DOI:
10.1039/D0RA06432G
(Paper)
RSC Adv., 2020,
10, 33417-33427
Bimetallic Au–Pd alloy nanoparticles supported on MIL-101(Cr) as highly efficient catalysts for selective hydrogenation of 1,3-butadiene†
Received
24th July 2020
, Accepted 2nd September 2020
First published on 10th September 2020
1. Introduction
Selective 1,3-butadiene (BD) hydrogenation is an important reaction in petroleum refining and in heterogeneous catalysis studies.1,2 In recent years, various catalysts, such as nanoparticle supported catalysts and single site catalysts, have been designed and used in diverse chemical reactions.3–15 Palladium and gold supported nanoparticle catalysts have been intensively investigated and extensively used in the selective BD hydrogenation because of their superior catalytic activity and selectivity.14–17 Previous studies on supported Pd-based nanoparticle catalysts have revealed that they have superior catalytic activity in the selective BD hydrogenation reaction.13–15 However, the supported Pd-based catalysts showed a strong affinity to the byproduct butane due to double bond and geometric isomerization, thereby lowering the purity of butenes.13–15 In particular, supported Au-based catalysts were highly selective for BD hydrogenation without overhydrogenation to butane.16,17 Recently, bimetallic nanoparticle catalysts have attracted much attention in heterogeneous catalysis.13,18–22 Bimetallic catalysts generally show better catalytic performance than the corresponding monometallic catalysts because catalytic activity could be enhanced by synergistic effects between different components.23 Hou et al. reported that the bimetallic PdNi/γ-Al2O3 catalyst shows higher BD hydrogenation activity and 1-butene selectivity than Pd/γ-Al2O3 and Ni/γ-Al2O3 catalysts.24 Aguilar-Tapia et al. found that the Au–Ni bimetallic nanoparticle catalyst supported on TiO2 with Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni mole ratio of 1:0.08 displays an excellent balance between the activity and butene selectivity for the selective hydrogenation of BD.25 Pd–Cu0.06/Mn2O3-SI shows a good balance between butene selectivity (92%) and BD conversion (99.1%) at 20 °C.13 Pattamakomsan et al. prepared Pd–Sn/MA and Pd/MA (MA = mixed phase alumina) catalysts through impregnation.26 For the bimetallic Pd–Sn/MA catalyst, the butene selectivity reached to 100% at 80% BD conversion, and butane formation was suppressed.26 By contrast, the butene selectivity was 94% even at about 70% BD conversion with the monometallic Pd catalysts.26 Ag–Pd alloy nanoparticles supported on γ-Al2O3 exhibit superior catalytic performance and high stability for the selective hydrogenation of BD.26 The conversion of BD and selectivity of butenes were 98.2% and 88.1% with the AgPd/γ-Al2O3 catalyst, which is remarkably enhanced compared with corresponding Ag/Pd monometallic catalysts.27 Therefore, the addition of amount of second metal to the Au/Pd-supported catalysts could enhance catalytic activity and butene selectivity. Bimetallic catalysts are suitable to lower the catalyst cost and maximize the efficiency of Au/Pd.
:Ni mole ratio of 1:0.08 displays an excellent balance between the activity and butene selectivity for the selective hydrogenation of BD.25 Pd–Cu0.06/Mn2O3-SI shows a good balance between butene selectivity (92%) and BD conversion (99.1%) at 20 °C.13 Pattamakomsan et al. prepared Pd–Sn/MA and Pd/MA (MA = mixed phase alumina) catalysts through impregnation.26 For the bimetallic Pd–Sn/MA catalyst, the butene selectivity reached to 100% at 80% BD conversion, and butane formation was suppressed.26 By contrast, the butene selectivity was 94% even at about 70% BD conversion with the monometallic Pd catalysts.26 Ag–Pd alloy nanoparticles supported on γ-Al2O3 exhibit superior catalytic performance and high stability for the selective hydrogenation of BD.26 The conversion of BD and selectivity of butenes were 98.2% and 88.1% with the AgPd/γ-Al2O3 catalyst, which is remarkably enhanced compared with corresponding Ag/Pd monometallic catalysts.27 Therefore, the addition of amount of second metal to the Au/Pd-supported catalysts could enhance catalytic activity and butene selectivity. Bimetallic catalysts are suitable to lower the catalyst cost and maximize the efficiency of Au/Pd.
Metal–organic frameworks (MOFs) are compounds built from metal ions or metal clusters with organic ligands and have been attracted a great deal of attention from scientists and engineers.28–44 In recent years, MOFs have been increasingly used as supports for heterogeneous catalyst due to their large surface areas, infinite synthetic and structural tunability, and well-defined open cavities.45–51 Among various MOF-types, MIL-101(Cr) has been shown to possess extremely large windows and surface areas (SLangmuir = 5900 m2 g−1), remarkable thermal stability (up to 300 °C), and excellent chemical stability toward many solvents.52–54 These properties have enabled MIL-101(Cr) to be successfully applied in various fields, including its particularly attractive use as a supports for heterogeneous catalyst.55–64 Recently, various metal nanoparticle (NP) catalysts supported on MIL-101(Cr) (NPs/MIL-101(Cr)), such as Au/MIL-101(Cr),59 Pd/MIL-101(Cr),65 Ru/MIL-101(Cr),66 and Pt/MIL-101(Cr),67,68 have been tested for the hydrogenation catalytic reaction. These researches have exhibited promising results in the hydrogenation of 4-nitrophenol,59 2-butyne-1,4-diol,65 levulinic acid,66 cinnamaldehyde,67 benzaldehydes,68 and nitrobenzenes68 with superior catalytic activity, selectivity, and stability. Few studies have been published on the application of catalysts supported on MIL-101(Cr) for the BD hydrogenation reaction. Herein, bimetallic Au–Pd nanoparticles were successfully immobilized on the MOF MIL-101(Cr) using the precursors of HAuCl4·4H2O and Pd(CH3COO)2 through coimpregnation and H2 reduction method. For comparison, monometallic Au/MIL-101(Cr) and Pd/MIL-101(Cr) catalysts were prepared using the similar approach, except using the corresponding pure Au3+ and Pd2+ salts. The catalytic performance of these catalysts was examined through the BD selective hydrogenation reaction under excess hydrogen. The bimetallic AuPd/MIL-101(Cr) presented improved catalytic performance in the BD hydrogenation reaction compared with the monometallic catalysts. The effect of reaction temperature, particle size, and Au:Pd mole ratio on catalytic activity and selectivity for the BD hydrogenation were investigated.
2. Experimental section
2.1 Materials
Cr(NO3)3·9H2O, HAuCl4·4H2O, Pd(CH3COO)2, and terephthalic acid (H2BDC) were bought from Macklin (China) and used as received. 1,3-Butadiene (1.0 vol% in N2), H2 (99.999%) and N2 (99.999%) were purchased from Anqiu Hengan Gas Manufacture Factory (Weifang, China).
