
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly sensitive fluorescent immunosensor for sensitive detection of nuclear matrix protein 22 as biomarker for early stage diagnosis of bladder cancer†
Hazha Omar Othman ab,
Foad Salehniab,
Neda Fakhric,
Rebwar Hassana,
Morteza Hosseini*dg,
Azad Faizullaha,
Mohammad Reza Ganjalibe and
Seyed Mohammad Kazem Aghamirf
ab,
Foad Salehniab,
Neda Fakhric,
Rebwar Hassana,
Morteza Hosseini*dg,
Azad Faizullaha,
Mohammad Reza Ganjalibe and
Seyed Mohammad Kazem Aghamirf
aChemistry Department, College of Science, Salahaddin University-Erbil, Iraq
bCenter of Excellence in Electrochemistry, Faculty of Chemistry, University of Tehran, Tehran 1417614418, Iran
cSchool of Chemical Engineering, College of Engineering, University of Tehran, Tehran 1417614418, Iran
dDepartment of Life Science Engineering, Faculty of New Sciences & Technologies, University of Tehran, Tehran 1417614418, Iran. E-mail: smhosseini@khayam.ut.ac.ir
eBiosensor Research Center, Endocrinology and Metabolism Molecular-Cellular Sciences Institute, Tehran University of Medical Sciences, Tehran 1417614418, Iran
fUrology Research Center, Tehran University of Medical Sciences, Tehran, Iran
gMedicinal Biomaterials Research Center, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran
First published on 4th August 2020
Abstract
A novel strategy is reported for highly sensitive, rapid, and selective detection of nuclear matrix protein NMP22 using two-color quantum dots based on fluorescence resonance energy transfer (FRET). Quantum dots (QDs) are highly advantageous for biological imaging and analysis, particularly when combined with (FRET) properties of semiconductor quantum dot (QDs) are ideal for biological analysis to improve sensitivity and accuracy. In this FRET system narrowly dispersed green emitting quantum dot CdTe core is used as a donor and labelled by monoclonal (mAb) antibody, while orange emitting quantum dot CdTe/CdS core shell is used as an accepter and labelled by polyclonal (pAb) antibody. The quantum dots are labelled by antibodies using EDC/NHS as crosslinking agent. Bovine serum albumin (BSA) solution was added to block nonspecific binding sites. The fluorescence intensity of QDs accepter decreased linearly with the increasing concentrations of NMP22 from 2–22 pg mL−1 due to FRET system and fluoroimmunoassay reaction. This method has good regression coefficient (R2 = 0.998) and detection limit was 0.05 pg mL−1. The proposed FRET-based immunosensor provides a quick, simple and sensitive immunoassay tool for protein detection, and can be considered as a promising approach for clinical applications. The proposed FRET-based immunosensor provides a quick, simple and sensitive immunoassay tool for protein detection, and can be considered as a promising approach for clinical applications.
1 Introduction
Bladder cancer (BC) is the second most prevalent genitourinary disease after malignant prostate cancer. Initial diagnosis and monitoring of BC are carried out by histopathologic investigation of biopsy specimens, which could be acquired by cystoscopy.1,2 Early screening of cancer is attractive as most tumors are detectable only when they achieve a specific size when they contain a large number of cells that may already have metastasized. Currently employed diagnostic techniques, for example, medical imaging, tissue biopsy and bioanalytical test of body fluids by enzyme-linked immunosorbent assay (ELISA) are insufficiently sensitive and specific to recognize most kinds of early-stage cancer. Besides, these assays are labor intensive, tedious, costly and do not have multiplexing capability.3Among the noninvasive techniques, urinary molecular of bladder malignant growth tests such as Nuclear Matrix Protein 22 (NMP22) have recently been introduced into clinical practice. NMP22 can recognize as the tumor.4 The clinical judgment value in urine which set by the US Food and Drug Administration is 6.5 U mL−1. Thus, the sensitive and precise detection of NMP22 is of significance to the early diagnosis of BC.1 Different kinds of biomarkers, for example, proteins, specific DNA or mRNA sequences, and circulating tumor cells, have been proposed for early cancer diagnosis.3
