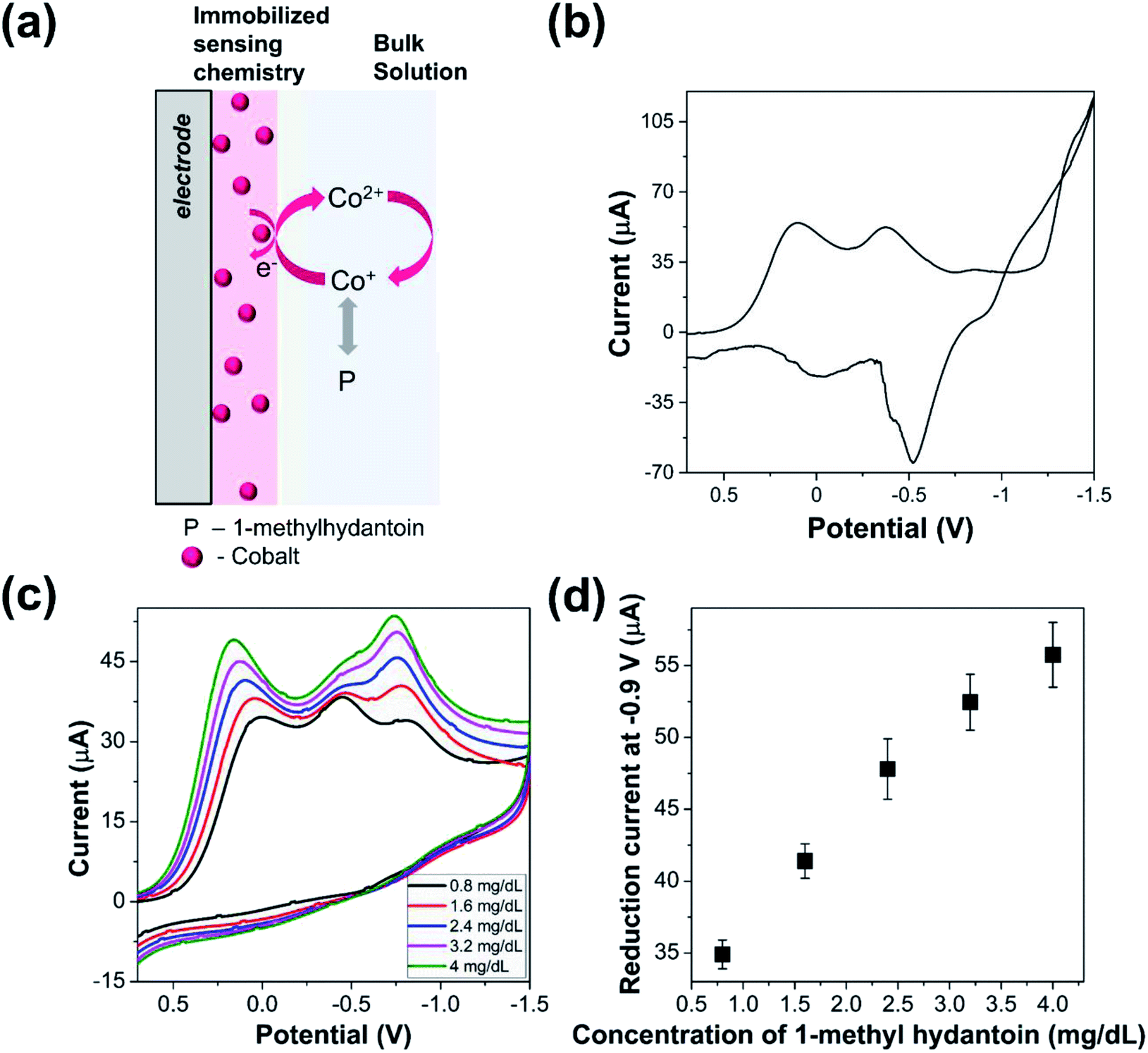 | ||
| Fig. 1 Electrochemical detection of 1-methylhydantoin. (a) Schematic of modified electrode for estimation of 1-methylhydantoin. Cobalt chloride is physisorbed on the electrode surface by drop-casting 4.5 mg cobalt chloride on the glassy carbon electrode. The electrode is then dried in a hot air oven at 45 °C for 20 min. The modified electrode is then placed in the electrochemical cell. Different concentrations of 1-methylhydantoin is then introduced in the cell that binds with the immobilized cobalt ions and thereby alters the current signal. (b) Cyclic voltammogram of cobalt-modified electrode in saline media. (c) Cyclic voltammograms of cobalt-modified electrode in presence of increasing concentration of 1-methylhydantoin from 0.8 to 4 mg dL−1. (d) Variation in reduction current signal of cobalt-modified electrode in presence of increasing concentration of 1-methylhydantoin from 0.8 to 4 mg dL−1. The error bars indicate the variability arising due to triplicated measurements for each concentration. | ||
Further, the cyclic voltammograms of cobalt modified electrode indicated a reduction peak during the reverse potential sweep and associated oxidation peaks. The residual reduction peak is indicative of cobalt deposition on the electrode surface, while the oxidation peak reflects the stepwise oxidation of Co(0) to Co(I) and Co(II). However, the signal suffered from noise. This may be attributed to the non-specific adsorption of accompanying ions, such as, chloride and sodium that increase the capacitive current and increase the energy barrier for oxidation of cobalt ions.
