
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ formation of Fe3O4/N-doped carbon coating on the surface of carbon fiber with improved electromagnetic wave-absorption property
Qilong Sun ab,
Yue Jia,
LiFen Hec and
Xiaoyun Long*ab
ab,
Yue Jia,
LiFen Hec and
Xiaoyun Long*ab
aCollege of Textiles and Clothing, Nantong University, China. E-mail: lxy1988@ntu.edu.cn
bNational & Local Joint Engineering Research Center of Technical Fiber Composites for Safety and Protection, Nantong University, China
cNantong Textile and Silk Industry Technology Research Institute, China
First published on 18th August 2020
Abstract
Carbon fiber is an absorbing material with high strength, acid and alkali resistance, high temperature resistance, flexibility, and processability and plays an important role in the electromagnetic (EM) wave absorption of civil buildings and military equipment. However, its EM wave-absorption performance is poor because of its large complex permittivity and no magnetic loss ability. In this study, dopamine hydrochloride and FeCl3 were used as precursors, and the Fe3O4/N-doped carbon coating was successfully grown in situ on the surface of short carbon fiber (SCF) via dopamine deposition, autopolymerization, FeCl3 solution immersion, and calcination at high temperature to improve its EM wave-absorption property. The obtained Fe3O4/N-doped carbon particles were uniformly attached to the SCF in the form of a thin layer to constitute a unique hierarchical structure. The Fe3O4/N-doped carbon coating/SCF displayed an excellent EM wave-absorption performance. An effective bandwidth of 8.64 GHz and lowest reflection loss of −31.38 dB at 3 mm were achieved because of the significant reduction in complex permittivity and improvement in complex permeability, wave impedance, and EM loss ability of the SCF. The Fe3O4/N-doped carbon coating is expected to show great potential in EM wave-absorption fields.
Introduction
With the advent of digital era, electronic products and equipment such as, mobile phones and computers, have significantly increased in our lives and provide us with great convenience. However, the electromagnetic (EM) radiation produced by these devices cause huge concerns, and may have adverse effects on human health.1,2 The surveillance of national defense secrets, important military targets, and war weapons have immensely increased because of the application of EM wave detection technology (electronic eavesdropping, radar detection, etc.).3,4 Hence, in recent years, EM wave-absorbing materials have been investigated by large number of researchers5–7 to avoid the serious threat of EM waves to human health and national defense security.Carbon Fiber (CF) is an electric loss-type EM wave-absorbing material with strong conductivity.8–10 It produces eddy current in an alternating electric field, thereby converting electric energy to heat energy to achieve EM wave attenuation. CF11–13 has high strength, good temperature and chemical resistance, flexibility, processability14 and plays an irreplaceable role in EM wave-absorbing materials of civil buildings and military equipment as compared to other EM wave-absorbing materials.15,16 However, the complex permittivity of CF is extremely large, resulting in extremely low wave impedance of CF and poor matching between the wave impedance of CF and free space,17 making the EM wave to be reflected on the CF surface. Moreover, the CF lacks magnetic loss capacity that is detrimental to rapid attenuation of EM wave inside the CF. Hence, the EM wave-absorption performance of CF is poor, and its application is limited.
Coating the surface by a carbonaceous or magnetic material is an effective method to improve the EM wave-absorption property of CF. Many studies11,12,17 have shown that the resistivity of CF is only (0.8–1.8) × 10−3 Ω cm because of its high graphitization degree and special graphite microcrystalline stacking structure. In accordance with the skin effect,18,19 the complex permittivity of CFs is usually regulated using carbonaceous material coatings, where their conductivity can be controlled,20,21 leading to reduction in complex permittivity and enhancement of wave impedance. In Zhou's study,20 silicon carbide nanofibers were in situ grown on the surface of CF by catalytic chemical vapor deposition. The results revealed that the wave impedance and EM wave-absorption property of CF were enhanced because of the reduction in the complex permittivity. However, the CF still lacks magnetic loss ability, and its EM loss ability is not improved after coating a carbonaceous material on its surface.22
Relevant studies have shown that by coating the CF with a magnetic material, the EM loss ability and the imaginary part of complex permittivity are improved because of the polarization effect of carbon/magnetic interface.23–25 Bostanabad26 used a multistep cathodic method to deposit magnetite on the CF surface through the reduction of an Fe(III)–triethanolamine complex. The results indicated that the EM wave-absorption properties of the CF were improved using a magnetite coating. However, compared to the other carbonaceous fibers (active carbon fiber,27 etc.), it is very difficult to effectively improve the EM wave-absorption property of CF by magnetic coating only, due to the high conductivity and complex permittivity of CF; that is, the wave impedance of CF can be enhanced slightly by the magnetic coating, which is unfavorable for the incidence of EM wave into the CF.
