
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple method for the synthesis of N-difluoromethylated pyridines and 4-pyridones/quinolones by using BrCF2COOEt as the difluoromethylation reagent†
Albert Gandioso‡
,
Mohamed El Fakiri‡,
Anna Rovira‡ and
Vicente Marchán *
*
Departament de Química Inorgànica i Orgànica, Secció de Química Orgànica, IBUB, Universitat de Barcelona, Martí i Franquès 1-11, E-08028, Barcelona, Spain. E-mail: vmarchan@ub.edu
First published on 13th August 2020
Abstract
We describe a novel transition metal-free method for the synthesis of N-difluoromethylated pyridines and 4-pyridones/quinolones by using readily available ethyl bromodifluoroacetate as a fluorine source. The formation of N-difluoromethylated pyridines involves a two-step process in which N-alkylation by ethyl bromodifluoroacetate is followed by in situ hydrolysis of the ester and decarboxylation. Besides optimizing the N-difluoromethylation conditions and assessing the influence of steric and electronic effects on the outcome of the reaction, we have synthesized the N-difluoromethylated analogues of two fluorophores and demonstrated that their spectroscopic properties can be improved through replacement of N-CH3 group by N-CF2H.
Introduction
The introduction of fluorinated scaffolds in organic compounds is a well-established approach in medicinal, agricultural and biomaterial sciences for the modification of biologically-relevant properties such as binding affinity, metabolic stability, lipophilicity, and bioavailability.1 Non-invasive diagnostic tools such as positron emission tomography (PET) and magnetic resonance imaging (MRI) are also based on compounds incorporating radioactive (18F) and non-radioactive (19F) fluorinated moieties, respectively.2 Remarkably, about half of the most successful drugs are based on fluorine-containing compounds and more than three out of ten drugs approved by the FDA in the last two years contain this atom.3 In this context, there is a clear demand for novel methods to expand the current fluorination toolbox in organic synthesis.Among fluorinated scaffolds, the difluoromethyl group (CF2H) has attracted great interest owing to its unique physicochemical properties. It is considered a lipophilic bioisostere of hydroxyl and thiol functional groups capable of hydrogen bonding,4 which has been exploited to improve membrane permeability and binding affinity of biologically-active compounds.5 Despite these promising properties, the chemistry of difluoromethylation has been less extensively studied compared with that of trifluoromethylation. The late-stage introduction of CF2H group into organic substrates has been mainly investigated by metal-mediated photocatalytic and thermal processes,6 and by transition-metal-free strategies.6a,b,7
Pyridine and its derivatives are found in natural products (e.g., vitamins, coenzymes and alkaloids) and in a wide number of drugs and agrochemicals.8 More than 100 pyridine-containing drugs have been approved by the FDA,9 making this nitrogen-containing heterocycle a privileged scaffold in medicinal chemistry. Remarkably, N-methylpyridinium moieties are found in several marketed drugs and in biologically active compounds such as antibacterials and anticancer agents and, consequently, replacement of the methyl group by CF2H offers an unprecedented opportunity to discover novel drug candidates with improved (or simply different) biological activities. Similarly, N-difluoromethylation of pyridine might have a strong impact in the bioimaging field since many fluorescent probes incorporate N-alkylpyridinium moieties.10
The quinolin-4(1H)-one is also a common structural motif in many bioactive compounds, including well-known antibacterial agents such as fluoroquinolones.11 Since many quinolone antibiotics are N-alkylated and incorporate at least one fluorine atom in their chemical structure, the N-difluoromethylation of the quinolin-4(1H)-one scaffold offers an exceptional opportunity to develop new antibiotics with an expanded spectrum and high efficacy.
Despite the enormous potential that the N-difluoromethylation reaction of pyridine and derivatives offers, only two methods have been reported to date (Scheme 1). In 1996, Röschenthaler and co-workers described the two-step N-difluoromethylation reaction of DMAP by using CF2Br2 in the presence of activated copper, followed by hydrogenation with tributyltin hydride.12 However, the ozone depletion potential of CF2Br2 and manipulation risks advises against the use of this reagent. More recently, Zafrani, Gershonov and collaborators have described the difluorocarbene-mediated N-difluoromethylation of trialkyl amines using diethyl bromodifluoromethylphosphonate and a fluoride source.13 Although the reaction was optimized for the synthesis of N-difluoromethyltrialkylammonium salts, the method proved to be adequate for the N-difluoromethylation of DMAP. On the other hand, the two examples reported in the literature for the synthesis of N-difluoromethylated 4-quinolones are based on the use of CHBrF2 as fluorinating reagent of a quinolin-4-ol derivative,14 and on the photogeneration of difluorocarbene by deiodination and deacetylation of ethyl difluoroiodoacetate under basic conditions.15 Examples on the synthesis of N-difluoromethyl-2-pyridone derivatives are based on the use of ClCF2COONa/18-crown-6 and BrCF2COOEt/K2CO3.16 Herein, we describe a new straightforward transition-metal-free method for the synthesis of N-difluoromethylated pyridines based on the use of ethyl bromodifluoroacetate (1), which is a cheap, safe and commercially available reagent (Scheme 1). Based on this reaction, a simple procedure for the synthesis of N-difluoromethylated 4-pyridones and 4-quinolones has been also discovered.