4.2 Detection of 1-methylhydantoin using cobalt-modified electrode
The cobalt modified electrode was utilized for electrochemical estimation of 1-methylhydantoin. Cyclic voltammograms of the modified electrode were recorded in presence of varying concentrations of 1-methylhydantoin, as shown in Fig. 1(c). A linearly increasing reduction current signal was obtained at 0 V and −0.9 V with increasing 1-methylhydantoin concentration from 0.8 to 4 mg dL−1 (Fig. 1(d)). The reduction peak at −0.58 V merged with the peak at −0.9 V in presence of increasing concentrations of 1-methylhydantoin. It can be inferred that the conjugated cobalt ions undergo similar electron transfer reactions as the adsorbed cobalt ions. The similarity lies in a similar change in oxidation state of cobalt ions. Furthermore, a minor shift of the peak potential in the positive direction was observed with increasing concentrations of 1-methylhydantoin. This also reaffirms the hypothesis of formation of an electroactive cobalt–hydantoin complex. This complex formation alters the redox peak potential, in accordance with the Nernst equation. However, the cyclic voltammograms did not indicate any oxidation peak in presence of varying concentrations of 1-methylhydantoin. This may be attributed to the irreversible redox transfer of cobalt–hydantoin complex and absence of free cobalt ions to undergo associated oxidation reactions.The linearly correlated reduction current signal increased with increasing reaction time. This indicates a positive correlation between the formation of an electroactive cobalt–hydantoin complex and the reaction time. Triplicated measurements indicated the coefficient of variation to be within ∼10%. This highlights the reliability of the cobalt modified electrode for quantification of 1-methylhydantoin.
4.3 Optimization of parameters of mediated enzymatic reaction
As indicated, the cobalt-modified electrode can quantify varying concentrations of 1-methylhydantoin. However, for the estimation of creatinine via 1-methylhydantoin, the enzymatic conversion of creatinine must be coupled with the electrochemical quantification of the generated 1-methylhydantoin. In accordance, the sensing chemistry now comprises of cobalt chloride, creatinine deiminase, and methylene blue. Hence, it is imperative to ascertain the influence of the varied components of the sensing chemistry and variation in ambient conditions that influence the electrochemical cell on the electrochemical estimation of creatinine. This is crucial to ensure the occurrence of simultaneous steps in presence of additional reagents and to ensure optimal conditions for similar activity of creatinine deiminase at all concentrations of creatinine. The sensor would then yield current signals that reflect varying concentrations of 1-methylhydantoin and thereby, creatinine.The factors that influence the electrochemical quantification by the mediated enzymatic reaction include the concentration of each component of the sensing chemistry—cobalt chloride, creatinine deiminase, and methylene blue—reaction time, pH, temperature, immobilization sequence. In this case, all the measurements were performed at room temperature and hence it was not varied. The reaction time was fixed at 1 minute based on experimental data observed with electrochemical measurements of 1-methylhydantoin from creatinine using the sensing chemistry and analyte in solution.
