
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot synthesis of formic acid via hydrolysis–oxidation of potato starch in the presence of cesium salts of heteropoly acid catalysts†
Nikolay V. Gromov *,
Tatiana B. Medvedeva,
Yulia A. Rodikova,
Dmitrii E. Babushkin,
Valentina N. Panchenko,
Maria N. Timofeeva,
Elena G. Zhizhina,
Oxana P. Taran and
Valentin N. Parmon
*,
Tatiana B. Medvedeva,
Yulia A. Rodikova,
Dmitrii E. Babushkin,
Valentina N. Panchenko,
Maria N. Timofeeva,
Elena G. Zhizhina,
Oxana P. Taran and
Valentin N. Parmon
Boreskov Institute of Catalysis SB RAS, Lavrentiev Av., 5, Novosibirsk, 630090, Russia. E-mail: gromov@catalysis.ru; Fax: +7-383-33-08-056; Tel: +7-383-32-69-591
First published on 4th August 2020
Abstract
Solid bifunctional catalysts based on cesium salts of V-containing heteropoly acids (CsHPA: Cs3.5H0.5PW11VO40, Cs4.5H0.5SiW11VO40, Cs3.5H0.5PMo11VO40) and Cs2.5H0.5PMo12O40 were used for studying one-pot hydrolysis–oxidation of potato starch to formic acid at 413–443 K and 2 MPa air mixture. It was shown that the optimum process temperature that prevents formic acid from destruction is 423 K. The studies were focused on the influence of the composition of heteropoly anions on the yield and selectivity of formic acid. Using W–V–P(Si) CsHPA results in the product overoxidation compared to Mo–V-containing CsHPA. The activity of Cs–PMo was significantly lower compared to Cs–PMoV. This may indicate that vanadium plays an important role in the oxidation process. The most promising catalyst was Cs3.5H0.5PMo11VO40 which provided the maximum yield of formic acid equal to 51%. Cs3.5H0.5PMo11VO40 was tested during nine cycles of starch hydrolysis–oxidation to demonstrate its high stability and efficiency.
Introduction
Formic acid (FA) is one of the main products that can be derived from plant biomass.1,2 It is applied for the production of medicaments, solvents, fragrances, and fibers, for conservation of forage, and also used in pulp and leather industries.3,4 Nowadays, FA is attracting more attention as a sustainable hydrogen source5,6 owing to its high volumetric hydrogen density of 53 g of H2 per liter.7 The main advantage of formic acid over molecular hydrogen is the simplicity and safety of its storage and transportation.2,8,9 Methods for substitution of formic acid for molecular hydrogen in the processes for biofuel production were suggested.9–14 Formic acid can be synthesized as a side product of glucose acidic dehydration into levulinic acid.15 However, the amount of thus produced formic acid is insufficient. Formic acid can be produced via peroxide oxidation of cellulose or glucose.16–18 However, low yields and low selectivity to the target product are characteristic of the process.18 Another way of FA production is oxidation of monosaccharides (glucose, xylose, etc.), polyols (sorbitol), alcohols, organic acids. When carrying out this reaction in the presence of H3+xPMo12−xVxO40 (where x = 1, 2, 5), yields of formic acid can reach 85% (Table S1, ESI†). Moreover, FA can be produced via one-pot hydrolysis–oxidation of plant biomass and its derivatives (disaccharides and polysaccharides) in the presence of bifunctional catalysts which have H+ and Ox–Red active sites (Scheme 1). | ||
| Scheme 1 One-pot hydrolysis–oxidation of plant biomass to formic acid. | ||
Soluble bifunctional catalysts based on vanadium-containing heteropoly acids (HPA) seem more promising for hydrolysis–oxidation of organic substrates, including polysaccharides, to formic acid than complexes of Fe(III), Ce(IV), Ru(II), Ru(III) metals and vanadium-containing compounds (VOSO4, [VO(acac)2], [n-Bu4N]VO3).4,19–25 Bifunctional HPA catalysts are advantageous due to their high Brønsted acidity and a high oxidation potential. HPA samples belong to “green chemistry” catalytic systems.22,23,26,27 Studies of cellulose hydrolysis–oxidation catalyzed by H3+xPMo12−xVxO40 were described previously.28–33 Using HPA catalysts makes possible FA production from various plant materials, such as disaccharides (cellobiose, sucrose) (Table S2, ESI†), polysaccharides (cellulose, xylan, arabinogalactan, starch) (Table S3, ESI†), lignin (Table S4, ESI†) and different types of biomass (Table S5, ESI†) via OxFA-process.20,29 It should be emphasized that all the processes described above were homogeneous. Zhang and co-authors reported the highest 68% yield of formic acid from cellulose polysaccharide.30 The reaction was carried out in the presence of H4PVMo11O40 HPA catalyst in pure water without any additives or co-solvents. Gromov et al. have shown earlier the possibility of FA synthesis from cellulose at the 66% yield in the presence of a Co0.6H3.8PMo10V2O40 solution in pure water without any additives.4 Moreover, Xu et al. demonstrated potentialities of phase-transfer catalysis for hydrolysis–oxidation of cellulose to FA.34 The authors used heteropoly anion-based ionic liquids with functionalized –SO3H cations and PMo11VO404− anions as catalysts to achieve 49.7% yield of formic acid.
An essential disadvantage of soluble HPA is the difficult separation of the catalyst from the reaction mixture and/or the main product. The problem may be solved using solid cesium salts of vanadium-containing heteropoly acids (CsHPA). CsHPA salts have high specific surface area up to 180 m2 g−1 unlike heteropoly acids (1–3 m2 g−1). At the same time, acidities of both CsPHA and HPA are close.35 As far as we know, hydrolysis–oxidation of starch, a natural polysaccharide, to formic acid catalyzed by vanadium-containing HPA has not been studied yet, and solid catalysts were not used for the process. Nevertheless, starch may be considered as a promising feedstock for FA production (Scheme 2).
 | ||
| Scheme 2 Formic acid production from starch. | ||
Starch is commercially produced during processing of cereal (mainly corn) and tuber (potato) crops; this is an abundant and inexpensive product to be easily extracted from agricultural crops, stored and transported. Conversion of the renewable and environmentally friendly starch feedstock to formic acid is particularly attractive from the point of intensive development of alternative non-fuel and non-coal technologies.36 Development of methods for synthesis of FA from starch is a promising area due to high potentialities and large scale of the production. However, as far as we know, an only work of hydrolysis–oxidation of starch to formic acid with molecular oxygen was reported.37 Tang et al. demonstrated hydrolysis–oxidation of starch to formic acid in the presence of soluble vanadyl sulfate VOSO4 as the catalyst to give 46% yield of FA.37
The present study was aimed at hydrolysis–oxidation of potato starch to formic acid in the presence of solid cesium salts of heteropoly acids (CsHPA: Cs3.5H0.5PW11VO40, Cs4.5H0.5SiW11VO40, Cs3.5H0.5PMo11VO40, Cs2.5H0.5PMo12O40) as bifunctional catalysts.