Until this point in time, the most commonly used detection technique of NMP22 in urine samples was enzyme-linked immunosorbent assay.1 Immunoassays depend on the reaction between antibody and antigen, and are mostly comprised of enzyme-linked immunosorbent assays (ELISA), immune chromatographic assays (ICAs), and other immunoassays joined with nanomaterials (gold nanoparticles (GNPs), quantum dots (QDs) and up-conversion materials) or technologies (surface enhanced Raman spectroscopy). These methods have been widely used in medical diagnostics, and have many advantages as they are fast, simple to operate, economical, and sensitive.4
Colloidal semiconductor nanocrystals, or “quantum dots” (QDs), are notable for their bright and narrow spectral photoluminescence (PL), which is additionally impervious to photobleaching.4,5 Organic fluorescent dyes have several drawbacks that have limited their usefulness as molecular imaging tags. Their low photobleaching threshold and broad absorption/emission peak width have hindered their use in long term imaging and multiplexing (detecting multiple labels simultaneously).6 QDs have properties that overcome these limitations of the organic fluorescent dyes including high resistance to photobleaching7 and broad-band absorption with narrow emission bands ranging from UV to NIR.8,9 In conclusion, semiconductor QDs are considered as another class of luminescent probes well suited for clinical medication and biological research.10,11 Luminescent nanocrystal QDs represent spherical particles with diameters in the range of 1–15 nm. Their typical core–shell structure and material composition reflect their impressive opto-chemical properties, such as size-tunable emission, excellent signal brightness, and nearly no photo-bleaching while charged ligands on the surface provide water solubility and biocompatibility. The capacity to link QDs with different bioactive molecules without losing the characteristics, as stated priory, offers wide-field applications.12–14
Adding to the utility of QDs is their capacity to take part in Förster resonance energy transfer (FRET).15,16 FRET is a valuable phenomenon which depends on the exchange of electromagnetic energy between two fluorescently labelled molecules (donor and acceptor), which are about 1–10 nm apart. The fluorescent pairs, termed as fluorophores or chromophores, are dipoles that facilitate the non-radiative transfer of energy. During FRET, an excited donor fluorophore transmits its excitation energy to the adjacent acceptor fluorophore through long-range intermolecular dipole–dipole interaction upon the overlap of their respective emission and absorption spectra.17–19
This study was conducted to develop a highly-sensitive and rapid FRET-based nano-biosensor to detect NMP22 in urine samples. The principle is based on energy transfer between two QDs, named as QD(D) (emission at 530) and QD(A) (emission at 610). The QD(D) conjugated anti-NMP22 monoclonal antibodies (mAb) and QD(A) conjugated with anti-NMP22 polyclonal antibodies (pAb) constructed the two moieties of the nano-biosensor. The QD(D)–mAb formed an immune complex with the QD(A)–pAb. The closeness of both of the QDs resulted in facilitated energy transfer from the donor to the acceptor. Existing of NMP22 in a specimen, the pAb competitively detached and replaced by NMP22 that led to reducing the fluorescence intensity of QD(A)–pAb due to the breaking of FRET mechanism (Fig. 1).
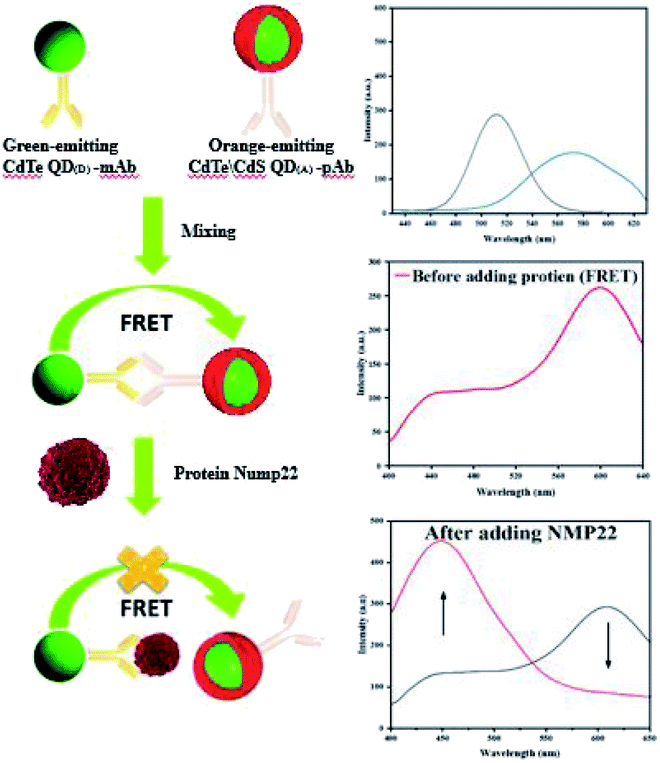 | ||
| Fig. 1 Illustration of FRET based detection of NMP22. | ||
2 Experimental section
2.1 Materials
NaBH4 (Merck, 99%), tellurium powder (Merck, 99%), CdCl2, thiourea, CdSO4, thioglycolic acid (TGA) (Merck, 98%), Na2S2O3 (Merck, 99%) were obtained from Merck Inc (Darmstadt, Germany, http://www.merck.de). Acetic acid, sodium acetate trihydrate, potassium dihydrogen phosphate, and sodium hydroxide were purchased from Sigma Aldrich (St. Louis, MO, USA, https://www.sigmaaldrich.com/). 1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) and N-hydroxysulfosuccinimide (sulfo-NHS) were also obtained from Sigma Aldrich.The NMP22 antibody (anti-NMP22) and NMP22 were purchased from DNA biotech Co. (Tehran, Iran, http://dnabiotech.ir/). Bovine serum albumin (BSA) was acquired from TAKARA Bio Inc. (http://www.takara-bio.com/). Ultrapure water was used in all syntheses. All reagents were of scientific reagent grade, and used as received without further purification. All experiments on the human samples were performed in accordance with the Declaration of Helsinki and approved by the ethics committee at the University of Tehran. Informed consents were obtained from human participants of this study.
