DOI:
10.1039/D0RA06357F
(Paper)
RSC Adv., 2020,
10, 33221-33226
Retracted Article: Functional disruption of staphylococcal accessory regulator A from Staphylococcus aureus by silver ions†
Received
21st July 2020
, Accepted 31st August 2020
First published on 8th September 2020
Abstract
Silver ions (Ag+) have attracted profound attention due to their broad-spectrum antimicrobial activities. Although the antibacterial properties of silver have been well known for many centuries, its mechanism of action is not fully understood and its protein targets remain largely unknown. Staphylococcus aureus (S. aureus) is the leading cause of hospital- and community-acquired infections. Staphylococcal accessory regulator protein family from Staphylococcus aureus has been found to play vital roles in the regulation of virulence genes. In this study, we demonstrated that silver ions bind to the staphylococcal accessory regulator A (SarA) of S. aureus via its cysteine residues. Importantly, binding of silver ions leads to functional disruption of SarA. In addition, qRT-PCR experiments showed that silver also significantly attenuated the mRNA transcription levels of the genes which SarA regulated. Overall, these results provide new insights into the antibacterial mechanism of silver ions.
Introduction
Staphylococcus aureus (S. aureus), a worldwide human pathogen, is the leading cause of hospital- and community-acquired infections. The pathogen causes a series of human diseases ranging from minor skin infections to life-threating sepsis.1 In particular, the emergence of drug-resistant strains of bacteria, such as methicillin-resistant and vancomycin-resistant S. aureus has posed a threat to public health worldwide.2 Worse yet, resistance to vancomycin, linezolid and daptomycin have already been reported in clinical MRSA strains, compromising the therapeutic alternatives for life-threatening MRSA infections.3–5 The most serious infections such as endocarditis, osteomyelitis, necrotizing pneumonia and sepsis occur on dissemination of the bacteria into the bloodstream.6 In S. aureus, SarA is a 14.7 kDa winged helix turn helix transcriptional activator and known to up-regulate the agr based quorum sensing system to elicit the exoprotein level.7,8 Simultaneously, the SarA indirect role on down-regulation of various other regulatory loci such as rot, sarS, sarV, sarT are also well documented.9 Furthermore, SarA is also involved in the agr-independent expression of several other virulence genes including fnbA (fibronectin binding protein A), TSS (toxic shock syndrome) and icaRA (coagulase) and bap (biofilm associated proteins).10–12 Recently, small molecule inhibitors targeting S. aureus virulence regulators SarA were also reported to be efficacious in animal models, indicating that targeting this regulator proteins might be a promising anti-bacterial strategy.10
Silver and silver-containing materials had been widely employed as antimicrobial agents due to their broad-spectrum bactericidal activity.13–17 Recent studies showed that silver ions could enhance the antibiotic activity against Gram-negative bacteria as well as restoring antibiotic susceptibility of the resistant bacterial strain, implying the potential application of silver in treatment of drug-resistant bacteria infection.18 Silver could also eliminate Gram-positive bacteria, such as S. aureus. However, up to now, few Ag+ protein targets have been identified and characterized. It is suggested that Ag+ could bind to thiol group (–SH) of bacterial enzymes and subsequently causing enzyme deactivation.19 Our previous study demonstrated that Ag+ binds to CcpA (catabolite control protein A) from S. aureus via the two Cys residues both in vitro and in vivo, leading to disruption of protein functions, thus attenuating the bacterial growth, bacterial toxin expression and biofilm formation. However, the decrease of erythrocyte lysis activity observed in ccpA::ccpA2CS mutant strain (in which Ag+ can't binding to CcpA) upon Ag+ treatment indicated that other hemolysin expression regulatory networks were also perturbed.20 Given the fact that the sole and conserved Cys residue in SarA acts as a redox switch to modulate the regulatory functions.21 We hypothesis that SarA also serves as a potential target of Ag+ in S. aureus. Herein, we investigated the interaction between staphylococcal accessory regulator A from Staphylococcus aureus and silver ions. In this study, we demonstrated that silver ion binds to the staphylococcal accessory regulator A (SarA) of S. aureus via its cysteine residues. Importantly, binding of silver ions leads to functional disruption of SarA.
Results and discussion
Ellman's assay
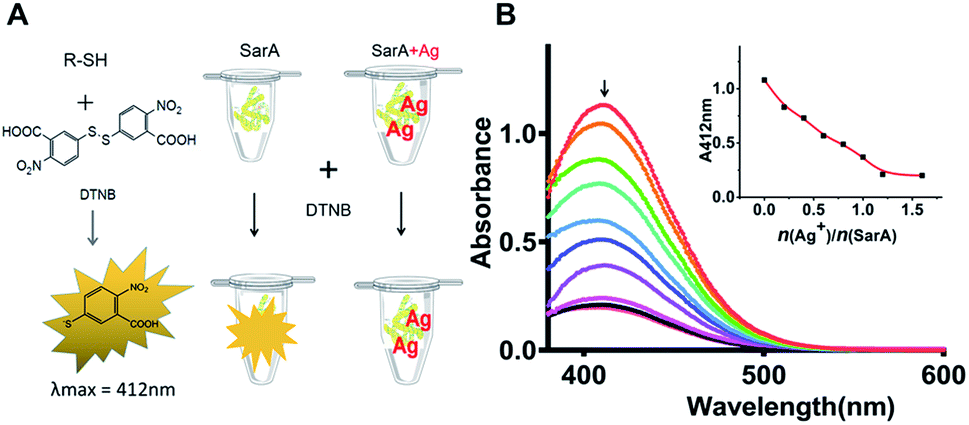
SarA is a 14.7 kDa winged helix turn helix transcriptional activator and has a conserved Cys residue (Cys9) (Fig. S1†). Given the highly thiophilic property of Ag+, the free cysteines of SarA could probably involve in Ag+ binding. At first, to test the hypothesis, we used the Ellman's assay to monitor the number of free sulfhydryl groups in SarA after pre-incubation with different molar equivalents of Ag+. The Ellman's reagent DTNB specifically reacts with the free sulfhydryl group to yield NTB, which exhibits maximum absorption at 412 nm in the UV-Vis spectra.22 As shown in Fig. 1, for SarA, the absorbance at 412 nm decreased with the increasing Ag+/protein ratios, indicative of the direct binding of Ag+ to the free sulfhydryl group. However, the absorbance at 412 nm levelled off after addition of around 1 molar equivalents of Ag+. This result suggested that silver ion could bind to the staphylococcal accessory regulator A (SarA) of S. aureus via its cysteine residues.
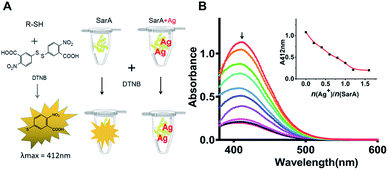 |
| | Fig. 1 Ag+-binding to SarA monitored by Ellman's assay (A). SarA was pre-incubated with various molar equivalents of Ag+ as indicated, the remained free thiols in protein samples were measured by Ellman's assay. The absorbance at 412 nm was plotted against Ag+/protein ratios (B). | |
Inductively coupled plasma mass spectrometry
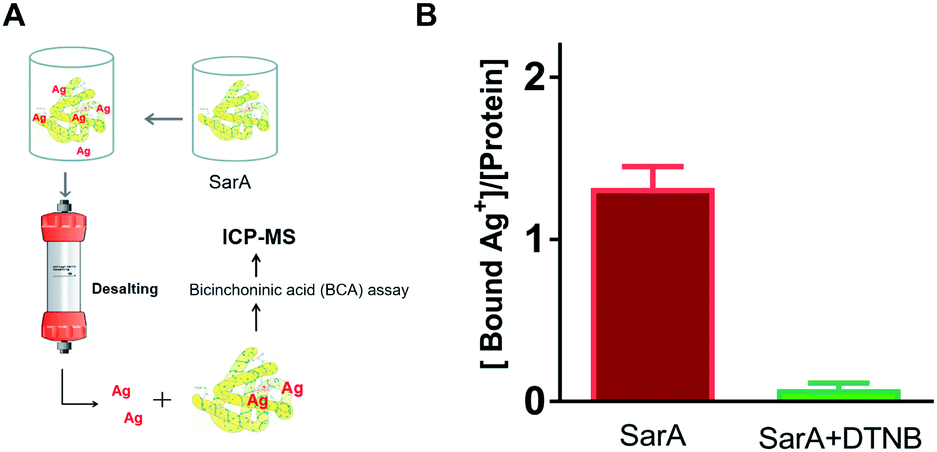
To further investigate whether S. aureus SarA can bind silver ions, inductively coupled plasma mass spectrometry (ICP-MS) was used to monitor the Ag+-binding capacities of SarA after pre-incubation with excess amounts of silver ions. In brief, different molar equivalents of Ag+ were incubated with SarA protein followed by desalting to remove unbound Ag+. The protein concentrations were measured by BCA assay and protein-bound Ag+ contents were quantified by ICP-MS. As the results showed in Fig. 2, SarA bound 1.32 ± 0.23 molar equivalents of Ag+, which consistent with the Ellman's assay. In contrast, after pre-incubation with three molar equivalents of DTNB, the Ag+-binding capabilities of the SarA was not detectable. In addition, the Cys mutant SarAC9S (cysteine was mutated to serine) were also expressed and purified. Once again, ICP-MS measurement showed that SarAC9S had no Ag+ binding capability (Fig. S3†). These results indicated again that silver ions bind to the SarA of S. aureus via its free sulfhydryl group.
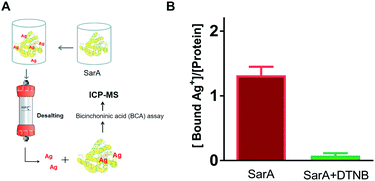 |
| | Fig. 2 Ag+-binding capability of SarA determined by ICP-MS (A). SarA or SarA pre-incubated with DTNB were treated with 3 molar equivalents of Ag+. Excess amounts of Ag+ were removed by a desalting column. The bound Ag+ contents were determined by ICP-MS and protein concentrations were measured by BCA assay (B). | |
Isothermal titration calorimetry (ITC)
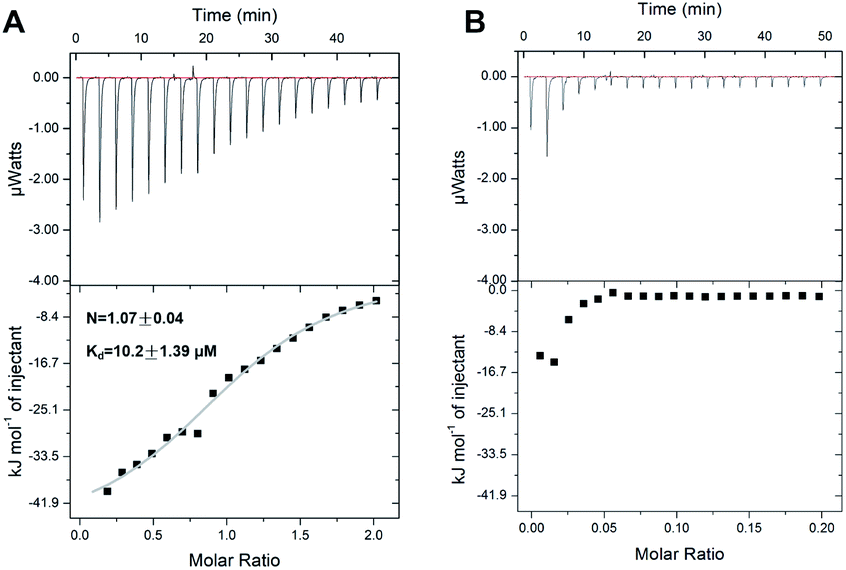
In order to further investigate the Ag+-binding properties of SarA, isothermal titration calorimetry (ITC) was applied to monitor the titration of Ag+ into SarA. As shown in Fig. 3, isothermal titration calorimetry (ITC) data confirmed that SarA could bind 1.07 ± 0.04 molar equivalents of Ag+ with an apparent dissociation constant (Kd) of 10.2 ± 1.39 μM. In addition, the binding of Ag+ to SarA which pre-incubation with three molar equivalents of DTNB was also investigated by ITC. Once again, SarA with no free sulfhydryls had no detectable Ag+-binding capability. These data are in consistent with the results obtained from ICP-MS and Ellman's assay. Collectively, these data demonstrate that SarA binds Ag+ via its conserved cysteine residues and SarA could bind 1 molar equivalents of Ag+ with an apparent dissociation constant (Kd) of 10.2 ± 1.39 μM.
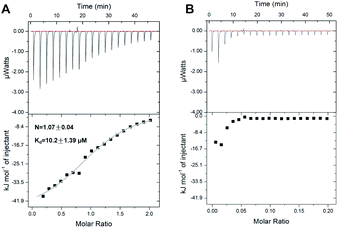 |
| | Fig. 3 Calorimetric titration of Ag+ to SarA (A) and SarA which pre-incubation with three molar equivalents of DTNB (B). All protein samples were prepared in 50 mM Tris–HNO3 buffer supplemented with 150 mM NaNO3, pH 7.4. The titrations were carried out at 25 °C. The titration curves were fitted to one-set of site binding model using the Origin software. | |
Ag+ binding decreases SarA thermal stability
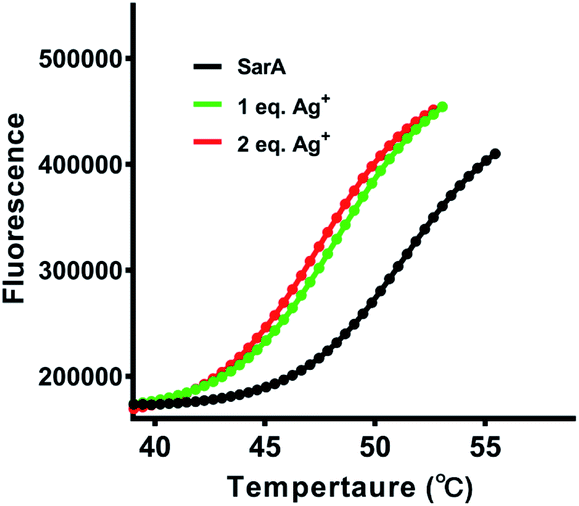
Previously study have showed that silver binding could decrease the thermal stabilization of target proteins.20 Therefore, the thermal stability of SarA was investigated upon silver binding. In brief, SarA was incubated with different molar equivalents of Ag+ and the apparent melting temperature (Tm) were determined by protein thermal shift assay as described in the Experimental section. As shown in Fig. 4, SarA exhibited typical S-shape thermal denaturation curve with a calculated Tm of 50.6 °C. Significant down shift of Tm was observed upon Ag+ binding, with Tm of 47.5 and 47.1 °C for 1 and 2 molar equivalents of Ag+, respectively. This results indicated that Ag+ substantially decreased SarA protein thermal stability.
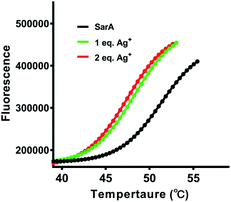 |
| | Fig. 4 Thermal shift analysis of SarA stability after Ag+ treatment. Fluorescence signals are plotted against temperature and presented as sigmoidal curves. The melting temperatures are calculated for each curve. | |
Ag+ binding abolishes SarA DNA-binding capability
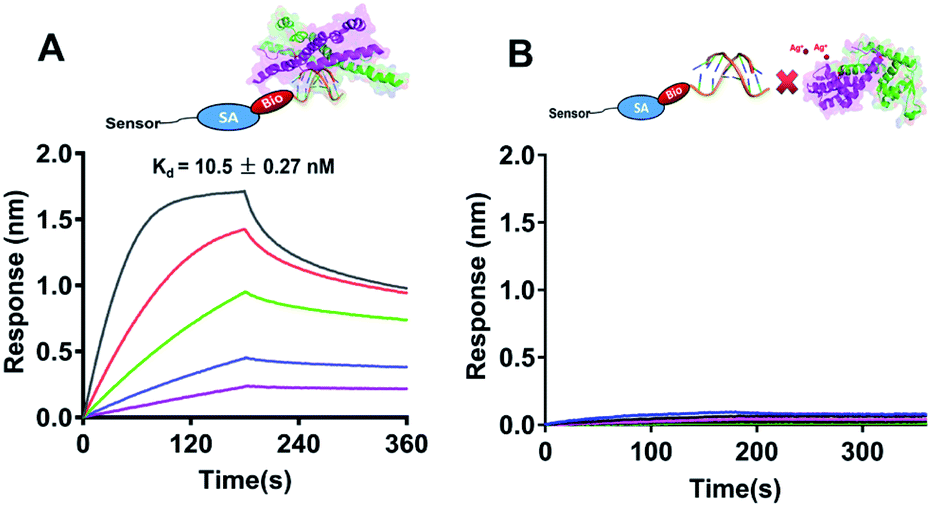
As a global transcription factor, SarA bind to the putative DNA sequences and regulates downstream gene expression. Recently study showed that the sole and conserved Cys residue in SarA acts as a redox switch to modulate the regulatory functions.21 Subsequently, we further measured the DNA binding capabilities of SarA by BioLayer Interferometry (BLI) with or without silver treated. Biotin-labeled hla DNA probe was immobilized on a streptavidin sensor to perform kinect analysis of SarA binding to hla promoter. All the data were collected and analyzed using the global fit binding model over five concentrations of SarA on a ForteBio Octet Red 96 system. As shown in Fig. 5A, SarA could specific binding to hla promoter sequence which in line with previous report.21 SarA possessed a strong binding affinity for hla with a Kd value of 10.5 ± 0.27 nM. However, after with Ag+ treated, the binding of SarA for hla is undetectable (Fig. 5B). In contrast, biolayer interferometry (BLI) assay also indicated that SarAC9S, which cysteine was mutated to serine, could binding to hla promoter even in the presence of silver ions (Fig. S4†). These results clearly demonstrated that Ag+ binding to the Cys residues of SarA and Ag+ binding disrupts its DNA binding capability.
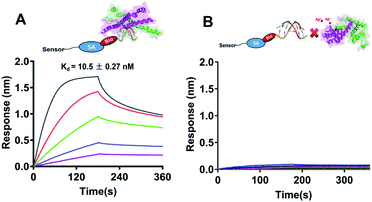 |
| | Fig. 5 The DNA binding capabilities of SarA (A), SarA with Ag+ (B) were measured by BioLayer Interferometry (BLI). Biotinylated hla (300 nM) were captured on pre-immobilized streptavidin Dip and Read sensor heads for 3 min. Association occurred from 0 to 180 s and dissociation was monitored thereafter up to 360 s. The Kd values are presented as the mean ± s.e.m. derived from a global fitting of all binding curves. | |
Quantitative real-time PCR analysis of genes regulated by SarA after Ag+ treatment
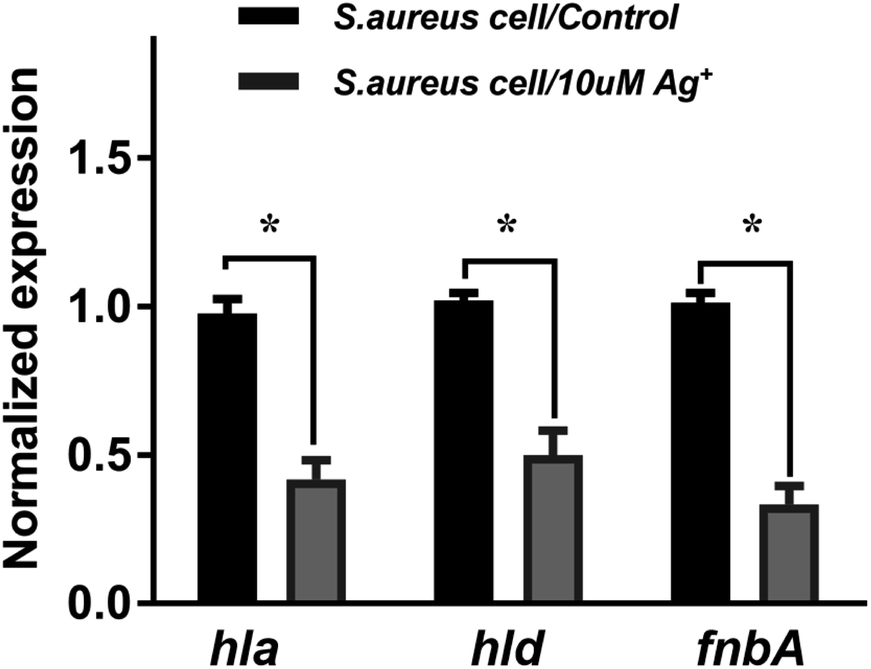
To further investigate the Ag+ effect on the SarA regulation function, the mRNA levels of three different SarA-regulated genes, including hla, hld and fnbA are examined by quantitative real-time PCR. Cytotoxic virulence factors like hemolysins are coded by hla and hld genes. The gene fnbA encodes fibronectin binding protein A, which mediates adherence to the host extracellular matrix and is important for bacterial virulence. As shown in Fig. 6, the mRNA transcription levels of all the three genes are significantly attenuated upon treatment of 10 μM silver nitrate (Ag+), indicating that SarA targeted by Ag+ would remarkably perturb its downstream regulated gene transcription. The results are consistent with a previous report that the SarA targeted inhibitor could down-regulated the expression of the hemolysin (hld, hla), and fibronectin-binding protein (fnbA).10,23
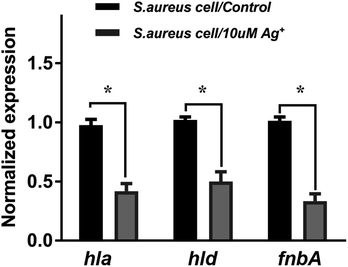 |
| | Fig. 6 Quantitative transcript analysis of hla, hld and fnbA of wide type S. aureus with or without Ag+ treatment. All experiments were performed with three biological replicates. The mean value of transcription level in wide type S. aureus control groups are set as 1. The Ag+ treated groups are normalized to that of control groups. Results are shown as mean ± sd. | |
Experimental
Protein expression and purification
The sarA genes were amplified by PCR using S. aureus Newman genomic DNA as template. The fragments and expression plasmid pET47b were double digested with the corresponding restriction enzymes and ligated by T4 ligase. The generated expression plasmids pET47b-sarA were verified by DNA sequencing (Sangon, China) and transformed into Escherichia coli (E. coli) BL21 (DE3) strain for protein expression. BL21 (DE3) cell, which harbored the expression plasmid, was grown in 1 L of Luria–Bertani (LB) medium for 3 h aerobically at 37 °C until OD600 reached 0.6. IPTG was added to a final concentration of 0.5 mM to induce protein expression. The bacteria were harvested after further incubation for 16 h at 25 °C by centrifugation (5000g for 20 min at 4 °C). The pellets were resuspended in resuspension buffer (20 mM Tris–HCl, 150 mM NaCl, pH 7.4). Cell pellets were lysed by sonication at 4 °C with 1 mM PMSF as protease inhibitor. The lysate was centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 10 min and the supernatant was collected. The supernatant was then dialyzed against 50 mM Tris–HCl (pH 8.0) buffer overnight and subsequently applied to a 5 mL HiTrap Q column (GE Healthcare). Purified protein was buffer-exchanged into Tris–HNO3 buffer (50 mM Tris–HNO3, 150 mM NaNO3, pH 7.4) by HiTrap desalting column (GE healthcare) to remove DTT and chloride ion immediately before use. Plasmids for SarAC9S mutant expression were generated via site-directed mutagenesis using Phusion high fidelity DNA polymerase. The expression and purification of SarA mutants were similar to wild-type SarA.
000g for 10 min and the supernatant was collected. The supernatant was then dialyzed against 50 mM Tris–HCl (pH 8.0) buffer overnight and subsequently applied to a 5 mL HiTrap Q column (GE Healthcare). Purified protein was buffer-exchanged into Tris–HNO3 buffer (50 mM Tris–HNO3, 150 mM NaNO3, pH 7.4) by HiTrap desalting column (GE healthcare) to remove DTT and chloride ion immediately before use. Plasmids for SarAC9S mutant expression were generated via site-directed mutagenesis using Phusion high fidelity DNA polymerase. The expression and purification of SarA mutants were similar to wild-type SarA.
Ellman's assay
Ellman's assays were carried out in Tris–HNO3 buffer. Different molar equivalents of Ag+ were added into 50 μM SarA and incubated for 20 min at room temperature (RT). Excess amounts of 5,5-dithio-bis-(2-nitrobenzoic acid) (Ellman's reagent, DTNB) were subsequently added with a final concentration of 160 μM. After further incubation of 20 min at RT, absorbance of each sample at 412 nm was measured by UV-Vis spectroscopy. The absorbance at 412 nm was plotted against Ag+/protein ratio.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) experiments were performed on a Malvern MicroCal iTC200 at 25 °C. The SarA were prepared in Tris–HNO3 buffer with concentration of 25 μM. The Ag+ titrant was prepared by dissolving AgNO3 in Tris–HNO3 buffer (0.25 mM). Typically, 40 μL of AgNO3 titrants were titrated into 200 μL protein sample with 150 s interval between each injection. AgNO3 was titrated into Tris–HNO3 buffer without protein for ligand-to-buffer subtraction. The ITC data were analyzed using the Origin software and fitted by one-set-of-site binding model.
Inductively coupled plasma mass spectrometry
All inductively coupled plasma mass spectrometry (ICP-MS) experiments were conducted on a Thermo Scientific iCAP Q ICP-MS spectrometer. Each sample was quantified three times and the average value was used. Around 3 molar equivalents of Ag+ were added into approximate 200 μM SarA in Tris–HNO3 buffer. After incubation for 30 minute at RT, excess amounts of Ag+ were removed by HiTrap desalting column. The eluted protein concentration was measured by bicinchoninic acid (BCA) assay and the bound Ag+ was determined by ICP-MS.
Protein thermal shift assay
Protein thermal stability of SarA were examined by thermal shift assay (TSA) using a StepOnePlus real-time PCR system (Life Technologies) as described previously.24 Prior to use, the environmentally sensitive fluorescent dye SYPRO orange stock solution in DMSO was diluted 1:125 in Tris–HNO3 buffer. The TSA samples were prepared in a 96-well plate in triplicate, which contained 3 μL of protein solution (1.0 mg mL−1), 2.5 μL of freshly diluted SYPRO orange dye and 19.5 μL of Tris–HNO3 buffer (20 mM Tris, 150 mM NaNO3, pH 7.5). The plate was sealed with optical quality sealing tape and centrifuged at 10000g for 5 min. The fluorescent intensity was monitored as the plate was heated from 298 to 368 K in an increment of 1 K min−1. The fluorescent data was analyzed using a Boltzmann model and the melting temperature (Tm) was calculated.
Bio-layer interferometry assay (BLI)
Interactions between purified SarA and the biotin labeled promoter region of hla were measured using biolayer interferometry (BLI) on an Octet Red96 system (ForteBio). Binding experiments were performed at 37 °C in binding buffer (25 mM Tris–HNO3, 80 mM NaNO3, 35 mM KNO3, 10 mM Mg(NO3)2, 0.1% Triton X-100, 10% glycerol, 0.1% BSA and 0.01% Tween 20, pH 7.5). Around 300 nM of biotinylated DNA probe was captured on pre-immobilized streptavidin Dip and Read sensor heads for 3 min. DNA-immobilized sensor heads were subsequently incubated in binding buffer for 30 s to reach equilibrium. The sensor was subsequently incubated in binding buffer containing various concentrations of purified SarA (100 nM to 6.25 nM in two-fold dilutions) with or without silver ions and the association of the protein with promoter was measured over 180 seconds. Dissociation was subsequently monitored over 180 seconds in binding buffer without protein. All data were normalized and analyzed using the global fit binding model over all concentrations using the software provide by Fortebio to derive kon, koff and KD values.
Measurement of gene expression by quantitative real time PCR
The S. aureus Newman strains were grown overnight in TSB medium. The next day, the cell cultures were 1:100 diluted into fresh TSB and grown until OD600 reached 0.6. Subsequently, the culture was treated with 10 μM AgNO3 at 37 °C for 1 hour. Total RNAs from both untreated (as control) and treated group were isolated with SV total RNA isolation system (Promega) according to the manufacturer's recommendations. cDNA was generated by reverse transcription using GoScript Reverse Transcriptase (Promega). The transcription level of detected gene was subsequently determined by real-time PCR using GoTaq qPCR Master Mix kit (Promega) on a StepOnePlus real-time PCR system (Life Technologies). The rrsA (16S rRNA) was used as an internal control. All of the experiments were conducted in triplicate and relative expression levels were measured using the 2−ΔΔCt method. The mean value before Ag+ treatment was set as 1 and data are presented as mean ± SD. The Ag+ treated groups were normalized to that of control groups. The following primers were used for real-time PCR: 16S rRNA-For and 16S rRNA-Rev, hla-For and hla-Rev, hld-For and hld-Rev, fnbA-For and fnbA-Rev (Table S1†).
Conclusion
Despite the broad-spectrum antimicrobial activities of silver, its molecular targets remain obscure. It reported that silver damages multiple enzymes in glycolysis and tricarboxylic acid (TCA) cycle, leading to the stalling of the oxidative branch of the TCA cycle and an adaptive metabolic divergence to the reductive glyoxylate pathway in E. coli.25 In addition, GAPDH also reported as one of the vital molecular targets of Ag+ against E. coli. Ag+ could binds to the three cysteine sites of GAPDH and inhibits the enzymatic activity of GAPDH.26 In S. aureus, the staphylococcal accessory regulator (SarA) protein is a global regulator that governs many of the virulence factors produced by S. aureus.27,28 In addition, SarA is also an important positive regulator of biofilm formation, in part because of its reciprocal repressive activity on protease and nuclease production.27 Recent studies reported that the SarA targeted inhibitor showed negligible antimicrobial activity but markedly reduced the minimum inhibitory concentration of conventional antibiotics when used in combination.23 Interestingly, silver ion could also enhance the antibiotic activity against bacteria as well as restoring antibiotic susceptibility of the resistant bacterial strain.18 Therefore, we assumed that S. aureus SarA might act as a putative target of Ag+. We showed clearly that Ag+ binds to SarA via the sole and conserved Cys residues in vitro. SarA could bind 1 molar equivalents of Ag+ with an apparent dissociation constant (Kd) of 10.2 ± 1.39 μM. Importantly, binding of silver ions leads to functional disruption of SarA. In addition, qRT-PCR experiments showed that silver also significantly attenuated the mRNA transcription levels of the genes which SarA regulated. Collectively, this work demonstrates that the SarA in S. aureus is a target of Ag+, which further expanded our understanding on the antibacterial mechanism of Ag+.
Conflicts of interest
The authors have declared that there is no conflict of interest.
Acknowledgements
We gratefully acknowledge the generous support provided by The Natural Science Foundation of Jiangxi, China (20192BAB213002); Department Education Science and Technology Research Project of Jiangxi, China (GJJ180634); Jiangxi Science & Technology Normal University (KFGJ18017).
References
- D. Franklin and F. D. Lowy, N. Engl. J. Med., 1998, 339, 520–532 CrossRef.
- R. M. Klevens, M. Morrison and J. Nadle, JAMA, 2007, 298, 1763–1771 CrossRef CAS.
- E. Nannini, B. E. Murray and C. A. Arias, Curr. Opin. Pharmacol., 2010, 10, 516–521 CrossRef CAS.
- M. Roch, P. Gagetti, J. Davis, P. Ceriana, L. Errecalde, A. Corso and A. E. Rosato, Front. Microbiol., 2017, 8, 2303 CrossRef.
- C. J. Chen, Y. C. Huang and S. S. Shie, Front. Microbiol., 2020, 11, 1414 CrossRef.
- O. M. El-Halfawy, T. L. Czarny, R. S. Flannagan, J. Day, J. C. Bozelli Jr, R. C. Kuiack, A. Salim, P. Eckert, R. M. Epand, M. J. McGavin, M. G. Organ, D. E. Heinrichs and E. D. Brown, Nat. Chem. Biol., 2020, 16, 143–149 CrossRef CAS.
- A. L. Cheung, A. S. Bayer, G. Y. Zhang, H. Gresham and Y. Q. Xiong, FEMS Immunol. Med. Microbiol., 2004, 1, 1–9 CrossRef.
- K. E. Beenken, L. N. Mrak, L. M. Griffin, A. K. Zielinska, L. N. Shaw, K. C. Rice, A. R. Horswill, K. W. Bayles and M. S. Smeltzer, PLoS One, 2010, 5, e10790 CrossRef.
- R. Arya and S. A. Princy, Future Microbiol., 2013, 10, 1339–1353 CrossRef.
- R. Arya, R. Ravikumar, R. S. Santhosh and S. A. Princy, Front. Microbiol., 2015, 6, 416 CrossRef.
- C. R. Arciola, D. Campoccia, P. Speziale, L. Montanaro and J. W. Costerton, Biomaterials, 2012, 33, 5967–5982 CrossRef CAS.
- D. O. Andrey, A. Renzoni, A. Monod, D. P. Lew, A. L. Cheung and W. L. Kelley, J. Bacteriol., 2010, 192, 6077–6085 CrossRef CAS.
- T. Bechert, P. Steinrucke and J. P. Guggenbichler, Nat. Med., 2000, 6, 1053–1056 CrossRef CAS.
- S. Chernousova and M. Epple, Angew. Chem., Int. Ed., 2013, 52, 1636–1653 CrossRef CAS.
- L. Rizzello and P. P. Pompa, Chem. Soc. Rev., 2014, 43, 1501–1518 RSC.
- S. Eckhardt, P. S. Brunetto, J. Gagnon, M. Priebe, B. Giese and K. M. Fromm, Chem. Rev., 2013, 113, 4708–4754 CrossRef CAS.
- T. Veranobraga, R. Miethlinggraff, K. Wojdyla, A. Rogowska-Wrzesinska, J. R. Brewer, H. Erdmann and F. Kjeldsen, ACS Nano, 2014, 8, 2161–2175 CrossRef CAS.
- J. R. Moronesramirez, J. A. Winkler, C. S. Spina and J. J. Collins, Sci. Transl. Med., 2013, 5, 190ra81 Search PubMed.
- S. Y. Liau, D. C. Read, W. J. Pugh and A. D. Russell, Lett. Appl. Microbiol., 1997, 25, 279–283 CrossRef CAS.
- X. Liao, F. Yang, R. Wang, X. He, H. Li, R. Y. T. Kao, W. Xia and H. Sun, Chem. Sci., 2017, 8, 8061–8066 RSC.
- F. Sun, Y. Ding, Q. Ji, Z. Liang, X. Deng, C. C. Wong, C. Yi, L. Zhang, S. Xie, S. Alvarez, L. M. Hicks, C. Luo, H. Jiang, L. Lan and C. He, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15461–15466 CrossRef CAS.
- W. L. Zahler and W. W. Cleland, J. Biol. Chem., 1968, 243, 716–719 CAS.
- P. Balamurugan, V. P. Krishna, D. Bharath, R. Lavanya, P. Vairaprakash and S. A. Princy, Front. Microbiol., 2017, 8, 1290 CrossRef CAS.
- F. F. Soon, K. M. Suino-Powell, J. Li, E.-L. Yong, H. E. Xu and K. Melcher, PLoS One, 2012, 7, e47857 CrossRef CAS.
- H. Wang, A. Yan, Z. Liu, X. Yang, Z. Xu, Y. Wang, R. Wang, M. Koohi-Moghadam, L. Hu, W. Xia, H. Tang, Y. Wang, H. Li and H. Sun, PLoS Biol., 2019, 17, e3000292 CrossRef.
- H. Wang, M. Wang, X. Yang, X. Xu, Q. Hao, A. Yan, M. Hu, R. Lobinski, H. Li and H. Sun, Chem. Sci., 2019, 10, 7193–7199 RSC.
- L. Li, A. Cheung, A. S. Bayer, L. Chen, W. Abdelhady, B. N. Kreiswirth, M. R. Yeaman and Y. Q. Xiong, J. Infect. Dis., 2016, 214, 1421–1429 CrossRef CAS.
- P. M. Dunman, E. Murphy, S. Haney, D. Palacios, G. Tucker-Kellogg, S. Wu, E. L. Brown, R. J. Zagursky, D. Shlaes and S. J. Projan, J. Bacteriol., 2001, 183, 7341–7353 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06357f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Guijuan Jiang,
Jing Wang and
Jintao Wang
*,
Guijuan Jiang,
Jing Wang and
Jintao Wang