
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow does the Li-distribution in the 16d sites determine the stability of A3(Li,Ti5)O12 (A = Li and Na)?†
Kohei Tada ,
Hiroyuki Ozaki*,
Tetsu Kiyobayashi,
Mitsunori Kitta and
Shingo Tanaka
,
Hiroyuki Ozaki*,
Tetsu Kiyobayashi,
Mitsunori Kitta and
Shingo Tanaka
Research Institute of Electrochemical Energy (RIECEN), Department of Energy and Environment, National Institute of Advanced Industrial Science and Technology (AIST), 1-8-31 Midorigaoka, Ikeda, Osaka 563-8577, Japan. E-mail: hiroyuki.ozaki@aist.go.jp
First published on 10th September 2020
Abstract
Li3(Li,Ti5)O12 (LTO) is a stable and safe negative electrode material for Li-ion batteries, and its Na substitute Na3(Li,Ti5)O12 (NTO) is a counterpart for the Na-ion battery. In LTO and NTO, a sixth of the Ti-sites (16d) in the spinel framework are replaced by Li: Li mixing in the 16d sites. For conducting theoretical studies on these materials, e.g., density functional theory (DFT) calculations, one has to confront the astronomical number of combinations of Li distribution in 16d sites to construct model structures, of which the size is sufficiently large to represent the bulk material properties. Only a limited number of models, whose structures are a priori specified by “researcher intuition,” have been examined thus far, and how Li-mixing determines the material stability has yet to be clarified. Herein, we statistically analyzed the DFT total energy of more than 2 × 104 model structures of LTO and NTO that were extracted from the 4 × 108 possible combinations of Li-mixing with computer-aided symmetry analysis and an automated model building system. The local energy analysis further revealed the local stability/instability of each structure. We found that LTO and NTO stability can be well explained by the apparent coulombic repulsion between Li+ in the 16d sites as if they were placed in a matrix of dielectric constants of 1.92 and 2.04 for LTO and NTO, respectively. That is, the sum of the inverse of the Li–Li distance (S) serves as a good descriptor in predicting the stability of these materials. The extent to which the O2− anions are displaced from the Wyckoff position (32e) is considered to differentiate NTO from LTO. However, the electronic structure of NTO does not significantly differ from that of LTO unless S exceeds a certain limit. These results suggest that the spinel framework tolerates the structural instability and variety to some extent, which is important in constructing a spinel structure with the mixing of other cations, thereby replacing the rare element Li.
1 Introduction
Secondary batteries are indispensable in daily life. Their recent widespread usage has necessitated further improvement in the safety of rechargeable Li-ion batteries.1–3 Li4Ti5O12 (LTO)4–14 is a negative electrode material for Li-ion secondary batteries and, compared to conventional carbon electrodes, is safer for use in electric and plug-in hybrid electric vehicles. The Li ion is inserted in and extracted from the LTO according to| (Li3)8a(Li1,Ti5)16d(O12)32e + 3Li+ + 3e− ⇌ (Li6)16c(Li1,Ti5)16d(O12)32e, | (1) |
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m symmetry (spinel). During the reaction, the Ti–O framework maintains its spinel structure, which is stabilized by the Li+ cations substituted for a sixth of Ti4+ cations situated in the 16d sites (referred to as “Li-mixing”). The material functions as a negative electrode for Li-ion batteries when Li ions move between 8a and 16c sites. Li insertion only slightly expands the spinel lattice ((Li3)8a(Li1,Ti5)16d(O12)32e: 8.36 Å, (Li6)16c(Li1,Ti5)16d(O12)32e: 8.35 Å), so that the charge and discharge process is strain-free. This structural robustness yields good cycling properties (durability) and renders LTO suitable as a safe negative electrode material.
m symmetry (spinel). During the reaction, the Ti–O framework maintains its spinel structure, which is stabilized by the Li+ cations substituted for a sixth of Ti4+ cations situated in the 16d sites (referred to as “Li-mixing”). The material functions as a negative electrode for Li-ion batteries when Li ions move between 8a and 16c sites. Li insertion only slightly expands the spinel lattice ((Li3)8a(Li1,Ti5)16d(O12)32e: 8.36 Å, (Li6)16c(Li1,Ti5)16d(O12)32e: 8.35 Å), so that the charge and discharge process is strain-free. This structural robustness yields good cycling properties (durability) and renders LTO suitable as a safe negative electrode material.
Our group has recently synthesized a Na substitute for LTO, Na3LiTi5O12 (NTO), in which all the Li+ cations situated in the 8a sites are replaced by Na and, as in LTO, a sixth of the 16d sites are occupied by Li.15–18 During the charge and discharge process with the Na counter electrode, NTO works as a negative electrode when Na ions in the 8a sites move to the 16c sites:
(Na3)8a(Li1,Ti5)16d(O12)32e + 3Na+ + 3e− ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ⇌ (Na6)16c(Li1,Ti5)16d(O12)32e, ⇌ (Na6)16c(Li1,Ti5)16d(O12)32e,
| (2) |
Computational calculations based on density functional theory (DFT) are widely applied to elucidate material functionalities and design new materials. Examples of battery materials examined by DFT can be found in ref. 19 and 20. As for LTO and NTO, the electronic states have been investigated to design high-performance materials.16–18,21–31 In a previous DFT study of LTO and newly discovered NTO, the electronic states and local atomic structures were examined. The O2− anion in LTO and NTO can flexibly move a distance from Ti, whereby it forms a coordinate bond with Ti and results in an analogous electronic state either for LTO or NTO with a local structural strain.17 The crystallographic structure of LTO and NTO in the present study is described as (Na or Li)8a(Li1/6Ti5/6)16dO4, in which a sixth of 16d sites in the spinel structure is occupied by Li. For LTO, the Li-substitution in the 16d sites was experimentally confirmed by the solid-state NMR.14 The presence of Li in the 16d sites has been experimentally observed by us in NTO as well.18 That is, the Li-substitution in the 16d sites is essential for either LTO or NTO from the viewpoint of crystallographic structure. Previous studies,16–18,21–29 however, have not examined how Li-mixing in the 16d sites influences the stability of the spinel framework and the relationship between the local structural strain and local energetic instability.
