
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen-free hydrogenation of nitrobenzene via direct coupling with cyclohexanol dehydrogenation over ordered mesoporous MgO/SBA-15 supported Cu nanoparticles†
Ravi Kumar Marella *abc,
Venkata Rao Madduluric,
Sivarama Krishna Lakkaboyana*d,
Marlia M. Hanafiahef and
Sarala Yaarathag
*abc,
Venkata Rao Madduluric,
Sivarama Krishna Lakkaboyana*d,
Marlia M. Hanafiahef and
Sarala Yaarathag
aDepartment of Chemistry (H & S), PACE Institute of Technology & Sciences, Ongole 523001, Andhra Pradesh, India. E-mail: ravikumarmarella@gmail.com
bDepartment of Chemical Engineering, University of Cape Town, Rondebosch 7701, Western Cape, South Africa
cCatalysis and Fine Chemicals Division, Indian Institute of Chemical Technology, Hyderabad 500007, Telangana, India
dSchool of Ocean Engineering, Universiti Malaysia Terengganu, 21030, Kuala Nerus, Terengganu Darul Iman, Malaysia. E-mail: svurams@umt.edu.my
eDepartment of Earth Sciences and Environment, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, 43600 UKM Bangi, Selangor, Malaysia
fCentre for Tropical Climate Change System, Institute of Climate Change, Universiti Kebangsaan Malaysia, 43600 UKM Bangi, Selangor, Malaysia
gDepartment of Chemistry, Chaitanya Bharathi Institute of Technology, Gandipet, Hyderabad 500 075, India
First published on 22nd October 2020
Abstract
Direct catalytic coupling of nitrobenzene hydrogenation and cyclohexanol dehydrogenation was studied in the gas phase over mesoporous MgO-SBA15 supported Cu nanoparticles. This approach avoids an external supply of H2 and utilizes the in situ liberated H2 from the dehydrogenation step of the first reactant for the hydrogenation reaction of the second reactant. A catalyst series consisting of four Cu/MgO-SBA15 mesoporous solids with varying Cu loadings (5–20 wt%) were prepared and systematically characterized by BET, ICP, XRD, TPR, TPD, FT-IR, SEM, XPS, and TEM. Among the series, the 15 wt% Cu catalyst exhibited the best performance with ≥82% conversion of nitrobenzene along with ≥89% cyclohexanol conversion. In addition, significantly higher yields of cyclohexanone (83%) and aniline (75%) could be achieved successfully over the same catalyst. Furthermore, the catalyst exhibited almost stable activity during 30 h time-on-stream with slow deactivation. The highly ordered mesoporous silica increases the metal–support interaction with smaller particles of Cu on the surface, and the synergism between acid–base sites is responsible for the improved catalytic activity.
Introduction
Aniline (AL) is used in rubber processing chemicals, herbicides, dyes, pigments, drugs and polyurethane plastics.1 Industrially, aniline is produced by the hydrogenation of nitrobenzene (NB) in the presence of metal catalysts. Most of the reports claimed that the selectivity for AL is still challenging due to the formation of secondary products.2 In recent years, aqueous phase hydrogenation using noble metal catalysts like Pd and Pt has attracted more attention.3,4 However, the noble metals are high cost-effective, less in abundant and industrially not viable for pilot-scale production of desired products.5 In this scenario, few reports are claimed for the hydrogenation of NB and its derivatives have been carried out using non-noble Ni and Co catalysts.6,7 Nevertheless, there are some disadvantages such as the usage of toxic organic chemicals, harsh reaction conditions, high reaction pressure, and tedious catalyst preparation method.Hydrogen represents the current inheritor of energy providers which is typically a clean, high potential green fuel and produce only H2O as a safe by-product.8 The global increasing demand for hydrogen energy makes researchers to develop remarkably efficient and innovative catalytic process to produce chemical feed stocks.9 Catalytic transfer hydrogenation (CTH) of nitro compounds using hydrazine, NaBH4, and HCOOH as H2 donors offers an alternative approach for the hydrogenation NB using molecular H2.10 However, the application is hampered due to the separation steps required for the catalyst reuse.
Alcohols also can be widely utilized as potential hydrogen donors. For specific instance, methanol acts as an outstanding H2 donor for the transfer hydrogenation of NB carried out over Pd-based catalysts.11 Similarly, isopropanol functions as a hydrogen donator for the transfer hydrogenation of various unsaturated compounds with a non-noble Co@C–N catalyst system.12 Likewise, glycerol employed both as a potential H2 donor and a green solvent to efficiently produce AL by transfer hydrogenation of NB.13 Nevertheless, CTH is limited to liquid phase batch catalytic process inevitably involves an excessive amount of alcohol reactants and base additives, therefore not suitable for industrial application. To overcome the above limitations, direct catalytic coupling process is appealing ways which alleviates the usage of flammable hydrogen, high pressure operation and enhance the simplicity of the synthetic procedure.14 In addition, direct dehydrogenation–hydrogenation coupling reactions can be carried out simultaneously in a single reactor on a single catalyst and under similar reaction conditions.15
In recent years, various hydrogenation reactions using alcohol dehydrogenation have been developed such as maleic anhydride (MA) and 1,4-butanediol (BDO),16 MA and n-butanol,17 NB and 2-propanol,18 MA and ethanol,19 furfural and BDO,20 furfural and cyclohexanol,21 NB and BDO,22 acetophenone and BDO,23 NB and cyclohexanol,24 benzaldehyde and BDO.25 Despite the advantages that direct coupling reactions can provide at the industrial scale, separation of organic products with close boiling points is tedious by normal separation methods. Therefore, alternative procedure like extractive distillation,26 and pervaporation27 are receiving potential industrial applications for the post-separation of reaction products.
The selective dehydrogenation of cyclohexanol to cyclohexanone (SDCC) is industrially significant due to the vital applications of cyclohexanone mainly in textile industries. To date, gas-phase dehydrogenation of cyclohexanol extensively studied on various copper catalysts including Cu/Al2O3,28 Cu/MgO,29 Cu/SiO2,30 and Cu/ZrO2.31 However, improvement of the catalyst with high activity and stability for the SDCC is quite a challenging task.
In this perspective few reports on direct coupling of cyclohexanol dehydrogenation and NB hydrogenation reactions overwhelmed the thermodynamic constraints and typically improve the product yield. In the open literature, Cu/MgO–Al2O3,8,32 Cu/SiO2,15,33 Cu/MgO34,35 catalysts have been tested for the hydrogenation of NB via coupling process. Highly dispersed metal nanoparticles such as Fe, V, Co supported on activated carbon acts as a promising catalyst for the coupling of ethylbenzene and NB.36 Similarly, hydrogenation of ortho chloro nitrobenzene is coupled with the dehydrogenation of BDO over Cu/MgO catalysts.37 Nevertheless, most reported catalysts have low surface areas, exhibited low catalytic activity and it can attribute to the deactivation of supported Cu catalysts.38
From the past two decades, mesoporous silica SBA-15 has received more attention in catalysis due to hexagonal ordered mesoporous structure.39 In addition, it has a uniform pore size and easy access for large organic reactants through mesoporous channels. The surface Si–OH groups and amorphous SBA-15 pore walls allow the unalterable surface modification of the framework. Therefore, the present study extensively focused on Cu nanoparticles supported MgO/SBA-15 catalytic systems for the coupling of cyclohexanol dehydrogenation and NB hydrogenation reaction at atmospheric conditions. Furthermore, we correlate the catalyst performance with the surface structural properties for the both independent and coupled reactions.