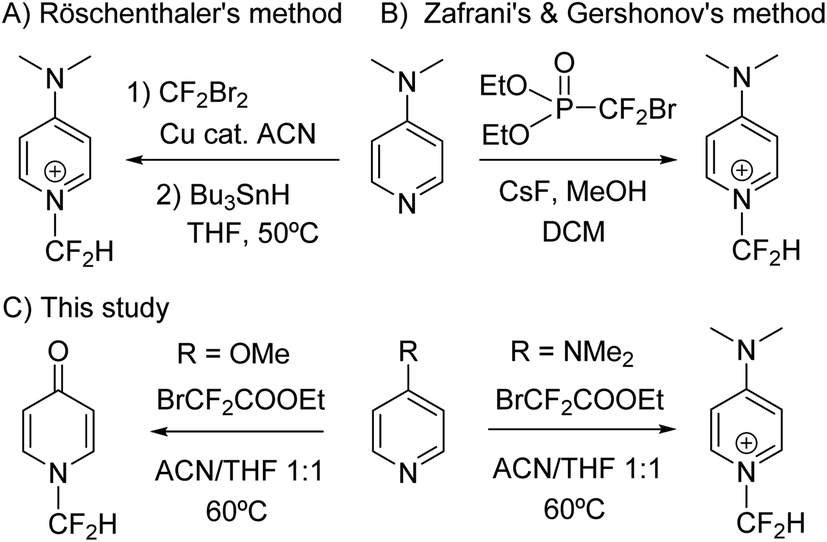 | ||
| Scheme 1 (A and B) Previously reported methods for the N-difluoromethylation of DMAP, and (C) synthesis of N-difluoromethylated pyridines and 4-pyridones described in this work. | ||
Results and discussion
Initially, DMAP was chosen as a model substrate to investigate the use of ethyl bromodifluoroacetate (1) as a potential N-difluoromethylating reagent of pyridine. First, DMAP was reacted with 1 (5 mol equiv.) in ACN as a solvent (HPLC quality) at 60 °C for 24 h. Reversed-phase UV-HPLC-MS analysis (Fig. S1, S2 and Table S1†) analysis revealed the presence of compound 2a (24%) resulting from the N-alkylation of the pyridine heterocycle together with a small amount of unreacted DMAP (4%) (Scheme 2). To our delight, both the carboxylic acid derivative 2b (32%) and the N-difluoromethylated pyridinium salt 3 (40%) were also identified in the crude, which indicates that hydrolysis of the ester and decarboxylation take place in situ in the reaction media affording the expected N-difluoromethylated derivative 3. Although halodifluoromethyl compounds have been extensively used as difluorocarbene synthons in difluoromethylation reactions,17 the presence of 2a and 2b supports a direct nucleophilic attack of the pyridine nitrogen on the electrophilic carbon of 1 rather than the generation of a difluorocarbene intermediate. In order to confirm this mechanism, compound 2b was isolated by reversed-phase HPLC and fully characterized by 1H, 13C and 19F-NMR, IR and HR ESI-MS (see ESI†). It is worth noting that the ester intermediate (2a) could not be isolated since it was completely transformed into 2b during the purification by silica column chromatography.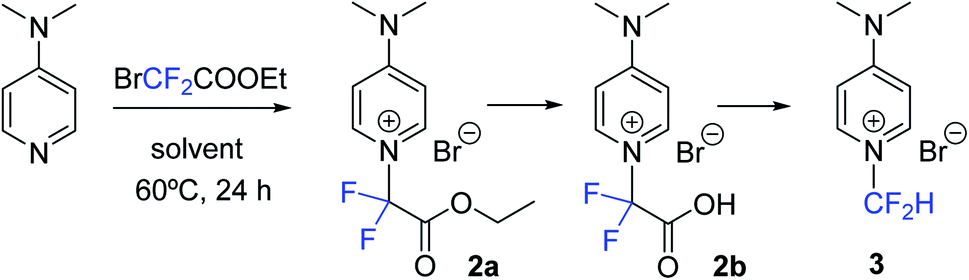 | ||
| Scheme 2 N-Difluoromethylation of DMAP using ethyl bromodifluoroacetate (1). | ||
Based on these results, we focused on the optimization of the reaction conditions for the N-difluoromethylation of DMAP, and the progress of the reaction was investigated by capillary electrophoresis analysis (Fig. S3 and S4†). As shown in Table 1 (entry 2), the amount of 3 was increased (83%) when THF (which was previously eluted through basic alumina) was used instead of ACN (56%). By contrast, the outcome of the reaction was negatively affected in distilled THF (Table 1, entry 3): the amount of N-alkylation did not significantly change but the transformation of the N-alkylated intermediates into 3 was considerably reduced. Very interestingly, the N-difluoromethylation of DMAP was quantitative (entry 5) when a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of neutralized THF and ACN was used as a solvent. Again, the transformation of the intermediates into 3 was reduced when distilled THF was combined with ACN (entry 6), suggesting that the presence of basic traces in alumina-neutralized THF facilitates the in situ hydrolysis of the ester and decarboxylation (see below). With the optimized conditions in hand, the reaction was repeated at large scale and the pure N-difluoromethylated compound was isolated (yield: 92%) and fully characterized by 1H, 13C and 19F-NMR and HR ESI-MS. Fig. 1 shows the aromatic region of the 1H-NMR of 3 where a characteristic triplet at 8.54 ppm resulting from the coupling of the fluorine atoms to the proton of the CF2H group is observed. Similarly, the carbon of the CF2H group appears as a triplet at 111 ppm in the 13C NMR spectrum (Fig. 1), and the fluorine atoms as a doublet in the 19F NMR spectrum (see ESI†).