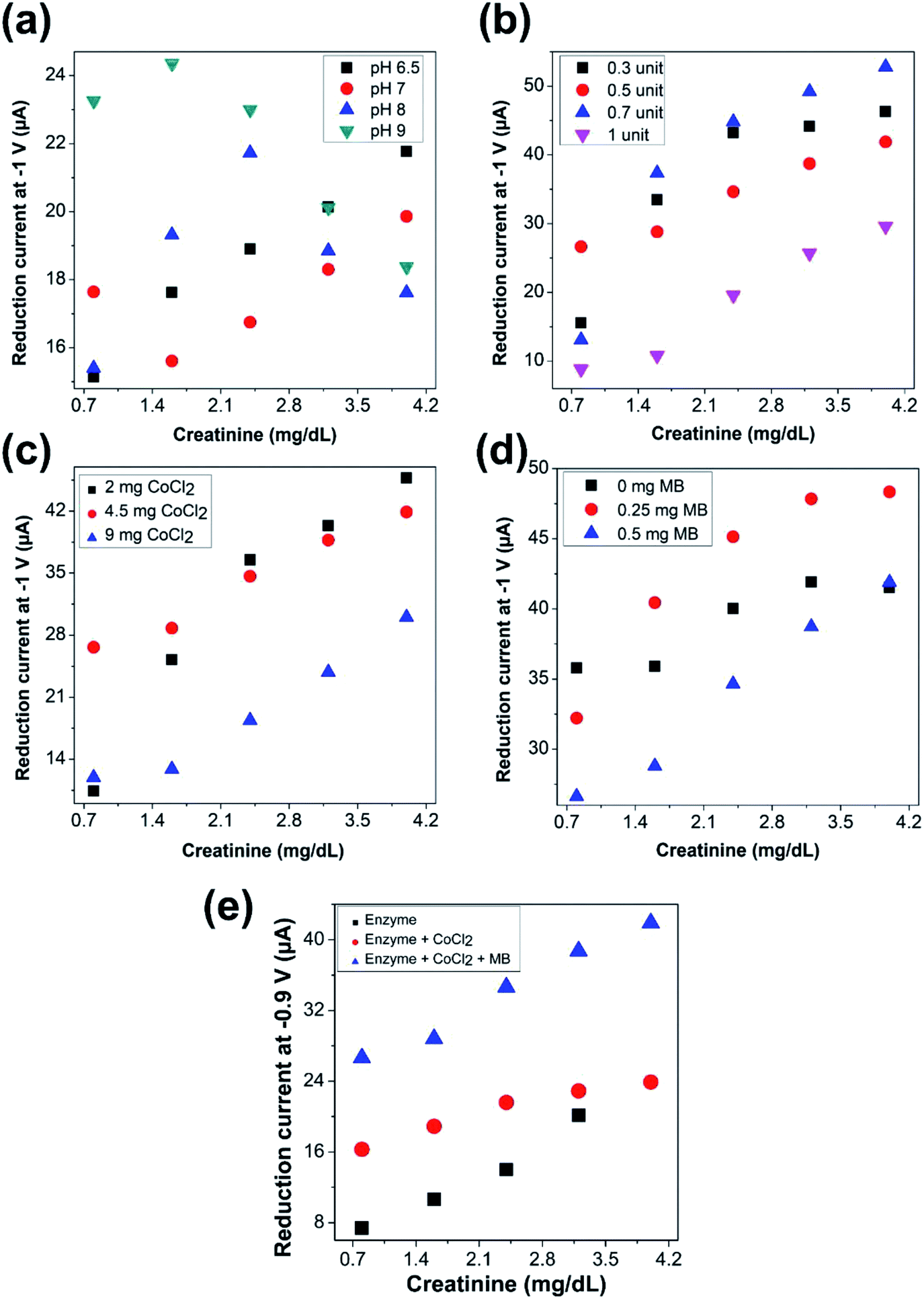 | ||
| Fig. 2 Optimization of parameters for detection of creatinine. Effect of (a) variation of pH of the test solution (with 4.5 mg CoCl2, 0.5 mg methylene blue, and 0.5 units of creatinine deiminase dispensed on the electrode and in presence of 3 g dL−1 albumin in test solution) (b) variation of concentration of the enzyme dispensed (with 4.5 mg CoCl2 and 0.5 mg methylene blue) on the electrode surface (c) variation of quantity of cobalt chloride dispensed (with 0.5 mg methylene blue and 0.5 units of creatinine deiminase) on the electrode surface (d) variation of quantity of methylene blue dispensed (with 4.5 mg CoCl2 and 0.5 units of creatinine deiminase) on the electrode surface, on the electrochemical estimation of 1-methylhydantoin from creatinine. (e) Effect of immobilization of different components of the sensing chemistry on the electrode surface—enzyme alone (0.5 units of creatinine deiminase), enzyme (0.5 units of creatinine deiminase) with cobalt chloride (4.5 mg), and enzyme (0.5 units of creatinine deiminase) with cobalt chloride (4.5 mg) along with methylene blue (0.5 mg). | ||
 in Fig. 2(e)), the enzyme along with cobalt chloride was immobilized on the electrode surface, while methylene blue was present in solution along with varying concentrations of the analyte. In the third case (as indicated by
in Fig. 2(e)), the enzyme along with cobalt chloride was immobilized on the electrode surface, while methylene blue was present in solution along with varying concentrations of the analyte. In the third case (as indicated by  in Fig. 2(e)), the entire sensing chemistry comprised of enzyme, cobalt chloride, and methylene blue was immobilized on the electrode surface, while the solution only composed of the intended physiological fluid with varying concentrations of the analyte.