2.2 Synthesis of CdTe core (QD(D))
QD formation contains the nucleation and growth process. In general, QDs core covered with inorganic materials to form core–shell structures.20 Supersaturating of monomers follows the burst of nucleation. Different concentration of monomer in the framework consumed then followed by growth of nuclei. Thermodynamics and kinetics contribute to nanocrystal growth and various shapes with different size distributions of QDs.21In this work, a CdTe core was synthesized to act as QD(D). For preparation of CdTe quantum dots, the thermochemical procedure was conducted in aqueous solution. The core CdTe QDs were synthesized using NaHTe and CdSO4 as the precursors and thioglycolic acid (TGA) as the capping agent. The NaHTe solution was prepared by a reaction between sodium borohydride (NaBH4) and tellurium powder. Briefly, 300 mg of sodium borohydride and 100 mL of deionized water were moved to a three-neck flask. Next, 100 mg of tellurium powder was added, and the solution was vigorously stirred under argon gas flow, which resulted in the formation of a pink solution. The prepared solution was stirred for 1 h until the pink solution became relatively colorless. In parallel, 300 mg CdSO4 was dissolved in 50 mL D.I. water, and 0.2 mL of TGA was added to this solution (to obtain Cd-TGA solution). Then, the solution pH was adjusted to 8.5 using a few drops of NaOH solution. At that point, the freshly prepared Ar-purged Cd-TGA solution was extracted to a syringe (50 mL) and was then injected quickly into the NaHTe solution (in the three-neck flask) and mixed for 2 h at room temperature. The obtained CdTe QD(D) we precipitated with cold ethanol for purifying QD(D). The precipitated separated by centrifuge 5000 rpm min−1 and re-dissolved in ultrapure water. The process repeated for three times to remove excess salts and the purified CdTe QD(D) were dried over night at room temperature in vacuum. The solution of 10 ppm is prepared in 50 mL and stored in refrigerator for further use.
2.3 Growth of CdS shell around CdTe core (QD(A))
A basic method for the development of CdS shell around a CdTe core was carried out to prepare QD(A). Thiourea is a heat sensitive material releasing sulfur species and free electron required for the CdS formation reaction. A solution of CdCl2, TGA, and thiourea was prepared as shell precursor, and NaOH was used to adjust the pH of the solution at 9. Initially, 45 mg CdCl2, 15 mg thiourea and 40 μL TGA were dissolved in 20 mL deionized water and stirred vigorously. Then, 6 mL of 10 ppm of as-prepared CdTe QDs added to the solution. The prepared solution was heated at 90 °C for 12 h to obtain CdTe/CdS core/shell structure.3 Results and discussion
3.1 Physical characterization
Science CdTe/CdS core/shell were prepared from the same batch of CdTe core QDs, the CdS shell thicknesses can be determined by subtraction of the core size from that of the prepared core/shell particles. HR-TEM images for the CdTe and CdTe/CdS are shown in Fig. 2(a) and (b) respectively. As its clear the particles monodispersed and spherical in shapes. The average diameter of CdTe core 3.5 nm. After shell growth processing, it can be clearly seen that a uniform shell formed around the CdTe cores to get CdTe/CdS core/shell. Spherical particles can also be observed, and the average diameter of the core/shell is about 5.4 nm. Fig. 2(c). Shows the XRD patterns of CdTe and CdTe/CdS core–shell QDs. The positions of the three main peaks for core correspond to crystalline planes of the cubic phase CdTe. Broadening of diffraction peaks demonstrates the development of the nanodot-sized CdTe. After shell growth processing, the focal peaks of CdS showing up in XRD patterns prove that the crystalline CdS shells have covered the CdTe cores appropriately.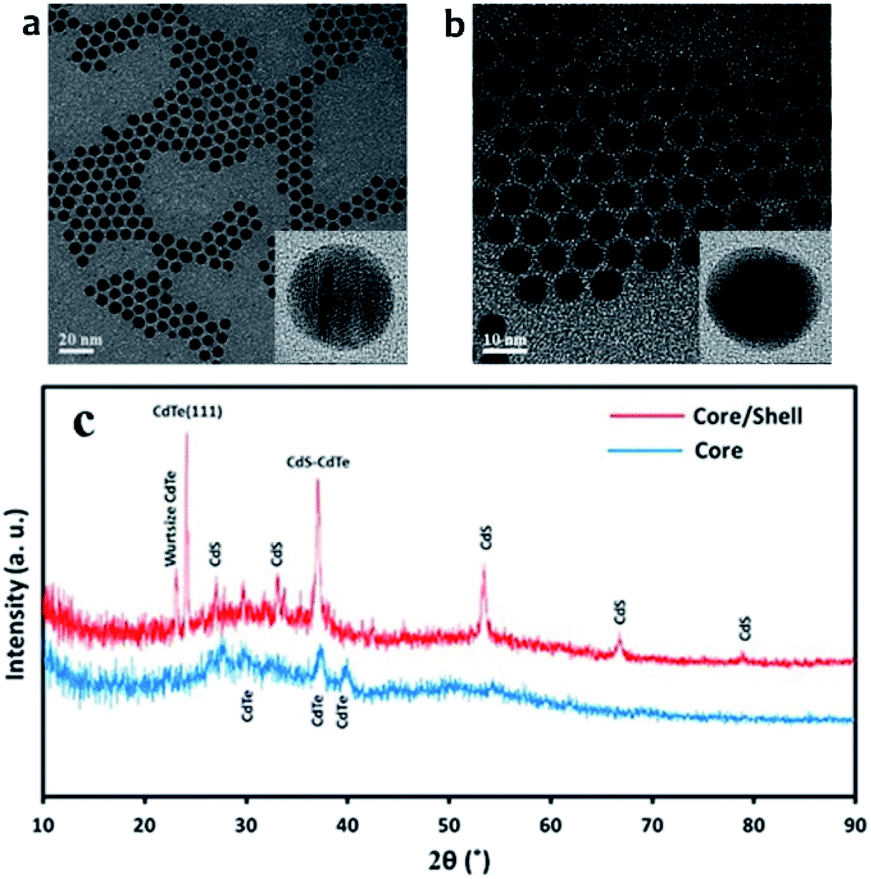 | ||
| Fig. 2 TEM image for (a) CdTe core quantum dot (b) CdTe/CdS core–shell quantum dot and (c) is X-ray diffraction (XRD) for CdTe core and CdTe/CdS core–shell. | ||
The as-synthesized TGA capped CdTe QDs and CdTe/CdS nanocomposites FT-IR spectra are shown in Fig. S1.† In both samples, a broad absorption band was observed around 3400 cm−1 corresponding to O–H vibrations of the aqueous solution. When we compare CH2 vibrations of capped layer of TGA with pure TGA we found a shift to lower frequency at 2940 cm−1 may be due to the field of solid surfaces which it means electrostatic interaction between TGA capping agent and electronic field around the core of CdTe and TGA ligand was grafted on the CdTe and CdTe/CdS. Pure TGA S–H vibrations were detected at 2565 cm−1, and 2665 cm−1, which totally vanished in the TGA capped CdTe and CdTe/CdS nanocomposites this was attributed to the thiol bound to the surface of QDs, which can dramatically remove the traps states, passivate the QDs surface and improve the PL efficiency. A significant shift in the asymmetric stretching vibration of the carboxyl group (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O = 1702 to
O = 1702 to 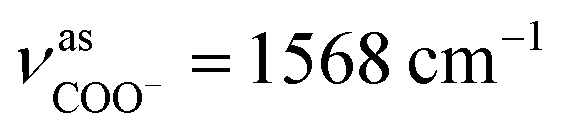 and
and 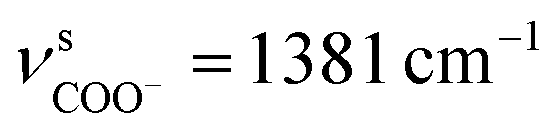 ) was also observed in the TGA capped CdTe QDs was strong evidences about presence of TGA molecules on CdTe surfaces. Methylene (CH2) scissoring vibration of the QDs is seen at around 750 cm−1 because of the bond formation between Cd2+ and Te2−, which affirms the development of CdTe QDs. For the higher refluxing time, a new peak was seen at around 1100 cm−1 because of carbonyl stretching (C–O) vibrations. This peak may be due to the development of CdTe
) was also observed in the TGA capped CdTe QDs was strong evidences about presence of TGA molecules on CdTe surfaces. Methylene (CH2) scissoring vibration of the QDs is seen at around 750 cm−1 because of the bond formation between Cd2+ and Te2−, which affirms the development of CdTe QDs. For the higher refluxing time, a new peak was seen at around 1100 cm−1 because of carbonyl stretching (C–O) vibrations. This peak may be due to the development of CdTe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CdS composites.22 The FTIR spectra of QD(D)–mAb demonstrated typical amide bond peaks at 1670 cm−1 (CO amide I) and 1481 cm−1 (N–CO amide II), while only the CO stretch was seen for QD(D). This peak explain formation of covalently amide bond between QD(D) with mAb. The same thing for bioconjugated QD(A)–pAb.23,24 Other peaks at 1225 cm−1 in labeled peak QD(A)–pAb (demonstrating the presence of pAb) and also peak at 1215 cm−1 in QD(D)–mAb (demonstrating the existence of mAb) were also observed.
:CdS composites.22 The FTIR spectra of QD(D)–mAb demonstrated typical amide bond peaks at 1670 cm−1 (CO amide I) and 1481 cm−1 (N–CO amide II), while only the CO stretch was seen for QD(D). This peak explain formation of covalently amide bond between QD(D) with mAb. The same thing for bioconjugated QD(A)–pAb.23,24 Other peaks at 1225 cm−1 in labeled peak QD(A)–pAb (demonstrating the presence of pAb) and also peak at 1215 cm−1 in QD(D)–mAb (demonstrating the existence of mAb) were also observed.