Furthermore, Li-mixing with Ti in the 16d sites in LTO (Li(Ti5/3,Li1/3)16dO4) and NTO (Na(Ti5/3,Li1/3)16dO4), which compensates for the charge of Ti to retain the neutrality of the entire system, is of interest from a crystallographic point of view. In LiTi2O4 (Li(Ti3+,Ti4+)16dO4)12,13,28,32–34 and LiMn2O4 (Li(Mn3+,Mn4+)16dO4),3,13,35–42 two other electrode materials with spinel structures, the 16d sites are occupied by identical cations with different valences. This contrasts with LTO and NTO, where the 16d sites are occupied by different cations, Li+ and Ti4+. For LiTi2O4, the difference in the ionic radius between Ti3+ (67 pm) and Ti4+ (60.5 pm) is 11%. For LiMn2O4, the ionic radius difference between Mn3+ (58 pm) and Mn4+ (53 pm) is 10%. On the other hand, radii of Li+ and Ti4+ (76 pm and 60.5 pm, respectively) differ by 26%. In addition, as for the stability of the spinel electrode materials other than LTO and NTO, the Li–Mn–O systems were examined in detail by Yabuuchi et al., who proved that the material Li8a(LixMn2−x)16dO4, in which a part of the 16d sites was occupied by Li, more stably retains its spinel structure than the regular composition LiMn2O4.43 Thus, it is anticipated that the significant difference in cation mixing in the 16d sites should manifest itself in the stability of the spinel structure of LTO and NTO.
As NTO is synthesized by substituting Na for Li at the 8a sites of LTO, the states of the LTO precursor must be reflected in the 16d sites of the NTO product.15,18 Examination of the circumstances of the 16d sites provides the key to understanding why only NTO can retain its spinel structure among sodium titanates.
As mentioned above, previous theoretical studies have focused only on a limited number of stable structure configurations.16–18,21–31 Some studies attempted to predict the most stable structure of the LTO using systematic models.30,31 However, the models used were non-stoichiometric compositions, of which the results could contain computational errors, such as the dynamic and static correlations as well as the spin contamination, affecting the results of electrode materials.25,44 To precisely correct the errors, the multi-referenced calculations should be performed, which is often prohibited by the computational cost. Although the genetic algorithm,45,46 which seeks the most stable structure among those considered, may be applied to investigate the stable configuration of the dopant, it is beyond the scope of this technique to quantitatively relate the configuration to the stability (or instability). That is, we still do not know how Li-mixing determines the stability of the spinel framework of LTO and NTO, despite its importance in determining material performance.
Herein, we analyzed the DFT total energy of a number of randomly generated structural models to elucidate how the Li configuration influences the stability of LTO and NTO. The recent growth of super-computing systems has aided in the construction of a database that allows for statistical treatment of these randomly generated models. We used a super-computing system provided by Kyushu University and an in-house coded tool,25,47 which generates numerous model structures and analyzes data obtained by the DFT calculation. The statistical method can provide the descriptors for machine-learning, whereby one can determine the most stable structure from numerous candidates through the interpolative scheme. In addition, local energy analysis48–57 of the obtained stable structures allowed for the evaluation of the influence of local strain caused by Li-mixing. This analysis revealed the difference between LTO and NTO with respect to their stability.
2 Computational procedure
To represent the chemical composition of LTO (Li:Ti:O = 4:5:12) and NTO (Na:Li:Ti:O = 3:1:5:12), we adopted a 1 × 1 × 3 supercell model in which the number of sites was 24(8a), 48(16d), and 96(32e). All 32e sites were occupied by O2−, and all 8a sites were occupied by Li+ in LTO or Na+ in NTO. The Li+ and Ti4+ cations are mixed in the 16d sites in such a way that Ti4+ occupy five-sixth of the sites (40 sites), and the remaining one-sixth (eight sites) is occupied by Li+. This results in 48C8 = 377, 348, 994 possible configurations if the symmetry is not taken into account. We first randomly generated 15000 configurations, out of which 8444 were extracted by eliminating the combinations of equivalent symmetry. The set of energies calculated for all 8444 configurations by DFT, without optimization, was statistically analyzed for LTO and NTO. From the regression line, 58 models considered relevant were selected, and these models were optimized by DFT to obtain stable structure and energy. Further analysis of these selected data yielded more reliable regression results. The atomic positions were optimized whereas the lattice parameters were not. The purpose of the present work is to clarify the stability of the spinel framework by the Li-mixing. To preserve the spinel framework structure, we fixed the lattice parameter to the experimental values. Unstable structures would be unable to keep the framework, and hence takes a long time for the lattice parameters to be optimized. The optimization per se and the final structure with which the unstable state would end up are beyond the scope of the present study.
The energy and electronic states were obtained by DFT using the ab initio calculation package VASP58–61 on the ITO super-computing system provided by Kyushu University. The structural models were generated, and the calculated data were managed using an in-house coded Python program.25,47 The structures were visualized using VESTA.62
GGA-PBE63 was adopted as the exchange and correlation functional of DFT. The electrons in the core region were treated with the projector-augmented plane-wave (PAW) method64,65 with an energy cutoff of 450 eV. The number of valance electrons was one, one, ten, and six for Li, Na, Ti, and O, respectively. The k-points were sampled on the Monkhorst mesh66 of 4 × 4 × 2. The calculation conditions were optimized in our previous study,17 which confirmed that the corrections of the on-site Coulomb and dispersion forces hardly influence the electronic states and structures.
The formal valence in X3LiTi5O12 is X = +1, Li = +1, Ti = +4 and O = −2, respectively, suggesting that these ions are in the closed shell structure. The experimental EELS (Electron Energy Loss Spectrums) study indeed corroborates Ti(IV) in X3LiTi5O12. We hence consider that, as a first approximation, it should be reasonable to calculate under the spin-restriction with excluding the open shell structure. In the stable structures, the closed shell configuration is the electronic ground state and Ti(III) will occur in the high energy excited state; therefore, the spin-unrestricted calculation is not required. As for the unstable structures, Ti(III) could partially occur, for which the spin-restricted calculation would entail the error associated with the static electronic correlation. We made a brief consideration in Results and discussion regarding this issue.
By analyzing the local energy of the optimized structures using the QMAS program,48 we investigated how the structural strain caused by the Li-mixing depends on the configurations as well as how it differs in LTO and NTO. Local energy/stress analysis using the QMAS code has been adopted successfully in grain boundary systems and surface systems.49–55 The local energy discussed in this paper was obtained by integrating the energy density in the local region into which the calculated cell is divided into centered atoms.56,57 This method can clarify how much each atom contributes to the total energy obtained in the calculated cell. When the local energy is small, it is relatively unstable. Conversely, when the local energy is significant, it is stable. Therefore, by comparing the same atomic species, we are able to analyze the effect of structural distortion on the stability of the spinel framework in LTO and NTO.