Experimental
Materials
The following chemicals were used for the studied without pretreatment: vanadium oxide V2O5 (99%, Vectron), molybdenum oxide MoO3 (99%), hydrogen peroxide H2O2 (high purity 8–4), phosphoric acid H3PO4 (99.4%, Akros), sulfuric acid H2SO4 (96%, Organic Acros), sodium hydroxide NaOH (98%, Panreac), hydrogen chloride HCl (36%, Reakhim), cesium carbonate Cs2CO3 (99.5%, Organic Acros), phosphatotungstic acid H3PW12O40·nH2O (83%, Vectron), D-glucose (98%, Alfa Aesar), formic acid HCOOH (98%, Panreac), acetic acid CH3COOH (99.5%, Reakhim), levulinic acid C5H8O3 (98%, Organic Acros). An air mixture of 20% oxygen and 80% nitrogen (National State Standard 5583-78) was used for oxidation. Distilled water after additional purification using a Milli-Q utility (Millipore, France) was used for preparation of all the solutions.Synthesis of heteropoly acids
A solution of H4PMo11VO40 was prepared using 85% H3PO4, V2O5, MoO3 and 30% H2O2 by the method described elsewhere.4,38,39 H4PW11VO40 and H5SiW11VO40 were prepared from H3PW12O40 or H4SiW12O40 solutions and V2O5, NaOH, HCl by adapted three-stage method.38,40,41 Synthesis of H4PW11VO40 and H5SiW11VO40 is given in ESI.†Synthesis of CsHPA salts
Cesium salts of heteropoly acids, such as Cs3.5H0.5PMo11VO40 (Cs–PMoV), Cs3.5H0.5PW11VO40 (Cs–PWV), Cs4.5H0.5SiW11VO40 (Cs–SiWV), were synthesized using stoichiometric quantities of cesium carbonate via partial neutralization of aqueous-alcoholic solutions of H4PMo11VO40, H4PW11VO40, H5SiW11VO40.Co0.6H3.8PMo10V2O40 (Co–PMoV2) was obtained by addition of stoichiometric quantities of cobalt carbonates to H5PMo10V2O40.4
Instrumental measurements
Textural properties of cesium salts were characterized using nitrogen adsorption isotherms at 73 K with a ASAP-2400 instrument (Micromeritics, USA). All the samples were pre-degassed in vacuum at 403–423 K. The BET model and STSA equation was used for calculating the surface area. Pore size distribution was estimated using QSDFT and NLDFT methods.Leaching the active component was investigated by ICP method using Inductively Coupled Plasma-Atomic Emission Spectrometry (ICP-AES, PerkinElmer Inc., USA).
The point of zero charge of the samples (pHPZC) was determined by mass titration described in the literature.42 A pH meter was calibrated using pH 1.68 and 6.86 buffers. All measurements were carried out using distilled–deionized water.
To study Brønsted and Lewis surface acidity by pyridine adsorption, samples were pretreated within the IR cell. Samples were pretreated by heating for 1 h in air and for 1 h under vacuum at 423 K. The samples were exposed to saturated pyridine vapors at room temperature for 10 min and were heated at 423 K for 15 min. Then pyridine was desorbed for 30 min under vacuum at 423 K. The strength of BAS was characterized by the proton affinity values (PA) according to Davydov.43 The amount of BAS and LAS was estimated from the intensity of the band of the stretching vibration of pyridinium ions with a maximum at 1540 cm−1 (BAS) and 1450 cm−1 (LAS) and using extinction coefficients of εLAS = 1.67 ± 0.1 and εBAS = 2.22 ± 0.1 cm μmol−1.44 FT-IR spectra were recorded on a Shimadzu FTIR-8300S spectrometer in the range of 400–6000 cm−1 with a resolution of 4 cm−1.
Gas chromatography was used to determine composition of gas products using a Kristall 2000M chromatograph (Chromatec, Russia) equipped with a flame-ionization detector, methanator, and a column 2 m × 2 mm filled with Chromosorb 102 sorbent. Argon was used as gas carrier.
The total yield of water-soluble products was determined using a total organic carbon (TOC) analyzer (Multi N/C 2100S TOC, Analytik Jena, Germany). The reaction mixture (500 μL) was injected into the analyzer. The amount of organic carbon (g L−1) was calculated based on the calibrations.