3.2 Optical characterization of QD(D) and QDs(A)
Fluorescence spectra of QD(D) and QD(A) were obtained. Different excitation tested first for each QD to estimate the maximum fluorescence intensities for QD(D) and QD(A) were measured at the excitation wavelength of 320 nm and 350 nm, respectively as its clear in inset photo in Fig. 3 each QD have different color under UV light. The Fig. 3 shows the fluorescence intensities for each quantum dot at 340 nm because it was the best excitation wavelength for FERT system. The widths of slits for both excitation and emission were 5.0 nm. All optical measurements were conducted at room temperature.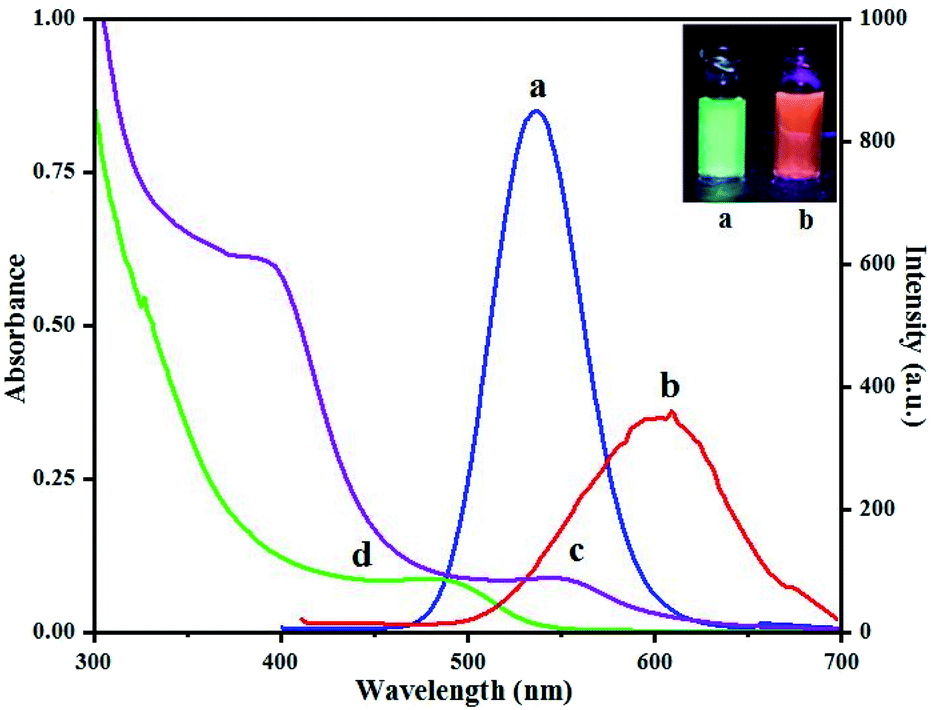 | ||
| Fig. 3 Fluorescence spectra (a) of green-emitting QD(D) and (b) of orange-emitting QD(A) absorption spectra (d) of green-emitting QD(D) and (c) of orange-emitting QD(A). | ||
Studying optical and electronic properties of amorphous germanium, Tauc et al., proposed and a technique for calculating the band gap utilizing optical absorbance data plotted appropriately in terms of energy.25 Later, Davis and Mott developed this method in a more general work on amorphous semiconductors.26,27 It was shown in their work that optical absorption strength relies upon the difference between the photon energy and the band gap. By using the Tauc's relationship in the high absorption zone of the semiconductor, the link between the absorption coefficient (a) and the incident photon energy (hν) can obtained as:
| αhν = α0(hν − Eg)n | (1) |
| (αhν)2 = α0(hν − Eg) | (2) |
Experimentally, to determine the optical energy gap, one can plot (αhν)2 versus the photon energy (hν) utilizing the information acquired from the optical absorption spectra, as presented in Fig. S2.† It reveals that the obtained plotting gives a straight line in a specific zone. To obtain the value of the direct allowed optical energy gap each QDs. The straight line can extend to intercept (hν)-axis at (αhν)2 = 0. The evaluated estimations of the energy gap for QD(A) and QD(D) were 2.34 eV and 2.6 eV, respectively.
3.3 Conjugating of monoclonal antibodies (mAb) with QD(D) and coupling of polyclonal antibody (pAb) with QD(A)
The QD(D) were firstly conjugated with mAb by carbodiimide. 10 μL (10 ppm) of QD(D) were activated by adding 10 μL (10 mg mL−1) of EDC and 10 μL (10 mg mL−1) of sulfo-NHS, respectively.26 The mixed solutions were shaken at room temp for 15 min and afterward reacted at room temperature for 30 min with 2.5 μL of mAb (1.12 mg mL−1). Using this procedure, QD(A) was conjugated with pAb. Based on the optimizations, 10 μL (10 ppm) of QD(A) was activated by adding 10 μL (10 mg mL−1) of EDC and 10 μL (10 mg mL−1) of sulfo-NHS. After 15 min shaking, 1 μL of pAb (0.198 mg mL−1) was added to the as-prepared solution and incubated for 30 min at room temperature. During this process, both QDs and antibodies were conjugated through stable covalent bonds, which were demonstrated by the change in absorption spectra and fluorescence intensity in Fig. S3† and DLS analysis in Fig. S4.† The last bioconjugated QDs were gathered and kept at 4 °C overnight.3.4 FRET system between bioconjugated QD(D) and QD(A)
Different ratio in microliter is examined between bioconjugated QDs. The ratio of QD(D)–mAb to QD(A)–pAb was (1:1, 1:2, 1:4, 2:1 and 4:1) μL in this experiment to help us choose best ratio for next steps in determination of NMP22. According to the spectra in Fig. 4(a) the spectra of 1:1 shows maximum quench for QD(D)–mAb and highest fluorescence intensity for QD(A)–pAb. According to this estimation the highest proportion was found to be 1:1 shown in Fig. 4(a). The freshly prepared bioconjugated 32.5 μL solution of QD(D) with mAb is mixed with 32.5 μL of QD(A) gave best FRET system between donor and acceptor Fig. 4(b).