3 Results and discussion
3.1 Li4Ti5O12 (LTO) system
Fig. 1 shows the total energy computation results of LTO. In Fig. 1(a), the y-axis is total energy E before the structure optimization, indicating the structural stability (i.e., the more negative, the more stable), and S in the x-axis is the sum of the reciprocal distance between Li in the 16d sites; namely,
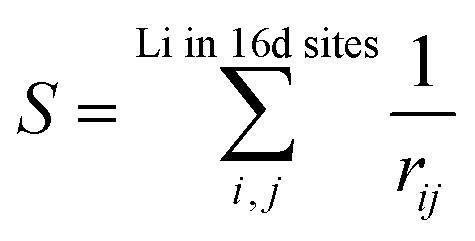 | (3) |
000 models, under the constraint S/Å−1 < 3.86, and the result of the corresponding DFT calculation is indicated by the red cross in Fig. 1(b). The stability of LTO with respect to Li-mixing positively correlates with the sum of the reciprocal distance between Li in the 16d sites.
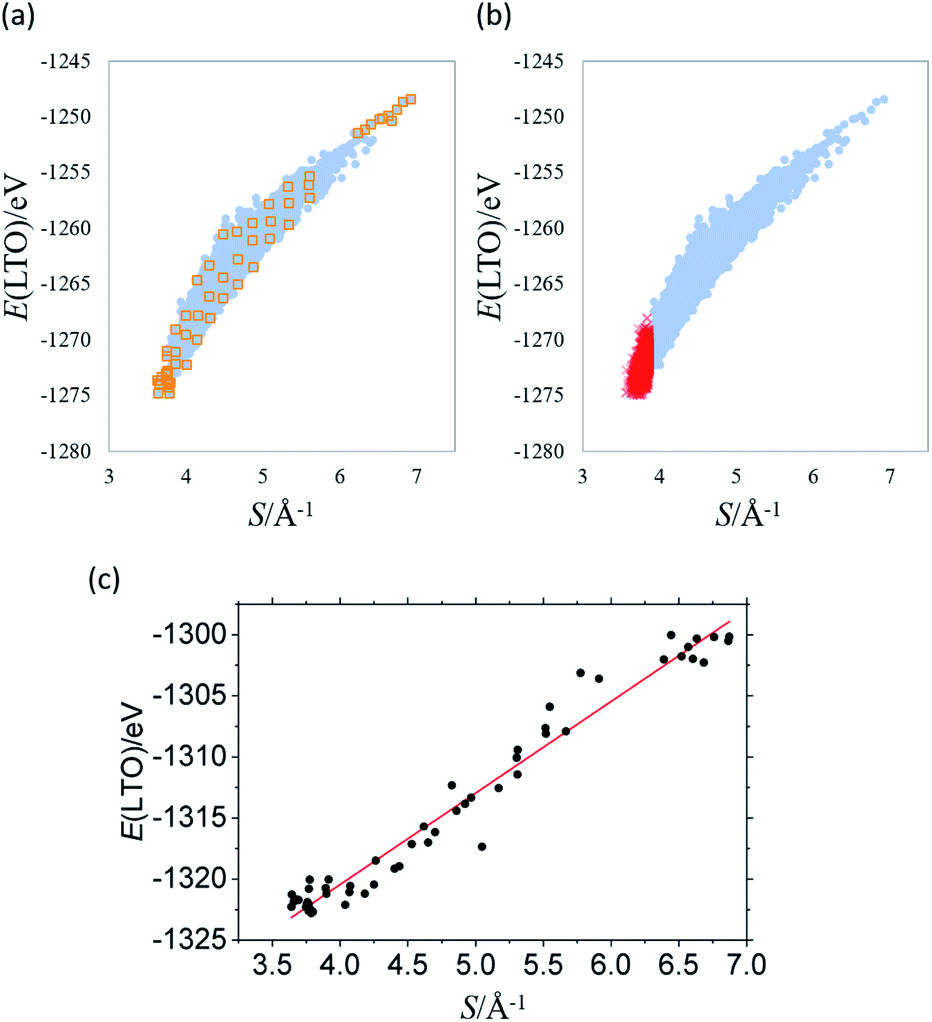 | ||
| Fig. 1 Total energy E(LTO) of Li4Ti5O12 as a function of the sum of the inverse distance between Li in the 16d sites S for (a) 8444 non-optimized structures, (b) additional 5000 structures (red cross) overlaid on (a), and (c) 58 optimized structures whose initial positions are indicated with orange squares in (a). | ||
The results shown in Fig. 1 were obtained by fixing all the ions at the ideal Wyckoff positions. However, both theoretical and experimental studies suggest that the O2− anions in LTO are slightly displaced from the 32e sites, the ideal position for the spinel (rock-salt) structure because O would be too far from Ti to form a sufficient coordinate Ti–O bond.17,18 To discuss the stable structures, structural optimization is required to consider the stabilization brought by the coordinate Ti–O bond properly. We optimized the structures of 58 models that were selected from those in Fig. 1(a), indicated by orange squares, as (1) 18 structures from the most stable (the top 18), (2) ten structures from the least stable (the bottom ten), and (3) 30 structures from the entire range of S with an equal interval. The result after the optimization is shown in Fig. 1(c), confirming the strong correlation between E and S. The regression line is given by:
| E/eV = a(S/Å−1) + b | (4) |
444 models insignificantly differ from that those in the previous studies, implying that the spin-unrestricted calculation is not necessary insofar as the electronic ground state of titania has the closed shell configuration.