Starch characterization
Commercial alimentary potato starch (Amiloros Co., Novosibirsk Region, Russia) of 99.2% purity and 15.0% moisture was used as the substrate. The moisture content was determined according to the Russian National State Standard 16932-82 procedure.45 A dry pure sample bottle (open, along with the cup) was dried in a drying oven at 376 ± 2 K to obtain constant weight. A starch sample (5 g) was loaded to the bottle and dried for 3 h. The bottle with the dried sample was covered and transferred to a desiccator, cooled and weighed. The 1 h procedure including drying, cooling and weighing was repeated until constant weight was achieved.The relative humidity, %, was calculated by eqn (1):
 | (1) |
The reaction of polysaccharide to glucose hydrolysis was used to detect impurities in the commercial starch. The reaction was conducted in an autoclave (Autoclave Engineers, USA) under vigorous stirring (1500 rpm) in an inert argon atmosphere. A starch sample (5 g of dry weight) was loaded to the autoclave and 100 mL of 2% HCl solution was added. The autoclave was closed and blown through with argon. Starch was hydrolyzed at 373 K for 3 h, then the autoclave was cooled to room temperature. No solid impurities in the reaction mixture indicated the complete substrate dissolution. High-effective liquid chromatography with a Shimadzu Prominence LC-20 (Japan) chromatograph equipped with a refractometric detector and a Rezex Organic Acids H+ (Phenomenex, 300 mm × 5.0 mm) thermostated at 40 °C was used for analysis of glucose in the reaction mixture. An aqueous solution of 2.5 mN sulfuric acid was used as the eluent, the flow rate was 0.6 mL min−1.4
Catalytic tests4
Hydrolysis–oxidation of potato starch was conducted in a high pressure autoclave (Autoclave Engineers, USA) under vigorous stirring (1500 rpm) under air mixture of 20% O2 and 80% N2 (artificial air) at 2 MPa and 413–443 K. The commercial starch (11.9 g L−1 containing 10 g L−1 of dry starch, 62 mM of glucose units) and a catalyst (1.25 g L−1) were loaded in the autoclave, and 60 mL of water added. The autoclave was closed, operation pressure set, and temperature started elevating. After the temperature stabilization (ca. 20 min), a reaction mixture sample to be analyzed was drawn; that was zero point of the process (zero sample). Reaction mixture samples were drawn for analysis during the reaction.Recycling test
After each cycle the catalyst was separated from the reaction mixture by centrifugation, washed with Milli-Q water (3 × 10 mL), dried in air for 12 h and 376 K for 3 h.Concentrations of water dissolved products were determined with the HPLC technique using a Shimadzu Prominence LC-20 chromatograph (Japan) equipped with a refractive index detector and Rezex Organic Acids H+ column (Phenomenex, 300 mm × 5.0 mm) thermostated at 40 °C. Aqueous solution of 2.5 mN sulfuric acid H2SO4 was used as the eluent at the flow rate of 0.6 mL min−1.
A spectrophotometer Uvikon 930 (wavelength 570 nm) was used for spectrophotometric detection of formaldehyde in the solution: 0.2 mL sample was mixed with 1.0 mL 0.5% of chromotropic acid, 0.8 mL of water, 8.0 mL of 81 v/v% H2SO4, the resulting mixture was heated for 20 min at 333 K, then cooled; the optical density of the solution was measured using a cell with 1 cm path length.
1H and 13C NMR spectra were recorded on a Bruker AVANCE-400 spectrometer in standard cylinder ampoules with a diameter of 5 mm at frequencies of 400.13 MHz (1H) and 100.61 MHz (13C) using one- and two-dimensional NMR methods. The reaction solutions were studied without dilution at 298 K, deuterium lock was not used. Quantitative analysis was performed using 1H and 13C NMR spectroscopy with reverse discontinuous suppression (INGAT) and numerous delays (30 s) between pulses, which ensured complete relaxation of nuclear 13C.
Each starch depolymerization experiment was repeated three times. Each analysis of the reaction mixtures was carried out three times. The standard deviation of the results was less 2%.
Product yields (mol%) of starch hydrolysis–oxidation were calculated by eqn (2):4
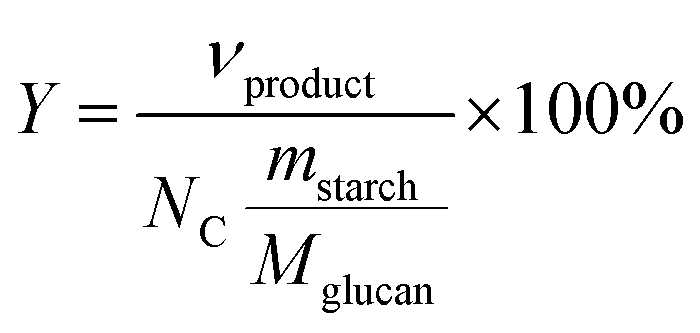 | (2) |
Results and discussion
Characterization of CsHPA salts
A number of physicochemical methods (nitrogen adsorption, IR spectroscopy with pyridine, ICP-AES, pHzero) were used for studying developed catalytic systems Cs–PMoV, Cs–PWV and Cs–SiWV. Low-temperature nitrogen adsorption was used for the texture characterization. It was established that the specific surface area, SBET, of CsHPA varies between 82 and 170 m2 g−1, micropore volume between 0.017 and 0.038 cm3 g−1 (Table 1). The specific surface area increased in the series Cs4.5H0.5SiW11VO40 < Cs3.5H0.5PMo11VO40 < Cs3.5H0.5PW11VO40. The largest surface area equal to 170 m2 g−1 was characteristic of Cs3.5H0.5PW11VO40.| Cs-salt of HPA | Textural propertiesa | Acidic propertiesb | |||||
|---|---|---|---|---|---|---|---|
| Abbreviation | SBET (m2 g−1) | VΣ, (cm3 g−1) | Vμ, (cm3 g−1) | BAS (μmol g−1) | LAS (μmol g−1) | pHPZC | |
| a SBET – specific surface area, Vμ – micropore volume.b pHPZC – point zero charge, amount of BAS and LAS determined by IR spectroscopy with pyridine as the probe molecule. | |||||||
| Cs3.5H0.5PW11VO40 | Cs–PWV | 170 | 0.118 | 0.038 | 32 | 57 | 6.7 |
| Cs3.5H0.5PMo11VO40 | Cs–PMoV | 139 | 0.087 | 0.035 | 204 | 40 | 3.4 |
| Cs4.5H0.5SiW11VO40 | Cs–SiWV | 82 | 0.075 | 0.017 | 31 | 54 | 6.7 |
| Cs2.5H0.5PMo12O40 | Cs–PMo | 125 | 0.111 | 0.052 | — | — | — |
Surface acidity of the samples was estimated by mass titration (pHzero) (Fig. S2, ESI†).42 Experimental data indicate equal surface acidities of Cs–PWV and Cs–SiWV, pHzero 6.7 (Table 1). The surface acidity of the Cs–PMoV catalyst is much higher of those of Cs–PWV and Cs–SiWV, pHzero 3.5. These data agree with the IR data obtained with pyridine used as the probe molecule (Fig. S3, ESI†): the highly acidic Cs–PMoV catalyst bears more Brønsted acid centers (204 μmol g−1) than Cs–PWV and Cs–SiWV (32 and 31 μmol g−1, respectively) (Table 1).
