
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntimicrobial peptide-modified silver nanoparticles for enhancing the antibacterial efficacy†
Wenxi Li‡
 b,
Yongchun Li‡a,
Pengchao Sund,
Nan Zhangac,
Yidan Zhaoa,
Shangshang Qin*a and
Yongxing Zhao*ac
b,
Yongchun Li‡a,
Pengchao Sund,
Nan Zhangac,
Yidan Zhaoa,
Shangshang Qin*a and
Yongxing Zhao*ac
aSchool of Pharmaceutical Science, Zhengzhou University, Zhengzhou, Henan 450001, PR China. E-mail: zhaoyx@zzu.edu.cn; Fax: +86 0371 67739546; Tel: +86037167739165
bZhengzhou Traditional Chinese Hospital of Orthopaedics, Zhengzhou, Henan 450004, PR China
cKey Laboratory of Advanced Pharmaceutical Technology, Ministry of Education of China, Zhengzhou, Henan 450001, PR China
dInstitute for Biological Interfaces 1, Karlsruhe Institute of Technology, 76344 Eggenstein-Leopoldshafen, Germany
First published on 22nd October 2020
Abstract
Antibiotic-resistant bacteria are becoming a serious threat to public health worldwide. To address this problem, we have developed multifunctional peptide (MFP)-coated silver nanoparticles (MFP@AgNPs) for antibacterial studies. MFPs, which can physically adsorb to AgNPs via electrostatic interactions are comprised of a matrix metalloproteinase (MMP) cleavable sequence (PVGLIG), an antimicrobial peptide (tachyplesin-1), and a target peptide (PGP-PEG). The resulting MFP@AgNPs were characterized by various technologies, including UV-vis spectrophotometry, zeta potential analyzer, circular dichroism (CD) spectroscopy, attenuated total reflection-Fourier-transform infrared spectroscopy (ATR-FTIR), and transmission electron microscopy (TEM). The MIC and MBC were investigated against both Gram-positive bacteria and Gram-negative bacteria. The antibacterial activity in vivo was evaluated on MDR-AB (multidrug-resistant Acinetobacter baumannii) infected mice. We found that MFP@AgNPs exhibited antibacterial activity against both Gram-positive bacteria and Gram-negative bacteria. Compared to bare AgNPs, MFP@AgNPs-1 killed MDR-AB faster and more efficiently. SEM images showed that MFP@AgNPs-1 induced cell disruption via cell membrane damage. In vivo studies further confirmed the enhanced antibacterial activity against MDR-AB infections. The developed MFP@AgNPs-1 reduced the cytotoxicity of AgNPs and enhanced the antibacterial activity against MDR-AB in vitro and in vivo, providing a possible solution against multidrug-resistant bacterial infections.
1. Introduction
Antibiotic-resistant bacteria are becoming a serious threat to public health worldwide.1,2 The infections caused by these bacteria are difficult to be cured by antibiotics that used to be efficacious. Solutions to this problem include controlling antibiotic use, developing new antibiotics and inhibiting evolution.3,4 With the development of nanotechnology, nanoparticles have drawn much attention in the antibacterial field and are promising as alternatives to new antibiotics. ZnO particles,5,6 chitosan nanoparticles,7,8 silver nanoparticles (AgNPs),9,10 graphene,11 and some composite particles12,13 have been synthesized and used for antibacterial studies. Among these particles, AgNPs have been intensively studied because of their excellent antibacterial activity.14 However, their applications are limited due to the poor activity against drug-resistant pathogens and their toxicity.14,15 To reduce toxicity, one possible solution is to modify the AgNPs surface with different biomolecules via covalent or non-covalent approaches.16Antimicrobial peptides (AMPs) are natural peptides that exhibit strong antibacterial activities.17 AMPs are usually positively charged and can interact with microbial membranes through electrostatic interactions, resulting in morphology damage.18 To increase the efficacy, AMPs are generally modified by adding some fragments. N-Acetylated Proline–Glycine–Proline (N-ac-PGP) is chemotactic for neutrophils in vitro as well as in vivo.19 N-ac-PGP exerts its chemotactic activity by mimicking key sequences found within classical neutrophil chemokines (such as CXCL8) and targets its receptor CXCR1/2 to alleviate chronic neutrophilic lung diseases.20 Matrix metalloproteases (MMPs), especially MMP2, are overexpressed in some types of human inflammation microenvironments.21 PVGLIG is an MMP-2-sensitive peptide and has been successfully modified on nanoparticle surfaces for targeted drug delivery systems.22,23 It is reported that tachyplesin-1 peptide is an AMP with a β-hairpin structure, and can induce cell apoptosis via a membranolytic mechanism;24,25 however, it is strongly hemolytic against mammalian erythrocytes,26 and also shows the disadvantages of poor stability and membrane penetration.27 To improve these drawbacks of tachyplesin-1 and reduce the cytotoxicity of AgNPs, we developed multifunctional peptide (MFP)-coated AgNPs (MFP@AgNPs) for antibacterial studies. The MFP is comprised of an MMP cleavable sequence (PVGLIG), an AMP (tachyplesin-1) and a target peptide (N-ac-PGP-PEG) (Scheme 1). PEGylation can enhance the circulation of AgNPs in vivo while it shields the targeting properties of ligands by weakening the intracellular trafficking of cellular uptake and endosomal escape.28 Together with PGP-PEG, PVGLIG can prevent tachyplesin-1 from degradation by enzymes and prolong its bioactivity. Once arriving at the slightly acidic (pH < 6.5) infection microenvironment, MFP can disassociate so that PVGLIG is cleaved by MMP, resulting in the exposure of tachyplesin-1 and further bioactivities. MFP@AgNPs were prepared by mixing the positively charged MFPs and the negatively charged AgNPs. The antibacterial activity of MFP@AgNPs was first investigated against both Gram-positive bacteria and Gram-negative bacteria, followed by in vivo studies on MDR-AB-infected mice. Our results suggest that surface modification with MFP enhanced the antibacterial activity of AgNPs against MDR-AB, providing a new solution to anti-MDR studies.
 | ||
| Scheme 1 Schematic illustration of (A) the multifunctional peptide (MFP) design, (B) the preparation of MFP@AgNPs and (C) the possible interactions between MFP@AgNPs and MRD-AB cells. | ||
2. Materials and methods
2.1 Materials
The tachyplesin-1 and MFP were synthesized by DGP Peptides Co. Ltd. (Hangzhou, China). Silver nanoparticles with an average size of 20 nm were purchased from Zhongke Leiming Co. Ltd. (Beijing, China). Ultrapure Milli-Q water (18 MΩ cm; Millipore-Q; EMD Millipore, Billerica, MA, USA) was used for solution preparation. The CCK-8 kit was purchased from Dojindo (Japan). TNF-α and IL-6 Elisa kits were purchased from Ikesai Biotechnology (Taicang, China). Amikacin was provided by Bidepharm (Shanghai, China). Propidium iodide (PI) and 2.5% glutaraldehyde were obtained from Legend Biotech (Beijing, China). The hematoxylin-eosin staining kit, vancomycin and phosphate-buffered solution were bought from Solarbio® Life Sciences (Beijing, China). Cyclophosphamide was purchased from Ryon Biological Technology (Shanghai, China). Mueller-Hinton broth (MHB) and Mueller–Hinton agar (MHA) were obtained from Aobox (Beijing, China). Acinetobacter chromogenic agar was provided by CHROMagar (Paris, France). Sheep red blood cells were purchased from Bianzhen Biotech (Nanjing, China). Saline solution was obtained from Kelun Pharmaceutical Co. (Henan, China). All the above-mentioned materials were used without any further modification.E. coli (Escherichia coli), MDR-AB (multidrug-resistant Acinetobacter baumannii bacteria), S. aureus (Staphylococcus aureus) and MRSA (Methicillin-resistant Staphylococcus aureus) were provided by Dr Shangshang Qin (Zhengzhou University, Henan, China). Female BALB/c mice were obtained from the laboratory animal center of Henan province [license number: SCXK (Yu) 2017-0001]. Animal care and experiments were carried out with the approval of the Animal Ethics Committee of Zhengzhou University (Zhengzhou, China) according to the requirements of the National Act on the Use of Experimental Animals (China).
2.2 Preparation of MFP@AgNPs conjugates
AgNPs (final concentration: 64 μg mL−1, final volume: 1 mL) were incubated with MFP at different mass ratios (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5, 1:1, and 1:1.5) in the dark at 37 °C. After 12 h, the particles were collected by centrifugation (10000 rpm, 30 min) and washed twice with water. The resulting particles were redispersed in 1 mL of water and denoted as MFP@AgNPs-0.5, MFP@AgNPs-1, and MFP@AgNPs-1.5, respectively. All the supernatants were collected and quantified by UV-vis spectroscopy (UV-2700, Shimadzu, China). The loading capacity of MFP was determined by the following equations:
:0.5, 1:1, and 1:1.5) in the dark at 37 °C. After 12 h, the particles were collected by centrifugation (10000 rpm, 30 min) and washed twice with water. The resulting particles were redispersed in 1 mL of water and denoted as MFP@AgNPs-0.5, MFP@AgNPs-1, and MFP@AgNPs-1.5, respectively. All the supernatants were collected and quantified by UV-vis spectroscopy (UV-2700, Shimadzu, China). The loading capacity of MFP was determined by the following equations: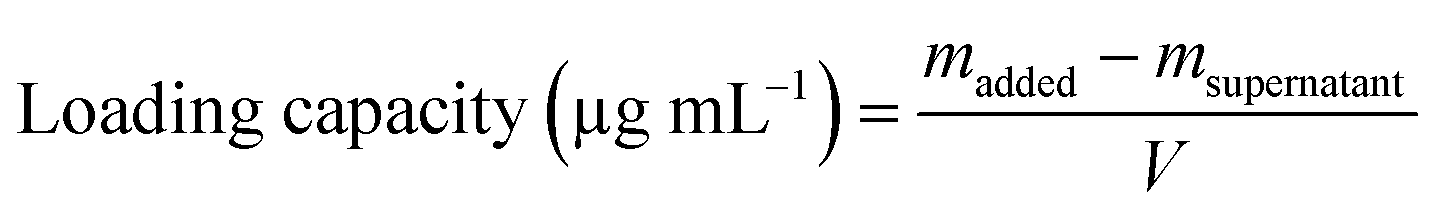
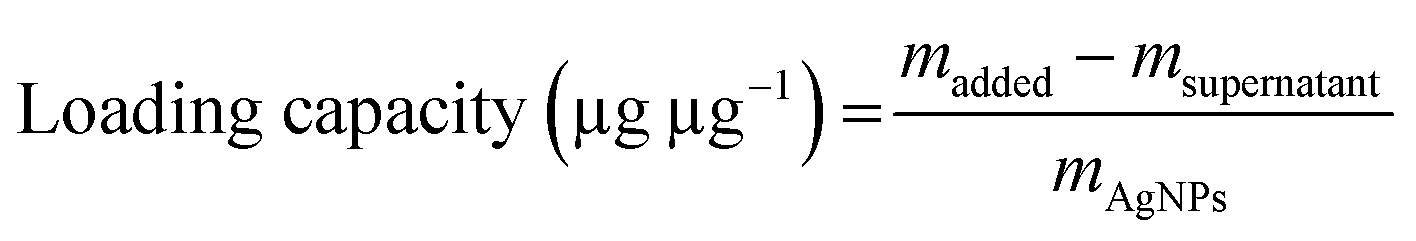
2.3 UV-vis spectroscopy
AgNPs, MFP and MFP@AgNPs were diluted with water and the UV-vis absorbance of the samples was recorded via UV-vis spectroscopy (UV-2700, Shimadzu, China).2.4 Zeta potential analysis
AgNPs and MFP@AgNPs were diluted with water followed by zeta potential analysis using a zeta potential analyzer (NGL-88Bano, Malvern, UK).2.5 Circular dichroism (CD) spectroscopy
The secondary structure of MFP on AgNPs was confirmed by CD spectroscopy (J-1500, JASCO, Japan). The samples were loaded into the cuvette (Hellma Analytics, Munich, Germany) with a light path of 1 mm and scanned from 190 to 260 nm with a resolution of 1 nm.2.6 Attenuated total reflection-Fourier-transform infrared spectroscopy (ATR-FTIR)
The infrared spectra of MFP@AgNPs were recorded by ATR-FTIR (6700, Nicolet, USA). For each sample, an accumulation of 100 interferograms was collected in single beam mode from 4000 cm−1 to 400 cm−1 with a resolution of 4 cm−1.2.7 Transmission electron microscopy (TEM)
The morphologies of AgNPs and MFP@AgNPs were determined by TEM (HT7700, Philips-FEI, Netherlands). Here, 10 μL of the sample was spotted on a discharged TEM grid and incubated for 20 s. After the removal of the excess sample, the grid was purified with 50 μL of water followed by staining with 2% of aqueous uranyl formate solution. After 30 s, the excess solution was removed by filter paper. The grid was dried at room temperature and prepared for TEM imaging.2.8 Cell culture
Both Gram-positive bacteria (S. aureus and MRSA) and Gram-negative bacteria (E. coli and MDR-AB) were used for in vitro antibacterial activity studies. All the bacteria were cultured in Nutrient broth at 37 °C on a shaker (200 rpm) or agar plates at 37 °C without shaking.2.9 Minimum inhibitory concentration test (MIC)
The MIC of MFP@AgNPs, AgNPs, MFP, and tachyplesin-1 were determined by the broth micro-dilution method.29 Briefly, E. coli, MDR-AB, S. aureus and MRSA were seeded in 96 well plates at a density of 5 × 103 CFU per well (1 × 105 CFU mL−1, 50 μL). Various concentrations (from 64 to 0.125 μg mL−1, 50 μL) of MFP@AgNPs, MFP, and AgNPs in MHB medium were added to each well. Amikacin and vancomycin were used as positive controls for the treatment of Gram-negative bacteria and Gram-positive bacteria, respectively. Bacteria without any treatment were used as negative controls. All the experiments were performed in triplicate. The bacteria were incubated under standard conditions for 16–20 h. The optical density at 600 nm was measured using a microplate reader (Powerwave XS2, BioTek, USA).2.10 Time-dependent killing
Time-dependent killing was performed according to the previous study.30 A single colony of MDR-AB was pre-cultured in MHB medium for 12 h at 37 °C in a shaker (200 rpm). The pre-culture was diluted 10000 times with MHB medium and aliquoted at 1.5 mL per tube. After incubation for 2 h, the bacteria were treated with MFP@AgNPs-1, AgNPs, MFP, and tachyplesin-1 at different concentrations (1× MIC, 3× MIC, and 6× MIC). Amikacin-treated bacteria were used as positive controls. Bacteria without any treatment were used as negative controls. Then, 100 μL of bacteria were collected at different time points (0, 0.5, 1, 2, 3, 4, 5, 6, 8, 10 and 24 h) and centrifugated at 4000 rpm for 3 min. The bacteria were redispersed in 1 mL of PBS and 10 μL of the resulting samples were seeded on an MHA plate and cultured at 37 °C overnight. Colonies were counted and the living bacteria in each sample were calculated.