in Fig. 2(e)), the entire sensing chemistry comprised of enzyme, cobalt chloride, and methylene blue was immobilized on the electrode surface, while the solution only composed of the intended physiological fluid with varying concentrations of the analyte.It was observed that a linearly increasing current signal was obtained with increasing creatinine concentration for all the cases. The current levels increased with cobalt chloride along with enzyme immobilization (second case) as compared to only enzyme immobilization (first case), as more cobalt ions were accessible to the electrode surface thereby overcoming limitation due to diffusion. Further, the even higher current levels observed with immobilization of the entire sensing chemistry (third case) confirms the accelerated electron transfer due to inclusion of the redox mediator. The intensity of the current signals was highest with maximum sensitivity, in case of immobilization of the entire sensing chemistry on the electrode surface. Hence, it was ascertained to be the optimal choice of immobilization sequence for electrochemical sensing of creatinine.
4.4 Electrochemical detection of creatinine in saline media
The proposed sensor with the optimized parameters was then utilized to investigate the electrochemical detection of creatinine, as iilustrated in Fig. 3(a). The concentration of creatinine was varied, and cyclic voltammograms were recorded with the modified electrode, as shown in Fig. 3(b). A linearly increasing reduction current signal for all the three peaks was observed with increasing creatinine concentration. However, we continue to monitor the intensity of the current signals derived from the reduction of cobalt–hydantoin complex at −0.85 V, as indicated in Fig. 3(c). Based on the previous results, this is intuitive as the measurements imply that increasing concentrations of creatinine is gradually converted to 1-methylhydantoin which then binds with cobalt to form an electroactive complex. The increased formation of the electroactive complex leads to increasing current signal with increasing concentration of creatinine. Triplicated measurements were performed for detection of creatinine with the modified electrode to assess its performance and the coefficient of variation was within ∼10%.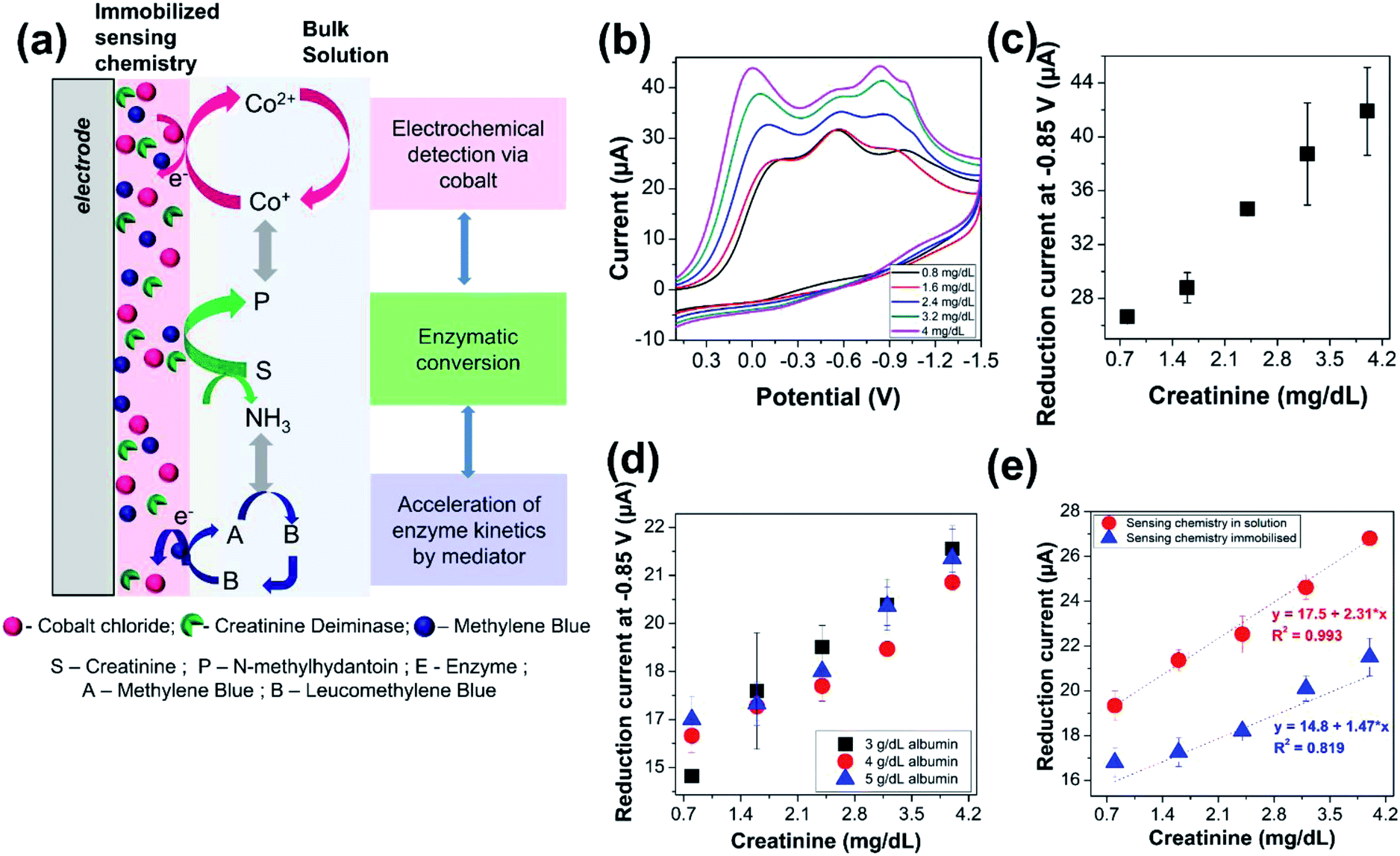 | ||