Hydrolysis–oxidation of starch in the presence of Cs–PWV catalyst
Catalytic properties of Cs–PWV systems were studied during one-pot hydrolysis oxidation of commercial potato starch. Typical kinetic curves are shown in Fig. 1. At the early period of the reaction, glucose and oligosaccharides are formed to be rapidly consumed for oxidation. In 7 h, formic acid is the main reaction product in the aqueous phase. The formation of side products (glucose, succinic, glycolic, and acetic acids, hydrated formaldehyde (methylene glycol)) was detected, and the formation of gas products (mainly CO2 and small quantity of CO) observed. The catalytic behavior of vanadium-containing CsHPA was confirmed by blank runs: in the absence of a catalyst yields of glucose and formic acid were 8 and 2 mol%, respectively, in 5 h of the reaction (Table 2, line 1). | ||
| Fig. 1 Kinetic curves of water-soluble products of the reaction accumulated during starch hydrolysis–oxidation (experimental conditions: 11.9 g L−1 of starch, 1.25 g L−1 of Cs–PWV, reaction volume 60 mL, 2 MPa of air mixture, 423 K). | ||
| No. | Catalyst | T (K) | τ (h) | X (mol%) | ∑Yg (mol%) | ∑YL (mol%) | Yield of product (mol%) | Maximum product yieldb (mol%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gaseous | Water-solublec | Glu | FA | |||||||||||||||
| CO2 | CO | Glu | SA | GA | FA | AA | FMA | τ (h) | Ymax (%) | τ (h) | Ymax (%) | |||||||
| a Experimental conditions: 11.9 g L−1 of starch, 1.25 g L−1 of catalyst, 60 mL of reaction volume, 2 MPa of air mixture; τ – reaction time, X – starch conversion, ∑Yg, – total yield of gaseous products, ∑YL, – total yield of water-soluble products.b Maximum yields of glucose and formic acid achieved during the experiment and the time of its achieving.c Glu – glucose, SA – succinic acid, GA – glycolic acid, FA – formic acid, AA – acetic acid, FMA – hydrated formaldehyde (methylene glycol).d Summary yield (glucose – 9%, fructose – 12%). | ||||||||||||||||||
| 1 | Blank | 423 | 5 | — | — | — | — | — | 8 | 0 | 2 | 0 | 0 | 5 | 8 | 5 | 2 | |
| 2 | Cs–PWV | 413 | 7 | 72 | 24 | 38 | 23 | 1 | 13 | <1 | <1 | 18 | 3 | <1 | 5 | 64 | 11 | 21 |
| 3 | Cs–PWV | 423 | 7 | 100 | 70 | 30 | 66 | 4 | <1 | 1 | 1 | 22 | 5 | 1 | 2 | 51 | 5 | 22 |
| 4 | Cs–PWV | 443 | 7 | 100 | 71 | 29 | 68 | 3 | 0 | 0 | 0 | 19 | 7 | <1 | 0.5 | 56 | 2 | 23 |
| 5 | Cs–SiWV | 423 | 7 | 100 | 70 | 30 | 66 | 4 | 0 | 2 | 5 | 18 | 5 | 0 | 3 | 39 | 7 | 18 |
| 6 | Cs–PMoV | 423 | 7 | 100 | 42 | 58 | 40 | 2 | 0 | <1 | 0 | 51 | 3 | 3 | 1 | 6 | 2 | 51 |
| 7 | Cs–PMo | 423 | 7 | 43 | 0 | 43 | n.d. | n.d. | 0 | 0 | 2 | 14 | 5 | 0 | 1 | 22d | 7 | 14 |
The process temperature influences considerably the process of hydrolysis–oxidation of polysaccharides.4 The optimum reaction temperature was determined in the presence of Cs–PWV. Starch was hydrolyzed–oxidized at 413–443 K at the air mixture pressure of 2 MPa. The kinetic accumulations of formic acid are shown in Fig. 2A. The results obtained show that temperature has a little influence of on the yield of the target product but affects the stability and rate of formic acid accumulation.
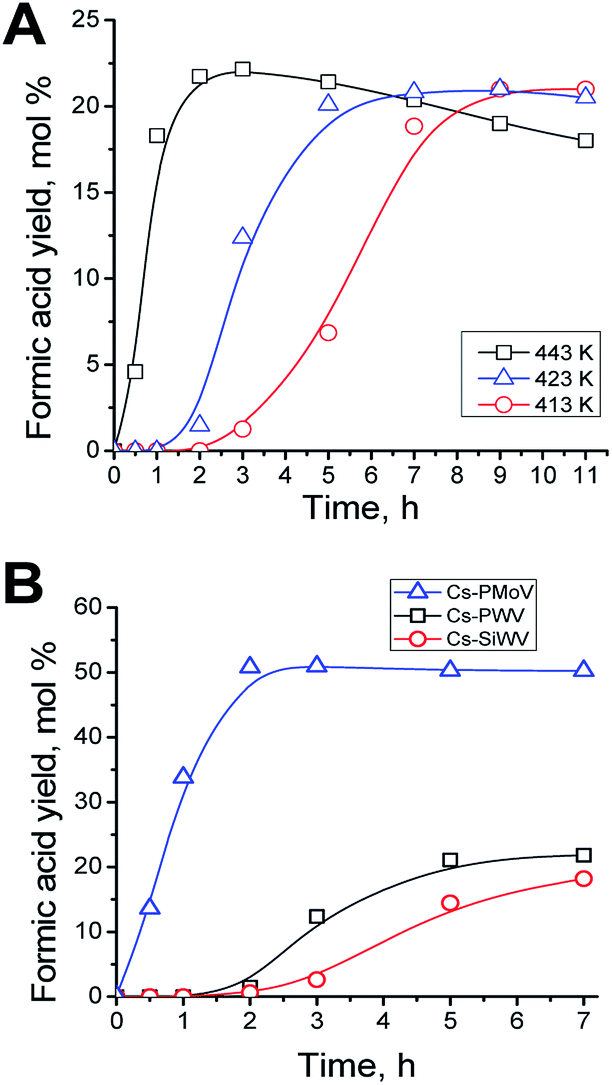 | ||
| Fig. 2 Formic acid kinetic curves in hydrolysis–oxidation of starch in the presence of Cs–PWV at 413–443 K (A) and in the presence of Cs–PMoV, Cs–PWV and Cs–SiWV at 423 K (B) (other experimental conditions: 11.9 g L−1 of starch, 1.25 g L−1 of a catalyst, 60 mL of reaction volume, 2 MPa of air mixture). | ||
The temperature of 423 K is optimum for the process and allows the maximum yield of formic acid to be reached in 5–7 h of the reaction. There are several reasons. First of all, the conversion of starch was only 72% in 7 h at 413 K in contrast to one at 423 and 443 K. Moreover, the temperature elevation up to 443 K does not allow the yield of the target product to be increased, while formic acid becomes unstable and is destructed to form gaseous side products (Fig. 2A).4,46,47 The sufficient stability of formic acid in the hydrothermal medium at 423 K was confirmed by 11 hour experiments (Fig. 2A). An apparent S-curve of accumulation of formic acid is observed at low temperature (413 K) but not a higher temperatures. At 413 K, formic acid starts accumulating not earlier than in 3 h of the reaction, and the maximum yield is reached in 11 h. Notice that 423–433 K also is the optimum temperature for hydrolysis–oxidation of cellulose to formic acid.4 The kinetic curves of formic acid accumulation were used to calculate the initial reaction rates. The initial reaction rates were plotted versus the inverse temperature of the reaction. The apparent activation energy of the process (Ea) equal to 47 ± 2 kJ mol−1 was revealed from the linear dependence. That was half of Ea of hydrolysis–oxidation of cellulose (78 ± 5 kJ mol−1).4
Anyway, 22% yield of formic acid was obtained in the presence of Cs–PWV at the optimum temperature of 423 K (Table 2, line 3). Acetic, glycolic, succinic acids and hydrated formaldehyde (methylene glycol) were side products in the solution in 7 h (Fig. S4–S6†). Thus, there was 73% of formic acid among the water dissolved reaction products. However, a total product yield in the liquid phase was no more than 30%, while the yield of gas products, CO2 and CO, was 70%. The considerable yields of carbon dioxide upon hydrolysis–oxidation of polysaccharides in the presence of HPA catalysts was observed earlier.29,30
Hydrolysis–oxidation of starch in the presence of different CsHPA catalysts
The influence of heteropoly anion composition on the catalytic properties of CsHPA was studied. Cs–PWV, Cs–SiWV, Cs–PMoV, and Cs–PMo were synthesized (Table 1). The catalytic properties of all the CsHPA samples were studied under conditions used for investigating Cs–PWV, 423 K and air mixture pressure of 2 MPa. The main results are summarized in Table 2.First of all, we investigated an effect of heteropoly anion central atom (P and Si) on CsHPA catalytic properties. Total yields of CO and CO2 were similar in the presence of Cs–SiWV and Cs–PWV (Fig. 3). However, in the presence of Cs–SiWV, the yield of glycolic acid (5%) was higher and one of formic acid was lower (18%) compared to Cs–PWV (1 and 22%, respectively). We assumed that was caused by different textural properties because amounts of acid sites were close (Table 1). Specific surface area of Cs–PWV was twice higher (170 m2 g−1) in compared with Cs–SiWV (82 m2 g−1) (Table 1). Therefore, the density of acid sites on the surface of Cs–SiWV was higher than that on Cs–PWV. Because size of glucose molecule (open chain form) is about 15 Å,48 and diameter of Keggin heteropoly anion is about 9 Å,49 we can assume that some of active sites are inaccessible for the reactants on the surface of Cs–SiWV. Based on the structure of vanadium active sites (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) VV–OH) the reaction mechanism can be described by Scheme 3:
VV–OH) the reaction mechanism can be described by Scheme 3:
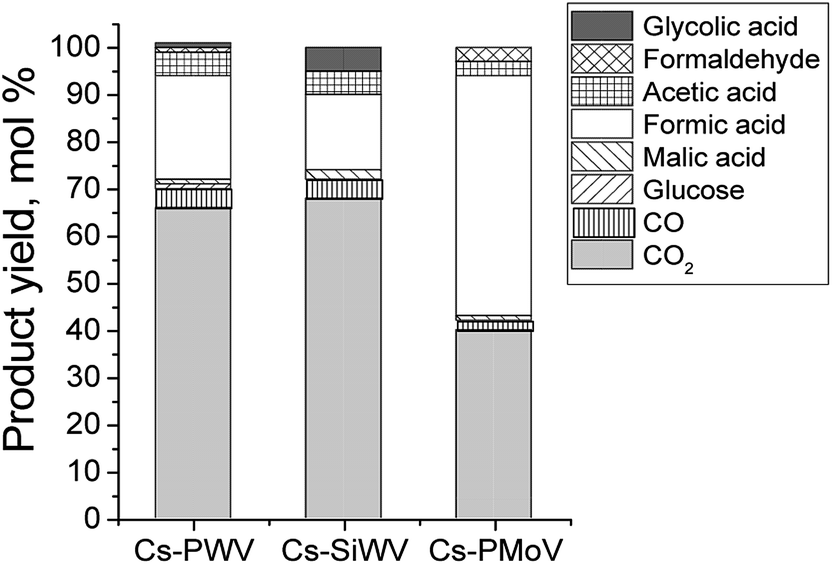 | ||
| Fig. 3 Product yields of starch hydrolysis–oxidation (experimental conditions: 11.9 g L−1 of starch, 1.25 g L−1 of Cs–PWV, 60 mL of reaction volume, 2 MPa of air mixture, 423 K). | ||
 | ||
| Scheme 3 Reaction mechanism of the oxidation on the vanadium active site. | ||
(1) absorption of glucose molecule onto a vanadium site via hydrogen bonding between –OH group of glucose and oxygen atom of –OH group in a vanadium site (step 1);
(2) formation of an intermediate having a ketone group (step 2);
(3) transformation of unstable intermediate to hydroxyacetone and glyceraldehyde, and regeneration of a vanadium site by molecular oxygen (step 3).
(4) formation of formic acid from hydroxyacetone and glyceraldehyde on V-centers (Scheme S1, ESI†).37
The investigation of external sphere chemical composition of heteropoly anion showed that the activity of Cs–PMo was lower compared to Cs–PMoV. Maximum yield of FA equal to 51% was observed in 2 h in the presence of Cs–PMoV. On the other hand, the yield of target product did nor exceed 14% (Table 2, lines 6 and 7). This differences can be explained by the high electron affinity Mo (71.9 kJ mol−1) than V (50.6 kJ mol−1). The low electron affinity of vanadium should favour increasing the reaction rate of the oxidation process.