2.11 Morphological characterization
Scanning electron microscopy (SEM) was used to investigate the morphological changes of MDR-AB that were treated with MFP@AgNPs-1, AgNPs, MFP, and tachyplesin-1 at 3× MIC. A single colony of MDR-AB cells was seeded in 1 mL of MHB medium and cultured under standard conditions for 5 h. After incubation, the bacteria were collected by centrifugation (4000 rpm, 10 min) and redispersed in 5 mL of PBS. The bacteria were aliquoted and treated with MFP@AgNPs-1, AgNPs, MFP, and tachyplesin-1, amikacin and MHB medium, respectively. After incubation for 30 min, the bacteria were collected by centrifugation (8000 rpm, 1 min) and washed three times with PBS. The bacteria were fixed with 2.5% glutaraldehyde at 4 °C overnight and dehydrated with various concentrations of ethanol (from 30% to 100%, incubation time: 15 min).31 To remove any moisture, the bacteria were further dehydrated in ethanol/tert-butanol (v/v, 50%) and tert-butanol (100%) for 15 min, respectively. The resulting samples were spotted on a monocrystalline silicon wafer and dried under vacuum at 37 °C followed by SEM imaging.2.12 Antibacterial mechanism
Propidium iodide (PI) was utilized to investigate the integrity of the cell membrane32. Bacteria were seeded in a black 96 well plate at a final concentration of 1.5 × 107 CFU per well. PI (10 μM, 50 μL) was added to each well and incubated under standard conditions for 30 min. Before treatment, the fluorescence intensity at 617 nm was determined every 2 min with a microplate reader (EnSpire, PerkinElmer, USA) using an excitation of 535 nm. After 8 min, MFP@AgNPs-1, AgNPs, MFP, tachyplesin-1, and amikacin were added. For the negative control, the same volume of water was added. The fluorescence intensity of each well was measured every 2 min using the same method.2.13 Cytotoxicity in vitro
The cytotoxicity of the developed materials was tested against the human umbilical vein endothelial cells (HUVEC) via CCK8 assay. HUVEC were seeded in a 96 well plate at a final density of 5 × 103 cells per well and cultured under standard conditions (37 °C, 5% CO2, humidity 100%) for 24 h. The digested medium was replaced with fresh medium containing MFP@AgNPs-1, AgNPs, and MFP (concentration various from 256 to 8 μg mL−1). After incubation for 24 h, the medium was removed and the cells were washed with PBS once. The cells were incubated with 100 μL of fresh medium containing 5% of CCK8 agent for 4 h followed by absorbance measurements with the microplate reader. The cytotoxicity was calculated by the following equation: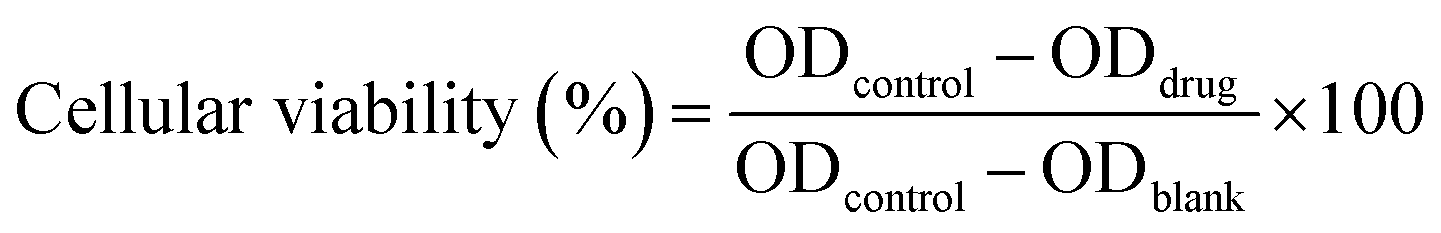
2.14 Hemolysis
Sheep red blood cells were used as model blood cells to evaluate the biocompatibility of MFP@AgNPs-1. Here, 2 mL of sheep blood were washed with PBS at least three times and redispersed in 20 mL of PBS to obtain 10% of blood cells. Various concentrations (from 256 to 8 μg mL−1) of MFP@AgNPs-1 were incubated with the same volume of 10% blood cells for at 37 °C for 1 h. Then, 0.1% of Triton X-100 treated blood cells were used as positive controls. After incubation, the cells were collected by centrifugation (1000 rpm, 15 min). The UV-vis absorbance at 540 nm of the samples' supernatant (150 μL) was determined by the microplate reader. The hemolysis rate was calculated via the following equation:where A1 is the OD of the sample, A0 is the OD of the negative control, and Atotal is the OD of the positive control.