Experimental
Preparation of Cu/MgO-SBA15 catalysts
A series of Cu catalysts supported on MgO-SBA15 was prepared by incipient wetness impregnation method using aqueous solution of Cu(NO3)2·3H2O as the metal precursor. The resulting powders then dried in vacuum and calcined at 723 K for 4 h with a heat ramp of 2 K min−1 in static air. In a representative catalyst xCu/MgO-SBA15, the prefixed number (x = 5 to 20) represents the percentage of Cu loading (by weight) on the MgO-SBA15 support.Catalyst characterization
Examination of the crystalline/amorphous phases as well as the typical crystallite size of calcined and reduced catalysts was determined by powder X-ray diffraction. The measurements were performed on a Bruker D8 Advance XRD instrument equipped with Ni filtered Cu Kα radiation (λ = 1.54056 A) operated at 40 kV and 25 mA. All diffraction patterns were recorded in the step-scan mode in the range of 2θ values from 0.7 to 80° with a step interval of 0.01° and a scanning speed of 1° min−1. The diffractograms were investigated using X'PertHighscore Plus software and the diffraction peaks of crystalline phases were compared with those of standard compounds reported in the JCPDS data file. The size of copper/copper oxide crystallites was measured from the peak width at half maxima adopting the Scherrer formula.BET surface area measurements (SBET) were performed on a Micrometrics ASAP 2020 instrument by N2 gas adsorption at 77 K. All the catalyst samples were outgassed at 423 K for 4 h prior to the N2 sorption studies. The pore-size distribution data can be obtained from the desorption branch of the isotherms using the BJH method. The total pore volume (Vtotal) of samples was calculated by considering the relative pressure (P/P0) at 0.99.
TPR and TPD studies were carried out in a U-shaped quartz reactor on a Micrometrics AutoChem2950 chemisorption analyzer. The reducibility of copper oxide species present in the calcined catalysts was studied using the H2-TPR. About 100 mg of the catalyst placed in the reactor and pre-treated at 493 K for 1 h in Ar flow (60 ml min−1). The catalyst was cooled down to 373 K under Ar and exposed to 5% H2 balance Ar flow followed by slow ramping up to 900 K at a heating rate of 10 K min−1. The exhaust gas stream was analyzed by a TCD detector to record the H2 consumption during the reduction process. The H2 consumption was calibrated with known amounts of Ag2O.
The distribution of basic sites on the catalyst surface was measured by the CO2-TPD technique using a 10% CO2/He gas mixture. Typically, about 100 mg of the sample was pre-treated in flowing helium gas at 773 K for 1 h and allowed to cool to 373 K. At this temperature, the sample was exposed to 10% CO2/He gas mixture for 30 min with a flow rate of 20 ml min−1. Subsequently, the catalyst was purged with He (99.999% pure) gas at 373 K for 1 h in order to remove the physisorbed CO2. The temperature of the sample increased from 373 to 1173 K by flowing helium gas with a flow rate of 20 ml min−1. The desorption of CO2 was done at a heat ramp of 5 K min−1 and it is monitored by online GC equipped with TCD. Similarly, the distribution of acidic sites on the catalyst surface was measured by the NH3-TPD technique using a 10% NH3/He gas mixture. The copper content present in all the catalysts was estimated using iCAP 6500 duo ICP-OES Analyzer (M/s. Thermo Fisher Scientific, USA). For the ICP sample preparation, catalyst was dissolved in a standard solution of aquaregia and diluted hydrofluoric acid. FT-IR patterns of the samples were documented on a Spectrum GX Spectrometer (M/s. PerkinElmer, Germany) within the range of 4000 to 400 cm−1. TG-DTA analysis of the oven-dried samples was performed in a TGA/SDTA 851e (Mettler Toledo AG) Thermogravimetric Analyzer. Thermograms were recorded while heating the samples under air from 300 K to 900 K with a heating rate of 10 K min−1.
N2O pulse chemisorption studies were carried out on a home-made micro reactor connected to an automatic gas sampling valve and a GC equipped with a TCD. Typically, about 100 mg of the catalyst sample was reduced at 523 K for 2 h under H2 (99.999% pure) followed by cooling to 363 K. Subsequently, 6% N2O/He mixture gas was injected in pulses at regular intervals through a 6-port valve until the saturation in the concentration of N2O at the outlet. The number of surface Cu atoms and its dispersion calculated by assuming the following stoichiometric equation.40
| N2O(g) + 2Cu(s) → N2(g) + Cu2O(s) |
The X-ray photoelectron spectroscopy measurements were recorded on a Kratos Axis 165 XPS Spectrometer operated with Mg Kα radiation (1253.6 eV) over the catalysts reduced under H2 flow at 523 K for 3 h. The SEM measurements were performed on a JEOL 840A microscope operated at 10 kV accelerating voltage and 10 mA emission current. Furthermore, the elemental composition is determined by using energy dispersive X-ray (EDX) analysis. The size distribution of Cu nanoparticles and morphology of the catalyst were examined by transmission electron microscopy. The TEM images of catalyst samples were recorded on an electron microscope FEI Tecnai G2 F20 operated at a voltage of 200 kV. The specimens were uniformly dispersed in ethanol and placed on carbon-coated holey Cu grids.
Results and discussion
Thermo gravimetric analysis
In the thermogravimetric (TG) curve of oven-dried MgO/SBA-15 under airflow, the weight losses were concentrated in three ranges (303–483 K, 484–643 K, and 644–723 K), as shown in Fig. 1. | ||
| Fig. 1 Thermogravimetric (TG) analysis of oven-dried MgO/SBA-15 sample. | ||
The broad differential TG (DTG) peak around 333 K was assigned to the loss of physical adsorbed H2O, and water produced by the condensation of Si–OH during heat treatment. The extra-large peak at 593 K might be fit to the thermal decomposition of Mg(CH3COO)2 into Mg(OH)2.41 Furthermore, a small peak at 699 K might be attributed to the pyrogenic decomposition of Mg(OH)2 into MgO. Interestingly, the DTG curve of MgO/SBA-15 containing several peak maxima below 723 K, indicating that the synthesis procedure requires a lower temperature for the complete decomposition of magnesium acetate into MgO.
The TG investigations of Cu/MgO-SBA15 samples have been studied and the results were presented in the (Fig. ESI 2†). Interestingly, it has been observed that TG curves of the series of Cu catalysts comprised two peaks: one large peak below 423 K and another small one in the temperature range of 523–723 K. The large peak might be due to the elimination of interstitial water during the heat treatment, whereas the small peak might be attributed to the thermal decomposition of copper nitrate. The TG curves of four Cu catalysts show similar thermal decomposition patterns during the heat treatment under airflow. It is noteworthy all the samples are completely decomposed below 723 K which is the optimum calcination temperature.
Powder X-ray diffraction
To check whether the functionalization makes any structural defects in the mesoporous silica, the Cu nanoparticles supported on MgO functionalized SBA-15 solids were characterized by low-angle XRD measurements.Fig. 2 displays the small-angle XRD of SBA-15 at the 2θ values of 0.92°, 1.59° and 1.83° corresponding to the lattice planes of (100), (110) and (200) respectively, with an ordered 2D hexagonal structure of P6mm symmetry. Considerably, it can be observed that the XRD patterns of as-synthesized MgO-SBA15 and Cu/MgO-SBA15 samples are almost similar. Hence it indicates the retention of the mesoporous structure after functionalization with MgO and subsequent loading of Cu metal ions. However, there is a small shift in the 2θ values along with a significant decrease in the intensity of diffraction patterns with increase in copper loading. This tendency might be due to the relative degree of structural order present in the samples after partial filling of metal into the pore channels of mesoporous SBA-15.42 From the above results, it is confirmed that during calcination process CuO is deposited inside the pore channels of SBA-15. The wide-angle XRD patterns of calcined samples of Cu/MgO-SBA15 were presented in the (Fig. ESI 3†). The broad peak at around 2θ value of 25° typically corresponds to amorphous silica.43 The direct XRD reflections of all copper-containing catalysts show peaks at 2θ values of 35.6, 38.8, 48.8, 58.1 and 66.3°, promptly confirming the active presence of CuO (JCPDS no. 05-661). 5Cu/SBA-15 catalyst exhibits very low intense diffraction peaks due to the more dispersion of copper. The formation of bigger copper oxide crystallites is illustrated by the nominal increase in crystallite sizes of CuO with copper loading. The above investigation typically implies that CuO is present in the extra-framework during calcination and thermal activation process.