Catalytic properties of Cs–SiWV and Cs–PWV are different of Cs–PMoV considerably. For example, Cs–PWV allows overoxidation of the hydrolysis products to provide far lower yield of water soluble products (∑YL = 30 mol%). On the other hand, a total of water soluble products is 58 mol% in the presence of Cs–PMoV (Fig. 3, Table 2 lines 3, 5 and 6). Such a difference in catalytic properties can be explained by an influence of W and Mo on electron density of V. It is confirmed by electron affinity (W (78.6 kJ mol−1) and Mo (71.9 kJ mol−1)).
The Cs–PMoV catalyst was more effective to hydrolysis–oxidation of starch to formic acid. The yield of the target product reached 51% as quickly as 2 h of the reaction (Fig. 2B, Table 2, line 6), starch being fully converted. A total product yield was 58% in the aqueous phase, among which there was 88% of formic acid. NMR spectroscopic studies revealed that only acetic, glycolic, succinic acids and hydrated formaldehyde (methylene glycol) were water-soluble side products when the reaction was completed (Fig. S4†).
Catalytic potential and stability of CsHPA systems
The most active sample Cs–PMoV was studied for nine successive cycles of starch hydrolysis–oxidation to demonstrate its high catalytic activity. The yield of formic acid slightly decreases from 51% in the first cycle to 48% in the ninth cycle (Fig. 4), while the yield of the side product (acetic acid) increases from 3 to 7%. High stability of heteropoly anion was confirmed by IR spectroscopy. IR spectra of Cs–PMoV salt in the ninth cycle of the reaction was similar to one of pristine salt (Fig. 5).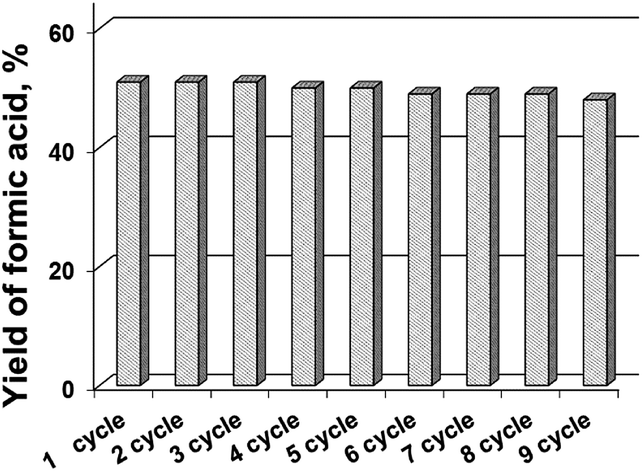 | ||
| Fig. 4 The yield of formic acid achieved during seven runs in the presence of Cs–PMoV catalyst (experimental conditions: 11.9 g L−1 of starch, 1.25 g L−1 of Cs–PWV, 60 mL of reaction volume, 2 MPa of air mixture, 423 K). | ||
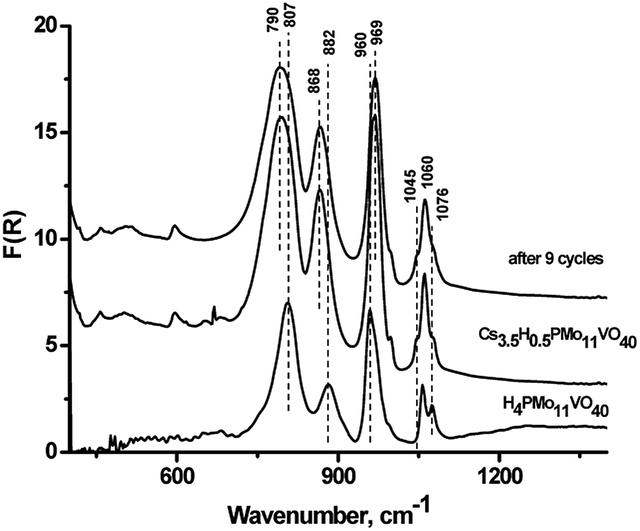 | ||
| Fig. 5 IR spectra of H4PMo11VO40 and Cs3.5H0.5PMo11VO40 pristine and after 9 cycles. | ||
ICP-AES analysis of the reaction mixture demonstrated a high stability of the catalyst under the reaction conditions; no more than 2% of the active component was leached to the reaction solution. Interestingly, leaching of the active component from cesium salts reached 10–42% during cellulose hydrolysis–dehydration.50 Leaching of HPA up to 70% was also observed in cellulose hydrolysis–hydrogenolysis.51 These results pointed that stability of CsHPA is designated by a type of reaction. Acidic products formed during the one-pot hydrolysis–oxidation led to high acidity of the medium, which inhibits the dissociation of HPA and, accordingly, its leaching into the solution.
We compared efficiencies of the best Cs–PMoV catalyst and of soluble HPA Co0.6H3.8PMo10V2O40 (Co–PMoV2) to hydrolysis–oxidation of starch (Table 3). Catalytic properties of Co–PMoV2 and Cs–PMoV were revealed under identical conditions at 423 K and air mixture pressure of 2 MPa. The similar FA yield (50%) was reached in one hour of the reaction at the acid concentration of 10 mM (20 mM of vanadium active sites). On the other hand, 51% yield of formic acid was observed in 2 hours of the reaction in the presence of Cs–PMoV at its concentration of 1.25 g L−1 (0.5 mM of the catalyst, 0.5 mM of vanadium active sites).