2.15 Antibacterial activity in vivo
MDR-AB-infected female BALB/c mice were used as pneumonia models for the antibacterial activity study of MFP@AgNPs-1 in vivo. The models were constructed according to the previous study.33 Here, 36 of MDR-AB-infected mice were divided into 6 groups and treated with MFP@AgNPs-1, AgNPs, MFP, tachyplesin-1, amikacin, and PBS for three days at a dosage of 0.1 mg per kg per day by intravenous injection, respectively. Six healthy mice were used as negative controls. The weights and temperatures of the mice were recorded every day.After three days' treatment, the blood of each mouse was collected via retro-orbital bleeding. The neutrophil-to-lymphocyte ratio (NLR) and neutrophils (NEUT) were determined by an automatic animal blood analyzer (XT-2000iv, SYSMEX, Japan).34,35 The right-upper lungs were removed from the bodies and washed with PBS. The weight of the lungs was recorded before and after drying. The weight ratio was calculated by the following equation:
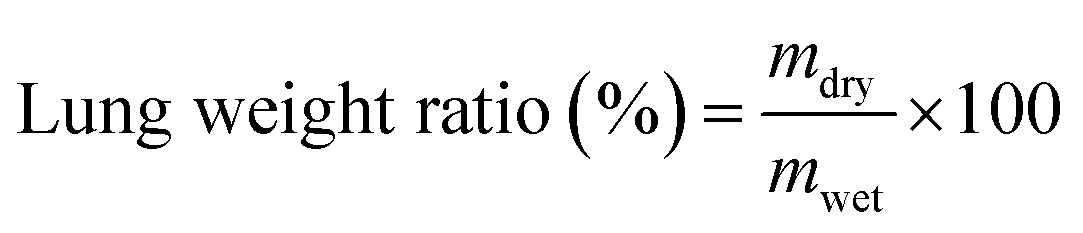
The distribution of bacteria in each organ of the mouse was also investigated. The hearts, livers, spleens, lungs, and kidneys were removed from the mouse bodies, washed with sterile saline solution and dried with filter paper, followed by homogenizing in ice-cold saline solution (9 mL g−1). The resulting homogenate of each organ was cultured on an acinetobacter chromogenic agar plate (CHOMagar, Paris, France) at 37 °C overnight. The colonies were counted and the density of the bacteria in each organ was calculated. The inflammatory factors, TNF-α and IL-6, in the lung tissue homogenate were determined via enzyme-linked immunosorbent assay (ELISA) using TNF-α ELISA kits and IL-6 ELISA kits, respectively. The experiments were carried out following the manual instructions without any modifications. For the histopathological study, the left lung of each mouse was stained by hematoxylin–eosin (HE) and imaged with optical microscopy. The lung tissue sections were prepared by the same method as the previous study.36 In brief, lungs were fixed in formalin (pH 7.0) overnight and embedded in paraffin, followed by HE staining. The HE stained lung sections were imaged with optical microscopy (Leica Biosystems Imaging Inc., GER) and the results were generated by Image Scope Software (Leica Biosystems Imaging Inc).
2.16 Statistical analysis
Statistical analysis was performed using SPSS 21.0. All experiments were repeated at least three times, the data are presented as mean ± SD. One-way analysis of variance (ANOVA) was used to compare to differences among three or more groups (statistical significance: p < 0.05).3. Results and discussion
3.1 Preparation and characterization of MFP@AgNPs conjugates
To prepare MFP@AgNPs conjugates, we incubated MFP with AgNPs at different mass ratios. The mass ratio and loading capacity are listed in Table 1. The loading capacity increased with the increase in the mass ratio.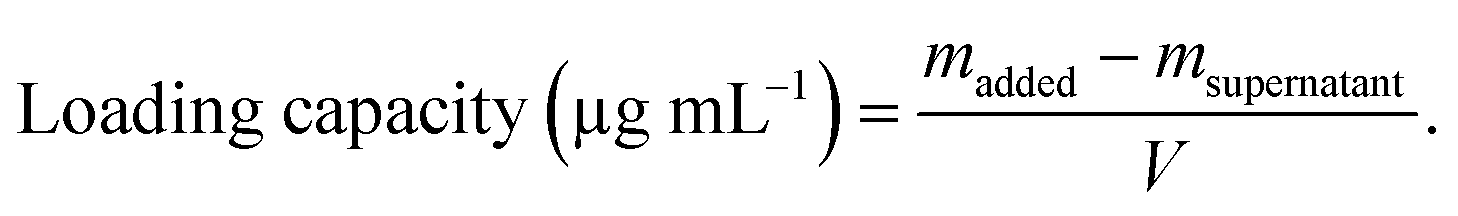

We then used different technologies, including UV-vis spectroscopy, zeta potential analyzer, ATR-FTIR (attenuated total reflection-Fourier-transform infrared spectroscopy), CD (circular dichroism) spectroscopy and TEM (transmission electron microscopy), to characterize the resulting MFP@AgNPs conjugates (Fig. 1). It is well-known that AgNPs show UV-vis absorption in the range of 400–500 nm because of strong surface plasmon resonance (SPR).37 The changes in the UV-vis absorption can be used to monitor the particle surface reactions.38,39 As shown in Fig. 1A, MFP@AgNPs conjugates exhibited the specific absorption of MFP at 280 nm and AgNPs at 410 nm. Furthermore, we observed a decrease in intensity at 410 nm and a redshift to 587 nm, in agreement with the previous study.40 With the surface coating of MFP, the SPR property of AgNPs decreased, resulting in a reduced absorbance at 410 nm. The redshift to 587 nm indicated that the surface of AgNPs changed due to MFP binding. The changes in UV-vis absorption can be explained by the MFP shell on AgNPs, which might affect the SPR. Compared to AgNPs, the zeta potential of the MFP@AgNPs conjugates decreased due to the positively charged MFP (theoretical isoelectric point: 10.1, calculated by Geneious 9.1.3) under neutral conditions (Fig. 1B). After coating with MFP, the zeta potential of AgNPs shifted from ∼−8 mV to ∼−4 mV (MFP@AgNP-0.5). With the increase in MFP, the zeta potential further decreased to ∼0 mV (MFP@AgNP-1). These results demonstrated that MFP was successfully added to the particle surface. The ATR-FTIR spectra of MFP@AgNPs conjugates exhibited variations in the Ag–O/Ag–Ag (∼565 cm−1 and 730 cm−1)41,42 and stretching variations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1650 cm−1) and bending variations of N–H (1540 cm−1 and 1515 cm−1)43 (Fig. 1C). CD spectroscopy is often used to investigate the secondary structures of peptides and proteins. As shown in Fig. 1D, the MFP and different MFP@AgNPs conjugates exhibited negative ellipticity with a significant peak in the range of 190–215 nm, which represents the random coil structure.44 These results demonstrate that the secondary structure of MFP was retained after conjugation to AgNPs. TEM images showed the spherical morphology of AgNPs and MFP@AgNPs-1 with an average size of 21.7 ± 2.5 nm and 25.7 ± 5.6 nm, respectively (Fig. 1E).