 | ||
| Fig. 2 Low angle X-ray diffraction patterns of ordered mesoporous SBA-15, MgO-SBA15 and Cu/MgO-SBA15 catalysts. | ||
All the reduced catalyst exhibits (Fig. 3), three well-resolved XRD reflections are observed at 2θ values of 43.3, 50.6 and 74.2°, unanimously confirming the presence of metallic Cu in accordance with the JCPDS no. 04-836. No XRD reflections precisely correspond to the MgO crystalline phase is observed, typically indicating the high dispersion of MgO on SBA-15 support.44 No other phases of copper are accurately detected; therefore the oxidation state of Cu is zero in all the reduced catalysts. The peak intensity of metallic Cu progressively increased with an increase in Cu loading. The crystallite size of copper particles was calculated by using the established Scherrer equation presented in the Table 1.
 | ||
| Fig. 3 Wide angle X-ray diffraction patterns of reduced samples of MgO-SBA15 and Cu/MgO-SBA15 catalysts. | ||
| S. no. | Catalyst | SBETa (m2 g−1) | Vtotalb (cc g−1) | Vmicroc (cc g−1) | Vmesod (cc g−1) | DBJHe (nm) | d(100)f (nm) | Dporeg (nm) | a0h (nm) | twalli (nm) | PCuj (nm) | H2 uptakek (μmol gcat−1) | Total basicityl (μmol gcat−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Specific surface area by BET method.b Total pore volume.c Micropore volume.d Mesopore volume.e Mesopore diameter (calculated by BJH method from desorption branch).f d spacing derived from low-angle XRD.g Average pore size.h Unit cell constant (corresponding to the hexagonal pore arrangement, a0 = 2d100/√3).i Mesopore wall thickness (twall = a0 − Dpore).j Average crystallite size of Cu (calculated from the Scherrer equation).k Determined from the H2-TPR analysis.l Measured from the CO2-TPD. | |||||||||||||
| 1 | SBA15 | 729 | 1.081 | 0.644 | 0.879 | 9.77 | 9.91 | 5.93 | 11.44 | 5.5 | — | — | — |
| 2 | MgO-SBA15 | 280 | 0.511 | 0.297 | 0.492 | 8.14 | 9.19 | 7.31 | 10.61 | 3.3 | — | — | 494 |
| 3 | 5Cu/MgO-SBA15 | 205 | 0.379 | 0.221 | 0.364 | 7.99 | 8.85 | 7.37 | 10.22 | 2.8 | — | 30.8 | 383 |
| 4 | 10Cu/MgO-SBA15 | 156 | 0.299 | 0.169 | 0.284 | 9.39 | 8.99 | 7.67 | 10.38 | 2.7 | 21.4 | 82.1 | 299 |
| 5 | 15Cu/MgO-SBA15 | 114 | 0.229 | 0.229 | 0.216 | 8.12 | 8.81 | 8.06 | 10.17 | 2.1 | 37.1 | 139.2 | 129 |
| 6 | 20Cu/MgO-SBA15 | 59 | 0.093 | 0.059 | 0.079 | 7.78 | 8.77 | 6.03 | 10.13 | 4.1 | 52.2 | 213.4 | 173 |
H2-TPR
The H2 reduction profiles of freshly calcined Cu/MgO-SBA15 catalysts with various copper loadings are typically displayed in Fig. 4. It has been extensively investigated that the precise nature of copper species could be undoubtedly influenced by the support, preparation method, metal precursor, activation treatment etc. For the 5% and 10% Cu/MgO-SBA15 catalysts, a broad reduction peak at 690 K (T1) significantly reveals the direct evidence of highly dispersed CuO particles. It was carefully observed that the massive shift in the reduction temperature (T1) to lower values with multiplying in Cu loading. This may be typically assessed to the unusual achievement of the bulk nature of confined CuO species.45 A second reduction peak that inevitably appears at a more elevated temperature (T2) in all the active Cu catalysts. It may merely represent the minor reduction of undesirable Cu2O species that aggregated outside the clogged pores. The abnormal results supposedly reveal that the unusual interaction between CuO and MgO-SBA15 support increases with Cu loading. Additionally, the less interacted CuO species are getting aggregated both in clogged pores and on the external surface of SBA-15.46 | ||
| Fig. 4 H2-TPR profiles of different loadings of Cu/MgO-SBA-15 catalysts. | ||
The obtained H2-TPR results are collated with the consistent XRD patterns where the crystallite size of copper increased progressively with loading. In addition, hydrogen consumption also increases gradually as shown in Table 1.
BET surface area and pore size distribution
The mesoporous nature of Cu/MgO-SBA15 samples was properly investigated using N2 sorption experiments. The N2 adsorption–desorption isotherms are shown in the Fig. 5, all the samples display type-IV isotherm with H1 hysteresis. The N2 sorption experiment results are consistent with the low-angle X-ray diffraction patterns, clearly indicating the preservation of mesoporous structure in all the samples.46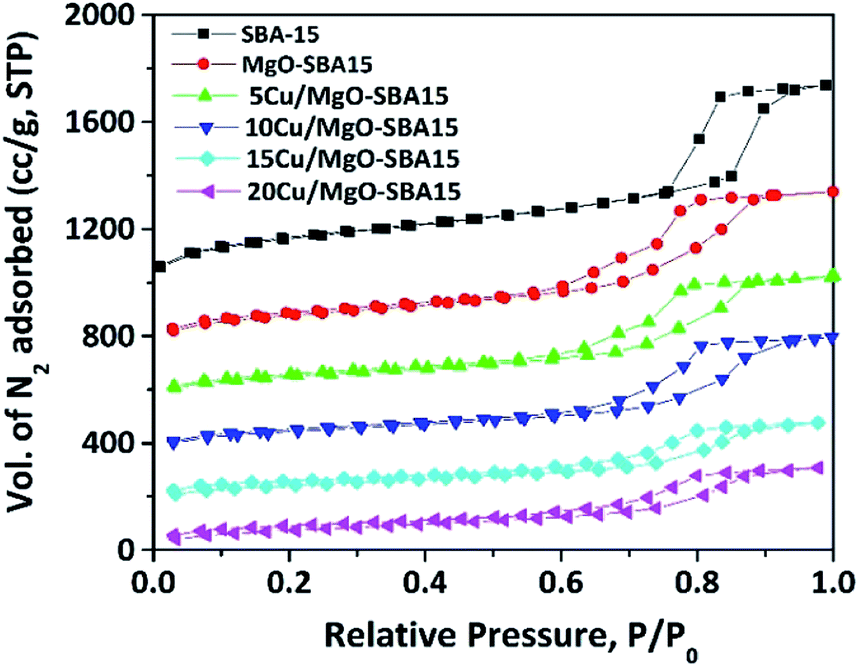 | ||
| Fig. 5 N2 adsorption–desorption isotherms of mesoporous SBA-15, MgO-SBA15 and Cu/MgO-SBA15 catalysts. | ||
The BJH pore size distributions (PSD) were acquired from the N2 exhibit a narrow pore size distribution in the range of 5–10 nm, which further confirms the existence of regularly ordered mesopores. As shown in the Fig. 6, the PSD curve of MgO-SBA15 comprises a bimodal mesoporous structure. This unusual PSD curve is rather different from that of MgO modified mesoporous silica materials prepared by one-pot synthesis.44
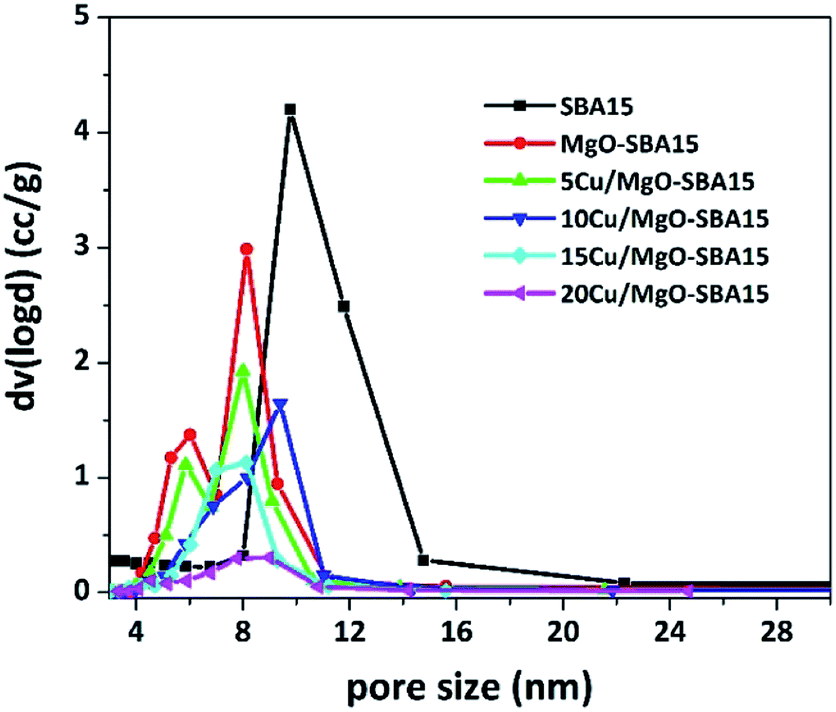 | ||
| Fig. 6 BJH pore size distribution patterns of mesoporous silica SBA-15, MgO-SBA15 and Cu/MgO-SBA15 catalysts. | ||
The addition of metal oxide, MOx (i.e., MgO, CuO) to SBA-15 silica forms a Si–O–M monolayer and the remaining metallic species convert into a uniform layer of MOx during thermal treatment in the catalyst preparation. This phenomenon results in the decrease of a surface area, mesopore size and volume of parent SBA-15.44 The relative decrease in BET surface area (SBET) of MgO-SBA15 supported copper catalysts in regard to the parent SBA-15, MgO-SBA15 support material is summarized in Table 1, together with pore diameter (DBJH) and pore volume (Vt), unit cell constant (a0) and wall thickness (tw) between the mesopores.
Temperature programmed desorption of CO2 and NH3
In order to further explore the surface acid–base nature, CO2-TPD and NH3-TPD of Cu/MgO-SBA15 catalysts were studied. As shown in Fig. 7, three CO2-TPD peaks were observed in the temperature ranges of 400–600, 600–750 and 750–900 K over MgO-SBA15. The CO2-TPD profile of the 5Cu/MgO-SBA15 catalyst was similar to that of MgO/SBA15, but the peak intensity at the 813 K (strong basic sites) was low. With a nominal increase in the copper loading, a further decrease in the peak intensity at 813 K was observed. Therefore, the strong basic sites are reduced by the acidic sites generated from the copper oxide. Zhang et al.47 have studied various alkali earth metal-oxide promoted on Cu/SiO2. Among them MgO doped Cu/SiO2 catalytic surface exhibit more basicity in contrary to the actual order of basicity (MgO < CaO < SrO < BaO < La2O3). The enhanced basicity of MgO promoted catalysts is due to the presence of Cu–O–Mg or Mg–O–Si bonds in addition to their corresponding oxide forms. | ||
| Fig. 7 CO2 TPD profiles of mesoporous silica SBA-15, MgO-SBA15 samples and Cu/MgO-SBA15 catalysts. | ||
To identify the distribution of the acid sites on the surface, NH3-TPD analysis was performed, and the direct results are as shown in Fig. 8. The incorporation of MgO generated weak and strong acid over the MgO-SBA15 support at around 513 K and 813 K respectively. MgO scarcely has Lewis acidic sites due to the anion vacancies created during the transformation of brucite to the MgO phase.48 The addition of copper to the MgO-SBA15 inevitably generated unknown acidic sites with medium strength (600 to 750 K).
 | ||
| Fig. 8 NH3 TPD profiles of mesoporous silica SBA-15, MgO-SBA15 samples and Cu/MgO-SBA15 catalysts. | ||
The intensity of the peak at 873 K was further increased with the addition of copper oxide. Meanwhile, copper oxides are crucial to the generation of solid acid sites, the Mg ions may interact with copper to deactivate the strong acid sites.
FT-IR analysis
The FT-IR spectra of SBA-15, MgO-SBA15, and 10Cu/MgO-SBA15 samples examined in the range of 400 to 4000 cm−1 and illustrated in the Fig. 9. For mesoporous SBA-15, the peaks in 1055 and 805 cm−1 were attributed to the asymmetric stretching and symmetric modes of Si–O–Si lattice vibrations, respectively. The peaks in 1640 and 970 cm−1 represent the characteristic peaks of Si–OH. After the assembly of MgO, the peak at 970 cm−1 became weak until it disappeared. But the characteristic peak of Si–OH at 1640 cm−1 became stronger with increasing MgO. However, after the incorporation of CuO into the MgO-SBA15 mesopores, this characteristic peak began to decrease. It can be ascribed to the release of the proton resulting in decreased amounts of the Si–OH groups. | ||
| Fig. 9 FTIR spectra of SBA-15, MgO-SBA15, 10Cu/MgO-SBA15 samples. | ||
In general, water and carbon dioxide molecules present in the air can be easily chemisorbed onto the MgO surface when exposed to the atmosphere. In the FT-IR spectrum of samples shown in Fig. 1b, broadband at 3440 cm−1 can be significantly attributed to the OH stretching vibrations of the surface adsorbed water molecules. In addition, an extremely weak band corresponding to the adsorption of gas-phase CO2 is observable around 2425 cm−1 and a band visible at 1438 cm−1 could be ascribed to the vibration of CO32− bonding.48 The increased intensity of both OH− and CO32− groups indicates precisely the enhanced basic strength of the MgO/SBA-15 sample when compared with parent SBA-15 silica. The bands appeared at a low frequency of 645 cm−1 and a peak complex at around 450 cm−1 corresponds to the stretching vibration of Mg–O–Mg bonding.38 The band in the range of 1055–1100 cm−1 can be assigned to the asymmetric and symmetric stretching vibrations of Si–O–Si bond. It is noteworthy the peak in the samples is somewhat red-shifted as the amount of added MgO. These remarkable results typically implied MgO didn't alter the ordered mesoporous structure of SBA-15 silica during the synthesis.
