DOI:
10.1039/D0RA06251K
(Paper)
RSC Adv., 2020,
10, 32927-32937
Retracted Article: Design and synthesis of magnetic Fe3O4@NFC-ImSalophCu nanocatalyst based on cellulose nanofibers as a new and highly efficient, reusable, stable and green catalyst for the synthesis of 1,2,3-triazoles†
Received
23rd July 2020
, Accepted 26th August 2020
First published on 4th September 2020
Abstract
The Fe3O4@NFC-ImSalophCu catalyst was used as a highly stable, reusable, active, green catalyst for the synthesis of 1,2,3-triazoles via one-pot three-component reaction of phenacyl bromides, sodium azide and alkynes. A Cu(II)–Schiff base complex containing an imidazolium ionic phase was prepared and decorated on core shell Fe3O4@NFC magnetic nanoparticles (Fe3O4@NFC-ImSalophCu) and was used as an efficient catalyst. The heterogeneous catalyst was characterized by FT-IR spectroscopy, FE-SEM, TEM, XRD spectroscopy, EDX spectroscopy, VSM, and ICP spectroscopy. This catalyst shows the dual function of the metal sites and imidazolium moieties. The catalytic system mentioned above also showed excellent activity in the synthesis of bis 1,4-disubstituted 1,2,3-triazoles. Moreover, the catalyst could be recycled and reused for four cycles without any decrease in its catalytic activity.
Introduction
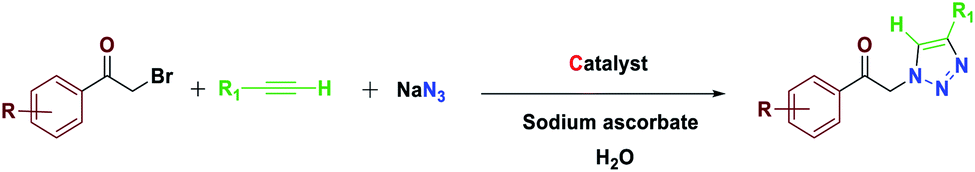
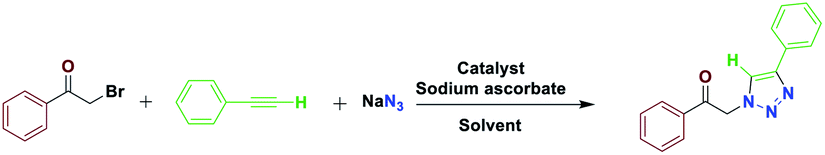
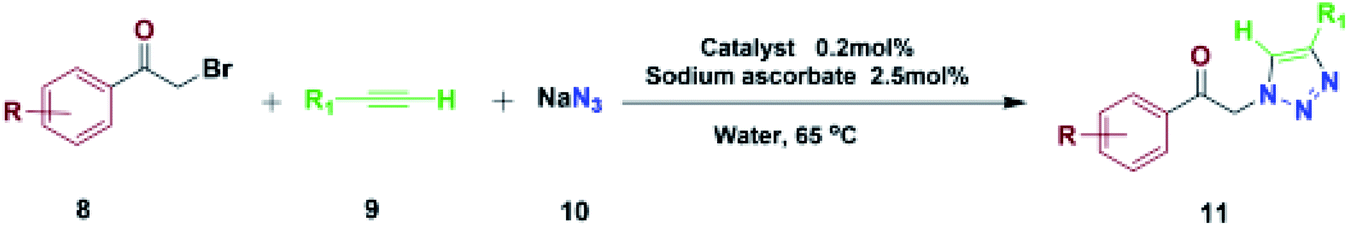
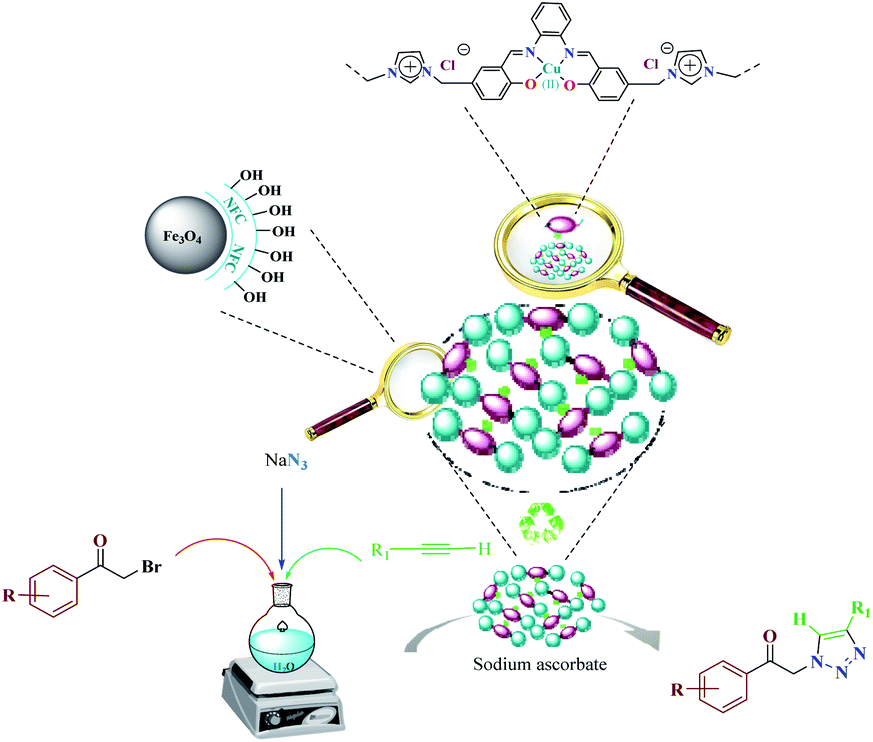
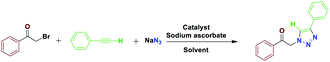
The most important advances in areas of green organic chemical reactions are the use of green catalysts. In green chemistry, the use of efficient and reusable catalysts, as well as the use of benign solvents (e.g. ionic liquids, water), is significant. Green chemistry is about designing a process that minimizes the use and production of dangerous substances.1 Iron oxide or Fe3O4 magnetite nanoparticles (MNPs) have grown increasing interest recently due to their unique features including high thermal and mechanical stability, low preparation cost and suitability for large-scale production. Also, their paramagnetic nature allows their simple and efficient separation from the reaction mixture required in the separation of heterogeneous catalysts. Magnetite nanoparticles (Fe3O4) can be easily separated from the reaction mixture by using just an external magnet.2–5 However, the serious drawback of magnetic nanoparticles (MNPs) is their nakedness. The naked, magnetic nanoparticles (MNPs) are mainly subject to aggregation in the reaction mixture due to the small particles spacing between them, high surface energy and the presence of van der Waals forces.6 This accumulation affects their unique properties, which are accompanied by catalytic reactions. To eliminate this problem, MNPs are repeatedly modified to preserve the properties of the nanoparticles and to maintain their scattered active sites using appropriate stabilizing coatings to prevent irreversible accumulation, the outer shells of the iron oxides being investigated, such as polymers,7 zeolites,8 titanium dioxide,9 silica10 and carbon.11 Recently, applications of biopolymer-based nanocomposites and their chemical properties have been developed in all fields of knowledge. In chemistry, due to their biocompatibility, catalytic degradability, thermal and chemical stability and some of the functional properties presented as solid support and or catalyst are widely used.12 Among the natural polymers, cellulose is the most abundant biodegradable material due to its non-petroleum based, natural carbon content and environmental health, humans and other organisms with many important commercial uses e.g., paper, food, fabric, cosmetics (Pharmaceuticals) are very important. Cellulose nanocomposites and their derivatives increase the attractiveness and usefulness of related materials in science and technology.13 Due to its unique surface property, it grouped its many hydroxyls, chemical, and thermal stability groups, toxicity, high catalytic activity, as well as excellent availability and average cost.14 Due to their high viscosity, ionic liquids (ILs) work only as a thin layer of them (diffusion layer) during the reaction processes and therefore their application in an organic reaction is limited. However, they are very expensive, and to overcome this obstacle, nano catalysts with ionic portions have been introduced which covers the immobilization and recycling concerns. Ionic liquids have been used as a solvent in the reaction medium with low vapor pressure.15–17 Accordingly, this strategy increases the “ionophilicity” of the reagent in the reaction medium (which is generally thought to facilitate the exchange of the counter ion with a combination of reagents).18,19 Also, catalysts having an imidazolium group can play a base role and also leach the metal in a reaction medium.20–22 In recent years, multi component reactions (MCRs) have gained more efficient as a synthetic strategy to assemble molecular complexity with attractive biological features through the simple and readily available materials in a one-pot operation.23 Among them, the Huisgen 1,3-dipolar cycloaddition of azides and alkynes resulting in 1,2,3-triazoles play urgent functions in organic chemistry, agrochemicals, medicinal chemistry, and dyes because of their ease of preparation by “click chemistry”.24,25 1,2,3-Triazoles are also attached with a wide range of biological and pharmacological properties such as antiviral,26 antitubercular,27 antifungal,28 anticancer29 and activities against Gram-positive bacteria.30 In general, these compounds are prepared through the coupling reaction between alkynes and azides at specified temperatures and by using catalysis.31 In many transition metals, recently copper has been used as a catalyst in organic synthesis, the groups of Sharpless and Meldal have reported the dramatic rate enhancement and improved regioselectivity of the Huisgen 1,3-dipolar cycloaddition reaction of a reaction of sodium azide with terminal alkynes to yield 1,4-disubstituted products.32–36 Among the solvents usually used in organic synthesis, water is most economical, quite non-toxic, safest and most environmentally friendly medium. Therefore, water because of its strong hydrogen-bonding potential shows a better selectivity and activity compared to the other conventional organic solvents.37 Herein, we report the preparation of novel and recyclable catalyst system composing of a magnetic Fe3O4 core and cellulose shell along with ligand. Use of readily available and economic nanofiber cellulose in water to promote the catalytic system because of ligand dual performance, ionic liquid/salophen–Cu and higher hippophilic character compared to the traditional Cu(II)/ascorbate system. In this study, the main goal we that a new class of Fe3O4@NFC-ImSalophCu catalysts could effectively promote triazoles with safe solvent. This catalyst greatly reduced the cost of this transformation, and the resulted operation and work-up procedures were greatly simplified. The catalyst Fe3O4@NFC-ImSalophCu showed high activity with the aid of ionic liquid and salophen/Cu at 65 °C temperature. Furthermore, the catalyst was examined in the reaction of acetylene, azide, and phenacyl bromide, in water as a model reaction, which proceeded smoothly to afford the corresponding, 1,4-substituted 1,2,3-triazole as the only regioselective product (Scheme 1).
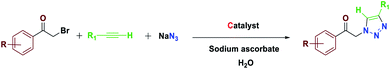 |
| | Scheme 1 Synthesis of 1,2,3-triazoles promoted by Fe3O4@NFC-ImSalophCu nanocatalyst. | |
Result and discussion
Catalyst characterization
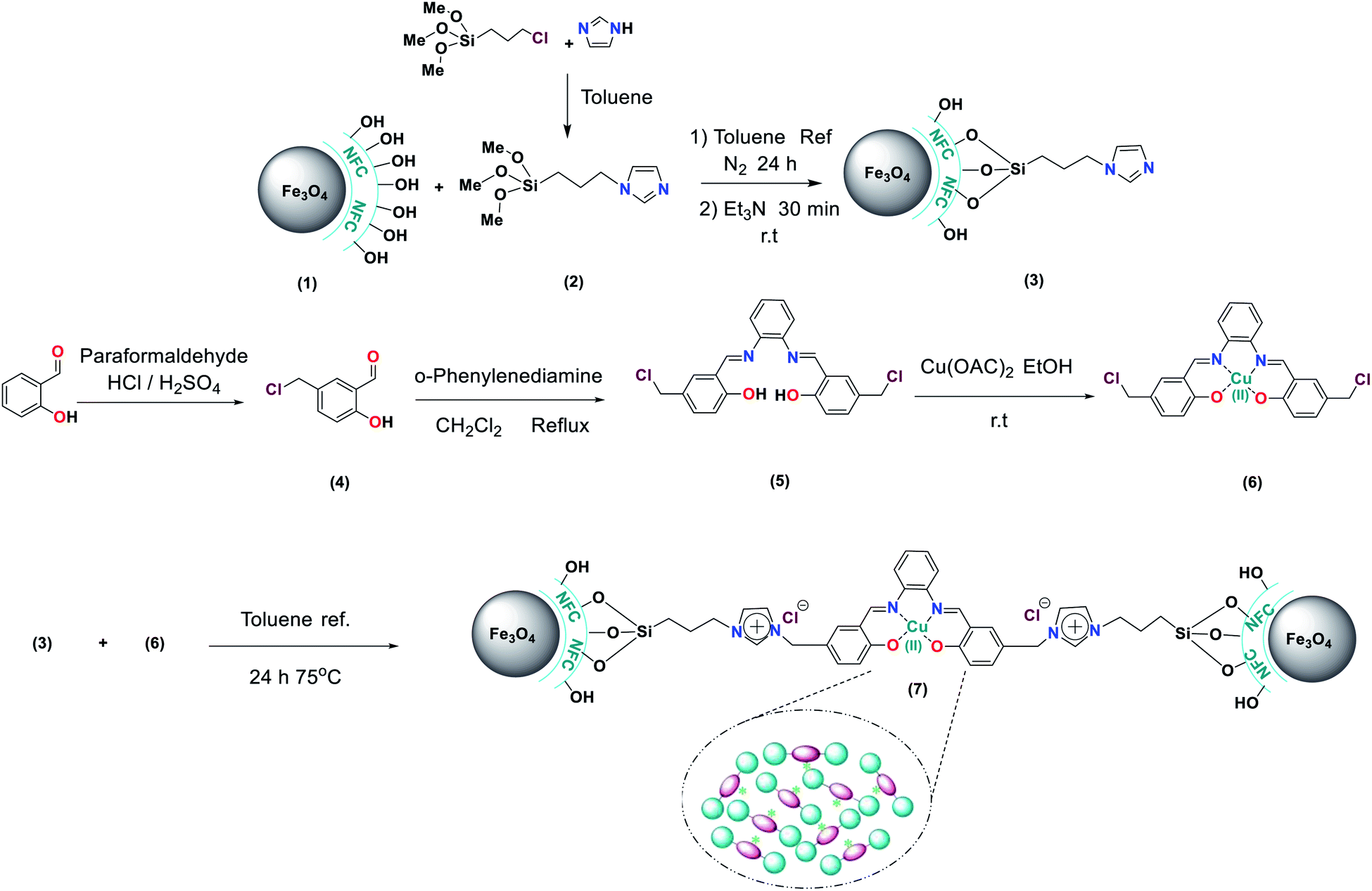
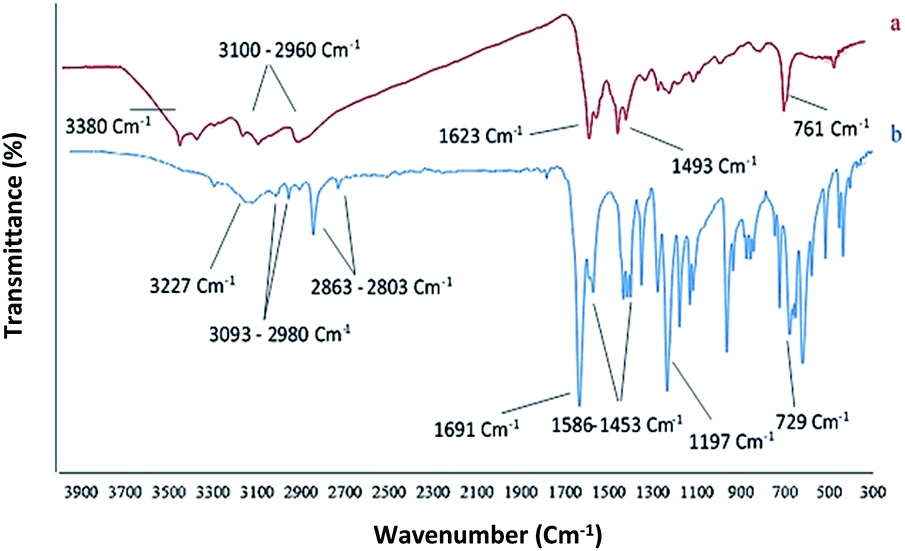
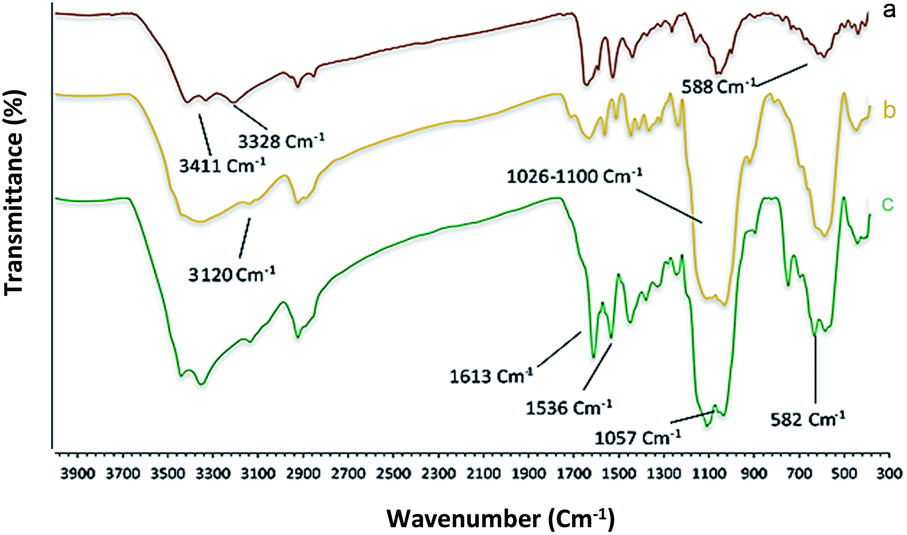
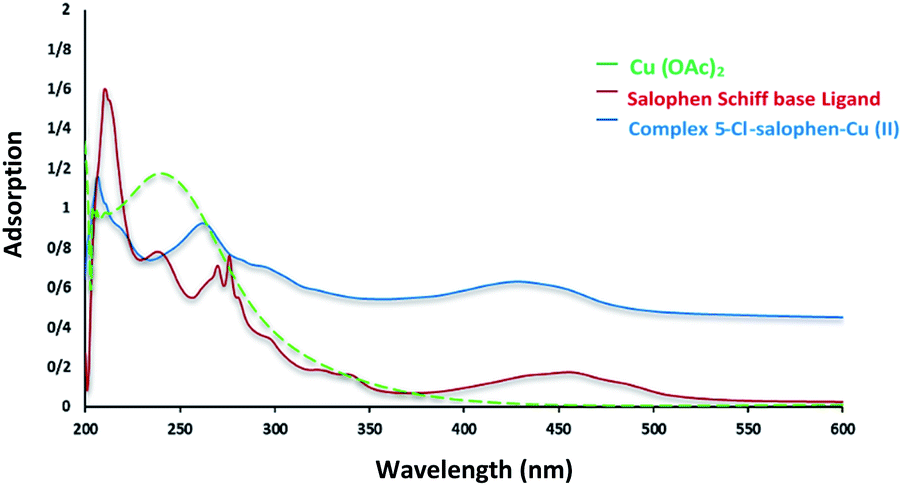
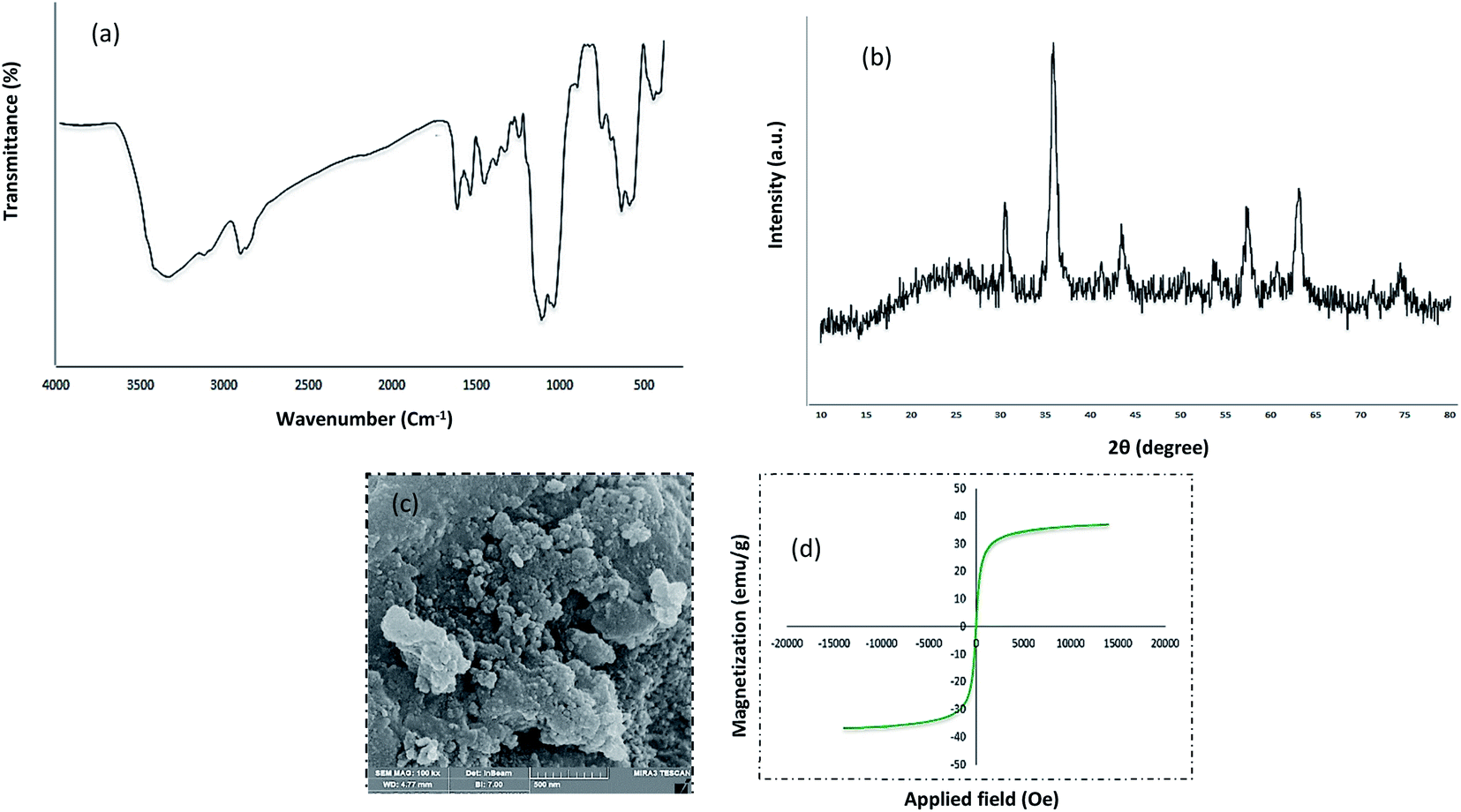
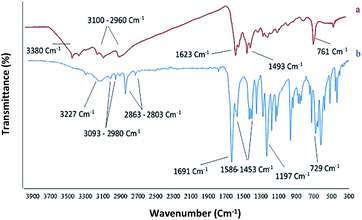
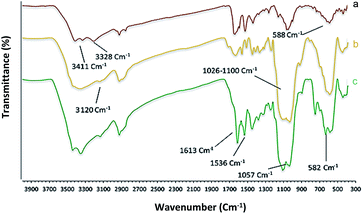
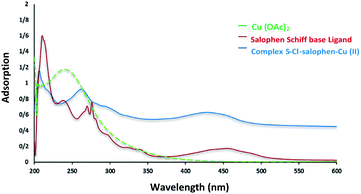
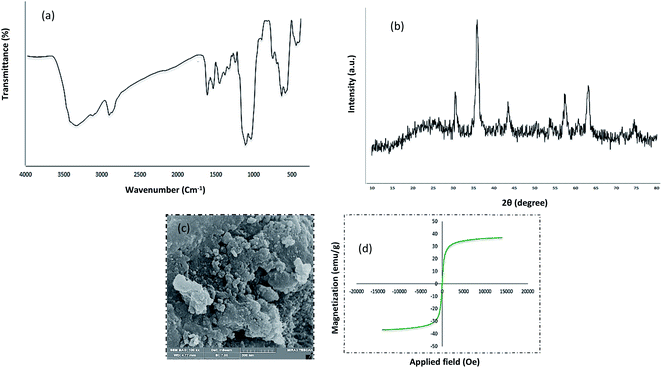
Chemistry is a very innovative and fundamental science that creates value-added materials that are referred to as green chemistry. So, we designed the core–shell nanocatalyst and was synthesized by the method outlined in Schemes 2 and 3. Given the importance of the cellulose, including its biodegradability and naturalness, we have made magnetic cellulose a base for stabilizing heterogeneous catalysts Scheme 3. In the second step, we prepared the salophen open shift and then placed on the magnetic cellulose substrate containing the imidazole group. Owing to the advisable need for clean and green recovery of the nanocatalyst, Fe3O4@NFC-ImSalophCu was used in a model reaction of α-bromo ketone, sodium azide, and phenylacetylene which led to a certain product called 1,2,3-triazole. With regarding the advantages of this catalyst such as easy recovery, non-toxicity, high catalytic efficiency, easy synthesizing and reusability, this catalyst can be a fit choice in catalysis. Finally, this catalyst was characterized by different techniques including ICP, XRD, EDX, IR, TEM, SEM, VSM and UV-Vis. FT-IR spectra of the compounds 1, 3, 4, 5 and Fe3O4@NFC-ImSalophCu are shown in Fig. 1a, b and 2c–e. As shown in Fig. 1a, the characteristic peaks at 729 cm−1 (C–Cl stretching vibration), 1197 cm−1 (C–O stretching vibration), 1453 cm−1 and 1586 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C aromatic stretching vibration), 1691 cm−1 (CO stretching vibration), and 2863–2803 cm−1 (C–H aldehyde stretching vibration), 2980 cm−1 and 3093 cm−1 (C–H aliphatic and aromatic stretching vibration), 3227 cm−1 (O–H stretching)confirms the structure of 1 (Fig. 1a).38,39 FT-IR spectrum of compound 4 (Fig. 1b), indicates the Schiff base reaction between o-phenyldiamine and 5-chlorosalicylaldehyde, the characteristic peaks at 761 cm−1 (C–Cl stretching), 1493 cm−1 (CC aromatic stretching vibration), 1623 cm−1 (CN stretching vibration) 2960–3100 cm−1 (C–H aliphatic and aromatic stretching vibration), and 3380 cm−1 (O–H stretching) confirms the structure of (Fig. 1b). Fig. 2c, shows the FT-IR spectrum of Fe3O4 nanoparticles with two characteristic peaks at 3411 cm−1 and 588 cm−1 related to O–H and Fe–O stretching bands, respectively, that approves the formation of Fe3O4 nanoparticles. The strong peak at 3328 cm−1 shows the (O–H stretching vibrations) of cellulose nanofibers (Fig. 2c). Fig. 2d, shows the FT-IR spectrum strong peak of Si–O at 1026 cm−1 and also a new sharp peak appeared at 1100 cm−1 was assigned to C–N stretching vibration and 3120 cm−1 C–H aromatic stretching vibration referring to the imidazolium linkage bond to the aromatic ring (Fig. 2d). These absorption bonds confirm the successful preparation of the catalyst. Complexation of copper ions to ligand 5 causes the imine bond absorption (1623 cm−1) to lower wavenumbers (1613 cm−1) by about 10 cm−1, indicating the participation of azomethine nitrogen in bonding with metal ion and confirming the coordination of ν(CV) stretch to the metal via a nitrogen atom. Also, two absorption bonds at 439 and 631 cm−1 could be assigned to Cu–O and Cu–N stretching, respectively (Fig. 2e). The main absorption bonds at 582 cm−1 (Fe–O), 1057 cm−1 (Si–O), 1536 cm−1 (CC), 1613 cm−1 (CN) in the FT-IR spectrum of Fe3O4@NFC-ImSalophCu, represent the functionalization of Fe3O4@NFC nanoparticles with Cu(II) complex 5 (Fig. 2e). The electronic spectra for samples were scrolled at a region of 200–600 nm in ethanol solvent (Fig. 3). Shows UV-Vis spectra for Cu(OAc)2, salophen Schiff base ligand, 5-Cl-salophen–Cu(II) complex. The band seen in the spectrum of the salophen was observed at higher energy 200–300 nm and 450 nm it was attributed to π–π* transitions associated with the benzene ring (intra ligand charge transfer) and n–π* transition CN bond for imine bond. In the UV-Vis spectrum the ligand λmax and adsorption intensity appear in 460 nm, while the 5-Cl-salophen–Cu(II) λmax is in region 430 nm, resulting in UV-Vis spectrum of Schiff base ligand to Cu(II) caused the reduction of absorption intensity of n–π* for CN bond and π–π* transitions for benzene ring (Fig. 3), which confirmed the successful chelation of Cu to the ligand salophen.
C aromatic stretching vibration), 1691 cm−1 (CO stretching vibration), and 2863–2803 cm−1 (C–H aldehyde stretching vibration), 2980 cm−1 and 3093 cm−1 (C–H aliphatic and aromatic stretching vibration), 3227 cm−1 (O–H stretching)confirms the structure of 1 (Fig. 1a).38,39 FT-IR spectrum of compound 4 (Fig. 1b), indicates the Schiff base reaction between o-phenyldiamine and 5-chlorosalicylaldehyde, the characteristic peaks at 761 cm−1 (C–Cl stretching), 1493 cm−1 (CC aromatic stretching vibration), 1623 cm−1 (CN stretching vibration) 2960–3100 cm−1 (C–H aliphatic and aromatic stretching vibration), and 3380 cm−1 (O–H stretching) confirms the structure of (Fig. 1b). Fig. 2c, shows the FT-IR spectrum of Fe3O4 nanoparticles with two characteristic peaks at 3411 cm−1 and 588 cm−1 related to O–H and Fe–O stretching bands, respectively, that approves the formation of Fe3O4 nanoparticles. The strong peak at 3328 cm−1 shows the (O–H stretching vibrations) of cellulose nanofibers (Fig. 2c). Fig. 2d, shows the FT-IR spectrum strong peak of Si–O at 1026 cm−1 and also a new sharp peak appeared at 1100 cm−1 was assigned to C–N stretching vibration and 3120 cm−1 C–H aromatic stretching vibration referring to the imidazolium linkage bond to the aromatic ring (Fig. 2d). These absorption bonds confirm the successful preparation of the catalyst. Complexation of copper ions to ligand 5 causes the imine bond absorption (1623 cm−1) to lower wavenumbers (1613 cm−1) by about 10 cm−1, indicating the participation of azomethine nitrogen in bonding with metal ion and confirming the coordination of ν(CV) stretch to the metal via a nitrogen atom. Also, two absorption bonds at 439 and 631 cm−1 could be assigned to Cu–O and Cu–N stretching, respectively (Fig. 2e). The main absorption bonds at 582 cm−1 (Fe–O), 1057 cm−1 (Si–O), 1536 cm−1 (CC), 1613 cm−1 (CN) in the FT-IR spectrum of Fe3O4@NFC-ImSalophCu, represent the functionalization of Fe3O4@NFC nanoparticles with Cu(II) complex 5 (Fig. 2e). The electronic spectra for samples were scrolled at a region of 200–600 nm in ethanol solvent (Fig. 3). Shows UV-Vis spectra for Cu(OAc)2, salophen Schiff base ligand, 5-Cl-salophen–Cu(II) complex. The band seen in the spectrum of the salophen was observed at higher energy 200–300 nm and 450 nm it was attributed to π–π* transitions associated with the benzene ring (intra ligand charge transfer) and n–π* transition CN bond for imine bond. In the UV-Vis spectrum the ligand λmax and adsorption intensity appear in 460 nm, while the 5-Cl-salophen–Cu(II) λmax is in region 430 nm, resulting in UV-Vis spectrum of Schiff base ligand to Cu(II) caused the reduction of absorption intensity of n–π* for CN bond and π–π* transitions for benzene ring (Fig. 3), which confirmed the successful chelation of Cu to the ligand salophen.
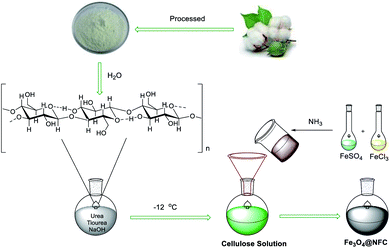 |
| | Scheme 2 Synthesis of the Fe3O4@NFC. | |
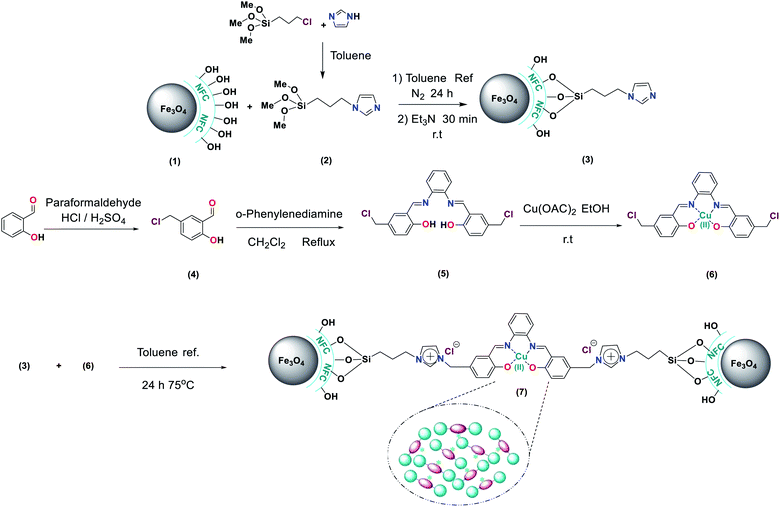 |
| | Scheme 3 Design and synthesis of the Fe3O4@NFC-ImSalophCu. | |
 |
| | Fig. 1 FT-IR spectra of (a) 4, (b) Schiff base. | |
 |
| | Fig. 2 (a) Fe3O4@NFC, (b) Fe3O4@NFC-Im (c) Fe3O4@NFC-ImSalophCu. | |
 |
| | Fig. 3 UV-Vis spectra of Cu(OAc)2, salophen Schiff base ligand, 5-Cl-salophen–Cu(II) complex in EtOH solvent. | |
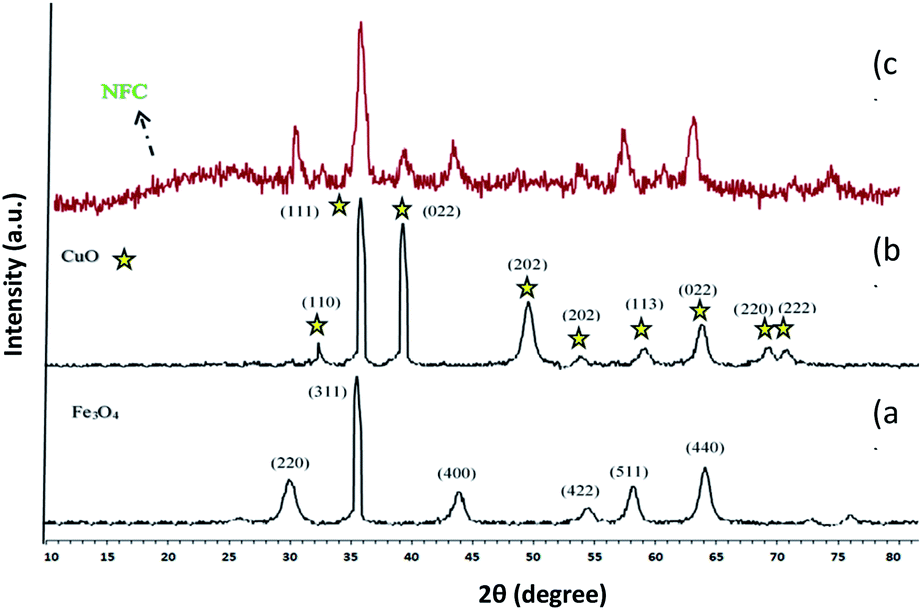
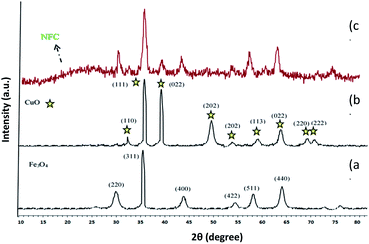
The XRD pattern of Fe3O4 shows six characteristic diffraction peaks at 2θ = 30.3°, 35.8°, 43.6°, 54.8°, 57.3°, and 63.2° corresponding to the (220), (311), (400), (422), (511), and (440) crystallographic phases, respectively, that were completely in agreement with the standard structure and the reported XRD pattern for magnetic (Fig. 4a).40 Also appear for the resultant composites, suggesting that Fe3O4 has been successfully prepared in the cellulose matrix. In addition, the XRD curve of the Fe3O4@cellulose nanoparticles composites of the same peaks was also observed in the Fe3O4@NFC XRD pattern, indicating retention of the crystalline spinel ferrite core structure during the cellulose coating process. Moreover, a diffraction peak at 2θ = 23.12 appeared in the Fe3O4@NCs MNPs, which could be related to the cellulose coating of Fe3O4 NPs.41 It is characteristic diffraction peaks of Cu nanoparticle were observed in the XRD pattern in (Fig. 4b). Weak and strong peaks at 2θ = 32.07°, 35.47°, 35.82°, 48.72°, 52.97°, 58.02°, 62.47°, 67.87° and 69.32° corresponding to the (110), (111), (022), (202), (202), (113), (022), (220) and (222) crystallographic phases in XRD pattern are related to CuO.42,43 The first part shows the XRD pattern of catalyst Fe3O4@NFC-ImSalophCu the amorphous structure which is related to the cellulose structure (Fig. 4c).
 |
| | Fig. 4 XRD spectra of (a) Fe3O4 (b) CuO (c) Fe3O4@NFC-ImSalophCu. | |
However, these results demonstrated that the functionalization of magnetic does not lead to its phase change and consequently confirms that the crystal structure of Fe3O4 remains intact during the functionalization.44
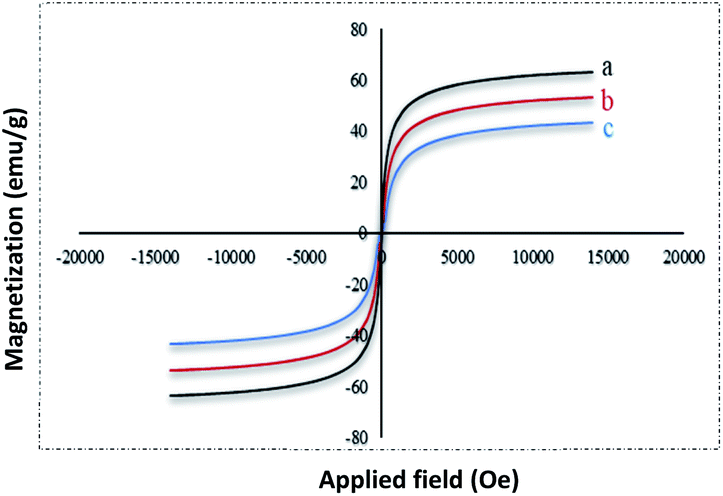
The magnetic properties of (a) Fe3O4 (b) Fe3O4@NFC (c) Fe3O4@NFC-ImSalophCu complex nanoparticles were studied by a vibrating sample magnetometer (VSM) at 300 K (Fig. 5). Fig. 3 shows the product has superparamagnetic at room temperature. The impregnation of magnetization values for Fe3O4, Fe3O4@NFC and Fe3O4@NFC-ImSalophCu nanoparticles were 63.238, 53.237 and 43.248 emu g−1.
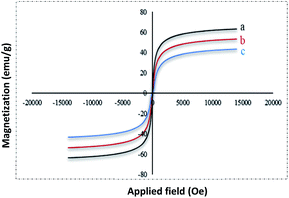 |
| | Fig. 5 VSM pattern Fe3O4 (a), Fe3O4@NFC (b) and (c) Fe3O4@NFC-ImSalophCu. | |
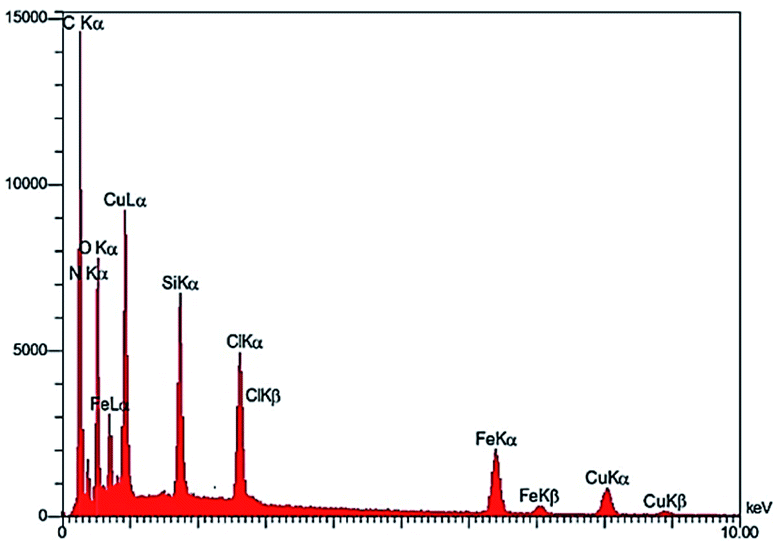
Scanning electron microscopy coupled with energy dispersive X-ray (FE-SEM-EDX): Fig. 6, shows energy-dispersive X-ray spectroscopy (EDX) of Fe3O4@NFC-ImSalophCu salen complex nanoparticles. It is recorded to investigate the elemental composition of nanoparticles. Also, the results demonstrate that Cu, Fe, Si, O, N and C appear in Fe3O4@NFC-ImSalophCu salen nanoparticles. Note that no impurity is present in the products. Fe-SEM analysis of the products (Fig. 7a and b) provided information on the size and morphology of Fe3O4@NFC-ImSalophCu.
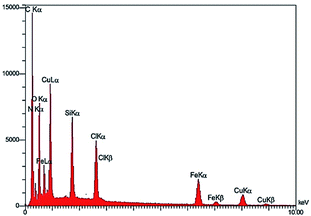 |
| | Fig. 6 EDX image of Fe3O4@NFC-ImSalophCu. | |
 |
| | Fig. 7 FE-SEM image (a) Fe3O4 and (b) Fe3O4@NFC-ImSalophCu. | |
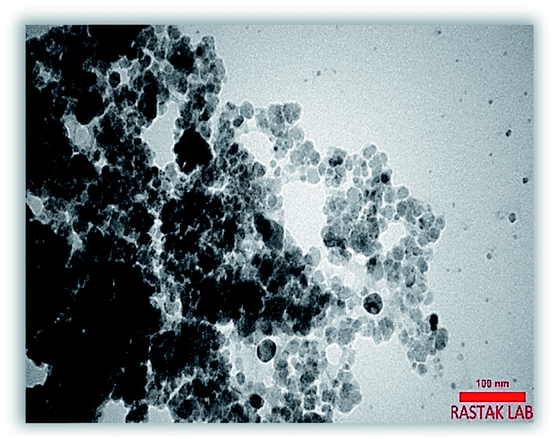
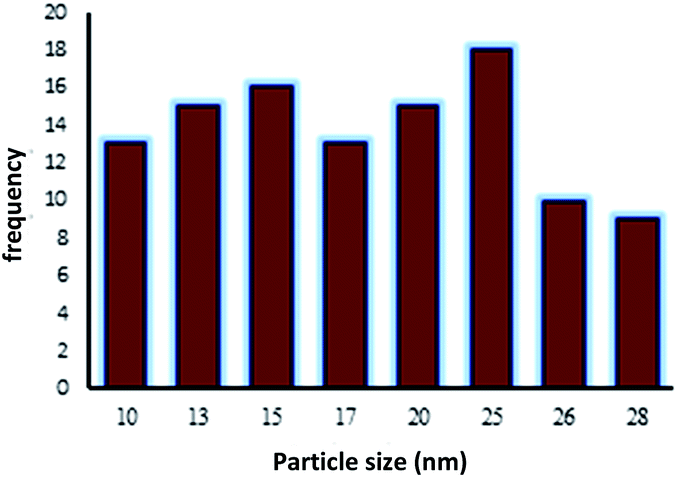
The TEM image of Fe3O4@NFC-ImSalophCu showed a mean diameter of 19.5 nm for the NPs (Fig. 8). However, TEM image shows spherical morphology (Fig. 9).
 |
| | Fig. 8 Transmission electron microscope image of the Fe3O4@NFC-ImSalophCu. | |
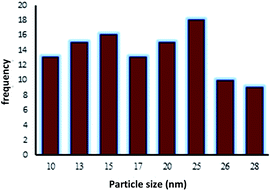 |
| | Fig. 9 Particle size distribution histogram of Fe3O4@NFC-ImSalophCu. | |
Fe3O4@NFC-ImSalophCu salen complex catalytic activity in the synthesis of triazole
Primary experiments were accomplished using the reaction of phenacyl bromide with sodium azide and phenylacetylene, as a model reaction, in the attendance of Fe3O4@NFC-ImSalophCu. The reactions were accomplished using various solvents such as H2O, EtOH, MeOH, CH3CN, DMF and in solvent-free conditions. The results obtained are summarized in Table 1. In solvents, the pKa (pKaH values) and pKb values is in reverse related to the acidity and basicity strength, respectively. The basicity order in water can differ significantly from the gas phase. pKa of methanol, water, and ethanol is 15.54, 15.74 and 15.9, respectively. Methanol more acidic than water, while ethanol is a weaker acid than water. Methanol is organic alcohol, which does have acidic hydrogen, and then there, is the water molecule that may not be considered as an organic one, still, it contributes to increasing the acidity and thus we can account for its acidity. All alcohols except for methanol are less acidic than water. A donor number (DN) is a quantitative measure of Lewis basicity. Lewis acids are characterized by acceptor numbers. The donation number has a reverse relationship with acidity. The donor number is expressed in kcal mol−1. If a solvent has a high donor number, making it a strong Lewis base and this solvent has a high dielectric constant (relative permittivity), making it a good solvent for ionic species. The DN of methanol, dimethylformamide (DMF) and ethanol 19 kcal mol−1 (79 kJ mol−1), 26.6 kcal mol−1 (111 kJ mol−1) and 31.5 kcal mol−1 (132 kJ mol−1), respectively.45 As can be seen in this table, the highest reaction yield was obtained when H2O was used as the solvent (Table 1, entry 1). To our surprise, the reaction gave lower yields when the reaction was performed under solvent-free or neat conditions (Table 1, entry 6). The conditions concerning the amount of catalyst were optimized, as shown in Table 1.
Table 1 Optimization of reaction conditions for synthesis of 1-phenyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)ethan-1-onea
|
| Entry |
Solvent |
Catalyst (mol%) |
Temp. (°C) |
Time (min) |
Yieldb (%) |
| Reaction conditions: acetophenone (1.0 mmol), alkyne (1.0 mmol), sodium azide (1.2 mmol), catalyst (0.2 mol%) sodium ascorbate (2.5 mol%) and H2O (3 ml), 65 °C. Isolated yield. |
| 1 |
Water |
0.2 |
80 |
30 |
98 |
| 2 |
EtOH |
0.2 |
Reflux |
30 |
80 |
| 3 |
MeOH |
0.2 |
Reflux |
30 |
75 |
| 4 |
CH3CN |
0.2 |
Reflux |
90 |
65 |
| 5 |
DMF |
0.2 |
80 |
90 |
70 |
| 6 |
Neat |
0.2 |
80 |
30 |
40 |
| 7 |
Water |
No catalyst |
80 |
180 |
30 |
| 8 |
Water |
0.1 |
80 |
60 |
80 |
| 9 |
Water |
0.3 |
80 |
30 |
97 |
| 10 |
Water |
0.2 |
25 |
90 |
45 |
| 11 |
Water |
0.2 |
40 |
60 |
55 |
| 12 |
Water |
0.2 |
50 |
45 |
75 |
| 13 |
Water |
0.2 |
60 |
30 |
92 |
| 14 |
Water |
0.2 |
65 |
30 |
97 |
| 15 |
Water |
0.2 |
70 |
30 |
97 |
| 16 |
Water |
0.2 |
80 |
30 |
98 |
It was found that 0.2 mol% of Fe3O4@NFC-ImSalophCu was the optimized situation (Table 1, entry 14). Increasing the amount of catalyst did not increase the yield (Table 1, entry 9). A low concentration of Copper resulted in a decreased yield (Table 1, entry 8). Besides, when the reaction was carried out in the lack of a catalyst, only a trace amount of the favorable product (Table 1, entry 7) was obtained, at low concentration the reaction continued for 3 hours but the reaction yield was decreased (Table 1, entry 7). Also, in Table 1 the effect of temperature on the reaction yield was checked, and the results obtained were in table. It is obvious that at room temperature, the low yield was obtained (Table 1, entry 10). Under the optimized reaction situation, the reaction was explored with various phenacyl bromides and alkynes aromatic/aliphatic. As shown in Table 2, the exchange of the phenacyl bromides did not have a clear steric effect on the reaction. The copper-catalyzed click reaction mechanism comprises the multi-general steps shown in Scheme 4.
Table 2 Synthesis of 1,4-disubstituted 1,2,3-triazoles catalyzed by Fe3O4@NFC-ImSalophCua
|
| Entry |
Product |
R |
R1 |
Time (min) |
Yieldb (%) |
Mp (°C) |
TON |
Ref. |
| Found |
Reported |
| Reaction conditions: acetophenone (1.0 mmol), alkyne (1.0 mmol), sodium azide (1.2 mmol), catalyst (0.2 mol%) sodium ascorbate (2.5 mol%) and H2O (3 ml), 65 °C. Isolated yield. |
| 1 |
11a |
Ph |
Ph |
30 |
97 |
169–170 |
169–171 |
485 |
46 |
| 2 |
11b |
4-NO2 |
Ph |
45 |
90 |
185–186 |
184–186 |
450 |
46 |
| 3 |
11c |
4-Br |
Ph |
45 |
95 |
168–169 |
169–170 |
475 |
49 |
| 4 |
11d |
Ph |
CH2OH |
45 |
90 |
115–116 |
115–117 |
475 |
47 |
| 5 |
11e |
4-NO2 |
CH2OH |
60 |
95 |
143–144 |
143–145 |
485 |
49 |
| 6 |
11f |
4-Br |
CH2OH |
60 |
85 |
157–158 |
157–159 |
425 |
45 |
| 7 |
11g |
Ph |
MeCH–OH |
30 |
94 |
128–129 |
127–129 |
470 |
49 |
| 8 |
11h |
Ph |
Me2C–OH |
45 |
87 |
97–98 |
96–98 |
435 |
48 |
| 9 |
11i |
4-NO2 |
Me2C–OH |
90 |
95 |
127–128 |
126–128 |
475 |
49 |
| 10 |
11j |
4-Br |
Me2C–OH |
60 |
75 |
169–171 |
170–171 |
375 |
47 |
 |
| | Scheme 4 The synthetic route for Fe3O4@NFC-ImSalophCu. | |
Recyclability and reusability of Fe3O4@NFC-ImSalophCu
Recycling of heterogeneous catalysts is very important, especially for commercial applications. Thus, the possibility of frequent use of Fe3O4@NFC-ImSalophCu was also inquired for the methods of synthesizing triazoles, using the reaction of phenacyl bromide with phenylacetylene (0.2 mol% of the catalyst), and one-pot three-component reaction from phenacyl bromide, phenylacetylene, sodium azide and sodium ascorbate (0.2 mol% of catalyst) and one-pot three-component reaction from phenacyl bromide, phenylacetylene, sodium azide and sodium ascorbate (0.2 mol% of catalyst). The catalyst was separated by the magnets after each experiment the reaction mixture, then washed with water and ethyl acetate, and dried carefully before being used in subsequent runs.
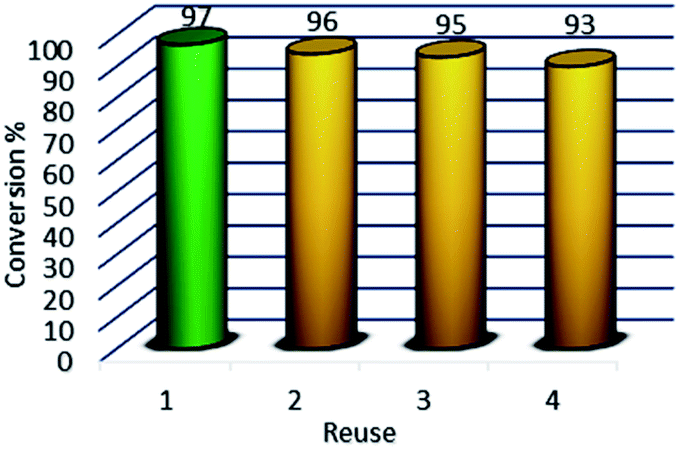
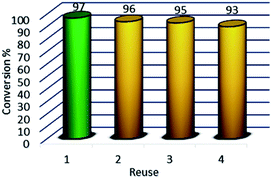
Due to the magnetic properties of the catalyst after the first use of the product catalyst, it is simply extracted by ethyl acetate while the catalyst remains in the aqueous phase. The catalyst showed good performance after four times of recycling. FT-IR, VSM, and XRD of the catalyst after four times reuse showed that the structure and morphology of the catalyst maintained unchanged during the recycling process (Fig. 10).
 |
| | Fig. 10 (a) VSM (b) Fe-SEM (c) FT-IR (d) XRD analysis of Fe3O4@NFC-ImSalophCu after four times. | |
Mechanism studies
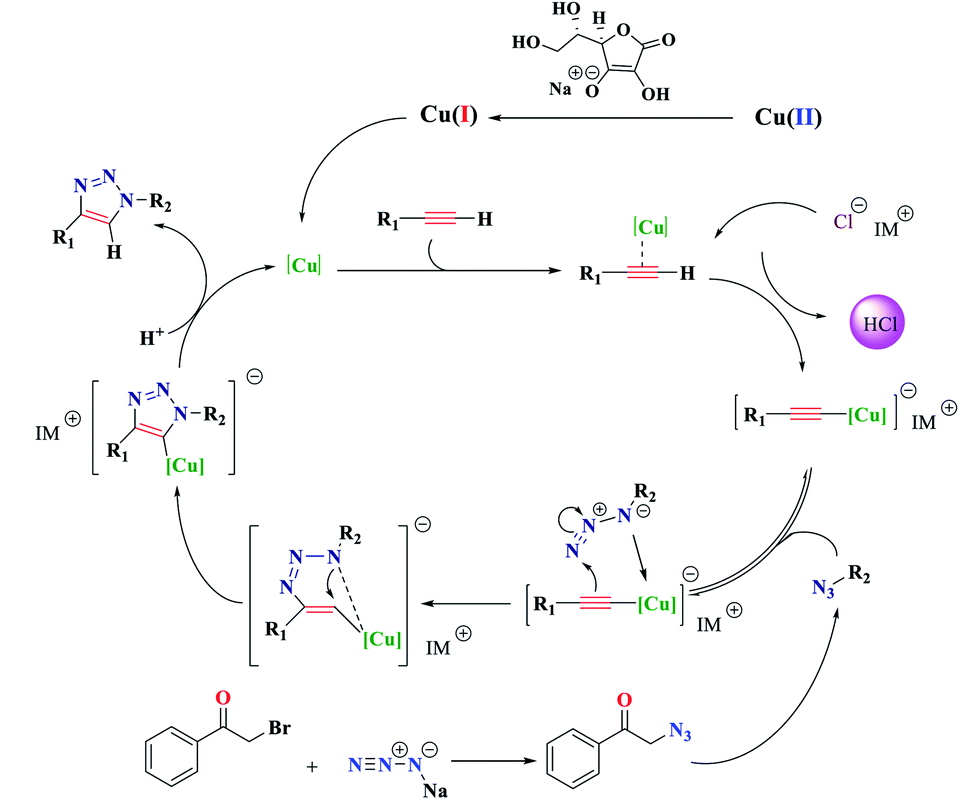
To illustrate the advantage and uniqueness of the Fe3O4@NFC-ImSalophCu catalyst as well as its application in the reaction of triazole, two homologs the Fe3O4@NFC-IM and salophen–Cu(II) of the catalyst Fe3O4@NFC-ImSalophCu were synthesized and used in the model reaction (Scheme 4). The two catalysts were compared in triazole reaction and the reaction did not progress much after 30 minutes whereas the salophen–Cu(II) catalyst showed 93% good yield after 90 minutes while the Fe3O4@NFC-IM catalyst did not yield after 90 minutes while the Fe3O4@NFC-IM catalyst did not perform well after three hours. However, it can be concluded that the imidazolium fraction plays a key role in the catalyst. We propose that the copper-catalytic cycle mechanism of the click reaction exhibits several steps50 shown in Scheme 4: (a) formation of copper(I) acetylide with the interference of imidazolium counter ion and generating of a HCl molecule, (b) the first C–N bond formation affords the six-membered ring copper metallacycle, (c) cyclization takes place to yield the copper triazole intermediate, (d) proteolysis of the Cu–C bond gives the triazole product and regenerates the catalyst. This catalyst constitutes the regioselective of product 1,4-triazole. To illustrate the advantage of Fe3O4@NFC-ImSalophCu as a heterogeneous catalyst in this reaction, our results and reaction conditions were compared with other conditions with those of this reaction using the heterogeneous catalysis in previous times (Table 3). The results obtained show that our proposed catalyst is quite comparable with those for the heterogeneous catalyst used regarding the yields (Fig. 11).
Table 3 Comparison of the catalytic activity of Fe3O4@NFC-ImSalophCu nanocomposites with literature samples reported for the 1,2,3-triazole (11a) reaction
| Entry |
Catalyst |
Solvent |
Time (min) |
Temp. (°C) |
Yield (%) |
Ref. |
| 1 |
Cu(II)Br2BTP@TMSP–nSiO2 |
EtOH/H2O |
25 |
85 |
98 |
51 |
| 2 |
Amberlyst A-21 CuI |
CH3CN |
60 |
89 |
89 |
52 |
| 3 |
PEG-400, CuI |
Water |
120 |
r.t. |
74 |
53 |
| 4 |
P4VPy–CuI |
Water |
25 |
Reflux |
89 |
54 |
| 5 |
CuI/AK |
Water |
20 |
Reflux |
84 |
55 |
| 6 |
Nano-Fe3O4@TiO2/Cu2O |
Water |
20 |
Reflux |
89 |
56 |
| 7 |
Fe3O4@NFC-ImSalophCu |
Water |
30 |
65 |
97 |
This work |
 |
| | Fig. 11 Recycling activity of the Fe3O4@NFC-ImSalophCu. | |
Experimental
Materials and instrumentation
The reagents and solvents used were supplied from Merck, Fluka or Aldrich and used without further purification. FT-IR spectra were obtained using a JASCO FT/IR 4600 spectrophotometer using KBr pellet. Melting points were determined using an electro-thermal C14500 apparatus. The 1HNMR and 13CNMR spectra were recorded on a Bruker Avance DPX-250 or a Bruker Avance 3–300 MHz spectrometer in CDCl3 and DMSO-d6 as a solvent and TMS as an internal standard. The reaction progress and the purity of compounds were monitored using TLC analytical silica gel plates (Merck 60 F250). Field emission scanning electron microscopy (FE-SEM) images were obtained on a Tescan MIRA3. EDX spectroscopy was performed using a field emission scanning electron microscope (JEOL 7600F), transmission electron microscopy (TEM) was performed on a Philips EM208 microscope operated at 100 kV. ICP experiments were accomplished using a Varian Vista Pro CCD simultaneous ICP-OES instrument. Lake Shore Cryotronics 7407 vibrating sample magnetometer (VSM) at room temperature.
Preparation of Fe3O4@NFC core–shell (NPs)
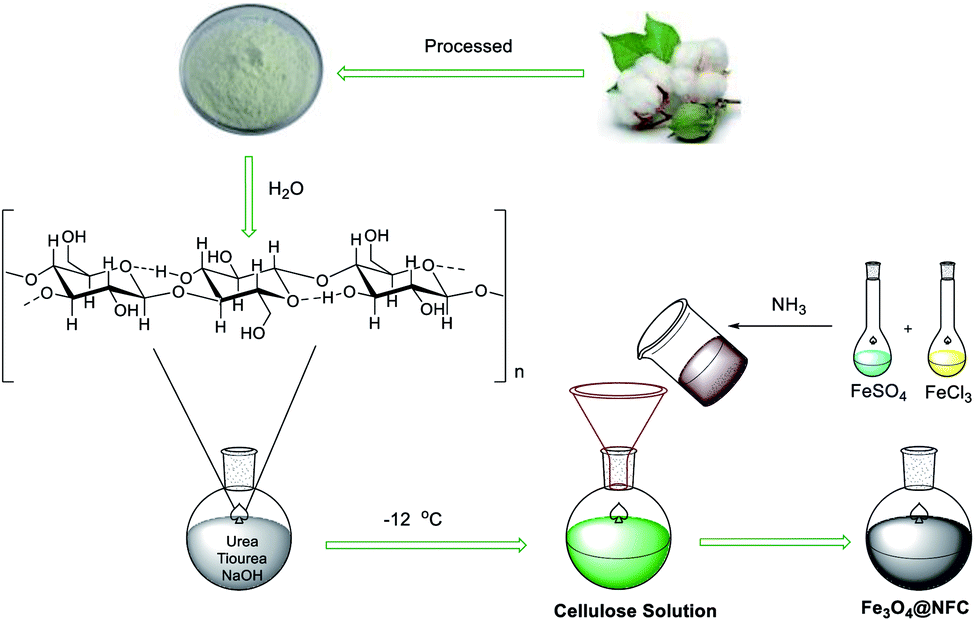
A solution with NaOH–thiourea–urea–H2O of 4.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.88:4.8:50 by weight was cooled to −12 °C. the cellulose of 1.1 g was immediately added into the above solution under vigorous stirring for 10 min to form a cellulose solution. FeCl3·6H2O and FeSO4·7H2O of 8.1 and 3.9 g, respectively, were dissolved in 25 ml distilled water with vigorous stirring at room temperature and reacting with NH3 of 13.3 ml after 1 hour, the suspension of magnetic nanoparticles (MNPs) and then the iron solution was added dropwise into the cellulose solution with vigorous stirring at 30 °C for 2.5 h. The suspension Fe3O4@NFC was separated with an external magnet, washed sequentially with distilled water, ethanol and acetone, and kept under vacuum.57,58
:3.88:4.8:50 by weight was cooled to −12 °C. the cellulose of 1.1 g was immediately added into the above solution under vigorous stirring for 10 min to form a cellulose solution. FeCl3·6H2O and FeSO4·7H2O of 8.1 and 3.9 g, respectively, were dissolved in 25 ml distilled water with vigorous stirring at room temperature and reacting with NH3 of 13.3 ml after 1 hour, the suspension of magnetic nanoparticles (MNPs) and then the iron solution was added dropwise into the cellulose solution with vigorous stirring at 30 °C for 2.5 h. The suspension Fe3O4@NFC was separated with an external magnet, washed sequentially with distilled water, ethanol and acetone, and kept under vacuum.57,58
Preparation of imidazole supported on Fe3O4@NFC (Fe3O4@NFC-Im) (3)
Firstly, (5.5 mmol) of imidazole was dissolved in (5 ml) of dry toluene and then (5.5 mmol) of 3-chloropropyl-trimethoxy silane was added slowly to the mixture. The mixture was then stirred for 3 h at room temperature. Then, compound 2 and (1.5 g) of Fe3O4@NFC sonicated in dry toluene (30 ml) for 30 min added slowly to the mixture. The reaction was performed for 24 h under N2 atmosphere at reflux conditions. The mixture was stirred with Et3N (0.5 ml) for 30 min at room temperature. After completion of the reaction, the reaction mixture was washed with toluene and diethyl ether and separated using an external magnet and dried at 70 °C in a vacuum.
Preparation of 5-(chloromethyl)-2-hydroxybenz aldehyde (4)
This compound was synthesized according to the procedure described in the literature.59 Firstly, (0.125 mmol) of salicylaldehyde were added to a mixture of 10 ml of concentrated hydrochloric acid 37% containing (0.205 mmol) of paraformaldehyde along with several drop of concentrated H2SO4 as a catalyst. The mixture was maintained at 70 °C under stirring for 24 hours until a precipitate is formed. The reaction mixture cooled to room temperature and then water (20 ml) was added to the mixture and the product was extracted into CH2Cl2 (20 ml). The solid product was collected by suction filtration, some anhydrous Na2SO4 was used for drying the organic phase. The CH2Cl2 was removed with a rotary evaporator and thick yellow oil was set aside overnight. Then a pale purple solid product was isolated. After preparation of 2-hydroxy 4-chloromethyl benzaldehyde (Scheme 3) (mp 86–87 °C, 98% isolated yield).
FT-IR (KBr): ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 3151 (O–H), 3033, 2975, 2865 (C–H aldehyde), 1665 (CO), 1434 (CC), 673 (CH2–Cl) cm−1.
= 3151 (O–H), 3033, 2975, 2865 (C–H aldehyde), 1665 (CO), 1434 (CC), 673 (CH2–Cl) cm−1.
1HNMR (DMSO, 300 MHz): δ = 4.80 (s, 2H, CH2), 7.56–7.60 (d,d, 2H, H-Ar), 7.72 (s, 1H, H-Ar), 10.06 (s, 1H, CH of aldehyde), 11.30 (s, 1H, O–H) ppm.
13CNMR (DMSO, 75 MHz): δ = 47.07, 117.81, 124.87, 131.18, 136.14, 136.47, 148.41, 163.27, 197.76 ppm.
Preparation complex N,N-bis(5-chloromethyl-salicylidene)-1,2-phenylenediamine by Cu(OAc)2 (5-Cl-salophen–Cu(II)) (5, 6)
5-Cl-salophen–Cu(II) was prepared and characterized as described in literature60 the solution of (2.5 mmol, 0.27 g) 1,2-phenylene diamine in 20 ml dichloromethane was added dropwise to the solution of 5-chloromethylsalicyaldehyde (5 mmol, 0.85 g) in 20 ml dichloromethane. The mixture was refluxed for 2 h. The orange colored precipitates were collected, and washed with reaction solvent and then dried in vacuum (4). Next step dissolving of Cu(OAc)2 (2 mmol) in EtOH (5 ml) by stirring at room temperature. Then the N,N-bis(5-chloromethylsalicylidene)-1,2-phenylenediamine (2 mmol) dissolved in a minimum volume of EtOH and added the above solution under stirring. The solid was filtered off, washed with EtOH and finally with diethyl ether and dried in vacuo (yield 75%, 6).
Synthesis of 5-Cl-salophCu supported on Fe3O4@NFC-Im (Fe3O4@NFC-ImSalophCu) (7)
The synthesized Fe3O4@NFC-Im (1.5 g) was sonicated in dry toluene (20 ml) for 30 min. 5-Cl-salophen–Cu(II) (5 mmol) was dissolved in dry toluene (20 ml) added dropwise to the dispersed mixture and stirred at 75 °C for 24 h. The solid was separated by an external magnet, washed with H2O and Et2O and dried at 70 °C in oven under vacuum.
General procedure for the one-pot synthesis of 1,4-disubstituted 1,2,3-triazoles from α-bromo ketones catalyzed by Fe3O4@NFC-ImSalophCu
A mixture of sodium ascorbate (2.5 mol%) and Fe3O4@NFC-ImSalophCu (0.2 mol%) in 3 ml of H2O was stirred for 2 min. Then the phenylacetylene (1 mmol), the α-bromo ketone (1 mmol), and sodium azide (1.2 mmol) were added, and the resulting mixture was stirred at 65 °C for 10–30 min. The progress of the reaction was monitored by TLC (eluting with n-hexane/ethyl acetate, 1:1). After completion of the reaction, the mixture was diluted with water and EtOAc and filtered. The organic layer was separated, and the aqueous layer was extracted with EtOAc (2 × 5 ml). The combined organic layers were dried over Na2SO4 and evaporated to afford the pure triazole in most cases. If necessary, the product was purified by recrystallization from n-hexane/EtOAc (Scheme 5).
 |
| | Scheme 5 Catalytic activity of the Fe3O4@NFC-ImSalophCu nanocomposite triazole reactions. | |
Selected spectral data
1-Phenyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)ethanone (11a). 1HNMR (DMSO, 300 MHz): δ = 6.29 (s, 2H, CH2), 7.35–7.40 (t, 1H, Hp-Ar), 7.46–7.68 (m, 4H, Hm-Ar), 7.75–7.81 (t, 1H, Hp-Ar), 7.88–7.92 (d, 2H, Ho-Ar), 8.11–8.15 (d, 2H, Ho-Ar), 8.55 (s, 1H, CH of triazole) ppm. 13CNMR (DMSO, 75 MHz): δ = 56.49, 123.53, 125.64, 128.36, 128.72, 129.45, 129.51, 131.24, 134.61, 134.79, 146.78, 192.70 ppm. Anal. calcd for C16H13N3O: C, 72.99; H, 4.98; N, 15.96; found: C, 72.67; H, 5.01; N, 15.94. Mp (°C): 169–170.
1-(4-Nitro phenyl)-2-(4-phenyl-1H-1,2,3-triazol-1-yl)ethanone (11b). 1HNMR (DMSO, 300 MHz): δ = 6.37 (s, 2H, CH2), 7.35–7.41 (t, 1H, Hp-Ar), 7.47–7.52 (t, 2H, Hm-Ar), 7.89–7.92 (t, 2H, Ho-Ar), 8.33–8.37 (d, 2H, Ho-Ar), 8.44–8.48 (d, 2H, Hm-Ar), 8.55 (s, 1H, CH of triazole) ppm. 13CNMR (DMSO, 75 MHz): δ = 56.47, 121.23, 123.54, 128.06, 128.76, 129.72, 129.80, 130.41, 140.58, 148.88, 150.51, 192.71 ppm. Anal. calcd for C16H12N4O3: C, 62.33; H, 3.92; N, 18.17; found: C, 61.94; H, 3.62; N, 18.21. Mp (°C): 185–186.
1-(4-Bromophenyl)-2-(4-phenyl-1H-1,2,3-triazol-1-yl)ethanone (11c). FT-IR (KBr): = 3152, 2950, 1700 (CO), 1462–1575 (CC) cm−1. 1HNMR (DMSO, 300 MHz): δ = 6.27 (s, 2H, CH2), 7.35–7.52 (t,t, 3H, Hm,p-Ar), 7.85–7.91 (m, 4H, H-Ar), 8.03–8.07 (d, 2H, H-Ar), 8.33–8.37 (d, 2H, H-Ar), 8.54 (s, 1H, CH of triazole) ppm. 13CNMR (DMSO, 75 MHz): δ = 56.47, 121.53, 127.54, 128.06, 128.86, 129.82, 129.85, 130.51, 131.54, 133.61, 148.78, 192.70 ppm. Anal. calcd for C16H12BrN3O: C, 56.16; H, 3.53; N, 12.28; found: C, 56.24; H, 3.55; N, 12.14. Mp (°C): 168–169.
2-(4-(Hydroxymethyl)-1H-1,2,3-triazol-1-yl)-1-phenylethanone (11d). 1HNMR (DMSO, 300 MHz): δ = 4.58–4.59 (d, 2H, CH2–OH), 5.23–5.27 (t, 1H, OH), 6.18 (s, 2H, CH2), 7.60–7.66 (t, 2H, Hm-Ar), 7.73–7.79 (t, 1H, Hp-Ar), 7.95 (s, 1H, CH of triazole), 8.08–8.11 (d, 2H, Ho-Ar) ppm. 13CNMR (DMSO, 75 MHz): δ = 55.57, 56.20, 124.87, 128.64, 129.47, 134.67, 148.41, 192.76 ppm. Anal. calcd for C11H11N3O2: C, 60.82; H, 5.10; N, 19.34; found: C, 60.79; H, 5.13; N, 19.37. Mp (°C): 115–116.
2-(4-(Hydroxymethyl)-1H-1,2,3-triazol-1-yl)-1-(4-nitrophenyl)ethanone (11e). FT-IR (KBr): = 3360 (OH), 3100, 2910, 1690 (CO), 1530 (CC) cm−1. 1HNMR (DMSO, 300 MHz): δ = 4.22 (s, 1H, OH), 4.65 (s, 2H, CH2–OH), 6.13 (s, 2H, CH2), 7.80 (s, 1H, CH of triazole), 8.35–8.40 (d,d, 2H, H-Ar) ppm. 13CNMR (DMSO, 75 MHz): δ = 55.56, 56.20, 121.52, 123.42, 129.46, 130.87, 139.91, 150.94, 191.52 ppm. Anal. calcd for C11H10N4O4: C, 50.38; H, 3.84; N, 21.37; found: C, 50.41; H, 3.55; N, 21.43. Mp (°C): 143–144.
1-(4-Bromophenyl)-2-(4-(hydroxymethyl)-1H-1,2,3-triazol-1-yl)ethanone (11f). 1HNMR (DMSO, 300 MHz): δ = 4.38 (s, 2H, CH2), 5.00 (s, 2H, CH2–OH), 5.75 (s, 1H, OH), 7.98 (s, 1H, CH of triazole), 8.33–8.37 (d, 2H, H-Ar), 8.44–8.48 (d, 2H, H-Ar) ppm. 13CNMR (DMSO, 75 MHz): δ = 55.57, 56.20, 122.77, 127.72, 129.47, 131.41, 134.67, 147.41, 192.76 ppm. Anal. calcd for C11H10BrN3O2: C, 44.62; H, 3.40; Br, 26.98; N, 14.19; found: C, 44.48; H, 3.44; N, 14.21. Mp (°C): 157–158.
2-(4-(1-Hydroxyethyl)-1H-1,2,3-triazol-1-yl)-1-phenylethanone (11g). FT-IR (KBr): = 3200 (OH), 3110, 2920, 1690 (CO), 1440–1580 (CC) cm−1. 1HNMR (CD3COCD3, 300 MHz): δ = 1.53–1.56 (d, 3H, j = 6.6, CH3), 4.44 (s, 1H, OH), 5.04 (q, 1H, j = 6.3, CH), 6.13 (s, 2H, CH2), 7.62 (t, 2H, j = 7.5, H-Ar), 7.2 (t, 1H, j = 7.2, H-Ar), 7.88 (s, 1H, CH of triazole), 8.13 (d, 2H, j = 8.4, H-Ar) ppm. 13CNMR (CD3COCD3, 75 MHz): δ = 23.28, 55.51, 62.67, 122.47, 128.12, 128.96, 134.04, 134.65, 152.99, 191.57 ppm. Anal. calcd for C12H13N3O2: C, 62.33; H, 5.67; N, 18.17; found: C, 62.21; H, 5.67; N, 18.31. Mp (°C): 128–129.
2-(4-(2-Hydroxypropan-2-yl)-1H-1,2,3-triazol-1-yl)-1-phenylethanone (11h). FT-IR (KBr): = 3310 (OH), 3056, 2990, 1690 (CO), 1450–1540 (CC) cm−1. 1HNMR (DMSO, 300 MHz): δ = 1.51 (s, 2 × 3H, CH3), 5.16 (s, 1H, OH), 6.15 (s, 2H, CH2), 7.61–7.66 (t, 2H, H-Ar), 7.73–7.79 (t, 2H, CH-Ar), 7.86 (s, 1H, CH of triazole), 8.08–8.11 (d,d, 2H, H-Ar) ppm. 13CNMR (DMSO, 75 MHz): δ = 31.30, 56.46, 78.32, 122.92, 128.75, 130.78, 133.14, 133.53, 134.53, 190.90 ppm. Anal. calcd for C13H15N3O2: C, 63.66; H, 6.16; N, 17.13; found: C, 63.58; H, 6.11; N, 17.27. Mp (°C): 97–98.
2-(4-(2-Hydroxypropan-2-yl)-1H-1,2,3-triazol-1-yl)-1-(4-nitrophenyl)ethanone (11i). FT-IR (KBr): = 3380 (OH), 3100, 2945, 1700 (CO), 1530 (CC) cm−1. 1HNMR (DMSO, 300 MHz): δ = 1.38 (s, 6H, CH3), 4.90 (s, 2H, CH2), 5.53 (s, 1H, OH), 7.75 (s, 1H, CH of triazole), 8.31–8.33 (d,d, 2H, H-Ar), 8.41–8.44 (d, 2H, j = 8.7, H-Ar) ppm. 13CNMR (DMSO, 75 MHz): δ = 31.18, 56.49, 78.29, 122.82, 123.83, 129.75, 131.24, 140.58, 152.58, 190.90 ppm. Anal. calcd for C13H14N4O4: C, 53.79, H, 4.86; N, 19.30; found: C, 53.63; H, 4.80; N, 19.39. Mp (°C): 127–128.
1-(4-Bromophenyl)-2-(4-(2-hydroxypropan-2-yl)-1H-1,2,3-triazol-1-yl)ethanone (11j). 1HNMR (DMSO, 300 MHz): δ = 1.51 (s, 6H, CH3), 5.16 (s, 1H, OH), 6.13 (s, 1H, CH2), 7.84–7.87 (d, 3H, CH of triazole and H-Ar), 8.00–8.03 (d, 2H, H-Ar) ppm. 13CNMR (DMSO, 75 MHz): δ = 31.29, 56.50, 78.29, 122.52, 127.32, 128.75, 129.88, 131.54, 133.53, 152.58, 190.91 ppm. Anal. calcd for C13H14BrN3O2: C, 48.17; H, 4.35; N, 12.96; found: C, 48.13; H, 48.19; N, 4.34. Mp (°C): 169–171.
Conclusions
We have synthesized an environmentally friendly catalyst. This catalyst is a new and reusable catalyst that has several advantages including such as a shorter reaction time, cleaner reaction, reusability of the catalyst, and high reaction yield. Moreover, this innovative heterogeneous catalyst could be easily separated and reused for at least four frequent cycles without a significant loss in its catalytic activity.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the financial support of the research Council of the University of Birjand and we also thank University of Ferdowsi.
References
-
(a) S. Shylesh, V. Schunemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428 CrossRef;
(b) Y. Zhu, L. P. Stubbs, F. Ho, R. Liu, C. P. Ship, J. A. Maguire and N. S. Hosmane, ChemCatChem, 2010, 2, 365 CrossRef.
- R. Mrówczyński, A. Nanb and J. Liebscher, RSC Adv., 2014, 4, 5927–5952 RSC.
- A. Kumar, R. Parella and S. A. Babu, Synlett, 2014, 25, 835–842 CrossRef CAS.
- S. Shylesh, V. Schuenemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS.
- M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC.
- R. Hudson, Y. Feng, R. S. Varma and A. Moores, Green Chem., 2014, 16, 4493–4505 RSC.
- X. Zhu, J. Niu, F. Zhang, J. Zhou, X. Li and J. Ma, New J. Chem., 2014, 38, 4622–4627 RSC.
- T. A. Salah El-Din, A. A. Elzatahry, D. M. Aldhayan, A. M. Al-Enizi and S. S. Al-Deyab, Int. J. Electrochem. Sci., 2011, 6, 6177–6183 Search PubMed.
- S. Xuan, W. Jiang, X. Gong, Y. Hu and Z. Chen, J. Phys. Chem. C, 2009, 113, 553–558 CrossRef.
- S. P. Pujari, L. Scheres, A. T. M. Marcelis and H. Zuilhof, Angew. Chem., Int. Ed., 2014, 53, 6322–6356 CrossRef CAS.
- Z. Chen, Z. Xue, L. Chen, Z. Geng, R. Yang, L. Chena and Z. Wang, New J. Chem., 2013, 37, 3731–3736 RSC.
- X. Y. Wei, J. H. Li and J. C. Chen, Adv. Mater. Res., 2013, 634, 2077–2080 Search PubMed.
- C. X. Lin, H. Y. Zhan, M. H. Liu, S. Y. Fu and L. A. Lucia, Langmuir, 2009, 25, 10116 CrossRef CAS.
- F. Nemati, M. M. Heravi and A. Elhampour, RSC Adv., 2015, 57, 45775–45784 RSC.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123 RSC.
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508 CrossRef CAS.
- W. Zheng, R. Tan, S. Yin, Y. Zhang, G. Zhao, Y. Chen and D. Yin, Catal. Sci. Technol., 2015, 5, 2092 RSC.
- A. Corma, H. García and A. Leyva, Tetrahedron, 2004, 60, 8553 CrossRef.
- A. S. Reddy and K. K. Laali, Tetrahedron Lett., 2015, 56, 4807 CrossRef.
- J. C. Xiao, B. Twamley and J. N. M. Shreeve, Org. Lett., 2004, 6, 3845 CrossRef.
- P. Nehra, B. Khungar, K. Pericherla, S. C. Sivasubramanian and A. Kumar, Green Chem., 2014, 16, 4266 RSC.
-
(a) C. Allais, J. M. Grassot, J. Rodriguez and T. Constantieux, Chem. Rev., 2014, 114, 10829–10868 CrossRef;
(b) H. G. O. Alvim, E. N. J. d. S. Jumior and B. N. D. Neto, RSC Adv., 2014, 4, 54282–54299 RSC;
(c) H. Y. Cho and J. P. Morken, Chem. Soc. Rev., 2014, 43, 4368–4380 RSC;
(d) V. Estevez, M. Villacampa and J. C. Menendez, Chem. Soc. Rev., 2014, 43, 4633–4657 RSC.
- G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278 CrossRef.
- H. Y. Guo, F. F. Yang, Z. Y. Jiao and J. R. Lin, Chin. Chem. Lett., 2013, 24, 450 CrossRef.
- D. Addla, A. Jallapally, D. Gurram, P. Yogeeswari, D. Sriram and S. Kantevari, Bioorg. Med. Chem. Lett., 2014, 24, 233 CrossRef.
- F. Mir, S. Shafi, M. S. Zaman, N. P. Kalia, V. S. Rajput, C. Mulakayala, N. Mulakayala, I. A. Khan and M. S. Alam, Eur. J. Med. Chem., 2014, 76, 274 CrossRef CAS.
- K. D. Thomas, A. V. Adhikari and N. S. Shetty, Eur. J. Med. Chem., 2010, 45, 3803 CrossRef CAS.
- K. Kushwaha, N. Kaushik and S. C. Lata Jain, Bioorg. Med. Chem. Lett., 2014, 24, 1795 CrossRef CAS.
- M. J. Genin, D. A. Allwine, D. J. Anderson, M. R. Barbachyn, D. E. Emmert, S. A. Garmon, D. R. Graber, K. C. Grega, J. B. Hester, D. K. Hutchinson, J. Morris, R. J. Reischer, C. W. Ford, G. E. Zurenko, J. C. Hamel, R. D. Schaadt, D. Stapert and B. H. Yagi, J. Med. Chem., 2000, 43, 953 CrossRef CAS.
- A. Maleki, M. Panahzadeh and R. Eivazzadeh-keihan, Green Chem. Lett. Rev., 2019, 4, 395–406 CrossRef.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef.
- I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337–2364 CrossRef.
- V. V. Fokin, ACS Chem. Biol., 2007, 2, 775–778 CrossRef.
- B. H. Lipshutz and Y. Yamamoto, Chem. Rev., 2008, 108, 2793–2795 CrossRef.
-
(a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef;
(b) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef.
- H. Torii, M. Nakadai, K. Ishihara, S. Saito and H. Yamamoto, Angew. Chem., 2004, 43, 1983–1986 CrossRef CAS.
- M. Kazemnejadi, A. Shakeri, M. Mohammadi and M. Tabefam, J. Iran. Chem. Soc., 2017, 14, 1917 CrossRef CAS.
- M. Kazemnejadi, A. Shakeri, M. Nikookar, M. Mohammadi and M. Esmaeilpour, Res. Chem. Intermed., 2017, 43, 6889 CrossRef CAS.
- B. Z. Tang, Y. Geng, J. W. Y. Lam, B. Li, X. Jing, X. Wang and X. X. Zhang, Chem. Mater., 1999, 11, 1581 CrossRef CAS.
- S. Azad and B. B. F. Mirjalili, RSC Adv., 2016, 6, 96928–96934 RSC.
- H. Esmaeili-Shahri, H. Eshghi, J. Lari and S. A. Rounaghi, Appl. Organomet. Chem., 2018, 32, e3947 CrossRef.
- H. Khanehzaei, M. B. Ahmad, K. Shameli and Z. Ajdari, Int. J. Electrochem. Sci., 2014, 9, 8189 Search PubMed.
- J. Chu, C. Lin and W. Leau, J. Electron. Mater., 2009, 38, 100–107 CrossRef CAS.
- R. W. Taft, N. J. Pienta, M. J. Hamlet and E. M. Arnett, J. Org. Chem., 1981, 46, 661–667 CrossRef CAS.
- Y. C. Duan, Y. C. Ma, E. Zhang, X. J. Shi, M. M. Wang, X. W. Ye and H. M. Liu, Eur. J. Med. Chem., 2013, 62, 11–19 CrossRef CAS.
- E. Hashemi, Y. S. Beheshtiha, S. Ahmadi and M. M. Heravi, Transition Met. Chem., 2014, 5, 593–601 CrossRef.
- M. Nasr-Esfahani, I. Mohammadpoor-Baltork, A. R. Khosropour, M. Moghadam, V. Mirkhani, S. Tangestaninejad and H. Amiri Rudbari, J. Org. Chem., 2014, 79, 1437–1443 CrossRef CAS.
- M. Bakherad, A. Keivanloo, A. H. Amin and P. Ghamari kargar, Iran. J. Catal., 2018, 3, 179–187 Search PubMed.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS.
- Z. Dehbanipour, M. Moghadam, S. Tangestaninejad, V. Mirkhani and I. Mohammadpoor-Baltork, Polyhedron, 2017, 138, 21–30 CrossRef CAS.
- J. Albadi and M. Keshavarz, Synth. Commun., 2013, 43, 2019–2030 CrossRef CAS.
- P. Kasiviswanadharaju, P. Khedar, B. Khungar and A. Kumar, Tetrahedron Lett., 2012, 53, 6761–6764 CrossRef.
- J. Albadi, M. Keshavarz, F. Shirini and M. Vafaie-Nezhad, Catal. Commun., 2012, 27, 17–20 CrossRef CAS.
- R. Mirsafaei, M. M. Heravi, Sh. Ahmadi, M. H. Moslemin and T. Hosseinnejad, J. Mol. Catal., 2015, 402, 100–108 CrossRef CAS.
- F. Nemati, M. M. Heravi and A. Elhampour, RSC Adv., 2015, 5, 45775–45784 RSC.
- X. Yu, S. Tong, M. Ge, J. Zuo, C. Cao and W. Song, J. Mater. Chem. A, 2013, 1, 959–965 RSC.
- F. F. Kaid, A. T. Totti Silveira Junior and H. E. Toma, ACS Appl. Nano Mater., 2019, 10(6), 530 Search PubMed.
- M. Kazemnejadi, Z. Rezazadeh, M. A. Nasseri, A. Allahresani and M. Esmaeilpour, Green Chem., 2019, 21, 1718–1734 RSC.
- A. Bezaatpour, S. Khatami and M. Amiri, RSC Adv., 2016, 6, 27452–27459 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06251k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ghodsieh Bagherzade
a,
Ghodsieh Bagherzade