2.2 Preparation of the catalysts
Chromium(III) terephthalate MOF (MIL-101(Cr)) was prepared and purified according to the procedures in the literature.69 The MIL-101(Cr)-supported catalysts were synthesized through coimpregnation followed by H2 reduction.52,70 First, 24.0 mg of HAuCl4·4H2O and 7.0 mg of Pd(CH3COO)2 were melted in 1 mL of ethanol. Subsequently, the mixture solution of HAuCl4·4H2O and Pd(CH3COO)2 was added slowly dropwise into MIL-101(Cr) (0.5 g). After sonicating for 1 h, the suspension liquid was sustained at 4 °C in darkness for 24 h. The final homogenous mixture was further dried in vacuum oven (0.1 Mpa) for 18 h at 40 °C. The catalyst was then heat-treated at 50 °C for 2 h in H2 flow (8.0 mL min−1). The Au and Pd weight percents in AuPd/MIL-101(Cr) were 2.03 and 0.58 wt%, respectively, as identified through inductively coupled plasma-atomic emission spectroscopy (ICP-AES), and the Au:Pd mole ratio was 2:1 (Au(2)Pd(1)/MIL-101) (Table S1,† entry 2). Au(3)Pd(1)/MIL-101(Cr), Au(1)Pd(2)/MIL-101(Cr), and Au(1)Pd(3)/MIL-101(Cr) were synthesized by the same method to determine the Au:Pd mole ratio with the best catalytic performance. Monometallic Au/MIL-101(Cr) and Pd/MIL-101(Cr) catalysts were also received using the similar method as above, except using the corresponding pure Au3+ and Pd2+ salts. The actual Au and Pd loadings on the MIL-101(Cr) were tested by ICP-AES (Table S1†).
2.3 Catalyst characterization
The structures of samples were determined by X-ray powder diffractometer (XRD, Brüker D8, Germany) using Cu Kα radiation (λ = 0.15406 nm). The samples were measured in the angle range (2θ) of 1°–10°. Transmission electron microscopy (TEM) was performed on a JEOL-2100F microscope (Tokyo, Japan) at 200 kV. Energy dispersive X-ray spectroscopy (EDS) was carried out utilizing the Oxford X-MaxN 80T IE250 instrument (UK). The valence states of Au and Pd were determined through X-ray photoelectron spectroscopy (XPS, AXIS ULTRADLD, Shimadzu, Japan). The each XPS spectrum binding energy was calibrated by the carbon element C 1s peak with carbon binding energy 284.8 eV. MIL-101(Cr) and Au(2)Pd(1)/MIL-101(Cr) were characterized through nitrogen sorption measurements at 77 K on an automatic physical adsorption instrument (Quantachrome, Boynton Beach, Florida, USA). MIL-101(Cr) and Au(2)Pd(1)/MIL-101(Cr) were outgassed at 150 °C for 24 h before analysis. The surface areas of MIL-101(Cr) and Au(2)Pd(1)/MIL-101(Cr) were obtained by Brunauer–Emmett–Teller (BET) method at the relative pressure (P/P0) range of 0.01–0.3. The pore volume and average pore size were obtained by using the Barrett–Joyner–Halenda equation based on the adsorption branch of the isotherm. ICP-AES was performed on an Optima 5300DV instrument (Perkin-Elmer, USA) to test the Au and Pd loadings of the as-synthesized bimetallic and monometallic catalysts.
2.4 Activity test
Selective hydrogenation of BD catalyzed by monometallic and bimetallic catalysts was carried out in a homemade continuous flow fixed bed quartz micro-reactor (i.d. = 6 mm) at 30–80 °C under atmospheric pressure. Typically, 2 mg of catalyst and 500 mg quartz sand (inert diluents, 40–80 mesh) were mixed and placed in tubular furnace, and the mixture was further pretreated in a flow of 8.0 mL min−1 N2 or H2 at 50–100 °C for 1 h. The reactant of 1.0 vol% BD in N2 (20.0 mL min−1) was premixed with H2 (6.5 mL min−1) and was flowed through the catalyst with space velocities of 795000 mL (h gcat)−1. The effluent from the reactor was collected and analyzed using an online gas chromatography (GC-6890, Purkinje General Instrument Co., Ltd., China) equipped with a 30 m × 0.53 mm × 10 μm Al2O3 capillary column and a hydrogen flame ionization detector.
3. Results and discussion
3.1 Synthesis strategy
Scheme 1 illustrates the synthetic strategy to synthesize the bimetallic AuPd/MIL-101(Cr) catalyst. First, MIL-101(Cr) was hydrothermally prepared using Cr(NO3)3·9H2O, H2BDC, and water at 218 °C for 18 h.69 The as-prepared MIL-101(Cr) was washed with hot ethanol at 80 °C in order to eliminate the unreacted H2BDC trapped in the pores of MIL-101(Cr).71 The particles of MIL-101(Cr) were vacuum-dried and activated at 150 °C overnight.69,71 Second, the solution of HAuCl4·4H2O and Pd(CH3COO)2 was added slowly dropwise into MIL-101(Cr). After sonicating for 1 h, the suspension was stored for 24 h and dried in vacuum oven at 40 °C for 18 h. Finally, the solid was reduced at 50 °C for 2 h under H2 flow at the rate of 8.0 mL min−1. The AuPd/MIL-101(Cr) catalysts with Au:Pd mole ratio ranging from 1:3 to 3:1 were prepared to determine the Au:Pd mole ratio with the best catalytic activity and selectivity. Monometallic Au/Pd supported catalysts were also synthesized using the similar method, except using the corresponding pure Au3+ and Pd2+ salts.
 |
| | Scheme 1 The synthetic strategy for the AuPd/MIL-101(Cr) catalyst by coimpregnation and the H2 reduction method. | |
3.2 XRD study
Fig. 1 shows the XRD patterns of MIL-101(Cr), Au(2)Pd(1)/MIL-101(Cr), Au/MIL-101(Cr), and Pd/MIL-101(Cr). The XRD diffraction peaks observed at 2θ of 1.82°, 2.89°, 3.37°, 3.52°, 4.05°, 4.41°, 4.95°, 5.27°, 5.72°, 5.99°, 8.55°, 8.73°, and 9.18°, which were similar to the reported data for MIL-101(Cr) in other studies, thereby indicating that pure MIL-101(Cr) crystals were successfully prepared.52,72 The X-ray diffraction relative intensities and the peak positions of Au(2)Pd(1)/MIL-101(Cr), Au/MIL-101(Cr), and Pd/MIL-101(Cr) were consistent with MIL-101(Cr).73 The results exhibited that the MIL-101(Cr) structure was kept well after loading of Au–Pd alloy, Au, and Pd nanoparticles.45
 |
| | Fig. 1 XRD patterns of MIL-101(Cr) (a), Au(2)Pd(1)/MIL-101(Cr) (b), Au/MIL-101(Cr) (c), and Pd/MIL-101(Cr) (d). | |
3.3 Microscopic study
TEM analysis was performed on the Au(2)Pd(1)/MIL-101(Cr), Au/MIL-101(Cr), and Pd/MIL-101(Cr) catalysts to investigate the dispersion and size of the nanoparticles loaded on MIL-101(Cr). The TEM micrograph and nanoparticle-size distribution histogram of Au(2)Pd(1)/MIL-101(Cr) were presented in Fig. 2a and b. The STEM-HAADF image and EDS mapping of elements are presented in Fig. 2c–f. The TEM micrograph revealed that Au–Pd nanoparticles were uniformly distributed on the MIL-101(Cr). The diameter of Au–Pd nanoparticles immobilized by MIL-101(Cr) spanning from 3 nm to 11 nm, and the mean particle size of Au–Pd nanoparticles in Au(2)Pd(1)/MIL-101(Cr) was 5.6 nm. The HAADF-STEM and EDS analyses of randomly chosen Au–Pd nanoparticles revealed that bimetallic Au–Pd alloys were formed. The TEM micrographs for Au/MIL-101(Cr) and Pd/MIL-101(Cr) catalysts were given in Fig. S1.† Au and Pd nanoparticles were uniformly immobilized on the MIL-101(Cr). The Au and Pd nanoparticle sizes for monometallic Au/MIL-101(Cr) and Pd/MIL-101(Cr) catalysts ranged from 3 nm to 15 nm and 2 nm to 14 nm, respectively. The mean diameters of Au and Pd nanoparticles were 6.6 nm and 4.6 nm for Au/MIL-101(Cr) and Pd/MIL-101(Cr), respectively. Obviously, the mean particle size of the Au(2)Pd(1)/MIL-101(Cr) catalyst was between that of Au/MIL-101(Cr) and Pd/MIL-101(Cr) catalysts. The bimetallic Au(2)Pd(1)/MIL-101(Cr) catalyst had a narrower particle size distributions compared with monometallic catalysts Au/MIL-101(Cr) and Pd/MIL-101(Cr).
 |
| | Fig. 2 (a) TEM micrograph and (b) the Au–Pd nanoparticle size distribution histogram of Au(2)Pd(1)/MIL-101(Cr); (c) HAADF-STEM image and (d–f) EDS elemental mapping images. | |
3.4 XPS study
The valence states of Au and Pd in the Au(2)Pd(1)/MIL-101(Cr), Au/MIL-101(Cr), and Pd/MIL-101(Cr) catalysts were received through XPS measurements. The Au 4f and Pd 3d XPS spectras were presented in Fig. 3. In recently published papers, the binding energy of Au0 4f5/2 and Au0 4f7/2 were 87.3 eV–87.8 eV and 83.6 eV–84.5 eV.74,75 Binding energy peaks of Pd 3d5/2 and 3d3/2 at 335.6 eV and 340.4 eV were assigned to metallic palladium (Pd0).76,77 For the bimetallic Au(2)Pd(1)/MIL-101(Cr) catalyst, two obvious peaks were obtained in the Au 4f spectra at the binding energy of 83.8 eV and 87.7 eV, respectively (Fig. 3a). These binding energy values were the characteristic peaks of metallic Au0 4f7/2 and Au0 4f5/2, respectively.74,75 The XPS spectrum of Pd 3d for Au(2)Pd(1)/MIL-101(Cr) catalyst exhibited two intense binding energy peaks at 341.2 eV and 335.9 eV, corresponding to the metallic palladium 3d3/2 and 3d5/2, respectively (Fig. 3b).76,77 As shown in the Au 4f spectra of Au/MIL-101(Cr) in Fig. 3c, two obvious binding energy peaks centered at 84.2 eV and 87.9 eV corresponded to metallic gold 4f7/2 and 4f5/2.74,75 For the Pd/MIL-101(Cr) catalyst, the binding energy peaks at 341.1 eV and 335.6 eV were attributed to Pd 3d3/2 and Pd 3d5/2, respectively, which is consistent with Pd0 (Fig. 3d).76,77 There was a negative shift (0.4 eV) of the Au0 4f7/2 binding energy centered at 83.8 eV for bimetallic catalyst Au(2)Pd(1)/MIL-101(Cr) compared with the monometallic catalyst Au/MIL-101(Cr) (84.2 eV). And there was a positive shift (0.3 eV) of the Pd0 3d5/2 binding energy of the Au(2)Pd(1)/MIL-101(Cr) catalyst centered at 335.9 eV compared with the Pd/MIL-101(Cr) catalyst (335.6 eV). The obvious binding energy shifts of Pd0 3d5/2 and Au0 4f7/2 between bimetallic and monometallic catalysts suggest the existence of charge transfer between Au and Pd, thereby confirming the formation of Au–Pd alloy.75,76 Cai et al. found that the Au 4f peaks of TiO2–Pd–Au shift toward higher binding energy and Pd 3d peaks toward lower binding energy compared to TiO2–Au and TiO2–Pd.75 Chen et al. reported that the apparent binding energy shifts of Au 4f7/2 and Pd 3d5/2 for PdAu/Fe3O4, Au/Fe3O4 and Pd/Fe3O4 catalysts.76
 |
| | Fig. 3 Au 4f and Pd 3d XPS of the catalysts (a and b) Au(2)Pd(1)/MIL-101(Cr); (c) Au/MIL-101(Cr); (d) Pd/MIL-101(Cr). | |
3.5 Nitrogen physisorption study
Fig. 4 presents the nitrogen physisorption isotherms of MIL-101(Cr) and Au(2)Pd(1)/MIL-101(Cr). The nitrogen physisorption isotherms of MIL-101(Cr) show typical type IV curves, with a rapid nitrogen adsorbed and a slight hysteresis loop in a P/P0 range of 0.9–1.0, thereby suggesting its mesoporous structure.78,79 After depositing the Au–Pd nanoparticles on the MIL-101(Cr), the bimetallic Au(2)Pd(1)/MIL-101(Cr) catalysts show a largely decreased in adsorption of nitrogen, but the type of nitrogen physisorption isotherms stays the same with mesoporous features. The BET specific surface areas calculated from isotherms are 1186.0 m2 g−1 and 991.9 m2 g−1 for MIL-101(Cr) and Au(2)Pd(1)/MIL-101(Cr), respectively. The pore volume and mean pore diameter of MIL-101(Cr) are 0.49 m3 g−1 and 2.8 nm, respectively, whereas Au(2)Pd(1)/MIL-101(Cr) possesses a pore volume of 0.40 m3 g−1 and a mean pore diameter of 2.7 nm. The BET surface area of bimetallic Au(2)Pd(1)/MIL-101(Cr) catalysts reduces compared with that of MIL-101(Cr), because of the immobilization of Au–Pd alloy nanoparticles inside the pores of MIL-101(Cr).45,52
 |
| | Fig. 4 Nitrogen physisorption isotherms of MIL-101(Cr) and Au(2)Pd(1)/MIL-101(Cr). | |
3.6 Catalytic performance
The as-synthesized catalysts were tested in the selective BD hydrogenation at 30–80 °C in a homemade continuous-flow fixed-bed quartz micro-reactor. Fig. 5a displays the BD conversions for Au(2)Pd(1)/MIL-101(Cr), Pd/MIL-101(Cr), Au/MIL-101(Cr), and MIL-101(Cr). The pure MIL-101(Cr) has very low catalytic activity with BD conversion less than 1% toward the selective BD hydrogenation because of lack of active sites. For the Au(2)Pd(1)/MIL-101(Cr) and Pd/MIL-101(Cr) catalysts, the BD conversion continuously increased with the enhancement of reaction temperature from 30 °C to 60 °C (Fig. 5a). The BD conversion was 10.8% at 30 °C and reached 98.8% at 60 °C over the Au(2)Pd(1)/MIL-101(Cr) catalyst. The BD conversions were 11.5%, 30.7%, and 99.9% on the Pd/MIL-101(Cr) catalyst at 30 °C, 40 °C, and 50 °C, respectively. At low temperature range (30–40 °C), no difference was observed between the activities of Pd/MIL-101(Cr) and Au(2)Pd(1)/MIL-101(Cr). The BD conversion on the Pd/MIL-101(Cr) catalyst increased faster than that on the bimetallic Au(2)Pd(1)/MIL-101(Cr) catalyst at high temperature (>40 °C). The Au/MIL-101(Cr) catalyst had poor catalytic activity for the selective BD hydrogenation. The maximum BD conversion of ca. 1.4% was found on the Au/MIL-101(Cr) catalyst at 30–70 °C. The BD hydrogenation could produces 1-butene and cis/trans-2-butenes by 1,2-hydrogenation and 1,4-hydrogenation, and butane could further obtain by the hydrogenation of butenes.14,80,81 The selectivities for BD hydrogenation for Au(2)Pd(1)/MIL-101(Cr) and Pd/MIL-101(Cr) catalysts are displayed in Fig. 5b. For the bimetallic Au(2)Pd(1)/MIL-101(Cr) catalysts, the main product detected was butenes (containing 1-butene and cis/trans-2-butenes), with a small amount of butane produced. At reaction temperatures from 30 °C to 60 °C, the percentages of different products were shown below: trans-2-butene (46–58%) > 1-butene (35–41%) > cis-2-butene (5–9%) > butane (0–5%). The high selectivities of butenes for Au(2)Pd(1)/MIL-101(Cr) at all reaction temperatures suggested that the hydrogenation of butenes to butane was prevented even at low BD concentrations.82–84 Highly preferential adsorption to BD was observed compare with other butenes on Au(2)Pd(1)/MIL-101(Cr) under catalytic reaction conditions.82–84 The results showed that the selectivities to 1-butene and cis/trans-2-butene slightly changed with the increase in BD conversion. The selectivities of 1-butene, trans-2-butene, and cis-2-butene for Au(2)Pd(1)/MIL-101(Cr) were 35.6%, 57.7%, and 5.9% at 31.1% BD conversion. The selectivities for 1-butene, trans-2-butene, and cis-2-butene were 40.4%, 47.7%, and 10.7%, respectively, when the conversions increased from 31.1% to 75.2%. The change in selectivities to different butenes indicated that some isomerizations occurred in the processing of BD hydrogenation.80 When the BD conversions lower than 30.7%, Pd/MIL-101(Cr) displayed the best butene selectivity of 99.7%. However, the selectivity of butenes quickly reduced at high conversion of 99.9%. The selectivity for trans-2-butene rapidly dropped with a quick increase in butane when the conversion was 99.9%. Secondary hydrogenation of trans-2-butene to butane occurred first with the increase in BD conversion.85 These results clearly showed that Au(2)Pd(1)/MIL-101(Cr) exhibited much better activity for the BD selective hydrogenation reaction than Au/MIL-101(Cr) but its activity was still lesser than Pd/MIL-101(Cr). The selectivity of butenes for bimetallic Au(2)Pd(1)/MIL-101(Cr) catalyst was higher than Pd/MIL-101(Cr) at near complete 1,3-butadiene conversion. By contrast to the Au/MIL-101(Cr) and Pd/MIL-101(Cr), the catalyst containing the Au–Pd alloy nanoparticles showed excellent balance between the activity (BD conversion) and selectivity to butenes for the BD selective hydrogenation. Table 1 displays the BD conversion and butene selectivity on different Au–Pd bimetallic catalysts in the selective BD hydrogenation reaction. Bachiller-Baeza et al. prepared bimetallic PdAu–ZnO-n and PdAu–ZnO-t catalysts with 0.3–0.4 Au:Pd mole ratio using two differently shaped ZnO supports (needles ZnO-n and tetrapods ZnO-t) by wet impregnation method.86 They found that both bimetallic PdAu–ZnO-n and PdAu–ZnO-t catalysts displayed the similar BD conversion and selectivity in the selective BD hydrogenation reaction.86 The BD conversions and butene selectivities over PdAu–ZnO-n and PdAu–ZnO-t were 92% and 23%, and 99% and 32%, respectively, at 40 °C.86 Hugon et al. compared the catalytic properties of Al2O3 supported bimetallic DP-Au/Pd(90) and monometallic DP-Au catalysts for the selective BD hydrogenation reaction.87 The results showed that bimetallic DP-Au/Pd(90) catalyst presented better catalytic active than the monometallic DP-Au catalyst: BD conversion is reached 100% at 120 °C and 170 °C, respectively.87 The production of butane increases more noticeably when 100% BD conversion was reached.87 The BD conversion (45–54%) of bimetallic Pd–Au/SiO2 microfabricated catalyst was lower than the Pd/SiO2 catalyst (60%) at 220 °C for the selective hydrogenation of BD.88 The bimetallic Pd–Au/SiO2 catalyst was more selective towards butenes (90–100%) than Pd/SiO2 (66%) at BD conversion of 55%.88 Thus, our Au(2)Pd(1)/MIL-101(Cr) catalyst (98.8% at 60 °C) displays higher catalytic activity (BD conversion) than Al2O3 supported bimetallic DP-Au/Pd(90) (100% at 120 °C) and Pd–Au/SiO2(45–54% at 220 °C). Although the catalytic activity (BD conversion) of Au(2)Pd(1)/MIL-101(Cr) is lower than those of PdAu–ZnO-n (92% at 40 °C) and PdAu–ZnO-t catalysts (99% at 40 °C), the bimetallic Au(2)Pd(1)/MIL-101(Cr) (95–100%) showed excellent butene selectivity than PdAu–ZnO-n (23%) and PdAu–ZnO-t (32%) catalysts. These results indicate that MIL-101(Cr) supported Au–Pd bimetallic catalyst is an excellent catalyst for selective BD hydrogenation reaction.
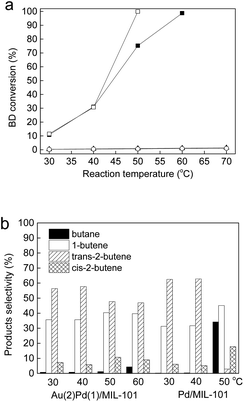 |
| | Fig. 5 (a) The BD conversion on Au(2)Pd(1)/MIL-101(Cr) (■), Pd/MIL-101(Cr) (□), Au/MIL-101(Cr) (▲), and MIL-101(Cr) (○) at different reaction temperature; (b) product selectivity on the Au(1)Pd(2)/MIL-101(Cr) and Pd/MIL-101(Cr) catalysts at different reaction temperature (the catalysts were pretreated in flowing N2 at 50 °C for 1 h before reaction). | |
Table 1 The BD conversion and butene selectivity on different Au–Pd bimetallic catalysts in the selective BD hydrogenation reaction
| Catalyst |
T (°C) |
Conv. (%) |
Sel.(%) |
Ref. |
| Au(2)Pd(1)/MIL-101(Cr) |
60 |
98.8 |
95.7 |
This work |
| PdAu–ZnO-n |
40 |
92 |
23 |
86 |
| PdAu–ZnO-t |
40 |
99 |
32 |
86 |
| DP-Au/Pd(90) |
120 |
100 |
0 |
87 |
| Pd–Au/SiO2 |
220 |
45–54 |
90–100 |
88 |
The particle size effect is the primary factor to enhance the catalytic activity of supported nanocatalysts.14,80,89 The bimetallic Au(2)Pd(1)/MIL-101(Cr) catalysts were pretreated at different temperature (50 °C, 80 °C, and 100 °C) and atmospheres (N2 and H2) to determine the influences of pretreatment temperatures and atmospheres on the Au–Pd nanoparticle size. Fig. S2† displays the TEM micrographs and nanoparticle size distribution of Au(2)Pd(1)/MIL-101(Cr) catalyst pretreated at different temperatures and atmospheres. The Au–Pd alloy nanoparticles were close to spherical with mean diameter of 5.6 nm for the as-synthesized Au(2)Pd(1)/MIL-101(Cr) (Fig. 2a and b). After pretreatment at 50 °C, 80 °C, and 100 °C in flowing N2, the Au–Pd alloy nanoparticles were close to spherical and their mean diameters were 6.2 nm, 8.2 nm, and 9.4 nm, respectively, which are larger than Au–Pd alloy nanoparticles (5.6 nm) in the as-synthesized Au(2)Pd(1)/MIL-101(Cr) (Fig. S2a–f†). The Au–Pd alloy nanoparticles appeared to agglomerate after pretreatment at 50–100 °C in flowing N2. After pretreating the Au(2)Pd(1)/MIL-101(Cr) catalyst at 50 °C in flowing H2, the Au–Pd alloy nanoparticles also appeared to agglomerate, and the mean diameters of Au–Pd alloy nanoparticles increased from 5.6 nm to 8.5 nm (Fig. S2g and h†). Fig. S3 and S4† present the XRD patterns and Au 4f and Pd 3d XPS spectras of Au(2)Pd(1)/MIL-101(Cr) with different Au–Pd nanoparticle sizes. The XRD results exhibited that the structure of Au(2)Pd(1)/MIL-101(Cr) with different Au–Pd nanoparticle sizes was similar to those of the MIL-101(Cr), thereby indicating that the integrity of Au(2)Pd(1)/MIL-101(Cr) with different Au–Pd nanoparticle sizes was maintained (Fig. S3†). The Au 4f and Pd 3d XPS spectras indicated that Au and Pd on Au(2)Pd(1)/MIL-101(Cr) with different Au–Pd nanoparticle sizes are in a zero valence state (Fig. S4†).74–77 The catalytic activity and selectivity of the bimetallic Au(2)Pd(1)/MIL-101(Cr) with Au–Pd nanoparticle sizes ranged from 6.2 nm to 9.4 nm were investigated in the BD selective hydrogenation (Fig. 6). The catalytic activity of BD hydrogenation was closely related to the Au–Pd nanoparticle size of the Au(2)Pd(1)/MIL-101(Cr) catalyst. The bimetallic Au(2)Pd(1)/MIL-101(Cr) catalyst with the smallest Au–Pd particle size showed higher catalytic activity (BD conversion). In particular, the BD conversions were 75.22%, 37.47%, 34.92%, and 27.55% at 50 °C on Au(2)Pd(1)/MIL-101(Cr) with Au–Pd nanoparticle size of 6.2 nm, 8.2 nm, 8.5 nm, and 9.4 nm, respectively. The bimetallic Au(2)Pd(1)/MIL-101(Cr) catalyst with an Au–Pd nanoparticle size of 6.2 nm presented the highest BD conversion of 98.75% at 60 °C. The catalytic results of Au(2)Pd(1)/MIL-101(Cr) on the hydrogenation of BD were consistent with those published in previous studies.14,80,89 Lozano-Martín et al. showed that the catalyst with the smallest particle size (AuFGO) exhibits high catalytic activity at high temperatures on the BD hydrogenation reaction.80 Liu et al. stated that the catalytic activity was dependent on the Au nanoparticle size of the Au NP-coated SiO2–N catalyst.89 They found that the catalyst with smaller nanoparticle size exhibits higher BD conversion.89 Decarolis et al. found that the particle size of Pd-supported catalyst affects the catalytic activity in BD hydrogenation reaction.14 Fig. 7 presents the product selectivities on Au(2)Pd(1)/MIL-101(Cr) with different Au–Pd particle sizes at reaction temperatures of 30–70 °C. Au(2)Pd(1)/MIL-101(Cr) with different particle sizes exhibited similar butane and butene selectivity. At low reaction temperatures, butane selectivity was close to 0%. Butane selectivity slightly increased with the increase in reaction temperature, but never exceeded 5%. The Au(2)Pd(1)/MIL-101(Cr) with different nanoparticle sizes of Au–Pd maintained the high selectivity of butenes (95–100%) at investigated temperature range (30–70 °C). The percentages of various butenes werevaried as follows: trans-2-butene (46–60%) > 1-butene (34–41%) > cis-2-butene (5–12%). The butene selectivities did not apparently variation on Au(2)Pd(1)/MIL-101(Cr) with different Au–Pd particle sizes, thereby indicating the lack of isomerization.80
 |
| | Fig. 6 The BD conversions on the Au(2)Pd(1)/MIL-101(Cr) catalyst with different Au–Pd particle size (■) 6.2 nm; (□) 8.2 nm; (●) 8.5 nm; and (○) 9.4 nm. | |
 |
| | Fig. 7 The product selectivities on the Au(2)Pd(1)/MIL-101(Cr) catalyst with different Au–Pd particle size: (a) 6.2 nm; (b) 8.2 nm; (c) 8.5 nm; (d) 9.4 nm. | |
The catalytic activity of Au–Pd-supported catalysts relied on the mole ratio of Au:Pd for the BD hydrogenation reaction. Au(3)Pd(1)/MIL-101(Cr), Au(2)Pd(1)/MIL-101(Cr), Au(1)Pd(2)/MIL-101(Cr), and Au(1)Pd(3)/MIL-101(Cr) catalysts with Au:Pd mole ratios varying from 3:1 to 1:3 for the BD hydrogenation were investigated with space velocities of 795000 mL (h gcat.)−1 under atmosphere pressure to determine the Au:Pd mole ratio with the best catalytic activity. Fig. 8 and S5† present the influence of Au:Pd mole ratio on BD conversions and product selectivities on Au–Pd-supported catalysts. The results showed that the BD conversion and product selectivity strongly depended on the mole ratio of Au:Pd for the BD hydrogenation reaction. The BD conversion increased with the decrease in Au:Pd mole ratio under identical reaction conditions. In particular, the BD conversion amounted to 10.71%, 31.06%, 65.21%, and 99.95% on Au(3)Pd(1)/MIL-101(Cr), Au(2)Pd(1)/MIL-101(Cr), Au(1)Pd(2)/MIL-101(Cr), and Au(1)Pd(3)/MIL-101(Cr), respectively, at 40 °C (Fig. 8). The BD conversions of 98.75% and 100% were obtained on Au(2)Pd(1)/MIL-101(Cr) and Au(3)Pd(1)/MIL-101(Cr) when the reaction temperatures were 60 °C and 80 °C, respectively. However, the BD conversion suddenly increased on Au(1)Pd(3)/MIL-101(Cr) and Au(1)Pd(2)/MIL-101(Cr) catalysts, and approximately 100% conversion was reached when the reaction temperature was 40 °C and 45 °C. The selectivity of butenes increased, whereas butane decreased with the enhancement of the Au:Pd mole ratio of Au–Pd-supported catalysts when the BD conversion was close to 100% (Fig. S5†). At conversions more than 99%, the selectivities to butane, 1-butene, trans-2-butene, and cis-2-butene were 2.54%, 38.78%, 51.98%, and 6.7% for Au(3)Pd(1)/MIL-101(Cr) catalysts, respectively (Fig. S5a†). The selectivity of butane, 1-butene, trans-2-butene, and cis-2-butene were 4.32%, 39.7%, 46.96%, and 9.01% on Au(2)Pd(1)/MIL-101(Cr), respectively, when the mole ratio of Au:Pd decreased from 3:1 to 2:1 (Fig. S5b†). For Au(1)Pd(2)/MIL-101(Cr), butene selectivity rapidly decreased (the 1-butene, trans-2-butene, and cis-2-butene selectivities were 48.45%, 4.54%, 19.99%), whereas the butane selectivity rapidly increased (27.03%) at conversion more than 99% (Fig. S5c†). The selectivities of butane, 1-butene, trans-2-butene, and cis-2-butene were 23.04%, 50.11%, 5.71%, and 21.15%, respectively, when the Au:Pd mole ratio decreased to 1:3 (Fig. S5d†). The bimetallic Au(2)Pd(1)/MIL-101 catalysts with an Au:Pd mole ratio of 2:1 showed the best balance between the activity and butene selectivity in the BD selective hydrogenation.
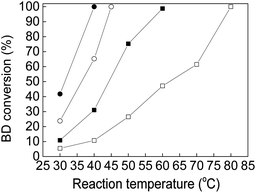 |
| | Fig. 8 The BD conversion over Au–Pd bimetallic catalysts with different Au:Pd mole ratio (□) Au(3)Pd(1)/MIL-101(Cr); (■) Au(2)Pd(1)/MIL-101(Cr); (○) Au(1)Pd(2)/MIL-101(Cr); (●) Au(1)Pd(3)/MIL-101(Cr) (the catalysts were pretreated in flowing N2 at 50 °C for 1 h before reaction). | |
3.7 Reusability of catalysts
Reusability studies of Au(2)Pd(1)/MIL-101(Cr) and Pd/MIL-101(Cr) were conducted on the BD selective hydrogenation reaction. The Au(2)Pd(1)/MIL-101(Cr) and Pd/MIL-101(Cr) were repeatedly used without any treatment (Fig. 9 and S6). The BD conversion of Au(2)Pd(1)/MIL-101(Cr) catalyst decreased slightly in the first run compared with the fresh catalysts (Fig. 9a). However, the BD conversion was greatly reduced in the second and third runs for the Au(2)Pd(1)/MIL-101(Cr) catalyst (Fig. 9a). Some differences were observed with the catalyst of Pd/MIL-101(Cr). The BD conversion in the first run of Pd/MIL-101(Cr) was almost the same as that of fresh catalyst (Fig. S6a†). No drop in conversion of BD was found on Pd/MIL-101(Cr) in the second run at low reaction temperatures (30 °C and 40 °C). Pd/MIL-101(Cr) featured a remarkable deactivation mode at high temperature (50 °C). The third run of Pd/MIL-101(Cr) displayed a rapid decrease in BD conversion. The product selectivities of recycled Au(2)Pd(1)/MIL-101(Cr) and Pd/MIL-101(Cr) catalysts were the same as that of as-synthesized catalysts at the similar 1,3-butadiene conversion (Fig. 9b–d and S6b–d†).
 |
| | Fig. 9 The reusability of Au(2)Pd(1)/MIL-101(Cr): (a) BD conversion; (b–d) Product selectivity. | |
The stability of bimetallic Au(2)Pd(1)/MIL-101(Cr) catalysts was studied by XRD and TEM. The XRD spectra of Au(2)Pd(1)/MIL-101(Cr) after three runs was similar to those of the as-synthesized catalyst, thereby indicating that the integrity of Au(2)Pd(1)/MIL-101(Cr) was maintained after three runs under the present reaction condition (Fig. S7†).70 The TEM micrograph and Au–Pd size distribution for the reused Au(2)Pd(1)/MIL-101(Cr) catalyst were presented in Fig. S8.† The mean size of Au–Pd particles for the reused Au(2)Pd(1)/MIL-101(Cr) catalyst was 8.9 nm. The mean particle diameter of reused Au(2)Pd(1)/MIL-101(Cr) (8.9 nm) was larger than that of the as-synthesized catalyst (5.6 nm) (Fig. 2b and S8b†). The particle size of Au(2)Pd(1)/MIL-101(Cr) takes a crucial role in the determination of catalytic activity. Considering that the Au–Pd nanoparticle sizes of reused Au(2)Pd(1)/MIL-101(Cr) catalyst were aggregated from 5.6 nm to 8.9 nm, the decrease in catalytic activity in the first, second, and third runs for the BD hydrogenation was because of the conglomeration of Au–Pd nanoparticles.
4. Conclusions
Bimetallic Au–Pd alloy nanoparticles on MIL-101(Cr) were successfully synthesized through coimpregnation and the H2 reduction. Au–Pd alloy particles were homogeneously distributed on the MIL-101(Cr) with mean particle size of 5.6 nm. The catalytic activity and selectivity of the as-synthesized AuPd/MIL-101(Cr) were evaluated on the BD hydrogenation reaction. Compared with monometallic catalysts, the AuPd/MIL-101(Cr) catalyst displayed improved catalytic properties for the BD selective hydrogenation. The bimetallic Au(2)Pd(1)/MIL-101(Cr) catalysts with mole ratio of Au:Pd of 2:1 showed the best balance between the activity and butene selectivity for the BD hydrogenation reaction. The catalytic activity of the BD hydrogenation reaction was significantly affected by the Au–Pd particle size. The bimetallic Au(2)Pd(1)/MIL-101(Cr) catalyst which possesses the smallest Au–Pd particle size, exhibited high BD conversion. The bimetallic Au(2)Pd(1)/MIL-101(Cr) catalysts can be reused repetitively at least three runs, and the selectivity to butenes remains basically unchanged. This work affords a reference of the utilization of MOF and alloy nanoparticles to develop high-efficiency catalysts.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the financial support of the National Natural Science Foundation of China (No. 21802104), the Natural Science Foundation of Shandong Province (No. ZR2017MB056), and High-tech Industrial Development Zone Science and Technology Huimin Plan of Weifang (No. 2019KJHM18).
Notes and references
- R. Hou, M. D. Porosoff, J. G. Chen and T. Wang, Appl. Catal., A, 2015, 490, 17–23 CrossRef CAS.
- L. Lozano, J. L. Brito, C. Olivera, J. Guerra and S. Curbelo, Fuel, 2013, 110, 76–82 CrossRef CAS.
- B. Song, Y. Zhou, H. M. Yang, J. H. Liao, L. M. Yang, X. B. Yang and E. Ganz, J. Am. Chem. Soc., 2019, 141, 3630–3640 CrossRef CAS.
- L. M. Yang, V. Bačić, I. A. Popov, A. I. Boldyrev, T. Heine, T. Frauenheim and E. Ganz, J. Am. Chem. Soc., 2015, 137, 2757–2762 CrossRef CAS.
- L. M. Yang, I. A. Popov, A. I. Boldyrev, T. Heine, T. Frauenheim and E. Ganz, Phys. Chem. Chem. Phys., 2015, 17, 17545–17551 RSC.
- L. M. Yang, I. A. Popov, T. Frauenheim, A. I. Boldyrev, T. Heine, V. Bačić and E. Ganz, Phys. Chem. Chem. Phys., 2015, 17, 26043–26048 RSC.
- L. M. Yang and E. Ganz, Phys. Chem. Chem. Phys., 2016, 18, 17586–17591 RSC.
- J. Liu, L. M. Yang and E. Ganz, ACS Sustainable Chem. Eng., 2018, 6, 15494–15502 CrossRef CAS.
- J. H. Liu, L. M. Yang and E. Ganz, J. Mater. Chem. A, 2019, 7, 3805–3814 RSC.
- J. H. Liu, L. M. Yang and E. Ganz, J. Mater. Chem. A, 2019, 7, 11944–11952 RSC.
- J. H. Liu, L. M. Yang and E. Ganz, RSC Adv., 2019, 9, 27710–27719 RSC.
- J. H. Liu, L. M. Yang and E. Ganz, Energy Environ. Mater., 2019, 2, 193–200 CrossRef CAS.
- T. Odoom-Wubah, Q. Li, M. Chen, H. Fang, B. B. A. Bediako, I. Adilov, J. Huang and Q. Li, ACS Omega, 2019, 4, 1300–1310 CrossRef CAS.
- D. Decarolis, I. Lezcano-Gonzalez, D. Gianolio and A. M. Beale, Top. Catal., 2018, 61, 162–174 CrossRef CAS.
- Y. Xu, D. Ma, J. Yu, X. Jiang, J. Huang and D. Sun, Ind. Eng. Chem. Res., 2017, 56, 10623–10630 CrossRef CAS.
- E. Castillejos, B. Bachiller-Baeza, E. Asedegbega-Nieto, A. Guerrero-Ruiz and I. Rodríguez-Ramos, RSC Adv., 2015, 5, 81583–81598 RSC.
- N. Masoud, L. Delannoy, H. Schaink, A. V. D. Eerden, J. W. D. Rijk, T. A. G. Silva, D. Banerjee, J. D. Meeldijk, K. P. D. Jong, C. Louis and P. E. D. Jongh, ACS Catal., 2017, 7, 5594–5603 CrossRef CAS.
- S. Kumar, D. Waller, H. Fjellvåg and A. O. Sjåstad, J. Alloys Compd., 2019, 786, 1021–1029 CrossRef CAS.
- L. Guo, J. Mao, S. Guo, Q. Zhang, S. Cai and W. He, Nano Res., 2019, 12, 1659–1662 CrossRef CAS.
- F. Lu, D. Sun and X. Jiang, New J. Chem., 2019, 43, 13891–13898 RSC.
- M. Duan, L. Jiang, G. Zeng, D. Wang, W. Tang, J. Liang, H. Wang, D. He, Z. Liu and L. Tang, Appl. Mater. Toda, 2020, 19, 100564–100582 CrossRef.
- P. Qu, S. Wang, W. Hu, Y. Wu, J. Chen, G. Zhang, P. Shen, Y. Chen and L. Zhong, Catal. Commun., 2020, 135, 105900–105904 CrossRef.
- L. Zhang, Z. Xie and J. Gong, Chem. Soc. Rev., 2016, 45, 3916–3934 RSC.
- R. Hou, W. Yu, M. D. Porosoff, J. G. Chen and T. Wang, J. Catal., 2014, 316, 1–10 CrossRef CAS.
- A. Aguilar-Tapia, L. Delannoy, C. Louis, C. W. Han, V. Ortalan and R. Zanella, J. Catal., 2016, 344, 515–523 CrossRef CAS.
- K. Pattamakomsan, E. Ehret, F. Morfin, P. Gélin, Y. Jugnet, S. Prakash, J. C. Bertolini, J. Panpranot and F. J. C. S. Aires, Catal. Today, 2011, 164, 28–33 CrossRef CAS.
- F. Lu, D. Sun, J. Huang, M. Du, F. Yang, H. Chen, Y. Hong and Q. Li, ACS Sustainable Chem. Eng., 2014, 2, 1212–1218 CrossRef CAS.
- L. M. Yang, V. Ponniah, R. Ponniah, H. Fjellvag and M. Tilset, Inorg. Chem., 2010, 49, 10283–10290 CrossRef CAS.
- L. M. Yang, G. Y. Fang, J. Ma, E. Ganz and S. S. Han, Cryst. Growth Des., 2014, 14, 2532–2541 CrossRef CAS.
- L. M. Yang, P. Ravindran, P. Vajeeston and M. Tilset, RSC Adv., 2012, 2, 1618–1631 RSC.
- L. M. Yang, E. Ganz, S. Svelle and M. Tilset, J. Mater. Chem. C, 2014, 2, 7111–7125 RSC.
- L. M. Yang, P. Ravindran, P. Vajeeston and M. Tilset, Phys. Chem. Chem. Phys., 2012, 14, 4713–4723 RSC.
- L. M. Yang, P. Ravindran, P. Vajeeston and M. Tilset, J. Mater. Chem., 2012, 22, 16324–16335 RSC.
- W. R. Lee, H. Jo, L. M. Yang, H. Lee, D. W. Ryu, K. S. Lim, J. H. Song, D. Y. Min, S. S. Han, J. G. Seo, Y. K. Park, D. Moone and C. S. Hong, Chem. Sci., 2015, 6, 3697–3705 RSC.
- K. Wang, L. M. Yang, X. Wang, L. Guo, G. Cheng, C. Zhang, S. Jin, B. Tan and A. Cooper, Angew. Chem., Int. Ed., 2017, 56, 14149–14153 CrossRef.
- L. M. Yang, P. Ravindran and M. Tilset, Inorg. Chem., 2013, 52, 4217–4228 CrossRef.
- L. M. Yang, P. Vajeeston, P. Ravindran, H. Fjellvg and M. Tilset, Phys. Chem. Chem. Phys., 2011, 13, 10191–10203 RSC.
- L. M. Yang, P. Ravindran, P. Vajeeston, S. Svelle and M. Tilset, Microporous Mesoporous Mater., 2013, 175, 50–58 CrossRef.
- L. M. Yang and R. Pushpa, J. Mater. Chem. C, 2014, 2, 2404–2416 RSC.
- L. M. Yang, Microporous Mesoporous Mater., 2014, 183, 218–233 CrossRef CAS.
- L. M. Yang, M. Dornfeld, P. M. Hui, T. Frauenheim and E. Ganz, J. Chem. Phys., 2015, 142, 244706–244717 CrossRef.
- L. M. Yang, E. Ganz, S. Wang, X. J. Li and T. Frauenheim, J. Mater. Chem. C, 2015, 3, 2244–2254 RSC.
- L. M. Yang, G. Y. Fang, J. Ma, R. Pushpa and E. Ganz, Phys. Chem. Chem. Phys., 2016, 18, 32319–32330 RSC.
- H. Zhang, L. M. Yang and E. Ganz, ACS Appl. Mater. Interfaces, 2020, 12, 18533–18540 CrossRef.
- L. L. Liu, X. J. Zhou, Y. M. Yan, J. Zhou, W. P. Zhang and X. S. Tai, Polymers, 2018, 10, 1089–1104 CrossRef.
- J. Tang, M. Cai, G. Xie, S. Bao, S. Ding, X. Wang, J. Tao and G. Li, Chem.–Eur. J., 2020, 26, 4333–4340 CrossRef.
- X. Fan, R. Vakili, E. K. Gibson, S. Chansai, S. Xu, P. Wells, C. Hardacre, A. Walton and N. Al-Janabi, ChemCatChem, 2018, 10, 4238–4242 CrossRef.
- X. Li, Q. Deng, L. Zhang, J. Wang, R. Wang, Z. Zeng and S. Deng, Appl. Catal., A, 2019, 575, 152–158 CrossRef.
- W. W. Jin, J. Z. Zou, S. Z. Zeng, S. Inguva, G. Z. Xu, X. H. Li, M. Peng and X. R. Zeng, Appl. Surf. Sci., 2019, 469, 404–413 CrossRef.
- Z. Jin, Z. Wang, H. Yuan and F. Han, Int. J. Hydrogen Energy, 2019, 44, 19640–19649 CrossRef.
- H. Alamgholiloo, S. Zhang, A. Ahadi, S. Rostamnia, R. Banaei, Z. Li, X. Liu and M. Shokouhimehr, Mol. Catal., 2019, 467, 30–37 CrossRef CAS.
- L. L. Liu, X. S. Tai, X. J. Zhou, J. X. Hou and Z. H. Zhang, J. Alloys Compd., 2019, 790, 326–336 CrossRef CAS.
- D. Ding, Z. Jiang, Q. Ouyang, L. J. Wang, Y. X. Zhang and L. Zan, J. Taiwan Inst. Chem. Eng., 2018, 82, 226–232 CrossRef.
- Q. L. Zhu, J. Li and Q. Xu, J. Am. Chem. Soc., 2013, 135, 10210–10213 CrossRef.
- Z. Lin, M. Luo, Y. Zhang, X. Wu, Y. Fu, F. Zhang and W. Zhu, Appl. Catal., A, 2018, 563, 54–63 CrossRef.
- G. Li, X. He, F. Yin, B. Chen and H. Yin, Int. J. Hydrogen Energy, 2019, 44, 11754–11764 CrossRef.
- N. Kamyar, Y. Khani, M. Amini, F. Bahadoran and N. Safari, ChemistrySelect, 2019, 4, 6113–6122 CrossRef.
- D. Gao, Y. Zhang and L. Zhou, Appl. Surf. Sci., 2018, 427, 114–122 CrossRef CAS.
- K. Wang, L. Xu, H. Z. Chen, H. Y. Qiao, C. Chen and N. Zhang, Chem. Res. Chin. Univ., 2016, 37, 723–727 CAS.
- P. Zhao, N. Cao, W. Luo and G. Cheng, J. Mater. Chem. A, 2015, 3, 12468–12475 RSC.
- Z. J. Liang, X. Z. Xiao, X. Y. Yu, X. Huang, Y. Q. Jiang, X. L. Fan and L. X. Chen, J. Alloys Compd., 2018, 741, 501–508 CrossRef CAS.
- S. P. Jian and Y. W. Li, Chin. J. Catal., 2016, 37, 91–97 CrossRef CAS.
- K. Z. Yang, L. Q. Zhou, X. Xiong, M. L. Ye, L. Li and Q. H. Xia, Microporous Mesoporous Mater., 2016, 225, 1–8 CrossRef CAS.
- L. Zhang, L. Q. Zhou, K. Z. Yang, D. D. Gao, C. Huang, Y. F. Chen, F. Zhang, X. Xiong, L. Li and Q. H. Xia, J. Alloys Compd., 2016, 677, 87–95 CrossRef CAS.
- D. D. Yin, C. Li, H. X. Ren, O. Shekhah, J. X. Liu and C. H. Liang, RSC Adv., 2017, 7, 1626–1633 RSC.
- Y. Guo, Y. Li, J. Chen and L. Chen, Catal. Lett., 2016, 146, 2041–2052 CrossRef CAS.
- N. Y. Lu, F. Zhou, B. B. Fan and R. F. Li, Chin. J. Inorg. Chem., 2015, 31, 2324–2330 CAS.
- H. Pan, X. Li, Y. Yu, J. Li, J. Hu, Y. Guan and P. Wu, J. Mol. Catal. A: Chem., 2015, 399, 1–9 CrossRef CAS.
- L. L. Liu, X. S. Tai and X. J. Zhou, Materials, 2017, 10, 99–111 CrossRef.
- L. L. Liu, X. J. Zhou, L. Liu, S. Jiang, Y. J. Li, L. X. Guo, S. J. Yan and X. S. Tai, Catalysts, 2019, 9, 538–555 CrossRef.
- J. W. Chen, X. L. Chen, Z. G. Zhang, Z. B. Bao, H. B. Xing, Q. W. Yang and Q. L. Ren, Mol. Catal., 2018, 445, 163–169 CrossRef CAS.
- H. Rajati, A. H. Navarchian and S. Tangestaninejad, Chem. Eng. Sci., 2018, 185, 92–104 CrossRef CAS.
- S. J. Zhao, J. Mei, H. M. Xu, W. Liu, Z. Qu, Y. Cui and N. Q. Yan, J. Hazard. Mater., 2018, 351, 301–307 CrossRef CAS.
- L. L. Liu, X. S. Tai, N. N. Zhang, Q. G. Meng and C. L. Xin, React. Kinet., Mech. Catal., 2016, 119, 335–348 CrossRef CAS.
- X. T. Cai, J. W. Wang, R. X. Wang, A. Wang, S. X. Zhong, J. R. Chen and S. Bai, J. Mater. Chem. A, 2019, 7, 5266–5276 RSC.
- X. Chen, D. J. Qian, G. D. Xu, H. Xu, J. T. Dai and Y. K. Du, Colloids Surf., A, 2019, 573, 67–72 CrossRef CAS.
- A. V. Bukhtiyarov, D. B. Burueva, I. P. Prosvirin, A. Y. Klyushin, M. A. Panafidin, K. V. Kovtunov, V. I. Bukhtiyarov and I. V. Koptyug, J. Phys. Chem. C, 2018, 122, 18588–18595 CrossRef CAS.
- Y. Q. Li, Q. Gao, L. J. Zhang, Y. S. Zhou, Y. X. Zhong, Y. Ying, M. C. Zhang, C. Q. Huan and Y. A. Wang, Dalton Trans., 2018, 47, 6394–6403 RSC.
- C. Sarkar, P. Koley, I. Shown, J. Lee, Y. F. Liao, K. An, J. Tardio, L. Nakka, K. H. Chen and J. Mondal, ACS Sustainable Chem. Eng., 2019, 7, 10349–10362 CrossRef CAS.
- M. C. Lozano-Martín, E. Castillejos, B. Bachiller-Baeza, I. Rodríguez-Ramos and A. Guerrero-Ruiz, Catal. Today, 2015, 249, 117–126 CrossRef.
- X. Zhang, Y. C. Guo, Z. C. Zhang, J. S. Gao and C. M. Xu, J. Catal., 2012, 292, 213–226 CrossRef CAS.
- D. Yardimci, P. Serna and B. C. Gates, ChemCatChem, 2012, 4, 1547–1550 CrossRef CAS.
- A. Corma, P. Serna, P. Concepcion and J. J. Calvino, J. Am. Chem. Soc., 2008, 130, 8748–8753 CrossRef CAS.
- P. Serna, M. Boronat and A. Corma, Top. Catal., 2011, 54, 439–446 CrossRef CAS.
- H. Yi, Y. Xia, H. Yan and J. Lu, Chin. J. Catal., 2017, 38, 1581–1587 CrossRef CAS.
- B. Bachiller-Baeza, A. Iglesias-Juez, G. Agostini and E. Castillejos-López, Catal. Sci. Technol., 2020, 10, 2503–2512 RSC.
- A. Hugon, L. Delannoy, J. M. Krafft and C. Louis, J. Phys. Chem. C, 2010, 114, 10823–10835 CrossRef CAS.
- A. C. Krauth, G. H. Bernstein and E. E. Wolf, Catal. Lett., 1997, 45, 177–186 CrossRef CAS.
- C. Liu, K. Yang, J. Zhao, Y. Pan and D. Liu, Catal. Commun., 2015, 67, 72–77 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06432g |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Xiaojing Zhou,
Luxia Guo,
Shijuan Yan,
Yingjie Li,
Shuai Jiang and
Xishi Tai*
*,
Xiaojing Zhou,
Luxia Guo,
Shijuan Yan,
Yingjie Li,
Shuai Jiang and
Xishi Tai*