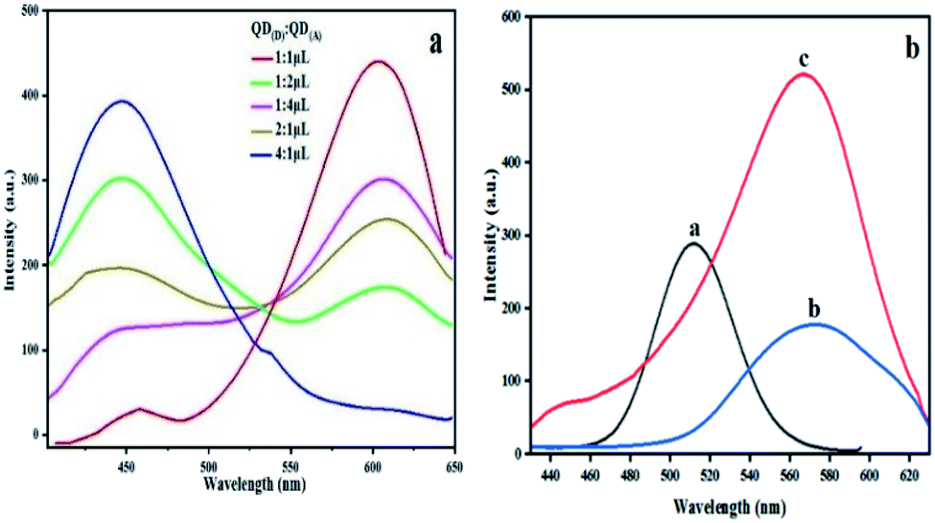 | ||
| Fig. 4 Fluorescence spectra of (a) different ratio (1:1, 1:2, 1:4, 2:1 and 4:1) μL QD(D)–mAb mixed QD(A)–pAb. (b) (a): PL spectra of QD(D)–mAb, (b): QD(A)–pAb and (c): QD(D)–QD(A) system λex = 340 nm. | ||
3.5 FRET measurement techniques
FRET is a non-radiative process. There is a transfer of dipole–dipole energy between two fluorophores. When the donor fluorophore is in its electronic excited state, its excited energy can be transferred to another neighboring fluorophore. Consequently, the acceptor FRET can be evaluated from fluorescence lifetime variations, steady-state intensities, or anisotropy.28 Transfer efficiency or the possibility of energy transfer de-excitation can be expressed in terms of the lifetime of the donor or the intensity of the emissions:15,29
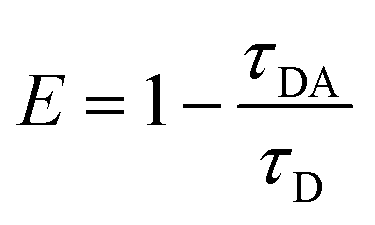 | (3) |
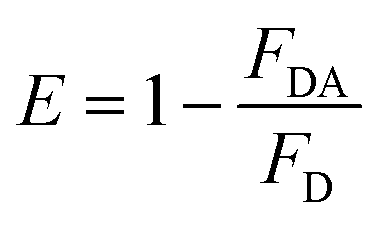 | (4) |
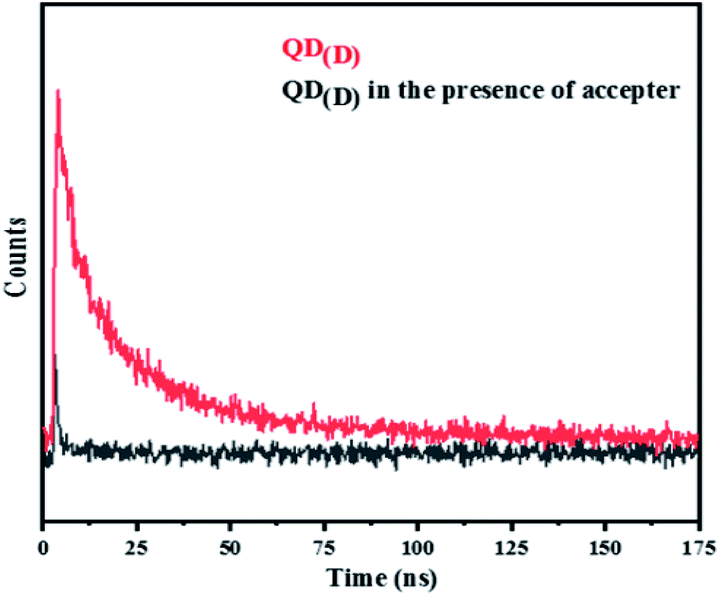 | ||
| Fig. 5 Time resolved of PL intensity of QD(D) and QD(D) in presence of acceptor (FRET system). | ||
3.6 FRET method for detecting NMP22
mAb as donor and the orange emitting QD(A)–pAb as acceptor. Fig. 1 demonstrates the principle of this protocol. Bio-affinity between mAb and pAb made these two colored-QDs close enough to start FRET. When the greater affinity NMP22 was added into the QD(D)–QD(A) system, they engaged the binding sites of the anti-NMP22 monoclonal antibody moiety immunized on QD(D) because of immunoassay between antigen and antibody. Therefore, the established FRET system broke, and the resonance energy transfer from QD(D) to QD(A) was blocked, which reduced the fluorescence from the QD(A) and resulted in quenching.In practice, to start their combination, orange-emitting QD(A)–pAb was firstly added into the green-emitting QD(D)–mAb in the initial step. During this process, the fluorescence of green-emitting QD(D) was quenched due to the transfer of fluorescence resonance energy between the two QDs. In the second step, for quantitative determination, the specific amounts of NMP22 sample from 2 pg mL−1 to 22 pg mL−1 were added into QD(D)–QD(A) system and kept at room temperature for 30 min. As shown in Fig. S5,† the system's fluorescence intensity became stable in 30 minutes. Therefore, all the intensities of fluorescence were registered after the 30 min incubation.