| No. | Catalyst | T (K) | P (MPa) | Time (h) | Starch/V atom (mmolglucose unit/mmolV) | Starch/V active site (mol/mol) | FA yield (%) | TOF molFA/(mol[V] h) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Cs–PWV | 423 | 2 | 5 | 62/0.4 | 155![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
22 | 6.8 | This work |
| 2 | Cs–SiWV | 423 | 2 | 7 | 62/0.4 | 155:1 |
16 | 4.0 | This work |
| 3 | Cs–PMoV | 423 | 2 | 2 | 62/0.5 | 124:1 |
51 | 32.0 | This work |
| 4 | Co–PMoV2 | 423 | 2 | 1 | 62/20 | 3.1:1 |
50 | 1.5 | This work |
| 5 | VOSO4 | 413 | 2 | 1.5 | 1/0.1 | 10:1 |
46 | 3.1 | Tang et al.37 |
We compared the effectiveness of the solid and soluble catalysts using TOF calculated by eqn (3):
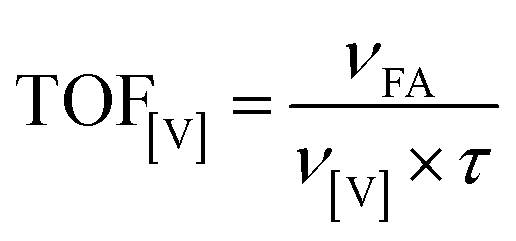 | (3) |
Comparative studies of the catalysts demonstrated that Cs–PMoV is 4–8 times as efficient as Cs–SiWV and Cs–PWV (Table 3, lines 1–3). The best solid catalyst Cs–PMoV is 21 times as efficient as the soluble catalytic system (Table 3, lines 3 and 4). We also compared the catalytic properties of CsHPA catalysts presented in our work and of the catalytic system VOSO4 reported previously.37 As far as we know, this is an only catalytic system for hydrolysis–oxidation of starch to formic acid with molecular oxygen without any additives or co-catalysts in water, which was studied before. TOF was higher in the presence of Cs–PMoV than in the presence of VOSO4. Again, much higher yield of formic acid (51 vs. 46%) at a higher starch:catalyst ratio (Table 3, lines 3 and 5) is reported in the present paper.
Conclusions
One-pot hydrolysis–oxidation of starch into formic acid was studied under hydrothermal conditions (413–443 K, 2 MPa air pressure) over solid bifunctional catalysts based on cesium salts of W(Mo)–V–P(Si) heteropoly acids (CsHPA). The optimum temperature (423 K) and the activation energy of the process (47 ± 2 kJ mol−1) were revealed. Catalyst composition influenced on formic acid yield and selectivity. The investigation of the chemical composition of heteropoly anion showed that the activity of Cs–PMo was significantly lower compared to Cs–PMoV. This may indicate that vanadium plays an important role in the oxidation process. W–V–P(Si) CsHPA results in the considerable formation of CO2 compared to Mo–V–P CsHPA. The most promising catalyst was Cs3.5H0.5PMo11VO40 as the highest formic acid yield reached 51%. High efficiency of Cs3.5H0.5PMo11VO40 was shown in nine cycles of the reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Science Foundation (Project 17-73-30032).References
- W. Deng, Q. Zhang and Y. Wang, Catal. Today, 2014, 234, 31–41, DOI:10.1016/j.cattod.2013.12.041.
- J. Li, D.-J. Ding, L. Deng, Q.-X. Guo and Y. Fu, ChemSusChem, 2012, 5, 1313–1318, DOI:10.1002/cssc.201100466.
- W. Reutemann and H. Kieczka, Formic Acid Ulmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005 Search PubMed.
- N. V. Gromov, O. P. Taran, I. V. Delidovich, A. V. Pestunov, Y. A. Rodikova, D. A. Yatsenko, E. G. Zhizhina and V. N. Parmon, Catal. Today, 2016, 278, 74–81, DOI:10.1016/j.cattod.2016.03.030.
- M. Zacharska, O. Y. Podyacheva, L. S. Kibis, A. I. Boronin, B. V. Senkovskiy, E. Y. Gerasimov, O. P. Taran, A. B. Ayusheev, V. N. Parmon, J. J. Leahy and D. A. Bulushev, ChemCatChem, 2015, 8, 2910–2917, DOI:10.1002/cctc.201500216.
- S. Zhang, Ö. Metin, D. Su and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 3681–3684, DOI:10.1002/anie.201300276.
- F. Valentini, V. Kozell, C. Petrucci, A. Marrocchi, Y. Gu, D. Gelman and L. Vaccaro, Energy Environ. Sci., 2019, 12, 2646–2664, 10.1039/C9EE01747J.
- M. Weber, J. T. Wang, S. Wasmus and R. F. Savinell, J. Electrochem. Soc., 1996, 143, L158–L160, DOI:10.1149/1.1836961.
- J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. A. Dumesic, Appl. Catal., B, 2010, 100, 184–189, DOI:10.1016/j.apcatb.2010.07.029.
- L. Deng, J. Li, D. M. Lai, Y. Fu and Q. X. Guo, Angew. Chem., Int. Ed., 2009, 121, 6651–6654, DOI:10.1002/ange.200902281.
- L. Deng, Y. Zhao, J. Li, Y. Fu, B. Liao and Q.-X. Guo, ChemSusChem, 2010, 3, 1172–1175, DOI:10.1002/cssc.201000163.
- S. G. Wettstein, D. M. Alonso, Y. Chong and J. A. Dumesic, Energy Environ. Sci., 2012, 5, 8199–8203, 10.1039/C2EE22111J.
- D. M. Alonso, S. G. Wettstein, J. Q. Bond, T. W. Root and J. A. Dumesic, ChemSusChem, 2011, 4, 1078–1081, DOI:10.1002/cssc.201100256.
- J.-H. Park, M.-H. Jin, D.-W. Lee, Y.-J. Lee, G.-S. Song, S.-J. Park, H. Namkung, K. H. Song and Y.-C. Choi, Environ. Sci. Technol., 2019, 53, 14041–14053, DOI:10.1021/acs.est.9b04273.
- X. Chen, Y. Liu and J. Wu, Mol. Catal., 2020, 483, 110716, DOI:10.1016/j.mcat.2019.110716.
- F. Jin, J. Yun, G. Li, A. Kishita, K. Tohji and H. Enomoto, Green Chem., 2008, 10, 612–615, 10.1039/B802076K.
- A. T. Quitain, M. Faisal, K. Kang, H. Daimon and K. Fujie, J. Hazard. Mater., 2002, 93, 209–220, DOI:10.1016/S0304-3894(02)00024-9.
- L. Calvo and D. Vallejo, Ind. Eng. Chem. Res., 2002, 41, 6503–6509, DOI:10.1021/ie020441m.
- J. Li, D.-J. Ding, L. Deng, Q.-X. Guo and Y. Fu, ChemSusChem, 2012, 5, 1313–1318, DOI:10.1002/cssc.201100466.
- J. Albert, R. Wolfel, A. Bosmann and P. Wasserscheid, Energy Environ. Sci., 2012, 5, 7956–7962, 10.1039/C2EE21428H.
- J. Zhang, M. Sun, X. Liu and Y. Han, Catal. Today, 2014, 233, 77–82, DOI:10.1016/j.cattod.2013.12.010.
- J.-M. Bregeault, Dalton Trans., 2003, 17, 3289–3302, 10.1039/B303073N.
- L. El Aakel, F. Launay, A. Atlamsani and J.-M. Bregeault, Chem. Commun., 2001, 21, 2218–2219, 10.1039/B106969A.
- T. Lu, Y. Hou, W. Wu, M. Niu and Y. Wang, Fuel Process. Technol., 2018, 171, 133–139, DOI:10.1016/j.fuproc.2017.11.010.
- I. V. Kozhevnikov and K. I. Matveev, Russ. Chem. Rev., 1982, 51, 1075, DOI:10.1070/RC1982v051n11ABEH002941.
- E. G. Zhizhina, K. I. Matveev and V. V. Russkikh, Chem. Sustainable Dev., 2004, 12, 47–51 CAS , https://sibran.ru/en/journals/issue.php?ID=119844%26ARTICLE_ID=124358.
- E. Rafiee and H. Jafari, Bioorg. Med. Chem. Lett., 2006, 16, 2463–2466, DOI:10.1016/j.bmcl.2006.01.087.
- A. A. Shatalov, D. V. Evtuguin and C. Pascoal Neto, Carbohydr. Polym., 2000, 43, 23–32, DOI:10.1016/S0144-8617(99)00195-2.
- J. Albert, R. Wolfel, A. Bosmann and P. Wasserscheid, Energy Environ. Sci., 2012, 5, 7956–7962, 10.1039/C2EE21428H.
- J. Zhang, M. Sun, X. Liu and Y. Han, Catal. Today, 2014, 233, 77–82, DOI:10.1016/j.cattod.2013.12.010.
- N. V. Gromov, T. B. Medvedeva, Y. A. Rodikova, A. V. Pestunov, E. G. Zhizhina and O. P. Taran, J. Sib. Fed. Univ., Chem., 2018, 11, 56–71, DOI:10.17516/1998-2836-0058.
- Y. Hou, Z. Lin, M. Niu, S. Ren and W. Wu, ACS Omega, 2018, 3, 14910–14917, DOI:10.1021/acsomega.8b01409.
- M. Niu, Y. Hou, S. Ren, W. Wang, Q. Zheng and W. Wu, Green Chem., 2015, 17, 335–342, 10.1039/C4GC00970C.
- J. Xu, H. Zhang, Y. Zhao, Z. Yang, B. Yu, H. Xu and Z. Liu, Green Chem., 2014, 16, 4931–4935, 10.1039/C4GC01252F.
- T. Okuhara, H. Watanabe, T. Nishimura, K. Inumaru and M. Misono, Chem. Mater., 2000, 12(8), 2230–2238, DOI:10.1021/cm9907561.
- P. Maki-Arvela, B. Holmbom, T. Salmi and D. Y. Murzin, Catal. Rev.: Sci. Eng., 2007, 49, 197–340, DOI:10.1080/01614940701313127.
- Z. Tang, W. Deng, Y. Wang, E. Zhu, X. Wan, Q. Zhang and Y. Wang, ChemSusChem, 2014, 7, 1557–1567, DOI:10.1002/cssc.201400150.
- V. F. Odyakov and E. G. Zhizhina, Russ. J. Inorg. Chem., 2009, 54, 361–367, DOI:10.1134/S003602360903005X.
- V. F. Odyakov and E. G. Zhizhina, React. Kinet. Catal. Lett., 2008, 95, 21–28, DOI:10.1007/s11144-008-5374-7.
- V. F. Odyakov, E. G. Zhizhina, Y. A. Rodikova and L. L. Gogin, Eur. J. Inorg. Chem., 2015, 3618–3631, DOI:10.1002/ejic.201500359.
- P. J. Domaille, G. Herevéa and A. Téazéa, Vanadium(V) substituted dodecatungstophosphates, in Inorganic Syntheses, ed. A. P. Ginsberg, John Wiley & Sons, Inc, 1990, vol. 27, pp. 96–104, DOI:10.1002/9780470132586.ch17.
- S. Subramanian, J. S. Noh and J. A. Schwarz, Determination of the point of zero charge of composite oxides, J. Catal., 1988, 114, 433–439, DOI:10.1016/0021-9517(88)90046-2.
- A. A. Davydov, Molecular spectroscopy of oxide catalyst surfaces, John Wiley & Sons, Inc, Chichester, 2003, DOI:10.1002/0470867981.
- E. A. Paukshtis, Infrakrasnaya spektroskopiya v geterogennom kislotno-osnovnom katalize (Infrared Spectroscopy in Heterogeneous Acid–Base Catalysis), Nauka, Novosibirsk, 1992, ISBN: 5-02-029281-8 Search PubMed.
- http://gostpdf.ru/gost-16932-82.
- S. Maerten, C. Kumpidet, D. Voß, A. Bukowski, P. Wasserscheid and J. Albert, Glucose oxidation to formic acid and methyl formate in perfect selectivity, Green Chem., 2020, 22, 4311–4320, 10.1039/d0gc01169j.
- Y. Hou, Z. Lin, M. Niu, S. Ren and W. Wu, Conversion of cellulose into formic acid by iron(III)-catalyzed oxidation with O2 in acidic aqueous solutions, ACS Omega, 2018, 3, 14910–14917, DOI:10.1021/acsomega.8b01409.
- D. Minoli, Nanotechnology Applications to Telecommunications and Networking, Wiley-Interscience, 1st edn, 2005, 512, ISBN: 978-0-471-71639-6 Search PubMed.
- M. Pope, Heteropoly and isopoly oxometalates, Springer-Verlag, Berlin Heidelberg, 1983, p. 180 Search PubMed.
- N. V. Gromov, T. B. Medvedeva, O. P. Taran, M. N. Timofeeva and V. N. Parmon, Katal. Prom-sti., 2020, 20(3), 234–242, DOI:10.18412/1816-0387-2020-3-234-242.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Green Chem., 2011, 13(8), 2167–2174, 10.1039/C1GC15350A.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05501h |
| This journal is © The Royal Society of Chemistry 2020 |