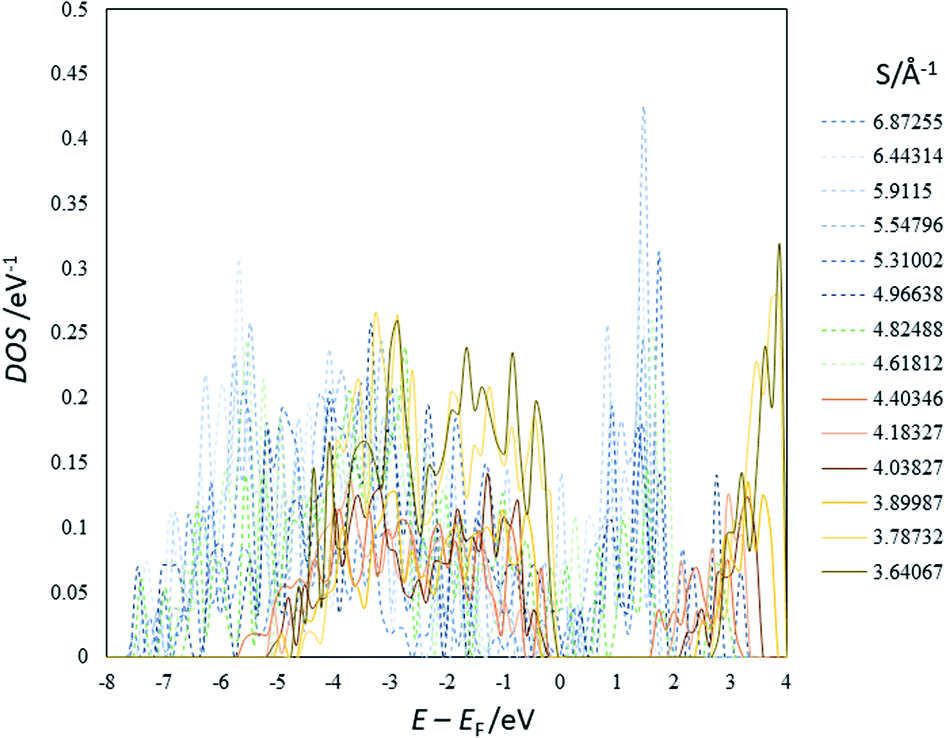 | ||
| Fig. 2 An example of the DOS of the optimized structures of Li4Ti5O12 (others are shown in ESI†). EF represents the Fermi energy. | ||
3.2 Na3LiTi5O12 (NTO) system
Fig. 3 summarizes the results for NTO. As in LTO (Fig. 1(a)), the total energy of NTO, E(NTO), is strongly correlated with the sum of the reciprocal distance between Li at the 16d sites, S in Fig. 3(a), wherein the results of DFT calculations for 8444 configurations are without structural optimization. In real NTO, however, the O2− anions are displaced from the ideal Wyckoff position (32e), the extent of which has been experimentally and theoretically confirmed to be more significant than in LTO. For example, the results of high-intensity X-ray diffraction are better fitted by placing O in the 192i (x,y,z) sites rather than in the 32e (x,x,x) sites.17,18 Structural optimization would hence influence NTO more than LTO. As for LTO, we optimized a total of 58 structures, indicated by orange squares in Fig. 3(a) that are (1) the top 18 stable, (2) the bottom ten unstable, and (3) 30 models taken out of Fig. 3(a) with an equal interval in S. The O-displacement in the optimized structures is consistent with the experimental observation (Fig. S32†). The E(NTO) versus S relation after the optimization and the DOS are summarized in Fig. 3(b) and 4, respectively.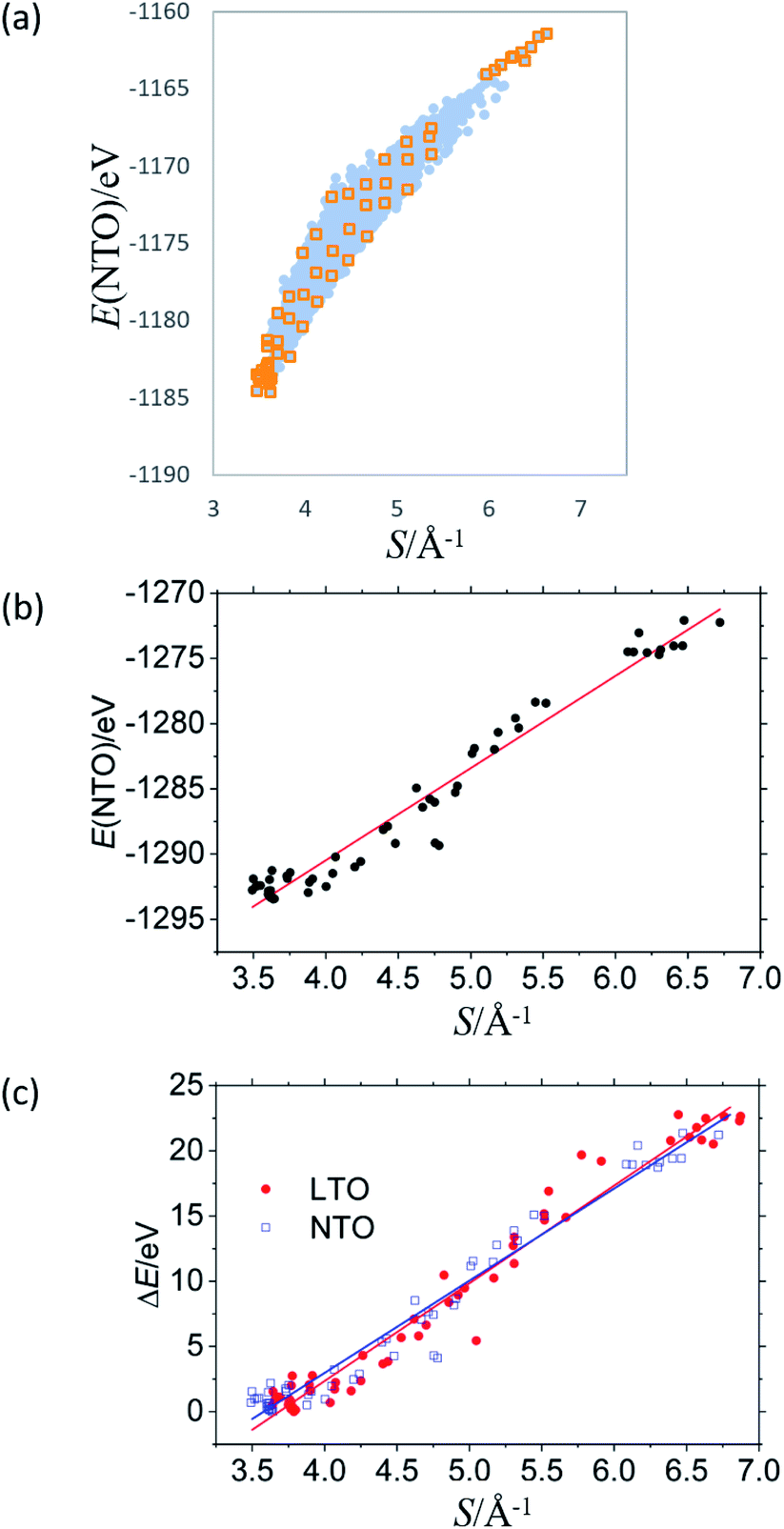 | ||
| Fig. 3 Total energy E(NTO) of Na3LiTi5O12 as a function of S for (a) 8444 non-optimized structures and (b) 58 optimized structures whose initial positions are indicated with orange squares in (a). (c) Energy difference from the most stable structure (ΔE) as a function of S for the optimized structures of Li4Ti5O12 and Na3LiTi5O12. | ||
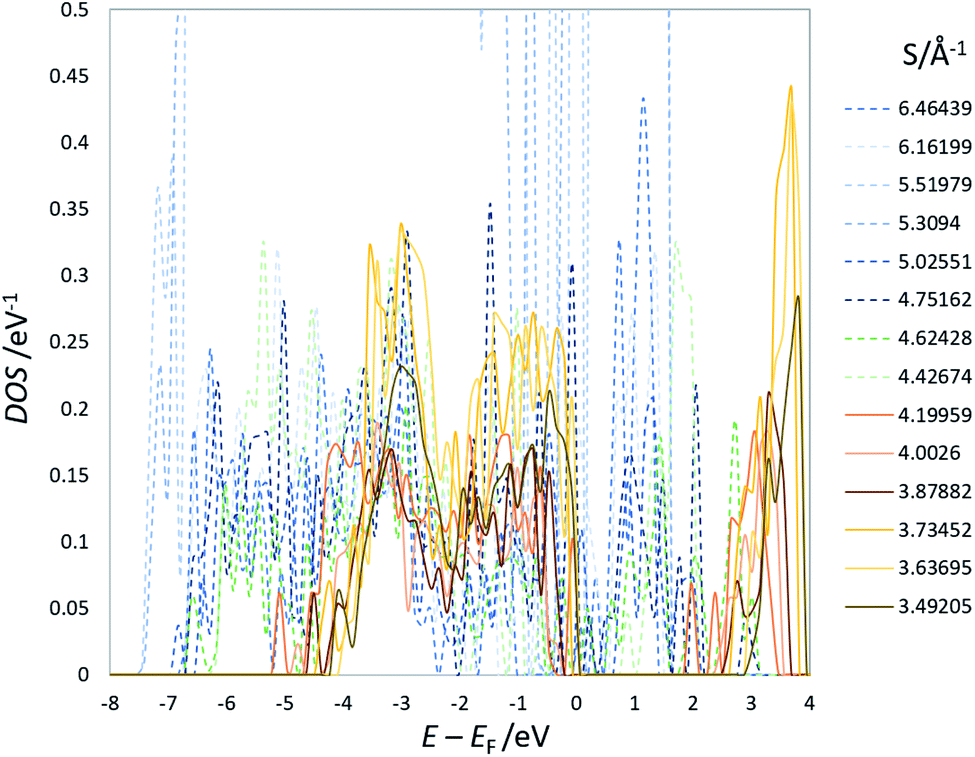 | ||
| Fig. 4 An example of the DOS of optimized structures of Na3LiTi5O12 (others are shown in the ESI†). EF represents the Fermi energy. | ||
The coordinate Ti–O bonds, achieved by the optimization, create a band gap (Fig. 4) and significantly lower the total energy. The strong correlation between E and S is nevertheless persistent (Fig. 3(b)). The linear optimization with the analogous equation in eqn (4) yields a/Å eV = 7.07 ± 0.36 and b/eV = −1318.7 ± 1.7 with R2 = 0.964. The band gap of structures with S less than 4.0–4.1 Å−1 is nearly constant at ca. 2 eV. These observations are in line with those of LTO. We can, therefore, conclude that, as in LTO, the apparent coulombic repulsion between Li+ in the 16d sites dominates the stability of the spinel framework of NTO with little orbital correlation between Ti and O. The analogous outcome drawn from LTO and NTO suggests that the 8a sites occupied by Li or Na are insignificant in determining the stability as far as the steady-state is concerned.
The present study implies that the stability of the (Ti, Li)–O spinel framework is dictated by the magnitude of the repulsion between Li, regardless of LTO or NTO. In effect, the models selected as the top 10 stable structures for LTO have identical Li configurations to those of NTO, and their order of stability is also identical for LTO and NTO (Fig. 5 and 6). A similar trend is observed for all selected (optimized) structures (see ESI†). The relationship between LTO and NTO is clearly seen in the overlapping data. However, there is a slight difference in