X-ray photoelectron spectra (XPS) of Cu
As shown in the Fig. 10, XPS of 15Cu/MgO-SBA15 reduced catalyst exhibit two distinct peaks at binding energy (B.E.) values of 932.9 and 953.8 eV respectively. The high-intensity peak corresponding to Cu 2p3/2 at 932.9 eV and the absence of satellite peak demonstrate the presence of either Cu+ or Cu0. As the B.E. values of Cu 2p3/2 for Cu+ and Cu0 are practically identical, it is challenging to distinguish them by XPS.49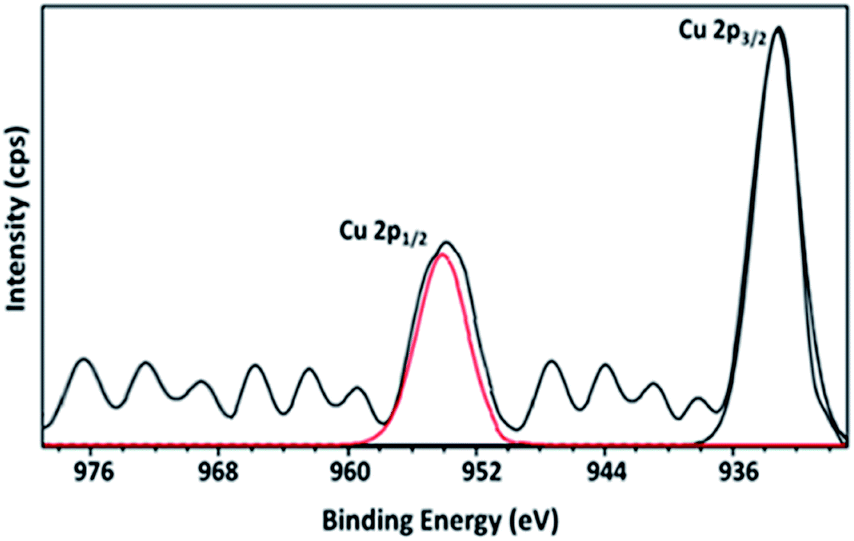 | ||
| Fig. 10 Cu 2p core-level XPS of 15Cu/MgO catalyst in situ reduced under H2. | ||
The partial reduction of Cu2+ ions which are typically interacting with silica SBA-15 can inevitably generate Cu+ species. These copper species still interact with the silica support even after minor reduction and has a high electron affinity towards SiO2. Therefore, the existence of Cu+ ions on the catalyst surface may be responsible for Lewis acid sites as shown in the NH3 TPD results. Further, the presence of MgO not only increases the surface Cu0 species in the reduced catalysts but also enhance the metal–support interaction.
Electron microscopy analysis
The TEM images of Cu/MgO-SBA15 catalysts are shown in Fig. 11, it can be evidenced that the parallel arrangement of tubular pores with long-range order is present among all the samples. The presence of 1D channel with ordered hexagonal mesoporous arrays confirms the existence of P6mm symmetry, which is in accordance with the low angle XRD results (Fig. 2). Furthermore, TEM images revealed the ordered structure of mesoporous silica SBA-15 is also preserved even after the functionalization of metal oxide. As mentioned earlier, the N2 adsorption–desorption isotherms and small-angle XRD patterns, in addition, provide strong evidence to these electron microscopy observations. The effective dispersion of Cu nanoparticles on the surface of MgO functionalized SBA15 support was evident from the TEM images. SEM images shown in the Fig. ESI 4.† | ||
| Fig. 11 TEM images of (a) 5Cu/MgO-SBA15 (b) 10Cu/MgO-SBA15 (C) 15Cu/MgO-SBA15 and (d) 20Cu/MgO-SBA15 reduced catalysts. | ||
In the case of 5Cu/MgO-SBA15 catalyst (Fig. 11a), TEM images display a little copper on the surface due to the lower loading. Hence, the sample does not show any reflections in the XRD patterns (Fig. 3).
Hydrogenation of nitrobenzene (NB)
In order to understand the reaction parameters for the coupled system, the individual reaction of nitrobenzene hydrogenation is studied at various Cu loadings and temperatures.As shown in the Scheme 1, NB hydrogenation is an exothermic reaction in which 1 mole of NB reacts with 3 moles of H2 to produce 1 mole of AN and 2 moles of H2O.
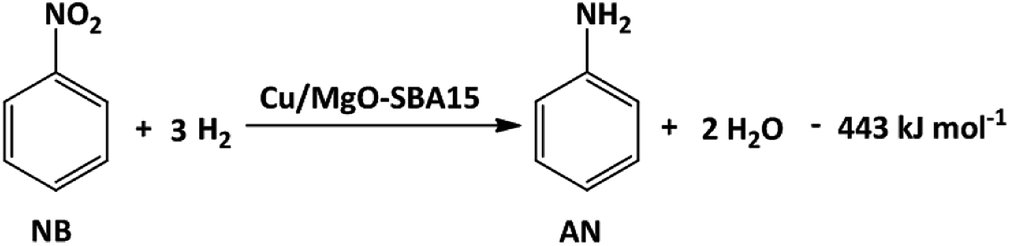 | ||
| Scheme 1 Nitrobenzene hydrogenation over Cu/MgO-SBA15 catalysts. | ||
To illustrate the influence of copper loading all the Cu/MgO-SBA15 catalysts were subjected to NB hydrogenation at atmospheric pressure. The activity results clearly indicate the exclusive production of aniline overall catalysts (5–20 wt%) in the temperature range of 473–573 K. Nevertheless, there is a considerable drop in the specific activity beyond 548 K due to the exothermic nature of the NB hydrogenation reaction. Industrially, the hydrogenation of NB over Ni, Pd, and Pt catalysts carried out in the liquid phase using molecular hydrogen produce AN at high pressure.3,4,6 The aniline selectivity typically depends on the catalyst composition as well as the reaction conditions.
From Fig. 12, at 548 K fractional conversion of NB is increased from 65 to 93% with copper loading from 5 to 15 wt% and then remains almost constant. However, the selectivity towards AL is almost the same among all the samples (ca. 99%). The present activity studies revealed that 548 K represents the optimum temperature to obtain the maximum conversion of NB (93%) and 15Cu/MgO-SBA15 could be a suitable catalyst. It was reported that gas-phase hydrogenation NB over Cu/MgO catalysts suffers rapid deactivation.34 Mohan et al. have studied the hydrogenation of NB over Ni/MgO catalysts and it is carefully observed that water adsorption on MgO support enables the brucite-periclase phase transformation. This kind of transformation results in a uniform layer of MgO or Mg(OH)2 over Ni particles and blocks adsorption of NB molecules on the active sites.50 To overcome this drawback, mesoporous silica is effectively employed as support which not only inhibits the phase change but also helps to improve the metal dispersion. For instance, Ni/SBA-15 activity is three times more than Ni/MgO catalyst. In another work, it was prominently mentioned that mesoporous silica with a 2D pore structure is more active than the 3D framework. Very recently, the Co/KIT-6 catalyst shows more activity than Co/SBA-16.51 The ordered mesoporous silica SBA-16 with the 3D-cubic structure containing inter-connected pores shows rapid deactivation. It can be ascribed to the pore blockage by coke deposition during the reaction pathway. The interconnected pores present in SBA-16 increase the residence time of reactant molecules, which leads to the formation of secondary products by condensation and enhances the coke formation. Nevertheless, COK-2 mesoporous silica with 2D-hexagonal pore structure exhibit stable activity and high selectivity to aniline. In the present study, the hydrophobic nature of SBA-15 enables stable activity by controlling the phase transformation of MgO. In addition, the high surface area SBA-15 can increase active metal dispersion. It can be observed that Cu particle size in 15Cu/MgO-SBA15 catalyst is bigger and it displays more significant activity compared to other Cu loaded catalysts. It may notice that more number of surface-active copper sites (Cu+/Cu0) is present in 15Cu/MgO-SBA15 catalyst evidenced from XPS results.
 | ||
Fig. 12 Effect of temperature on NB conversion over Cu/MgO-SBA15 catalysts (conditions: Vcat = 1 ml, T = 473–573 K, P = 1 atm, GHSV = 1620 h−1, total flow = 1.62 l h−1, NB = 2.5 mmol h−1, NB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 = 1:29). :H2 = 1:29). | ||
Dehydrogenation of cyclohexanol (CHol)
According to the reaction Scheme 2, one mole of cyclohexanone produced from one mole of cyclohexanol by the liberation of 1 mole of H2 as the by-product. | ||
| Scheme 2 Cyclohexanol dehydrogenation over Cu/MgO-SBA15 catalysts. | ||
Dehydrogenation of cyclohexanol (in N2) using various loadings of Cu/MgO-SBA15 catalysts resulted in the selective production of cyclohexanone with no detectable dehydration reaction (step (II) in Fig. 1). The exclusive production of cyclohexanone obtained in this study can be associated with the lower reaction temperature employed. The selective dehydrogenation of cyclohexanol to cyclohexanone is challenging due to competing reactions such as aromatization, dehydration, and condensation. Several reports are available on various copper-based catalysts like Cu/Al2O3,28 Cu/MgO,29 Cu/ZrO2 (ref. 31) and Cu/SBA-15.30 Nevertheless, there is a significant ambiguity that exists about the CHol dehydrogenation mechanism regarding the active phase of Cu. The Cu+ active sites are accountable for the formation of cyclohexanone without any by-products. Whereas, Cu0 active species can produce phenol along with cyclohexanone.52 The CHol dehydrogenation activity also depends on the dispersion of active copper sites, surface acid–base sites and the nature of support material. For instance, Cu/ZrO2 catalyst afforded benzene and cyclohexanone as possible products, which are major, depends on the active phase of supported zirconium oxide material.31
Initially, a blank test was typically conducted without the active catalyst using quartz pieces as an inert medium in the reactor, and no specific products were accurately detected. Therefore, a homogeneous surface/volume effect was ruled out. Later on, an in situ activity test was made operating a micro-reactor connected to a GC equipped TCD as reported elsewhere49 to confirm the formation of hydrogen along with cyclohexanone. To properly investigate the optimum loading of Cu on the MgO-SBA15 support, the dehydrogenation of cyclohexanol was conducted on all the four catalysts in the gas-phase at 523 K in the inert atmosphere using N2 (99.999% pure), and the data are displayed in Table 2.
| S. no. | Catalyst | Cu loadinga (wt%) | SCub (m2 g−1) | DCuc (%) | PCud (nm) | Surface Cu atomse (μmol gcat−1) | Particle size of Cuf (nm) | Reaction rateg (mol gcat−1 s−1) | TOFh (s−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Cu loading determined from ICP-OES.b Cu metal surface area.c Dispersion.d Cu particle size.e No. of surface Cu atoms obtained from N2O chemisorption.f Avg. particle size of Cu obtained from TEM analysis.g The reaction rates were measured by controlling the conversion of cyclohexanol below 20%.h Turnover frequency (TOF) was obtained from N2O chemisorption, based on the number of moles of cyclohexanol converted per one mole of surface copper atoms per second. | |||||||||
| 1 | 5Cu/MgO-SBA15 | 4.1 | 17.22 | 12.6 | 3.9 | 99.2 | 4.2 | 2.01 × 10−6 | 24.71 × 10−3 |
| 2 | 10Cu/MgO-SBA15 | 9.3 | 14.61 | 5.3 | 4.6 | 83.4 | 9.3 | 2.92 × 10−6 | 37.62 × 10−3 |
| 3 | 15Cu/MgO-SBA15 | 13.2 | 10.34 | 2.7 | 6.5 | 63.2 | 14.6 | 3.41 × 10−6 | 60.76 × 10−3 |
| 4 | 20Cu/MgO-SBA15 | 17.8 | 7.22 | 1.8 | 9.3 | 38.7 | 21.5 | 2.87 × 10−6 | 56.88 × 10−3 |
It can be carefully observed that the increase in reaction rate from 2.01 × 10−6 mol g−1 s−1 to 3.41 × 10−6 mol g−1 s−1 with the increase in Cu loading from 5 to 15 wt% at 523 K. However, the reaction rate on the 15Cu/MgO-SBA15 catalyst was considerably higher than that on the 20Cu/MgO-SBA15 catalyst. This is likely due to the considerable increase in the specific number of active sites and a high density of moderate basic sites on the catalyst surface (Fig. 8). The reaction rate also strongly depended on the optimal size of the active Cu particles involved in CHol dehydrogenation. When increasing the size from 3.9 to 6.5 nm, the turnover frequency (TOF) increased dramatically from 24.71 × 10−3 s−1 to 60.76 × 10−3 s−1 at 523 K, but further increasing the size of the copper particle to 9.3 nm only moderately lowered the TOF to 56.88 × 10−3 s−1 (entry 4, Table 2).
After recognizing the best catalyst (i.e., 15Cu/MgO-SBA15) of the Cu/MgO-SBA15 catalyst series, the influence of reaction temperature on the catalytic activity of this catalyst is studied and the results are displayed in Fig. 13. The conversion at 523 K is about 78%, which increased gradually with the rising in reaction temperature and attained a maximum of 98% at 598 K. Selectivity of cyclohexanone (S-CHone) maintained at ca. 96% up to 548 K beyond which the S-CHone decreased significantly. For instance, the selectivity of cyclohexene (S-CHene) increased at the expense of S-CHone due to the reason that the dehydration seems to become predominant at higher reaction temperatures.
 | ||
| Fig. 13 Effect of temperature on CHOL conversion over Cu/MgO-SBA15 catalysts (catalyst: 15Cu/MgO-SBA15, Vcat = 1 ml, T = 523–598 K, P = 1 atm, total flow = 1.62 l h−1, GHSV = 1620 h−1, CHOL = 7.5 mmol h−1, CHOL:N2 = 1:10). | ||
Direct coupling of dehydrogenation–hydrogenation of NB and CHol over Cu/MgO-SBA15 catalysts
The gas phase coupled dehydrogenation–hydrogenation reaction of cyclohexanol and nitrobenzene (3:1 mole ratio) has been carried out under N2 flow in the presence of Cu/MgO-SBA15 catalysts at atmospheric pressure.
Influence of cu loading on the catalytic performance
Blank tests (in absence of the catalyst) presented no conversion of both the educts at 548 K. The effect of Cu loading on the catalytic performance for the coupling reaction is depicted in Fig. 14. | ||
| Fig. 14 Copper loading vs. activity on the direct coupling reaction of NB and CHol (conditions: Vcat = 1 ml, T = 548 K, P = 1 atm, total flow = 1.86 l h−1, GHSV = 1860 h−1, CHOL = 7.5 mmol h−1, NB = 2.5 mmol h−1, CHOL:NB:N2 = 3:1:30). | ||
It is evident from the figure that the Cu loading has shown a profound influence on catalytic performance. The conversion of CHol (X-CHol) rose from 66 to 89% with the increase in Cu loading from 5 to 15 wt% and then decreased to 72%. While X-NB attenuated from 52 to 82% up to 15 wt% then decreased to 67%. Likewise, S-CHone and S-AN also increased in a similar fashion with Cu loading from 5 to 15% and then levelled-off. The activity of 5% Cu/Mgo-SBA15 catalyst is very poor and the formation of by-products can be clearly seen. The major condensation products are N-cyclohexylideneaniline (CDA), N-cyclohexylaniline (CHA) and diphenylamine (DPA) respectively. As shown in the scheme, Schiff's base (CDA) results from the AL condensation with CHone over the strong Lewis basic sites.
The production of CHA in the course of the reaction is due to the catalytic hydrogenation of CDA by excess hydrogen involved in the reaction. We can suggest two various schemes for the unique formation of diphenylamine (DPA) namely, AN auto condensation upon acid sites and CHA dehydrogenation over basic sites of the catalyst surface. These reactions are extremely undesirable because CDA and the intermediate hydrogenolysis products are accountable for the coke formation. A similar situation is typically observed for cyclohexylamine dehydrogenation. As evident from the TPD analysis in Fig. 7 and 8, the presence of moderate and strong acid/basic sites for the lower Cu loadings influences the condensation reactions. From this study, it is evident that 15%Cu is the best catalyst between others in terms of enhanced activity and selectivity. It can be attributed to the presence of more number of surface-active Cu species and dynamic metal–support interaction. Similar behavior in the specific activity of Cu catalysts supported on MgO and Mg–Al hydrotalcite was reported previously (Scheme 3).24,34 In the case of individual NB hydrogenation, there was no auto-condensation of AN or condensation between cyclohexanone and aniline, so high selectivity of AN was obtained. However, a prompt condensation phenomenon was observed between existed products in the coupling reaction. Consequently, the competitive adsorption between the two reactants has certainly effected the AN selectivity. It is noteworthy that the X-NB is invariably lower than X-CHOL because of the reason that it typically depends on the amount of H2 released in situ from CHOL dehydrogenation. In other words, the higher the H2 released in situ from the dehydrogenation of CHOL, the greater the conversion of NB. The formation of CHENE is expected from the dehydration of CHOL.
 | ||
| Scheme 3 Direct coupling of cyclohexanol dehydrogenation and nitrobenzene hydrogenation over Cu/MgO-SBA15 catalysts. | ||
Effect of temperature on the activity/selectivity
Effect of reaction temperature on the catalytic performance of the best catalyst is presented in Fig. 15. | ||
| Fig. 15 Effect of temperature on the activity and selectivity on the coupling of NB hydrogenation and CHol dehydrogenation (conditions: catalyst = 15Cu/MgO-SBA15, Vcat = 1 ml, T = 523–598 K, P = 1 atm, GHSV = 1860 h−1, total flow = 1.86 l h−1, CHOL = 7.5 mmol h−1, NB = 2.5 mmol h−1, CHOL:CAL:N2 = 3:1:30). | ||
With the rise in T from 523 to 598 K, X-CHol increased continuously from 63 to 93% while X-NB attenuated from 51 to 82% up to 548 K and then declined to 33% when T is further elevated to 593 K. Selectivity of all products is maintained up to 548 K and then there is a substantial change in their distribution with a further rise in T beyond 548 K. For instance, the desired products such as S-CHone and S-NB are almost equal and maintained at ca. 90% up to 548 K beyond which the selectivity decreased significantly. On the whole, 548 K is the optimum temperature to get high activity and selectivity. On the other hand, the selectivity of by-products such as S-CHene and S-DPA increased beyond 548 K. For instance, S-CHene rose at the expense of S-CHone due to the reason that the dehydration seems to become predominant at higher reaction temperatures. Identically, S-DPA rose at the expense of S-AN. One possible reason could be an enhanced carbon deposition at elevated temperature, as a result; some of the Cu species might have blocked by coke. At such high reaction temperature, Schiff base typically undergoes thermolysis to DPA under the influence of coke formation (Scheme 4).53 It is traditionally believed that CHene is inevitably produced on the acid sites of the catalyst surface at elevated temperatures.28
 | ||
| Scheme 4 Plausible reaction pathway for the direct catalytic coupling of NB hydrogenation and CHol dehydrogenation over Cu/MgO-SBA15 samples. | ||
Long-term stability of 15 wt% Cu/MgO-SBA15 catalyst
The time-on-stream study for the coupling reaction was properly conducted on the active catalyst at 548 K for 30 h. It is clearly evident from the figure that the X-CHol decreased from 89 to 74% in similar fashion X-NB also reduced from 82 to 64%. The previous results hint that the conversion of NB typically depends on the amount of available H2 which is liberated via in situ phenomenon in the process of dehydrogenation of CHol. Therefore, either an increase in the conversion of CHol or an enhanced concentration of CHone could be helpful to release more H2 into the reaction media. This specific aspect, in fact, prompted us to change the CHol/NB mole ratio from 1:1 to 3:1, in such a way that can enhance the conversion of NB. Of course, this can also be accomplished if used more CHOL in the reactant feed mixture.
As shown in Fig. 16, the catalytic activity majorly relies on uniform distribution of metallic Cu species on the highly ordered MgO-SBA15 with a high surface area. Further, excellent thermal stability Cu/MgO-SBA15 catalyst would not allow agglomeration of active Cu particles during the prolonged study. In addition, the synergistic acidic–basic properties of Cu/MgO-SBA15 catalyst surface favor less accumulation of coke. The present catalytic system could get almost steady activity up to 10 h in terms of conversion and selectivity in the coupling reaction as shown in Fig. 16. MgO with intrinsic basicity properly tunes the surface acidic environment and allow strong interaction with active Cu species represents the key driving force for remarkable catalytic activity.
 | ||
| Fig. 16 TOS study on direct coupling of NB and CHol over 15Cu/MgO-SBA15 catalyst (reaction conditions: Vcat = 1 ml, T = 548 K, P = 1 atm, total flow = 1.86 l h−1, GHSV = 1860 h−1, CHOL = 7.5 mmol h−1, NB = 2.5 mmol h−1, CHOL:CAL:N2 = 3:1:30). | ||
Conclusions
In summary, the present study showed the possibility of coupling two industrially important reactions in gas phase, in one step, one reactor and under identical reaction conditions. Through these investigations reaction conditions were optimized and also the potential catalyst composition is identified. In situ liberated H2 can be efficiently used for the hydrogenation of second reactant. Cu loading has shown a strong influence on the catalytic performance. 15 wt% Cu/MgO-SBA15 exhibited the best performance among others. Remarkably, high yields of cyclohexanone (78%) and aniline (75%) could be successfully achieved. To be specific, excellent thermal stability Cu/MgO-SBA15 catalyst would not allow agglomeration of active Cu particles and the base-functionalized mesoporous silica favor less accumulation of coke. Hence, there is a lot of scope for the direct coupling of hydrogenation and dehydrogenation reactions over ordered mesoporous catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. K. Marella sincerely acknowledges the R & D support by the Department of Chemtry (H & S), PACE Institute of Technology & Sciences, Ongole, Andhra Pradesh, India. R. K. Marella gratefully acknowledges for the award of postdoctoral fellowship by the Department of Chemical Engineering, University of Cape Town, South Africa. V. R. Madduluri acknowledges UGC, India for the research support. Marlia M. Hanafiah was funded by the UKM research grants (DIP-2019-001; GUP-2020-034).Notes and references
- M. Irfan, T. N. Glasnov and C. O. Kappe, ChemSusChem, 2011, 4, 300–316 CrossRef CAS.
- G. D. Yadav and R. K. Mewada, Chem. Eng. J., 2013, 221, 500–511 CrossRef CAS.
- Y. Lei, Z. Chen, G. Lan, R. Wang and X.-Y. Zhou, New J. Chem., 2020, 44, 3681–3689 RSC.
- C. Li, S. Liu, F. Yang, Y. Zhang, Z. Gao, X. Yuan and X. Zheng, Mater. Chem. Phys., 2020, 240, 122146 CrossRef CAS.
- A. A. Al-Raad, M. M. Hanafiah, A. S. Naje and M. A. Ajeel, Environ. Pollut., 2020, 265, 115049 CrossRef CAS.
- C. S. Budi, D. Saikia, C.-S. Chen and H.-M. Kao, J. Catal., 2019, 370, 274–288 CrossRef CAS.
- A. Hu, X. Lu, D. Cai, H. Pan, R. Jing, Q. Xia, D. Zhou and Y. Xia, Mol. Catal., 2019, 472, 27–36 CrossRef CAS.
- N. A. Bahari, W. N. R. Wan Isahak, M. S. Masdar and Z. Yaakob, Int. J. Energy Res., 2019, 43, 5128–5150 CrossRef CAS.
- A. S. Hashimi, M. A. N. M. Nohan, S. X. Chin, P. S. Khiew, S. Zakaria and C. H. Chia, Nanomaterials, 2020, 10, 1153 CrossRef CAS.
- J. Song, Z.-F. Huang, L. Pan, K. Li, X. Zhang, L. Wang and J.-J. Zou, Appl. Catal., B, 2018, 227, 386–408 CrossRef CAS.
- Y. Xiang, X. Li, C. Lu, L. Ma and Q. Zhang, Appl. Catal., A, 2010, 375, 289–294 CrossRef CAS.
- J. Long, Y. Zhou and Y. Li, Chem. Commun., 2015, 51, 2331–2334 RSC.
- D. Tavor, I. Gefen, C. Dlugy and A. Wolfson, Synth. Commun., 2011, 41, 3409–3416 CrossRef CAS.
- M. R. Rahimpour, M. R. Dehnavi, F. Allahgholipour, D. Iranshahi and S. M. Jokar, Appl. Energy, 2012, 99, 496–512 CrossRef CAS.
- C. V. Pramod, C. Raghavendra, K. H. P. Reddy, G. V. R. Babu, K. S. R. Rao and B. D. Raju, J. Chem. Sci., 2014, 126, 311–317 CrossRef CAS.
- Y.-L. Zhu, H.-W. Xiang, G.-S. Wu, L. Bai and Y.-W. Li, Chem. Commun., 2002, 254–255 RSC.
- R. Zhang, H. Yin, D. Zhang, L. Qi, H. Lu, Y. Shen and T. Jiang, Chem. Eng. J., 2008, 140, 488–496 CrossRef CAS.
- M. Tamura, K. Kon, A. Satsuma and K.-i. Shimizu, ACS Catal., 2012, 2, 1904–1909 CrossRef CAS.
- D. Gao, H. Yin, Y. Feng and A. Wang, Can. J. Chem. Eng., 2015, 93, 1107–1118 CrossRef CAS.
- Y.-L. Zhu, H.-W. Xiang, Y.-W. Li, H. Jiao, G.-S. Wu, B. Zhong and G.-Q. Guo, New J. Chem., 2003, 27, 208–210 RSC.
- H.-Y. Zheng, Y.-L. Zhu, Z.-Q. Bai, L. Huang, H.-W. Xiang and Y.-W. Li, Green Chem., 2006, 8, 107–109 RSC.
- K. H. P. Reddy, N. Anand, P. S. S. Prasad, K. S. R. Rao and B. D. Raju, Catal. Commun., 2011, 12, 866–869 CrossRef CAS.
- H. P. R. Kannapu, Y.-W. Suh, A. Narani, V. Vaddeboina, D. R. Burri and R. R. Kamaraju Seetha, RSC Adv., 2017, 7, 35346–35356 RSC.
- C. V. Pramod, M. Suresh, V. Mohan, B. Sridevi, B. David Raju and K. S. Rama Rao, Curr. Catal., 2012, 1, 140–148 CrossRef CAS.
- J. T. Bhanushali, D. Prasad, K. N. Patil, K. S. Reddy, K. S. Rama Rao, A. H. Jadhav and B. M. Nagaraja, Int. J. Hydrogen Energy, 2020, 45, 12874–12888 CrossRef CAS.
- P. Lek-utaiwan, B. Suphanit, P. L. Douglas and N. Mongkolsiri, Comput. Chem. Eng., 2011, 35, 1088–1100 CrossRef CAS.
- B. Smitha, D. Suhanya, S. Sridhar and M. Ramakrishna, J. Membr. Sci., 2004, 241, 1–21 CrossRef CAS.
- N. P. Tangale, P. S. Niphadkar, S. S. Deshpande and P. N. Joshi, Appl. Catal., A, 2013, 467, 421–429 CrossRef CAS.
- B. M. Nagaraja, V. Siva Kumar, V. Shashikala, A. H. Padmasri, S. Sreevardhan Reddy, B. David Raju and K. S. Rama Rao, J. Mol. Catal. A: Chem., 2004, 223, 339–345 CrossRef CAS.
- B. Sridevi, P. Nagaiah, A. H. Padmasri, B. David Raju and K. S. Rama Rao, J. Chem. Sci., 2017, 129, 601–608 CrossRef CAS.
- P. Nagaiah, C. V. Pramod, M. Venkata Rao, B. David Raju and K. S. Rama Rao, Catal. Lett., 2018, 148, 3042–3050 CrossRef CAS.
- C. V. Pramod, V. Mohan, B. D. Raju and K. S. R. Rao, Catal. Lett., 2013, 143, 432–437 CrossRef CAS.
- M. Li, Y. Hao, F. Cárdenas-Lizana, H. H. P. Yiu and M. A. Keane, Top. Catal., 2015, 58, 149–158 CrossRef CAS.
- H. P. R. Kannapu, C. K. P. Neeli, K. S. Rama Rao, V. N. Kalevaru, A. Martin and D. R. Burri, Catal. Sci. Technol., 2016, 6, 5494–5503 RSC.
- H. P. R. Kannapu, Y.-W. Suh, A. Narani, D. R. Burri and R. R. Kamaraju Seetha, Catal. Lett., 2017, 147, 90–101 CrossRef CAS.
- A. Malaika, J. Gertig, P. Rechnia, A. Miklaszewska and M. Kozłowski, Arabian J. Chem., 2019, 12, 4947–4956 CrossRef CAS.
- K. H. P. Reddy, Y.-W. Suh, N. Anand, B. D. Raju and K. S. R. Rao, Catal. Commun., 2017, 95, 21–25 CrossRef CAS.
- M. V. Twigg and M. S. Spencer, Appl. Catal., A, 2001, 212, 161–174 CrossRef CAS.
- R. Ryoo, C. H. Ko, M. Kruk, V. Antochshuk and M. Jaroniec, J. Phys. Chem. B, 2000, 104, 11465–11471 CrossRef CAS.
- O. Hinrichsen, T. Genger and M. Muhler, Chem. Eng. Technol., 2000, 23, 956–959 CrossRef CAS.
- S.-W. Bian, J. Baltrusaitis, P. Galhotra and V. H. Grassian, J. Mater. Chem., 2010, 20, 8705–8710 RSC.
- C. K. P. Neeli, A. Narani, R. K. Marella, K. S. Rama Rao and D. R. Burri, Catal. Commun., 2013, 39, 5–9 CrossRef CAS.
- H. Misran, M. A. Yarmo and S. Ramesh, Ceram. Int., 2013, 39, 931–940 CrossRef CAS.
- Y. M. Wang, Z. Y. Wu, Y. L. Wei and J. H. Zhu, Microporous Mesoporous Mater., 2005, 84, 127–136 CrossRef CAS.
- Y. Lwin, A. B. Mohamad, Z. Yaakob and W. R. W. Daud, React. Kinet. Catal. Lett., 2000, 70, 303–310 CrossRef CAS.
- A. Narani, R. K. Marella, P. Ramudu, K. S. Rama Rao and D. R. Burri, RSC Adv., 2014, 4, 3774–3781 RSC.
- B. Zhang, Y. Zhu, G. Ding, H. Zheng and Y. Li, Appl. Catal., A, 2012, 443–444, 191–201 CrossRef CAS.
- J. A. Wang, O. Novaro, X. Bokhimi, T. López, R. Gómez, J. Navarrete, M. E. Llanos and E. López-Salinas, J. Phys. Chem. B, 1997, 101, 7448–7451 CrossRef CAS.
- R. K. Marella, C. K. Prasad Neeli, S. R. Rao Kamaraju and D. R. Burri, Catal. Sci. Technol., 2012, 2, 1833–1838 RSC.
- V. Mohan, C. V. Pramod, M. Suresh, K. H. Prasad Reddy, B. D. Raju and K. S. Rama Rao, Catal. Commun., 2012, 18, 89–92 CrossRef CAS.
- M. Kondeboina, S. S. Enumula, V. R. B. Gurram, J. Yadagiri, D. R. Burri and S. R. R. Kamaraju, New J. Chem., 2018, 42, 15714–15725 RSC.
- V. Z. Fridman, A. A. Davydov and K. Titievsky, J. Catal., 2004, 222, 545–557 CrossRef CAS.
- V. D. Stytsenko, T. D. Hyu and V. A. Vinokurov, Kinet. Catal., 2007, 48, 292–297 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06003h |
| This journal is © The Royal Society of Chemistry 2020 |