O (1650 cm−1) and bending variations of N–H (1540 cm−1 and 1515 cm−1)43 (Fig. 1C). CD spectroscopy is often used to investigate the secondary structures of peptides and proteins. As shown in Fig. 1D, the MFP and different MFP@AgNPs conjugates exhibited negative ellipticity with a significant peak in the range of 190–215 nm, which represents the random coil structure.44 These results demonstrate that the secondary structure of MFP was retained after conjugation to AgNPs. TEM images showed the spherical morphology of AgNPs and MFP@AgNPs-1 with an average size of 21.7 ± 2.5 nm and 25.7 ± 5.6 nm, respectively (Fig. 1E).
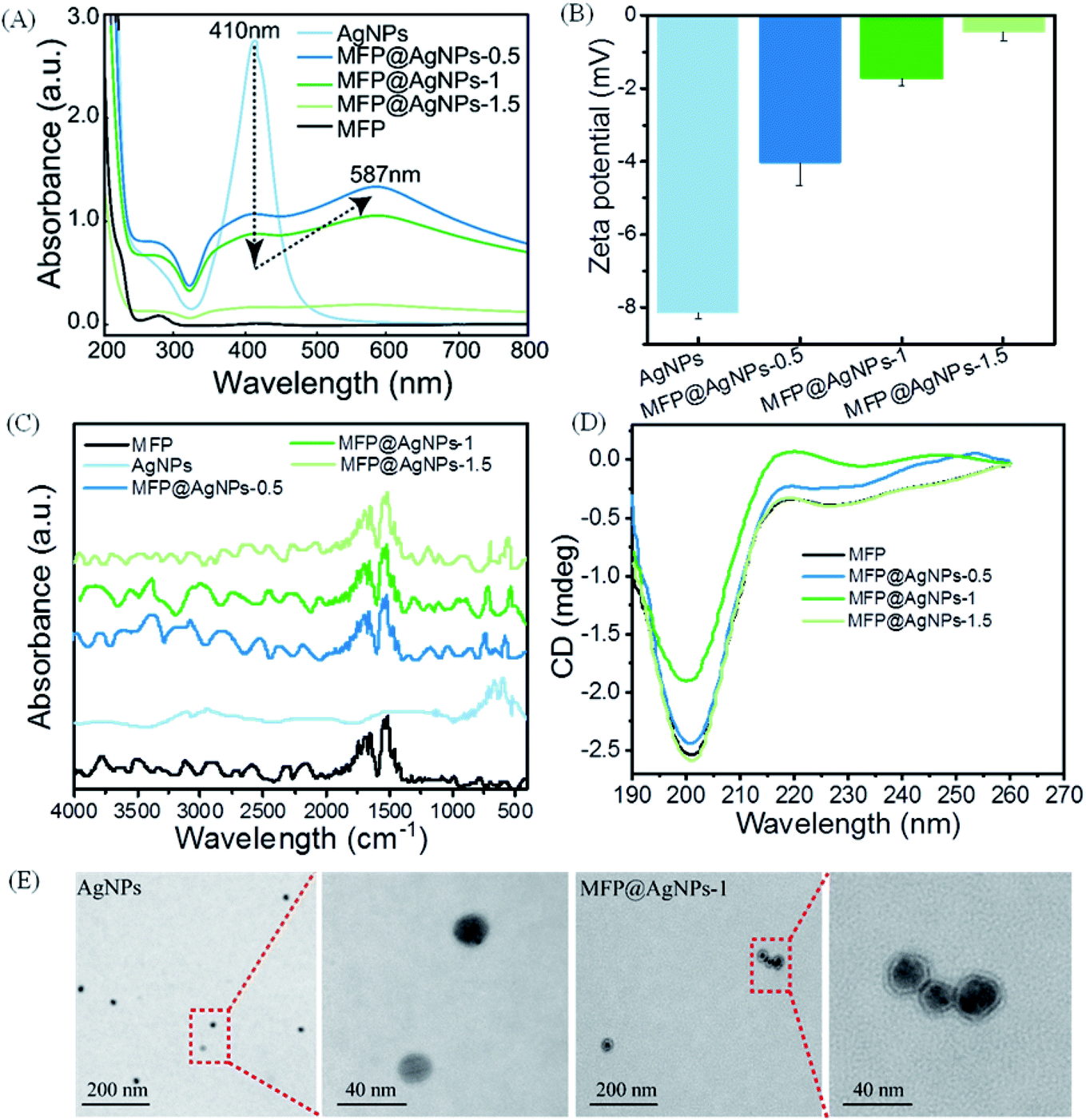 | ||
| Fig. 1 Characterization of MFP@AgNPs conjugates. (A) UV-vis spectra, (B) zeta potential, (C) ATR-FTIR spectra, (D) CD spectra of AgNP, MFP and MFP@AgNPs conjugates. (E) Representative TEM images of AgNPs and MFP@AgNPs-1. | ||
3.2 In vitro antibacterial activity
The antibacterial activity in vitro of MFP@AgNPs was investigated against Gram-positive bacteria (S. aureus and MRSA) and Gram-negative bacteria (E. coli and MDR-AB). We tested both the MIC (minimum inhibitory concentration) and the MBC (minimum bactericidal concentration). The results are listed in Table 2. In agreement with previous studies,45,46 AgNPs had an effect on both Gram-positive bacteria and Gram-negative bacteria. However, AgNPs were more effective towards Gram-positive bacteria than Gram-negative bacteria. The antibacterial peptide tachyplesin-1 and MFP were able to inhibit and kill E. coli, and MDR-AB at lower concentrations. Compared to MFP and AgNPs, MFP@AgNPs conjugates exhibited stronger antibacterial activity towards Gram-negative bacteria and overcame the drug resistance of AgNPs against MDR-AB. The antibacterial activity of MFP@AgNPs-1 against MDR-AB was almost equivalent to the positive control amikacin. Considering the cost of the experiments, MFP@AgNPs-1 was used for further studies. To better understand the antibacterial activity of MFP@AgNPs-1 against MDR-AB, we investigated the time-kill kinetics at 3× MIC and 6× MIC (Fig. S1†).| Items | MIC* (μg mL−1) | MBC* (μg mL−1) | ||||||
|---|---|---|---|---|---|---|---|---|
| S. aureus | MRSA | E. coli | MDR-AB | S. aureus | MRSA | E. coli | MDR-AB | |
| a N.A.: not applicable.*: the concentrations of MFP@AgNPs conjugates were calculated according to AgNPs. | ||||||||
| Amikacin | N.A. | N.A. | 2 | 1 | N.A. | N.A. | 2 | 1 |
| Vancomycin | 2 | 2 | N.A. | N.A. | 2 | 2 | N.A. | N.A. |
| AgNPs | 2 | 2 | 16 | 32 | 2 | 8 | 16 | 32 |
| Tachyplesin-1 | 16 | 2 | 0.125 | 1 | 16 | 2 | 0.125 | 1 |
| MFP | 4 | 16 | 2 | 4 | 16 | 16 | 2 | 4 |
| MFP@AgNPs-0.5 | 16 | 16 | 8 | 8 | 16 | 16 | 8 | 8 |
| MFP@AgNPs-1 | 8 | 16 | 2 | 2 | 16 | 16 | 8 | 4 |
| MFP@AgNPs-1.5 | 8 | 16 | 4 | 2 | 16 | 16 | 4 | 2 |
As shown in Fig. 2A, MFP@AgNPs-1 exhibited the greatest kill efficacy at 3× MIC. MFP@AgNPs-1 could kill 99.9% of MDR-AB within 0.5 h, which was much faster than the positive control amikacin. On the contrary, the other groups were not able to effectively kill MDR-AB within 24 h. When the concentration was increased to 6× MIC, MFP@AgNPs-1, MFP and tachyplesin-1 showed very similar kill efficacies (Fig. 2B). They killed MDR-AB within 0.5 h, while AgNPs took 2 h. We further used SEM to observe the morphology of MDR-AB. As shown in Fig. 2C, MFP, tachyplesin-1 and AgNPs slightly affected the cell morphology. However, MFP@AgNPs-1 completely destroyed the morphology of MDR-AB, which was even stronger than the positive control amikacin. These results were further