the slope in which the overlay is the energy difference (ΔE) from the most stable structure versus S for LTO and NTO (Fig. 3(c)). It can thus be concluded that, for either LTO or NTO, the stability of the spinel framework is determined by the distance between Li in the 16d sites, and their configuration hardly influences the electronic states from these results. This accompanies the fact that the optimized DOS is independent of the configuration if S is sufficiently small (Fig. 2 and 4) and is identical in LTO and NTO.17
 | ||
| Fig. 5 Perspective views of the optimized top ten stable structures and their ΔE values (the energy difference in the total energy from the most stable structure) for Li4Ti5O12. In the left panels, spheres in green, blue, and red represent Li, Ti, and O atoms, respectively; spheres in dark and light green represent Li atoms at the 16d and 8a sites, respectively. In right panels, the Li atoms in the 16d sites are shown as the green spheres, and the other atoms are shown as the sticks. | ||
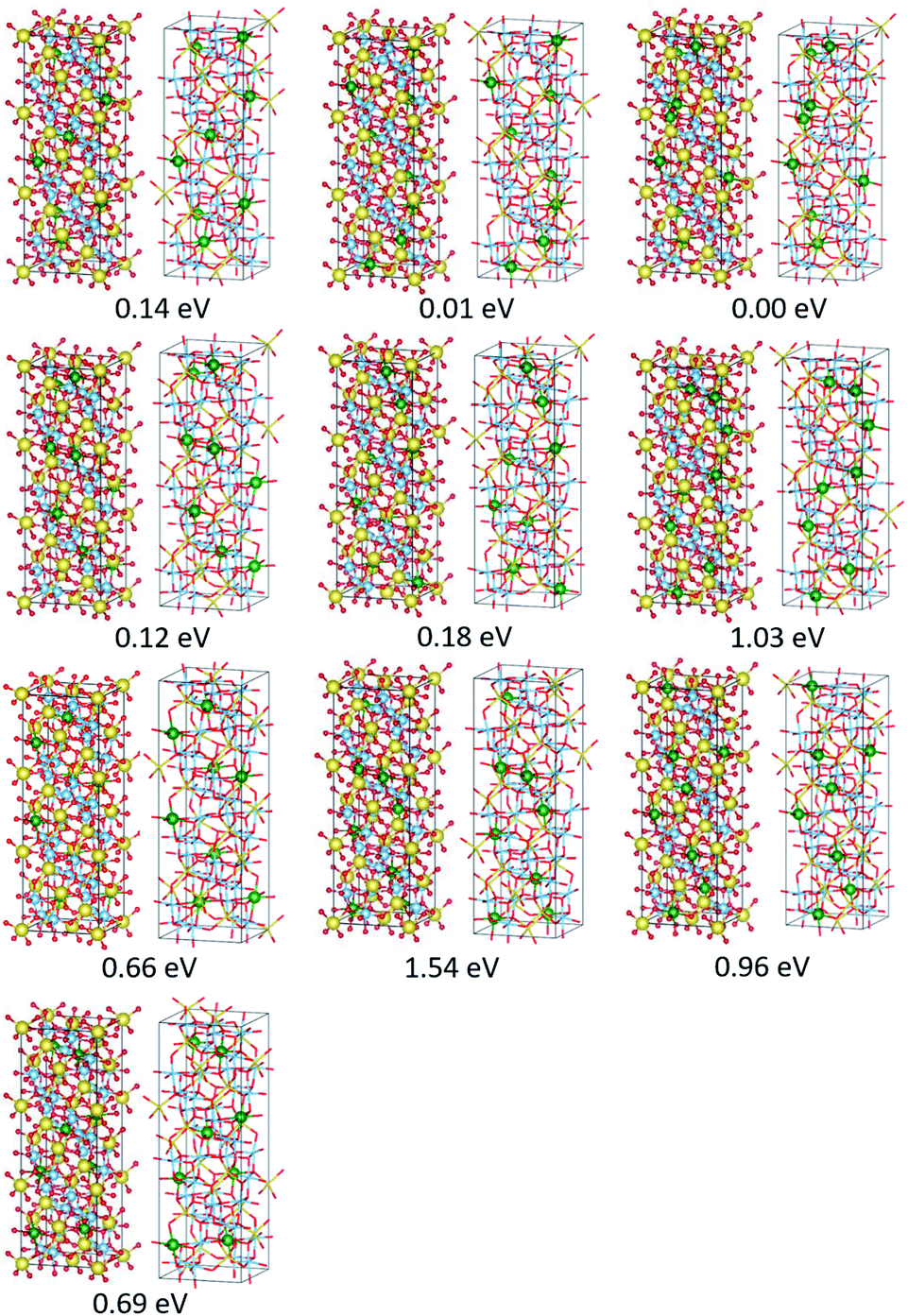 | ||
| Fig. 6 Perspective views of the optimized top ten stable structures and their ΔE values (the energy difference in the total energy from the most stable structure) for Na3LiTi5O12. In the left panels, spheres in yellow, green, blue, and red represent Na, Li, Ti, and O atoms, respectively In right panels, the Li atoms in the 16d sites are shown as the green spheres, and the other atoms are shown as the sticks. | ||
The strong ionicity of an alkali metal, as mentioned in a previous study,17 is attributed to the stability and is well supported by the coulombic repulsion between Li at the 16d sites. Spinel titanates can be viewed as ionic crystals comprised of alkali metals (Li+, Na+) at the 8a sites and a complex spinel framework formed by the coordinate Ti–O bonds. The stability of the spinel framework is determined by the coulombic repulsion between cations (Li+) in the 16d sites. In other words, the more homogeneous the Li-mixing, the more stable the framework. It can be expected from the present calculations that the spinel network is retained as a stable one as long as the substituting cation compensates the charge and has the ionic radius that the 16d sites can accommodate. Currently, only LTO and NTO are available as alkali spinel titanates, in which Li, a rare element, is used as the mixing cation. A further benefit is gained from the viewpoint of resource issues and industrial cost if we succeed in replacing Li with Na, an abundant element. The DFT calculation (Fig. S31†) suggests that a titanate, in which Na (ionic radius 102 pm) is mixed in the 16d sites, forms the spinel framework. Replacing Li+ in the 8a sites with Cu+ (60 pm) or Ag+ (100 pm) can also retain the spinel structure. On the other hand, inserting K+ (137 pm) in the 8a sites destroys the structure, indicating that the maximum ionic radius that can be accommodated in the 8a sites lies somewhere between the size of Ag+ and K+. Examining the stability of the spinel framework in this way, one can explore and design new functional materials.
The difference in the slope between LTO and NTO shown in Fig. 3(c) is explained as follows. The coulombic repulsion potential is expressed as:
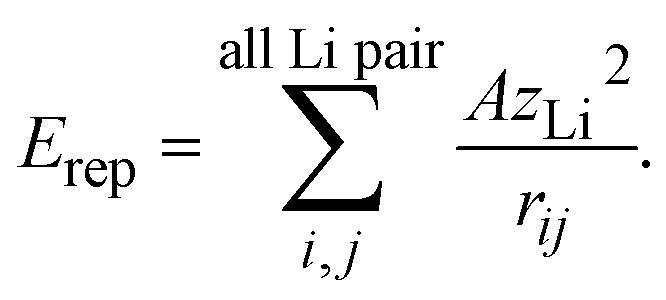 | (5) |
3.3 Local energies of LTO and NTO