:1 (v/v) mixture of neutralized THF and ACN was used as a solvent. Again, the transformation of the intermediates into 3 was reduced when distilled THF was combined with ACN (entry 6), suggesting that the presence of basic traces in alumina-neutralized THF facilitates the in situ hydrolysis of the ester and decarboxylation (see below). With the optimized conditions in hand, the reaction was repeated at large scale and the pure N-difluoromethylated compound was isolated (yield: 92%) and fully characterized by 1H, 13C and 19F-NMR and HR ESI-MS. Fig. 1 shows the aromatic region of the 1H-NMR of 3 where a characteristic triplet at 8.54 ppm resulting from the coupling of the fluorine atoms to the proton of the CF2H group is observed. Similarly, the carbon of the CF2H group appears as a triplet at 111 ppm in the 13C NMR spectrum (Fig. 1), and the fluorine atoms as a doublet in the 19F NMR spectrum (see ESI†).
| Entry | Compound 1 equiv. | Solvent | Conversiond [%] |
|---|---|---|---|
| a Reactions were performed on a 0.08 mmol scale in 2 mL of the solvent at 60 °C for 24 h.b THF was eluted through basic alumina.c THF was distilled over sodium and benzophenone.d Conversion yield was determined by capillary electrophoresis analysis. The percentage value corresponds to the amount of 3 in the reaction mixture and the value in parenthesis to the sum of the amount of 3 and 2a + 2b.e Reaction was carried out under microwave irradiation during 2 h at 60 °C. | |||
| 1 | 5 | ACN | 56 (95) |
| 2 | 5 | THFb | 83 (86) |
| 3 | 5 | THFc | 30 (93) |
| 4 | 5 | Dioxane | 25 (56) |
| 5 | 5 | ACN/THFb (1:1) |
100 (100) |
| 6 | 5 | ACN/THFc (1:1) |
43 (100) |
| 7 | 3 | ACN/THFb (1:1) |
100 (100) |
| 8 | 1.5 | ACN/THFb (1:1) |
100 (100) |
| 9 | 1.1 | ACN/THFb (1:1) |
84 (84) |
| 10e | 5 | ACN/THFb (1:1) |
100 (100) |
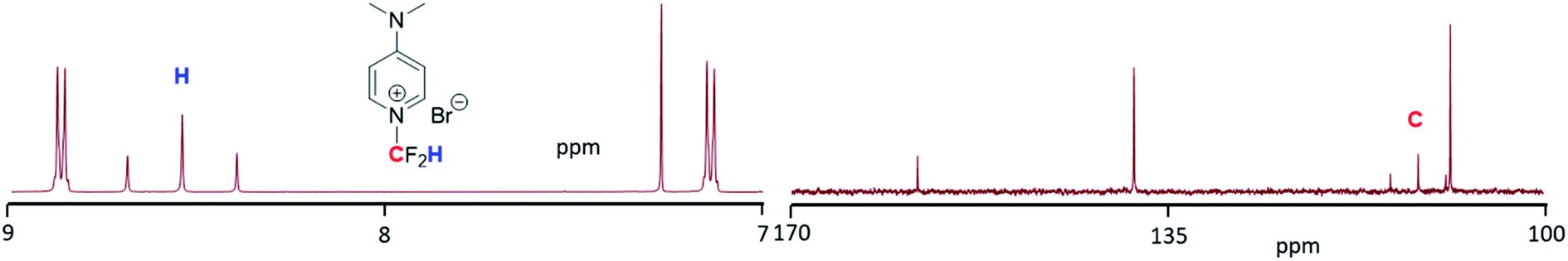 | ||
| Fig. 1 Expansions of the 1H (left) and 13C NMR (right) spectra of 3. | ||
Further evidence of the two-step process for the N-difluoromethylation of pyridine and of the source of proton in the final N-difluoromethylated product (3) was obtained by investigating the reaction of DMAP with 1 by NMR in a 98:2 (v/v) mixture of CD3CN and D2O. Very interestingly, a deuterated analogue of 3 (compound 3-d) was obtained as a major product (97%). Indeed, the CF2D group of 3-d appears in the 19F NMR spectrum (Fig. S5†) as a triplet, which results from the coupling of the fluorine atoms to deuterium. The presence of a doublet (3% relative to the triplet) in the 19F NMR spectrum and of a small triplet in the 1H NMR spectrum (Fig. S6†) confirmed the formation of compound 3, which can be attributable the residual non-deuterated water present both in D2O and in CD3CN. As expected, HR ESI-MS analysis (Fig. S7†) showed that the mass of 3-d was one unit higher than that of 3 (Table S2†) These results confirmed that hydrolysis of the ester intermediate 2a to the carboxylic acid 2b can be mediated by traces of water present in the solvent, which would also be source of the proton of the CF2H group in the final product. The fact that around 20% of compound 2b was spontaneously transformed into the N-difluoromethylated pyridinium salt (3) after standing in CD3CN solution at room temperature (see 1H NMR in Fig. S8†), also confirms the ease with which decarboxylation takes place from the carboxylic acid derivative. This is in contrast with the slow decarboxylation rate described for 2-aryl-2,2-difluoroacetic acid derivatives which require the addition of additives and high temperature heating.17,18
Once established the best combination of solvents, we investigated the effect of reducing the amount of 1 on the reaction efficiency. As shown in Table 1, the reaction between DMAP and 1 still gave good conversion yields at almost equimolar ratios between both reactants (entry 9), being quantitative with 1.5 mol equiv. of 1 after 24 h at 60 °C (entry 8). In all cases, a single product (3) was always detected in the reaction crude by capillary electrophoresis (Fig. S4†). Importantly, the reaction time can be reduced (up to 2 h) by using microwave irradiation (entry 10).