In this study, dopamine hydrochloride (DA) and ferric chloride (FeCl3) were used as precursors, and an Fe3O4/N-doped carbon coating was successfully grown in situ on the SCF surface through dopamine deposition and autopolymerization, FeCl3 solution immersion, and high-temperature calcination to prepare a carbon magnetic heterogenous coating with synergistic absorbing effect on the CF surface. In this N-doped carbon coating, pyridinic-N can lead to defect polarization, whereas pyrrolic-N can act as an active center for dipole polarization due to the different electronegativities of C and N; moreover, they can cause high electric loss factor and rapid attenuation of the EM wave in the SCF.28 X-ray diffraction (XRD) patterns and X-ray photoelectron spectroscopy (XPS) spectra indicate that the coating is composed of Fe3O4 and N-doped carbon along with a small amount of Fe2O3. The Fe3O4/N-doped carbon coating/SCF displayed excellent EM wave-absorption property. An effective bandwidth (reflection loss (RL) ≤ −10 dB) of 8.64 GHz and lowest RL of −31.38 dB at 3 mm were achieved because of the large reduction in the complex permittivity and improvement in the complex permeability, wave impedance, and EM loss ability of SCF. The Fe3O4/N-doped carbon coating/SCF is expected to show great potential in EM wave absorption fields. This study provides a wide reference for the development of many types of carbon magnetic heterogenous coatings on the SCF surface.
Experimental
Materials
Unmodified SCF with size (T700, 12 K, 4 mm) was purchased from Lianyungang Zhongfu Carbon Fiber Co., Ltd, China. DA, FeCl3, and trimethyl-amino-methane (Tris) were obtained from Nanjing Chemical Reagent Co., Ltd, China. Epoxy resin was purchased from Nantong Huacheng New Material Co., Ltd, China.Preparation of SCF with carbon/magnetic heterogeneous coating
The formation process of SCF with Fe3O4/N-doped carbon coating (modified SCF) is described as follows (Fig. 1).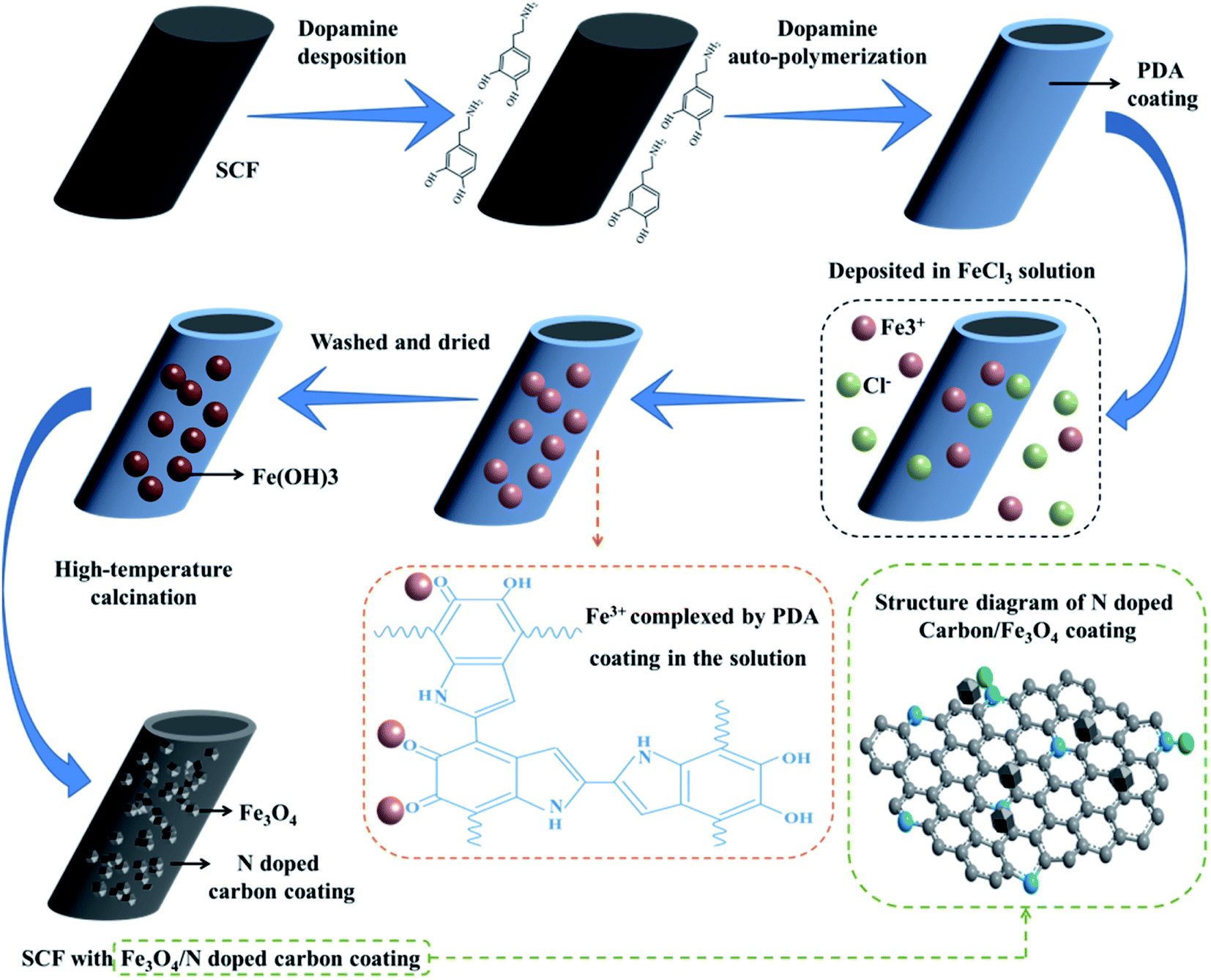 | ||
| Fig. 1 Schematic of the formation of Fe3O4/N-doped carbon/SCF. | ||
Step 1: DA (2 g) was added to 1 L deionized water followed by quick stirring for 5 min; then, 1.2 g Tris was slowly added to the abovementioned DA solution followed by continuous stirring until the pH of the solution was 8.5. SCF (1 g) was dispersed in a 1 L DA solution using an ultrasound bath for 20 min. The dopamine solution with SCF was oscillated (oscillating frequency was 40 times per min, and temperature was 30 °C) using an oscillating instrument for 50 h. In this process, a polydopamine (PDA) coating was formed on the SCF surface by the autopolymerization of dopamine.
Step 2: FeCl3 (0.6 mol L−1) was dispersed in 1 L deionized water to form an FeCl3 solution. PDA/SCF (1 g) was added to the FeCl3 solution followed by oscillation (oscillating frequency was 120 times per min and temperature was 30 °C) using the oscillating instrument for 8 h. The PDA/SCF was then washed three times with deionized water and dried. In this process, the Fe3+ ion complexed by PDA combined with (OH)− in water to form Fe(OH)3 on the SCF surface.
Step 3: Fe(OH)3/PDA/SCF (1 g) was calcined at high temperatures using a tube resistance furnace. The process parameters were as follows: 5 L min−1 nitrogen rate, 5 °C min−1, 750 °C, and 2 h holding time. After the internal temperature of the tube resistance furnace dropped to room temperature, the fibers were removed, washed with deionized water, and dried to obtain the modified SCF.
Measurements and characterization
A scanning electron microscope (SEM, Scios DualBeam) equipped with an energy dispersive spectroscopy (EDS) apparatus was used for morphology observation and elemental analysis. Before SEM imaging, the samples were sputter coated with a thin layer of Au nanoparticles to reduce charging effects.The surface chemical composition of the sample was analyzed via XPS (250Xi, Thermo ESCALAB, USA) with a soft Al Kα X-ray source (hv = 1486.6 eV) at 150 W.
Phase structure analysis of the modified SCF was performed using XRD (Rigaku D/max-2500PC, with CuKα).
The EM parameters of the SCF were measured using a vector network analyzer. Prior to measurement, the sample was ground into a fine powder, which was then mixed with paraffin wax to prepare a ring with a thickness of 3 mm (the mass fraction of the powder was 20%).
The electric and magnetic loss factors were calculated using eqn (1) and (2), respectively.29
| tg δε = ε′′/ε′, | (1) |
| tg δμ = μ′′/μ′, | (2) |
The wave impedance and EM wave-absorption property of the SCF were evaluated using eqn (3) and (4), respectively.30,31
 | (3) |
 | (4) |
Results and discussion
Surface phase composition analysis
The samples were examined by XRD to investigate the changes in the surface phase composition of the unmodified and modified SCFs. Fig. 2(a) shows the characteristic peaks of the unmodified SCF; these peaks are in good agreement with those reported in previous studies.32,33 As shown in Fig. 2(b), Fe3O4 (JCPDS 26-1134), which is a commonly used magnetic material and has good ferromagnetism, was detected on the surface of the modified SCF during surface modification. The characteristic peaks at 18.99°, 31.24°, 36.88°, 44.72°, 55.62°, and 62.64° correspond to the (111), (220), (222), (400), (422), and (440) crystal planes. The formation mechanism of Fe3O4 can be explained by the following equations:| Fe3+ + H2O → Fe(OH)3 + H+, | (5) |
 | (6) |
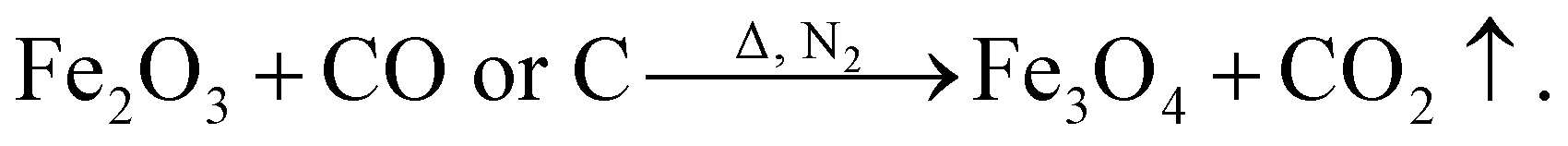 | (7) |
 | ||
| Fig. 2 (a) XRD survey spectra of the unmodified SCF and (b) XRD survey spectra of the modified SCF. | ||
At first, FeCl3 was dispersed in deionized water and hydrolyzed to Fe3+ and Cl−; then, Fe3+ in a certain amount was complexed by the oxygen of the PDA coating because of the excellent complexation ability of the oxygen atoms of PDA. Then, Fe3+ reacted with H2O on the SCF surface to form Fe(OH)3 when Fe3+/SCF was washed and dried. Finally, Fe(OH)3/SCF was calcined at high temperatures under nitrogen protection, and Fe(OH)3 reacted to form Fe2O3 and H2O. Fe2O3 was reduced to Fe3O4 under the action of C or CO, which was released by the carbonization of the PDA coating.
Fe2O3 (JCPDS 73-2234) was also identified on the surface of the modified SCF because of its inadequate reduction and considered as an impurity in this study.
Surface morphology analysis
SEM was used to investigate the surface morphology of the unmodified and modified SCFs. As shown in Fig. 3(a) and (b), the surface of the unmodified SCF was coarse, and several gullies were observed on it, as shown in the high-magnification (3000× and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×) SEM images of the unmodified SCF, respectively. High-magnification (3000× and 10000×) SEM images of the modified SCF were also obtained, as shown in Fig. 3(d) and (e), respectively. The gullies disappeared, and a nitrogen-doped carbon-loaded Fe3O4 coating was noticed on the SCF surface, which consisted of a carbonized PDA coating (marked by a yellow circle) and flaky Fe3O4 (marked by a white circle). The iron oxide coating was discontinuous because the solution was oscillated at a high speed (oscillating frequency was 120 times per min). This condition prevented a strong current on the surface of the modified SCF, leading to large complex permittivity of the modified SCF. Fig. 3(c) and (d) show the high-magnification (10000× and 3000×) SEM images of the cross-section of the modified SCF, respectively. No evident boundaries were observed between the nitrogen-doped carbon coating and SCF. This condition indicated that the nitrogen-doped carbon coating formed by PDA closely combined with SCF during high-temperature calcination.
000×) SEM images of the unmodified SCF, respectively. High-magnification (3000× and 10000×) SEM images of the modified SCF were also obtained, as shown in Fig. 3(d) and (e), respectively. The gullies disappeared, and a nitrogen-doped carbon-loaded Fe3O4 coating was noticed on the SCF surface, which consisted of a carbonized PDA coating (marked by a yellow circle) and flaky Fe3O4 (marked by a white circle). The iron oxide coating was discontinuous because the solution was oscillated at a high speed (oscillating frequency was 120 times per min). This condition prevented a strong current on the surface of the modified SCF, leading to large complex permittivity of the modified SCF. Fig. 3(c) and (d) show the high-magnification (10000× and 3000×) SEM images of the cross-section of the modified SCF, respectively. No evident boundaries were observed between the nitrogen-doped carbon coating and SCF. This condition indicated that the nitrogen-doped carbon coating formed by PDA closely combined with SCF during high-temperature calcination.
 | ||
| Fig. 3 (a) Surface morphology of the unmodified SCF (3000×); (b) surface morphology of the unmodified SCF (10000×); (c) surface morphology of the cross-section of the modified SCF (10000×); (d) surface morphology of the modified SCF (3000×); (e) surface morphology of the modified SCF (10000×); (f) surface morphology of the cross-section of the modified SCF (30000×); (g) SEM map scanning topography of the C element; (h) SEM map scanning topography of the N element; (i) SEM map scanning topography of the Fe element; and (j) SEM map scanning topography of the O element. | ||
To perform a detailed analysis of the main element distribution on the surface of the modified SCF, EDS surface scan of C, N, Fe, and O was conducted, as shown in Fig. 3(g), (h), (i), and (j), respectively. The N atoms (marked by dark cyan points) were uniformly doped into the carbon coating, the Fe and O atoms (marked by purple and green points, respectively) were uniformly dispersed on surface of the fiber, and their distribution maps approximately overlapped; this proved that these elements were combined via a series of reactions to generate Fe3O4.
Surface chemical component analysis
The changes in the surface chemical components of the unmodified and modified SCFs were evaluated through XPS. The dark cyan curve in Fig. 4(a) shows the characteristic peaks of the unmodified SCF, and the peaks at 285.08 and 533.08 eV correspond to the C 1s and O 1s elements of the sample, respectively. The concentrations of these elements were 96.6 and 4.4 at%. This finding is in good agreement with the characteristic peaks of the CF reported in a previous study.34 The orange curve in Fig. 4(a) shows the characteristic peaks of the modified SCF, and in addition to the peaks of the C 1s and O 1s elements, the characteristic peaks of Fe 2p were found at 709.9 and 728.2 eV, indicating that the Fe 2p element was also present on the surface of the sample. The concentrations of C, O, and Fe were 52.8, 19.7, and 22.4 at%, respectively. The N element was detected on the surface of the modified SCF, and its concentration was 5.1 at%, implying the formation of a nitrogen-doped carbon coating on the SCF surface. | ||
| Fig. 4 (a) XPS survey spectra of the unmodified and modified SCFs; (b) fitting result of the C 1s peak of the unmodified SCF; (c) fitting result of the C 1s peak of the modified SCF; (d) fitting result of the N1s peak of the modified SCF; (e) fitting result of the Fe 2p peak of the modified SCF; and (f) fitting result of the O 1s peak of the modified SCF. | ||
To perform a detailed analysis of the chemical components of SCF, the XPS survey spectra of the C 1s (unmodified SCF), C 1s, N 1s, Fe 2p, and O1s elements of the modified SCF were obtained, as shown in Fig. 4(b–f). As shown in Fig. 4(b), the C 1s peak of the unmodified SCF was successfully fitted into four peaks. The lowest-binding-energy peak at 284.24 eV was attributed to the C–C bond. The peaks of the C–N, C–O, and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds were found at the binding energies of 284.47 eV, 286.14 eV, and 287.25 eV, respectively.35 Fig. 4(c) shows the fitting result of the C 1s peak of the modified SCF. The peaks at the binding energies of 283.6, 284.26, 284.88, and 286.02 eV were attributed to the C–C, C–N, C–O, and O–CO bonds, respectively. The π–π shake-up peak (π–π*) originating from the conjugated aromatic ring of PDA was found at the binding energy of 289.18 eV,36 implying that the structural integrity of PDA was maintained even after calcination at high temperatures. Fig. 4(d) shows the fitting result of the N 1s peak of the modified SCF. The N 1s peak was fitted into three peaks. The peaks of graphitic-N, pyrrolic-N, and pyridinic-N were obtained at the binding energies of 398.01, 399.73, and 400.49 eV, respectively.29 The doping of three types of nitrogen species into carbon could ensure the conductivity and stability of the coating. Fig. 4(e) shows the fitting result of the Fe 2p peak of the modified SCF. The spectrum was successfully fitted into three main peaks and two satellite peaks in the 2p3/2 region. The lowest-binding-energy peak at 710.69 eV was attributed to Fe2+, with a corresponding satellite at 716.01 eV. The peak of the Fe3+ octahedral species was found at the binding energy of 711.04 eV, and the peak of the Fe3+ tetrahedral species was obtained at the binding energy of 711.97 eV, with a corresponding satellite at 720.25 eV. In accordance with the XPS survey spectra of Fe 2p, the calculated Fe2+/Fe3+ ratio was approximately 0.35, which was slightly lower than the theoretical value (0.5) of Fe3O4.37 Fig. 4(f) shows the fitting result of the O1s peak of the modified SCF. The lowest-binding-energy peak at 529.79 eV was attributed to the O–Fe bond (lattice O). The peak of the O–C bond was found at the binding energy of 531.81 eV, and the peak of the O–CO bond was obtained at the binding energy of 536.2 eV.38
O bonds were found at the binding energies of 284.47 eV, 286.14 eV, and 287.25 eV, respectively.35 Fig. 4(c) shows the fitting result of the C 1s peak of the modified SCF. The peaks at the binding energies of 283.6, 284.26, 284.88, and 286.02 eV were attributed to the C–C, C–N, C–O, and O–CO bonds, respectively. The π–π shake-up peak (π–π*) originating from the conjugated aromatic ring of PDA was found at the binding energy of 289.18 eV,36 implying that the structural integrity of PDA was maintained even after calcination at high temperatures. Fig. 4(d) shows the fitting result of the N 1s peak of the modified SCF. The N 1s peak was fitted into three peaks. The peaks of graphitic-N, pyrrolic-N, and pyridinic-N were obtained at the binding energies of 398.01, 399.73, and 400.49 eV, respectively.29 The doping of three types of nitrogen species into carbon could ensure the conductivity and stability of the coating. Fig. 4(e) shows the fitting result of the Fe 2p peak of the modified SCF. The spectrum was successfully fitted into three main peaks and two satellite peaks in the 2p3/2 region. The lowest-binding-energy peak at 710.69 eV was attributed to Fe2+, with a corresponding satellite at 716.01 eV. The peak of the Fe3+ octahedral species was found at the binding energy of 711.04 eV, and the peak of the Fe3+ tetrahedral species was obtained at the binding energy of 711.97 eV, with a corresponding satellite at 720.25 eV. In accordance with the XPS survey spectra of Fe 2p, the calculated Fe2+/Fe3+ ratio was approximately 0.35, which was slightly lower than the theoretical value (0.5) of Fe3O4.37 Fig. 4(f) shows the fitting result of the O1s peak of the modified SCF. The lowest-binding-energy peak at 529.79 eV was attributed to the O–Fe bond (lattice O). The peak of the O–C bond was found at the binding energy of 531.81 eV, and the peak of the O–CO bond was obtained at the binding energy of 536.2 eV.38
EM parameter and wave-absorbing mechanism analysis
To analyze the EM wave-absorption properties of the unmodified and modified SCFs, the RLs were calculated based on the complex permittivity and permeability using eqn (1) and (2).The RL curve and the corresponding 3D image of the RL curve of the unmodified SCF are shown in Fig. 5(a) and (b), respectively. A frequency range with an RL value less than −10 dB is defined as the effective absorption bandwidth (fE), where 90% of the EM wave energy can be dissipated. We have marked the values corresponding to −10 dB with a dark cyan line in the 2D image. The unmodified SCF test sample with different thicknesses showed poor EM wave-absorption properties, and the fE of all the tested samples with thickness in the range from 1.5 mm to 3.5 mm was 0 GHz. As shown in Fig. 5(c) and (d), the modified SCF showed excellent EM wave-absorption properties, the fE of the samples was 3.57, 6.66, 7.67, 8.64, and 12.13 GHz at the low thicknesses of 1.5, 2, 2.5, 3, and 3.5 mm, and the lowest RLs were −16.67, −23.9, −32.5, −31.38, and −25.9 dB, respectively.
 | ||
| Fig. 5 (a) RL curve of the unmodified SCF as a function of test sample thickness; (b) 3D image of the RL curve of the unmodified SCF as a function of test sample thickness; (c) RL curve of the modified SCF as a function of test sample thickness; (d) 3D image of the RL curve of the modified SCF as a function of test sample thickness; (e) complex permittivity of the unmodified and modified SCFs; (f) complex permeability of the unmodified and modified SCFs; (g) Zin of the unmodified SCF as a function of test sample thickness; (h) Zin of the modified SCF as a function of test sample thickness; and (i) EM loss factor of the SCF. | ||
To illustrate the intrinsic wave-absorption mechanism of Fe3O4/N-doped carbon/SCF, the EM parameters, wave impedance, and EM wave loss factor were investigated. To compare the complex permittivities of the unmodified and modified SCFs, the samples were measured using the vector network analyzer in the frequency range of 2–18 GHz. The real part of the complex permittivity was represented by ε′, and the imaginary part of the complex permittivity was represented by ε′′. As shown in Fig. 5(e), the ε′ (presented by the orange curve) and ε′′ of the unmodified SCF (presented by the dark cyan curve) were 7.39–23.8 and 17.97–36.95, respectively, because of the excellent electrical conductivity, high graphitization degree, and special graphite microcrystalline stacking structure of the unmodified SCF. However, extremely high complex permittivity will cause the reflection of the EM waves on the surface of the unmodified SCF.15 After surface modification, the ε′ of the SCF (shown by the blue curve) decreased from 7.39–23.8 to 0.6–7.79 and the ε′′ of the SCF (shown by the red curve) decreased from 17.97–36.95 to 0.06–6.69.
This phenomenon can be explained as follows (Fig. 6): the resistivity of the unmodified SCF was extremely small, which was only 0.8–1.8 × 10−3 Ω cm, because of the high graphitization degree and special graphite microcrystalline stacking structure of the unmodified SCF, leading to its large complex permittivity. In accordance with the skin effect, in the EM field, the closer to the surface of unmodified SCF, the greater the current density, that is to say, the current is concentrated on the surface of the SCF, and the thickness of the area where the charge propagates represents the skin depth. Hence, the complex permittivity was significantly reduced after surface modification because of the formation of the nitrogen-doped carbon-loaded Fe3O4 coating, whose conductivity could be regulated based on its nitrogen content, on the surface of the SCF.
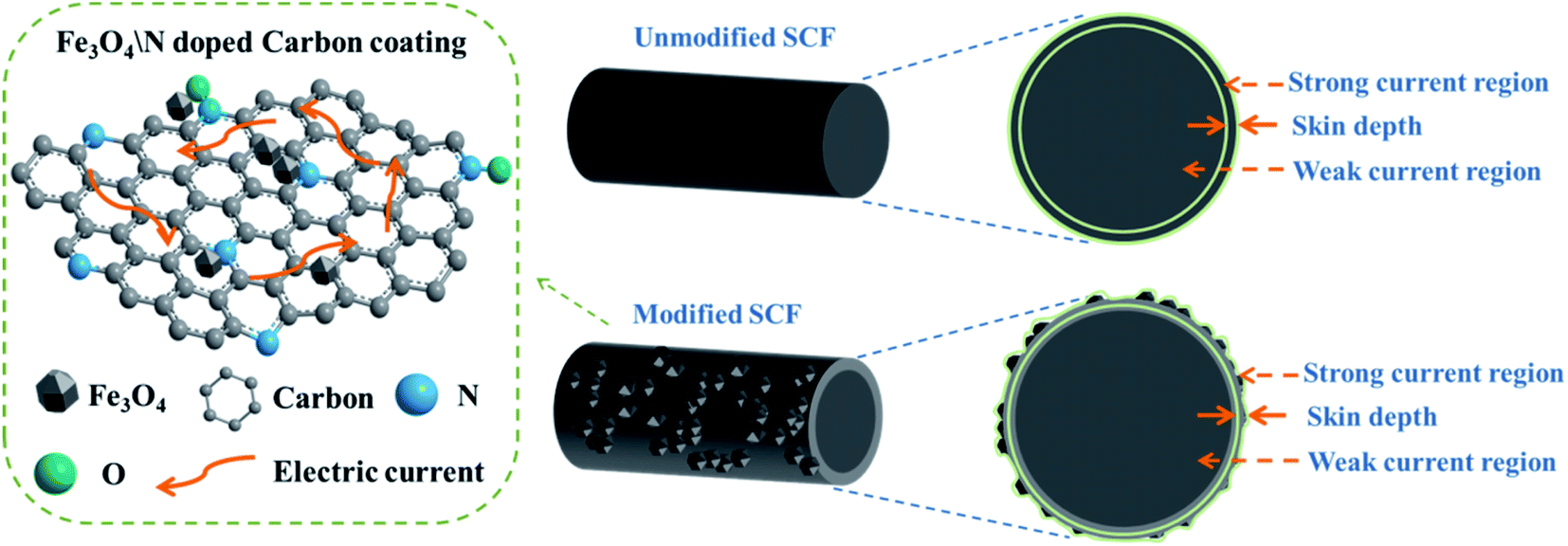 | ||
| Fig. 6 Schematic of the skin effect of the SCF. | ||
The complex permeabilities of the unmodified and modified SCFs were measured using the vector network analyzer in the frequency range of 2–18 GHz. The real part of the complex permeability is represented by μ′, and the imaginary part of the complex permeability is represented by μ′′. As shown in Fig. 5(f), the μ′ of the unmodified SCF (shown by the orange curve) was approximately one and the unmodified SCF had no magnetic loss ability, whereas the μ′′ of the unmodified SCF (shown by the dark cyan curve) was approximately zero. After surface modification, the μ′ of the modified SCF (presented by the blue curve) was 1.28–2.64, larger than the μ′ of the unmodified SCF. The μ′′ of the modified SCF (presented by the red curve) was 0.01–1.86, and its curve showed a downward trend with an increase in frequency from 2 to 18 GHz. This finding implied that Fe3O4 provided magnetic loss ability to the SCF after surface modification, which was beneficial for improving the wave impedance and EM loss ability of the SCF.
Fig. 5(g) shows the wave impedances of the unmodified and modified SCFs. The wave impedances of the unmodified SCF were 2.37–35.37, 3.17–48.4, 2.96–62.09, 4.75–77.56, and 5.55–94.87 Ω at the low thicknesses of 1.5, 2, 2.5, 3, and 3.5 mm, respectively, because of the large complex permittivity of the unmodified SCF. As shown in Fig. 5(h), the wave impedances of the modified SCF were 2.65–66.62, 3.54–83.34, 4.43–106.16, 5.31–130.62, and 6.65–156.62 Ω at the low thicknesses of 1.5, 2, 2.5, 3, and 3.5 mm, respectively, because of the significant reduction in the complex permittivity and improvement in the complex permeability of the SCF. Compared with the case of the unmodified SCF, the matching between the wave impedance of the modified SCF and free space (377 Ω) was better, thereby making the entry and attenuation of the EM wave inside the SCF easy.
Fig. 5(i) shows the calculation results of the electric loss and magnetic loss factors of the unmodified and modified SCFs. The electric loss factor of the unmodified SCF (shown by the orange curve) was 0.84–2.77, and the magnetic loss factor of the modified SCF (shown by the dark cyan curve) was 0. The electric loss factor of the modified SCF (presented by the blue curve) was 0.1–7.55. The magnetic loss factor of the modified SCF (presented by the red curve) was 0.1–1.01. After surface modification, the EM loss factor (sum of the electric loss and magnetic loss factors) of the modified SCF was larger than that of the unmodified SCF, causing rapid attenuation of the EM wave inside the SCF.
Conclusion
Herein, an Fe3O4/N-doped carbon coating was fabricated in situ on the surface of SCF for the first time and the formation of this coating was confirmed by SEM, XRD, XPS, and EDS. The results showed that the complex permittivity of the SCF was substantially reduced and the complex permeability was improved by the Fe3O4/N-doped carbon coating, resulting in enhanced impedance matching and EM loss ability of the SCF. This condition enabled easy entry and rapid attenuation of the EM inside the SCF. Thus, the Fe3O4/N-doped carbon/SCF showed excellent wave-absorption performance with a fE of 8.64 GHz and lowest RL of −31.38 dB at 3 mm. This novel method can provide a new way to improve the EM wave-absorption properties of SCFs.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This study was financially supported by the Open Project Program of Key Laboratory of Eco-textiles, Ministry of Education, Jiangnan University (No. KLET1812).References
- Z. Shen, H. Xing, Y. Zhu, X. Ji and L. Wang, J. Mater. Sci.: Mater. Electron., 2017, 28, 1–9 CrossRef.
- W. Ye, Q. Sun and G. Zhang, Ceram. Int., 2018, 45(4), 5093–5099 CrossRef.
- J. Li, Y. Xie, W. Lu and T. W. Chou, Carbon, 2017, 129, 76–84 CrossRef.
- Y. I. Lee, D. H. Jang and Y. H. Choa, J. Nanosci. Nanotechnol., 2016, 16, 5190–5194 CrossRef CAS PubMed.
- D. Kim, J. Korean Phys. Soc., 2010, 56(4), 237–241 Search PubMed.
- B. Qu, C. Zhu, C. Li, X. Zhang and Y. Chen, ACS Appl. Mater. Interfaces, 2016, 8(6), 3730–3735 CrossRef CAS PubMed.
- X. Hong, Q. Wang, Z. Tang, W. Q. Khan and T. Feng, J. Phys. Chem. C, 2015, 120, 317–322 Search PubMed.
- L. Huang, D. Chen, Y. Ding, S. Feng and M. Liu, Nano Lett., 2013, 13, 3135–3139 CrossRef CAS PubMed.
- K. Li, C. Wang, H. Li, L. Guo and J. Lu, Int. J. Miner., Metall. Mater., 2006, 15, 808–815 Search PubMed.
- J. Ren, Z. Y. Feng, T. C. Zou and H. L. Mou, Adv. Mater. Res., 2006, 676, 17–21 Search PubMed.
- L. I. Wei and Q. F. Guo, Chin. J. Opt., 2011, 4, 201–212 Search PubMed.
- D. M. Devine, J. Hahn, R. G. Richards, H. Gruner and S. G. Pearce, J. Biomed. Mater. Res., Part B, 2013, 101, 591–598 CrossRef PubMed.
- Y. Z. Wan, Y. L. Wang, K. D. Yao and G. X. Cheng, J. Appl. Polym. Sci., 2015, 75, 994–998 CrossRef.
- Q. Li, X. Tian, Z. Chen, J. Wu, Y. Li and Y. Li, J. Mater. Sci., 2018, 53, 1–9 CrossRef.
- Y. Jing, H. Ying, X. Chen and W. Chao, Synth. Met., 2016, 221, 291–298 CrossRef.
- K. Yu, Z. Min, Y. Yin, X. Zeng and R. Yu, Adv. Eng. Mater., 2017, 20, 1700543 CrossRef.
- Y. Fan, H. Yang, X. Liu, H. Zhu and G. Zou, J. Alloys Compd., 2008, 461, 0–494 CAS.
- F. Mitschke, Skin Effect, Springer Berlin Heidelberg, 2009 Search PubMed.
- V. Alessandrini, H. Fanchiotti, C. A. García Canal and H. Vucetich, Int. J. Electron., 1976, 40, 57–63 CrossRef.
- W. Zhou, L. Long, P. Xiao, Y. Li, H. Luo, W.-d. Hu and R.-m. Yin, Ceram. Int., 2017, 43, 5628–5634 CrossRef CAS.
- X. Z. Zhang and W. Sun, Adv. Mater. Res., 2009, 79–82, 1843–1846 CAS.
- Y. Cheng, P. Hu, S. Zhou, L. Yan, B. Sun, X. Zhang and W. Han, Carbon, 2018, S0008622318302148 Search PubMed.
- Y. Liu, X. X. Liu, X. Chen and X. J. Wang, J. Inorg. Mater., 2013, 28, 1328–1332 CrossRef CAS.
- F. He, J. Li, L. Chen, L. Chen and Y. Huang, Surf. Rev. Lett., 2015, 22, 150201225054003 Search PubMed.
- H. Salimkhani, F. Movassagh-Alanagh, H. Aghajani and K. Osouli-Bostanabad, Procedia Mater. Sci., 2015, 11, 231–237 CrossRef CAS.
- K. O. Bostanabad, H. Aghajani, E. Hosseinzade, H. Hossain and S. Maleki-Ghaleh, Mater. Manuf. Processes, 2016, 31(10), 1351–1356 CrossRef.
- Q. Sun, L. Sun, Y. Cai, T. Ji and G. Zhang, RSC Adv., 2018, 8, 35337–35342 RSC.
- W. Yang, L. Hou, X. Xu, Z. Li, X. Ma, F. Yang and Y. Li, Carbon, 2018, S0008622318300320 Search PubMed.
- Z. Zhao, K. Kou and H. Wu, J. Colloid Interface Sci., 2020, 574, 1–10 CrossRef CAS PubMed.
- P. Marin, D. Cortina and A. Hernando, IEEE Trans. Magn., 2008, 44, 3934–3937 CAS.
- M. J. Youh, H.-C. Wu, W.-H. Lin, S.-C. Chiu, C.-F. Huang, H.-C. Yu, J.-S. Hsu and Y.-Y. Li, J. Nanosci. Nanotechnol., 2011, 11(3), 2315–2320 CrossRef CAS PubMed.
- E. I. Moreno-Valencia, S. P. Paredes-Carrera, J. C. Sánchez-Ochoa, S. O. Flores and J. R. Avendaño-Gómez, Mater. Res. Express, 2017, 4(11), 115026 CrossRef.
- C. Deng, C. Hu and J. Huo, Iron Steel Vanadium Titanium, 2016 Search PubMed.
- W. H. Lee, J. G. Lee and P. J. Reucroft, Appl. Surf. Sci., 2001, 171, 136–142 CrossRef CAS.
- V. Calderon, S. A. Cavaleiro and S. Carvalho, Appl. Surf. Sci., 2015, 346, 240–247 CrossRef.
- Y. Chen, S. Zhao, M. Chen, W. Zhang, J. Mao, Y. Zhao, M. F. Maitz, N. Huang and G. Wan, Corros. Sci., 2015, 96, 67–73 CrossRef CAS.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- Y. C. G. Kwan, G. M. Ng and C. H. A. Huan, Thin Solid Films, 2015, 590, 40–48 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |