
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn easy synthesis of nitrogen and phosphorus co-doped carbon dots as a probe for chloramphenicol†
Pengli Zuo *a,
Zhongguang Chena,
Fengling Yua,
Jinyu Zhanga,
Wei Zuoa,
Yanli Gaoa and
Qingyou Liub
*a,
Zhongguang Chena,
Fengling Yua,
Jinyu Zhanga,
Wei Zuoa,
Yanli Gaoa and
Qingyou Liub
aCentral Laboratory, Linyi Central Hospital, Linyi 276400, China. E-mail: zuo2258960@126.com; Fax: +86 539 2256919; Tel: +86 539 2256919
bLinyi Center for Disease Prevention and Control, Linyi, 276000, China
First published on 4th September 2020
Abstract
Heteroatom doping in carbon dots (CDs) was found to be an efficient way to regulate the structure of electronic energy levels and enhance the fluorescence characteristics of CDs. Nevertheless, most reported fabrication processes of heteroatom-doped CDs are rigorous and complex. Herein, a facile and novel strategy was developed to rapidly prepare nitrogen and phosphorus co-doped CDs (N,P-CDs) using acetic acid as the carbon source, and arginine, 1,2-ethylenediamine (EDA) and diphosphorus pentoxide as the dopants, respectively. The optical, morphological and structural characterizations of the synthesized N,P-CDs were investigated via UV and photoluminescence spectroscopy, X-ray photoelectron spectroscopy, TEM, and FT-IR spectroscopy. The N,P-CDs display outstanding fluorescence stability under high ionic strength (1.6 M KCl), and long time UV irradiation, indicating that they can be used as favorable candidates for fluorescent probes. The fluorescence of N,P-CDs was selectively quenched by chloramphenicol (CAP) with a short response time. The linear range of the response to CAP was from 0.8 to 70 μM with a limit of detection of 0.36 μM (S/N = 3). Notably, the fabricated N,P-CDs were employed for the highly selective and sensitive detection of CAP in milk samples, indicating their potential applications in biologically related areas.
1. Introduction
Chloramphenicol (CAP) is a kind of broad-spectrum antibiotic, and it exhibits antibacterial activity against rickettsia, Gram-negative bacteria, Gram-positive bacteria, and some large viruses. The common side effects of the CAP treatment are non-regenerative anaemia and myelosuppression, and its most serious side effect is aplastic anemia.1 Owing to the adverse health effects, most countries have banned the use of CAP in food-producing animals. In addition, it must not be detected in food at present. However, it has still been detected in egg, honey, milk, cosmetics, shrimp, and other aquaculture products because of the illegal use of CAP. This not only makes the environment deteriorated, but also poses a potential health hazard to humans.2 Therefore, the determination at low concentration levels and control of CAP is far more important than that of other antibiotics. A great number of detection assays have been successfully utilized for CAP analysis in some biological matrices, including liquid chromatography-tandem mass spectrometry (LC-MS/MS),3 microbiological method,4 enzyme immunoassay (ELISA),5 radioimmunoassay (RIA),6 gas chromatographic method (GC),7 capillary electrophoresis (CE)8 and others.However, the existing determination methods for CAP in a complex matrix often need expensive equipment, complicated sample extraction and separation, which are time consuming and laborious.9 A microbiological assay is facile and the costs are minimal, but it suffers from its inferior specificity and sensitivity. Thus, there is great demand to exploit a cost-effective, fast, convenient and sensitive method for on-site detection of CAP in biological samples. Owing to the special advantages of fluorescent nanosensors, such as excellent sensitivity and easy fabrication, a nanosensor assay on the basis of the fluorescence changes for the determination of CAP residue should be developed as the circumstances demand.
Because of the remarkable photochemical and physicochemical characteristics of fluorescent carbon dots, such as low toxicity, robust chemical inertness, high resistance to photobleaching, fairly good cell permeability and biocompatibility, the past two decades have witnessed a revolution in fluorescent carbon nanomaterials. Furthermore, carbon dots have the merits of cheap raw materials and ease of synthetic methods.10 These advantages make them have found broad potential applications, such as sensing,11 catalysis, biomedicine, drug delivery and optoelectronic devices,12 as well as some important burgeoning applications in the field of photoscience13–15 and energy conversion.16 To date, many fabrication methods of carbon dots have been developed using numerous organic materials or natural products as precursors, and they can usually be categorized as two types, “bottom-up” methods and “top-down” approaches.17
In order to further regulate and improve the inherent properties of carbon dots, including optical properties, surface activity, structural defects, and local chemical properties,18 some effective ways (such as heteroatom doping and surface modification) have been developed and verified. As a more convenient and general strategy, doping with heteroatoms has also shown great potential to significantly enhance the quantum yield of CDs, such as P, N, S, Si, B, Se and Mg.19–23 Because a nitrogen atom has five valence electrons, and closely resembles a carbon atom, it was found to be the most prominent dopant of carbon nanomaterials, resulting in some new phenomena and properties.24 In light of the fact that the electron-rich phosphorous (P) atom can act as an n-type donor and form substitutional defects in diamond sp3 thin films, doping with P atoms can improve the fluorescence and electronic properties of CDs, and contribute to the electron and hole transfer processes, opening up novel applications.25
Co-doping N and P atoms into CDs, the synergistic effect of both benefits is expected to facilitate the formation of band gaps and provide more active sites in CDs, resulting in unanticipated characterizations and high quantum yield (QY). To date, a number of approaches for the fabrication of N, P co-doped CDs (N,P-CDs) have been developed. For example, Xu et al.26 produced N,P-CDs through a low-cost hydrothermal method. They fabricated high photoluminescence quantum yield (PLQY) CDs with independent emission behavior using sodium citrate and diammonium phosphate as precursors at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. Spectroscopic investigations revealed that P tuned the band gap and led to the high PLQY of 53.8%. The excellent fluorescence properties of the N,P-CDs can be selectively and effectively quenched by Hg2+ ions with a linear response over the concentration range of 0–900 nM with a short response time. Sun et al.27 reported a rapid microwave-assisted pyrolysis method for the synthesis of N,P-CDs. A homogeneous aqueous solution containing m-phenylenediamine (mPDA), ortho-phosphoric acid and ethylenediamine (EDA) at the stoichiometric ratio of 1:3.2:1.6 was heated in a domestic 800 W microwave for 40 s. The obtained N,P-CDs exhibit very bright blue and green dual emissions, and nearly pH-independent emission properties. The dual emission properties lay the foundation to enlarge the use of N,P-CDs in ratiometric sensing or other applications. Chen et al.28 reported a simple method for the preparation of N,P-CDs by the carbonization of adenosine triphosphate (ATP) powder within Ar atmosphere at 900 °C. Subsequently, this was followed with chemical exfoliation by refluxing in HNO3 (6 M) for 24 h. The obtained ATP-CDs showed a high fluorescence quantum yield, strong two-photon upconversion, high photostability, and good biocompatibility. Most notably, they were successfully employed to image and real-time track transferrin receptors in live cells. Nevertheless, the preparation process of most N,P-CDs often requires the use of organic solvents or high temperature and long reaction times. Therefore, it is desired to find a facile method to synthesize high-quality N,P-CDs.
:1 molar ratio. Spectroscopic investigations revealed that P tuned the band gap and led to the high PLQY of 53.8%. The excellent fluorescence properties of the N,P-CDs can be selectively and effectively quenched by Hg2+ ions with a linear response over the concentration range of 0–900 nM with a short response time. Sun et al.27 reported a rapid microwave-assisted pyrolysis method for the synthesis of N,P-CDs. A homogeneous aqueous solution containing m-phenylenediamine (mPDA), ortho-phosphoric acid and ethylenediamine (EDA) at the stoichiometric ratio of 1:3.2:1.6 was heated in a domestic 800 W microwave for 40 s. The obtained N,P-CDs exhibit very bright blue and green dual emissions, and nearly pH-independent emission properties. The dual emission properties lay the foundation to enlarge the use of N,P-CDs in ratiometric sensing or other applications. Chen et al.28 reported a simple method for the preparation of N,P-CDs by the carbonization of adenosine triphosphate (ATP) powder within Ar atmosphere at 900 °C. Subsequently, this was followed with chemical exfoliation by refluxing in HNO3 (6 M) for 24 h. The obtained ATP-CDs showed a high fluorescence quantum yield, strong two-photon upconversion, high photostability, and good biocompatibility. Most notably, they were successfully employed to image and real-time track transferrin receptors in live cells. Nevertheless, the preparation process of most N,P-CDs often requires the use of organic solvents or high temperature and long reaction times. Therefore, it is desired to find a facile method to synthesize high-quality N,P-CDs.
In this research work, we report a facile spontaneous fabrication method to synthesize N,P-CDs through the use of acetic acid as the C source, and arginine, 1,2-ethylenediamine, and diphosphorus pentoxide as the N and P sources, respectively. The approach can swiftly synthesize a large amount of N,P-CDs without the need for high temperature, long reaction times and sophisticated manipulations. The synthesized N,P-CDs exhibited outstanding stability under the circumstances of UV illumination, high ionic strengths and long time storage. The intense fluorescence quenching of N,P-CDs by CAP has been explored and utilized for the selective determination of CAP. By comparison to conventional fluorescent probes, the developed nanosensors were more selective and sensitive, making them appropriate for the determination of CAP in biological samples with satisfactory results.
2. Experimental details
2.1. Raw materials
Chloramphenicol (CAP), streptomycin, florfenicol, arginine (Arg), ascorbic acid (AA), glucose, H2O2, melamine, uric acid, adenosine triphosphate (ATP) and quinine were purchased from Aladdin Chemistry Co., Ltd. (Shanghai, China). Gentamicin was obtained from Sigma-Aldrich, China. Cefuroxime axetil (CFA) was supplied by Titan Pharmaceutical Co., Ltd., Guangdong, China. Ethanol, methanol, 1,2-ethylenediamine (EDA), acetic acid, potassium hydrogen phthalate, sodium acetate, citric acid, sodium citrate, AgNO3, NaHCO3, CaCl2, MgCl2, CoCl2, BaCl2, NiCl2·6H2O, Zn(OOCCH3)2, CuSO4, HgCl2, Pb(NO3)2, FeCl3, AlCl3, KCl, NaCl, Na2SO4, NaNO3, Na2CO3, Na2S, Na2SO3, KSCN, KF, KCl, KBr, Na2HPO4, and diphosphorus pentoxide (P2O5) were acquired by Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). All reagents were of at least analytical grade, and used as received without any further purification treatment. Milk was obtained from the local supermarket. Purified water was deionized by a Milli-Q water purification system, and was used throughout this study.2.2. Instrumentations
Transmission electron microscopy (TEM) observations were performed on a JEM-2100 (HR) transmission electron microscope (TEM) (JEOL, Japan). Fourier transform infrared (FT-IR) spectra were obtained on a Shimadzu IRAffinity-1S FT-IR spectrometer (Kyoto, Japan) with the KBr pellet technique. Fluorescence excitation and emission spectra were measured on a RF-6000 spectrofluorophotometer (Shimadzu Corporation, Japan) equipped with a Xe lamp at ambient conditions. UV-Vis absorption spectra were recorded on a UV1800 UV-Vis spectrophotometer (Shimadzu Corporation, Japan). XPS measurements were carried out using an X-ray photoelectron spectrometer (K-Alpha, Thermo Fisher). Elemental analysis was conducted using an Elementar Vario EL III. The zeta potential was measured on a Zetasizer Nano ZS90 (Malvern, U.K.). Fluorescence photographs were taken using a Canon camera under excitation of 365 nm by a hand-held UV lamp.2.3. Synthesis of nitrogen and phosphorus co-doped carbon dots
Fluorescent N,P-CDs were synthesized as follows: arginine (0.125 g) was mixed with 2 mL of acetic acid under ultrasonication to form a homogeneous solution, and P2O5 (3.5 g) was heated to 100 °C and mixed with the solution. Then, 0.5 mL of 1,2-ethylenediamine was quickly added to the mixture in a 50 mL boiling flask without stirring. Because of the spontaneous heat of the acid–base neutralization, the mixture automatically reacted and the color changed to brown, and then the volume of the mixture quickly swelled by foaming. The reaction lasted for about 5 min, and gradually cooled to room temperature. The dark mixture was added with water to release the CDs and neutralized by NaHCO3, and then centrifuged at 12000 rpm for 20 min to collect the supernatant. The crude product was dialyzed in deionized water for 3 days to remove residual acetic acid and ions. Finally, the fluorescent CDs were concentrated in vacuum at 45 °C overnight.
2.4. Quantum yield measurements
Quinine sulfate (dissolved in 0.1 M H2SO4; QY = 0.54) was selected as a reference standard. The QY of N,P-CDs (in aqueous solution) was obtained using a slope method,29 comparing the gradient of the integrated fluorescence intensity vs. the absorbance value of the samples (<0.05 at λex = 350 nm) with that of the reference.The QY of the sample can be determined using the following equation:
| ϕx = ϕst (Kx/Kst) (ηx/ηst)2 |
2.5. Fluorescence detection of chloramphenicol
The fluorescence emission intensity was recorded at the emission peak at about 440 nm (excitation wavelength, 349 nm). The width of the excitation slit was adjusted to 3.0 nm, while the emission slit was set to 5.0 nm. First, a stock solution of N,P-CDs (100 μL) was pipetted into 3.0 mL phosphate buffer solution (pH = 5.0, 50 mM). Secondly, an appropriate aliquot of the CAP work solutions was successively injected. The final volume was adjusted to 5 mL with phosphate buffer. Subsequently, the mixture was vortexed and shaken for about 4 min, then subjected to fluorescence determination at room temperature. All intensity measurements were repeated three times so as to acquire an average value for quantitative analysis. F0 and F signified the fluorescence intensities of the N,P-CDs in the absence and presence of CAP, respectively. To investigate the specificity of the sensor, some small molecules, ions and antibiotics were separately added into the probe mixture, instead of CAP with the same experimental conditions.2.6. Detection of chloramphenicol in milk samples
Milk samples were obtained from the local supermarket. First, a known amount of CAP was spiked into the fresh milk solution and the mixture was vortexed for 5 min to prepare a homogeneous solution. Then, for the protein precipitation, 2 mL of the milk mixture was blended with 8 mL acetonitrile and ultrasonically treated for 20 min. Afterwards, the sample was centrifuged at 10000 rpm for 15 min to get rid of the denatured protein. The collected supernatant was filtered through a 0.22 μM hydrophobic microporous membrane to eliminate suspended particulate particles. At last, the treated milk sample was 10-fold diluted using phosphate buffer (50 mM, pH 5.0). Milk samples containing different concentrations of CAP were prepared and stored at 4 °C. Then, the concentrations of the added CAP in milk were detected following the identical procedure in the buffer solution, and were determined using the calibration curve that was acquired in buffer.
3. Results and discussion
3.1. Material characterization
The N,P-CDs were prepared by an automatic method thermally initiated by acid–base reaction. In brief, 0.125 g arginine was mixed with 2.0 mL acetic acid. The homogeneous mixture solution was added to 3.5 g of heated P2O5 in a 50 mL boiling flask without stirring, and then 0.5 mL EDA was quickly added to the flask to fabricate N,P-CDs. EDA swiftly reacted with acetic acid. Because this reaction is exothermic, the released heat was utilized to initiate the carbonization reaction. Owing to the boiling acetic acid from the self-heating, the volume of the foaming mixture inflated extremely rapidly. The color of the reaction mixture changed to become brownish black after about 20 s. The reaction lasted for about 5 min until the acetic acid was depleted. During the carbonization, many ultrafine bubbles were self-engendered to get rid of the redundant heat, but they were also exploited as the templates to control the volume of N,P-CDs. A comprehensive comparison on the synthesis methods of the heteroatom-doped carbon dots between the recently reported technologies and this study is summarized in Table 1. As seen therein, in contrast to the other fabrication methods of fluorescent carbon nanoparticles that required high temperature, long reaction times, or complicated operations, this is an automatic method to rapidly produce large quantities of CDs by simply mixing the acetic acid solution of arginine, P2O5, and EDA. The morphology and the structure of N,P-CDs were investigated by transmission electron microscopy (TEM). The TEM characterization of the fabricated N,P-CDs is depicted in Fig. 1a. It was found that the N,P-CDs were torispherical nanodots, and distributed evenly on the surface of a grid. A size histogram was obtained by plotting the statistics of roughly 90 particles (Fig. 1b). The Gaussian fitting curves indicated that N,P-CDs possessed a size distribution within the range of 3.6–7.2 nm with an average diameter of 5.4 nm. The high-resolution TEM (HRTEM) of N,P-CDs showed the presence of amorphous carbon, which possessed well-defined lattice fringes. The average in-plane lattice spacing was 0.20 nm (Fig. 1c), which resembles the (102) diffraction planes within graphitic (sp2) carbon.37| Synthesis method | Doped CDs | Solvent | Temperature | Reaction time | Ref. |
|---|---|---|---|---|---|
| Solvothermal | P-CDs | N-Methyl-2-pyrrolidone | 140 °C | 6 h | 30 |
| Hydrothermal | P,N-CDs | Water | 250 °C | 2 h | 31 |
| Solvothermal | N,S-CDs | Formamide | 160 °C | 4 h | 32 |
| Acid oxidation | N-CDs | Formic acid, H2O2 | Room-temperature | 20 h | 33 |
| Microwave | N-CDs | Formamide | 160 °C | 1 h | 34 |
| Pyrolysis | Cu-CDs | None | 250 °C | 2 h | 35 |
| Carbonization | Mo-CDs | [BHDABCO][Tf2N] | 200 °C | 20 min | 36 |
| Spontaneous heat | N,P-CDs | Solvent-free | Room-temperature | 5 min | This work |
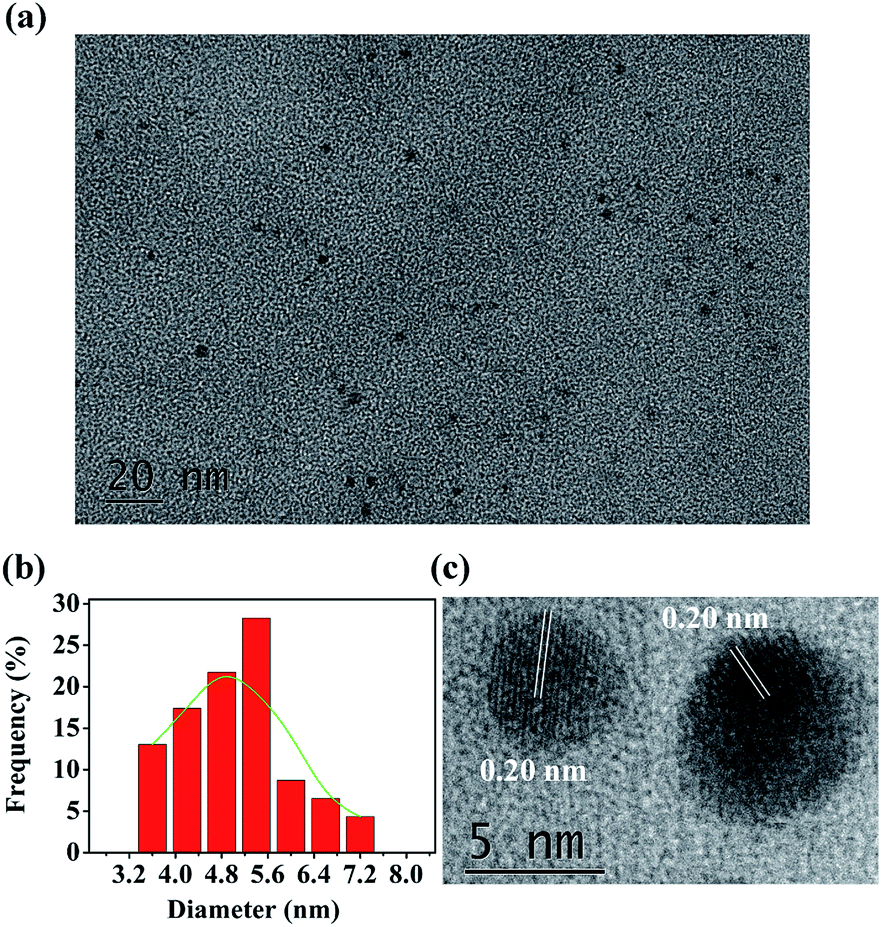 | ||
| Fig. 1 (a) TEM images of N,P-CDs. (b) The particle size distribution of the N,P-CDs. (c) HRTEM. | ||
3.2 Optical properties
The UV-Vis absorption of the fabricated N,P-CDs was investigated, and the spectra are shown in Fig. 2a. It was found that the N,P-CDs displayed two absorption peaks at 278 and 338 nm, which were possibly due to the n → π* transition of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band and the excited defect surface states induced by the heteroatoms of P/N, respectively.38 The PL spectra of the prepared N,P-CDs exhibited the excitation and emission peaks at 349 nm and 441 nm, respectively. The photograph of N,P-CDs in aqueous solution under 365 nm UV light showed bright blue fluorescence (inset of Fig. 2a).
O band and the excited defect surface states induced by the heteroatoms of P/N, respectively.38 The PL spectra of the prepared N,P-CDs exhibited the excitation and emission peaks at 349 nm and 441 nm, respectively. The photograph of N,P-CDs in aqueous solution under 365 nm UV light showed bright blue fluorescence (inset of Fig. 2a).
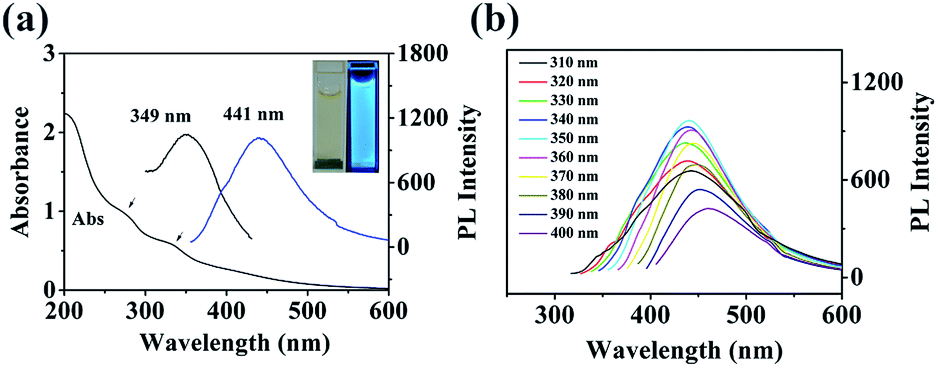 | ||
| Fig. 2 (a) UV-Vis absorption, maximum fluorescence excitation (349 nm), and emission (441 nm) spectra of N,P-CDs in aqueous solution. Insets are the photographs of the N,P-CDs in aqueous solution under visible light (left) and 365 nm UV light (right). (b) Excitation-dependent PL behavior of N,P-CDs. | ||
In order to further explore the optical characteristics of the synthesized N,P-CDs, the excitation-dependent PL behavior was examined and is demonstrated in Fig. 2b. Under the excitation wavelength from 310 to 350 nm, the maximum emission wavelength continued to be unvaried. With the excitation wavelength increasing from 350 to 400 nm, the emission wavelength of the N,P-CDs showed a red-shift and the peak intensity constantly declined. This behavior was also reported by Wang et al.39 The excitation-dependent fluorescence behavior was ascribed to the different surface-emissive states, size impact, morphologies, and sluggish solvent relaxation,40 and it was utilized in biological multicolor imaging. In addition, the QY of the prepared N,P-CDs was calculated to be 8.7%. The photostability of the prepared N,P-CDs was examined by constantly monitoring the fluorescence intensity at 440 nm. The results (Fig. S1†) demonstrated that there was no obvious photo-bleaching of N,P-CDs under constant UV irradiation (365 nm) for 4 h. The PL stability of the N,P-CDs was also investigated under various ionic strengths and pH. There was no obvious variation in the PL intensity and peak position in the presence of different concentrations of KCl (Fig. S2†). These results endow the practical applications of N,P-CDs under physiological conditions and high ionic strength conditions. The fabricated N,P-CDs illustrated pH-dependent PL behavior (Fig. S3†). At high pH (>8), the PL intensity decreased significantly with increasing pH. Conversely, at the pH range of 1–8, the PL intensities remained relatively constant, demonstrating its potential application under a wide pH environment. The dry powder of the N,P-CDs can be repeatedly redispersed in water, indicating its convenience for transportation and preservation. There was no precipitation observed in the homogeneous N,P-CDs aqueous solution even after several months under ambient conditions, demonstrating the long term stability of the colloidal suspension. The excellent PL properties of the N,P-CDs endow them with the advantages of the potential applications.
FTIR spectroscopy was performed to compare the functional groups of N,P-CDs with arginine. As shown in Fig. 3a, the differences between the spectra of the N,P-CDs and arginine were obvious. A wide absorption peak appeared at 3405 cm−1, which is attributed to the stretching vibrations of the O–H group, suggesting that the N,P-CDs possess a lot of hydroxyl groups.41 Sharp and high-intensity peaks emerged, which are assigned to phosphorus and/or oxygen-containing groups, such as P–O–C (1002 cm−1),31 hydrogen-bonded PO (1165 cm−1), CO and CC stretching vibrations (1662 cm−1).42 The characteristic peak at 1406 cm−1 is ascribed to the stretching vibration of C–N. The zeta potential (ζ) of N,P-CDs was measured to be −9.6 mV. As a matter of fact, the plentiful functional groups on the surface of N,P-CDs not only can make N,P-CDs negatively charged, but also enhance the stability and hydrophilicity in aqueous solution.
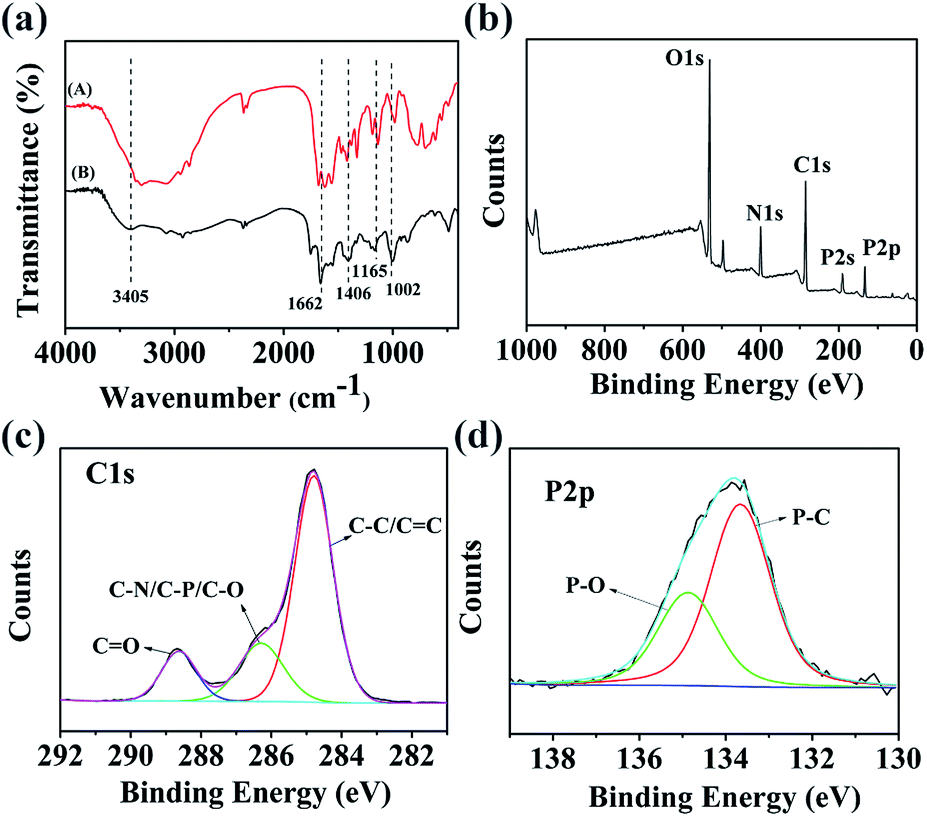 | ||
| Fig. 3 (a) FTIR spectra of arginine (A) and N,P-CDs (B). (b) XPS survey spectrum of N,P-CDs. High-resolution XPS spectra of (c) C 1s, (d) P 2p. | ||
In order to scrutinize the composition and chemical environment of the synthesized N,P-CDs, elemental analysis and X-ray photoelectron spectroscopy (XPS) were carried out. As shown in Fig. 3b, the XPS spectrum of N,P-CDs displays five peaks at 531, 400, 285, 191 and 133 eV, corresponding to O 1s, N 1s, C 1s, P 2s and P 2p, respectively. The XPS analysis result revealed that the N,P-CDs mostly comprised oxygen, carbon and nitrogen, with a small amount of phosphorus. The high resolution XPS spectrum of C 1s (Fig. 3c) was deconvoluted into three surface components, which can be attributed to C 1s states in CO, C–N/C–P/C–O and sp2 CC/C–C. This is in agreement with the binding energies of 288.68 eV, 286.28 eV and 284.78 eV, respectively.42 The high-resolution P 2p spectrum (Fig. 3d) points to the presence of two major phosphorus groups differentiated by their binding energies: P–O type groups (134.87 eV) and P–C groups (133.67 eV).43 The elemental analysis results reveal that the N,P-CDs were composed of C 43.62 wt%, H 2.01, N 13.15 wt%, P 7.75 wt% and O (calculated) 33.47 wt%, suggesting that they are nitrogen and phosphorus dual-doped CDs. Therefore, all data demonstrate the successful doping of N and P into the structure of the N,P-CDs.
3.3. Analytical performance of the N,P-CDs for chloramphenicol
Because the N,P-CDs possess strong fluorescence, unique optical characteristics, and good water solubility, they were expected to be utilized as a fluorescent sensor for the rapid and selective detection of CAP. Several influencing factors, such as the pH, temperature, buffer medium, volume of the mixture solution, and fluorescence quenching time of the N,P-CDs were examined. As shown in Fig. S3,† the fluorescence intensity of N,P-CDs is dependent on pH and comparatively low on alkali circumstances. In contrast, the fluorescence intensity of N,P-CDs remained high and stable under acidic conditions, so the subsequent experiments were carried out under pH = 5. The strongest fluorescence quenching was obtained in phosphate buffer solution (50 mM) in comparison to other buffer solutions, including potassium hydrogen phthalate, HAc–NaAc, and citric acid–sodium citrate. Thus, phosphate buffer solution was chosen in this study. The optimized reaction time, volume of buffer medium and incubation temperature were set at 4 min, 3 mL, and room temperature (25 °C), respectively.Under the above optimized conditions, CAP samples of variable concentrations were added to the N,P-CDs solution to investigate their analytical capabilities. As presented in Fig. 4a, with the increase of the CAP concentration from 0 to 160 μM, the fluorescence intensity of the N,P-CDs gradually declines, indicating the fluorescence quenching of N,P-CDs toward CAP. Fig. 4b depicts the relationship between the concentration of CAP and the fluorescence quenching efficiency, which reached a platform at higher concentrations, and a good linear curve was acquired ranging from 0.8 to 70 μM (inset of Fig. 4b). The equation of the fitted curve can be described as y = 0.011x + 0.016 with R2 = 0.994 (where y represents the fluorescence quenching efficiency, and x denotes the concentration of CAP). The limit of detection was calculated to be 0.36 μM based on a signal/noise ratio of 3. This developed fluorescent sensor rapidly and conveniently determined CAP, indicating that N,P-CDs could successfully be utilized to supervise CAP on real-time.
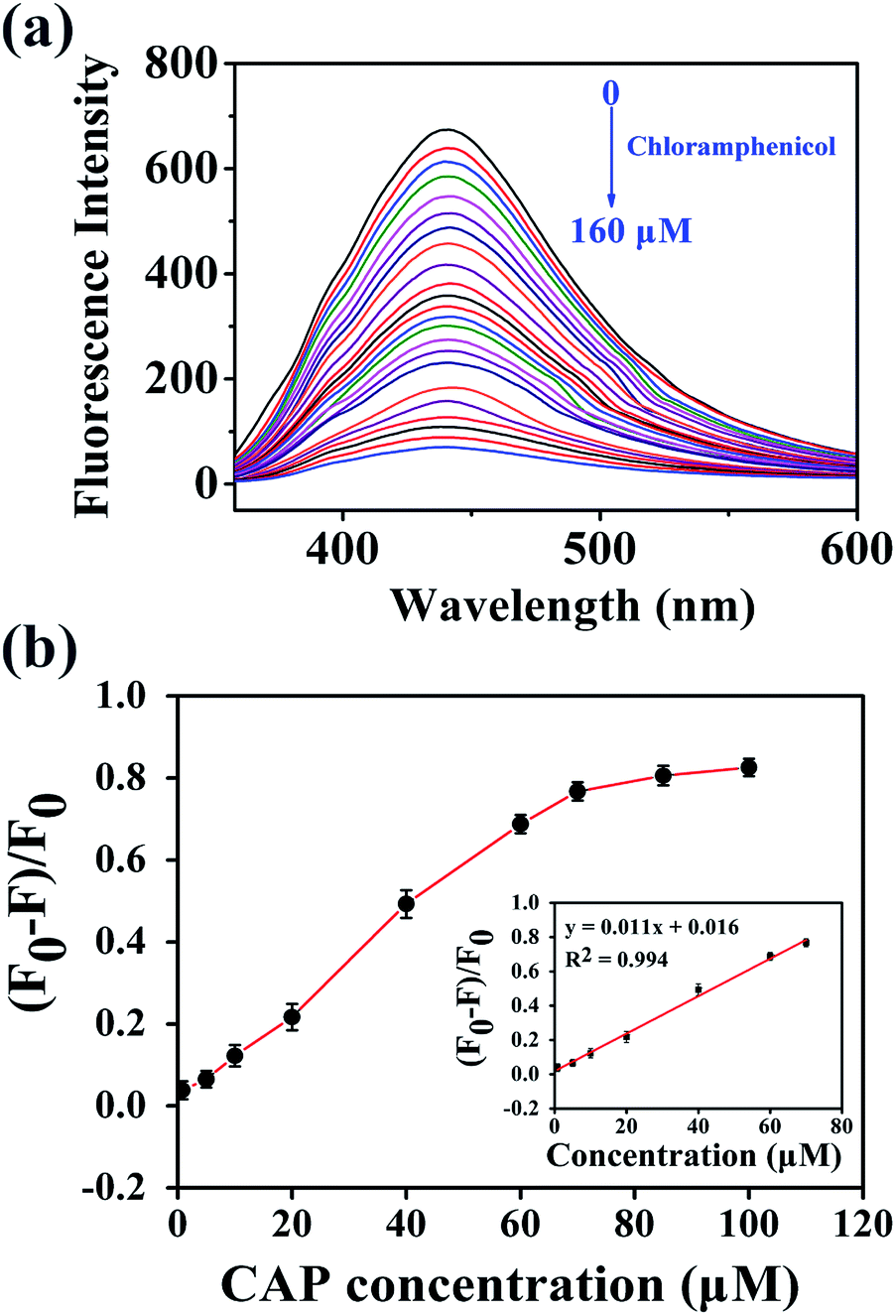 | ||
| Fig. 4 (a) Fluorescence responses of N,P-CDs after injection of various concentrations of chloramphenicol. (b) Fitted curve between the fluorescence quenching efficiency ((F0 − F)/F0) and chloramphenicol concentration. Inset: linear relationship between the fluorescence quenching efficiency and chloramphenicol. | ||
3.4. Mechanism of fluorescence response of the N,P-CDs toward chloramphenicol
At present, various fluorescence-probing systems have been designed and applied. The fluorescence quenching process is usually related to the following principles, such as dynamic quenching effect (DQE), electron transfer (ET), inner filter effect (IFE), static quenching effect (SQE), fluorescence resonance energy transfer (FRET), and intramolecular charge transfer (ICT).44 In order to explore the principle of the CAP-induced fluorescence quench of N,P-CDs, the emission and excitation spectra of the fluorescent N,P-CDs and UV-Vis absorption of CAP were examined. As demonstrated in Fig. S4,† no overlap was observed between the fluorescence emission spectrum of the N,P-CDs and UV-Vis absorption spectrum of CAP. FRET will occur only under the condition that there is a huge overlap between the emission spectrum of the fluorophore (donor) and the absorption spectrum of the quencher (acceptor). Meanwhile, the distance between them (fluorescence donor and acceptor) is not more than 10 nm.45 Therefore, FRET does not exist in this fluorescence quenching process. Furthermore, there is little overlap between the UV absorption of CAP in pH 5.0 phosphate buffer solution and the fluorescence excitation spectrum of N,P-CDs (Fig. S4†). This indicated that the predominant quenching mechanism could not be attributed to IFE in this probing system.In order to explore the probable mechanism of fluorescence quenching, the FTIR spectra of N,P-CDs and N,P-CDs-CAP were also investigated and compared. It was noticed that the peak position at 3456 cm−1 of the hydroxyl stretching vibration (Fig. S5,† red line) in the N,P-CD–CAP system was remarkably red-shifted by 51 cm−1, compared with that of the original N,P-CDs (3405 cm−1, Fig. S5,† black line), suggesting that the strong hydrogen bonding interactions between the N,P-CDs and CAP were successfully formed. Based on the experimental results, we propose that the N,P-CDs can react with CAP based on two types of interactions: one is hydrogen-bonding interactions between hydroxyl groups on the surface of N,P-CDs and CAP; the other is π–π stacking between the phenyl group of CAP and the aromatic heterocyclic part of N,P-CDs. Through these particular interactions, N,P-CDs firmly target CAP to act as nanoprobes for the detection of CAP. The high affinity between N,P-CDs and CAP could facilitate ICT, leading to the fluorescence quenching of N,P-CDs.46 Therefore, the quenching mechanism was regarded to be predominantly ICT during this fluorescence quenching process.
3.5. Selectivity of the N,P-CDs probe for chloramphenicol analysis
The complexity of biological and environmental samples remains a tremendous challenge for us concerning the determination of therapeutic drugs. Therefore, the selectivity of the probe was investigated during this research. The fluorescence intensities of N,P-CDs were measured under the same conditions in the presence of the potential interferent components and several antibiotics as follows: some metal ions (Na+, K+, Ag+, Ca2+, Cu2+, Mg2+, Zn2+, Co2+, Ba2+, Pb2+, Hg2+, Ni2+, Fe3+, and Al3+), anions (NO3−, CO32−, S2−, SO32−, SO42−, HPO42−, SCN−, Br−, Cl−, and F−), some small molecules (AA, glucose, H2O2, melamine, uric acid, and ATP), and antibiotics (streptomycin, gentamicin, CFA, florfenicol). As shown in Fig. 5a, it was found that most anions and metal cations did not have an obvious effect on the fluorescence intensity of N,P-CDs, even when the concentrations of the ions were as high as that of CAP. Usually, the concentration of the interferent chemicals is comparatively low in the tested samples. Hence, the above mentioned substances cannot bring about any interference to the CAP detection. As illustrated in Fig. 5b, only CAP can induce an obvious fluorescence quenching of N,P-CDs, whereas other small molecules and antibiotics (100 μM) only render a negligible fluorescence variation of N,P-CDs. These results suggest that the N,P-CDs-based fluorescent nanosensors exhibit obvious superiorities in their rapid response and specificity, so it can be potentially utilized for CAP analysis in biological and environmental samples.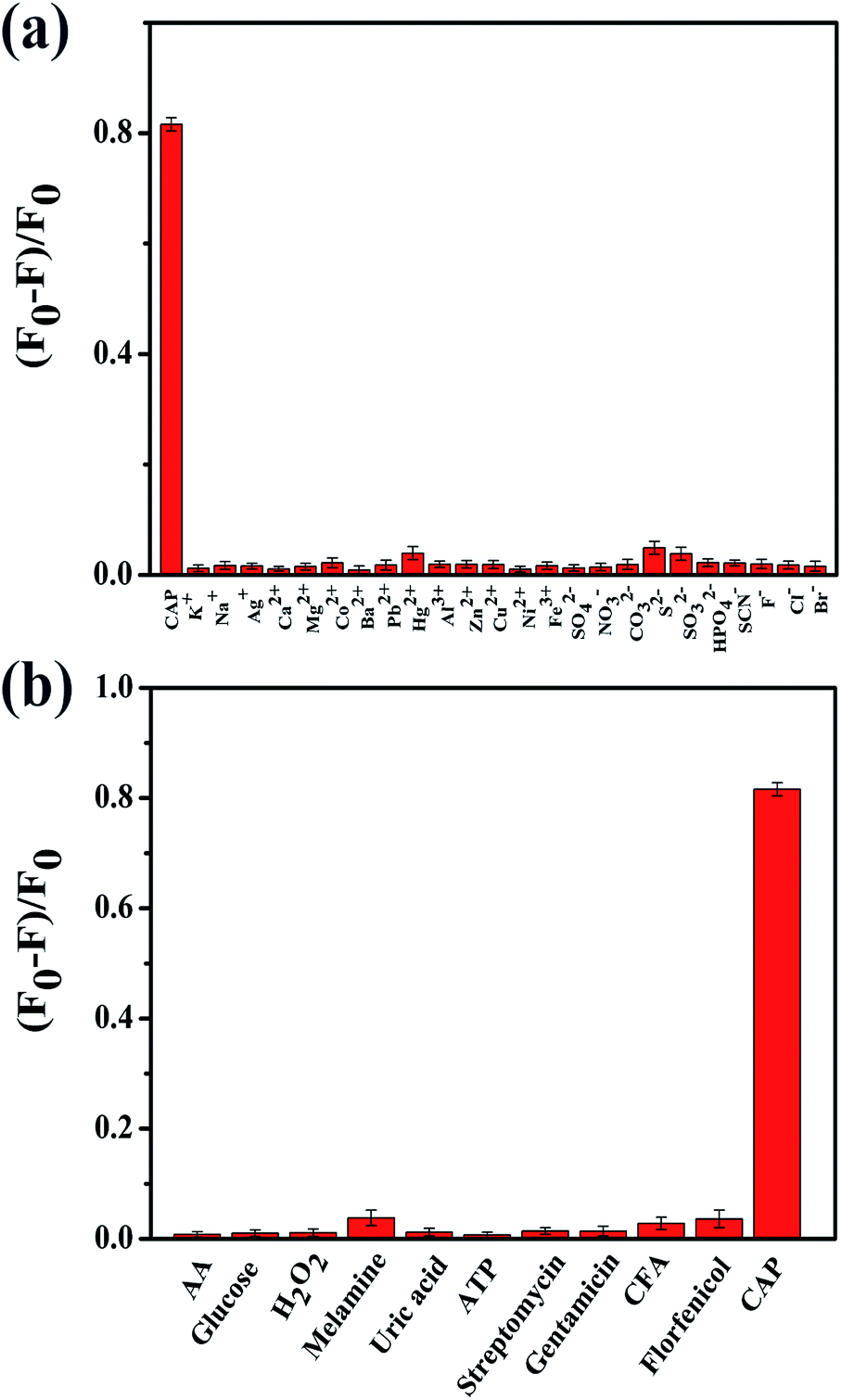 | ||
| Fig. 5 (a) Fluorescence quenching efficiency of CAP (100 μM) and some inorganic ions (100 μM) toward N,P-CDs. (b) Fluorescence quenching efficiency of small molecules and antibiotics (100 μM) toward N,P-CDs. The experiments were performed in pH 5.0 phosphate buffer. | ||
3.6. Chloramphenicol detection of real samples
Owing to the high efficiency of the probe conducted in phosphate buffer, the developed method was employed for the analysis of CAP in real samples. Four individual milk samples were detected, and the results are listed in Table S1.† As shown therein, CAP was not detected in the investigated samples. Then, the milk sample was spiked with standard solution of CAP and mixed. After protein denaturation, precipitation, and filtration, the pre-treated milk samples were diluted 10-fold using phosphate buffer solution and subjected to analysis in the probe solution. As shown in Table S1,† the obtained recoveries of the samples added with different amounts of standard CAP solution vary from 95.60% to 104.32%. The relative standard deviations (RSD) of the samples are from 2.13% to 4.42%, suggesting high precision and repeatability. Therefore, the developed assay is acceptable for the determination of CAP in biological and environmental samples.4. Conclusions
In summary, nitrogen and phosphorus co-doped CDs were successfully fabricated through an automatic route using acetic acid as the carbon source, and arginine, 1,2-ethylenediamine (EDA) and P2O5 as the dopants, respectively. This method can rapidly produce large quantities of N,P-CDs, in contrast to other synthesis methods that require high temperature, long reaction times, or complicated operations. The morphologies, optical properties, and chemical structures have been investigated by TEM, UV-Vis absorption and photoluminescence spectroscopy, FTIR, and XPS. N,P-CDs were successfully used as a fluorescent sensor to assay CAP based on the ICT mechanism. The constructed nanoprobes show high sensitivity and excellent selectivity, and have been successfully utilized for CAP determination in milk samples. The results of the recoveries and RSD were acceptable, indicating that the developed assay was capable of CAP determination in complicated samples. Considering its rapid response and facile manipulation, it is promising that the N,P-CDs would find increasing utilizations in a broad range of fields, for example, food monitoring and environmental controlling.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the Key Research & Development Program of Shandong Province (2018GGX109006), China Scholarship Council.Notes and references
- G. Lofrano, G. Libralato, R. Adinolfi, A. Siciliano, P. Iannece, M. Guida, M. Giugni, A. Volpi Ghirardini and M. Carotenuto, Ecotoxicol. Environ. Saf., 2016, 123, 65–71 CrossRef.
- H. Zhang, Y. Zhou, Y. Huang, L. Wu, X. Liu and Y. Luo, Chemosphere, 2016, 152, 229–237 CrossRef.
- A. F. Forti, G. Campana, A. Simonella, M. Multari and G. Scortichini, Anal. Chim. Acta, 2005, 529, 257–263 CrossRef.
- A. Parry-Hanson Kunadu, M. Holmes, E. L. Miller and A. J. Grant, Int. J. Food Microbiol., 2018, 277, 41–49 CrossRef.
- S. Wu, H. Zhang, Z. Shi, N. Duan, C. Fang, S. Dai and Z. Wang, Food Control, 2015, 50, 597–604 CrossRef.
- L. Lynas, D. Currie, C. T. Elliott, J. D. G. McEvoy and S. A. Hewitt, Analyst, 1998, 123, 2773–2777 RSC.
- H.-Y. Shen and H.-L. Jiang, Anal. Chim. Acta, 2005, 535, 33–41 CrossRef.
- C. Zhang, S. Wang, G. Fang, Y. Zhang and L. Jiang, Electrophoresis, 2008, 29, 3422–3428 CrossRef CAS.
- L. R. Guidi, P. A. Tette, C. Fernandes, L. H. M. Silva and M. B. A. Gloria, Talanta, 2017, 162, 324–338 CrossRef CAS.
- O. S. Wolfbeis, Chem. Soc. Rev., 2015, 44, 4743–4768 RSC.
- S. Sun, Q. Guan, Y. Liu, B. Wei, Y. Yang and Z. Yu, Chin. Chem. Lett., 2019, 30, 1051–1054 CrossRef CAS.
- S. Y. Lim, W. Shen and Z. Gao, Chem. Soc. Rev., 2015, 44, 362–381 RSC.
- S. Lu, G. Xiao, L. Sui, T. Feng, X. Yong, S. Zhu, B. Li, Z. Liu, B. Zou, M. Jin, J. S. Tse, H. Yan and B. Yang, Angew. Chem., Int. Ed., 2017, 56, 6187–6191 CrossRef CAS.
- S. Lu, L. Sui, J. Liu, S. Zhu, A. Chen, M. Jin and B. Yang, Adv. Mater., 2017, 29, 1603443 CrossRef.
- Q. Wang, S. Zhang, B. Wang, X. Yang, B. Zou, B. Yang and S. Lu, Nanoscale Horiz., 2019, 4, 1227–1231 RSC.
- W. Li, Y. Liu, M. Wu, X. Feng, S. A. T. Redfern, Y. Shang, X. Yong, T. Feng, K. Wu, Z. Liu, B. Li, Z. Chen, J. S. Tse, S. Lu and B. Yang, Adv. Mater., 2018, 30, 1800676 CrossRef.
- P. Zuo, X. Lu, Z. Sun, Y. Guo and H. He, Microchim. Acta, 2016, 183, 519–542 CrossRef.
- B.-P. Qi, H. Hu, L. Bao, Z. L. Zhang, B. Tang, Y. Peng, B. S. Wang and D. W. Pang, Nanoscale, 2015, 7, 5969–5973 RSC.
- P. Zuo, J. Liu, H. Guo, C. Wang, H. Liu, Z. Zhang and Q. Liu, Anal. Bioanal. Chem., 2019, 411, 1647–1657 CrossRef.
- G. Gao, Y. W. Jiang, H. R. Jia, J. Yang and F. G. Wu, Carbon, 2018, 134, 232–243 CrossRef.
- Y. Jia, Y. Hu, Y. Li, Q. Zeng, X. Jiang and Z. Cheng, Microchim. Acta, 2019, 186, 84 CrossRef.
- F. Li, T. Li, C. Sun, J. Xia, Y. Jiao and H. Xu, Angew. Chem., Int. Ed., 2017, 56, 9910–9914 CrossRef CAS.
- A. Bhati, S. R. Anand, G. Kumar, A. K. Garg, P. Khare and S. K. Sonkar, ACS Sustainable Chem. Eng., 2018, 6, 9246–9256 CrossRef.
- W. Wei, H. Liang, K. Parvez, X. Zhuang, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2014, 126, 1596–1600 CrossRef.
- F. Yan, Z. Sun, H. Zhang, X. Sun, Y. Jiang and Z. Bai, Microchim. Acta, 2019, 186, 583 CrossRef.
- Q. Xu, B. Li, Y. Ye, W. Cai, W. Li, C. Yang, Y. Chen, M. Xu, N. Li, X. Zheng, J. Street, Y. Luo and L. Cai, Nano Res., 2018, 11, 3691–3701 CrossRef.
- X. Sun, C. Brückner and Y. Lei, Nanoscale, 2015, 7, 17278–17282 RSC.
- A. Ananthanarayanan, Y. Wang, P. Routh, M. A. Sk, A. Than, M. Lin, J. Zhang, J. Chen, H. Sun and P. Chen, Nanoscale, 2015, 7, 8159–8165 RSC.
- J. Zhou, H. Zhou, J. Tang, S. Deng, F. Yan, W. Li and M. Qu, Microchim. Acta, 2017, 184, 343–368 CrossRef CAS.
- M. Wang, Y. Liang, Y. Liu, G. Ren, Z. Zhang, S. Wu and J. Shen, Analyst, 2018, 143, 5822–5833 RSC.
- N. Parvin and T. K. Mandal, Microchim. Acta, 2017, 184, 1117–1125 CrossRef CAS.
- S. Qian, L. Qiao, W. Xu, K. Jiang, Y. Wang and H. Lin, Talanta, 2019, 194, 598–603 CrossRef.
- X. Meng, Q. Chang, C. Xue, J. Yang and S. Hu, Chem. Commun., 2017, 53, 3074–3077 RSC.
- L. Pan, S. Sun, A. Zhang, K. Jiang, L. Zhang, C. Dong, Q. Huang, A. Wu and H. Lin, Adv. Mater., 2015, 27, 7782–7787 CrossRef.
- W. Wu, L. Zhan, W. Fan, J. Song, X. Li, Z. Li, R. Wang, J. Zhang, J. Zheng, M. Wu and H. Zeng, Angew. Chem., Int. Ed., 2015, 54, 6540–6544 CrossRef.
- T. Cai, Y. Zhang, D. Liu, D. Tong and S. Liu, Mater. Lett., 2019, 250, 20–24 CrossRef.
- Y. Fang, S. Guo, D. Li, C. Zhu, W. Ren, S. Dong and E. Wang, ACS Nano, 2012, 6, 400–409 CrossRef CAS.
- X. Gong, W. Lu, M. C. Paau, Q. Hu, X. Wu, S. Shuang, C. Dong and M. M. F. Choi, Anal. Chim. Acta, 2015, 861, 74–84 CrossRef CAS.
- M. Wang, Y. Liu, G. Ren, W. Wang, S. Wu and J. Shen, Anal. Chim. Acta, 2018, 1032, 154–162 CrossRef CAS.
- J. Schneider, C. J. Reckmeier, X. Yuan, M. V. Seckendorff, A. S. Susha, P. Kasak and A. L. Rogach, J. Phys.Chem. C, 2017, 121, 2014–2022 CrossRef CAS.
- Y.-C. Lu, J. Chen, A.-J. Wang, N. Bao, J.-J. Feng, W. Wang and L. Shao, J. Mater. Chem. C, 2015, 3, 73–78 RSC.
- X. Gong, Q. Zhang, Y. Gao, S. Shuang, M. M. F. Choi and C. Dong, ACS Appl. Mater. Interfaces, 2016, 8, 11288–11297 CrossRef CAS.
- L. Lin, Y. Wang, Y. Xiao and W. Liu, Microchim. Acta, 2019, 186, 147 CrossRef.
- J. Zhang, F. Cheng, J. Li, J.-J. Zhu and Y. Lu, Nano Today, 2016, 11, 309–329 CrossRef CAS.
- Y. S. Ma, Y. Cen, M. Sohail, G. H. Xu, F. D. Wei, M. L. Shi, X. M. Xu, Y. Y. Song, Y. J. Ma and Q. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 33011–33019 CrossRef CAS.
- W. Shi, F. Guo, M. Han, S. Yuan, W. Guan, H. Li, H. Huang, Y. Liu and Z. Kang, J. Mater. Chem. B, 2017, 5, 3293–3299 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04228e |
| This journal is © The Royal Society of Chemistry 2020 |