confirmed by monitoring the fluorescence intensity of MDR-AB using PI (propidium iodide) staining. Eight minutes later, bacteria (MDR-AB and PI mixture) were treated with AgNPs, MFP, tachyplesin-1, MFP@AgNPs-1 and amikacin, respectively. The results are shown in Fig. 2D. Before treatment, the fluorescence intensity of each group showed no differences. After treatment, the fluorescence intensity increased significantly in MFP@AgNPs-1 and tachyplesin-1 groups. The increasing fluorescence intensity suggested that PI combined with DNA, indicating that the cell wall and cell membrane of MDR-AB were destroyed. On the contrary, the fluorescence intensity only increased slightly in the AgNPs and MFP groups. The constant fluorescence intensity in the amikacin group was attributed to the strong interaction between amikacin and nuclear DNA, which weakened the interaction between PI molecules and DNA molecules.47,48
 | ||
| Fig. 2 Time-kill efficacy and antibacterial mechanism against MDR-AB. Time-kill kinetic assay at (A) 3× MIC and (B) 6× MIC. (C) Morphology changes after treatment. (D) The integrity of the cell membrane by monitoring the fluorescence intensity of PI overtime. | ||
3.3 In vivo antibacterial activity
We evaluated the antibacterial activity of MFP@AgNPs-1 in vivo using MDR-AB-infected mice models. Before administration, we confirmed the good biocompatibility of MFP@AgNPs-1 with a cytotoxicity assay (Fig. S2†) and hemolysis assay (Fig. S3†). As shown in Fig. S3,† the hemolytic rate of MFP@AgNPs-1 increased to 8% at the concentration of 256 μg mL−1. However, the therapeutic concentration of MFP@AgNPs-1 was only 0.1 mg per kg per day (blood concentration: ∼20 μg mL−1). Therefore, the hemolysis assay was not determined at higher concentrations. The infected mice were treated with PBS, AgNPs, MFP, tachyplesin-1, MFP@AgNPs-1 and amikacin, respectively. The treatment started on the fifth day and lasted for 3 days. The weights of the mice decreased after the treatment with AgNPs, MFP, tachyplesin-1 and amikacin. The constant weight and temperature of the mice in the MFP@AgNPs-1 group indicated the good biocompatibility of MFP@AgNPs-1 (Fig. S4†). Besides, 100% of mice survived after treatment with MFP@AgNPs-1 (Fig. S5†). To better understand the antibacterial therapy, we executed all the mice and analyzed MDR-AB distribution in blood and different organs after treatment. MDR-AB occurred in the liver, lungs and heart of the infected mice (Fig. 3A). After treatment, the number of MDR-AB significantly decreased in these organs (AgNPs vs. MFP@AgNPs-1, p < 0.001). The neutrophil-to-lymphocyte ratio (NLR) and neutrophils (NEUT) are often used to evaluate systemic inflammation and infection.49,50 Compared to the model group, both NLR and NEUT increased after therapy (Fig. 3B and C). The numbers of NLR and NEUT in MFP@AgNPs-1 and amikacin groups were equivalent to the control group, indicating the best recovery of these mice.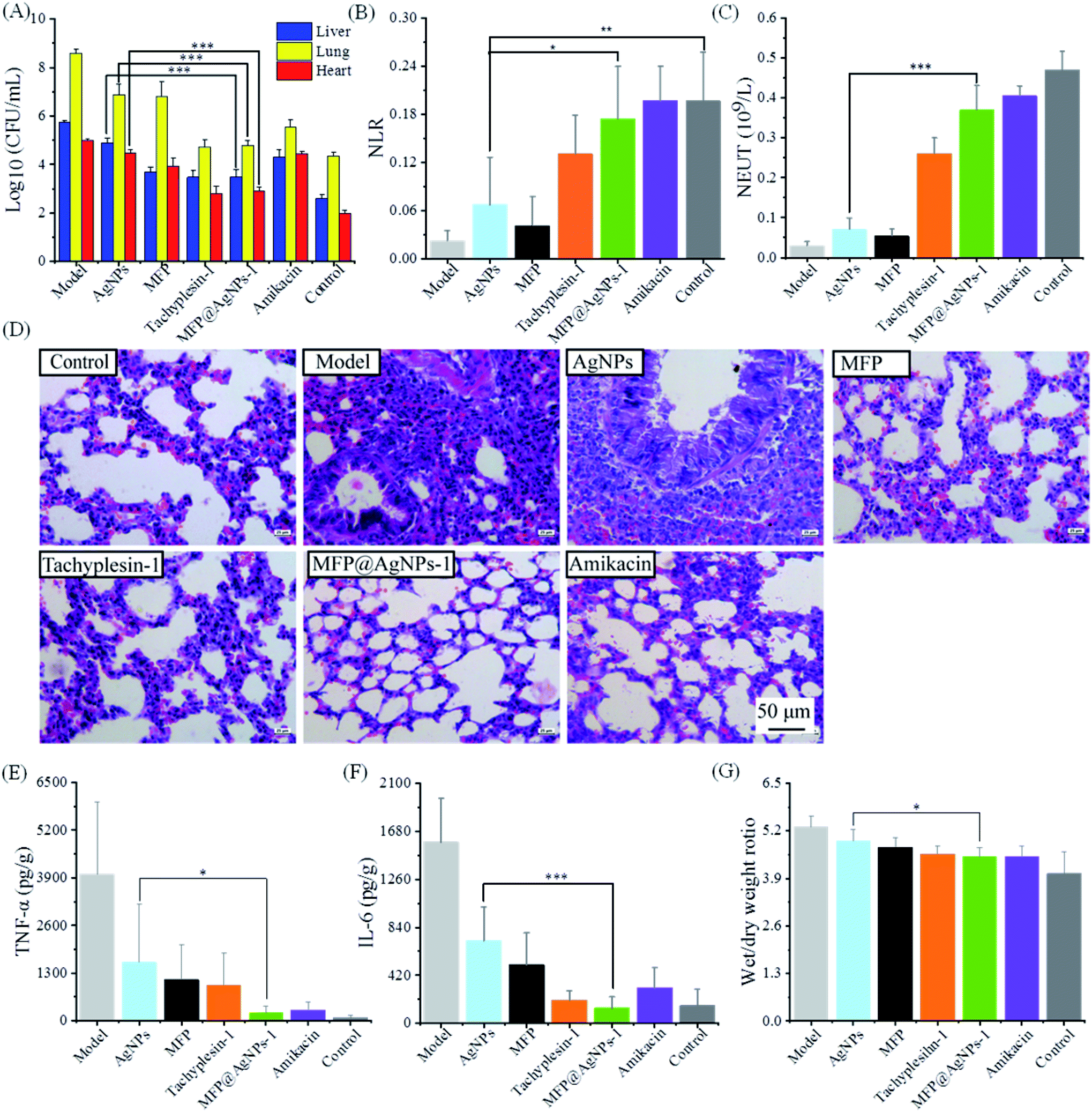 | ||
| Fig. 3 Antibacterial activity in vivo. (A) Bacterial distribution in the liver, lung and heart; (B) NLR; (C) number of NEUT. (D) Representative images of the histological analysis of the lung tissues. (E) Levels of IL-6 in lung homogenate. (F) Levels of TNF-αin lung homogenate, (G) lung edema assessment. | ||
We next investigated the morphology of the lung tissues. As shown in Fig. 3D, MDR-AB-infected mice showed obvious pathological changes with alveolar edema, swelling capillaries and narrow bronchi.51 After therapy with MFP, tachyplesin-1 MFP@AgNPs-1 and amikacin, the structure of the lung tissues improved with reduced inflammation. However, inflammatory cell infiltration was still observed in the AgNPs group (Fig. 3D). Inflammatory stimulus can increase the Interleukin-6 (IL-6) concentrations and tumor necrosis factor-a (TNF-a).52 To further evaluate the therapy of each group, we determined the concentrations of IL-6 and TNF-α in lung tissues. As shown in Fig. 3E and F, the concentrations of IL-6 and TNF-α decreased after treatment, indicating reduced inflammation. Compared to the AgNPs group and MFP group, the MFP@AgNPs-1 group showed better activity with anti-inflammation therapy. Pulmonary edema also improved after the antibacterial therapy with tachyplesin-1, MFP@AgNPs-1 and amikacin (Fig. 3G).
4. Conclusion
We have successfully developed a designed multifunctional peptide-coated AgNPs for antibacterial study. The bioactivity of the designed MFP was maintained after binding to the particle surface. Compared to the bare AgNPs, MFP@AgNPs exhibited excellent antibacterial activity against MDR-AB, which was further confirmed by the in vivo study on MDR-AB-infected mice. The developed MFP@AgNPs provides a possible solution against multidrug-resistant bacterial infections.Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
This work was supported by Henan Technology Innovation Guidance project (Industry-University Research Collaboration, No. 182107000051).References
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini and W. H. O. P. P. L. W. Group, Lancet Infect. Dis., 2018, 18, 318–327 CrossRef.
- C. L. Ventola, P T, 2015, 40, 277–283 Search PubMed.
- M. N. Ragheb, M. K. Thomason, C. Hsu, P. Nugent, J. Gage, A. N. Samadpour, A. Kariisa, C. N. Merrikh, S. I. Miller, D. R. Sherman and H. Merrikh, Mol. Cell, 2019, 73, 157–165 e155 CrossRef CAS.
- H. C. Neu, Science, 1992, 257, 1064–1073 CrossRef CAS.
- K. R. Raghupathi, R. T. Koodali and A. C. Manna, Langmuir, 2011, 27, 4020–4028 CrossRef CAS.
- A. Sirelkhatim, S. Mahmud, A. Seeni, N. H. M. Kaus, L. C. Ann, S. K. M. Bakhori, H. Hasan and D. Mohamad, Nano-Micro Lett., 2015, 7, 219–242 CrossRef CAS.
- L. Qi, Z. Xu, X. Jiang, C. Hu and X. Zou, Carbohydr. Res., 2004, 339, 2693–2700 CrossRef CAS.
- A. Anitha, V. V. D. Rani, R. Krishna, V. Sreeja, N. Selvamurugan, S. V. Nair, H. Tamura and R. Jayakumar, Carbohydr. Polym., 2009, 78, 672–677 CrossRef CAS.
- C. N. Lok, C. M. Ho, R. Chen, Q. Y. He, W. Y. Yu, H. Sun, P. K. Tam, J. F. Chiu and C. M. Che, J. Biol. Inorg Chem., 2007, 12, 527–534 CrossRef CAS.
- N. Duran, M. Duran, M. B. de Jesus, A. B. Seabra, W. J. Favaro and G. Nakazato, Nanomedicine, 2016, 12, 789–799 CrossRef CAS.
- H. W. Ji, H. J. Sun and X. G. Qu, Adv. Drug Delivery Rev., 2016, 105, 176–189 CrossRef CAS.
- D. W. Wei, W. Y. Sun, W. P. Qian, Y. Z. Ye and X. Y. Ma, Carbohydr. Res., 2009, 344, 2375–2382 CrossRef CAS.
- D. Pathania, M. Kumari and V. K. Gupta, Mater. Des., 2015, 87, 1056–1064 CrossRef CAS.
- A. Panacek, L. Kvitek, M. Smekalova, R. Vecerova, M. Kolar, M. Roderova, F. Dycka, M. Sebela, R. Prucek, O. Tomanec and R. Zboril, Nat. Nanotechnol., 2018, 13, 65–71 CrossRef CAS.
- C. Recordati, M. De Maglie, S. Bianchessi, S. Argentiere, C. Cella, S. Mattiello, F. Cubadda, F. Aureli, M. D'Amato, A. Raggi, C. Lenardi, P. Milani and E. Scanziani, Part. Fibre Toxicol., 2016, 13, 12 CrossRef.
- A. Ravindran, P. Chandran and S. S. Khan, Colloids Surf. B Biointerfaces, 2013, 105, 342–352 CrossRef CAS.
- H. G. Boman, J. Intern. Med., 2003, 254, 197–215 CrossRef CAS.
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS.
- S. Braber, S. A. Overbeek, P. J. Koelink, P. A. J. Henricks, G. J. R. Zaman, J. Garssen, A. D. Kraneveld and G. Folkerts, Eur. J. Pharmacol., 2011, 668, 443–449 CrossRef CAS.
- R. J. Snelgrove, Eur. J. Pharmacol., 2011, 667, 1–5 CrossRef CAS.
- W.-H. Chen, G.-F. Luo, Q. Lei, H.-Z. Jia, S. Hong, Q.-R. Wang, R.-X. Zhuo and X.-Z. Zhang, Chem. Commun., 2015, 51, 465–468 RSC.
- Y. Chau, R. F. Padera, N. M. Dang and R. Langer, Int. J. Cancer, 2006, 118, 1519–1526 CrossRef CAS.
- F. E. T. Ying Chau and R. Langer, Bioconjugate Chem., 2004, 15, 931–941 CrossRef.
- I. A. Edwards, A. G. Elliott, A. M. Kavanagh, M. A. T. Blaskovich and M. A. Cooper, ACS Infect. Dis., 2017, 3, 917–926 CrossRef CAS.
- I. A. Edwards, A. G. Elliott, A. M. Kavanagh, J. Zuegg, M. A. Blaskovich and M. A. Cooper, ACS Infect. Dis., 2016, 2, 442–450 CrossRef CAS.
- I. A. Edwards, A. G. Elliott, A. M. Kavanagh, M. A. T. Blaskovich and M. A. Cooper, ACS Infect. Dis., 2017, 3, 917–926 CrossRef CAS.
- V. Teixeira, M. J. Feio and M. Bastos, Prog. Lipid Res., 2012, 51, 149–177 CrossRef CAS.
- H. Hatakeyama, H. Akita and H. Harashima, Adv. Drug Deliv. Rev., 2011, 63, 152–160 CrossRef CAS.
- I. Wiegand, K. Hilpert and R. E. Hancock, Nat. Protoc., 2008, 3, 163–175 CrossRef CAS.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schaberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS.
- D. Knafl, S. Tobudic, S. C. Cheng, D. R. Bellamy and F. Thalhammer, Eur. J. Clin. Microbiol. Infect. Dis., 2016, 36, 677–680 CrossRef.
- P. Stiefel, S. Schmidt-Emrich, K. Maniura-Weber and Q. Ren, BMC Microbiol., 2015, 15, 36 CrossRef.
- A. C. Jacobs, M. G. Thompson, C. C. Black, J. L. Kessler, L. P. Clark, C. N. McQueary, H. Y. Gancz, B. W. Corey, J. K. Moon, Y. Si, M. T. Owen, J. D. Hallock, Y. I. Kwak, A. Summers, C. Z. Li, D. A. Rasko, W. F. Penwell, C. L. Honnold, M. C. Wise, P. E. Waterman, E. P. Lesho, R. L. Stewart, L. A. Actis, T. J. Palys, D. W. Craft and D. V. Zurawski, mBio, 2014, 5, e01076-01014 CrossRef.
- D. B. DeNicola, Top. Companion Anim. Med., 2011, 26, 52–61 CrossRef.
- P. Froom, A. Diab and M. Barak, Am. J. Clin. Pathol., 2013, 140, 828–830 CrossRef.
- R. D. Cardiff, C. H. Miller and R. J. Munn, Cold Spring Harbor protocols, 2014, 2014, 655–658 Search PubMed.
- M. R. Das, R. K. Sarma, S. Borah, R. Kumari, R. Saikia, A. B. Deshmukh, M. V. Shelke, P. Sengupta, S. Szunerits and R. Boukherroub, Colloids Surf. B Biointerfaces, 2013, 105, 128–136 CrossRef CAS.
- M. Sastry, K. S. Mayya and K. Bandyopadhyay, Colloid. Surface. Physicochem. Eng. Aspect., 1997, 127, 221–228 CrossRef CAS.
- Z. Khan, S. A. Al-Thabaiti, A. Y. Obaid and A. O. Al-Youbi, Colloids Surf., B, 2011, 82, 513–517 CrossRef CAS.
- M. Arakha, S. M. Borah, M. Saleem, A. N. Jha and S. Jha, Free Radicals Biol. Med., 2016, 101, 434–445 CrossRef CAS.
- G. I. N. Waterhouse, G. A. Bowmaker and J. B. Metson, Phys. Chem. Chem. Phys., 2001, 3, 3838–3845 RSC.
- L. Gharibshahi, E. Saion, E. Gharibshahi, A. H. Shaari and K. A. Matori, Materials, 2017, 10, 402 CrossRef.
- K. Bakshi, M. R. Liyanage, D. B. Volkin and C. R. Middaugh, Methods Mol. Biol., 2014, 1088, 255–269 CrossRef CAS.
- M. D. Adhikari, G. Das and A. Ramesh, Chem. Commun., 2012, 48, 8928–8930 RSC.
- E. Pazos-Ortiz, J. H. Roque-Ruiz, E. A. Hinojos-Márquez, J. López-Esparza, A. Donohué-Cornejo, J. C. Cuevas-González, L. F. Espinosa-Cristóbal and S. Y. Reyes-López, J. Nanomater., 2017, 2017, 4752314 Search PubMed.
- A. Abbaszadegan, Y. Ghahramani, A. Gholami, B. Hemmateenejad, S. Dorostkar, M. Nabavizadeh and H. Sharghi, J. Nanomater., 2015, 2015, 720654 Search PubMed.
- M. S. Ramirez and M. E. Tolmasky, Molecules, 2017, 22, 2267 CrossRef.
- X. Long, Q. Miao, S. Bi, D. Li, C. Zhang and H. Zhao, Talanta, 2004, 64, 366–372 CrossRef CAS.
- O. Kartal and A. T. Kartal, Bratisl. Lek. Listy, 2017, 118, 513–516 CAS.
- Y. Huang, A. Liu, L. Liang, J. Jiang, H. Luo, W. Deng, G. Lin, M. Wu, T. Li and Y. Jiang, Int. Immunopharm., 2018, 64, 10–15 CrossRef CAS.
- K. Dietert, B. Gutbier, S. M. Wienhold, K. Reppe, X. Jiang, L. Yao, C. Chaput, J. Naujoks, M. Brack, A. Kupke, C. Peteranderl, S. Becker, C. von Lachner, N. Baal, H. Slevogt, A. C. Hocke, M. Witzenrath, B. Opitz, S. Herold, H. Hackstein, L. E. Sander, N. Suttorp and A. D. Gruber, PloS One, 2017, 12, e0188251 CrossRef.
- M. R. Bacci, R. C. Leme, N. P. Zing, N. Murad, F. Adami, P. F. Hinnig, D. Feder, A. C. Chagas and F. L. Fonseca, Braz. J. Med. Biol. Res., 2015, 48, 427–432 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05640e |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2020 |