In brief, NMP22 would quickly and specifically be attached by monoclonal anti-NMP22 antibodies through higher affinity immuno-recognition. Then, the combination of the two QDs via the binding site on mAb was detached, thus prohibiting the FRET between them Fig. 6a. The fluorescence intensity of both QD(D)–QD(A) systems was recorded Fig. 6b. The change of fluorescence intensity was used for quantification of NMP22.
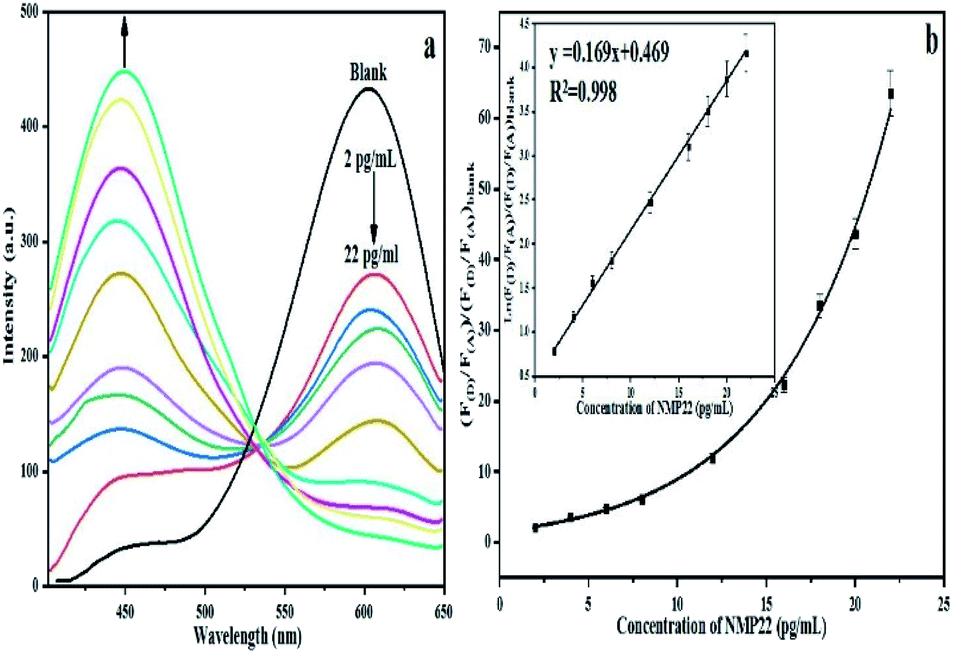 | ||
| Fig. 6 (a) Fluorescence spectra of FRET mechanism form 2 pg mL−1 to 22 pg mL−1, (b) calibration curve for detection of NMP22 from 2 pg mL−1 to 22 g mL−1. | ||
In the five repetitive assays, the relative standard deviations of FRET peaks for three different concentrations (2, 12, 20 pg mL−1) were seen to be 3.02%, 2.14%, 1.86%, respectively. The Table S1† shown the comparison of our method with other methods for detection of NMP22.
3.7 Interference
The interference of some common ions, K+, Na+, Cl− and common molecules like BSA, glucose, vitamin C and cytochrome c was examined as shown in Fig. S6.† The study was performed within an error range of ±5%. The concentration of NMP22 was 12 pg mL−1 is mixed with 0.05 mmole mL−1 for K+, Na+, Cl− interferences and 100 ng mL−1 molecules the fluorescence intensity is studied. The results showed that most of the previously mentioned ions and molecules did not interfere with the determination under the chosen experimental conditions. These observations showed that in this FRET system, anti-NMP22 antibodies exhibited a favorable selectivity and higher affinity for NMP22 compared with the other various coexisting species. Therefore, the FRET immunoassay technique based on QD for the identification of NMP22 proved to be selective, reliable and simple.3.8 Determination of NMP22 in urine samples by FRET method
The urine samples utilized in the investigation were obtained from a local hospital in Tehran, Iran. Four samples of patients diagnosed with bladder cancer were used in this experiment.23 Each specimen pre-treated firstly and then they were filtrated by using Whatman filter paper for removing any solid particles. The filtrated urine solution centrifuged at 10000 rpm 10 min, then 5 μL of each sample is used for detection of NMP22 according to the previously discussed procedure. Different samples were added to the FRET system. Subsequent to incubating for 30 min, the fluorescence spectra were recorded immediately. To validate the data, the concentration of NMP22 was also determined by ELISA and the data are presented in Table S2.† The recoveries were between 100% to 105%, and relative standard deviations were all less than 3%, indicating feasibility of the proposed method.
4 Conclusion
The FRET system relying on two colored QDs and combined with immunoassay reaction makes the NMP22 detection method sensitive, fast and specific. In the established FRET system, the addition of NMP22 inhibits the transition of resonance energy between the two QDs, thus resulting in elevated fluorescence of green-emitting bio-conjugated QD(D) conjugates and reduced fluorescence for orange-emitting bio-conjugated QD(A) conjugates. This method gives a quantification protocol for NMP22, and a linear relationship can be set up. There is excellent consistency between the results of our methodology and ELISA standard method utilized for detection of NMP22 in the urine of real patient samples, which indicates that this competitive FRET assay is feasible, quick, sensitive and specific. Moreover, the QDs used in this paper were simple and low cost. The method may be applied extensively in fundamental and clinical studies and diagnostics because of the simplicity and sensitivity of immunosensor.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research was sponsored by the research council of the University of Tehran, Salahaddin University-Erbil and Iran National Science Foundation (INSF98000848). The authors are grateful for their commercial support.References
- T. Han, X. Li, Y. Li, W. Cao, D. Wu, B. Du and Q. Wei, Sens. Actuators, B, 2014, 205, 176 CrossRef CAS.
- M. H. He, L. Chen, T. Zheng, Y. Tu, Q. He, H. L. Fu, J. C. Lin, W. Zhang, G. Shu, L. He and Z. X. Yuan, Front. Pharmacol., 2018, 9, 745 CrossRef PubMed.
- S. K. Vashist, R. Tewari, R. P. Bajpai, L. M. Bharadwaj and R. Raiteri, AZojono J. Nanotechnology Online, 2006, 2, 1 Search PubMed.
- L. Guo, X. Wu, L. Liu, H. Kuang and C. Xu, Small, 2018, 14, 1701782 CrossRef PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763 CrossRef CAS PubMed.
- E. Schröck, S. du Manoir, T. Veldman, B. Schoell, J. Wienberg, M. A. Ferguson-Smith, Y. Ning, D. H. Ledbetter, I. Bar-Am, D. Soenksen, Y. Garini and T. Ried, Science, 1996, 273, 494 CrossRef PubMed.
- W. C. Chan, D. J. Maxwell, X. Gao, R. E. Bailey, M. Han and S. Nie, Curr. Opin. Biotechnol., 2002, 13, 40 CrossRef CAS PubMed.
- R. E. Bailey and S. Nie, J. Am. Chem. Soc., 2003, 125, 7100 CrossRef CAS PubMed.
- S. Kim and M. G. Bawendi, J. Am. Chem. Soc., 2003, 125, 14652 CrossRef CAS PubMed.
- Y. S. Borghei and M. Hosseini, RSC Adv., 2018, 53, 30148 RSC.
- Y. S. Borghei, M. Hosseini and M. R. Ganjali, Microchim. Acta, 2017, 184, 4713 CrossRef CAS.
- V. Dvorakova, M. Cadkova, V. Datinska, K. Kleparnik, F. Foret, Z. Bilkova and L. Korecka, Anal. Methods, 2017, 9, 1991 RSC.
- T. L. Nguyen, P. Spizzirri, G. Wilson and P. Mulvaney, Chem. Commun., 2008, 2, 174–176 RSC.
- J. M. Pietryga, R. D. Schaller, D. Werder, M. H. Stewart, V. I. Klimov and J. A. Hollingsworth, J. Am. Chem. Soc., 2004, 126, 11752 CrossRef CAS PubMed.
- M. Wu, M. Massey, E. Petryayeva and W. Russ Algar, J. Phys. Chem. C, 2015, 119, 26183 CrossRef CAS.
- A. Shamirian, A. Ghai and P. Snee, Sensors, 2015, 15, 13028 CrossRef PubMed.
- E. M. Obeng, E. C. Dullah, M. K. Danquah, C. Budiman and C. M. Ongkudon, Anal. Methods, 2016, 8, 5323 RSC.
- S. R. Inamdar, G. Pujar and M. J. Sannaikar, Luminescence, 2018, 203, 67 CrossRef CAS.
- H. Mattoussi, I. L. Medintz, A. R. Clapp, E. R. Goldman, J. K. Jaiswal, S. M. Simon and J. Matthew Mauro, JALA, 2004, 9, 8 Search PubMed.
- J. J. Lu, S. G. Ge, F. W. Wan and J. H. Yu, Adv. Mater. Res., 2011, 306–307, 1350 CAS.
- M. Z. Hu and T. Zhu, Nanoscale Res. Lett., 2015, 10, 469 CrossRef PubMed.
- A. Arivarasan, G. Sasikala and R. Jayavel, Mater. Sci. Semicond. Process., 2014, 25, 238 CrossRef CAS.
- H. Safarpour, H. Safarpour, M. R. Safarnejad, M. Tabatabaei, A. Mohsenifar, F. Rad, M. Basirat, F. Shahryari and F. Hasanzadeh, Can. J. Plant Pathol., 2012, 34, 507 CrossRef.
- H. Zhang, T. Xu, L. Gao, X. Liu, J. Liu and B. Yu, Molecules, 2017, 22, 1250 CrossRef PubMed.
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi, 1966, 15, 627 CrossRef CAS.
- E. Davis and N. Mott, Philos. Mag., 1970, 22, 0903 CrossRef CAS.
- N. F. Mott, E. Davis and K. Weiser, Phys. Today, 1972, 25, 55 CrossRef.
- C. Gayrard and N. Borghi, Methods, 2016, 94, 33 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer Science and Business Media, 2013 Search PubMed.
- K. Chou and A. Dennis, Sensors, 2015, 15, 13288 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06191c |
| This journal is © The Royal Society of Chemistry 2020 |