To gain insight into the scope and limitations of this novel synthetic method, the N-difluoromethylation of different pyridine-containing substrates was examined. First, the influence of the steric hindrance around the heterocyclic nitrogen of pyridine was investigated by evaluating the N-difluoromethylation of pyridine (4), 2-picoline (5) and 2,6-lutidine (6) (Fig. 2). All the reactions were carried out under the DMAP-optimized conditions (Table 1, entry 5), and the conversion yield was determined by 1H NMR (Fig. S9–S11†) by comparing the integration of diagnostic peaks in the starting compounds (4–6) and in the N-difluoromethylated analogues (4a–6a). The conversion yield of pyridine and 2-methylpyridine into their N-difluoromethylated derivatives was found 63% and 81%, respectively. On the one hand, the reduced reactivity of pyridine towards 1 compared with DMAP seems to be a consequence of the absence of an electron-donating group that activates the electron-deficient pyridine heterocycle. On the other hand, the electron-donating effect of the methyl group in 2-picoline seems to compensate the increased steric hindrance around the nitrogen atom, leading to a higher conversion into the N-difluoromethylated compound compared with pyridine. In the case of 2,6-lutidine, NMR spectra of the reaction crude showed the lowest conversion yield among the three pyridine derivatives investigated (<5%), which confirms that in this case steric effects prevail over the electron-donating effect of the two methyl substituents. In good agreement with these results, the conversion yield for the N-difluoromethylation of ethyl isonicotinate was found very low (<5%), both under the DMAP-optimized conditions and under microwave irradiation, which indicates that the ethyl bromodifluoroacetate-based method does not tolerate the presence of electron-withdrawing groups in the pyridine.
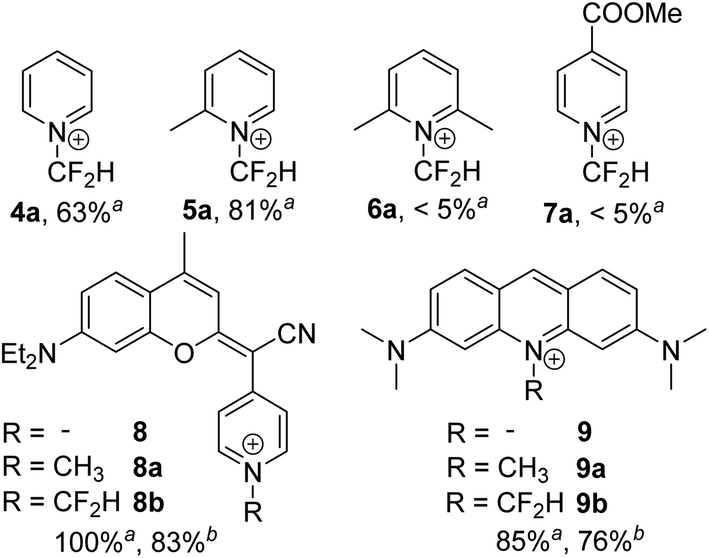 | ||
| Fig. 2 N-Difluoromethylation of pyridine derivatives (4–7) and pyridine-containing fluorophores (8–9). a Conversion yield was determined by 1H NMR (4a–6a) or HPLC-MS (7a, 8b–9b) analysis. b Isolated yield. | ||
N-Alkylation of heterocycles has been widely exploited to modulate the photophysical and physicochemical properties of organic fluorophores,10 and many fluorescent mitochondrial probes incorporate lipophilic positively charged moieties such as N-methyl pyridinium.19 In this context, we envisaged that replacement of the N-methyl group in pyridine-containing fluorophores by N-CF2H could have a strong impact on the spectroscopic properties of the compounds. Our group has recently described a new class of push–pull fluorophores, nicknamed COUPYs, based on the N-alkylation of a pyridine moiety in a novel coumarin scaffold (e.g., compound 8 in Fig. 2) which exhibit several attractive photophysical properties, including emission in the far-red/NIR region.20 Taking into account that absorption and emission maxima can be red-shifted through the incorporation of strong electron-withdrawing groups via N-alkylation of the pyridine heterocycle,20a,b here we focused on investigating the N-difluoromethylation of the parent COUPY scaffold (8) to further explore the scope of the methodology. To our delight, 8 was efficiently N-difluoromethylated by using 1 and the expected COUPY derivative (8b) was isolated by silica column chromatography as a dark blue solid (yield: 83%) and fully characterized by NMR and HRMS. Similarly, N-difluoromethylation of acridine orange (9) with 1 provided a novel N-difluoromethylated analogue (9b) of this commercially available dye. Having at hand compounds 8b and 9b, we investigated the effect of replacing the N-CH3 group in the parent compounds (8a and 9a, respectively; see Fig. 2) by N-CF2H on their spectroscopic properties (Fig. 3). Remarkably, the absorption maximum of 8b (λabs = 580 nm) was considerably red-shifted (ca. 32 nm) with respect to the parent compound 8a (λabs = 548 nm), and the same tendency was found with the acridine derivatives (λabs = 497 nm for 9a and λabs = 532 nm for 9b). The emission maxima of the compounds were also red-shifted with respect to the parent compounds (λem = 638 nm for 8b vs. λem = 603 nm for 8a, and λem = 565 nm for 9b vs. λem = 524 nm for 9a), which reproduces the effect of replacing the N-CH3 group with N-CF2H on the compounds' absorption maxima.
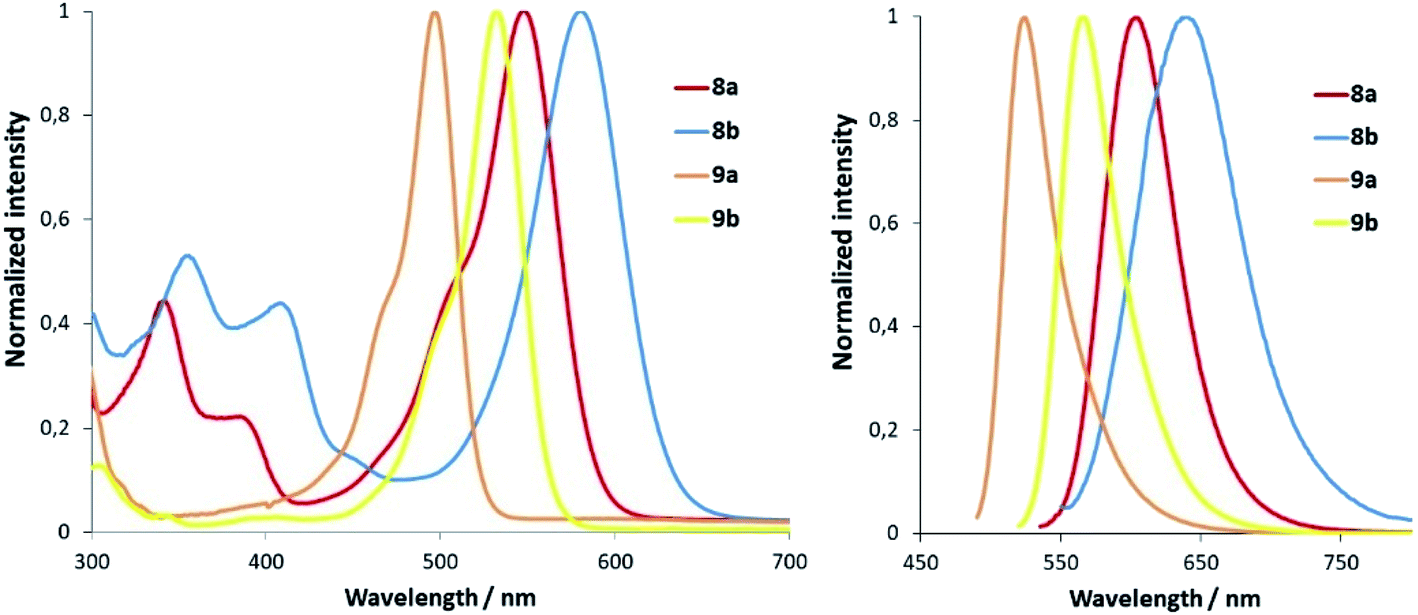 | ||
| Fig. 3 Comparison of the normalized absorption (left) and fluorescence (right) spectra of N-CH3 fluorophores (8a–9a) and their N-CF2H analogues (8b–9b) in ACN. | ||
Finally, it is worth noting that during the evaluation of the substrate scope of the N-difluoromethylation method with pyridine derivatives containing electron-donating groups, an unexpected result was revealed. HPLC-MS analysis for the reaction of 4-methoxypyridine (10) with 1 showed the disappearance of 10 but neither the major product (83%) nor the minor product (17%) were the expected N-difluoromethylated salt (10a) (Scheme 3). MS data of the major compound was consistent with the formation of the N-difluoromethylated pyridin-4-one (10b), and the minor product was characterized as the N-methylated derivative of 4-methoxypyridine (10c). As a possible mechanism, we postulate the nucleophilic attack of the bromide anion on the methoxy substituent of 10a, which would generate 10b and CH3Br. The formation of 10c can be explained by N-methylation of unreacted 4-methoxypyridine with methyl bromide (Scheme 3). A similar mechanism has been proposed by Ma and collaborators for the conversion of 4-methoxypyridines into N-methyl-4-pyridones.21 Compound 10b was isolated by silica column chromatography (55% yield) and fully characterized by NMR spectroscopy and HR ESI-MS. 1H and 19F-NMR spectra showed the expected multiplicities for the proton and fluorine atoms of the CF2H group (triplet and doublet, respectively), and 13C-NMR spectrum provided further evidence of the structure of the pyridin-4-one moiety: in addition to the expected triplet for the CF2H group, a characteristic peak due to the carbonyl function was observed around 180 ppm. Similarly, reaction of 4-methoxyquinoline (11) with ethyl bromodifluoroacetate provided the corresponding N-difluoromethylated 4-quinolin-4-one (11a) with a 60% yield after purification (Scheme 3).
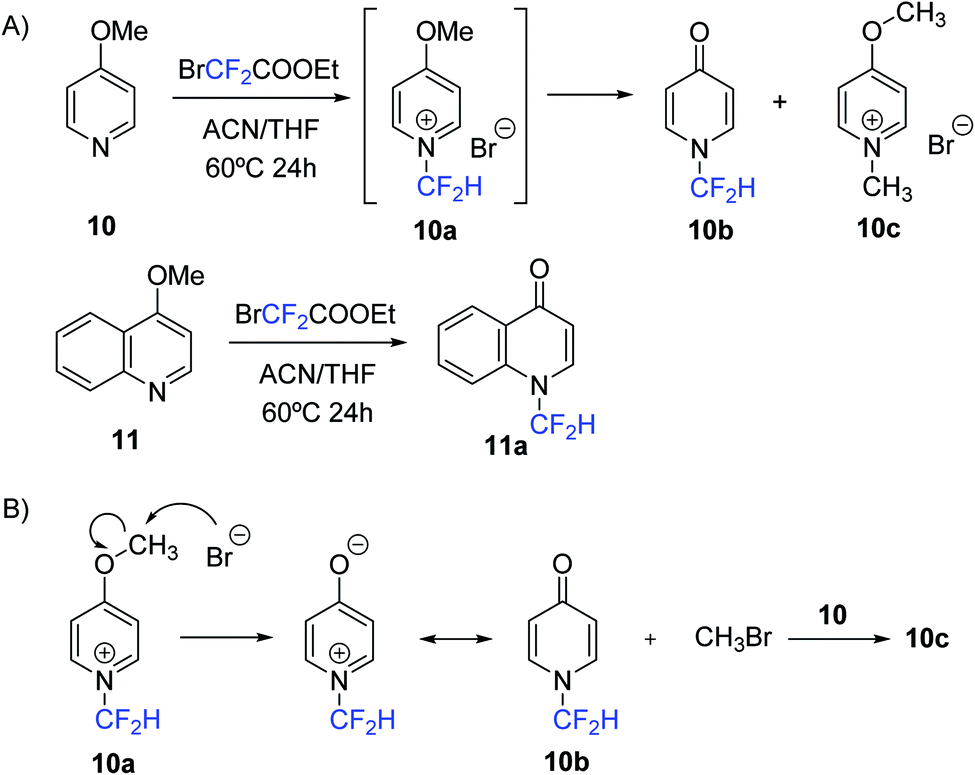 | ||
| Scheme 3 Synthesis of N-difluoromethylated 4-pyridone and 4-quinolone (A) and proposed pathway for the formation of compound 10b and 10c in the reaction of 4-methoxypyridine with 1 (B). | ||
Conclusions
In summary, a novel transition metal-free synthetic method for the N-difluoromethylation of pyridine-containing substrates has been developed. The procedure involves the use of a cheap, safe and readily available reagent, ethyl bromodifluoroacetate (1), which provides the corresponding N-difluoromethylated pyridinium salts in moderate to good yields after reaction with the parent pyridine compounds. The formation of N-difluoromethylated pyridines involves a two-step process in which N-alkylation by ethyl bromodifluoroacetate is followed by in situ ester hydrolysis and decarboxylation By investigating the substrate scope and limitations of the method with a series of pyridine-containing molecules, we have observed that steric and electronic effects of the substituents in the heterocycle play an important role both on the effectiveness of the reaction and on the structure of the final compound. Remarkably, the presence of a methoxy group at the para position relative to the nitrogen atom of the pyridine ring results in the formation of a N-difluoromethylated pyridin-4-one structure, while the presence of electron-withdrawing groups difficult the reaction. This new facile N-difluoromethylating method should allow the synthesis of novel biologically active compounds in which the N-methyl group in N-methyl pyridinium and N-methyl-4-pyridone/quinolone moieties could be easily replaced by N-CF2H. In addition, we anticipate that N-difluoromethylation of conventional pyridine-containing organic fluorophores will allow to improve their spectroscopic properties, opening the door to novel fluorescent probes for bioimaging applications. Work is in progress in our laboratory to apply this new method to the synthesis of other N-difluoromethylated heterocycles, including imidazole, indole and pyrrol.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funds from the Spanish Government (MCIU/AEI/FEDER, UE; grant CTQ2017-84779-R) and the Generalitat de Catalunya (2017 DI 072). The authors acknowledge helpful assistance of Josep Martí (capillary electrophoresis), Dr Francisco Cárdenas (NMR) and Dr Irene Fernández and Laura Ortiz (MS) from CCiTUB. A. R. and A. G. were recipient fellows of the University of Barcelona.Notes and references
- (a) T. Liang, C. N. Neumann and T. Ritter, Introduction of Fluorine and Fluorine-Containing Functional Groups, Angew. Chem., Int. Ed., 2013, 32, 8214 CrossRef PubMed; (b) N. A. Meanwell, Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design, J. Med. Chem., 2018, 61, 5822 CrossRef CAS PubMed.
- (a) A. F. Brooks, J. J. Topczewski, N. Ichiishi, M. S. Sanford and P. H. J. Scott, Late-stage [18F] fluorination: new solutions to old problems, Chem. Sci., 2014, 5, 4545 RSC; (b) I. Tirotta, V. Dichiarante, C. Pigliacelli, G. Cavallo, G. Terraneo, F. B. Bombelli, P. Metrangolo and G. Resnati, 19F Magnetic Resonance Imaging (MRI): From Design of Materials to Clinical Applications, Chem. Rev., 2015, 115, 1106 CrossRef CAS PubMed.
- (a) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed; (b) H. Mei, J. Han, S. Fustero, M. Medio-Simon, D. M. Sedgwick, C. Santi, R. Ruzziconi and V. A. Soloshonok, Fluorine-Containing Drugs Approved by the FDA in 2018, Chem.–Eur. J., 2019, 25, 11797 CrossRef CAS PubMed; (c) B. G. de la Torre and F. Albericio, The Pharmaceutical Industry in 2019. An Analysis of FDA Drug Approvals from the Perspective of Molecules, Molecules, 2020, 25, 745 CrossRef CAS PubMed.
- C. D. Sessler, M. Rahm, S. Becker, J. M. Goldberg, F. Wang and S. J. Lippard, CF2H, a Hydrogen Bond Donor, J. Am. Chem. Soc., 2017, 139, 9325 CrossRef CAS PubMed.
- (a) N. A. Meanwell, Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed; (b) Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, Difluoromethyl Bioisostere: Examining the “Lipophilic Hydrogen Bond Donor” Concept, J. Med. Chem., 2017, 60, 797 CrossRef CAS.
- (a) Y. Lu, C. Liu and Q.-T. Chen, Recent Advances in Difluoromethylation Reaction, Curr. Org. Chem., 2015, 19, 1638 CrossRef CAS; (b) D. E. Yerien, S. Barata-Vallejo and A. Postigo, Difluoromethylation Reactions of Organic Compounds, Chem.–Eur. J., 2017, 23, 14676 CrossRef CAS PubMed; (c) Z. Feng, Q.-Q. Min, X.-P. Fu, L. An and X. Zhang, . Chlorodifluoromethane-triggered formation of difluoromethylated arenes catalysed by palladium, Nat. Chem., 2017, 9, 918 CrossRef CAS PubMed; (d) Z. Feng, Y.-L. Xiao and X. Zhang, Transition-Metal (Cu, Pd, Ni)-Catalyzed Difluoroalkylation via Cross-Coupling with Difluoroalkyl Halides, Acc. Chem. Res., 2018, 51, 2264 CrossRef CAS PubMed; (e) X.-P. Fu, X.-S. Xue, X.-Y. Zhang, Y.-L. Xiao, S. Zhang, Y.-L. Guo, X. Leng, K. N. Houk and X. Zhang, Controllable catalytic difluorocarbene transfer enables access to diversified fluoroalkylated arenes, Nat. Chem., 2019, 11, 948 CrossRef CAS PubMed.
- (a) P. Dai, X. Yu, P. Teng, W.-H. Zhang and C. Deng, Visible-Light- and Oxygen-Promoted Direct Csp2-H Radical Difluoromethylation of Coumarins and Antifungal Activities, Org. Lett., 2018, 20, 6901 CrossRef CAS PubMed; (b) A. Lemos, C. Lemaire and A. Luxen, Progress in Difluoroalkylation of Organic Substrates by Visible Light Photoredox Catalysis, Adv. Synth. Catal., 2019, 361, 1500 CrossRef CAS; (c) Y. Huang, Z. Lin, Y. Chen, S. Fang, H. Jiang and W. Wu, Transition-metal-free N-difluoromethylation of hydrazones with TMSCF2Br as the difluoromethylation reagent, Org. Chem. Front., 2019, 6, 2462 RSC; (d) X. Xu and F. Liu, Transition-metal-free radical tri-/difluoromethylation of N,N-dialkylhydrazones with sodium sulfinates, Org. Chem. Front., 2017, 4, 2306 RSC.
- Y. Higashio and T. Shoji, Heterocyclic compounds such as pyrrole, pyridines, pyrrolidine, piperidine, indole, imidazol and pyrazines, Appl. Catal., A, 2004, 260, 251 CrossRef CAS.
- E. Vitaku, D. T. Smith and J. T. Njardarson, Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceutical, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- (a) Q. Zheng and L. D. Lavis, Development of photostable fluorophores for molecular imaging, Curr. Opin. Chem. Biol., 2017, 39, 32 CrossRef CAS PubMed; (b) L. G. Freidus, P. Pradeep, P. Kumar, Y. E. Choonara and V. Pillay, Alternative fluorophores designed for advanced molecular imaging, Drug Discovery Today, 2018, 23, 115 CrossRef CAS PubMed.
- (a) T. D. M. Pham, Z. M. Ziora and M. A. T. Blaskovich, Quinolone antibiotics, MedChemComm, 2019, 10, 1719 RSC; (b) P. Ghosh and S. Das, Synthesis and Functionalization of 4-Quinolones – A Progressing Story, Eur. J. Org. Chem., 2019, 4466 CrossRef CAS.