| Fig. 3 Electrochemical detection of creatinine. (a) Schematic of the modified electrode and electrochemical quantification of creatinine. The electrode surface is modified by dispensing 4.5 mg cobalt chloride, 0.5 mg methylene blue, and 0.5 units of creatinine deiminase on the electrode surface. The electrode is subsequently dried in a hot air oven at 45 °C for 20 min. It is then introduced into the electrochemical cell. Electrochemical estimation of creatinine is then performed by varying the concentration of creatinine in the cell. The creatinine in the test solution undergoes enzymatic conversion to form 1-methylhydantoin by the enzyme immobilized on the electrode surface. It then forms a complex with the immobilized cobalt ions to yield the corresponding current signal correlated to its concentration. (b) Cyclic voltammograms of the modified electrode in presence of increasing concentration of creatinine from 0.8 to 4 mg dL−1. (c) Variation in reduction current of the modified electrode in presence of increasing concentration of creatinine from 0.8 to 4 mg dL−1. (d) Variation in reduction current of the modified electrode due to increasing concentration of creatinine from 0.8 to 4 mg dL−1 in presence of physiological levels of serum albumin, i.e., 3–5 g dL−1 albumin. (e) Comparison of electrochemical estimation of creatinine with sensing chemistry comprising cobalt chloride, creatinine deiminase, and methylene blue present in a homogenous solution with creatinine and albumin against immobilization of the sensing chemistry on the electrode surface. | ||
4.5 Electrochemical quantification of creatinine in presence of serum proteins
The reliability and selectivity of the sensor was then evaluated in presence of serum proteins that constitute a primary source of interference for majority of the biosensors. Serum proteins are predominated by albumin, whose concentration ranges from 3 to 5 g dL−1. Hence, electrochemical measurements of 1-methylhydantoin from creatinine in presence of physiological levels of serum albumin, i.e., 3–5 g dL−1, was performed. A linearly increasing current signal was obtained with increasing creatinine concentration from 0.8 to 4 mg dL−1, as shown in Fig. 3(d). Triplicated measurements were performed, and the coefficient of variation was found to be within 10%, as indicated in Table 1. This confirms that the interference from serum proteins is negligible, and hence, the electrochemical quantification of creatinine can be performed directly from serum without any pre-processing. This is a distinct advantage as it meets the goal of POC estimation of creatinine by eliminating the need of any pre-treatment procedures for removal of any possible interferents from the complex matrix of serum or even whole blood.| Creatinine (mg dL−1) | Concentration of albumin (g dL−1) | |||||||
|---|---|---|---|---|---|---|---|---|
| 0 | 3 | 4 | 5 | |||||
| Average current (μA) | Coefficient of variation (%) | Average current (μA) | Coefficient of variation (%) | Average current (μA) | Coefficient of variation (%) | Average current (μA) | Coefficient of variation (%) | |
| 0.8 | 26.6 | 0.88 | 15.1 | 0.96 | 16.3 | 3.08 | 16.8 | 3.90 |
| 1.6 | 28.8 | 3.90 | 17.6 | 9.63 | 17.2 | 2.39 | 17.3 | 3.69 |
| 2.4 | 34.6 | 0.65 | 18.9 | 3.33 | 17.8 | 2.51 | 18.2 | 0.78 |
| 3.2 | 38.7 | 9.77 | 20.1 | 3.70 | 18.9 | 0.41 | 20.1 | 2.81 |
| 4 | 41.9 | 7.78 | 21.8 | 3.09 | 20.8 | 0.37 | 21.5 | 3.95 |
The current signals arising from the electrochemical estimation of 1-methylhydantoin from creatinine with immobilized sensing chemistry is slightly lower compared to electrochemical detection with sensing chemistry in solution, as shown in Fig. 3(e). This is attributed to a delay or variation in enzymatic conversion of creatinine to 1-methylhydantoin followed by electrochemical detection. However, the immobilized dry chemistry gives as good a linearity of detection as the liquid chemistry. This is crucial for the POC diagnostic applications.
4.6 Electrochemical quantification of creatinine in real samples
Electrochemical measurements were then performed in whole blood samples that were spiked with different concentrations of creatinine by standard addition (Fig. 4(a)). An image of the modified electrode is indicated in Fig. 4(b). A volume of 300 μL of whole blood sample was spiked with creatinine and the total volume was maintained at 10 mL by 0.085 M sodium chloride. The pH of the spiked blood samples was measured and was observed to be within 6.5–7, which is the optimum pH for the required enzyme activity for the electrochemical measurements. The fluid in the electrochemical cell containing blood sample was spiked with increasing concentrations of creatinine from 0.8 to 4 mg dL−1. In accordance with the measurements in saline media, a linearly increasing reduction current signal was observed with increasing concentrations of creatinine (from 13.6 μA corresponding to spiked creatinine concentration of 0.8 mg dL−1 to 23 μA corresponding to spiked creatinine concentration of 4 mg dL−1) even in whole blood sample using glassy carbon electrode modified with the sensing chemistry, as shown in Fig. 4(c). This indicates that electrochemical quantification of creatinine is feasible directly from whole blood samples without any sample pre-treatment or data processing post measurement. It provides a path for future development of a POC device for creatinine measurement from whole blood and serum.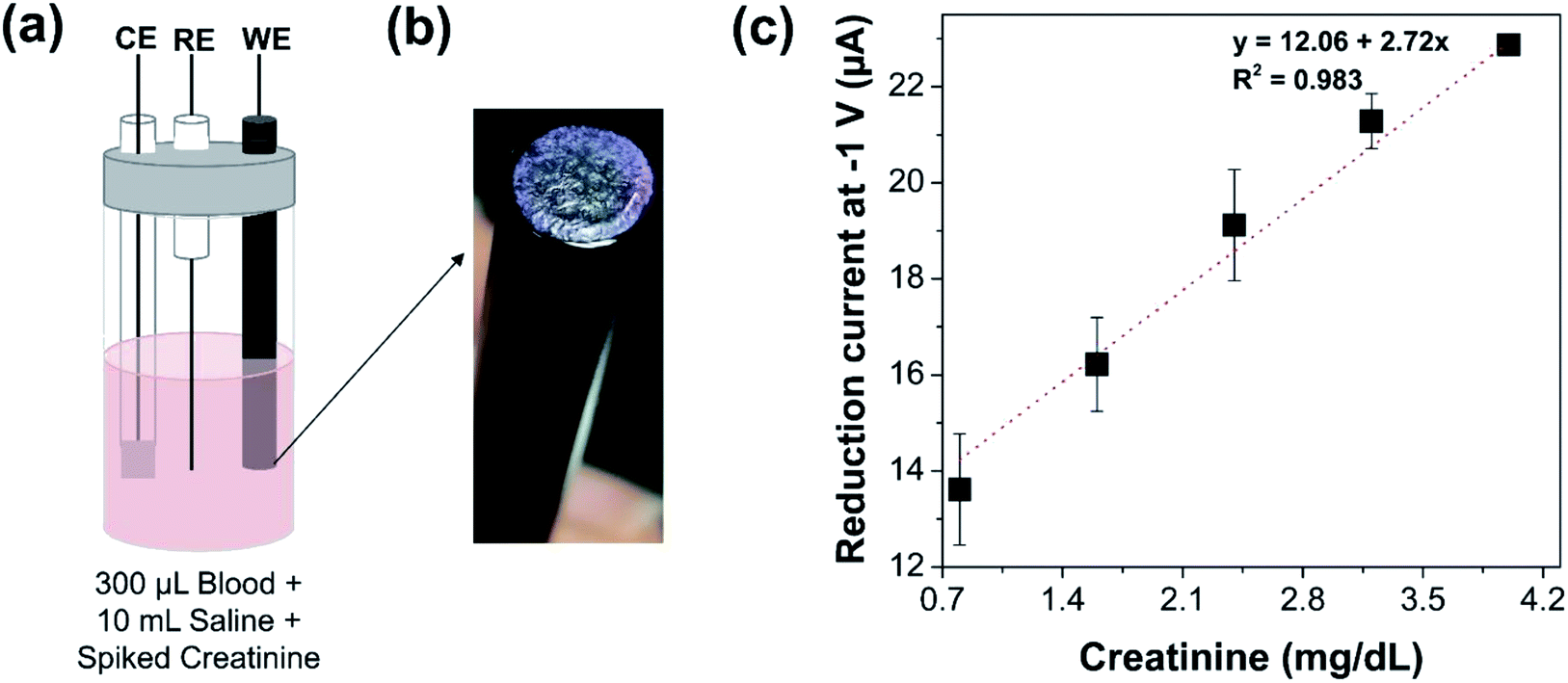 | ||
| Fig. 4 Electrochemical detection of creatinine from whole blood samples. (a) Schematic of the electrochemical cell with the modified electrode. The electrochemical cell contains 300 μL of whole blood sample spiked with different concentrations of creatinine from 0.8 to 4 mg dL−1. The total volume in the electrochemical cell is maintained at 10 mL by 0.085 M sodium chloride. (b) Image of the electrode modified by physisorption of cobalt chloride, methylene blue, and creatinine deiminase. Photo credit: Pallavi Dasgupta, Indian Institute of Science (c) Variation in reduction current of the modified electrode due to whole blood sample spiked with varying concentration of creatinine from 0.8 to 4 mg dL−1. | ||
5 Conclusions
This work presents a novel electrochemical route for estimation of 1-methylhydantoin using cobalt based redox probe. In addition, the mono-enzymatic conversion of creatinine by creatinine deiminase is coupled to the developed electrochemical route for measurement of 1-methylhydantoin, produced in the enzymatic reaction. It then leads to indirect quantification of creatinine. The electrode surface is modified with the sensing chemistry—creatinine deiminase for conversion of creatinine, methylene blue for acceleration of the enzyme activity, and cobalt chloride for biorecognition of the analyte—by simple physical adsorption. The influence of several parameters for optimal immobilization has been investigated. The proposed sensor can reliably measure creatinine in serum from 0.8 to 4 mg dL−1. The developed sensor can quantify creatinine from whole blood samples reliably as well, without interference from serum proteins. A sensitivity of 2 μA mg−1 dL−1 over a range of 0.8–4 mg dL−1 of creatinine concentration was observed. The sensor provides distinct advantages of the absence of any sample pre-processing, low reaction time, confinement of sensing chemistry on electrode surface providing ease of use, and accurate estimation. In conclusion, the results reported in this study, pave the way for realizing low cost, easy to use serum creatinine biosensors with disposable screen-printed electrodes.Conflicts of interest
The authors are inventors on two patent applications related to this work filed by Indian Institute of Science (Indian Patent Application No: 201941007991, 24 February 2020; US 16/802,179, 26 February 2020). The authors declare no other competing interests regarding the publication of this paper.Acknowledgements
We thank Visvesvaraya Fellowship Program offered by Ministry of Electronics and Information Technology, Ministry of Human Resource Development, and Department of Science and Technology, Government of India for funding this research. We thank the technical staff members at Micro and Nano Characterization Facility, Centre for Nanoscience and Engineering, Indian Institute of Science, and PathShodh Healthcare Pvt. Ltd.Notes and references
- R. D. Perrone, N. E. Madias and A. S. Levey, Serum creatinine as an index of renal function: new insights into old concepts, Clin. Chem., 1992, 38(0009–9147), 1933–1953 CrossRef CAS.
- S. Narayanan and H. D. Appleton, Creatinine: A review, Clin. Chem., 1980, 26(8), 1119–1126 CrossRef CAS.
- M. Peake and M. Whiting, Measurement of Serum Creatinine – Current Status and Future Goals, Clin. Biochem. Rev., 2006, 27(4), 173–184 Search PubMed.
- R. Cánovas, M. Cuartero and G. A. Crespo, Modern creatinine (Bio)sensing: Challenges of point-of-care platforms [Internet], Biosens. Bioelectron., 2019, 130, 110–124 CrossRef , available from: https://www.sciencedirect.com/science/article/pii/S0956566319300776?dgcid=rss_sd_all.
- D. A. Walsh and E. Dempsey, Comparison of electrochemical, electrophoretic and spectrophotometric methods for creatinine determination in biological fluids, Anal. Chim. Acta, 2002, 459(2), 187–198 CrossRef CAS.
- W. O. Ho, S. Krause, C. J. McNell, J. A. Pritchard, R. D. Armstrong and D. Athey, et al., Electrochemical sensor for measurement of urea and creatinine in serum based on ac impedance measurement of enzyme-catalyzed polymer transformation, Anal. Chem., 1999, 71(10), 1940–1946 CrossRef CAS.
- Y. T. Shih and H. J. Huang, A creatinine deiminase modified polyaniline electrode for creatinine analysis, Anal. Chim. Acta, 1999, 392(2–3), 143–150 CrossRef CAS.
- O. A. Zinchenko, S. V. Marchenko, T. A. Sergeyeva, A. L. Kukla, A. S. Pavlyuchenko and E. K. Krasyuk, et al., Application of creatinine-sensitive biosensor for hemodialysis control, Biosens. Bioelectron., 2012, 35(1), 466–469, DOI:10.1016/j.bios.2012.02.062.
- M. J. Ruedas-Rama and E. A. H. Hall, Analytical nanosphere sensors using quantum dot-enzyme conjugates for urea and creatinine, Anal. Chem., 2010, 82(21), 9043–9049, DOI:10.1021/ac101838n.
- B. Ozansoy Kasap, S. V. Marchenko, O. O. Soldatkin, S. V. Dzyadevych and B. Akata Kurc, Biosensors Based on Nano-Gold/Zeolite-Modified Ion Selective Field-Effect Transistors for Creatinine Detection, Nanoscale Res. Lett., 2017, 12(1), 162 CrossRef , available from: http://nanoscalereslett.springeropen.com/articles/10.1186/s11671-017-1943-x.
- M. Braiek, M. A. Djebbi, J.-F. Chateaux, A. Bonhomme, R. Vargiolu and F. Bessueille, et al., A conductometric creatinine biosensor prepared through contact printing of polyvinyl alcohol/polyethyleneimine based enzymatic membrane, Microelectron. Eng., 2018, 187–188, 43–49 CrossRef CAS , available from: https://linkinghub.elsevier.com/retrieve/pii/S0167931717303945.
- S. Georgieva, P. Todorov, A. Bezfamilnyi and A. Georgiev, Coordination behavior of 3-amino-5,5′-dimethylhydantoin towards Ni(II) and Zn(II) ions: Synthesis, spectral characterization and DFT calculations, J. Mol. Struct., 2018, 377–387 CrossRef CAS.
- S. Georgieva and P. Todorov, Spectroscopic and voltamperometric studies of Cu(II) complex with 3-amino-5,5-diphenylhydantoin, J. Chem. Technol. Metall., 2018, 465–472 CAS.
- S. Georgieva, P. Todorov and D. Wesselinova, Synthesis, characterization and cytotoxic activity of novel Cu(II) and Co(II) complexes with 3-amino-5,5-dimethylhydantoin, C. R. Chim., 2014, 1212–1220 CrossRef CAS.
- G. Z. Pavlovich and R. G. Luthy, Complexation of metals with hydantoins, Water Res., 1988, 22(3), 327–336 CrossRef CAS.
- M. Puszyńska-Tuszkanow, M. Daszkiewicz, G. Maciejewska, A. Adach and M. Cieślak-Golonka, Interaction of hydantoins with transition metal ions: Synthesis, structural, spectroscopic, thermal and magnetic properties of [M(H2O)4(phenytoinate)2] M = Ni(II), Co(II), Struct. Chem., 2010, 21(2), 315–321 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |