
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnpredicted photocatalytic activity of clinoptilolite–mordenite natural zeolite†
Edith A. Alvarez-Aguiñaga a,
María P. Elizalde-González*a and
Sergio A. Sabinas-Hernándezb
a,
María P. Elizalde-González*a and
Sergio A. Sabinas-Hernándezb
aCentro de Química, Instituto de Ciencias, Benemérita Universidad Autónoma de Puebla, Ciudad Universitaria, Edif. IC7, Puebla, Pue. 72570, Mexico. E-mail: maria.elizalde@correo.buap.mx
bInstituto de Física, Benemérita Universidad Autónoma de Puebla, PO Box J-48, Puebla, Pue. 72570, Mexico
First published on 26th October 2020
Abstract
Zeolites are not often used directly as photocatalysts. Their framework and nanocavities have served as support or hosts for photoactive materials or traces of transition metals functioning as photoactive sites for catalysing decomposition and oxidation reactions in the gas phase. Research in this area has been limited to a few synthetic zeolites and in this context, efforts are directed to the preparation of new zeolite-based photocatalysts, when in nature there is an abundance of materials with properties yet to be discovered. We report the application of a natural clinoptilolite–mordenite zeolite as an efficient self-photocatalytic material for the decomposition of caffeine in aqueous solution. Adsorption experiments, combined with textural, crystallographic, and spectroscopic characterization were performed comparatively for the natural zeolite, a synthetic homologue, and the iron-exchanged zeolite. The neat zeolite containing 1.2 wt% of endogenous iron exhibited 99% decomposition of caffeine after 4 h irradiation and a faster reaction rate, followed by the synthetic sample. In contrast, the iron-loaded sample was the less effective zeolite because of pore blocking. Caffeine adsorption occurred on the outer zeolite surface and the photoproducts were hydroxylated pyrimidine rings and linear amide derivatives.
1. Introduction
For a heterogeneous photocatalytic reaction to take place, the participation of three protagonists is necessary: a photon absorbed at a given wavelength, the oxidizing reactant (most frequently the hydroxyl radical), and a solid catalyst (recurrently a semiconductor), which gives the process its heterogeneous character. Although the term photocatalysis was introduced by Plotnikow and more specifically heterogeneous photocatalysis by Landau more than 100 years ago,1 the formation of photoproducts was revealed by Kestner and Hume in their biological and biochemical experiments in which they mentioned the production of “some compounds” affecting respectively, blood regeneration2 and the growth of rats.3 At the same time, the generalized interest in titanium dioxide has persisted now for 50 years since the pioneering work carried out by Fujishima and Honda.4 Therefore, photocatalysis is primarily a change in the speed of a chemical reaction under the influence of photons. If the catalyst is photoactive, this means that under the action of electromagnetic radiation energy, the solid transfers electrons from its full band to the conduction band, matching and/or overcoming the so-called band gap (Eg) and leaving a “positive hollow”. In addition, other chemical events arise: the couple electron–hole promotes (i) oxidation of a substrate via the formation of OH˙ radicals coming from water, and (ii) reduction by capturing electrons, creating superoxide anions O2− coming from oxygen and hydrogen peroxides H2O2. Moreover, the magnitude of the electron wavelength is comparable to the size of the crystal when the semiconductor particles are nanometric and the charge carriers can be considered at a mechanical-quantum level, as particles contained in a box with crystal dimensions and having a higher kinetic energy than non-nanometric materials.Zeolites are materials known for their multifunctional properties as: ion exchangers, molecular sieves, sorbents, supports of catalytic metals, and catalysts in acid–base, oxidation, and hydrogenation reactions. For photocatalytic applications, their two most important properties are their transparency to UV-Vis radiation and the framework-nanocavities couple. While the framework participates in the photoinduced electron transfer, the cavities provide confinement and hydrophilic character.5,6 In contrast, microporous carbons exhibit a combined hydrophobic/hydrophilic surface and despite being strong light absorbing materials, have demonstrated photoactivity due to the interaction of photons with molecules adsorbed in the micropores.7,8
Zeolites photochemistry currently encompasses the use of zeolites as a host rather than a photocatalyst and research is deployed by encapsulating a semiconductor or a photoactive guest in the zeolitic nanopores for the preparation of the so called “zeolite-based photocatalysts”. Zeolite-semiconductor hybrids and composites involve both the framework and cavities of zeolites that function more as a support or matrix for semiconductor clusters.9,10 Relevant research on zeolite-based photocatalysts has been devoted mainly to synthetic powdered zeolites and gas phase reactions.11,12 Authors focused their attention on the effect of both Ti and Fe species as impurities or on what has been called photoactive single sites implanted in the zeolitic structure.10–17 Although much important work has been carried out on the siliceous materials (MCM41,18 silicalite19) and zeolitic catalysts such as NaA,16 faujasite,10 mordenite,10 and HZSM-5,12,13,17,20–22 the photoactivity of neat natural zeolites has rarely been explored before.23 On the other hand, research of zeolite photoreactions in aqueous phase is scarce.14,16,21 Photocatalytic reactions of dissolved dyes, studied by Vis-spectroscopy without the demonstration of photoproducts, is a distinctive feature of such studies. In the past, the ability of natural clinoptilolites to transform some organoarsenicals, among others, arsenobetaine (C5H11O2As) and trimethylarsinic oxide (C3H9OAs) in aqueous medium under natural light has been demonstrated.24,25
Caffeine represents a good model of an organic chemical compound because of its fused heteronuclear ring structure, size, and chemical properties. It is also considered an emerging pollutant26,27 and a marker of human activity, for example storing a concentration of 8.5 μg L−1 in the hydrologic unit of Tijuana in 2015.28 The investigations devoted to photocatalytic transformations of caffeine in water have used TiO2 particles,29,30 doped TiO2,31,32 and TiO2/carbon composites33,34 under UV-Vis,29,31,34,35 and solar irradiation.30 Photolysis of caffeine has also been addressed in a few of these publications.29,30 Carbon has been widely used for the adsorption of caffeine for removal purposes, whereas inorganic sorbents like bentonite,26 sepiolite,36 and montmorillonite are used less frequently.37 There are isolated reports of caffeine retention by zeolitic materials: zeolite NaY38 and natural clinoptilolite from Bulgaria.39
In our study, the photocatalytic decomposition of caffeine under UV-light irradiation was observed for the first time on natural clinoptilolite–mordenite zeolite in form of millimetric granules sieved from the native tuff, and in the absence of a semiconductor. To reveal the photocatalytic activity of pristine zeolitic structures, a mixture of synthetic clinoptilolite and mordenite was studied too. Adsorption of caffeine from aqueous solution was also measured to examine the role of the zeolites framework and nanopores. Zeolite samples were characterized by XRD, XRF, UV-Vis DRS, nitrogen adsorption, and DTG. The photocatalytic pathway of the caffeine decomposition over natural and synthetic zeolites was followed by UV-spectroscopy, DAD-HPLC, and LC-QToF/MS. We found that the photocatalytic activity was associated with adsorption and iron moieties on the natural and synthetic clinoptilolite–mordenite blends. Additionally, photolysis products were involved in the photocatalytic process. Results exhibit an unexpected feature of natural zeolites by themselves as unspoiled photocatalysts for organic aromatic molecules such as caffeine.
2. Experimental
2.1 Chemicals
Hydrochloric acid (37%), ferrous chloride tetrahydrate and caffeine were purchased from Merck. Ammonium hydroxide (28.8%) and formic acid (≥95%) were provided by Sigma-Aldrich. The HPLC grade methanol was purchased from Burdick & Jackson. The deionized water (pH 5.5) used in all experiments was obtained from a Milli-Q purification system.2.2 Zeolites
The natural zeolitic tuff employed in this study was obtained from the location Carranco Blanco, municipality Villa de Reyes, San Luis Potosí 21°50′33′′N 101°4′25′′W (1940 mamsl). The tuff is tertiary volcanic rock that has been formed from deposits of Tophaceous rocks from the upper Mesozoic and the Cenozoic with ignimbrite composition.40 The rock was ground and sieved to obtain a grain size within 2.38 and 3.36 mm. The particles exhibit a rough surface morphology with some clefts of different depth (Fig. S1†). The selected particles were washed for 8 h with deionized water and dried in air at 150 °C to obtain NZ. After that, 1 g of zeolite was suspended in 10 mL of an aqueous hydrochloric acid solution (1 mol L−1) and was stirred for 8 h at 60 °C to produce NH. Also, NZ was ion-exchanged to produce NFe. First, 1 g of NZ and 10 mL of an ammonium hydroxide solution (1 mol L−1) were stirred for 8 h at room temperature. Then, the resulting material was put in contact with 10 mL of a ferrous chloride solution (0.25 mmol L−1) and was stirred for 8 h at room temperature. The prepared NH and NFe were filtered, washed with deionized water and dried in air at 150 °C overnight.With the exception of scanning electron microscopy (SEM), nitrogen adsorption, and potentiometric titration measurements, all characterization analyses were performed with ground samples. Photocatalytic experiments were carried out with grains.
The synthetic clinoptilolite (C) and synthetic Na-mordenite (M) were powders supplied from ZeoSynth (CBV10A) and Zeolyst, respectively. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of C and M was obtained mixing 0.5 g (dry mass) of each. This blend was washed with HCl under the conditions above described for NH. This mixture of synthetic zeolites was referred to as SH.
:1 mixture of C and M was obtained mixing 0.5 g (dry mass) of each. This blend was washed with HCl under the conditions above described for NH. This mixture of synthetic zeolites was referred to as SH.
2.3 Characterization of zeolites
SEM micrographies were obtained using a JSM-6610 LV scanning electron microscope from JEOL. X-ray diffraction (XRD) patterns of the zeolites were obtained using a D8 Discover diffractometer from Bruker with Cu Kα radiation (α = 1.5406 Å). The data were collected over the 2θ range of 5–65°. The bulk content of chemical elements was determined in duplicate by wavelength dispersive X-ray fluorescence using a Bruker S8 TIGER (Billerica, MA, USA) spectrometer. The particles were ground and mixed with 20% Hoechst wax C micro-powder to form tablets.Diffuse reflectance (DRS-UV-Vis-NIR) measurements were performed without dilution on a Cary 5000 spectrometer coupled to an internal diffuse reflectance accessory, both from Agilent-Varian. The band gap energy Eg of synthetic clinoptilolite was estimated using the Kubelka–Munk formalism and the Tauc plot in the high energy range.
Nitrogen adsorption was measured at −196 °C using an Autosorb-1 equipment from Quantachrome. Before analysis, the samples were outgassed at 300 °C for 12 h. The specific surface area (SL) obtained from the monolayer capacity was calculated using the Langmuir equation. The micropore surface (Smicro), micropore volume (Vmicro), and average pore diameter (dp) were calculated from the application of the Dubinin–Radushkevich (D–R) equation and also by the density functional theory (NLDFT) applying the cylindrical pore model to the adsorption isotherm.
The thermal stability of the zeolites structures was studied with a thermogravimetric analyzer TGA Q500 from TA Instruments in nitrogen atmosphere in a temperature range of 50 to 900 °C. The point of zero charge (pHpzc) was determined by potentiometric titration according to the reported methodology.41
2.4 Adsorption experiments
Adsorption tests were performed in aqueous medium (pH 5.5) at 21 °C. The mass-volume ratio was 33.3 g L−1. Polycarbonate cells were used in all batch adsorption experiments. The initial and final concentrations of caffeine in the adsorption experiments were determined at 274 nm with a UV-Vis HACH spectrophotometer.The kinetic curves of adsorption were obtained by contacting of the NH and NFe zeolites with a caffeine solution (50 mg L−1). An aliquot of 50 μL was taken at time intervals up to 71 h. The equilibrium curves of adsorption were measured in the concentration range within 1 and 60 mg L−1 (309 μmol L−1). The system was stirred for 1 h and then the caffeine solution was left in contact with the zeolite sample for 48 h.
2.5 Photocatalytic activity
The photocatalytic degradation of caffeine was performed under nitrogen flow in a glass reactor equipped with a water jacket to maintain the temperature at 21 °C. A short wavelength (λ = 254 nm) mercury lamp (UPV®) was used as radiation source and it was introduced in a quartz jacket. Then, 0.1 g of zeolite and 10 mL of caffeine solution (50 mg L−1) were added into the reactor. The system was kept in the dark for 30 minutes to allow adsorption. Aliquots of 120 μL were collected at −0.5, 0, 0.5, 1, 1.5, 2, and 4 hours of irradiation.A liquid chromatograph Series 1200 Infinity coupled with a diode array detector (DAD) Series 1260 Infinity from Agilent was used to determine the caffeine concentration and to detect photoproducts. The analyses were conducted at 25 °C using an Ultrasphere ODS (4.6 × 75 mm, 3 μm) column from Beckman. The flow rate was 1 mL min−1 and the mobile phase was water (A) and methanol (B), both containing 0.1% formic acid. At time 0, the eluent composition was 90% (A) and 10% (B), being 10% (A) and 90% (B) after 20 min. The injection volume was 10 μL. The detection was performed at 236 and 274 nm. The identification of photoproducts was performed with an Agilent 1260 HPLC system and a ESI-QToF-MS spectrometric detector (6520) from Agilent. For the chromatographic separation, a LC column from Phenomenex (Synergi Fusion-RP, 4.6 × 150 mm, 80 Å, 4 μm) operating at 25 °C was used. The injection volume was 20 μL of each sample. The mobile phase consisted of water containing 0.1% of formic acid (A) and methanol (B) with 0.1% formic acid, which were delivered at 1 mL min−1. At time 0, the eluent composition was 100% of (A); being 60% of (A) and 40% of (B) after 20 min. Electrospray ionization (ESI) was used as source operating in positive mode. The MS scanned from 100 to 1700 m/z was obtained with a fragmentor voltage of 175 V. Ultrahigh pure nitrogen was used as drying and nebulizer gas at temperatures of 200 and 350 °C, respectively.
3. Results and discussion
3.1 Photocatalytic decomposition of caffeine by clinoptilolite–mordenite zeolite
In our research, the native tuff NZ corresponded to a mixture (1:1) of natural zeolites identified and quantified by XRD as clinoptilolite and mordenite (Fig. S2†). Other identified minerals were calcite and quartz. No hematite (Fe2O3) and ilmenite (FeTiO3) were found. NZ was converted to its hydrogenated form NH to remove impurities and, additionally, for three more purposes: (i) to minimize interferences of electrostatic fields caused by extra-framework cations, (ii) to release cavity volume occupied by water coordinated to cations, and (iii) to favour the electron-donating ability of the zeolite by creating more Lewis basic sites. Caffeine decomposition in aqueous solution was evaluated over NH under UV-light irradiation (Fig. 1). The formation of the main photoproducts will be discussed later. Under the same conditions, the iron-exchanged sample NFe prepared ex-profeso from NZ showed less photoactivity than NH with a respective decrease of the caffeine concentration of 55% (NFe), and 99% (NH) after 4 hours irradiation.
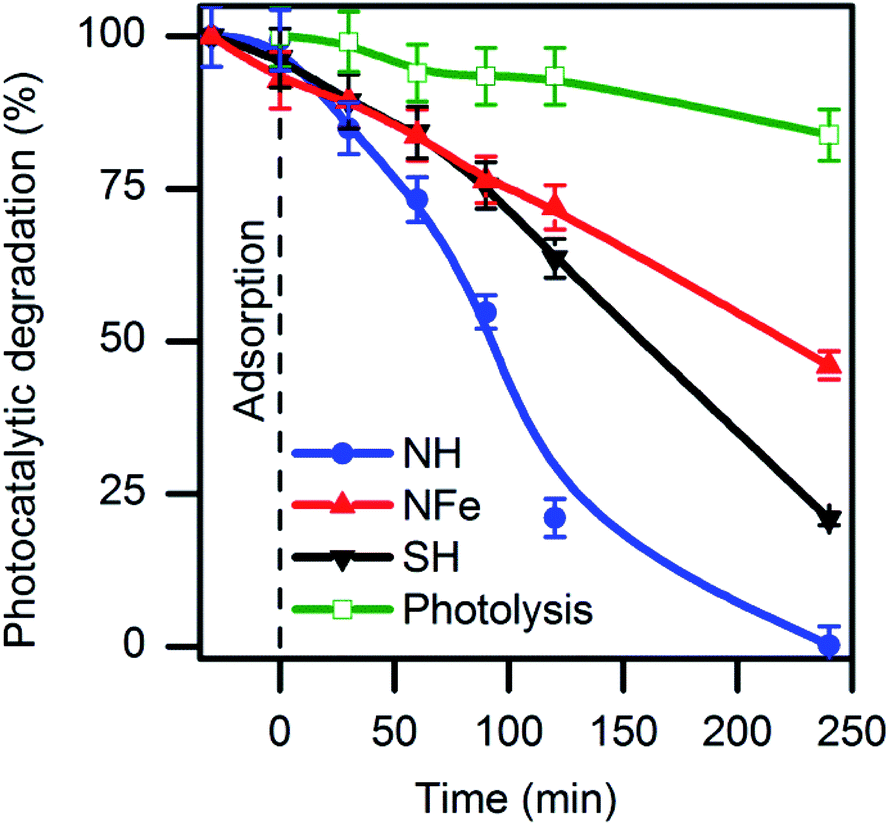 | ||
| Fig. 1 Decomposition of caffeine in aqueous solution by photolysis and photocatalysis on natural (NH, NFe) and synthetic (SH) clinoptilolite–mordenite. | ||
As reference, TiO2 particles needed the same time for the total decomposition of caffeine under similar conditions (Fig. S3†). Mineralization of caffeine with initial concentration of 10 mg L−1 was achieved by the 2.33% Fe–MFI zeolite but after 22 hours of an oxidative degradation with H2O2.19 It can be seen in Fig. 1 that photolysis produces a 16% decrease of the caffeine concentration after 240 minutes and ∼5% after 1 hour in agreement with reports for the same caffeine concentration.27,29
Among the studied samples, NH was the most efficient zeolite for the decomposition of caffeine with a rate of 12.2 × 10−5 s−1 (Table 1). This magnitude implies that the photocatalytic reaction over the zeolite NH occurred ten times faster than photolysis following at 1.2 × 10−5 s−1.
| Zeolite | Langmuir–Hinshelwood parameters | ||
|---|---|---|---|
| kobs × 105 (s−1) | k′ × 108 (s−1 m−2 L−1) | R2 | |
| a k′ = (V/wSL)kobs (pseudo-first order rate constant normalized per square meter, where V, w, and S are the irradiated volume, photocatalyst mass and specific surface area SL, respectively). | |||
| Photolysis | 1.2 | — | 0.94 |
| NH | 12.2 | 7.1 | 0.93 |
| NFe | 4.3 | 5.3 | 0.98 |
| SH | 4.7 | 2.5 | 0.94 |
In order to confirm the photoactivity of the clinoptilotite- and mordenite-type zeolite structures, the mixture (1:1) of synthetic clinoptilotite and mordenite was tested under identical conditions as the natural sample. The synthetic hydrogenated blend SH showed photoactivity in caffeine degradation, but the reaction rate was almost one third (kobs = 4.7 × 10−5 s−1) in comparison with the natural NH. The answer to this question will be sought when analysing the composition and textural features of SH and NH. While the observed rate kobs was similar over NFe and SH, the normalized k’ value is higher for NFe than that for SH due to the fact that NFe might have contracted its surface area during the ion-exchange procedure. Coincidentally, the value of 4.3 × 10−5 s−1 we obtained with NFe (1.57% iron content) was equal to that obtained by Motuzas et al. with 2.33% Fe–MFI for the same caffeine concentration in presence of H2O2 though the different mass to volume ratio (w/V) used by the authors (0.33 g L−1).19 The w/V ratio set by us (10 g L−1) is justified by the fact that we use millimetric particles of zeolite, which at the same time facilitates their recovery after the experiment in comparison with nanometric powders that must be centrifuged and filtered.
3.2 Adsorption of caffeine by the framework of clinoptilolite–mordenite natural zeolite
Most studies on supported photoactive semiconductors have put forward the explanation that during adsorption the reactant directly contacts photoactive sites on the surface of the photocatalyst. An alternative view is the formation of intermediates nearby the sites. To get a deeper insight into the self-photoactivity of the natural clinoptilolite–mordenite, we examined the adsorption of caffeine from aqueous solutions on NH and its iron-exchanged form NFe (Fig. 2).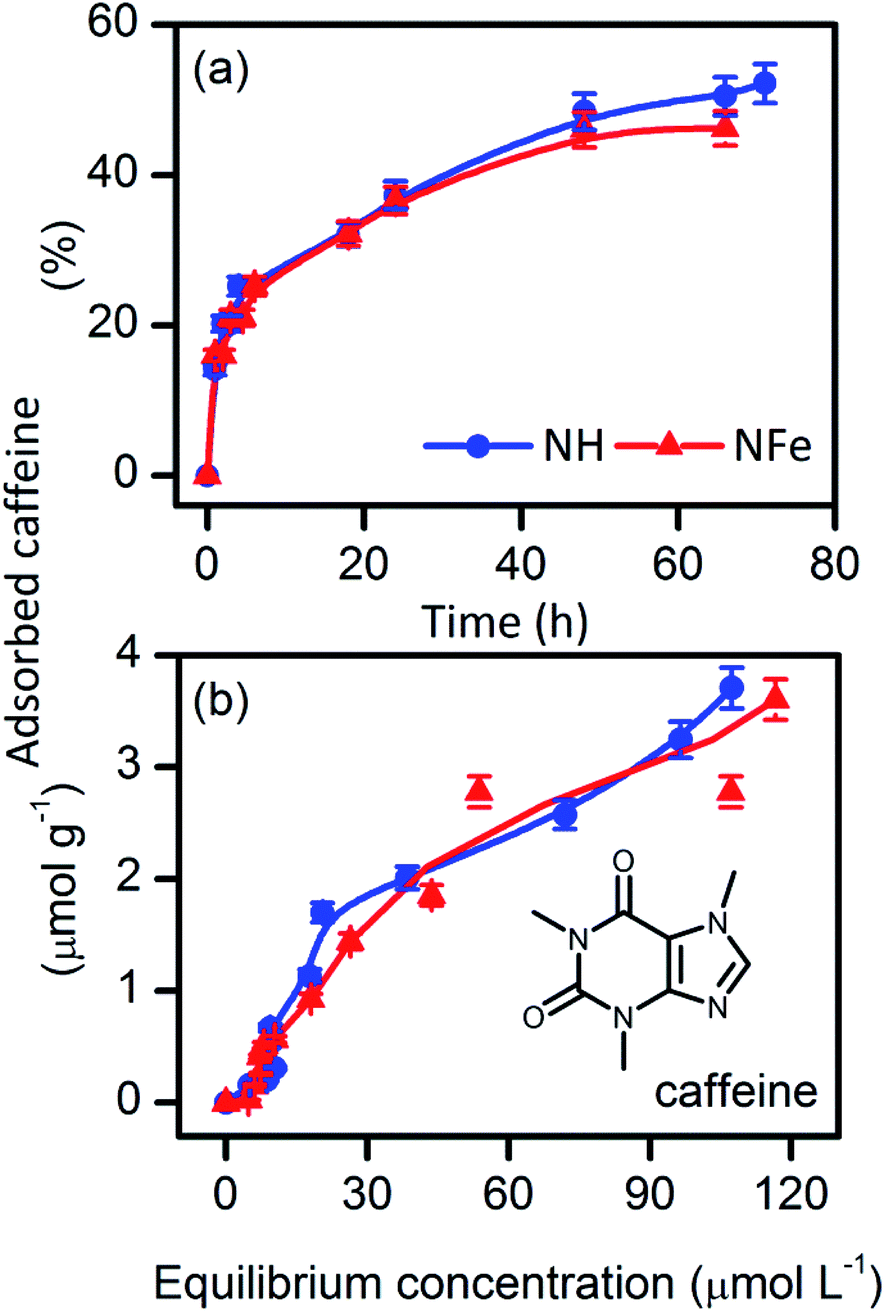 | ||
| Fig. 2 (a) Adsorption kinetics, and (b) adsorption equilibrium of caffeine from water on the natural clinoptilolite–mordenite zeolites NH and NFe. | ||
Table 2 shows that the rate of caffeine adsorption is similar on NH (k2 = 13.5 × 10−6 g mol−1 s−1) and NFe (k2 = 14.2 10−6 g mol−1 s−1). Comparison of these magnitudes with those obtained by Yaneva et al.39 for Bulgarian clinoptilolite is not possible since the authors performed their experiments with stirring at temperature and pH values different from ours. The adsorption equilibrium parameters KLF, nm, and β are also comparable in NH and NFe. This suggests that the interaction between caffeine and the two zeolite samples is alike. At the highest concentration used in our study (C0 = 50 mg L−1 to 0.25 mmol L−1), caffeine can be still considered a monomer.42 The molecular diameter of caffeine (Dmolec. = 0.752 nm (ref. 39)) is beyond the large 12-member ring window (0.65 × 0.70 nm)43 of mordenite and also of the two 8-member ring windows of clinoptilolite, those sized (0.26 × 0.47 nm), and (0.33 × 0.46 nm).24 With a calculated molecular height of 0.21 nm, caffeine would penetrate only the large, 10-member ring window of channel A (0.30 × 0.76 nm) of clinoptilolite.
| Zeolite | Pseudo-second order kinetic parameters | Langmuir–Freundlich adsorption parameters | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| neq,exper × 106 (mol g−1) | neq,calc × 106 (mol g−1) | k2 × 106 (g mol−1 s−1) | R2 | χ2 | KLF (L g−1) | nm × 106 (mol g−1) | β | R2 | χ2 | |
| a neq: maximum adsorbed amount: experimental (exper) and by calculation (calc) applying a pseudo-second order kinetic equation, KLF: Langmuir–Freundlich adsorption constant, nm: maximum monolayer adsorption capacity, β: adjustment coefficient related with surface heterogeneity, R2: determination coefficient, χ2: Pearson's distribution. | ||||||||||
| NH | 4.2 | 4.5 | 13.6 | 0.98 | 0.0019 | 0.11 | 4.6 | 1.3 | 0.96 | 0.0027 |
| NFe | 3.6 | 3.9 | 14.2 | 0.96 | 0.0025 | 0.13 | 4.0 | 1.4 | 0.96 | 0.0023 |
If caffeine is oriented in perfect correspondence with this window, its diffusion into the zeolite cavity could be unrestricted and this only in case the molecule is not solvated, which is difficult to achieve in aqueous solution. Thus, Yaneva's et al.39 graphical relationship between the caffeine dimensions and its encapsulation in clinoptilolite cavities needs to be clarified because the authors disregarded the window size of the channels in the crystalline structure of the zeolite. Furthermore, they assumed that the tuff micro- and mesopore sizes measured by nitrogen adsorption corresponded to the zeolite cavity. Regardless of these conceptual errors, the amount of caffeine adsorbed by the 87%-clinoptilolite39 at saturation matches with the nm magnitude (Table 2) obtained by us for NH (50%-clinoptilolite). As the framework chemical nature of clinoptilolite and mordenite is similar, the magnitude of nm does not reflect the difference in the clinoptilolite content and another would have been the result in case of the intra-cavity interaction of caffeine with the clinoptilolite portion of samples with different clinoptilolite content. Taking into account that caffeine does not protonate in water at pH 5.5 and is stable (Fig. S4†) during adsorption on NH (pHpzc 3.0), and NFe (pHpzc 3.0), we can conclude that it adsorbs on the framework external surface of the natural zeolite under study.
3.3 Relationship between zeolites nanoporosity and adsorption of caffeine
The nitrogen adsorption isotherms in Fig. 3 show type Ib adsorption isotherms according to the refined IUPAC classification44 and demonstrate that all zeolites are microporous. Analysis of the data obtained by the Langmuir equation (Table 3), applicable to type I isotherms indicates that the specific surface area decreases in the order: SH ∼ NH > NFe. The synthetic M and C used as precursors of SH present the upper-lower extreme positions in Fig. 3.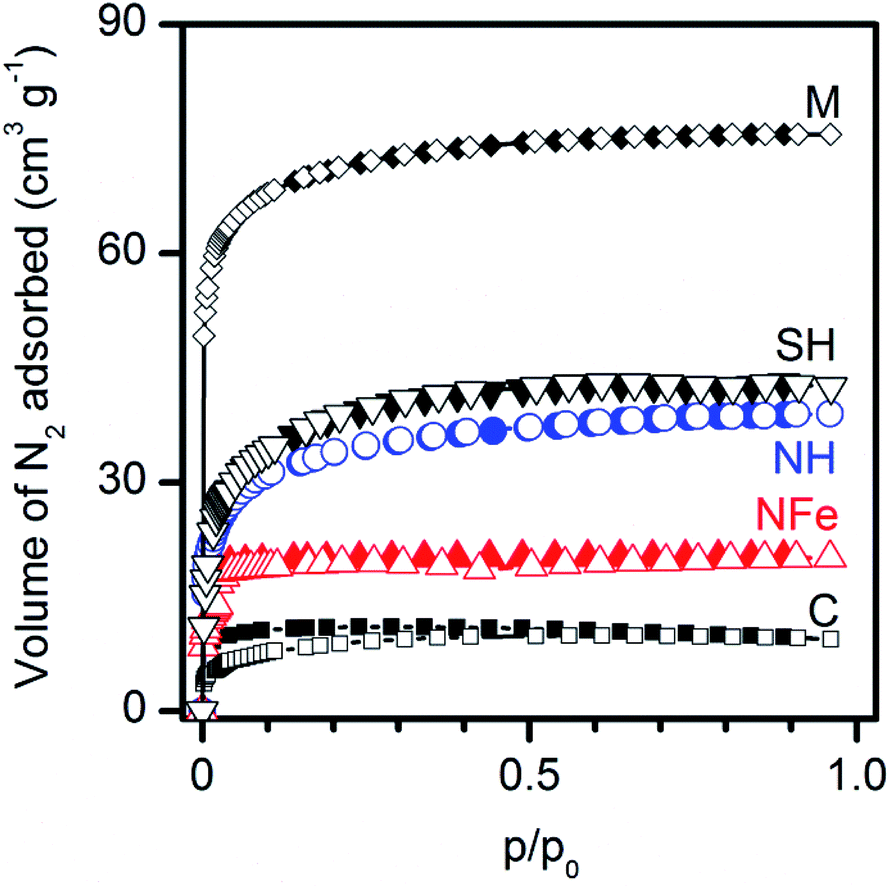 | ||
| Fig. 3 Microporosity of natural (NH, NFe) and synthetic (M, C, SH) clinoptilolite–mordenite zeolites assessed by nitrogen adsorption–desorption isotherms at 77 K. | ||
| Zeolite | SLa (m2 g−1) | Vtotalb (cm3 g−1) | Dubinin–Radushkevich (D–R) equation | DFT analysis | ||||
|---|---|---|---|---|---|---|---|---|
| Smicro (m2 g−1) | Vmicro (cm3 g−1) | Vmeso (cm3 g−1) | dp (nm) | Vtotal (cm3 g−1) | dp (nm) | |||
| a Langmuir specific surface area.b Total adsorbed volume at p/p0 = 0.95. | ||||||||
| NH | 171 | 0.060 | 159 | 0.057 | 0.003 | 2.6 | 0.057 | 1.4 |
| NFe | 87 | 0.031 | 85 | 0.030 | 0.001 | 2.4 | 0.030 | 1.6 |
| SH | 191 | 0.066 | 186 | 0.066 | 0 | 3.0 | 0.063 | 1.6 |
The pore volumes obtained by the D–R equation indicate that mesoporosity is negligible. The pore sizes obtained by the DFT method correspond to wide-microsize (dp < ∼2.5 nm) pores in all cases. The most remarkable result in the series of samples is the decrease, by half in the pore volume of NFe with respect to NH, which indicates the blockage of pores with iron species. The calculation of the surface portion of the zeolites occupied by the caffeine molecule (ωm = 0.65 nm2 molec.)45 in Fig. 2b at saturation values (nm magnitudes in Table 2) allows us to observe that only a small fraction of the zeolite external surface was covered: 1.8 m2 g−1 for NH and 1.6 m2 g−1 for NFe. These values match approximately the magnitude order of the resulting area difference (SL − Smicro) and indicate that adsorption of caffeine from water in the dark occurs only on external specific sites of the framework as discussed in the previous section.
3.4 Correlation of photoactivity with zeolites composition
The DTG curves of the zeolites (Fig. S5†) show a sharp weight loss centred at 84 °C, which does not depend on the form of the zeolites and corresponds to the loss of physically adsorbed water in the zeolite cavities. A shoulder in NH at 150–250 °C represents the removal of hydroxyl species by associative desorption.46 The minor and broad peak within 300 and 500 °C is probably due to loss of water coordinated to cations. The differential thermogravimetric profiles of the samples NH, NFe, and SH are similar and do not present any weight loss which could correspond to organic moieties. The inorganic composition of the native tuff NZ (Table 4) includes elements typical of the natural Mexican clinoptilolites/mordenites and those from other regions.46,47 The preparation of NH from NZ by acid treatment resulted in dealumination (16% in NH and 23% in NFe) and the desired decationation concerning Na+, although NFe did not become homoionic. Among the elements present in the native tuff, iron, and titanium are especially important due to the role they can play in photocatalysis. While titanium is practically negligible in the tested photoactive zeolites, the iron content increases in the series: SH < NH < NFe and does not linearly correlate either with the rate kobs (R2 = 0.75) and k′ (R2 = 0.59) in Table 1, nor with the zeolites photoactivity in the caffeine decomposition reaction in Fig. 1. This finding is not consistent with the results found mainly with the synthetic zeolite Fe-ZSM-5 when it contained trace quantities (0.043 wt%) of iron impurities13,20 or framework iron deliberately introduced up to values of 0.089 wt% Fe.12 The authors found that the rate of photocatalytic reactions in the gas phase depended linearly on the concentration of the iron species in the zeolite, which acted as photoactive centres excited by UV light to form a charge-transfer excited state.| Zeolite | Si/Alat | TEC (meq. g−1) | Weight percent (%) | |||||
|---|---|---|---|---|---|---|---|---|
| Na | Mg | Al | K | Fe | Ti | |||
| a bdl: below detection limit. | ||||||||
| NZ | 6.0 | 2.9 | 1.3 | 0.15 | 4.14 | 2.9 | 1.21 | 0.07 |
| NH | 6.3 | 1.1 | 0.2 | 0.14 | 3.49 | 2.6 | 1.22 | bdl |
| NFe | 5.3 | 4.8 | 0.8 | 0.15 | 3.20 | 2.4 | 1.57 | 0.12 |
| SH | 5.7 | 2.2 | 2.3 | 0.05 | 5.53 | 4.5 | 0.01 | 0.01 |
The nature of iron species is complex. It has been demonstrated that ca. 95% of the iron in Fe-ZSM5 corresponds to mononuclear sites only when Fe ≤ 0.3 wt% (ref. 48) and iron ions must be intra-framework.20 The diffusion through the channels and the occupation of the cavities in an aqueous medium are determined by the hydration energy of the species. In addition, the occupiable volume in zeolite ZSM5 (framework type MFI) is much larger than that of clinoptilolite–mordenite (framework type heulandite and mordenite, respectively).43 If we consider that because of the genesis of the tuff it is unavoidable47 to find iron (1.2% in NZ), which is also conserved after acid treatment (1.2% in NH), the gain of iron by cation exchange in NFe corresponds to 0.4%, which is a high value and improves the Fe/Al ratio from 0.3 in NH to 0.5 in NFe. Thus, and as previously demonstrated by us for the same natural zeolite with a 0.4 Fe/Al ratio,49 the following additional extra-framework iron species are present in the studied NFe: hydrated cations Fe(OH)2+, mono- and dinuclear aqua-hydroxo iron cations [Fe(OH)2(H2O)4]+, [Fe2(OH)2(H2O)8]4+, and FexOy clusters, of which only some would be in channel A and other would block the zeolite windows with a half decrease of the pore volume as already discussed (Table 3). According to Yan's et al. investigation, both ion-exchanged and outside-framework iron species do not represent photoactive sites.20
In our study, despite the small number of studied self-photoactive zeolites, we sought to correlate the rate k′ with some physicochemical feature that could play a role in the photocatalytic decomposition of caffeine. In the absence of a photogenerated electron–hole pair, a charge-transfer excited state in an oxygen-bonded metal impurity or another active site may be responsible for a photoreaction. In the first case, Anpo and other authors11–13,20 have verified the function of transition metal impurities and in the second case, Kato50 and Wang et al.22 attributed photoactivity of ZSM5 to highly isolated framework Al–O units interacting with no metal cations. In our case, the values of k′ in Table 1 correlate poorly with the ratio Fe/Al (R2 = 0.52) and with the aluminium percent (R2 = 0.69). We did not consider the Lewis acid–base property (Si/Al ratio) of the zeolites to correlate with the reaction rate because in natural zeolites a fraction of Si can come from quartz.
Pair–wise comparisons allow combined effects of zeolite performance to be seen. If, due to the magma origin of the natural zeolites,47 the iron in the tuff NZ and the sample NH is endogenous, it could be considered a framework element. In Fig. 1 we observe that the reaction over NH (1.2 wt% Fe, Fe/Al 0.3) is faster than over the synthetic homologue SH (0.1 wt% Fe, Fe/Al 0.009) due to a lower charge-transfer capacity in SH since the textural parameters are similar. The position of the curve for the synthetic zeolite SH in Fig. 1 is important. It shows that despite the low iron content (0.1 wt%), as reported, it is sufficient to generate a photoexcited charge-transfer effect and a photoreaction.12,13,20,48 It would also be an indirect indication of the presence of framework iron.20 In contrast, NFe with high iron concentration (1.6 wt% Fe = 1.2 framework + 0.4 iron-exchanged iron) presents a similar value of kobs (Table 1) as the synthetic SH reflecting the lack of access to all framework photoactive sites of NFe. On the other side, the decomposition of caffeine occurs faster over the natural NH in comparison to the iron-exchanged NFe carrying intra- and extra-framework iron. This indicates that not only the photo-excited charge transfer of [M(n−1)+–O−]* species is responsible for the photoactivity, but also diffusion and stabilization of the photogenerated charge carriers in the micropores volume (Vmicro in NH ∼ 2Vmicro in NFe) upon irradiation. It is also important to mention the possibility that intermediary photoproducts are confined, increasing the interaction with radicals and that the recombination rate is slower in micropores.
3.5 Electronic structure and framework iron in natural zeolites
The natural NH zeolite, ion exchanged NFe, synthetic clinoptilolite C, and synthetic mordenite M were studied by DRS. The lamp used in photocatalytic experiments is suitable for the excitation of natural and synthetic zeolites since the lamp's most intense emission line is at 254 nm (∼4.88 eV) (Fig. S6†) matches with one of the most intense absorption bands of the zeolites. The excitation of different species of aluminium, iron and titanium present in zeolites is also possible because the lamp presents other emission lines, whose intensity has a ∼3:100 relationship with the line at 254 nm (manufacturer's information).
DRS spectra (Fig. S7†) show that all zeolites absorb light in the UV region. The absorption of the synthetic C and M extends up to 350 nm, while the natural NH and NFe absorb up to 550 nm and more intensively. In the range 240–260 nm the bands corresponding to oxygen-bonded tetrahedral framework aluminium and charge transfer processes from aluminium to oxygen are described.51 Octahedral extra-framework aluminium produces a band within 280–370 nm with a width depending on the environment.52,53 In M this band appears at 318 nm.
Transitions associated with iron are relevant to the natural zeolites NH (with endogenous iron) and NFe (endogenous + exchanged iron). The presence of the element iron in natural zeolites (Table 4) may explain the higher light absorption in the UV region and also the presence of the absorption bands in the visible region compared to the synthetic zeolites C and M. Charge transfer transitions dπ–pπ between iron and oxygen appear within 200 and 300 nm as intense bands and correspond to framework iron and oxygen.52 Both in NH and NFe this band is centered around 262 nm. The transition associated with extra-framework iron (Fe3+) octahedrally coordinated is presented around 360 nm also in both samples. Additionally, low intensity bands assigned to spin and symmetry forbidden d–d transitions of Fe3+ appear between 350 and 500 nm.54 The band at 504 nm corresponds to the forbidden transition d–d, while at 408 nm it is attributed to octahedral Fe3+ ions in small oligomeric groups (clusters) (Fe3+xOy), according to Pérez-Ramírez et al.54 The band at 749 nm is only present in the natural zeolites NH and NFe. It may be due to Fe2+ ion transitions or Fe2+–Fe3+ intervalence charge transfer in sites of near-octahedral coordination.55,56 The broad band centred at 1047 nm is assigned to transitions of the Fe2+ ion,56,57 whose existence in this zeolite we demonstrated in the past.49
In NFe, a wide band appears at 2115 nm. This may be due to vibrations from different species of Al–OH or Mg–OH,58 generated in the ion-exchange procedure. The wide bands (1416, 1465, 2185–2210 nm) are due to metal–OH and OH vibrations from water (1910 nm).56,58 Transitions associated with titanium in the NFe, C and M samples were not visible due probably to the low titanium concentrations. Thus, we infer that the photoinduced species that can play the role of photoactive species in the framework of the iron-containing samples, NH and NFe under UV irradiation, are: 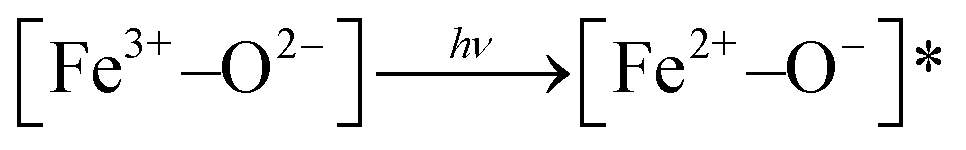 . In case of SH, practically lacking iron, the photoactivity of Al–O species could be considered. Furthermore, for NFe and SH containing traces of titanium, the interaction
. In case of SH, practically lacking iron, the photoactivity of Al–O species could be considered. Furthermore, for NFe and SH containing traces of titanium, the interaction 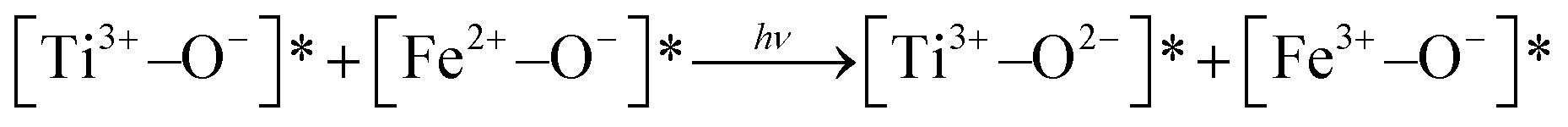 would facilitate migration and charge separation by increasing the life of the charge carriers17 for the degradation of caffeine.
would facilitate migration and charge separation by increasing the life of the charge carriers17 for the degradation of caffeine.
3.6 Experimental band gap energy of synthetic clinoptilolite and mordenite
By applying the Kubelka–Munk function to the diffuse reflectance spectra (Fig. 4) we observe that the absorption edge of the natural zeolites NH and NFe extended to lower energy (wider wavelength) in comparison to the mixture SH and its precursors, synthetic clinoptilolite C and synthetic mordenite M. According to spectroscopic studies of the zeolite ZSM-5, only iron species in different states originate bands in the 200−500 nm (6.19–2.48 eV) range and iron is unavoidable in synthetic zeolites.12 So as mentioned in the previous sub-section, the iron species in the synthetic zeolites possibly contribute to absorption in the weak bands of Fig. 4, despite the low iron content: 0.04 wt% in C, 0.07 wt% in M, and 0.01% in SH. The framework of typical aluminosilicate zeolites has a band gap of about 7 eV.12 Results from natural mordenite registering a band gap estimation of 2.63 eV suggest that this value can be attributed to the high iron (10.2 wt%) and titanium (3.5 wt%) content in the not calcined zeolitic tuff.23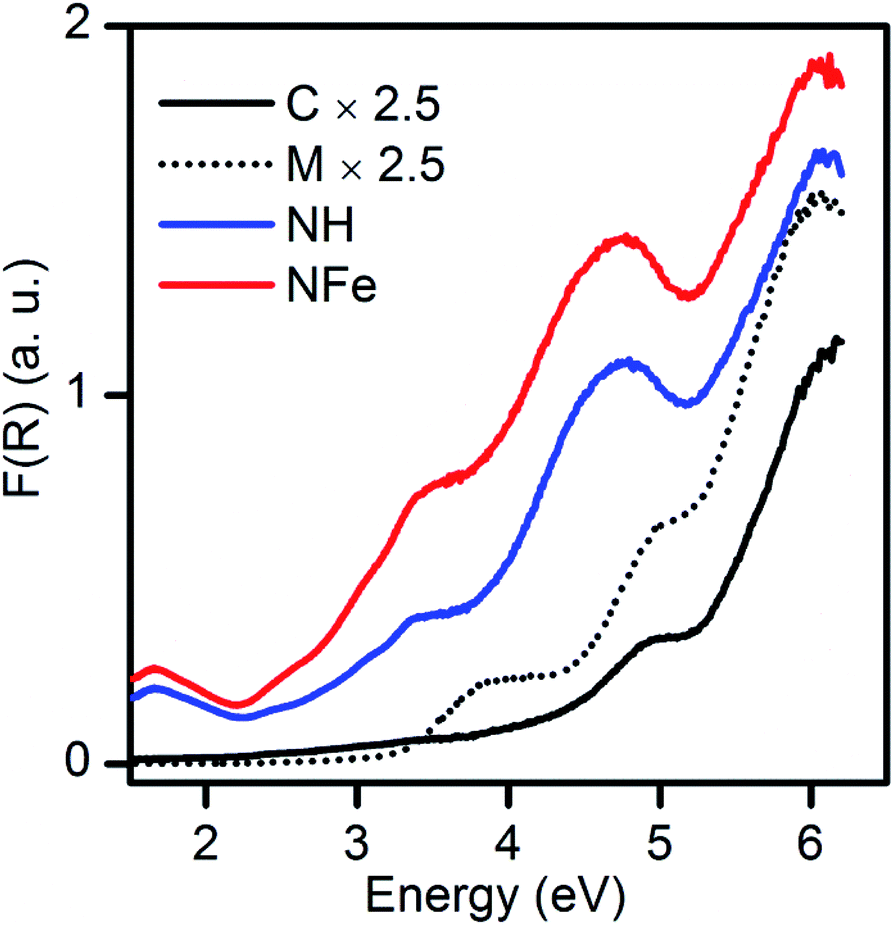 | ||
| Fig. 4 Electronic evidence of iron species in natural and synthetic zeolites. | ||
Since very little has been written about self-photoactivity of zeolites, there has been no examination of the optical properties of natural zeolites, such as the abundant clinoptilolite. Although the type of transition of the zeolites is unknown, it was interesting to construct the Tauc plot to determine the band gap energies by using our experimental spectra and considering both a direct and an indirect transition. For the synthetic clinoptilolite C used in our study, band gap energies of 4.26 and 4.46 eV (291.0 and 278.0 nm) were determined for the indirect and direct transitions, respectively (Fig. S8b and c†). A theoretical value found for an idealized clinoptilolite consisting of 44 silicon and 76 oxygen atoms, with space group C2/m was 5.593 eV (221.7 nm).59 This value is very close to one of the values found, E1D = 5.53 eV (∼224 nm), that we obtained for a direct transition in Fig. S8c†, being thus related to transitions in Si and O atoms. This cut-off wavelength value does not mean that synthetic clinoptilolite does not absorb at longer wavelengths as it can be seen in Fig. 4. Additionally, two values of band gap energies were determined for the synthetic mordenite M, 3.26 and 3.45 eV (380.3 and 359.4 nm), for indirect and direct transitions (Fig. S8e and f†), respectively. Summarizing, if we compare the band gap energy of the two synthetic materials, mordenite has lower values than clinoptilolite whatever the type of transition.
3.7 Hydroxylated, demethylated and amine photoproducts of caffeine
When the formation of photoproducts is not clearly demonstrated and only the decrease in concentration of the reagent is registered, the continuation of adsorption during irradiation may be suspected. Photocatalytic decomposition of caffeine (Fig. 1) over NH, NFe, and SH was followed by DAD-HPLC and LC-QTOF/MS (Fig. 5 and S9†). The two natural zeolites NH and NFe show notable differences in photocatalytic performance and texture properties although they adsorb similarly caffeine, whereas the natural clinoptilolite-mordenite sample NH differs from its homologue synthetic blend SH in the photocatalytic performance and iron concentration. Once NH has proved effectivity in the decomposition of caffeine, we were interested in obtaining information about the reaction path and possible product selectivity. Thus, we performed the identification of the photoproducts of caffeine produced under UV-light (Fig. 5). Monitoring the reaction during four hours we observed the formation of the same products over the three zeolites (Fig. S9†) with the exception of the products that emerged as peaks 6 (diamino pyrimidine compound) and 7 (amino pyrimidine) in the trial with NFe. They were found throughout the whole reaction (Fig. S9†). However, we cannot conclude that both compounds were formed only by NFe because the products could have been degraded further or adsorbed on NH and SH, but not on NFe which is the slowest zeolite. We compared the kinetic curves of the products obtained over NFe with those achieved in photolysis (Fig. S10†). The graph corresponds to a complex set of consecutive reactions, being 1,3,7-trimethyl-2,3,6,7,8,9-hexahydro-1H-purine-2,6-diol (peak 12 in Fig. 5) one of the main intermediates. Also two open chain nitrogen-containing compounds (peaks 1 and 4) were found in photolysis and photocatalytic reactions as depicted in the general decomposition path of caffeine (Fig. S11†). As already discussed, NH and NFe exhibit different rates despite having similar photoactive charge carriers promoted by iron species. The results call for some explanation and comment: we can briefly say that the reaction pathway is accompanied by the photolysis of caffeine in solution, whose solvated molecule does not penetrate the pores of the zeolites but gives rise to smaller intermediates that do not have access to the blocked pores of NFe, but diffuse into the NH nanopores, where they are confined for further interaction with the charge carriers.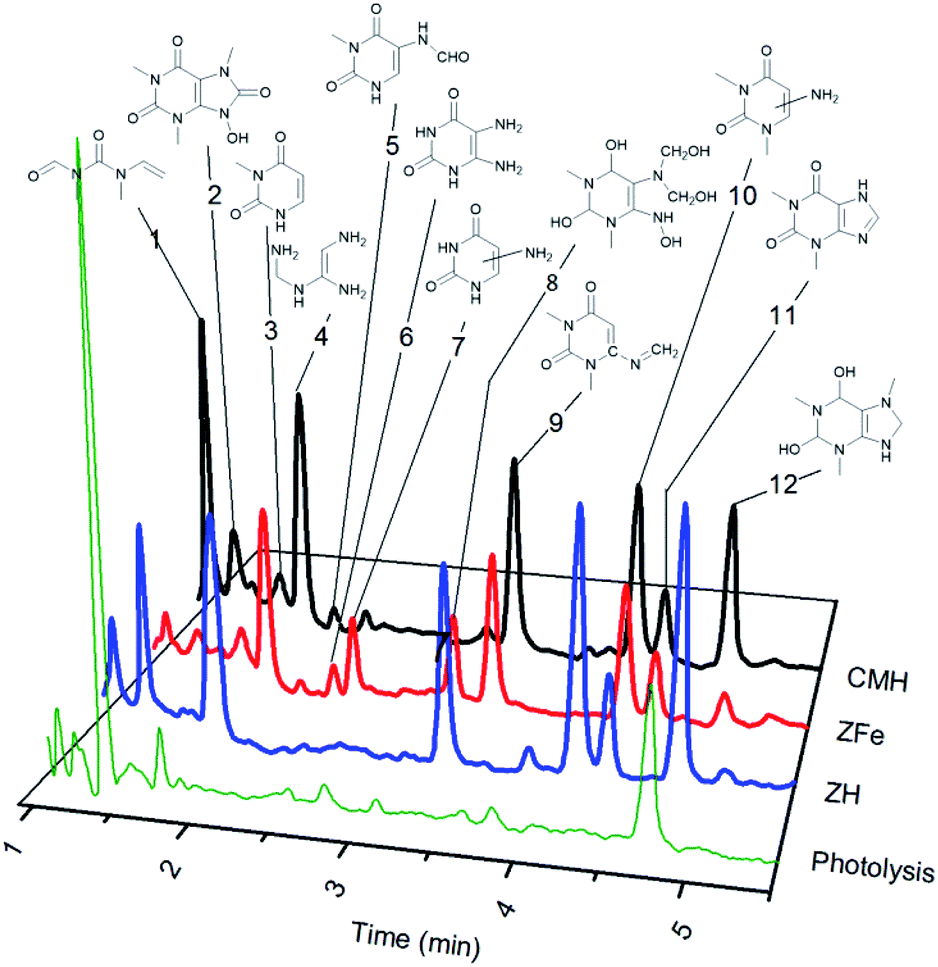 | ||
| Fig. 5 Photoproducts of caffeine formed under UV irradiation over natural and synthetic clinoptilolite–mordenite zeolites. | ||
Twelve compounds were identified in different extents over the three zeolites, while photolysis produced small amounts of the same intermediates (Fig. 5) and mainly N-methyl-N-(methyl(vinyl)carbamoyl)formamide (peak 1), (Z)-N′-(((aminomethyl)amino)methyl)formimidamide (peak 4), and trimethyl-hexahydro-1H-purine-diol (peak 12). Theophylline (peak 11) was detected in all the experiments and the other photoproducts correspond mainly to pyrimidine rings (peaks 3, 5–10). Some compounds remained as dione or transformed into diol groups. Demethylation at position 1 and 3 of the pyrimidine ring was also evidenced.
Some authors were not successful in mineralizing caffeine (50 mg L−1) by a Fenton-like heterogeneous reaction using a MFI zeolite with 1.2 wt% Fe. Their results would have been very useful for comparison with NH (1.2 wt% Fe) if the identity of the by-products had been reported.19 Ten of the products detected by us agree with those detected by Barrocas et al.60 in photocatalytic reactions using titanate nanostructures and UV-Vis irradiation. In contrast, only one photoproduct agreed with the set detected by Qi et al. over Co-MCM41.61
4. Conclusions
By using a Mexican natural zeolite consisting of clinoptilolite and mordenite (1:1), the photocatalytic decomposition of caffeine by irradiation at 254 nm was possible. The natural decationized zeolite NH presented the highest rate constant value in the decomposition of the studied stimulant compound. This zeolite contains small quantities of iron (1.2 wt% Fe), which could be the origin of different photoinduced species responsible of the photocatalytic performance. It is important to mention, that although Al–O species can originate active photocatalytic sites, iron can give origin to more effective photocatalytic species under our experimental conditions. Despite the similar textural properties of NH and SH, the rate constant is higher in NH due to its larger iron content. Additionally, a higher presence of Al–OH species as shown by DRS-UV-Vis-NIR in NFe does not increase the rate constant. It is possible that even though some of the aluminium was replaced by iron in the zeolite framework, there is an important detrimental effect in the textural properties showing a negative consequence on the kinetics of the photocatalytic reaction.
Although the mineralization of the solution was not measured, the identification of several photocatalytic intermediates showed that a natural clinoptilolite–mordenite, long considered non-photoactive, was able to decompose caffeine even involving the ring opening of the molecular structure.
This blend of grain-particulated decationized zeolites (clinoptilolite + mordenite) has self-photocatalytic activity and therefore can function as a single site photocatalyst for the decomposition of caffeine in aqueous solution using UV irradiation. The occurrence of the radiation interaction with matter and the formation of reactive radicals coming from photoactive iron species are committed, at the same time with the zeolites' nanoporosity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Patricia Amador (FCQ, BUAP) for the DTG experiments, Justo Miguel Gracia y Jiménez (IFUAP) for the measurement of the emission spectrum of the UV lamp, and Primavera López (CDS, ICUAP) for the X-ray fluorescence analysis. This work was supported by the following research projects: CONACyT INFR-123779-2009, INFRA-01-252847-2015, and VIEP-00344-2018. EAAA is grateful for the fellowship (445400) received from CONACyT. SASH thanks to the PRODEP-SEP (Mexico) program for a post-doctoral grant (511-6/2019-4353).References
- N. Serpone, A. V. Emeline, S. Horikoshi, V. N. Kuznetsov and V. K. Ryabchuk, Photochem. Photobiol. Sci., 2012, 11, 1121–1150 RSC.
- O. Kestner, Z. Biol., 1921, 73, 7–9 CAS.
- E. M. Hume and H. H. Smith, Biochem. J., 1923, 17, 364–372 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- S. Hashimoto, J. Photochem. Photobiol., C, 2003, 4, 19–49 CrossRef CAS.
- A. Corma and H. Garcia, Chem. Commun., 2004, 1443–1459 RSC.
- L. F. Velasco, I. M. Fonseca, J. B. Parra, J. C. Lima and C. O. Ania, Carbon, 2012, 50, 249–258 CrossRef CAS.
- C. O. Ania, P. A. Armstrong, T. J. Bandosz, F. Beguin, A. P. Carvalho, A. Celzard, E. Franckowiak, M. A. Gilarranz, E. Frackowiak, K. László, J. Matos and M. F. R. Pereira, Carbon, 2020, 164, 69–84 CrossRef CAS.
- Y. V. Meteleva, F. Roessner and G. F. Novikov, J. Photochem. Photobiol., A, 2008, 196, 154–158 CrossRef CAS.
- K. Guesh, Á. Mayoral, Y. Chebude, M. J. López-Muñoz, C. Márquez-Álvarez and I. Diaz, New J. Chem., 2018, 42, 12001–12007 RSC.
- M. Anpo and J. M. Thomas, Chem. Commun., 2006, 3273–3278 RSC.
- Y. Tong, Y. Zhang, N. Tong, Z. Zhang, Y. Wang, X. Zhang, S. Zhu, F. Li and X. Wang, Catal. Sci. Technol., 2016, 6, 7579–7585 RSC.
- G. Yan, X. Wang, X. Fu and D. Li, Catal. Today, 2004, 93–95, 851–856 CrossRef CAS.
- N. Dubey, S. S. Rayalu, N. K. Labhsetwar, R. R. Naidu, R. V. Chatti and S. Devotta, Appl. Catal., A, 2006, 303, 152–157 CrossRef CAS.
- H. Yamashita and K. Mori, Chem. Lett., 2007, 36, 348–353 CrossRef CAS.
- D. I. Petkowicz, R. Brambilla, C. Radtke, C. D. S. da Silva, Z. N. da Rocha, S. B. C. Pergher and J. H. Z. dos Santos, Appl. Catal., A, 2009, 357, 125–134 CrossRef CAS.
- Y. Tong, L. Chen, S. Ning, N. Tong, Z. Zhang, H. Lin, F. Li and X. Wang, Appl. Catal., B, 2017, 203, 725–730 CrossRef CAS.
- M. A. Zanjanchi and S. Asgari, Solid State Ionics, 2004, 171, 277–282 CrossRef CAS.
- J. Motuzas, M. Drobek, D. L. Martens, C. Vallicari, A. Julbe and J. C. Diniz da Costa, Environ. Sci. Pollut. Res., 2018, 25, 3628–3635 CrossRef CAS.
- G. Yan, J. Long, X. Wang, Z. Li, X. Wang, Y. Xu and X. Fu, J. Phys. Chem. C, 2007, 111, 5195–5202 CrossRef CAS.
- M. M. Mohamed, F. I. Zidan and M. Thabet, Microporous Mesoporous Mater., 2008, 108, 193–203 CrossRef CAS.
- L. Wang, W. Wang, M. Liu, H. Ge, W. Zha, Y. Wei, E. Fei, Z. Zhang, J. Long, R. Sa, Y. jie Wang, X. Fu and R. Yuan, J. Catal., 2019, 377, 322–331 CrossRef CAS.
- K. Guesh, M. J. López-Muñoz, C. Márquez-Álvarez, Y. Chebude and I. Diaz, Bull. Chem. Soc. Ethiop., 2015, 29, 431–440 CrossRef CAS.
- M. P. Elizalde-González, J. Mattusch and R. Wennrich, Appl. Organomet. Chem., 2002, 16, 9–16 CrossRef.
- M. P. Elizalde-González, J. Mattusch and R. Wennrich, Microchim. Acta, 2005, 151, 257–262 CrossRef.
- W. A. Cabrera-Lafaurie, F. R. Román and A. J. Hernández-Maldonado, J. Colloid Interface Sci., 2012, 386, 381–391 CrossRef CAS.
- R. R. N. Marques, M. J. Sampaio, P. M. Carrapiço, C. G. Silva, S. Morales-Torres, G. Dražić, J. L. Faria and A. M. T. Silva, Catal. Today, 2013, 209, 108–115 CrossRef CAS.
- L. Busse and C. Nagoda, Detection of caffeine in the streams and rivers within the San Diego region - Pilot study, California Regional Water Quality Control Board, San Diego, CA, 2015 Search PubMed.
- M. Krivec, R. A. Segundo, J. L. Faria, A. M. T. Silva and G. DraŽić, Appl. Catal., B, 2013, 140–141, 9–15 CrossRef CAS.
- L. Rimoldi, D. Meroni, E. Falletta, V. Pifferi, L. Falciola, G. Cappelletti and S. Ardizzone, Photochem. Photobiol. Sci., 2017, 16, 60–66 RSC.
- M. B. Tahir, M. Sagir and K. Shahzad, J. Hazard. Mater., 2019, 363, 205–213 CrossRef CAS.
- A. Gil, A. M. García, M. Fernández, M. A. Vicente, B. González-Rodríguez, V. Rives and S. A. Korili, J. Ind. Eng. Chem., 2017, 53, 183–191 CrossRef CAS.
- S. M. Miranda, G. E. Romanos, V. Likodimos, R. R. N. Marques, E. P. Favvas, F. K. Katsaros, K. L. Stefanopoulos, V. J. P. Vilar, J. L. Faria, P. Falaras and A. M. T. Silva, Appl. Catal., B, 2014, 147, 65–81 CrossRef CAS.
- B. Czech and K. Tyszczuk-Rotko, J. Photochem. Photobiol., A, 2019, 376, 166–174 CrossRef CAS.
- M. Krivec, K. Žagar, L. Suhadolnik, M. Čeh and G. Dražić, ACS Appl. Mater. Interfaces, 2013, 5, 9088–9094 CrossRef CAS.
- J. L. Sotelo, G. Ovejero, A. Rodríguez, S. Álvarez and J. García, Water, Air, Soil Pollut., 2013, 224, 1466–1481 CrossRef.
- K. Yamamoto, T. Shiono, R. Yoshimura, Y. Matsui and M. Yoneda, Adsorpt. Sci. Technol., 2018, 36, 967–981 CrossRef CAS.
- T. P. J. Izod, European Patent Office, EP0013451A1, 1980, pp. 1–22.
- Z. L. Yaneva, M. S. Staleva and N. V. Georgieva, Eur. J. Chem., 2015, 6, 169–173 CrossRef CAS.
- J. L. Costafreda Mustelier, D. A. Martín Sánchez and J. L. Costafreda Velázquez, Las zeolitas naturales de Iberoamérica, Fundación Gómez-Parde, 2018 Search PubMed.
- P. C. C. Faria, J. J. M. Orfão and M. F. R. Pereira, Water Res., 2004, 38, 2043–2052 CrossRef CAS.
- S. Banerjee, P. K. Verma, M. R. Kumar and P. S. Kumar, J. Fluoresc., 2012, 22, 753–769 CrossRef CAS.
- International Zeolite Association, Database zeolite Struct, http://www.iza-structure.org/databases/ Search PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- F. Stoeckli, T. A. Centeno, J. B. Donnet, N. Pusset and E. Papirer, Fuel, 1995, 74, 1582–1588 CrossRef CAS.
- O. Korkuna, R. Leboda, J. Skubiszewska-Zięba, T. Vrublevs'ka, V. M. Gun'ko and J. Ryczkowski, Microporous Mesoporous Mater., 2006, 87, 243–254 CrossRef CAS.
- L. De Pablo Galan, M. Doval, A. La Iglesia, J. Soriano and L. Chavez, Am. Mineral., 2013, 98, 977–985 CrossRef CAS.
- M. Schwidder, M. S. Kumar, K. Klementiev, M. M. Pohl, A. Brückner and W. Grünert, J. Catal., 2005, 231, 314–330 CrossRef CAS.
- M. M. Dávila-Jiménez, M. P. Elizalde-González, J. Mattusch, P. Morgenstern, M. A. Pérez-Cruz, Y. Reyes-Ortega, R. Wennrich and H. Yee-Madeira, J. Colloid Interface Sci., 2008, 322, 527–536 CrossRef.
- Y. Kato, H. Yoshida, A. Satsuma and T. Hattori, Microporous Mesoporous Mater., 2002, 51, 223–231 CrossRef CAS.
- E. D. Garbowski and C. Mirodatos, J. Phys. Chem., 1982, 86, 97–102 CrossRef CAS.
- Y. Garcia-Basabe, I. Rodriguez-Iznaga, L. C. De Menorval, P. Llewellyn, G. Maurin, D. W. Lewis, R. Binions, M. Autie and A. R. Ruiz-Salvador, Microporous Mesoporous Mater., 2010, 135, 187–196 CrossRef CAS.
- M. A. Zanjanchi and A. Razavi, Spectrochim. Acta, Part A, 2001, 57, 119–127 CrossRef CAS.
- J. Pérez-Ramírez, M. S. Kumar and A. Brückner, J. Catal., 2004, 223, 13–27 CrossRef.
- D. Hradil, T. Grygar, M. Hrušková, P. Bezdička, K. Lang, O. Schneeweiss and M. Chvátal, Clays Clay Miner., 2004, 52, 767–778 CrossRef CAS.
- G. R. Rossman and B. L. Ehlmann, in Remote Compositional Analysis: Techniques for Understanding Spectroscopy, Mineralogy, and Geochemistry of Planetary Surfaces, 2019, pp. 3–20 Search PubMed.
- L. E. Iton, R. B. Beal and D. T. Hodul, J. Mol. Catal., 1983, 21, 151–171 CrossRef CAS.
- J. L. Bishop, in Remote Compositional Analysis: Techniques for Understanding Spectroscopy, Mineralogy, and Geochemistry of Planetary Surfaces, 2019, pp. 68–101 Search PubMed.
- F. Dong, L. Bian, M. Song, W. Li and T. Duan, Appl. Clay Sci., 2016, 119, 74–81 CrossRef CAS.
- B. Barrocas, M. C. Neves, M. Conceição Oliveira and O. C. Monteiro, Environ. Sci.: Nano, 2018, 5, 350–361 RSC.
- F. Qi, W. Chu and B. Xu, Appl. Catal., B, 2013, 134–135, 324–332 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06421a |
| This journal is © The Royal Society of Chemistry 2020 |