The O2− anions are more displaced from the 32e sites in NTO than in LTO, and the trend is independent of Li-mixing at the 16d sites. While the net stability of the spinel framework is discussed in Sections 3.1 and 3.2, we now deal with the local stability resulting from the oxygen position. The local energy of the structures belonging to the top ten (Fig. 5 and 6) was analyzed, and the results are shown in Fig. 7. The difference in the local energy between LTO and NTO is insignificant, which is surprising when considering the greater local distortion and the greater variance in the bond length in NTO caused by the oxygen displacement. However, as discussed in Section 3.2, the stability of the spinel structure is explicable in terms of the repulsion between Li+ in the 16d sites, and the coordinate Ti–O bonds are deemed identical in LTO and NTO. Although the structure (or symmetry) is more disrupted in NTO than LTO, the coordinate bonds nearly equally stabilize their structure, of which the total stability is determined by the inter-ionic interaction so that the energetics are insensitive to local disruption. The oxygen position in LTO and NTO has a high degree of freedom, serving as an absorber against structural distortion. The cycle stability of LTO as an active electrode material for the Li-ion batteries was described in the previous study.11 Our previous study examined the stability of NTO as an active material for the Na-ion batteries.18 This stability stems from the spinel structure of LTO and NTO. As for the stability of the spinel electrode materials other than LTO and NTO, the material Li8a(LixMn2−x)16dO4, in which a part of the 16d sites was occupied by Li, more stably retains its spinel structure than the regular composition LiMn2O4.43 We have recently suggested the importance of the Li-substitution in the 16d sites for NTO to retain the spinel structure.16 The partial substitution of Li in the 16d sites in the spinel network thus plays an important role in maintaining the structural stability of the electrode during the charge and discharge cycles. The statistical analysis of the Li-substituted models in the present study is relevant to considering its stability as an active electrode material. In the present study, we examined the spinel Ti–O framework in the material to find that the difference in its stability between LTO and NTO is insignificant. Hence the difference in the redox potential of Li and Na, the former is lower than the latter, should be almost directly reflected in the electrode potential of LTO and NTO.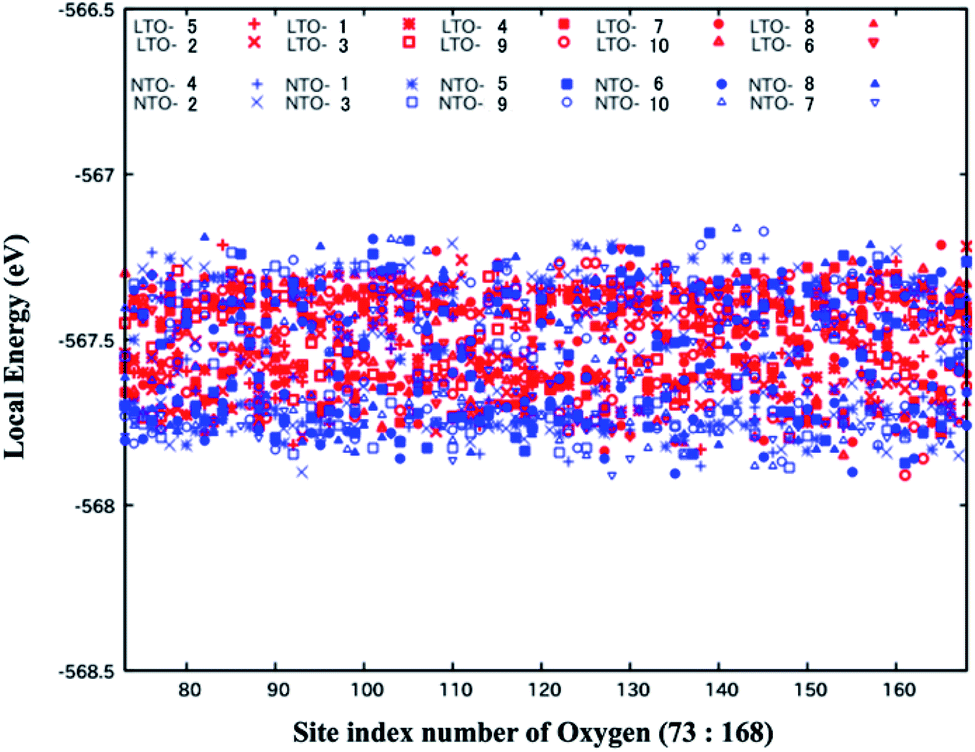 | ||
| Fig. 7 Local energy of oxygen atoms for the optimized top ten stable structures of Li4Ti5O12 and Na3LiTi5O12. LTO-1 and NTO-1 (LTO-10 and NTO-10) indicate the most stable (the 10-th stable) models. The abscissa is the site index of oxygen (73 to 168) of the total 168 sites in the superlattice of Li4Ti5O12 and Na3LiTi5O12 used in the present study. | ||
4 Conclusion
We investigated how Li configurations partially replacing Ti in the 16d sites of alkali titanates (LTO and NTO) influence the structural stability based on DFT calculations, statistical treatment, and local energy analysis. The stability (total energy) of alkali titanates is linearly well-correlated with the sum of the reciprocal distance between Li in the 16d sites with R2 > 0.96. This suggests that the stability of the spinel titanate framework is described by the coulombic repulsion between the mixing Li+; hence, the sum of the inverse of Li–Li distance is a good descriptor for predicting the stability of these materials. The distinction between LTO and NTO shows up in the apparent relative permittivity from the slope of the linear relationship and in the matrix where Li+ repels each other, as 1.92 and 2.04 for LTO and NTO, respectively, in which the greater displacement of O from the 32e sites in the latter is presumably reflected. The local energy analysis suggests, however, that there is no significant difference between LTO and NTO. Thus, it can be concluded that the spinel framework of alkali titanates tolerates a certain degree of destabilization, alluding to a diversity in stable spinel structures. In effect, a material in which Na, instead of Li, is partially substituted for Ti is calculated such that it should retain its structure. The present study proves that investigating the stability of the spinel framework is crucial for replacing the rare element Li with an abundant element.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Computations described in this work were carried out using the computer facilities at the Research Institute for Information Technology, Kyushu University. This research was funded by JSPS KAKENHI (grant number 20K05696). The authors would like to thank Editage (http://www.editage.jp) for English language editing.Notes and references
- S. Abada, G. Marlair, A. Lecocq, M. Petit, V. Sauvant-Moynot and F. Huet, J. Power Sources, 2016, 306, 178–192 CrossRef CAS.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS.
- T. Kim, W. Song, D.-Y. Son, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2019, 7, 2942–2964 RSC.
- N. Takami, H. Inagaki, T. Kishi, Y. Harada, Y. Fujita and K. Hoshina, J. Electrochem. Soc., 2009, 156, A128–A132 CrossRef CAS.
- S. Scharner, W. Weppner and P. Schmid-Beurmann, J. Electrochem. Soc., 1999, 146, 857–861 CrossRef CAS.
- K. Ariyoshi, R. Yamato and T. Ohzuku, Electrochim. Acta, 2005, 51, 1125–1129 CrossRef CAS.
- T. Ohzuku, A. Ueda and N. Yamamoto, J. Electrochem. Soc., 1995, 142, 1431 CrossRef CAS.
- K. Zaghib, M. Simoneau, M. Armand and M. Gauthier, J. Power Sources, 1999, 81–82, 300–305 CrossRef CAS.
- A. Guerfi, S. Sévigny, M. Lagacé, P. Hovington, K. Kinoshita and K. Zaghib, J. Power Sources, 2003, 119–121, 88–94 CrossRef CAS.
- E. Ferg, R. J. Gummow, A. de Kock and M. M. Thackeray, J. Electrochem. Soc., 1994, 141, L147–L150 CrossRef CAS.
- N. Takami, H. Inagaki, Y. Tatebayashi, H. Saruwatari, K. Honda and S. Egusa, J. Power Sources, 2013, 244, 469–475 CrossRef CAS.
- K. Colbow, J. Dahn and R. Haering, J. Power Sources, 1989, 26, 397–402 CrossRef CAS.
- M. M. Thackeray, J. Electrochem. Soc., 1995, 142, 2558 CrossRef CAS.
- W. Schmidt and M. Wilkening, J. Phys. Chem. C, 2016, 120, 11372–11381 CrossRef CAS.
- M. Kitta and M. Kohyama, Electrochemistry, 2018, 86, 194–197 CrossRef CAS.
- M. Kitta, R. Kataoka, S. Tanaka, N. Takeichi and M. Kohyama, ACS Appl. Energy Mater., 2019, 2, 4345–4353 CrossRef CAS.
- K. Tada, M. Kitta, H. Ozaki and S. Tanaka, Chem. Phys. Lett., 2019, 731, 136598 CrossRef CAS.
- M. Kitta, T. Kojima, R. Kataoka, K. Yazawa and K. Tada, ACS Appl. Mater. Interfaces, 2020, 12, 9322–9331 CrossRef CAS.
- M. S. Islam and C. A. J. Fisher, Chem. Soc. Rev., 2014, 43, 185–204 RSC.
- A. Urban, D.-H. Seo and G. Ceder, npj Comput. Mater., 2016, 2, 16002 CrossRef CAS.
- S. Tanaka, M. Kitta, T. Tamura, Y. Maeda, T. Akita and M. Kohyama, J. Mater. Sci., 2014, 49, 4032–4037 CrossRef CAS.
- T. D. H. Nguyen, H. D. Pham, S.-Y. Lin and M.-F. Lin, RSC Adv., 2020, 10, 14071–14079 RSC.
- M. Li, T. Gould, Z. Su, S. Li, F. Pan and S. Zhang, Appl. Phys. Lett., 2019, 115, 073902 CrossRef.
- Y. Tanaka, M. Ikeda, M. Sumita, T. Ohno and K. Takada, Phys. Chem. Chem. Phys., 2016, 18, 23383–23388 RSC.
- H. Ozaki, K. Tada and T. Kiyobayashi, Phys. Chem. Chem. Phys., 2019, 21, 15551–15559 RSC.
- F. Ning, H. Wang, B. Xu and C. Ouyang, Solid State Ionics, 2017, 312, 17–20 CrossRef CAS.
- K. S. Exner, ChemElectroChem, 2017, 4, 3231–3237 CrossRef CAS.
- Y. Liu, J. Lian, Z. Sun, M. Zhao, Y. Shi and H. Song, Chem. Phys. Lett., 2017, 677, 114–119 CrossRef CAS.
- L. Wang, Y. Zhang, H. Guo, J. Li, E. A. Stach, X. Tong, E. S. Takeuchi, K. J. Takeuchi, P. Liu, A. C. Marschilok and S. S. Wong, Chem. Mater., 2018, 30, 671–684 CrossRef CAS.
- P. chun Tsai, W.-D. Hsu and S. kang Lin, J. Electrochem. Soc., 2014, 161, A439–A444 CrossRef.
- S. Zahn, J. Janek and D. Mollenhauer, J. Electrochem. Soc., 2016, 164, A221–A225 CrossRef.
- Y. Okada, Y. Ando, R. Shimizu, E. Minamitani, S. Shiraki, S. Watanabe and T. Hitosugi, Nat. Commun., 2017, 8, 15975 CrossRef CAS.
- D. C. Johnston, J. Low Temp. Phys., 1976, 25, 145–175 CrossRef CAS.
- R. W. McCallum, D. C. Johnston, C. A. Luengo and M. B. Maple, J. Low Temp. Phys., 1976, 25, 177–193 CrossRef CAS.
- M. Yonemura, A. Yamada, H. Kobayashi, M. Tabuchi, T. Kamiyama, Y. Kawamoto and R. Kanno, J. Mater. Chem., 2004, 14, 1948–1958 RSC.
- A. S. Wills, N. P. Raju and J. E. Greedan, Chem. Mater., 1999, 11, 1510–1518 CrossRef CAS.
- Y. Shimakawa, T. Numata and J. Tabuchi, J. Solid State Chem., 1997, 131, 138–143 CrossRef CAS.
- A. Yamada and M. Tanaka, Mater. Res. Bull., 1995, 30, 715–721 CrossRef CAS.
- M. Kitta, T. Akita and M. Kohyama, J. Power Sources, 2013, 232, 7–11 CrossRef CAS.
- T. Ohzuku, J. Electrochem. Soc., 1990, 137, 769 CrossRef CAS.
- M. M. Thackeray, P. J. Johnson, L. A. de Picciotto, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1984, 19, 179–187 CrossRef CAS.
- M. Kitta, T. Akita and M. Kohyama, AIP Adv., 2016, 6, 115216 CrossRef.
- N. Yabuuchi, M. Yano, S. Kuze and S. Komaba, Electrochim. Acta, 2012, 82, 296–301 CrossRef CAS.
- K. Tada, T. Kawakami, S. Tanaka, M. Okumura and K. Yamaguchi, Adv. Theory Simul., 2020, 3, 2000050 CrossRef CAS.
- J. H. Holland, Sci. Am., 1992, 267, 66–73 CrossRef.
- D. Whitley, Stat. Comput., 1994, 4, 65–85 CrossRef.
- K. Tada, Y. Maeda, H. Ozaki, S. Tanaka and S. Yamazaki, Phys. Chem. Chem. Phys., 2018, 20, 20235–20246 RSC.
- S. Ishibashi, T. Tamura, S. Tanaka, M. Kohyama and K. Terakura, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 153310 CrossRef.
- Y. Shiihara, M. Kohyama and S. Ishibashi, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 075441 CrossRef.
- S. K. Bhattacharya, S. Tanaka, Y. Shiihara and M. Kohyama, J. Phys.: Condens. Matter, 2013, 25, 135004 CrossRef.
- S. K. Bhattacharya, S. Tanaka, Y. Shiihara and M. Kohyama, J. Mater. Sci., 2014, 49, 3980–3995 CrossRef CAS.
- Y. Ohno, K. Inoue, K. Fujiwara, K. Kutsukake, M. Deura, I. Yonenaga, N. Ebisawa, Y. Shimizu, K. Inoue, Y. Nagai, H. Yoshida, S. Takeda, S. Tanaka and M. Kohyama, Appl. Phys. Lett., 2017, 110, 062105 CrossRef.
- S. K. Bhattacharya, M. Kohyama, S. Tanaka, Y. Shiihara, A. Saengdeejing, Y. Chen and T. Mohri, Mater. Res. Express, 2017, 4, 116518 CrossRef.
- T. Tamura, M. Karasuyama, R. Kobayashi, R. Arakawa, Y. Shiihara and I. Takeuchi, Modell. Simul. Mater. Sci. Eng., 2017, 25, 075003 CrossRef.
- Z. Xu, S. Tanaka and M. Kohyama, J. Phys.: Condens. Matter, 2019, 31, 115001 CrossRef.
- N. Chetty and R. M. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 6074–6088 CrossRef.
- M. Yu, D. R. Trinkle and R. M. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 115113 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- S. Roberts, Phys. Rev., 1947, 71, 890–895 CrossRef CAS.
- S. Roberts, Phys. Rev., 1949, 76, 1215–1220 CrossRef CAS.
- A. Devonshire, London, Edinburgh Dublin Philos. Mag. J. Sci., 1949, 40, 1040–1063 CrossRef CAS.
- G. H. Haertling, J. Am. Ceram. Soc., 1999, 82, 797–818 CrossRef CAS.
- J. Rödel, W. Jo, K. T. P. Seifert, E.-M. Anton, T. Granzow and D. Damjanovic, J. Am. Ceram. Soc., 2009, 92, 1153–1177 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06125e |
| This journal is © The Royal Society of Chemistry 2020 |