- A. Kolomeitsev, R.-M. Schoth, E. Lork and G.-V. Röschenthaler, Facile new method for synthezising N-polyfluoroalkylated heterocycles – molecular structure of N-(bromodifluoromethyl)-4-dimethylaminopyridinium bromide, Chem. Commun., 1996, 335 RSC.
- Y. Zafrani, D. Amir, L. Yehezkel, M. Madmon, S. Saphier, N. Karton-Lifshin and E. Gershonov, Chemoselective N-Difluoromethylation of Functionalized Tertiary Amines, J. Org. Chem., 2016, 81, 9180 CrossRef CAS.
- J. Tani, Y. Mushika and T. Yamaguchi, Studies on biologically active halogenated compounds. IV. Synthesis and antibacterial activity of fluorinated quinoline derivatives, Chem. Pharm. Bull., 1982, 30, 3530 CrossRef CAS PubMed.
- Y. Li, M. Rao, Z. Fan, B. Nian, Y. Yuan and J. A. Cheng, A visible-light-irradiated electron donor-acceptor complex-promoted radical reaction system for the CH perfluoroalkylation of quinolin-4-ols, Tetrahedron Lett., 2019, 60, 151046 CrossRef.
- (a) J. Hu, W. Zhang and F. Wang, Selective difluoromethylation and monofluoromethylation reactions, Chem. Commun., 2009, 7465 RSC; (b) M. Ando, T. Wada and N. Sato, Facile One-Pot Synthesis of N-Difluoromethyl-2-pyridone Derivatives, Org. Lett., 2006, 8(17), 3805 CrossRef CAS PubMed.
- (a) X. Ma, S. Mai, Y. Zhou, G.-J. Cheng and Q. Song, Dual role of ethyl bromodifluoroacetate in the formation of fluorine-containing heteroaromatic compounds, Chem. Commun., 2018, 54, 8960 RSC; (b) Z. Feng, Q.-Q. Min and X. Zhang, Access to Difluoromethylated Arenes by Pd-Catalyzed Reaction of Arylboronic Acids with Bromodifluoroacetate, Org. Lett., 2019, 18, 44 CrossRef PubMed.
- K. Fujikawa, Y. Fujioka, A. Kobayashi and H. Amii, A New Method for Aromatic Difluoromethylation: Copper-Catalyzed Cross-Coupling and Decarboxylation Sequence from Aryl Iodides, Org. Lett., 2011, 13, 5560 CrossRef CAS PubMed.
- W. Xu, Z. Zeng, J.-H. Jiang, Y.-T. Chang and L. Yuan, Discerning the Chemistry in Individual Organelles with Small-Molecule Fluorescent Probes, Angew. Chem., Int. Ed., 2016, 55, 13658 CrossRef CAS PubMed.
- (a) A. Gandioso, R. Bresolí-Obach, A. Nin-Hill, M. Bosch, M. Palau, A. Galindo, S. Contreras, A. Rovira, C. Rovira, S. Nonell and V. Marchán, Redesigning the Coumarin Scaffold into Small Bright Fluorophores with Far-Red to Near-Infrared Emission and Large Stokes Shifts Useful for Cell Imaging, J. Org. Chem., 2018, 83, 1185 CrossRef CAS PubMed; (b) A. Gandioso, M. Palau, R. Bresolí-Obach, A. Galindo, A. Rovira, M. Bosch, S. Nonell and V. Marchán, High Photostability in Nonconventional Coumarins with Far-Red/NIR Emission through Azetidinyl Substitution, J. Org. Chem., 2018, 83, 11519 CrossRef CAS PubMed; (c) A. Rovira, A. Gandioso, M. Goñalons, A. Galindo, A. Massaguer, M. Bosch and V. Marchán, Solid-Phase Approaches for Labeling Targeting Peptides with Far-Red Emitting Coumarin Fluorophores, J. Org. Chem., 2019, 84, 1808 CrossRef CAS PubMed; (d) V. Novohradsky, A. Rovira, C. Hally, A. Galindo, G. Vigueras, A. Gandioso, M. Svitelova, R. Bresolí-Obach, H. Kostrhunova, L. Markova, J. Kasparkova, S. Nonell, J. Ruiz, V. Brabec and V. Marchán, Towards Novel Photodynamic Anticancer Agents Generating Superoxide Anion Radicals: A Cyclometalated IrIII Complex Conjugated to a Far-Red Emitting Coumarin, Angew. Chem., Int. Ed., 2019, 58, 6311 CrossRef CAS PubMed; (e) A. Rovira, M. Pujals, A. Gandioso, M. López-Corrales, M. Bosch and V. Marchán, Modulating Photostability and Mitochondria Selectivity in Far-Red/NIR Emitting Coumarin Fluorophores through Replacement of Pyridinium by Pyrimidinium, J. Org. Chem., 2020, 85, 6086 CrossRef CAS.
- X. Yi, J. Chen, X. Xu and Y. Ma, Solvent and substituent effects on the conversion of 4-methoxypyridines to N-methyl-4-pyridones, Synth. Commun., 2017, 47, 872 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of all compounds. See DOI: 10.1039/d0ra06322c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |