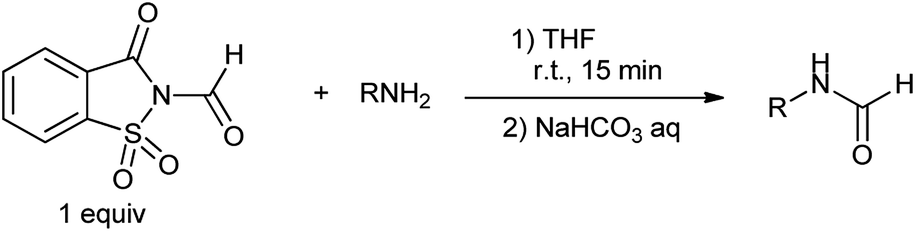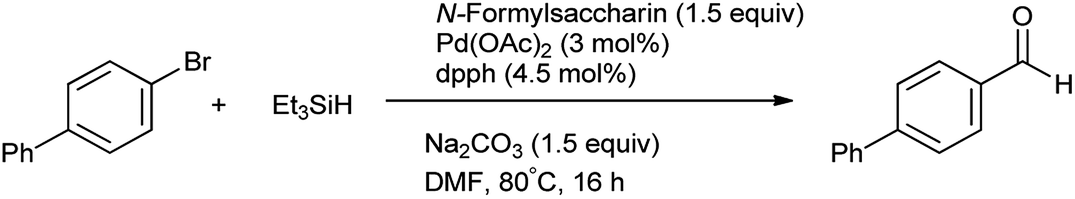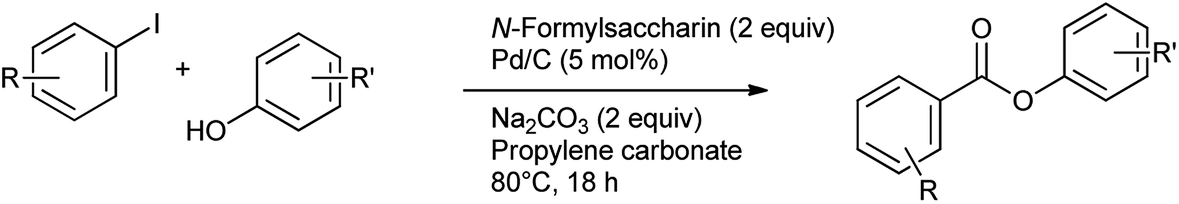DOI:
10.1039/D0RA05974A
(Review Article)
RSC Adv., 2020,
10, 36571-36608
Artificial sugar saccharin and its derivatives: role as a catalyst
Received
8th July 2020
, Accepted 24th September 2020
First published on 6th October 2020
Abstract
The primary objective of this review was to demonstrate the significance of artificial sugar saccharin and its derivatives as catalysts for a wide variety of organic transformations. The application of saccharin and its derivatives represents a greener and superior catalytic approach for reactions. In particular, we were interested in bringing together the literature pertaining to these saccharin derivatives from a catalysis perspective. The present review reports synthesis of saccharin and its derivatives such as saccharin-N-sulfonic acid, sodium saccharin, N-halo saccharin, saccharin lithium-bromide, N-formyl saccharin, N-acyl saccharin, N-nitrosaccharin, N-SCF3 saccharin, N-fluorosultam, N-phenylselenosaccharin, N-thiocyanatosaccharin palladium saccharin, DMAP–saccharin, and [Bmim]Sac. This catalytic application of saccharin and its derivatives includes reactions such as the Biginelli reaction, Paal–Knorr pyrrole synthesis, azo-coupling reaction, halogenations, domino Knoevenagel, Michael, deoximation reaction, catalytic condensation, functional group protection and oxidation etc. Also, these saccharin derivatives act as a source of CO, NH2, SCN, SCF3 and nitro groups. We reported all the available data on saccharin and its derivatives acting as a catalyst from 1957 to date.
 Kamalpreet Kaur | Kamalpreet Kaur is a Project Assistant in National Institute of Technology, Delhi, India under Dr Suman Srivastava Assistant Professor, NIT, Delhi. She received her Master of Science degree in Chemistry under the supervision of Dr Sonia Kaushal Assistant Chemist, Punjab Agricultural University, Ludhiana, India in 2019. |
 Suman Srivastava | Dr Suman Srivastava studied chemistry at Banaras Hindu University (Varanasi), where she got her Master's degree in chemistry (2006). She completed her Ph.D. thesis in 2013 under the supervision of Professor Ranjana S. Khanna (Banaras Hindu University) and co-supervision of Dr Atul Kumar (CSIR-Central Drug Research Institute). She went to Indian Institute of Technology Mandi as a postdoc, and then she started her independent research as a DST Inspire faculty at National Institute of Technology Delhi. Her research interests include Development of green methodology for bioactive heterocycle synthesis. |
1. Introduction
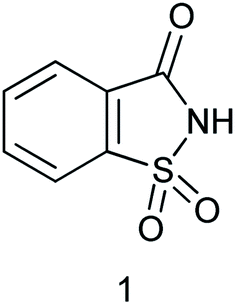
The design and application of new catalysts and catalytic systems are simultaneously achieving the dual goals of green chemistry. Catalysis offers numerous green chemistry benefits including lower energy requirements, achieving reaction faster, increased selectivity, and decreased use of processing and separation agents. The development of efficient green catalysts for the synthesis of various bioactive moieties has become a major focus for researchers. Developing green chemistry methodologies is a challenge that may be viewed through the framework of the “Twelve Principles of Green Chemistry”. These principles identify catalysis as one of the most important tools for implementing green chemistry. Natural sugars and unnatural sweeteners are becoming employed as reagents and catalysts in organic functional group transformations. Recently, β-cyclodextrin, a cyclic oligosaccharide containing seven glucose units in the molecule has been introduced as a catalyst for the synthesis of tryptanthrin.1 Saccharin is a non-nutritive sweetener2 and it is 300 times sweeter than the sucrose.3 Saccharin (1,2-benzisothiazol-3(2H)-one-1,1-dioxide) was discovered in 1878 by Remson and Fahlberg.4 Saccharin is a Brønsted acid with moderate acidity (pKa 2.32). This inexpensive, edible, and benign chemical has been used as a catalyst for several organic functional group transformations.
Taking wide application of saccharin and its derivatives for use as catalyst and in continuation of our ongoing research towards development of novel and green reaction media,5 we have summarized the catalytic applicability of saccharin and its derivatives on various organic transformations. This review article highlights studies of saccharin and its derivatives as a catalyst for synthesis of a diverse range of a broad area of molecules. A review published by Banerjee et al. in 2019 (ref. 6) which includes saccharin and its derivatives such as saccharin sulfonic acid, sodium saccharin, tetrazole-amino-saccharin and 4-N,N-dimethylaminopyridine (DMAP)–saccharin as catalyst only. We have covered every derivatives of saccharin as catalyst discovered so far. The present review reports synthesis of saccharin and its derivatives such as saccharin-N-sulfonic acid, sodium saccharin, N-halo saccharin, saccharin lithium-bromide, N-formyl saccharin, N-acyl saccharin, N-ntrosaccharin, N-SCF3 saccharin, N-fluorosultam, N-phenylselenosaccharin, N-thiocyanatosaccharin, palladium saccharin, DMAP–saccharin, tetrazole-amino-saccharin and [Bmim]Sac. Along with these, this review also reports references where saccharin derivative acts as a source of CO, NH2, SCN, SCF3 and nitro group.
2. Saccharin as catalyst
2.1. Quinoxalines and pyrido[2,3-b]pyrazines synthesis
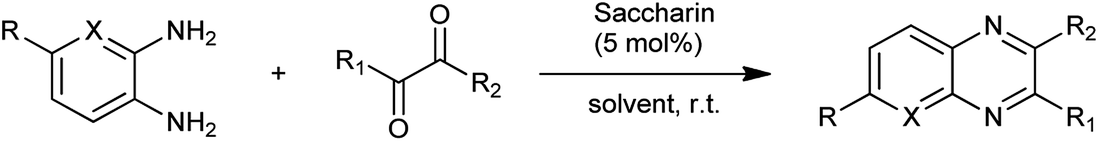
Organic compounds which contain nitrogen heterocyclic rings are important compounds in medicinal chemistry as quinoxalines are polynitrogen heterocyclic compounds possessing diverse range of biological activities and used as therapeutics.7 The most common synthetic pathway to reach these scaffolds is the condensation of 1,2-diketones with 1,2-arylenediamines in refluxing ethanol or acetic acid.8 These processes suffer from several limitations such as high cost, pollution, long reaction time, waste treatment, and poor chemical yields. Lassagne et al. evaluated the saccharin as a organocatalyst for synthesis of pyrido[2,3-b]pyrazines and quinoxalines through cyclocondensation of various 1,2-arylenediamines with different 1,2-dicarbonyl compounds at room temperature shown in Scheme 1.9
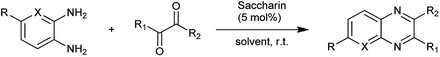 |
| | Scheme 1 Quinoxalines and pyrido[2,3-b]pyrazines synthesis. | |
It was found that it is effective method to synthesize quinoxalines with 97% of yield and saccharin as a organocatalyst presents advantages such as reusability, availability easy to handle, absence of toxicity, environmental acceptability with mild reaction conditions, and short reaction times.
2.2. Paal–Knorr pyrroles synthesis
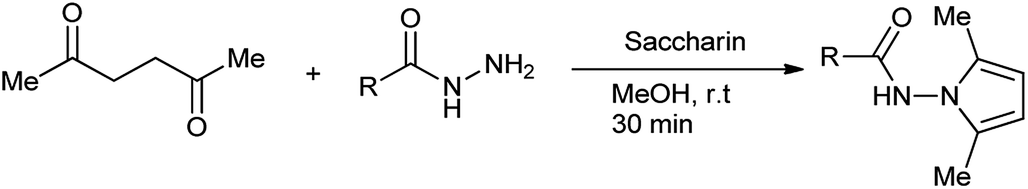
Pyrrole ring is core unit of important naturally occurring compounds, such as vitamin B12, heme, and chlorophyll.10 The ring closure reaction between amines and open chain carbonyl compounds forms compounds of a class pyrrole called Paal–Knorr pyrroles synthesis.11 Bhandari et al. synthesized N-substituted 2,5-dimethylpyrroles by using saccharin as a green catalyst (Scheme 2).12
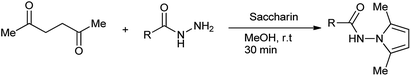 |
| | Scheme 2 Paal–Knorr pyrroles synthesis. | |
The method proposed is effective, proficient and ecological safe with 86% of yield of desired product.
2.3. Multicomponent synthesis of nitrogen heterocycles
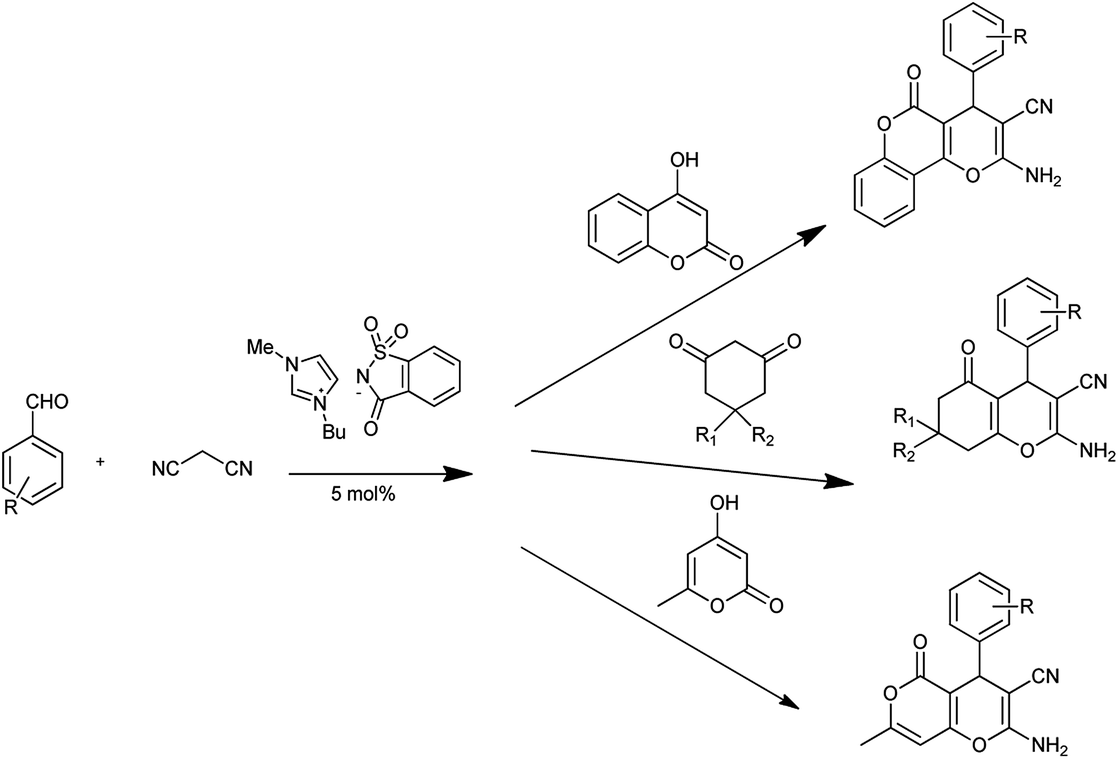
Recently, substantial researches have been accomplished on synthesis of bioactive nitrogen heterocycles using multi-component reaction.13 Mohamadpour et al. reported that saccharin act as green and effective catalyst for one pot multicomponent synthesis of substituted dihydro-2-oxypyrrole, 1H-pyrazolo[1,2-b]phthalazine-5,10-dione and 3,4-dihydropyrimidin-2-(1H)-one derivatives as shown in Schemes 3–5 respectively.14
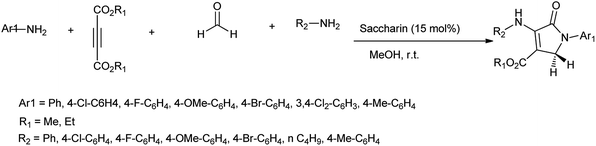 |
| | Scheme 3 Synthesis of dihydro-2-oxypyrrole. | |
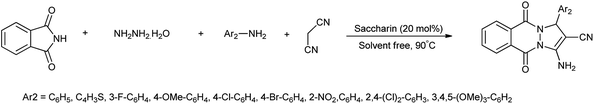 |
| | Scheme 4 Synthesis of 1H-pyrazolo[1,2-b]phthalazine-5,10-dione. | |
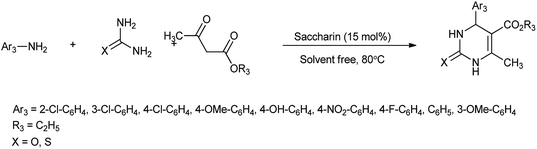 |
| | Scheme 5 Synthesis of 3,4-dihydropyrimidin-2-(1H)-one. | |
Saccharin has important benefits such as non-toxic, eco-friendly, green, high efficiently, environmentally benign nature, high catalytic activity, and low cost.
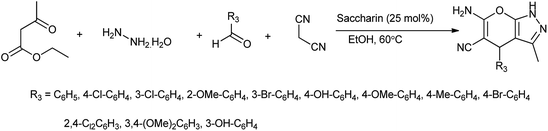
In another attempt they have developed the procedure for the one pot multi-component synthesis of pyrano[2,3-d]pyrimidinone, dihydropyrano[2,3-c]pyrazole and tetrahydrobenzo[b]pyran scaffolds by saccharin catalyzed Knoevenagel cyclocondensation reaction shown in Schemes 6–8 respectively15 where the desired product were produced with excellent yield of 86%.
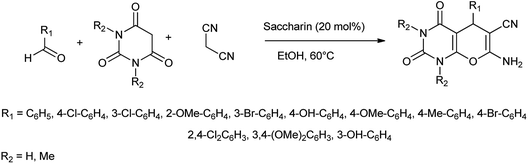 |
| | Scheme 6 Synthesis of pyrano[2,3-d]pyrimidinone scaffolds. | |
 |
| | Scheme 7 Synthesis of dihydropyrano[2,3-c]pyrazole. | |
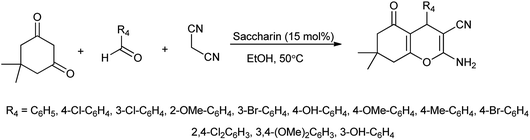 |
| | Scheme 8 Synthesis of tetrahydrobenzo[b]pyran. | |
This method found to be convenient and green and overcome the several disadvantages such as long reaction times, low yields, tedious workup, harsh conditions and requirement of excess amounts of catalyst and reagents.
2.4. Azo coupling reaction
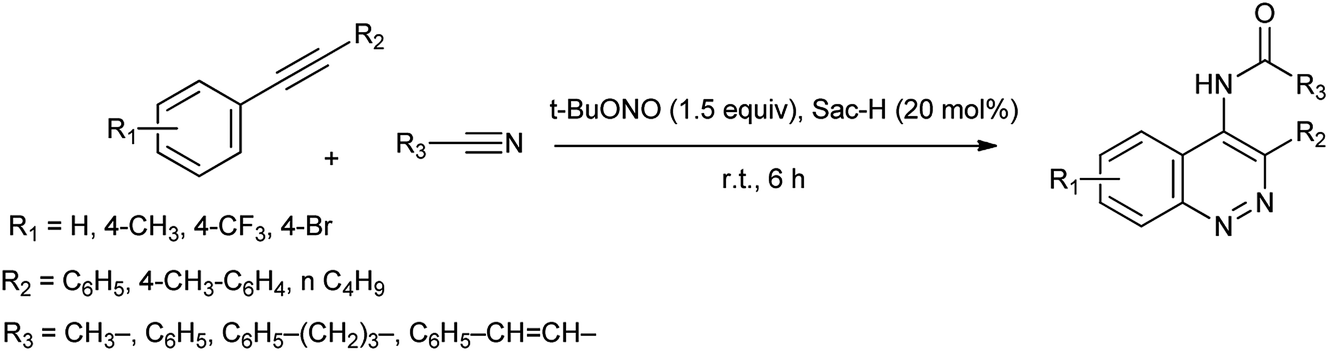
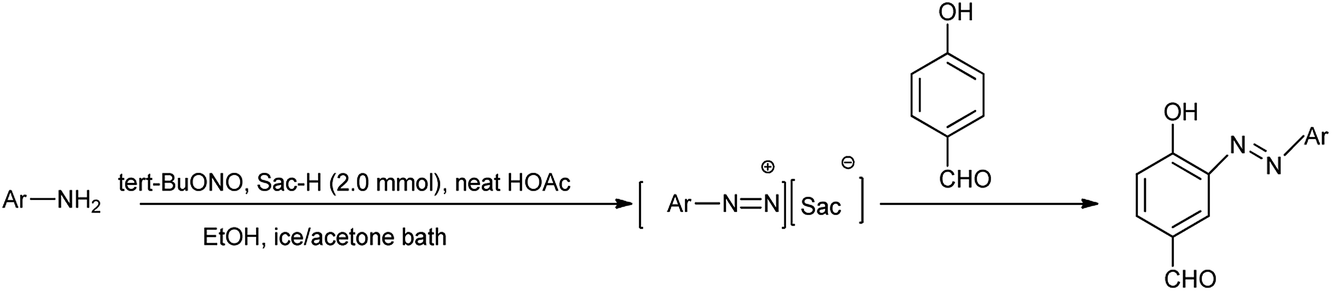
The azo coupling reaction is an important procedure for the synthesis of azo compounds because the variety of the coupling and diazo components can be used and prodigious range of viable azo compounds can be synthesized.16 Cinnolines and their derivatives are a great important aza-heterocycles of pharmaceutical cores17 and also have been widely utilized in organic synthesis as well as they displayed optical and luminescent properties.18 By becoming more concerned, Khaligh et al. reported one-pot synthesis of 4-amidocinnolines by using saccharin as a cheap and ecofriendly organocatalyst under halogen and metal free condition shown in Scheme 9.19
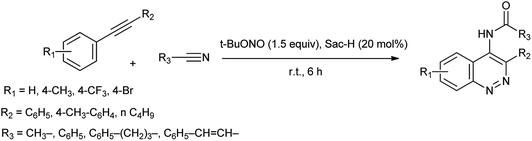 |
| | Scheme 9 Synthesis of 4-amidocinnoline derivatives. | |
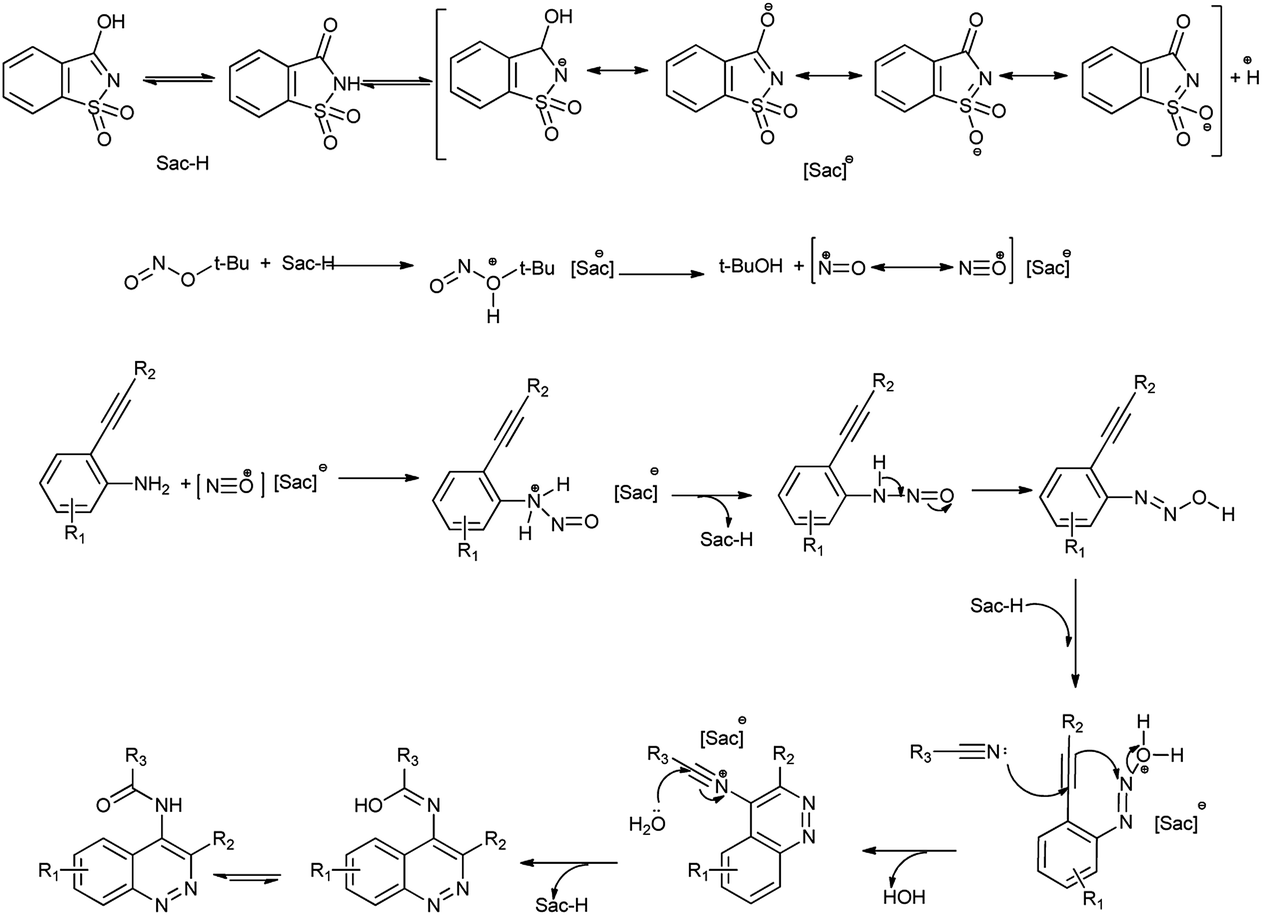
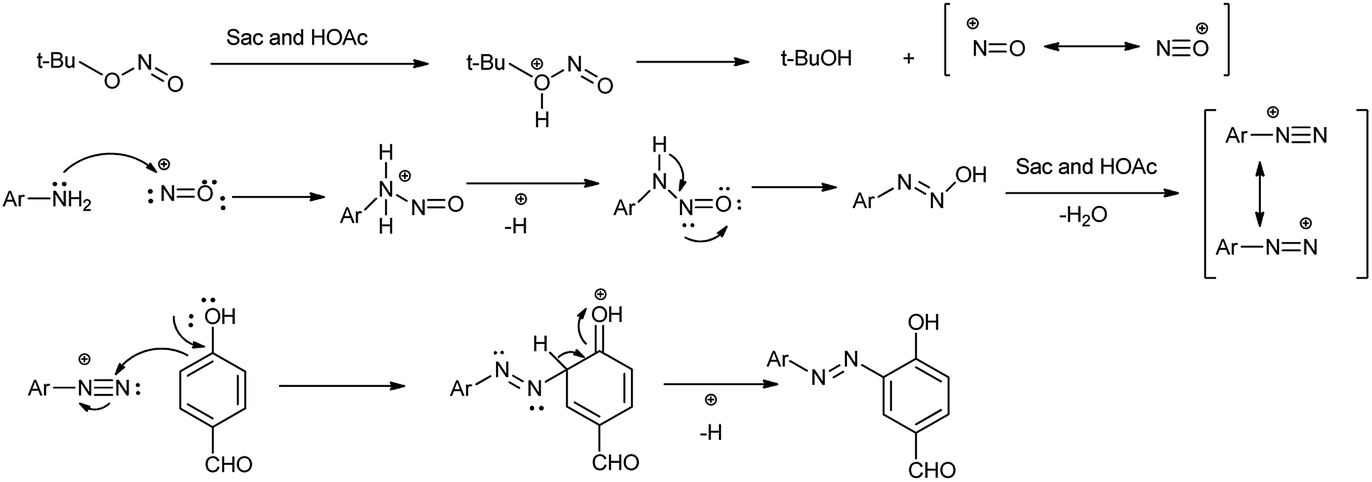
This method has advantages such as reduced waste, cost-effectiveness, simple experimental and environmentally benign procedure, and good yield of the products. Further the proposed mechanism is shown in Scheme 10 which showed that mild electrophile nitroso was formed from saccharin and tert-butyl nitrite.
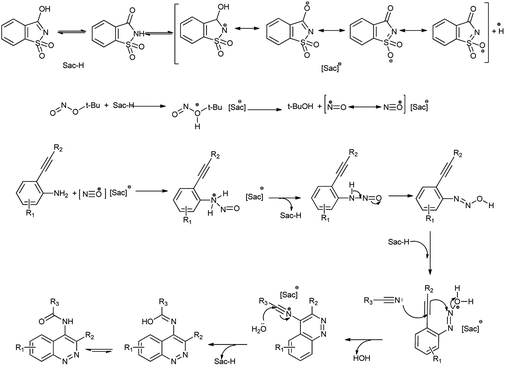 |
| | Scheme 10 Proposed mechanism for the synthesis of 4-amidocinnolines derivatives. | |
Amine nitrogen of aryl amine reacts with nitroso electrophile to form o-alkynylphenyldiazonium saccharinate. Then simultaneously the nitrile addition to o-alkylphenyldiazonium and cyclization followed by hydrolysis gives the desired 4-amidocinnoline and saccharin return back to the reaction cycle.
Similarly, they also proposed a procedure for cost effective, simple and energy efficient synthesis procedure for the synthesis of 1,2,3-benzotriazine-4-(3H)-ones by using saccharin as catalyst and tert-butyl nitrite as a diazotization reagent (Scheme 11).20
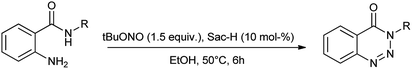 |
| | Scheme 11 Synthesis of 1,2,3-benzotriazine-4-(3H)-ones using saccharin as catalyst. | |
Here saccharin is described as safe and cheap organocatalyst for one-pot synthesis under mild reaction condition. Khaligh et al. further showed the synthesis of azo dyes by using saccharin as a catalyst via telescope dediazotization (Scheme 12). Synthesis of azo compounds was developed by using tert-butyl nitrite and a mixture of saccharin and glacial acetic acid in ethanol mixed solvent followed by the azo coupling reaction with 4-hydroxybenzaldehyde.
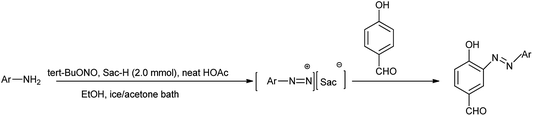 |
| | Scheme 12 Synthesis of 4-hydroxy-3-[(E)-aryldiazenyl]-benzaldehyde derivatives via stable arene diazonium saccharinates. | |
Arenediazonium saccharinates intermediates were used in the azo coupling reaction with 4-hydroxybenzaldehyde via telescoped dediazotization. Arenediazonium saccharinates obtained as stable intermediate can be stored for longer time and can be used again without any loss of activity (Scheme 13).21 The current method has advantages such as reduced waste by avoiding solvent for the purification of intermediate in diazotization step, cost-effectiveness, simple experimental procedure, good yield of azo dyes, metal-free waste, and environmentally benign conditions. An interesting aspect of this study is the recovery of saccharin from the reaction, which could be reused.
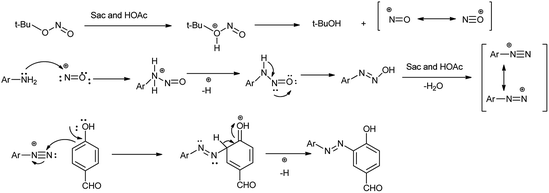 |
| | Scheme 13 Mechanism of diazotization process. | |
3. Saccharin-N-sulfonic acid
3.1. Synthesis of saccharin-N-sulfonic acid
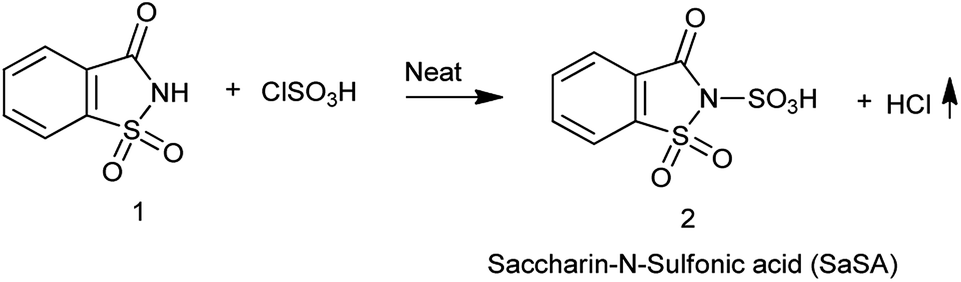
Sulfonic acid is strong acid used as sulfonating agent since ages. Though it is vary carcinogenic and not easy to handle scientist are now moving towards ecofriendly approach as Zare et al. in 2019,22 Shirini et al. in 2009 (ref. 23) and Shirini et al. in 2010 (ref. 24) reported the procedure for the synthesis of saccharin-N-sulfonic acid (SaSA) where the neat chlorosulfonic acid was added dropwise into the flask containing the saccharin with continuous stirring and no special workup is needed as HCl is removed as gas shown in Scheme 14.
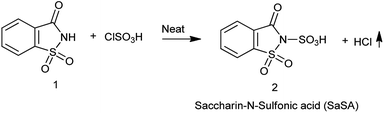 |
| | Scheme 14 Synthesis of saccharin-N-sulfonic acid (SaSA). | |
3.2. Alcohols protection
Protection and deprotection of organic functional groups are important processes during multi-step organic synthesis.25 The procedure which followed for the functional group transformations depends on its short reaction times, simplicity, low cost of the process, high yields of the desired products and ease of the work-up methods.
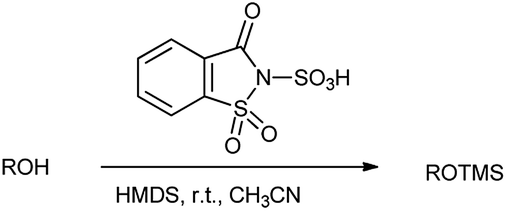
Earlier, trimethylsilylation is one of prime and approved methods for protecting the alcoholic hydroxyl group.26 Generally, the formation of silyl ether is carried out by reacting alcohols with silyl triflates and silyl chlorides in the presence of stoichiometric amounts of a base.27 However, Shirini et al. in 2009 reported the chemoselective trimethylsilylation of alcohols catalyzed by saccharin sulphonic acid with hexamethyldisilazane (HMDS) in the presence of thiols and amines as shown in Scheme 15.23 Here, the method resulted in the good product yield with 95% and also short reaction times, easy workup and reagent availability.
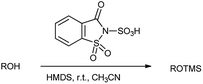 |
| | Scheme 15 Trimethylsilyation of alcohol is carried out in the presence of SaSA with HDMS. | |
3.3. Aldehyde protection
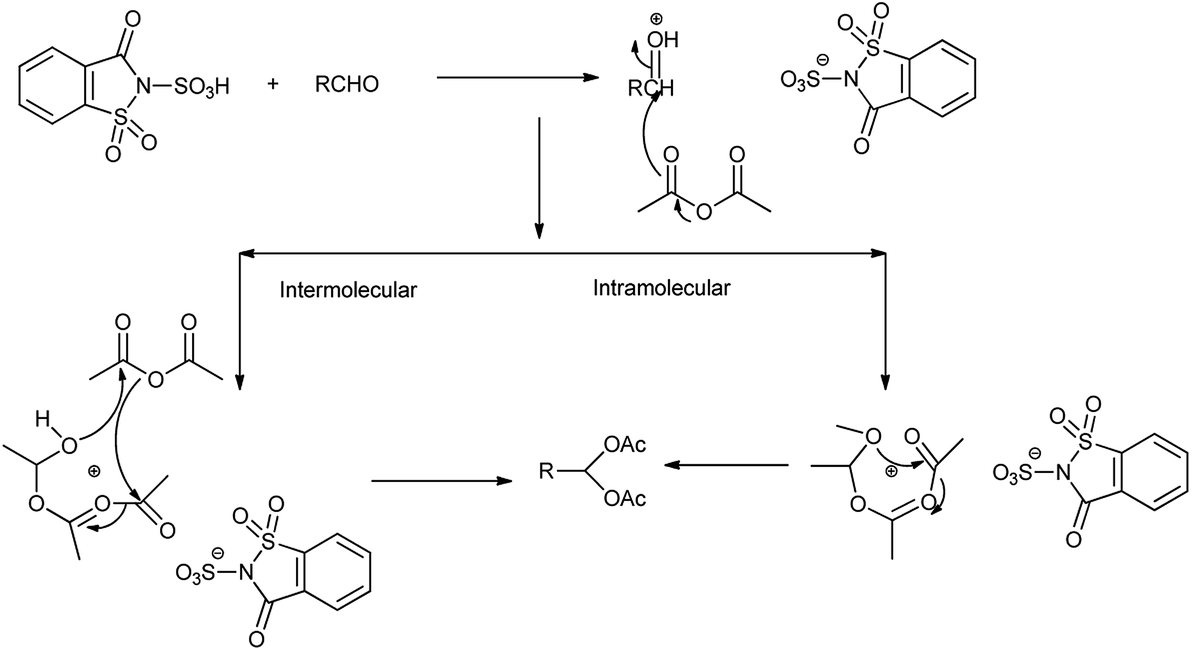
Among the several methods available for the protection of aldehydes, acylal formation is often preferred due to the ease of preparation and the stability of the produced 1,1-diacetate towards basic and neutral conditions28 but Shirini et al. 2010 showed the saccharin sulphonic acid as an effective catalyst for preparation and deprotection of 1,1-diacetates by reacting aldehyde with acetic anhydride (Scheme 16). The reaction took place by two different mechanisms shown in Scheme 17.24
 |
| | Scheme 16 Conversion of aldehyde to their corresponding 1,1-diacetate with acetic anhydride by using SaSA as catalyst. | |
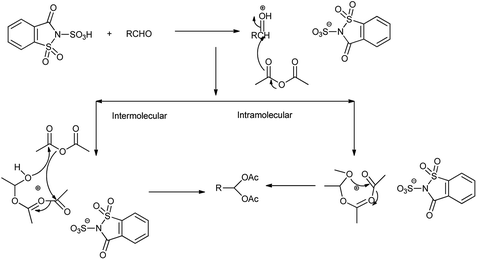 |
| | Scheme 17 Mechanism for the synthesis of 1,1-diacetate. | |
This method can be useful for the chemoselective acylation of aldehydes in the presence of ketones under mild, solvent-free reaction condition, high selectivity and yield of product followed by easy work up and further also found suitable for the regeneration of aldehydes from the related acylals in the presence of wet SiO2. This method is also very useful for the protection of aliphatic aldehydes.
3.4. Acetylation of alcohols, phenols, and amines
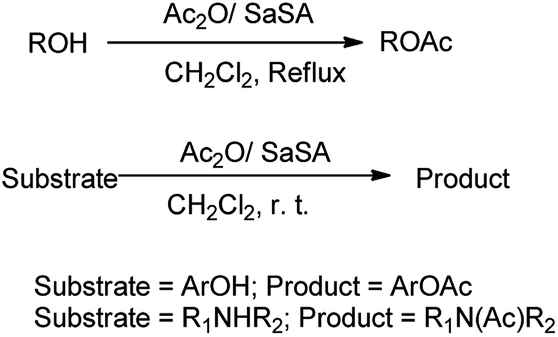
Acetylation is one of the most important methods widely used for protection of the alcoholic hydroxyl group. This method is important because of the ease of introduction of the acetyl group, the stability of the product to acidic conditions, and ease of removal of the protecting group by alkaline hydrolysis. The acetylation of alcohols, phenols, and amines with Ac2O is efficiently catalyzed in the presence of SaSA (Scheme 18).29 This method was found to be efficient due to ease of preparation, reusability and stability of reagent, high yields of the products, relatively short reaction times, and easy work-up addition to the methods available.
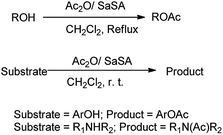 |
| | Scheme 18 Acetylation of alcohols, phenols and amines in presence of SaSA as catalyst. | |
3.5. N-tert-Butyloxycarbonyl (N-Boc) protection
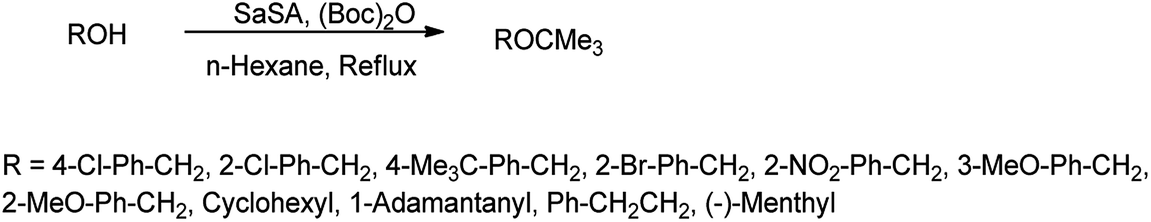
N-tert-Butyloxycarbonyl protection (Boc) has attracted much attention of many organic chemists. Di-tert-butyldicarbonate [(Boc)2O] as a stable and commercially available reagent is widely used for this purpose.30 Shirini et al. (2010) reported saccharin sulphonic acid as a catalyst for the Boc protection of amines using di-tert-butyldicarbonyl [(Boc)2O] shown in Scheme 19 with 95% yield and this catalyst was also suitable for the formation of tert-butyl ethers from the related alcohols (Scheme 20) with 90% of yield.31
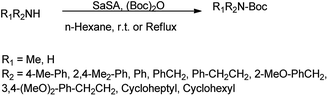 |
| | Scheme 19 Formation of N-Boc protected amines. | |
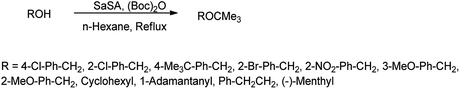 |
| | Scheme 20 Formation of tert-butyl ether by using SaSA as a catalyst. | |
This procedure found to be effective due to its high yield of product, easy work-up, and mild reaction condition. Further, this catalyst is also suitable for the synthesis of tert-butyl ethers from the related alcohols.
3.6. Multicomponent reactions
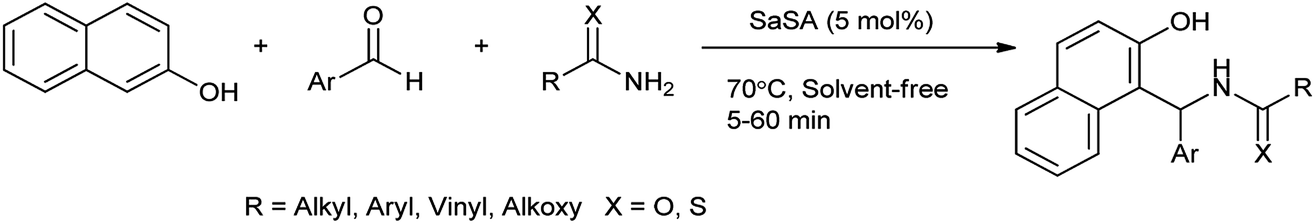
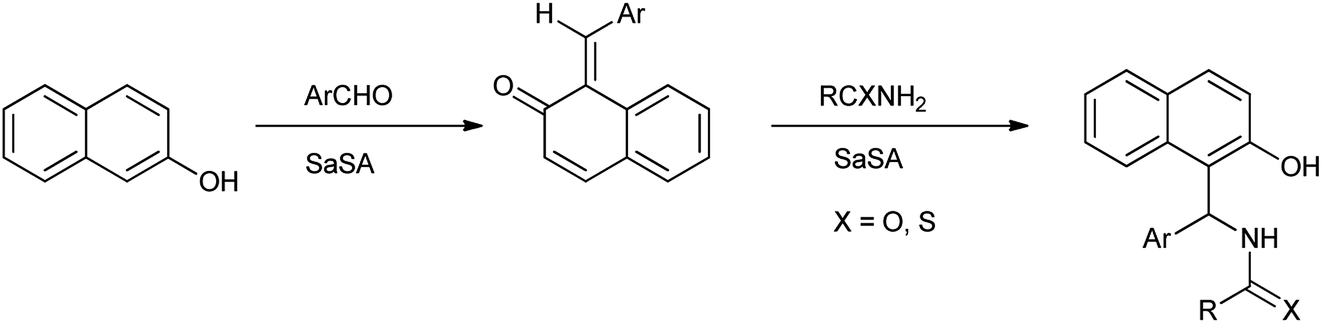
2-Naphthol derivatives are found to be valuable pharmacologic compounds possessing various important biological properties such as antiinflammatory, antibacterial, hypotensive, and bradycardia activities.32 Zare et al. (2013) determined the one-pot multicomponent synthesis of 1-carbamatoalkyl-2-naphthols, 1-amidoalkyl-2-naphthols, bis(1-amidoalkyl-2-naphthol)s, 1-thioamidoalkyl-2-naphthols and bis(1-carbamatoalkyl-2-naphthol)s by condensation reaction of 2-naphthol with amides (thioamides or alkyl carbamates) and arylaldehydes under solvent free, mild and green condition by using recyclable catalyst as saccharin sulfonic acid (Scheme 21). The mechanism followed for the formation of 2-naphthol derivatives shown in Scheme 22.33
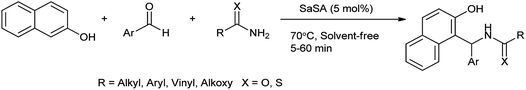 |
| | Scheme 21 The condensation of 2-naphthol with the arylaldehyde and amides. | |
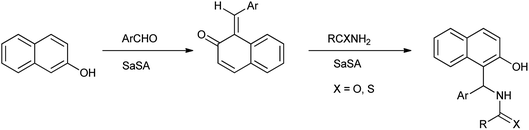 |
| | Scheme 22 The mechanism for the formation of 1-amidoalkyl-2-naphthol. | |
This procedure was inexpensive, nontoxic, recyclable, and use green catalyst; moreover it overcome all the limitations earlier such as toxic catalyst, the use of large amount of catalyst, high reaction time, low product yield, usage of an additional energy (ultrasound or microwave), harsh reaction conditions, and no specificity.
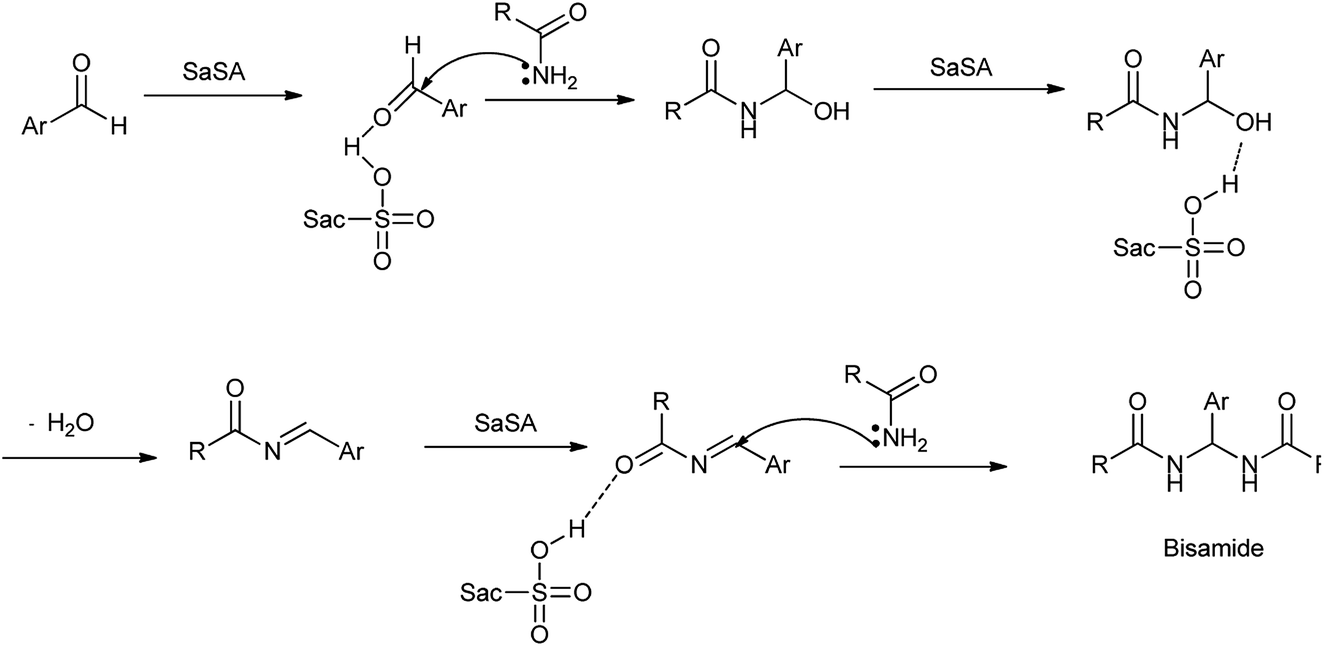
In another study by Zare et al. showed the synthesis of N,N′-alkylidene bisamides using saccharin-N-sulfonic acid as solid-acid catalyst in solvent free condition by condensation between chlorobenzaldehyde and acetamide shown in Scheme 23.22 Mechanism has been proposed in Scheme 24 showed that the acidic hydrogen of SaSA activates the aldehyde carbonyl which is followed by nucleophilic attack of the amide. Then water is removed to yield intermediate which is activated by the SaSA and second molecule of amide attacked resulted in the yield of alkylidene bisamide. This method has advantages over the reported methods such as milder condition, easy workup procedure, effectiveness, short reaction time, generality, high yields, solvent free and easy synthesis of the catalyst.
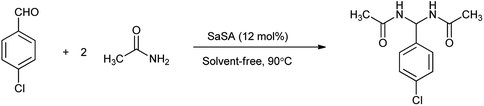 |
| | Scheme 23 Synthesis of N,N′-alkylidene bisamides. | |
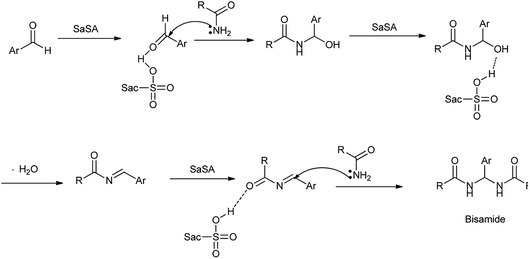 |
| | Scheme 24 The mechanism proposed for the synthesis of N,N′-alkylidiene bisamide. | |
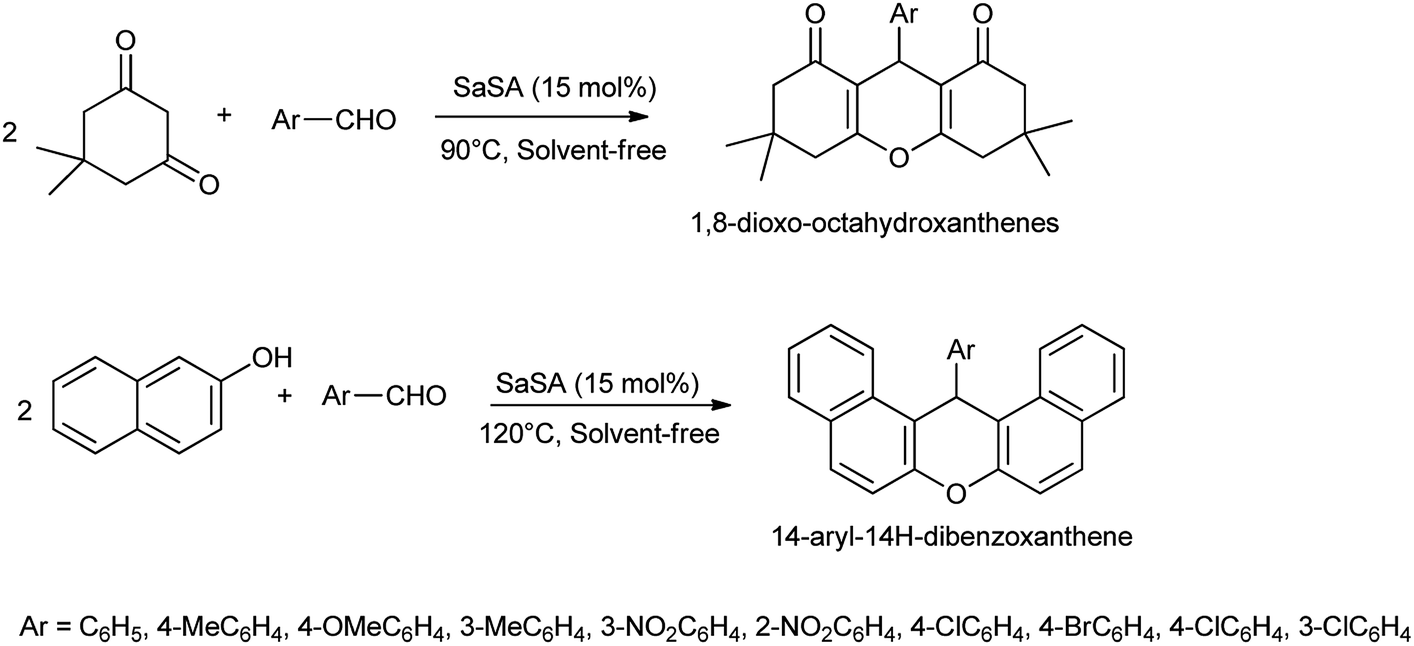
Saccharin sulfonic acid (SaSA) was also reported as effective and inexpensive catalyst for one pot multi-component solvent-free organic transformations for the synthesis of 1,8-dioxooctahydroxanthenes, and 14-aryl-14H-dibenzoxanthenes derivatives (Scheme 25)34 with 90% of yield.
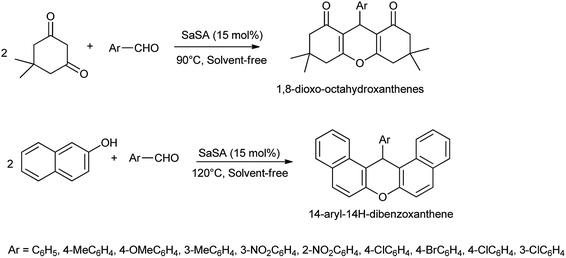 |
| | Scheme 25 Synthesis of 1,8-dioxo-octahydroxanthenes and 14-aryl-14H-dibenzoxanthene derivatives. | |
This catalyst found to be effective due to its generality, shorter reaction time, high yield, no side products, simplicity, easy work up, ease of preparation of the catalyst, and compliance with the green chemistry protocols.
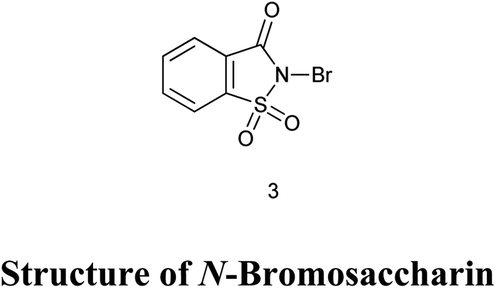
4. N-Bromosaccharin
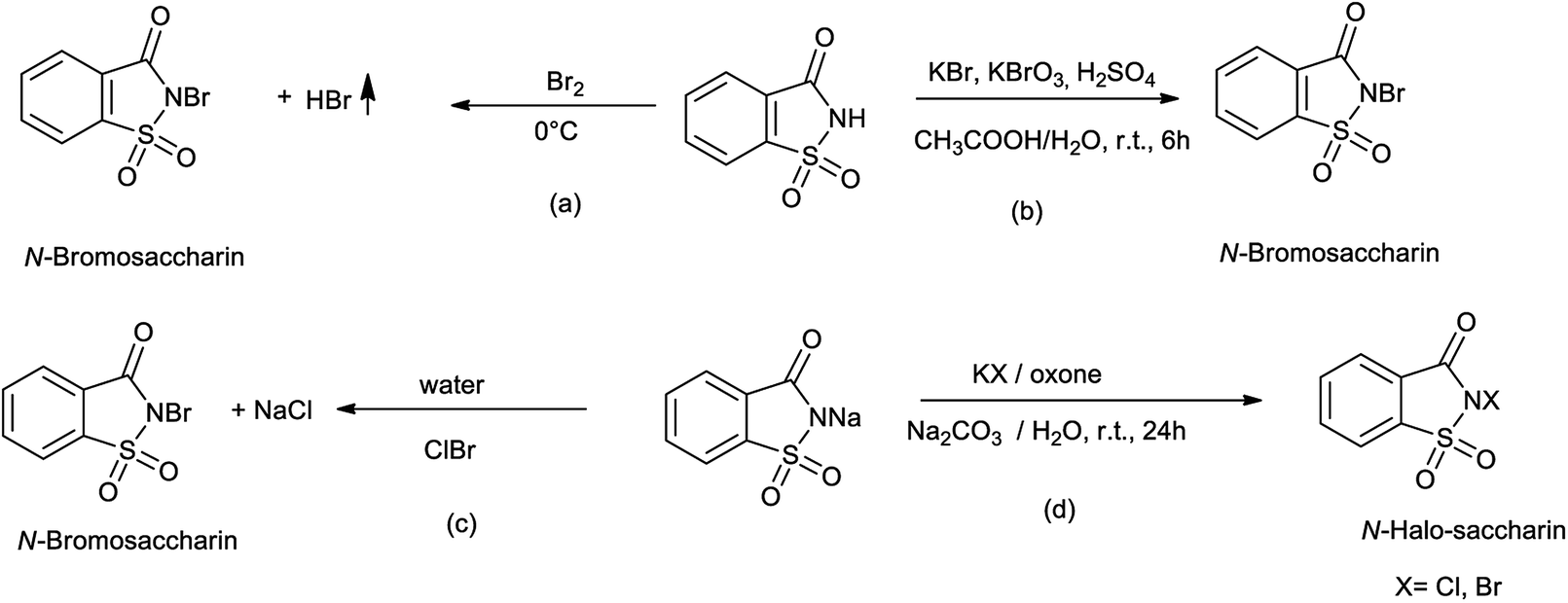
4.1. Synthesis of N-bromosaccharin
Bachhawat et al. (1971) reported the synthesis of N-bromosaccharin as shown in Scheme 26a35 Zajc reported synthesis of N-bromosaccharin from saccharin by the reaction of KBr and KBrO3 as shown in Scheme 26b.36 Sanchez et al. reported the synthesis of N-bromosaccharin by reacting sodium saccharin with bromine chloride as shown in Scheme 26c.37 De Souza et al. also reported the synthesis of N-bromo and N-chloro saccharin from sodium salts of saccharin in presence of potassium bromide or chloride and Oxone in water at the room temperature as shown in Scheme 26d.38,39
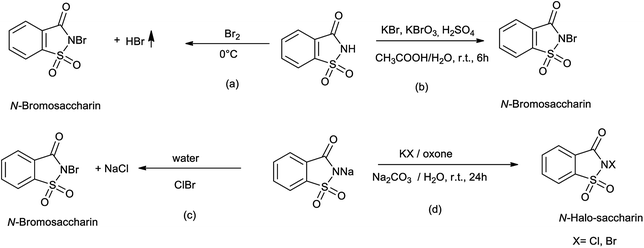 |
| | Scheme 26 Synthesis of N-bromosaccharin. | |
4.2. As a brominating agent
Earlier, diverse range of brominating agents40 has been reported for the monobromination of heteroaromatic and aromatic compounds. The direct bromination carried by the molecular bromine as a basic electrophilic brominating reagent has various drawbacks arising out of its corrosive and toxic nature, high reactivity, and difficult handling, which leads to highly nonselective and exothermic reactions.
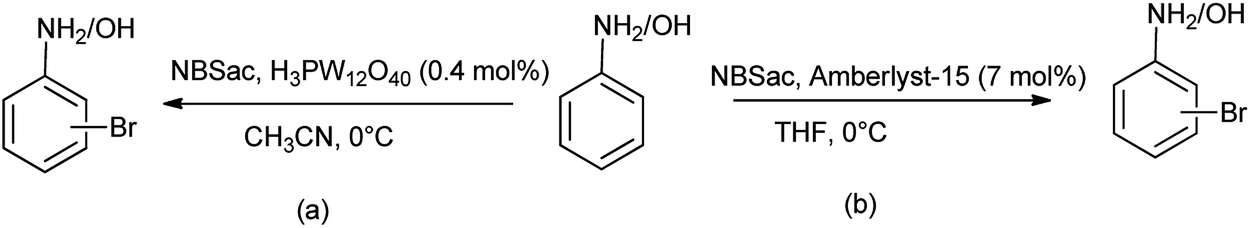
Alinezhad et al. in 2010 evaluated the N-bromosaccharin (NBSac) as heterogeneous recyclable catalyst for bromination of anilines and phenols with tungstophosphoric acid (H3PW12O40) (Scheme 27a).41 This procedure found to be effective over other due its environmental compatibility, greater selectivity, reusability, nontoxicity, ease of isolation, operational simplicity and noncorrosiveness, whereas Baharfar and coworkers evaluated the bromination of anilines and phenols using N-bromosaccharin in catalytic amount of Amberlyst-15 as catalyst (Scheme 27b).42 This protocol was found to be efficient due to mild reaction conditions, high regioselectivity, rapid conversion, good to excellent yields, and reusability of the catalyst.
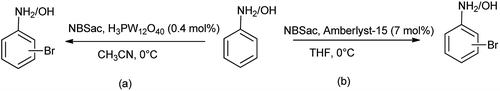 |
| | Scheme 27 Bromination of phenols and anilines using N-bromosaccharin. | |
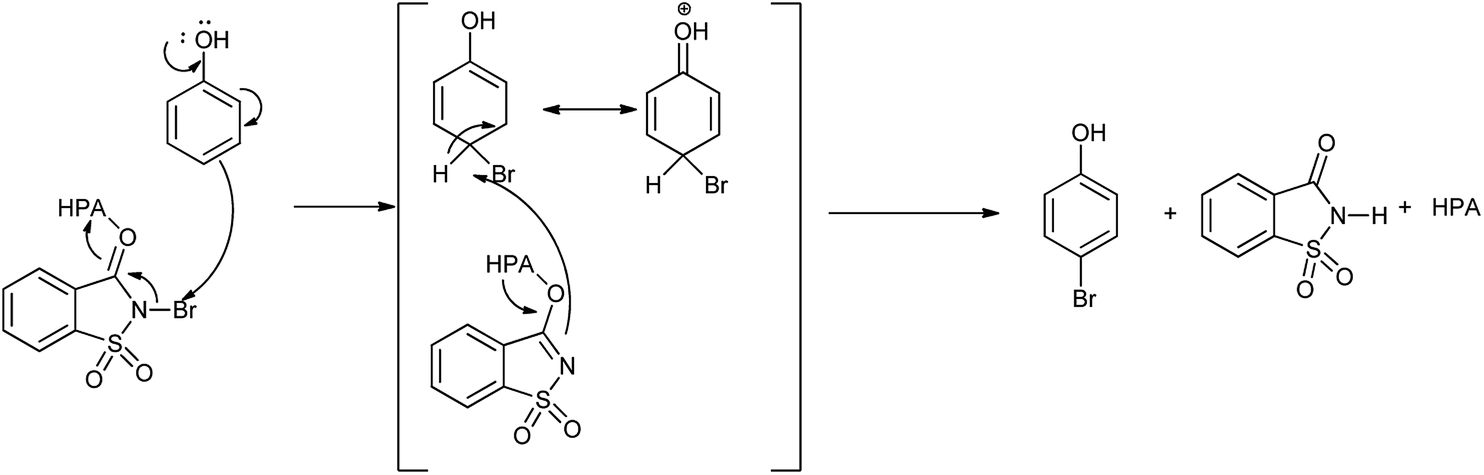
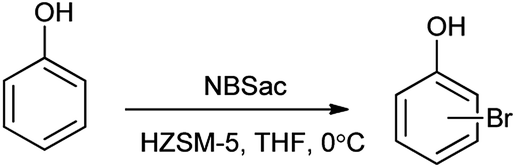
Mechanism for the reaction for bromination of anilines and phenols with tungstophosphoric acid (H3PW12O40) is shown in Scheme 28. Abrishamkar et al. reported the bromination of phenol in presence of N-bromosaccharin and ZSM-5 zeolite as a catalyst as shown in the Scheme 29.43 Some notable advantages of this protocol are mild reaction conditions, rapid conversion, simple experimental procedure, excellent yields and reusability of the catalyst.
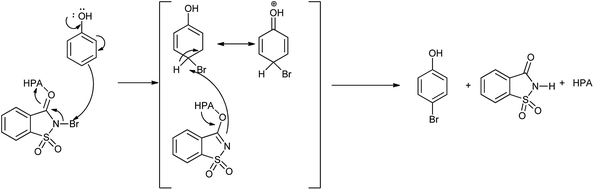 |
| | Scheme 28 Mechanism for the synthesis of bromo aniline and phenol using NBSac–H3PW12O40. | |
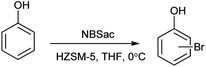 |
| | Scheme 29 Bromination of phenol with NBSac over ZSM-5 zeolite. | |
In another study, by Alinezhad et al. (2011) showed that monobromination of 1,3-diones by using N-bromosaccharin/Mg(ClO4) without ring bromination (Scheme 30).44 NBSac is found as alternative and efficient reagent to carryout bromination of 1,3-diones without the ring bromination and is synthesized easily by commercial sodium saccharin and reaction conditions are safe, easy, and fast.35
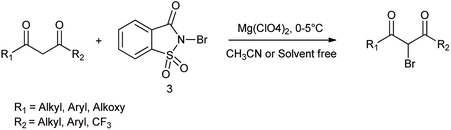 |
| | Scheme 30 Monobromination of 1,3-diones using N-bromosaccharin/Mg(ClO4). | |
Sănchez et al. reported the comparative study of bromination at α-carbonylic and benzylic position by using N-bromosaccharin (NBSac) and N-bromosuccinimide (NBS) as brominating agent. It was found that NBSac resulted in the excellent yield in both α-carbonylic and benzylic position in comparison to NBS which produces slightly lower yields with the carbonylic compounds.45
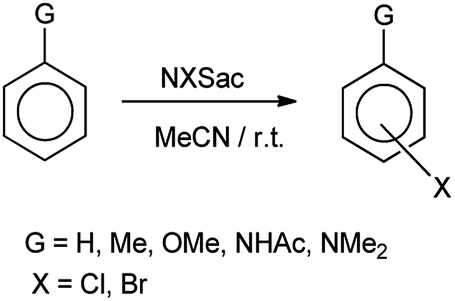
De Souza et al. 2003 showed that N-halosaccharin helps in the cohalogenation of alkenes and halogenation of electron rich aromatics. Results showed that N-bromosaccharin only gave the para-substituted product whereas N-chlorosaccharin also gave both ortho and para-substituted mixture as shown in Scheme 31. The reaction of halosaccharin with alkenes resulted in respective halohydrins (Scheme 32).39
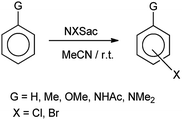 |
| | Scheme 31 Halogenation of aromatic ring. | |
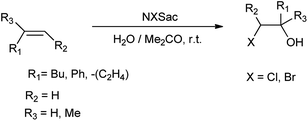 |
| | Scheme 32 Cohalogenation of alkenes. | |
Both NBSac and NClSac are effective and alternative reagents to carryout halogenation of electron rich aromatic compounds and cohalogenation of alkenes. As this method showed more efficacies due to the reaction conditions which are safe, simple, and these reagents are easily prepared by commercial sodium saccharin.
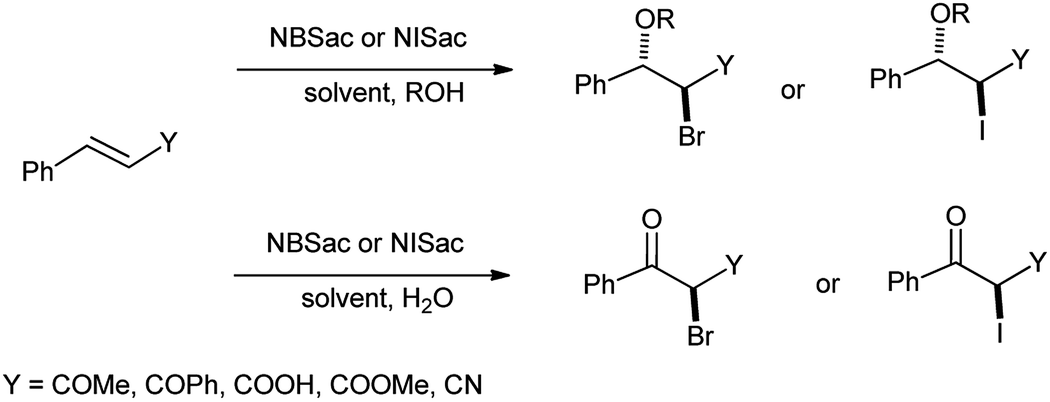
Urankar et al. demonstrated the synthesis of halohydrins from corresponding α,β-unsaturated ketones, nitriles, esters and acids in presence of N-iodo and N-bromosaccharin in aqueous organic solvents (Scheme 33).46 Herein, the reaction procedure was effective as reaction took place at room temperature, in short time and highly anti stereoselective.
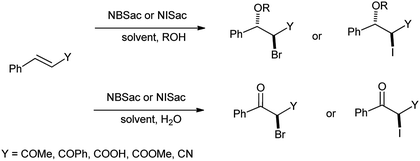 |
| | Scheme 33 Halogenation of α,β-unsaturated ketones, nitriles, esters and acids in presence of N-iodo and N-bromosaccharin. | |
4.3. As an oxidizing agent
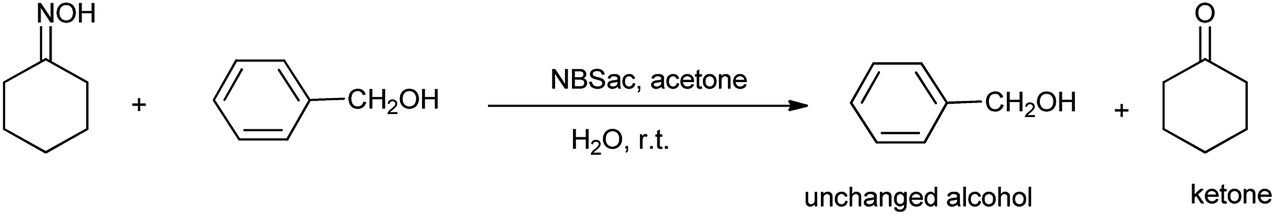
Earlier different methods were used to deoximation using several oxidizing agents such as manganese triacetate, pyridinium chlorochromate, dimethyl dioxirane and periodic acid.47 There were some drawbacks which includes long reaction times, formation of over oxidation product with low yield and difficulties in isolation of products.48 So, Khazaei et al. described N-bromosaccharin as effective oxidative reagents for regenerating carbonyl compound from oximes (Scheme 34). They reported the direct conversion of oximes to corresponding aldehyde and ketones in presence of N-bromosaccharin as shown in Scheme 34.
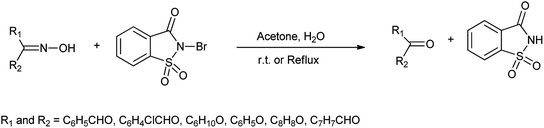 |
| | Scheme 34 Conversion of oximes to corresponding ketones or aldehydes in presence of N-bromosaccharin. | |
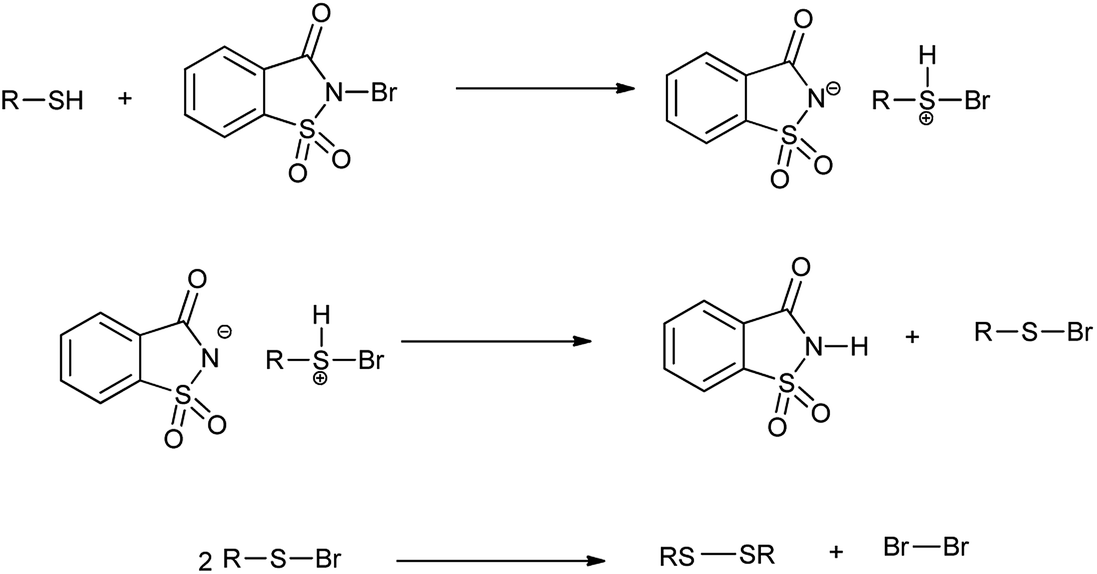
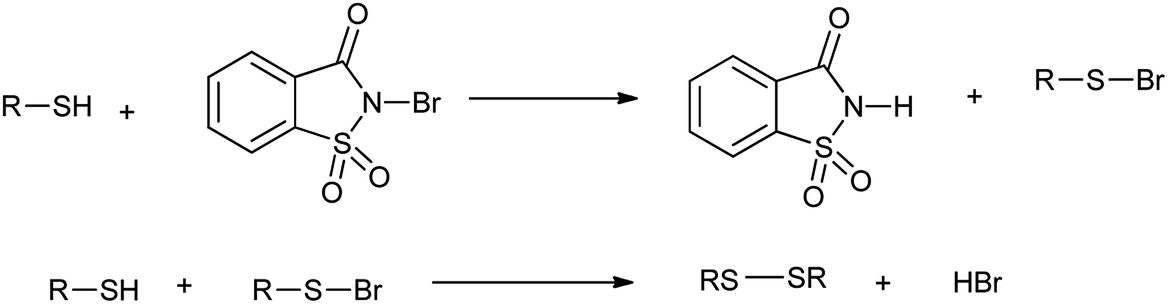
However the same protocol was used to carry out deoximation of oximes in presence of alcohols as shown in Scheme 35. This method found to be convenient took place at room temperature, under mild conditions; no formation of over oxidation products, high yields, easy workup procedure, and the oxidative reagent (NBSac) can be recovered and reused many times. Results showed that saccharin was formed after the completion of the reaction which can be isolated and can be brominated for the further used as deoximation reaction.49 Further Khazaei et al. 2006 showed that N-bromosaccharin act as an effective oxidative reagent and chemoselective under the microwave irradiation for oxidation of thiols to disulfides (Scheme 36).50 The two possible mechanisms for this reaction are shown in Schemes 37 and 38. The procedure offers various positive points such as simplicity of the reaction conditions, ease of isolation of product and reagent, selectivity and applicable to aryl, alkyl, and the hetrocyclic thiols. Further catalyst is recovered and reused many times without affecting the yield.
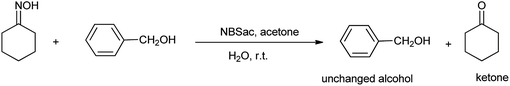 |
| | Scheme 35 Deoximation of oxime. | |
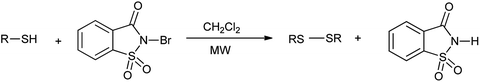 |
| | Scheme 36 Oxidation of thiols to the disulfides. | |
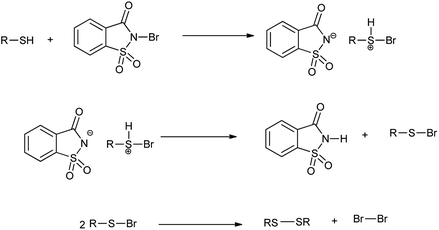 |
| | Scheme 37 Possible mechanisms for the oxidation of thiols to disulfides. | |
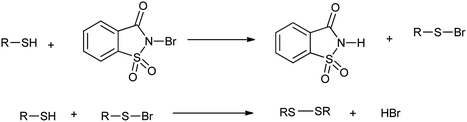 |
| | Scheme 38 Possible mechanisms for the oxidation of thiols to disulfides. | |
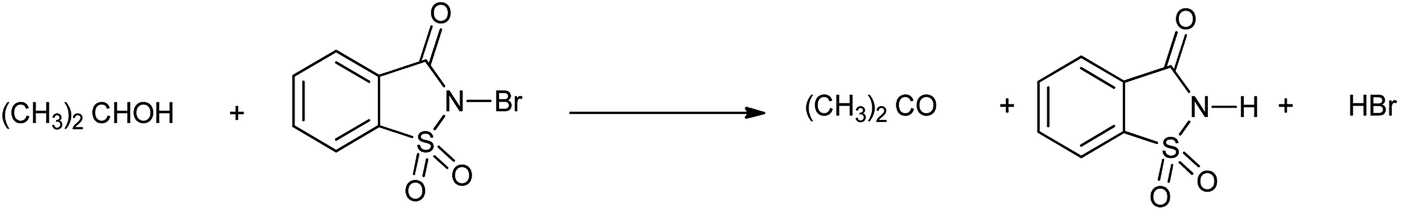
Jain et al. in 2016 investigated the kinetic study of N-bromosaccharin oxidation of 2-propanol in aqueous acetic acid medium in absence and presence of micelles sodium lauryl sulphate (NaLS) and cetyltrimethyl ammonium bromide (CTAB). The results showed that NaLS is more effective then CTAB as in Scheme 39.51
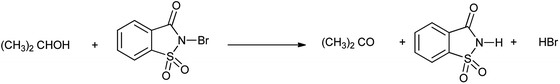 |
| | Scheme 39 Oxidation of alcohol to corresponding carbonyl compound by NBSac. | |
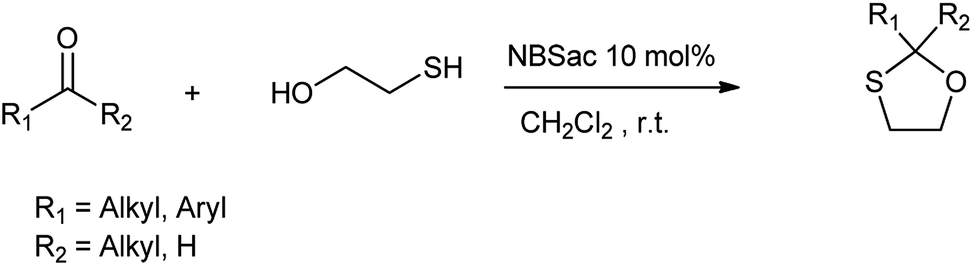
4.4. Conversion of carbonyl groups to their corresponding oxathioacetals
Conversion of carbonyl groups to their respective oxathioacetals is important from many aspects as the main reasons for the synthesis of 1,3-oxathiolanes are their significant stability under different reaction conditions, ease of formation and removal and equality to acyl carbanions in C–C bond forming reactions.52 Alinezhad et al. (2012) reported the synthesis of 1,3-oxathiolane derivatives from 2-mercaptoethanol and carbonyl compound using N-bromosaccharin as efficient catalyst in dichloromethane as a solvent at room temperature and catalyst was recovered at the end of reaction (Scheme 40).53
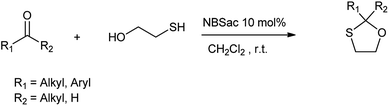 |
| | Scheme 40 Synthesis of 1,3-oxathiolane. | |
4.5. As a source of halogen and oxygen/nitrogen
Song et al. in (2016) reported that N-bromosaccharin act as a source of halogen and oxygen/nitrogen source for the haloetherification and haloamination of alkenes. The reaction of styrene with NBSac under the visible light promoted catalyst free condition (Scheme 41).54 N-Bromo and N-chlorosaccharins, endowed with a weakened N–X bond, are amenable to visible light promoted homolytic cleavage, facilitating reliable and a wide range of olefins, including propene, ethylene, were tolerated to give haloetherification and haloamidation products in high yields.
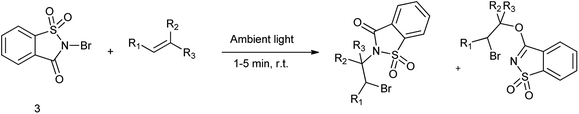 |
| | Scheme 41 Bromoamidation and bromoether adduct formation. | |
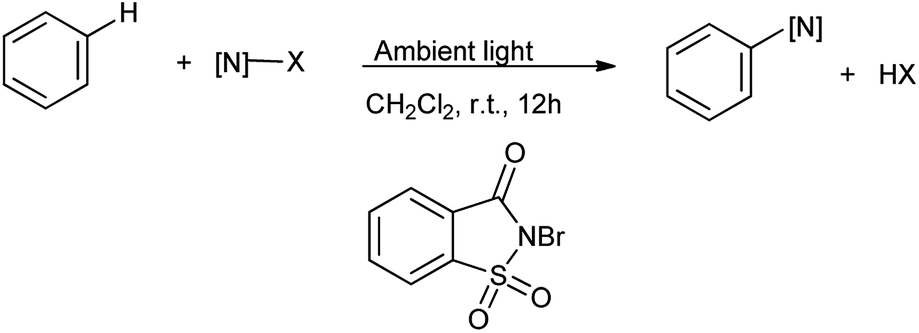
Song et al. in 2014 reported the catalyst-free visible-light photolytic imidation of heteroarenes and arenes. Herein the N-bromosaccharin act as chemoselective nitrogen radical precursor which undergoes homolytic cleavage under ambient light radiation (Scheme 42).55 This was elegant method for imidation of arenes.
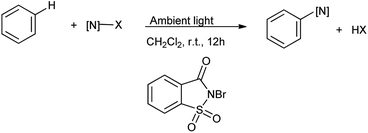 |
| | Scheme 42 Visible light photolytic imidation with N-bromosaccharin. | |
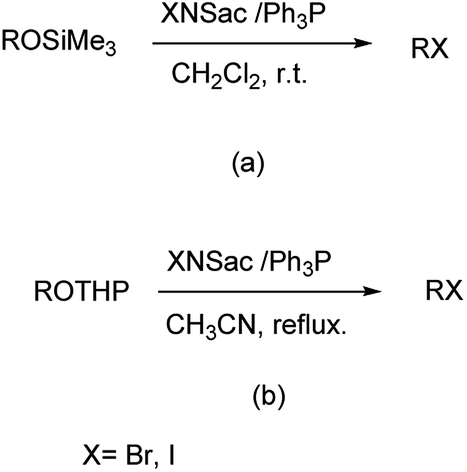
Firouzabadi et al. reported the synthesis of trimethylsilyl and tetrahydropyranyl ethers to their corresponding bromides and iodides with high selectivity and efficiency by N-iodo and N-bromosaccharin in the presence of triphenylphosphine under neutral conditions as showm in Scheme 43.56 Saccharin is easily available, cheap, non-toxic and recovered from the reaction mixture and halogenated easily for reusing in the similar reactions.
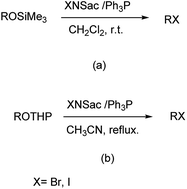 |
| | Scheme 43 (a) Conversion of trimethylsilyl ether and (b) conversion of tetrahydropyranyl ethers in presence of N-iodo and N-bromo saccharin in presence of Ph3P. | |
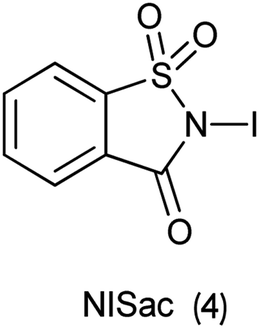
5. N-Iodosaccharin as iodinating agent
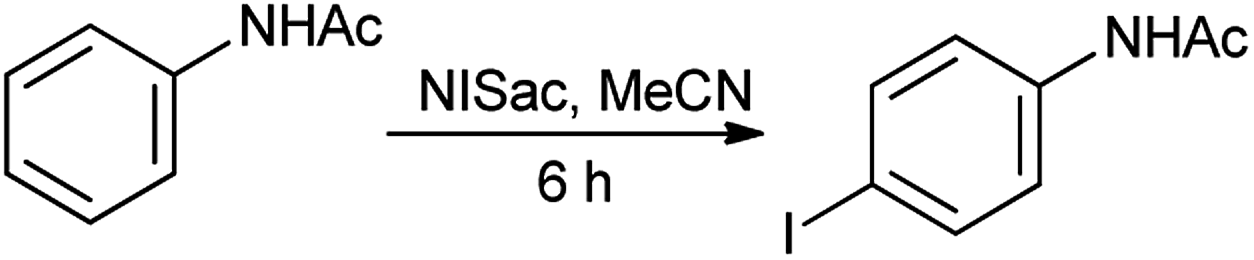
Organic iodine compounds are always of interest as iodine is an excellent leaving group and replaced with other groups in copious reactions. Introduction of an iodine atom within an organic molecule is difficult due to the weakness of the carbon–iodine bond, so various iodinating reagents has been introduced from last few years. To carry out electrophilic iodination, more reactive reagents are those in which the iodine atom is made more electropositive.57 N-Haloamides were used as electrophilic halogenating reagents. The most commonly used reagents are succinimide (i.e. N-bromosuccinimide, N-iodosuccinimide). N-Iodosaccharin found to be more potent source of I+ than succinimide.58 The alkenes were transformed to the corresponding adducts of iodine and nucleophile as by N-iodosaccharin reported by Dolenc. The synthesis of N-iodosaccharin (NISac) was achieved by the reaction of a silver salt of saccharin with iodine (Scheme 44).59 Additions follow the Markovnikov rule with high regioselectivity. Reaction with disymmetric alkenes results in the formation of a single diastereomer as a result of anti addition. Iodination by NISac was found to be 500 times faster than N-iodosuccinimide (NIS) as shown in Scheme 45. When the aromatic ring was substituted with the deactivating group it resulted in the mono-iodination (Scheme 46) while phenols under goes di-iodination.59
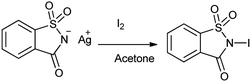 |
| | Scheme 44 Synthesis of N-iodosaccharin. | |
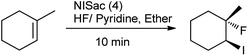 |
| | Scheme 45 Iodination of aromatic ring. | |
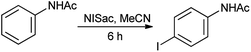 |
| | Scheme 46 Iodination of substituted ring. | |
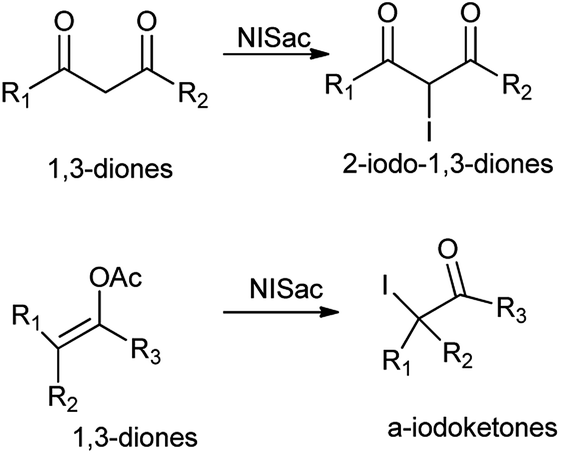
Further, iodination of 1,3-diones and enol acetates was reported by using N-iodosaccharin resulting in corresponding 2-iodo-1,3-diones and α-iodoketones at room temperature under netural conditions (Scheme 47).60 The reagent does not affect oxidizable groups, such as hydroxyl or aldehyde and took place under mild and fast condition without heavy metals and strong acids.
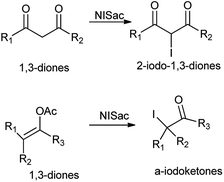 |
| | Scheme 47 Iodination of 1,3-diones and enol acetate. | |
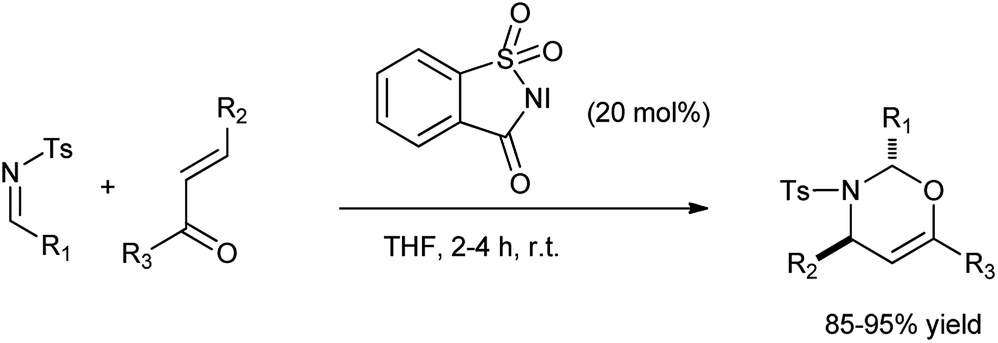
Rai et al. reported the synthesis of 1,3-oxazine via [2 + 4] cycloaddition of enones and imines in the presence of N-iodosaccharin as a catalyst (Scheme 48).61 The reagent was easily isolated and reused without the loss of its efficacy and reactivity.
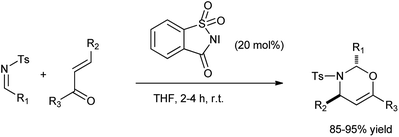 |
| | Scheme 48 Synthesis of 1,3-oxazine. | |
6. N-Chlorosaccharin as chlorinating agent
However, N-chlorosaccharin has little use as a chlorinating agent in water because of its poor solubility and slow rate of dissolution. Whereas, it can be readily soluble in various organic solvents and expected to act as chlorinating agent, because it can easily releases its positive chlorine. The reactions of N-chlorosaccharin in aqueous hypochlorite solution or methanol do not reduce the availability of positive chlorine appreciably and these solutions also should be useful chlorinating systems. N-Chlorosaccharin was found to be strong chlorinating reagent as shown by its low chlorine potential (pKcb = 4.85 at 25 °C) and oxidizing reagent than commonly used N-chlorosuccinimide or chloramine-T in water. It's possible used as a chlorinating or detoxifying reagent.62
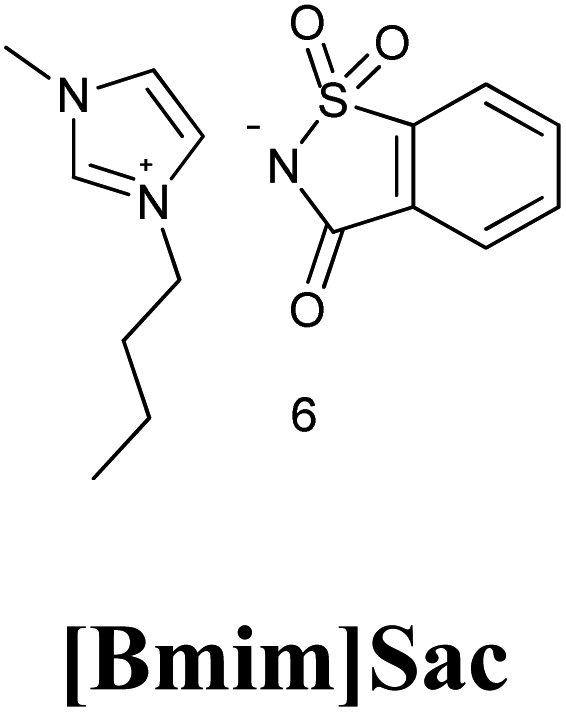
7. Saccharin based ionic liquid [Bmim]Sac
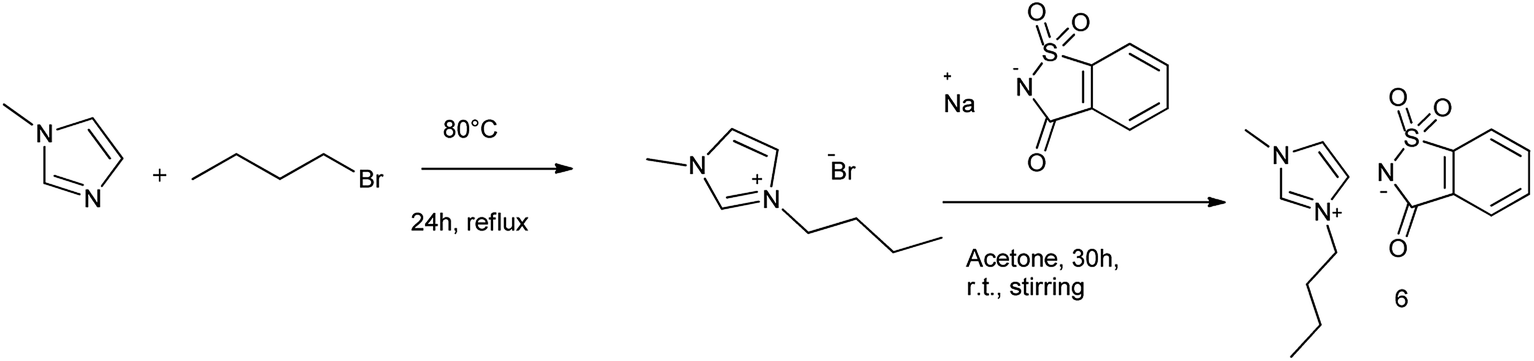
7.1. Synthesis of [Bmim]Sac
Currently, ionic liquids have attracted significant attention in green organic synthesis owing to their unique properties such as low vapor pressure, wide liquid range, good conductivity and large electrochemical window.63 In addition to these, the gold benchmark for green chemistry is functional ionic liquid mediated synthesis (FILMs). Nowadays, FILMs has become a novel approach representing an attempt to describe “design capacity of ionic liquids”, which makes them an accurate working system rather than simply novel media, and their properties can be altered to suit the requirement of a particular process.64
[Bmim]Sac was synthesized in two steps as the procedure given by Nockemann et al. (2007)65 shown in Scheme 49.
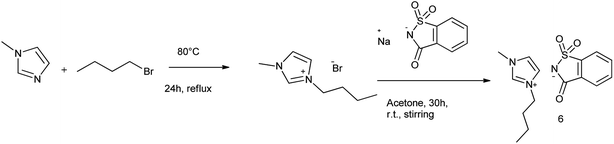 |
| | Scheme 49 Synthesis of [Bmim]Sac. | |
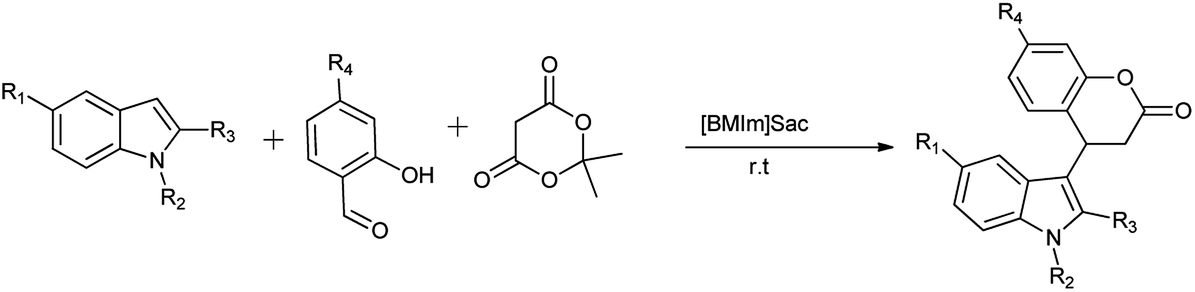
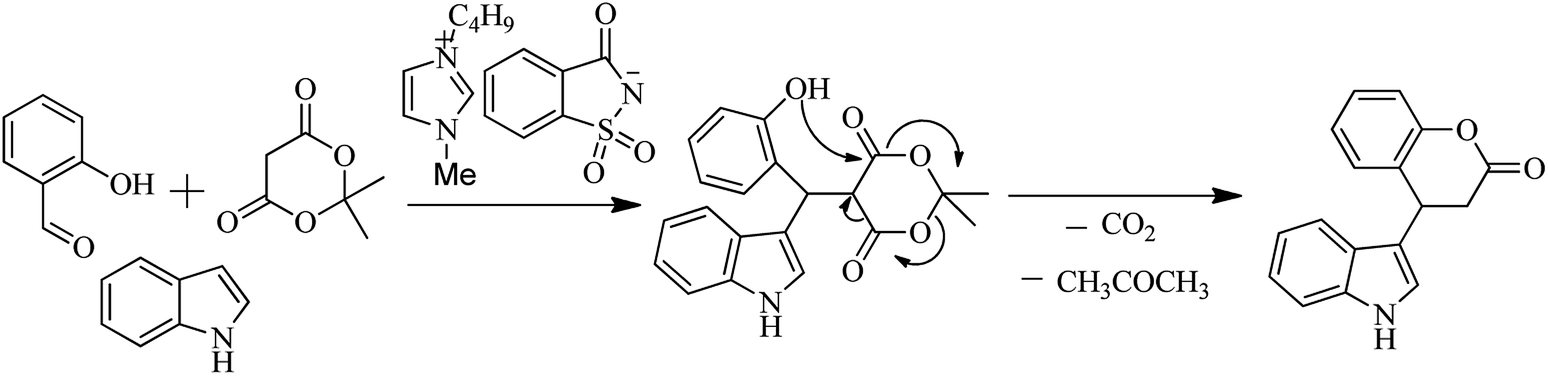
7.2. Domino Knoevenagel–Michael reaction
Saccharin derivatives are also useful in carrying out named reaction as Kumar et al. (2012) evaluated the saccharin based functional ionic liquids [Bmim]Sac for the synthesis of indole-3-dihydrocoumarin by coupling of salicylaldehyde and meldrum's acid followed by a Michael type reaction with indole shown in Schemes 50 and 51. Results showed that [Bmim]Sac gave effective results at the room temperature and convenient for use and gave good yield.66
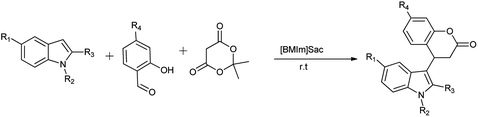 |
| | Scheme 50 Synthesis of indole-3-dihydrocoumarin. | |
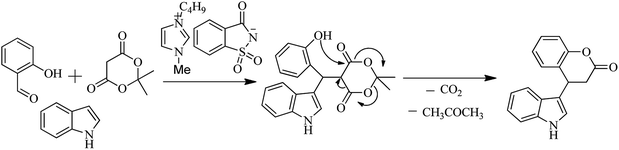 |
| | Scheme 51 Mechanism for synthesis of indole-3-dihydrocoumarin. | |
Sharma et al. showed that a saccharin-based ionic liquid [Bmim]Sac as a catalyst for the synthesis of 4,5-dihydropyrano[4,3-b]pyran, tetrahydrobenzo[b]pyrans scaffolds and 3,4-dihydropyrano[c]chromenes through Domino Knoevenagel–Michael reaction (Scheme 52).
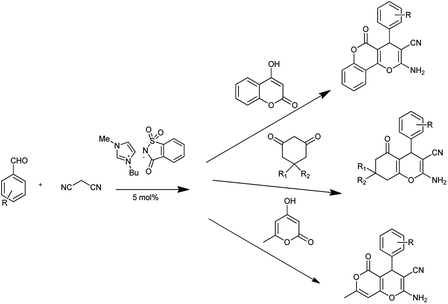 |
| | Scheme 52 3,4-Dihydropyrano[c]chromenes, 4,5-dihydropyrano[4,3-b]pyran, and tetrahydrobenzo[b]pyrans scaffolds. | |
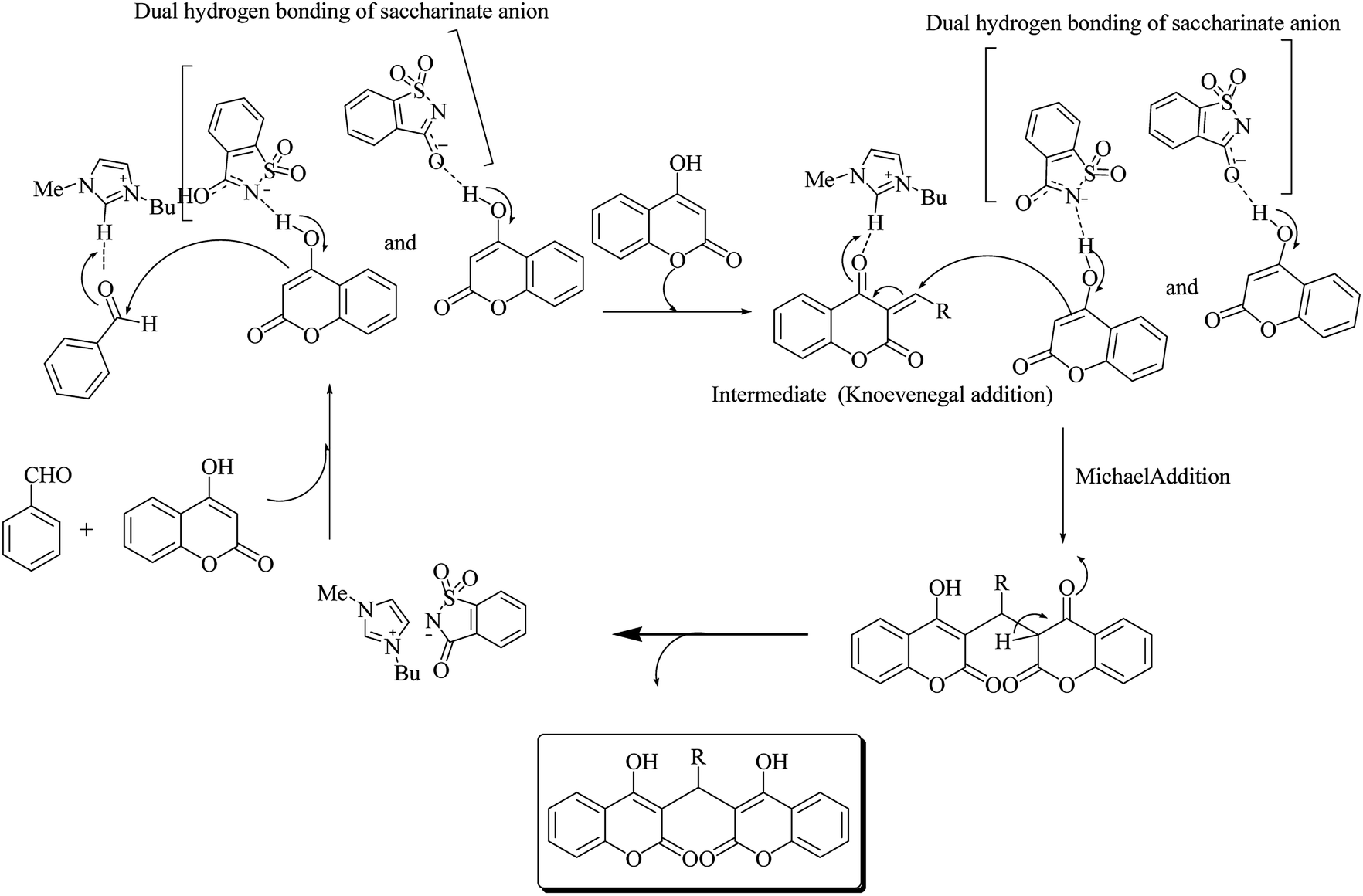
The reaction occurred in three steps: a Knoevenagel condensation, a Michael addition, and then an intramolecular cyclization (Scheme 53). Results showed that [Bmim]Sac gave the highest yield (95%) in aqueous medium than in the non-aqueous medium (82%).67
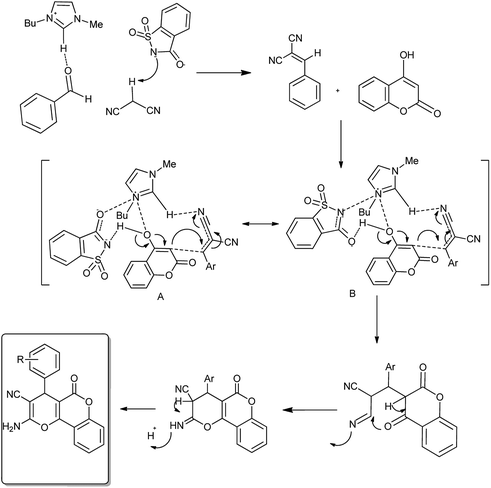 |
| | Scheme 53 Mechanism proposed for the dual activation of IL. | |
Sharma et al.68 also reported the synthesis of bis-enol derivative with the substituted aldehyde catalyzed by [Bmim]Sac (Scheme 54). The reaction mechanism is shown in Scheme 55.
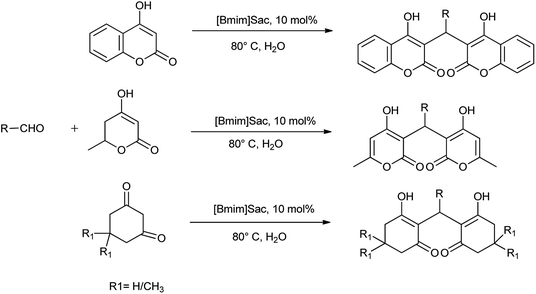 |
| | Scheme 54 Domino Knoevenagel–Michael reaction of substituted aldehydes catalyzed by [Bmim]Sac. | |
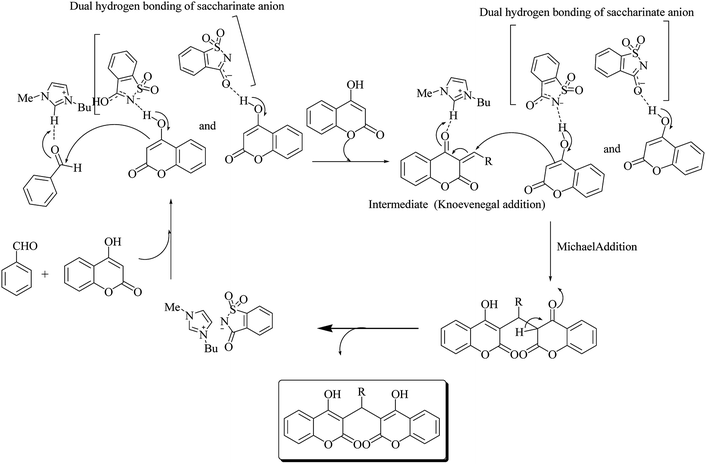 |
| | Scheme 55 Mechanism via dual of hydrogen bonding of saccharinate anion and dual activation of [Bmim]Sac. | |
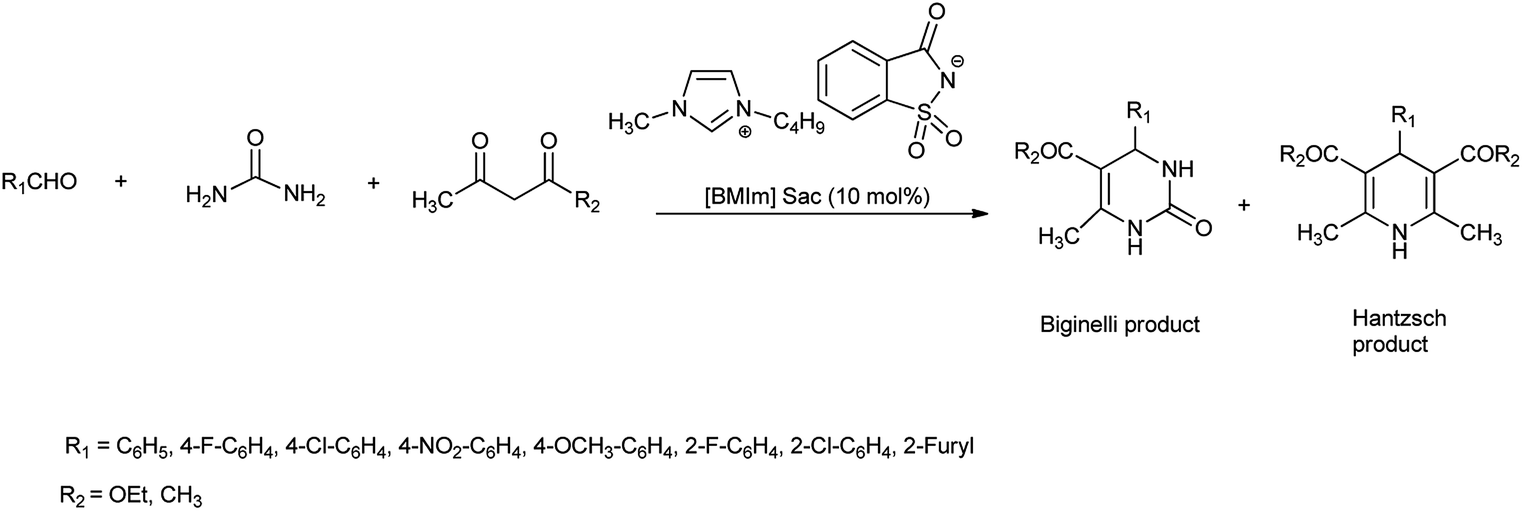
This method includes higher yields, milder and cleaner reaction condition, purity of product, and easy workup. Further, ionic liquid catalyst is easily synthesized and highly biodegradable, biocompatible, non toxic, green and safe. Ming et al. in 2006 reported the synthesis of 3,4-dihydropyrimidinones (Biginelli products) and 1,4-dihydropyridinones (Hantzsch products) at room temp (Scheme 56)69 with 80% of yield. Mild reaction conditions, non-toxic catalyst and environmental benign are the advantages of this new procedure.
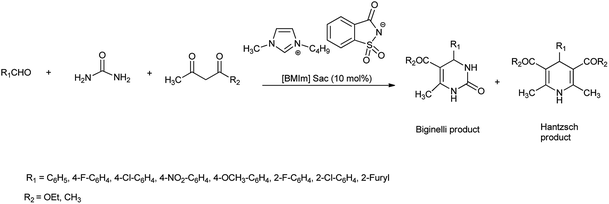 |
| | Scheme 56 Synthesis of Bignelli and Hantzach product using [Bmim]Sac. | |
7.3. Michael addition of thiol to ferrocenyl enone
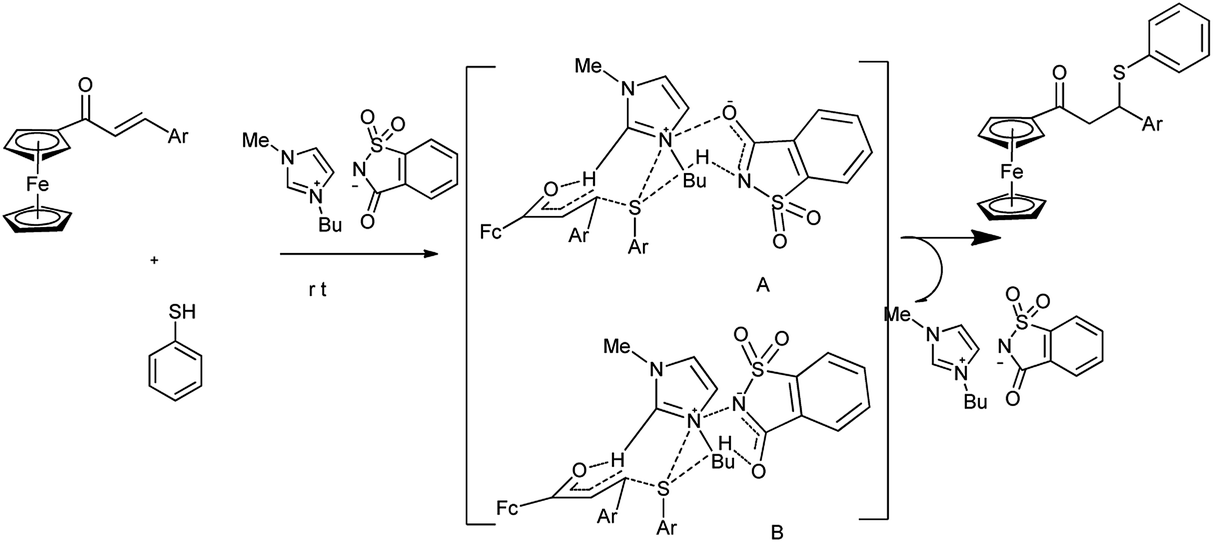
Ferrocene derivatives have acquired a growing significance in applications such as pharmaceuticals, material sciences, biosensors, as well as molecular receptors, which necessitate a more economical and efficient synthesis. It is interesting to apply the anion–cation cooperative interaction of functional ionic liquids for the greener synthesis of bioactive organometallics. Kumar et al. reported the synthesis of ferrocenyl thiopropanones under solvent free condition by Michael addition of thiol to ferrocenyl enone by using saccharin based functional ionic liquid [Bmim]Sac as a catalyst (Scheme 57)70 and also evaluated their anti-proliferative activity.
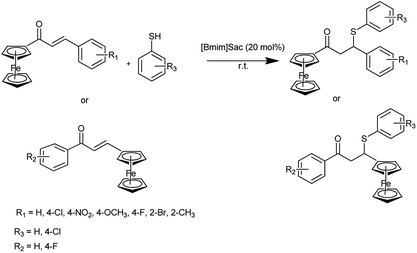 |
| | Scheme 57 Synthesis of ferrocenyl thiopropanones. | |
The catalytic efficiency of most of the ionic liquids is strongly influenced due to C-2 hydrogen of the Bmim and counter anion of the ionic liquid. In case of saccharinate, negative charge is delocalized and thus both nitrogen and carbonyl oxygen bears partial negative charge. Due to delocalization of negative charge both amidic nitrogen and amidic oxygen form hydrogen bond with thiol S–H abbreviated as ([Bmim]Sac A and [Bmim]Sac B) respectively and thus activate thiol (dual nucleophilic activation) (Scheme 58).
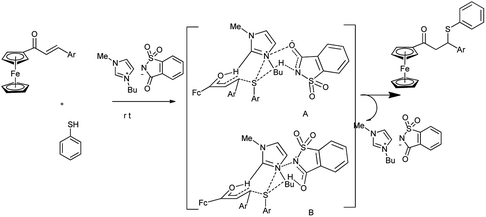 |
| | Scheme 58 Role of [Bmim][Sac] in the thia Michael addition of ferrocenyl enone. | |
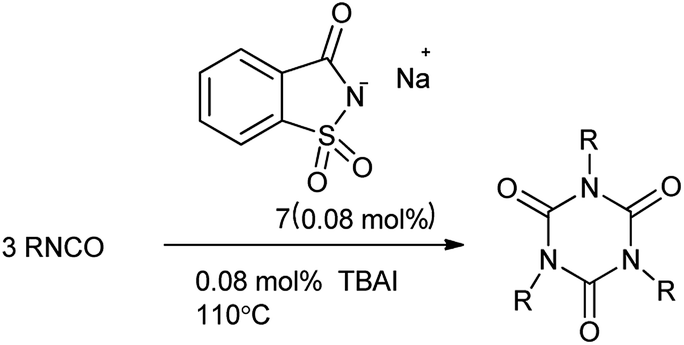
8. Sodium saccharin as a catalyst for multicomponent reaction
Saccharin, is generally used in the form of sodium or calcium salt. Recently, sodium saccharin as a basic green and easy available compound was used as catalyst. 1,3,5-Triazinane-2,4,6-trione, commonly known as isocyanurate, amend the properties of polyurethane foams and resins.71 Triaryl isocyanurates are useful activators for the continuous anionic polymerization and postpolymerization of ε-caprolactam to nylon-6, insecticides and found interesting application in the drug desinging.72 Moghaddam et al. 2004 reported the symmetrical synthesis of isocyanurates under solvent free condition using combined catalyst i.e. sodium saccharin and tetrabutylammonium iodide (Scheme 59).73 This anionic catalyst has been found for the efficient under solvent-free and eco-friendly conditions.
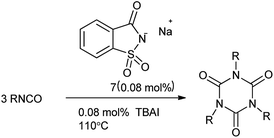 |
| | Scheme 59 Synthesis of isocyanurates. | |
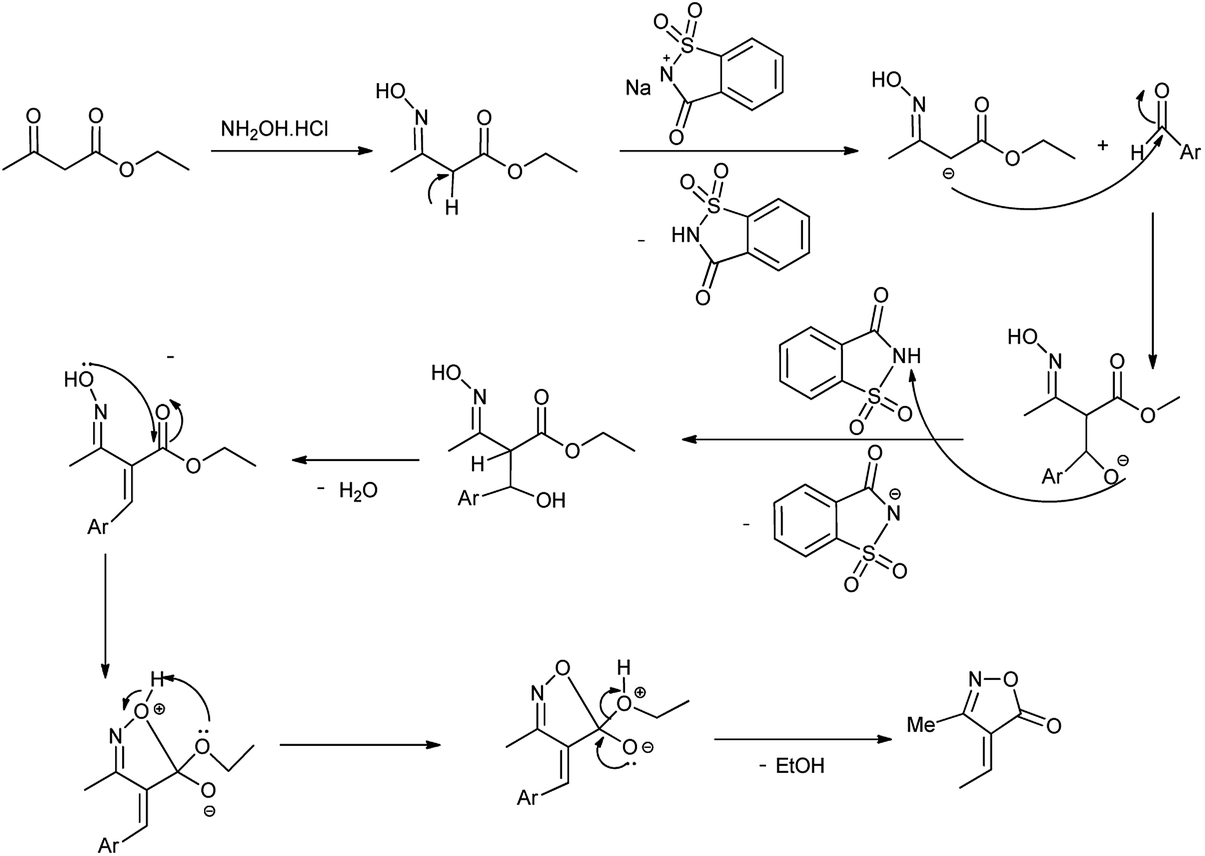
Isoxazole ring is one of natural and medicinal active molecules and their derivatives show interesting biological and pharmaceutical activity.74 Kiyani et al. showed the synthesis of 4-arylidene-3-methylisoxazol-5(4H)-ones by condensation of aryl aldehyde, hydroxylamine hydrochloride and ethyl acetoacetate at room temperature by using sodium saccharin as a catalyst (Scheme 60) and mechanism of reaction is shown in Scheme 61.75 The reaction procedure carried out under room temperature and water as solvent at optimal conditions.
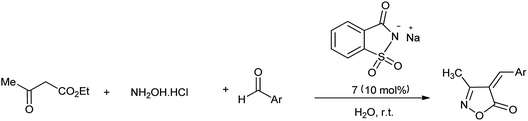 |
| | Scheme 60 Synthesis of 4-arylidene-3-methylisoxazol-5(4H)-ones. | |
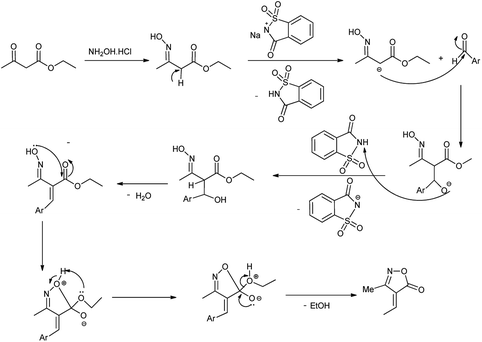 |
| | Scheme 61 Mechanism followed for the synthesis of 4-arylidene-3-methylisoxazol-5(4H)-one. | |
Moradi et al. (2017) reported the synthesis of dihydropyrano[2,3-g]chromenes under microwave irradiation by using sodium saccharin a catalyst (Scheme 62). Results showed that sodium saccharin act as effective catalyst than the saccharin as it results in 90% yield in 8 minutes. Mechanism for the formation of desired product is shown in Scheme 63.76 The reaction procedure carried out in microwave irradiation, easy workup and short reaction condition with high yields of products.
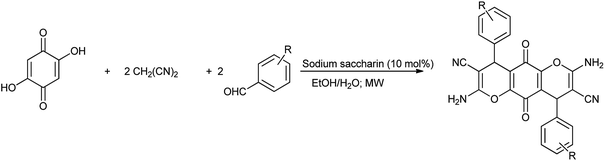 |
| | Scheme 62 Synthesis of dihydropyrano[2,3-g]chromenes. | |
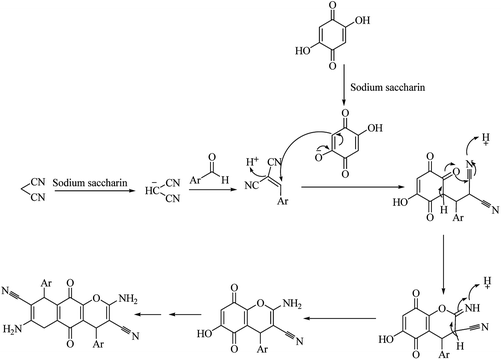 |
| | Scheme 63 Mechanism purposed for synthesis of dihydropyrano[2,3-g]chromenes. | |
9. N-Formyl saccharin
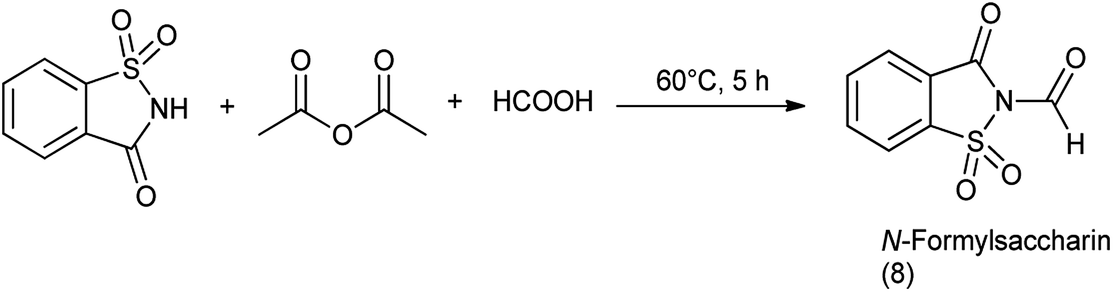
9.1. Synthesis of N-formylsaccharin
N-Formylsaccharin was synthesized by the formylation of saccharin with the in situ generated acetic formic anhydride (Scheme 64).77
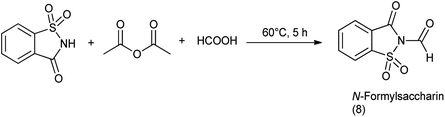 |
| | Scheme 64 Synthesis of N-formylsaccharin. | |
9.2. As a source of CO
Earlier CO gas was used for the reductive carbonylation of aryl halides in comparison to amino- and alkoxycarbonylations.78 Over the last few decades, CO-free carbonylation chemistry has been the focus of extensive research for alternative to toxic CO gas. Various compounds such as formic acid derivatives, metal carbonyl compounds and acetic formic anhydride has been developed as a CO source.79 Cochet et al. proposed chemoselective formylation of primary amines using N-formylsaccharin (Scheme 65).80 N-Formylsaccharin generates CO and it is an alternative method to carry out halogen metal exchange with stoichiometric amount of metal. This process offers an alternative to the conventional methods involving using toxic CO gas.
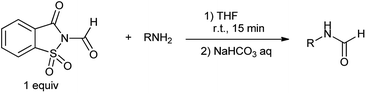 |
| | Scheme 65 Formylation of primary amines. | |
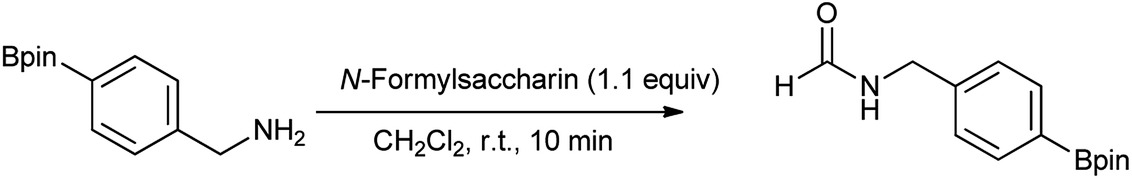
Fang et al. proposed that N-formylation of benzyl amines was compatible with a boron substituent either in the para or meta position (Scheme 66).81
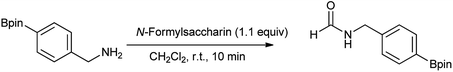 |
| | Scheme 66 N-Formylation of benzyl amines. | |
Synthesis of aldehydes from aryl halides in the presence of palladium as a catalyst reported by Ueda et al.82 N-Formylsaccharin act as a good source of CO in presence of Pd(OAc)2, diphosphine ligand (dppb), Na2CO3, whereas Et3SiH act as a hydride donor (Scheme 67). It has significant advantages such as stability, ease of handling, low cost, good availability and high reactivity as a CO source.
 |
| | Scheme 67 Synthesis of aldehydes from aryl halides. | |
They also reported the synthesis of acyl flouride from aryl bromide in presence of N-formylsaccharin as CO source with catalytic amount of Pd(OAc)2 and xantphos, KF as an activator as well as nucleophile (Scheme 68).83 This method provides convenient access to esters and aldehydes without concerns of toxic CO handling. It has significant advantages such as stability, ease of handling, low cost, good availability and high reactivity as a CO source.
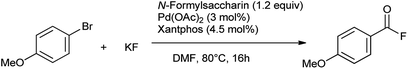 |
| | Scheme 68 Flourocarbonylation of aryl halides. | |
The synthesis of aromatic esters has been reported from aryl iodides and phenols using N-formylsaccharin as a CO source and Pd/C as catalyst by Gautam et al.84 The reaction was performed under the ligand free conditions in propylene carbonate as eco-friendly and sustainable solvent (Scheme 69). Only iodobenzene yield the corresponding esters.
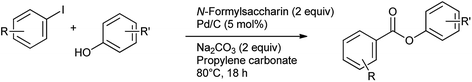 |
| | Scheme 69 Phenylcarbonylation to synthesize aryl esters. | |
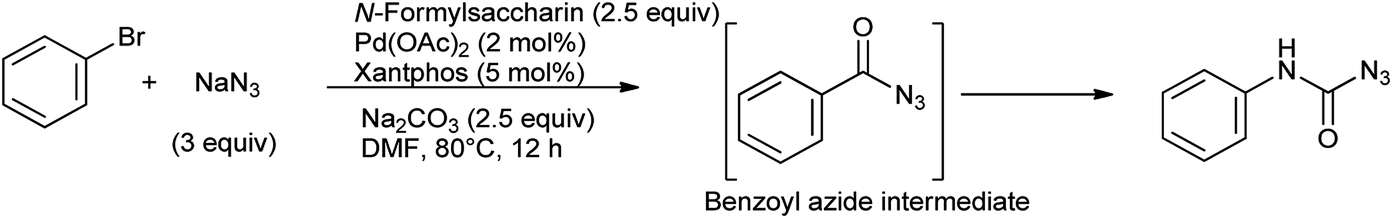
The direct synthesis of aromatic carbamoyl azides from aryl bromides and NaN3 has been evaluated using N-formylsaccharin/Na2CO3 in the presence of Pd(OAc)2/xantphos as a catalyst reported by Yadav et al.85 In this study bromobenzene and NaN3 react to synthesize the benzoyl azide intermediate which further undergoes an in situ Curtius rearrangement to generate phenylcarbamoyl azide (Scheme 70). This protocol offers efficient alternative to access carbamoyl amides, as it is used as solid, easy to handle as CO source and easily accessible.
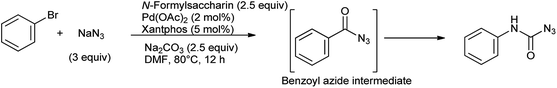 |
| | Scheme 70 Direct synthesis of aromatic azides from aryl bromides and NaN3. | |
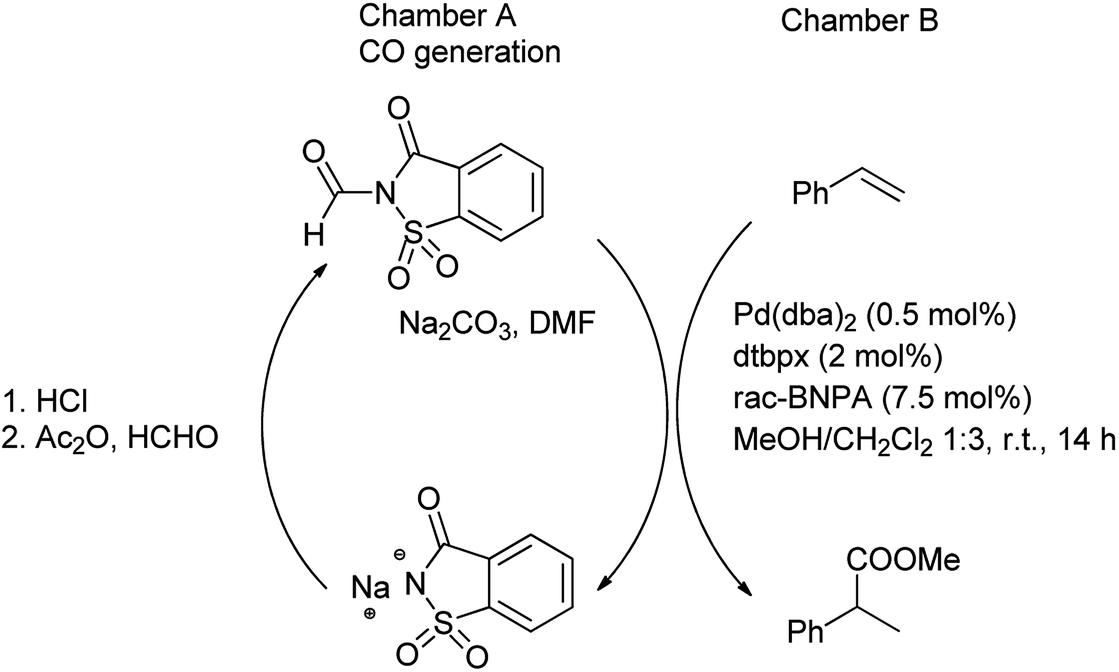
Further, Gehrtz et al. reported methoxycarbonylation of styrene was catalyzed by the palladium and N-formylsaccharin and Na2CO3 were used for the generation of ex situ CO in DMF providing the branched ester with regioselectivity and good yield.77 The reaction was carried out under mild conditions with low catalyst loading at room temperature. The procedure was carried out in two-chambered pressure vessel (chamber A, chamber B) for generation of CO (Scheme 71). It was seen that saccharin was recovered and recycled after the treatment of sodium saccharinate with HCl in chamber A (CO generation) and further used for preparation of N-formylsaccharin. However, the ex situ generation of CO was used for the synthesis of branched thioester through thiocarbonylation of styrene with aliphatic thiol using Pd(dba)2 as catalyst in combination of bidentant ligand and diphenylphosphoric acid (dppa) as cocatalyst which enables high regioselectivity reported by Hirschbeck et al. (Scheme 72).86
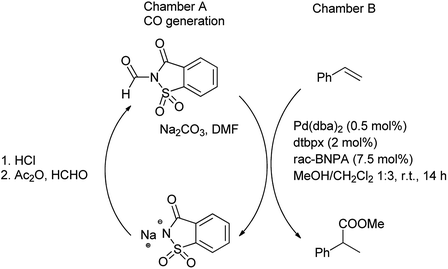 |
| | Scheme 71 Methoxycarbonylation of styrene. | |
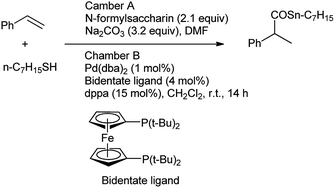 |
| | Scheme 72 Thiocarbonylation of styrene with thiol. | |
Fan and coworkers too reported palladium-catalyzed carbonylative and reductive cyclization of ortho-iodo-tethered methylenecyclopropanes (MCPs) using N-formylsaccharin as CO source affording the desired indanone derivatives with good yields (Scheme 73).87 This method used as a carbonylative cyclization and tandem reductive of MCPs using N-formylsaccharin as CO source under the catalysis of palladium via a new ring-opening pattern.
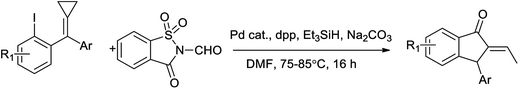 |
| | Scheme 73 A palladium-catalyzed carbonylative and reductive cyclization. | |
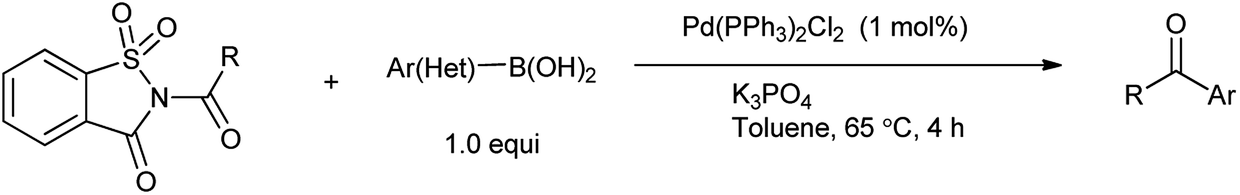
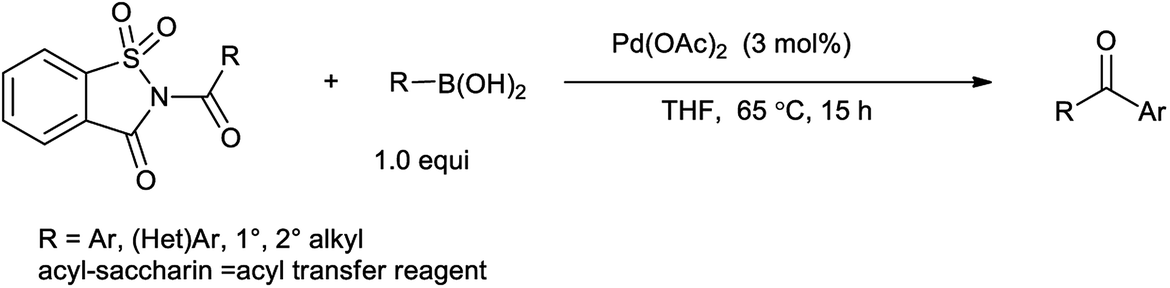
10. N-Acyl saccharin
10.1. Synthesis of N-acylsaccharin
Perez et al. reported the synthesis of N-alkyl and N-acyl saccharin derivatives by the reaction of sodium saccharin with alkyl and acyl halide in toluene in the presence of catalytic amount of tetrabutylammonium bromide (TBAB) under phase transfer condition (Scheme 74).88 Phase-transfer catalysis has been significantly used for the alkylation of NH groups in which acidity is enhanced by neighboring electron-withdrawing substituents.
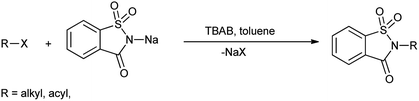 |
| | Scheme 74 Synthesis of N-acyl and N-alkyl saccharin derivatives. | |
Further Ramegowda et al. reported the synthesis via reaction of acid chloride and sodium salt of saccharin.89 Micheel et al. also showed the synthesis of N-acyl saccharin using γ-saccharin chloride and carboxylic acid.90
10.2. N-Acylsaccharin as a catalyst
Ramegowda et al. in year 1973 reported the synthesis different aldehydes from carboxylic acids via reduction of N-acyl saccharin using sodium dihydro bis-(2-methoxyethoxy)aluminate.89 This protocol is useful for the reduction of variety of aliphatic, aromatic, alicyclic, and α,β-unsaturated acids to the corresponding aldehydes with satisfactory amount of yield. Wu et al. reported that Suzuki coupling of amides with arylboronic acid in presence of palladium catalyzed C–N cleavage of N-acylsaccharin (Scheme 75).91 This method has application over the other reported methods due to less reaction time, excellent yield, mild conditions and low catalyst loading. Similarly Liu and coworkers (2016) reported the Suzuki–Miyaura coupling of N-acylsaccharin with boronic acid catalyzed in the presence of Pd-catalyst (Scheme 76).92 This protocol enables to synthesize the wide range of ketones with wide range of functional group tolerance and high yield.
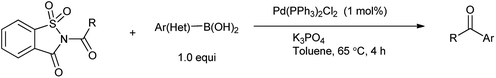 |
| | Scheme 75 Pd-catalyzed acylative Suzuki coupling of amides. | |
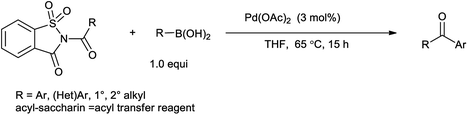 |
| | Scheme 76 Pd-catalyzed Suzuki–Miyaura coupling of N-acylsaccharin. | |
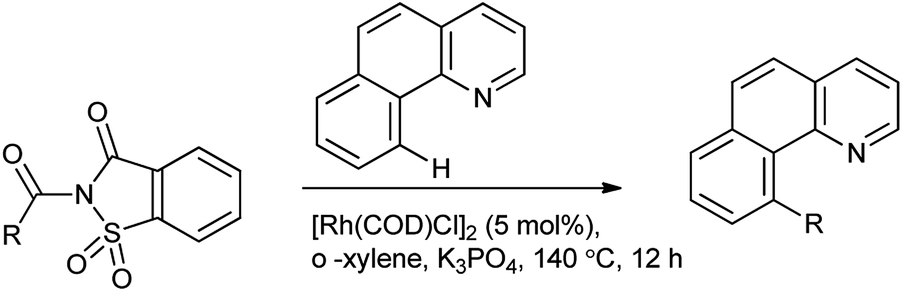
Wu et al. reported the arylation of amides which are activated by rhodium catalyst. C–H arylation was employed by N-acylsaccharin (Scheme 77).93 This protocol provides an alternative route in C–H functionalization and a new activation model of C–N amide bonds.
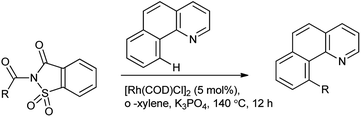 |
| | Scheme 77 Diamination of amide. | |
Liu and coworkers elucidated the Heck reaction of amides catalyzed by Pd catalyst and N-acylsaccharin is used as coupling partner to promote N–C cleavage (Scheme 78).94 This procedure was found to first for sequential Heck coupling of C–Br and C–N bonds and it is suitable for the umpteen of amides and olefin substrate. N-Acylsaccharin serve as selective bench-stable aryl transfer reagents, air-stable, crystalline solids which show comparable reactivity to N-glutarimide amides and may enable the development of a broad range of novel metal-catalyzed transformations.
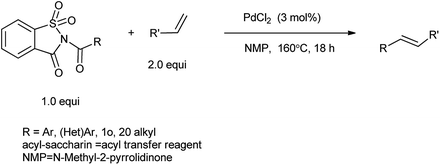 |
| | Scheme 78 Decarbonylative Heck reaction of amides via C–N activation. | |
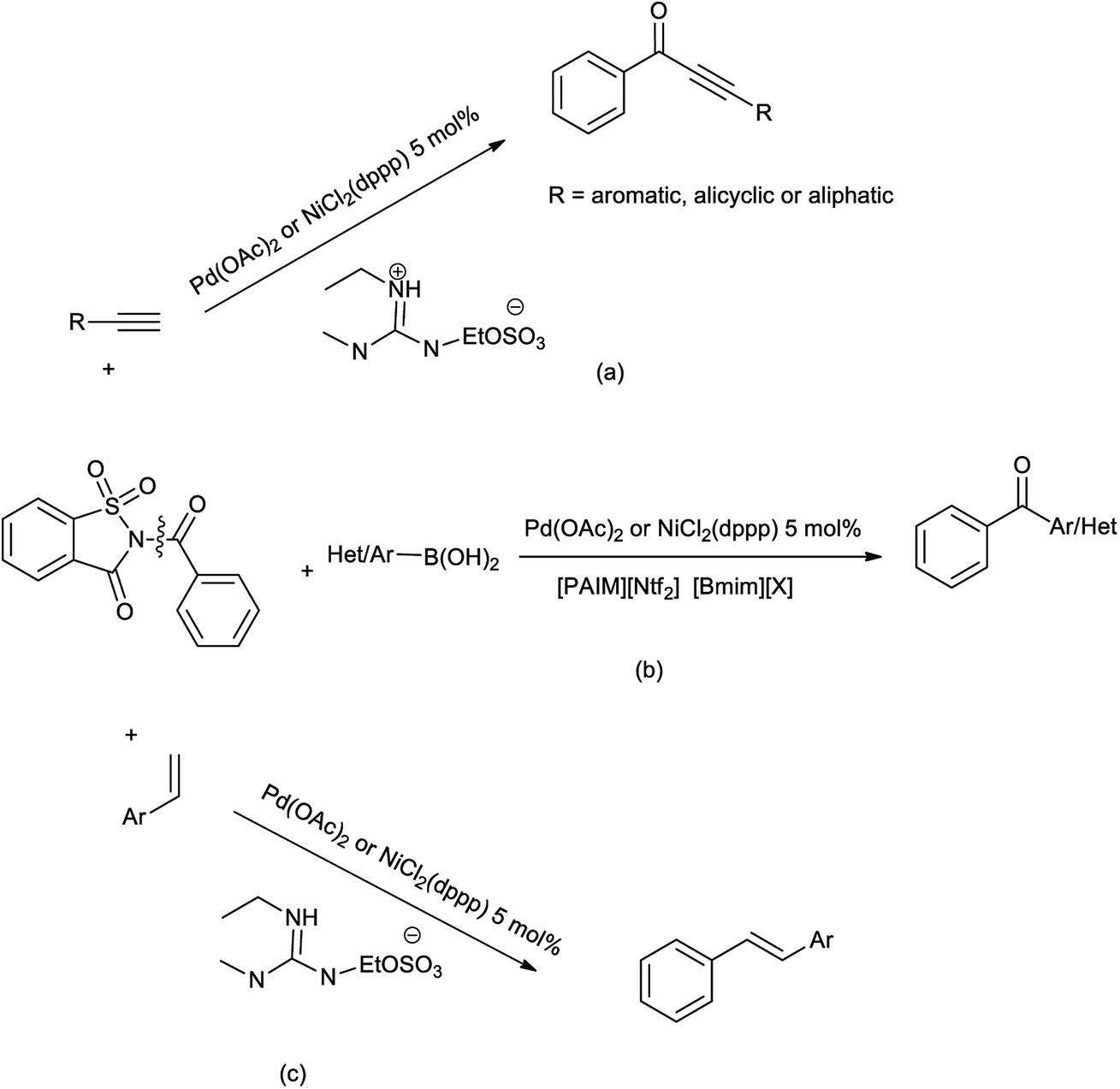
Malunavar et al. collectively evaluated the ionic liquid mediated benzoyl transfer-coupling in Sonogashira and Suzuki reaction in guanidinium-IL (GIL) as solvent, and Bmim-IL/[PAIM][NTf2] as solvent/base respectively. GIL was used as solvent in decarbonylative aryl transfer-coupling occurs in the Heck reaction (Scheme 79).95 In these reactions N-benzoylsaccharin was used as reagent and were catalyzed by the NiCl2(dppp) or Pd(OAc)2. This protocol was performed under mild condition with good yield and the ionic liquids are recycled and reused for 2 or 3 cycles further in reaction. While this helped to synthesize the wide range of keto-ethynes, diarylketones, and diarylethenes.
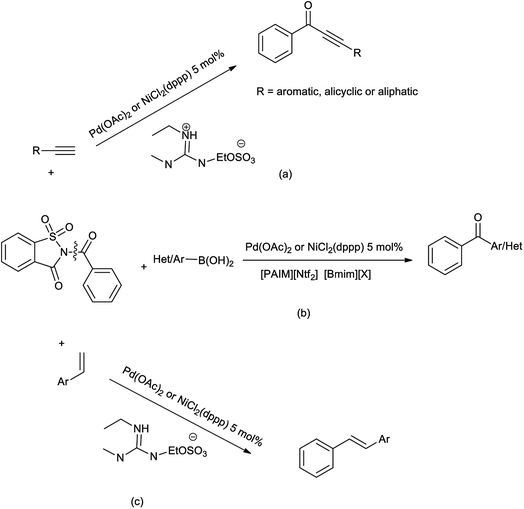 |
| | Scheme 79 (a) trans-Benzoylation Sonogashira cross-coupling in GIL using N-benzoylsaccharin, (b) trans-benzoylation Suzuki cross-coupling using N-benzoylsaccharin, and (c) decarbonylative Heck coupling in GIL using N-benzoylsaccharin. | |
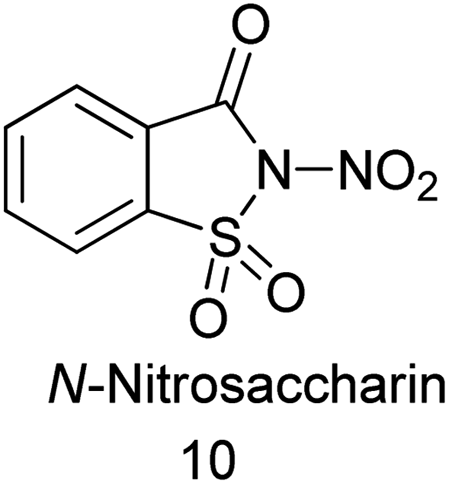
11. N-Nitro saccharin as a source of NO2
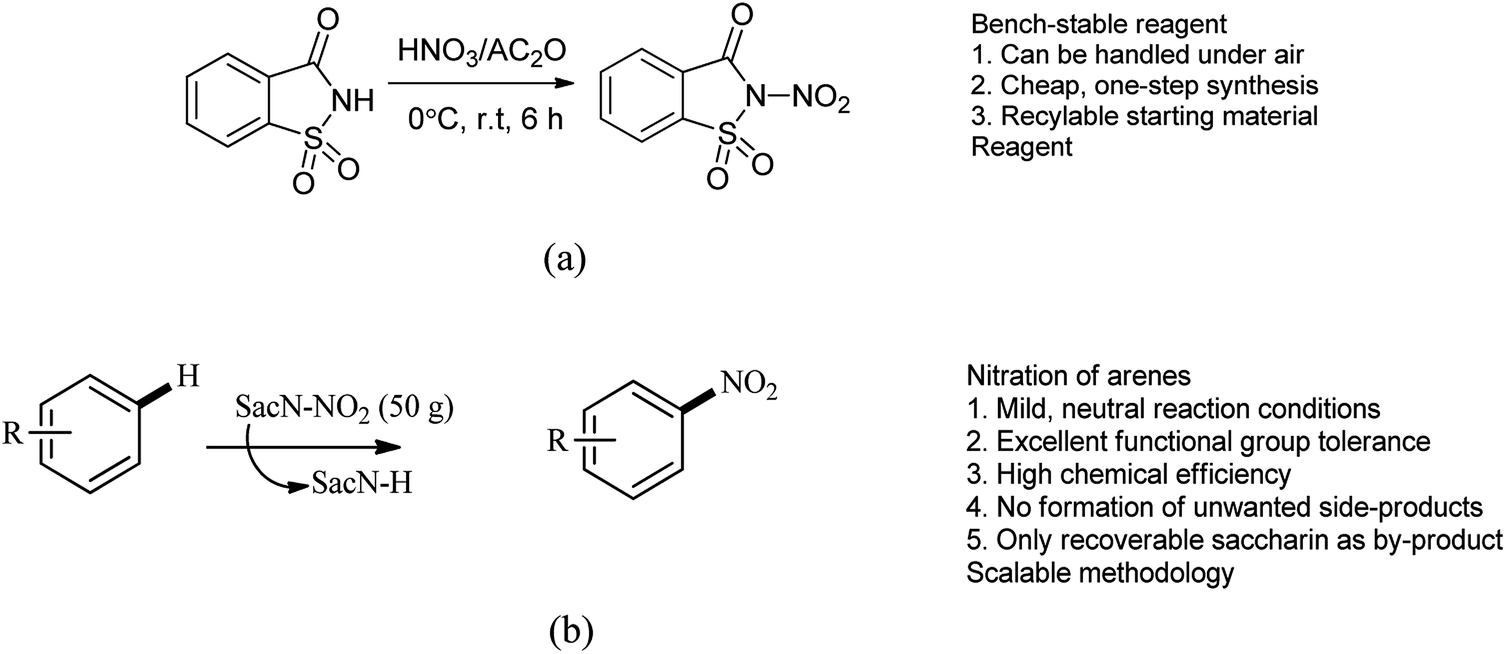
Till date ‘mixed acid’ such as mixture of nitric acid (HNO3) and sulfuric acid (H2SO4) as a source of active electrophilic nitronium (NO2+) species used for the production of nitroaromatics on both industrial and laboratory scales.96 This method includes several disadvantages which includes poor regioselectivity and functional group tolerance of the protocol, side products resulting from hydrolysis or oxidation are often obtained and it is unamenable to acid-sensitive groups. Here to overcome the limitation, Calvo et al. reported the synthesis of N-nitrosaccharin and its application in nitration of arenes (Scheme 80a and b).97 This study also gives acid-free methodology for the electrophilic nitration of a variety of heteroarenes and arenes displaying an exceptional functional group.
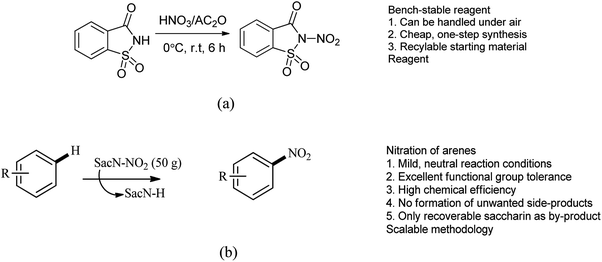 |
| | Scheme 80 (a) Synthesis of N-nitro saccharin. (b). Nitration of arenes. | |
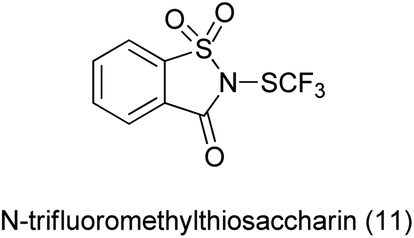
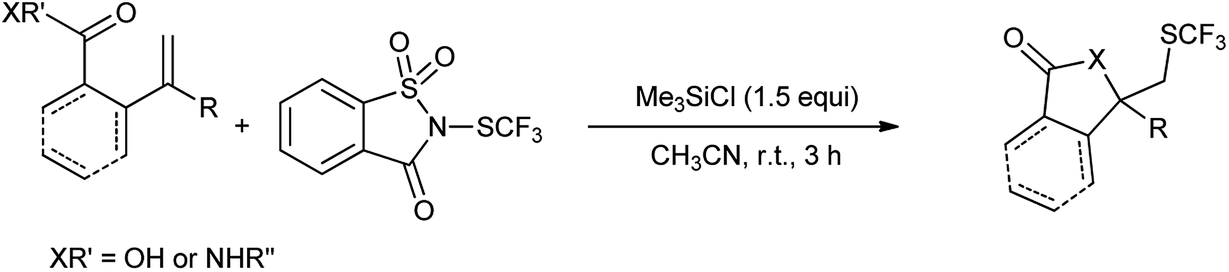
12. N-SCF3 saccharin as a source of N-SCF3
The simplest electrophilic trifluoromethylthiolating reagent was trifluoromethylsulfenyl chloride (CF3SCl). The use of this reagent was restricted due to its toxic and gaseous nature.98 The development of a readily accessible, easy-to-handle, electrophilic trifluoromethylthiolating reagent, which is effective for a broad substrate scope under relatively mild reaction conditions is highly desirable.
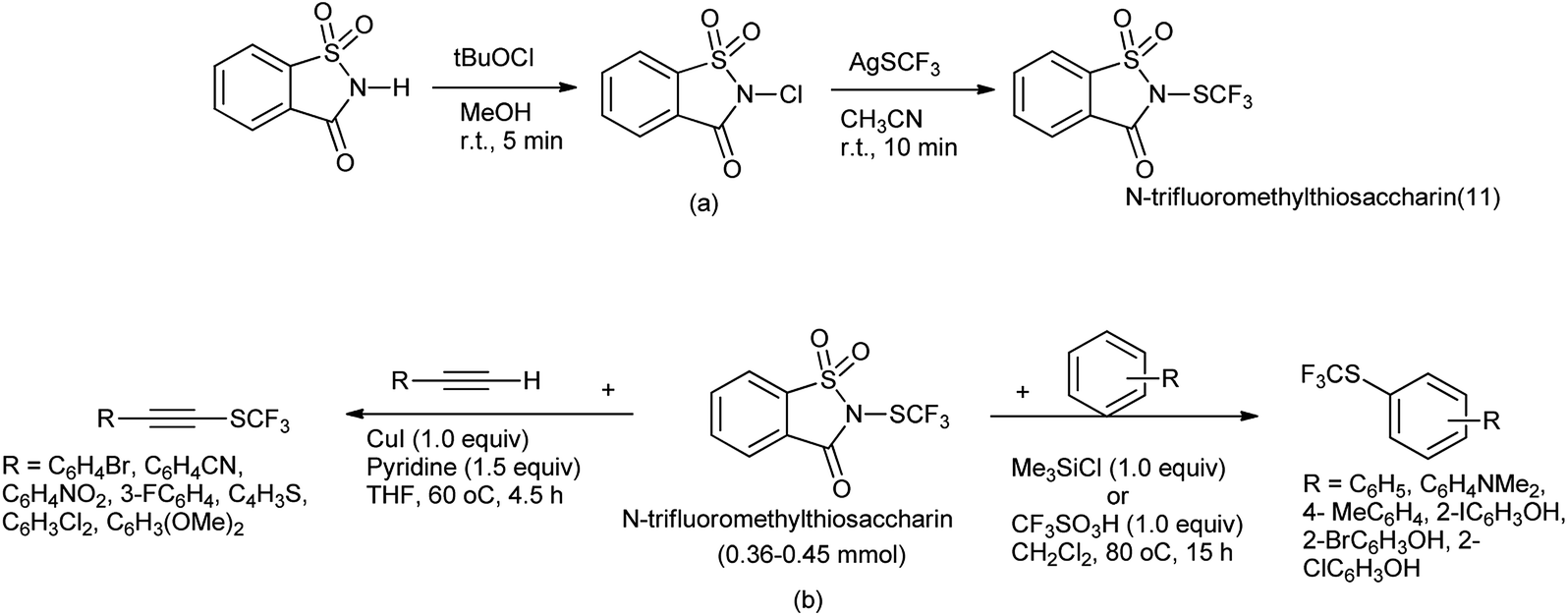
N-Trifluoromethylthiosaccharin was synthesis from saccharin (Scheme 81a) and further used as trifluoromethylthiolating reagent for variety of nucleophile such as thiols, amines, electron-rich arenes, alcohols, aldehyde, alkynes and acyclic β-ketoesters under mild conditions by Xu et al. (Scheme 81b).99
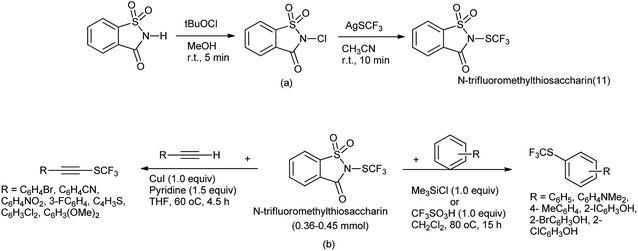 |
| | Scheme 81 (a) Synthesis of N-trifluoromethylthiosaccharin (b) trifluorothiolating reagent for various nucleophile. | |
Xu et al. also reported the lactomization/lactonization of olefins with highly selective Lewis acid mediated trifluoromethylthio-saccharin (Scheme 82).100 The reaction proceeds through the formation of thiiranium ion as intermediate, which further attacked by the amide or acid to generate the corresponding trifluoromethylthiolated lactam/lactone as shown in Scheme 83.
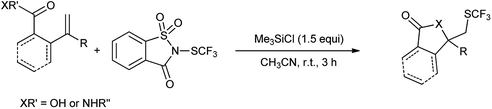 |
| | Scheme 82 Synthesis of trifluoromethylthiolated lactam/lactone. | |
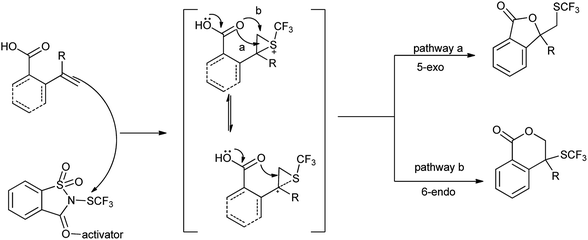 |
| | Scheme 83 Proposed mechanism. | |
Liu et al. reported enantioselective trifluoromethylthiolation of alkenes catalyzed by an indane-based bifunctional chiral sulfide with N-trifluoromethylthiosaccharin101 with 80% of yield. It was the first enantioselective trifluoromethylthiolation of alkenes that was enabled by a catalyst with a Lewis basic sulfur center (Scheme 84). The reagent is highly reactive and shelf-stable electrophilic effective in other trifluoromethylthiolation processes. This chiral bifunctional sulfide catalyst was also designed and delivered excellent enantioselectivities for this transformation, which should inspire the design of other Lewis basic catalysts based on this scaffold.
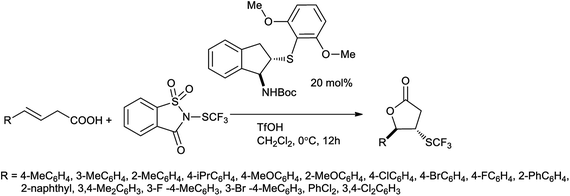 |
| | Scheme 84 Enantioselective trifluoromethythiolation lactonization of alkenes. | |
Carthy et al. demostrated that N-trifluoromethylthiosaccharin was used to activate triglycosides in presence of catalytic amount of TMSOTf.102 It acts as powerful reagent to activate a range of ethyl thiogylcosides which underwent glycosidation reactions with various glycosyl acceptors to produce the desired disaccharides with good yield (Scheme 85). However this new promoter system could selectively activate ethyl thioglycoside in the presence of phenyl thioglycoside with the same protecting group pattern. So, this orthogonal activation, in combination with thioperoxide promoter system, can be utilized to design iterative one-pot oligosaccharide assembly.
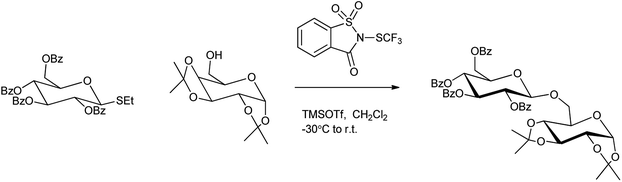 |
| | Scheme 85 Activation of triglycosides by trifluoromethylthiosaccharin. | |
13. N-Fluorosultam as a fluorinating agent
Before the advancement in fluorinating reagent hazardous fluorooxy or perchloryl fluoride compounds were used for fluorination of carbanions.103
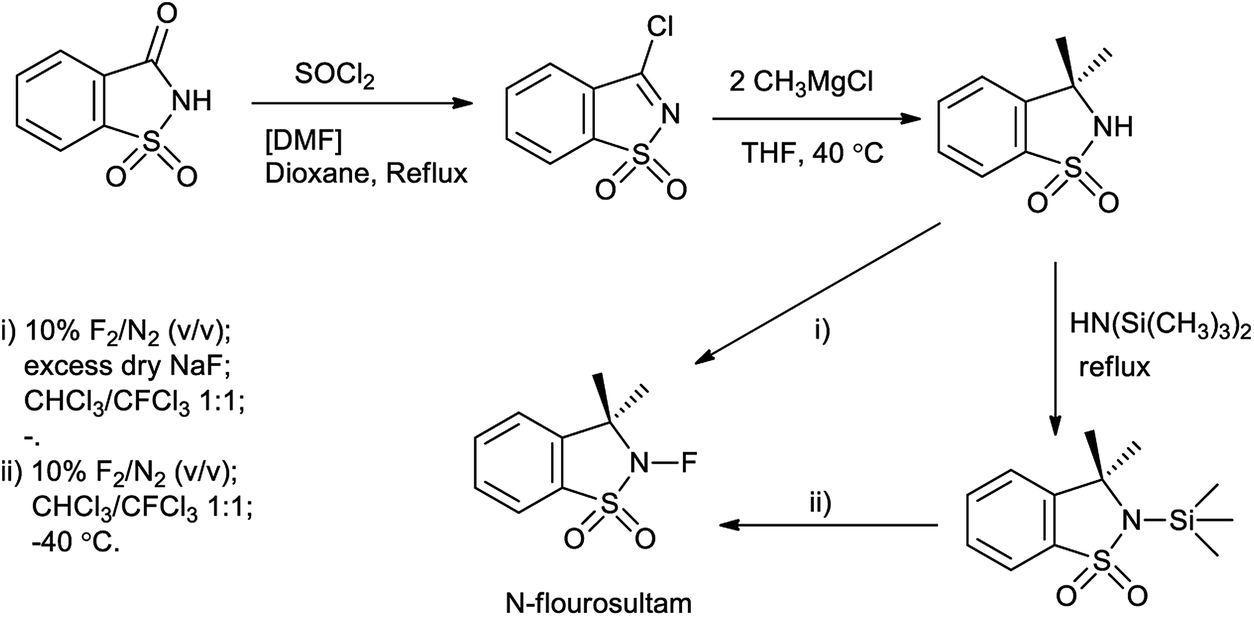
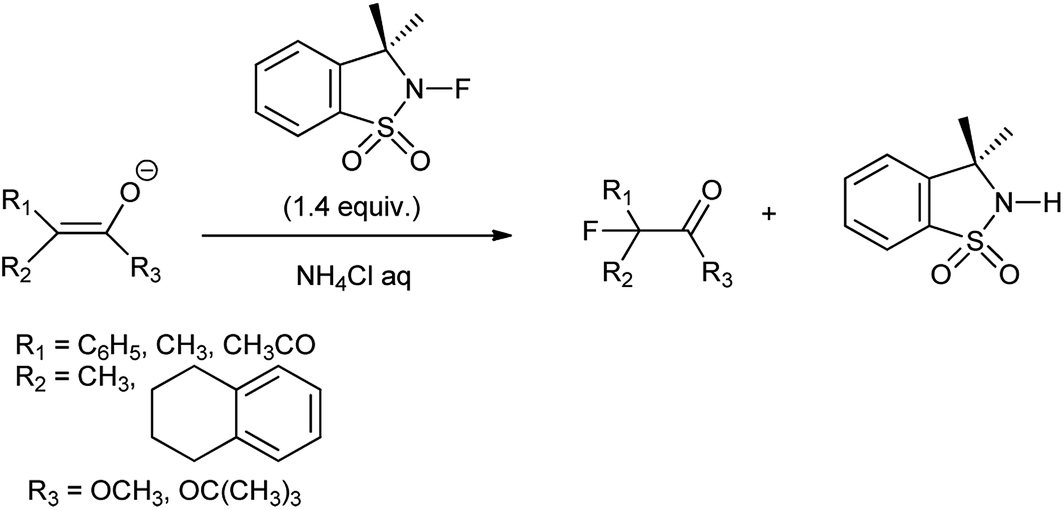
N-Fluorosultam was proven to be versatile, efficient, and easily available fluorinating reagent. N-Flourosultam was synthesized from the saccharin by Differding et al. (Scheme 86). The carbonyl compound undergoes fluorination in presence of N-fluorosultam (Scheme 87).104 N-Fluorosultam found to be versatile, an efficient, and easily accessible fluorinating reagent. It has remarkable advantages over commercially available N-alkyl-N-fluorosulfonamides, in particular due to absence of H-atoms in the α-position of the N-atom and, thus, preventing base-induced HF elimination.
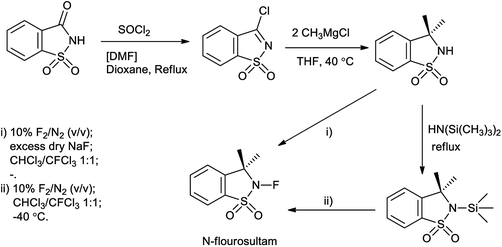 |
| | Scheme 86 Synthesis of N-flourosultam from saccharin. | |
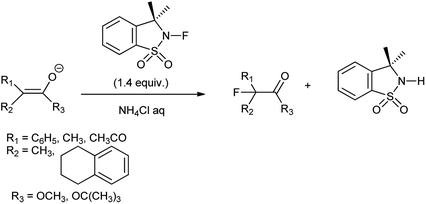 |
| | Scheme 87 Fluorination of carbonyl compound. | |
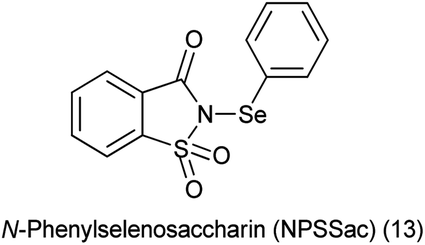
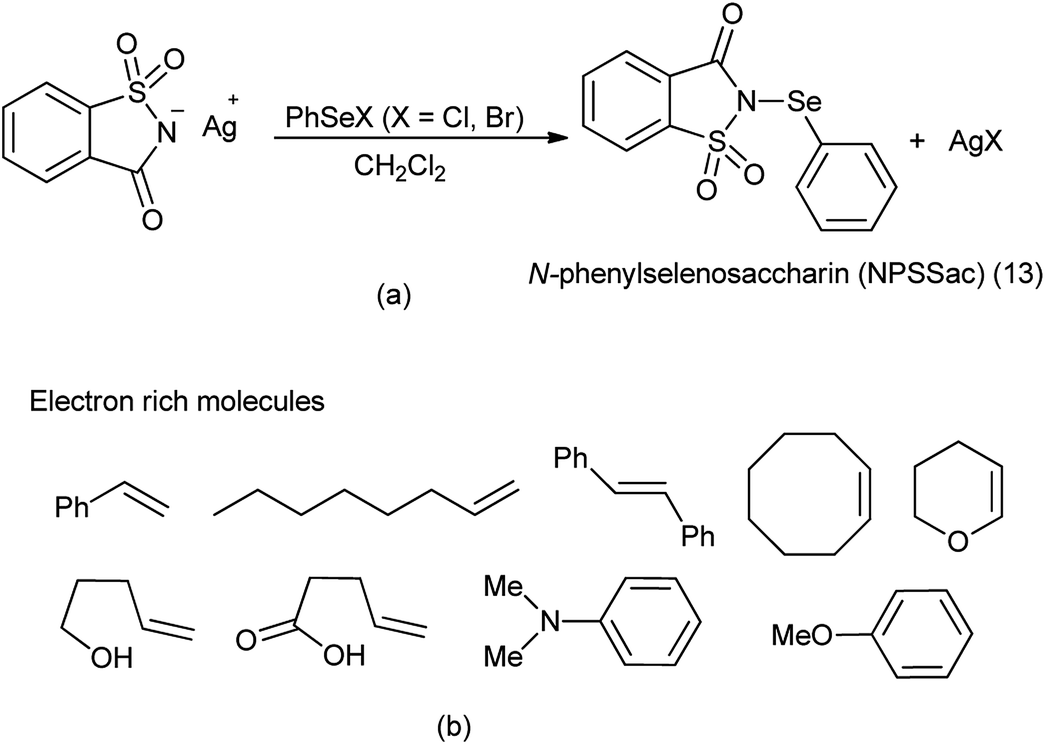
14. N-Phenylselenosaccharin (NPSSac)
From last two decades focused was to find better sources of electrophilic selenium species. For direct sources of the PhSe group, N-phenylselenophthalimide (NPSP) and phenylselenenyl chloride or bromide (PhSeX, X = Cl, Br) was used.105 There is other method to insert the electrophilic phenylselenium reagent by the oxidation of PhSeSePh with several oxidizing agents like hypervalent iodine,106 ammonium persulfate,107 or metal-containing oxidizing agents.
A new reagent N-phenylselenosaccharin (NPSSac) was synthesized and used as a source of the electrophilic phenylselenyl group by Tingoli et al. (Scheme 88a). This relatively stable new compound was able to react with electron rich organic molecules like alkenes in the presence of internal or external nucleophiles, enolizable carbonyl derivatives, or activated aromatic substrates, under very mild conditions (Scheme 88b).108
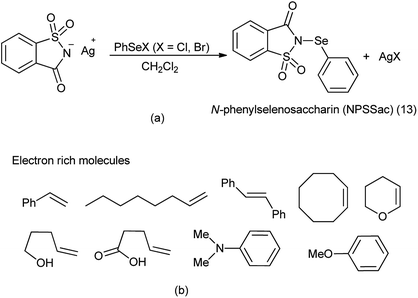 |
| | Scheme 88 (a) Synthesis of N-phenylselenosaccharin (b) phenylselenylation of electron rich molecules. | |
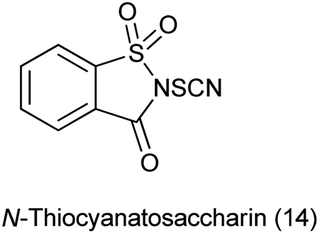
15. N-Thiocyanatosaccharin as a source of SCN
Thiocyanato functional group is of great important as it is synthetic intermediate to access sulfur-containing compounds109 and widely present in bioactive natural products.110
Recently, N-thiocyanatosaccharin was prepared from saccharin in two steps (Scheme 89a). This new reagent was used as novel electrophilic thiocyanation to diverse nucleophiles such as indoles, benzothiophenes, oxindoles, phenols, aromatic amines, aromatic ketones and β-keto carbonyl compounds by Wu and coworkers (Scheme 89b).111 The potential recycling of saccharin, the wide scope of substrates, and the mild reaction conditions made this protocol much more practical.
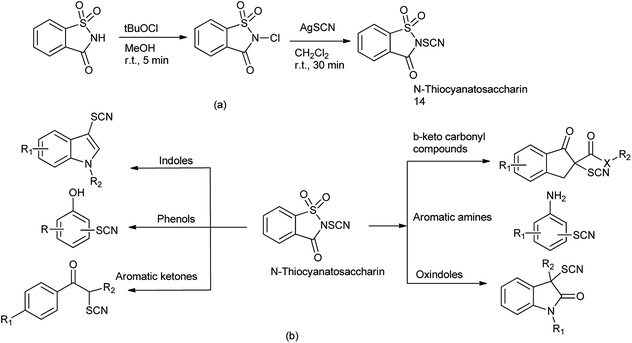 |
| | Scheme 89 (a) Synthesis of N-thiocyanatosaccharin (b) reaction of N-thiocyanatosaccharin with different nucleophile. | |
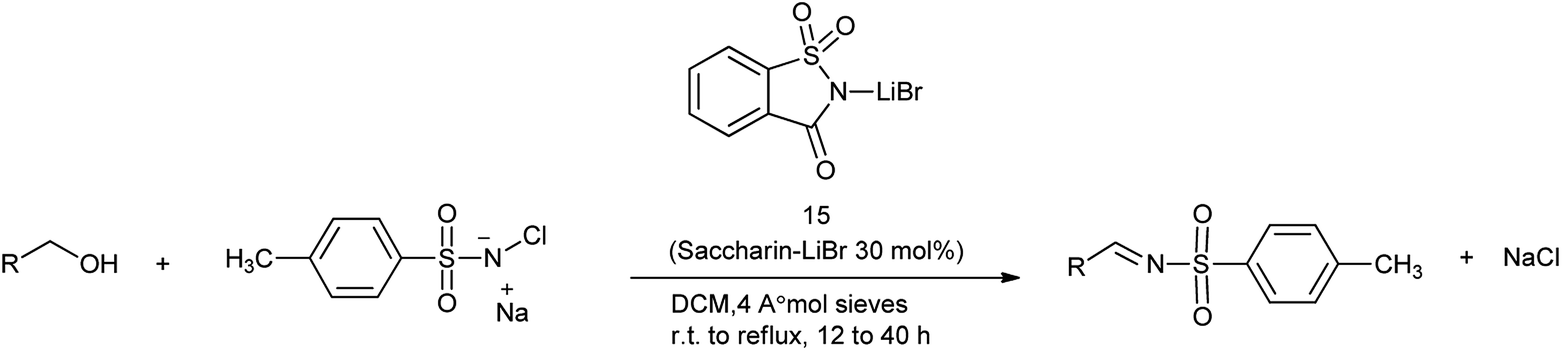
16. Saccharin-lithium bromide
N-Sulfonylimines are one of the few types of electron-deficient imines that are stable enough to be isolated but reactive enough to undergo addition reactions.112 The direct condensation of carbonyl compounds with sulfonamides is used as the straightforward route for the preparation of N-sulfonylimines usually requires harsh acidic conditions.113 Patel et al. reported the saccharin-lithium bromide catalyzed synthesis of N-tosylimines directly from alcohols under the neutral and mild conditions (Scheme 90).114 The protocol involves oxidation of alcohols to aldehydes or ketones with chloramine-T followed by their condensation with the in situ formed oxidation by-product p-toluenesulfonamide to afford N-tosylimines in a one-pot operation (Scheme 91). The present work opens up a new and efficient one pot synthetic route to N-sulfonylimines directly from alcohols.
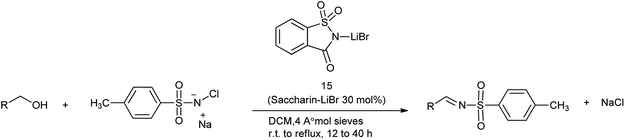 |
| | Scheme 90 Synthesis of N-tosylimines directly from alcohols. | |
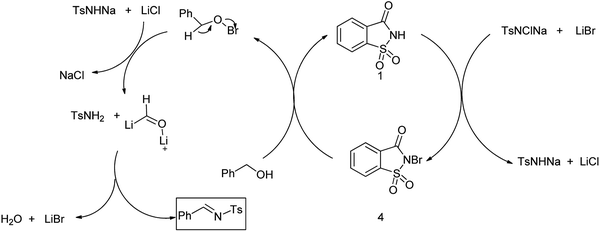 |
| | Scheme 91 Mechanism followed for the formation of N-sulfonylimines. | |
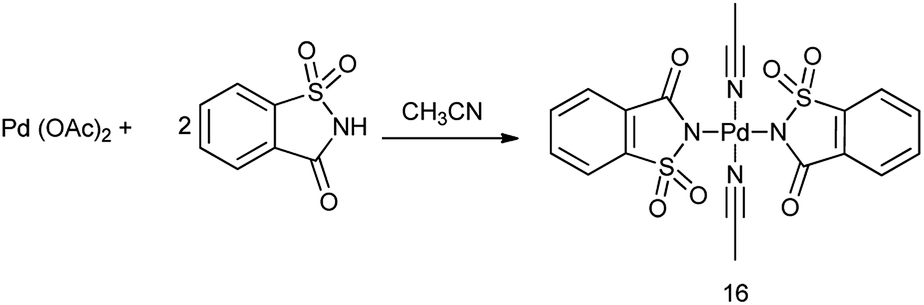
17. Palladium saccharin
Bisacetonitrile palladium disaccharide complex (16) was synthesized by treatment of acetonitrile solution of palladium diacetate with saccharin (Scheme 92).
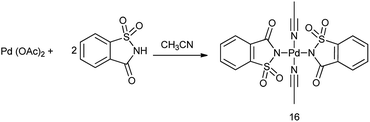 |
| | Scheme 92 Synthesis of palladium saccharin. | |
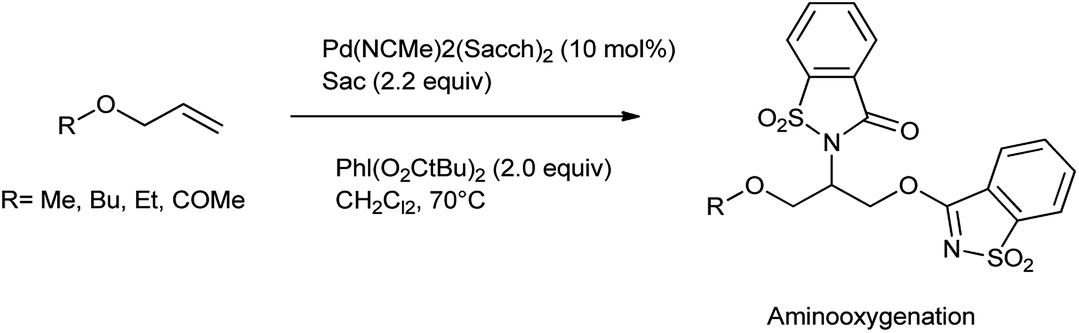
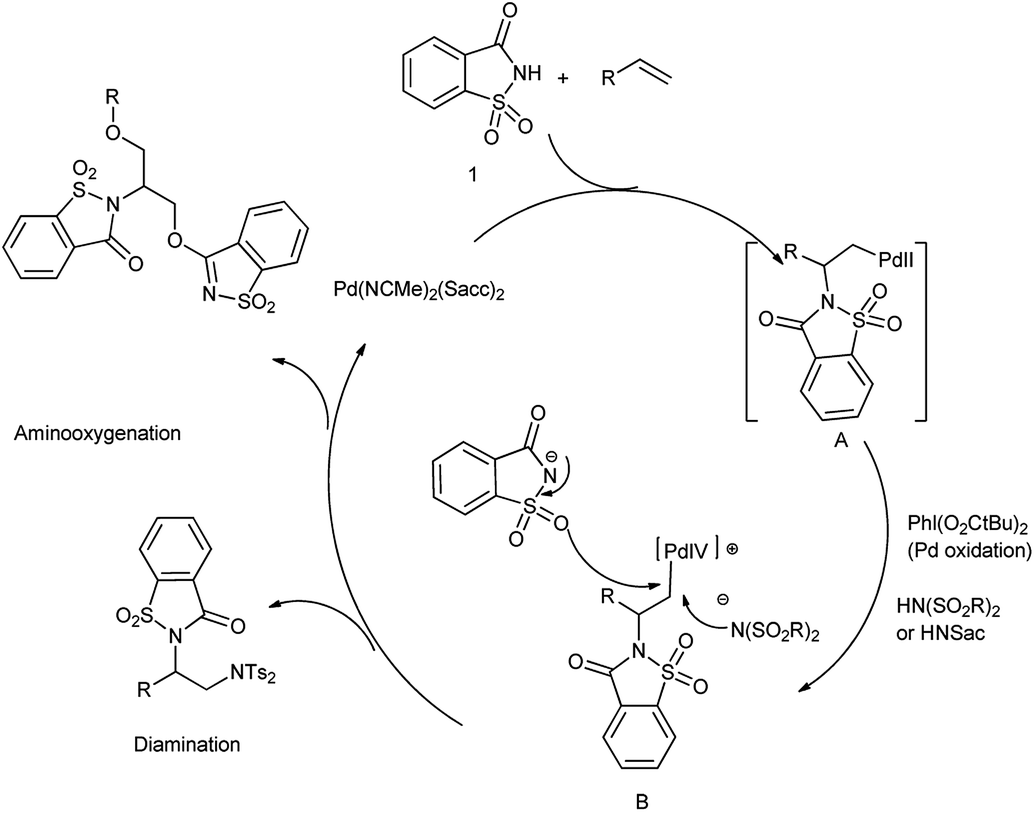
The oxidative difunctionalization of alkenes represents a powerful tool for the efficient 1,2-introduction of heteroatoms into organic frameworks and thus for structural diversification from common hydrocarbon groups. Within such vicinal difunctionalization, palladium catalysis constitutes a particularly effective approach.115 Martínez et al. reported the formation of bis-saccharido palladium(II) complexes by the addition of acetonitrile solution of palladium diacetate in saccharin, a bisacetonitrile palladium saccharide complex Pd(NCMe)2(Sacch)2 is formed which further on crystallization gave trimeric palladium complex. The aminooxygenation or diamination of alkenes took place in the presence of Pd(NCMe)2(Sacch)2 as a nitrogen source shown in Schemes 93 and 94 respectively116 The mechanism proceed through the formation of aminopalladium intermediate A when palladium catalyst engage in aminometalation with saccharin further metal oxidation to higher oxidation state palladium(IV) intermediate B is accomplished with the hypervalent iodine reagent shown in Scheme 95.
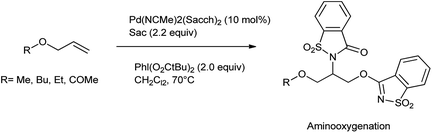 |
| | Scheme 93 Aminooxygenation catalyzed by palladium catalyzed of allyl ether or ester. | |
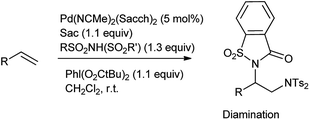 |
| | Scheme 94 Palladium-catalyzed diamination of terminal alkenes with Pd(NCMe)2(Sacch)2. | |
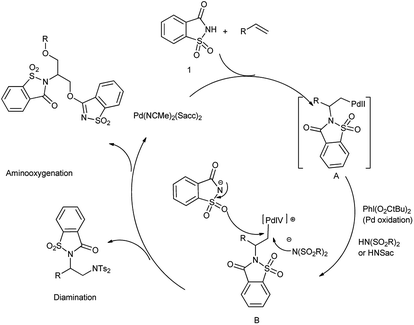 |
| | Scheme 95 Mechanism of palladium-catalyzed difunctionalization reaction with saccharin. | |
The diamination pathway proceeds through nucleophilic attack of bissulfonimide at the α-carbon of the σ-alkylpalladium(IV) leading to diamination products but in case of saccharin, the nucleophilic displacement takes place through oxygenation to provide aminooxygenation products. Saccharin owes its attractiveness as a nitrogen source to its commercial availability and low price.
Iglesias et al. also reported the intramolecular diamination of alkene catalyzed by palladium chloride. Both bistosylimide and saccharin act as the nitrogen source (Scheme 96).117 This method employs two commercially available nitrogen sources with high regioselectivity under very mild conditions and morever no intramolecular reaction step is required.
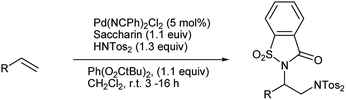 |
| | Scheme 96 Palladium catalyzed diamination of alkene. | |
18. DMAP–saccharin
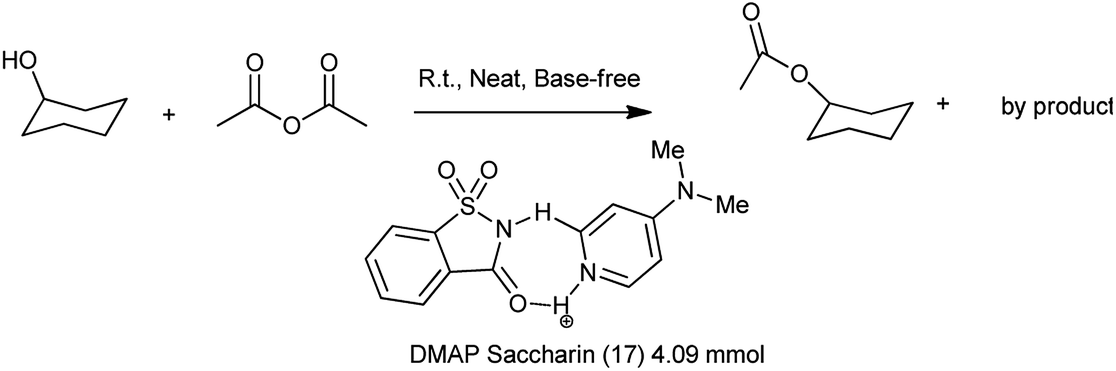
Recently, it has been reported that 4-(N,N-dimethylamino)pyridine (DMAP) is an effective homogeneous catalyst for the esterification of alcohol with acid anhydride, the ring-opening polymerization, the silylation of alcohol. The homogeneous nature of DMAP catalyzed acylation still suffers from the problem of separation. Lu et al. 2011 showed the catalytic condensation reaction of alcohol with equimolar amount of acid anhydride under solvent free and base free conditions by using 4-(N,N-dimethylamino)pyridine (DMAP) DMAP saccharin catalyst shown in Scheme 97. The DMAP–saccharin catalyzed acylation of alcohol was efficiently carried out more than 8 times without losing the activity118 which might efficient and reliable process in industry.
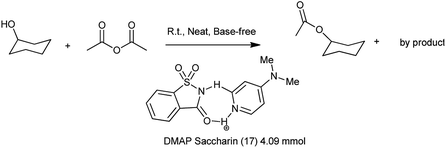 |
| | Scheme 97 DMAP–Saccharin catalyzed acylation of 1-cyclohexanol. | |
Lu et al. (2016) reported four pyridinium saccharinate salt including pyridinium saccharinate (I), 4-N,N-dimethylaminopyridinium saccharinate (II), 2-N,N-dimethylaminopyridinium saccharinate (III) and 4-(1-pyrrolidinyl)pyridinium saccharinate (IV) as effective as recyclable homogeneous acylation catalyst for alcohols as shown in Scheme 98. These salts (I), (III) and (IV) contain very interesting seven membered synthon showing multiple H-bonding interactions for pair of pyridinium cation and saccharinate anion in the solid state. The salt (II), exhibits H-bonding interaction of N(sac). HeN(py) in the solid state, instead of seven-membered synthon (Scheme 99). Salt (III) was a poor catalyst for esterification due to its steric hindrance and salt (IV) without donating substituent on pyridine ring was also proved a poor catalyst. The catalytic reactivity studies show that salts (I), and (II), are both very effective, with salt (II), even better in reactivity, and both salts could be recycled for 10 times without decreasing catalytic activity in the esterification of a variety of alcohols, under solvent-free and base-free conditions at room temperature.
 |
| | Scheme 98 Acylation reaction of alcohol with acid anhydride catalyzed by pyridinium saccharin salt. | |
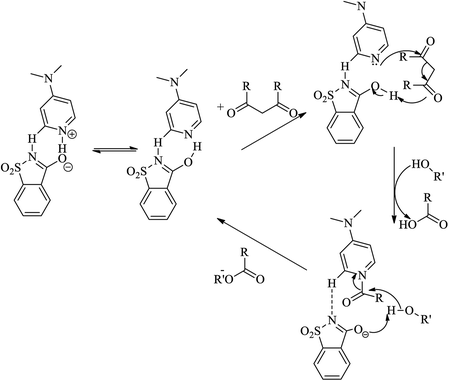 |
| | Scheme 99 Mechanism of DMAP catalyzed acylation reaction based on synthon connectivity as the resting state with O(sac)⋯HeN(py)/O(sac)–H. N(py) for propagation. | |
Salts (I) and (II) whose kinetics have been studied, are very efficient recyclable catalysts for the acylation of a variety of 2° and 3° alcohols, from the kinetic studies, salt (II) turned out to be the best catalyst, even better than salt (I) in this esterification.119
Lal et al. 1957 reported the polymerization of methyl methacrylate with different amines and salts of saccharin. Results showed that only aromatic tertiary amines in admixture with saccharin were able to polymerize the monomer i.e., salts of N,N-dimethyltoluidine (o-, m-, and p-) or N,N-dimethylaniline with saccharin catalyzed the polymerization whereas polymerization was not favoured by the secondary or primary amines.120 Thus, reagent is indeed an inexpensive, efficient, and recoverable catalyst for benign esterification methods.
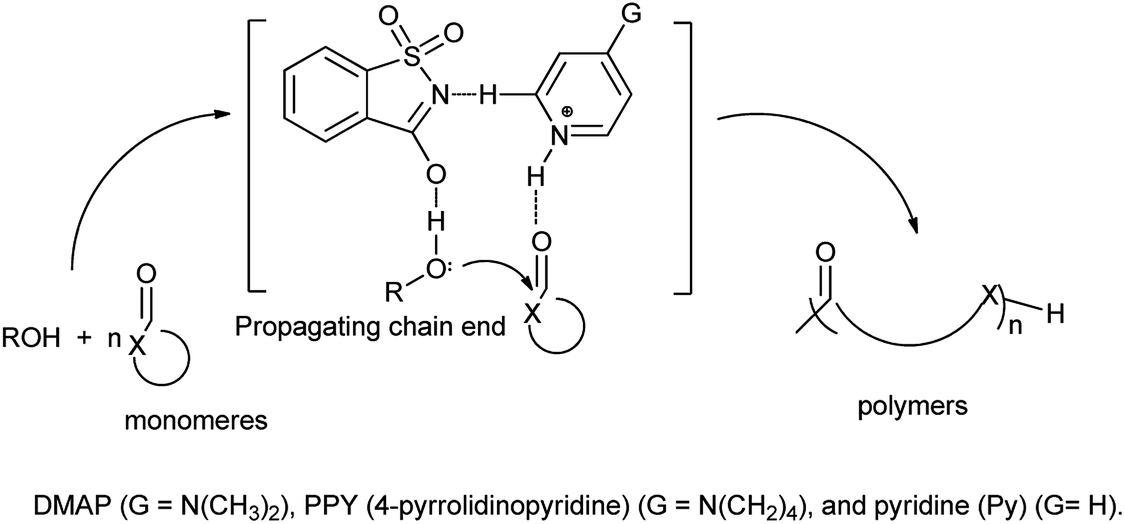
Wei et al. also reported that pyridinium saccharinate promotes ring-opening polymerizations (ROP) of typical cyclic ester monomers like trimethylene carbonate (TMC), δ-valerolactone (VL), L-lactide (LLA), and ε-caprolactone (CL) as the monomers, and used three pyridines of pyridine (Py), DMAP and 4-pyrrolidinopyridine (PPY) as Bronsted part of pyridinium saccharinate co-catalysts. Study showed that DMAP·saccharinate (DMAP·Sac) catalyzed ROP of LLA at a fast rate (Scheme 100).121
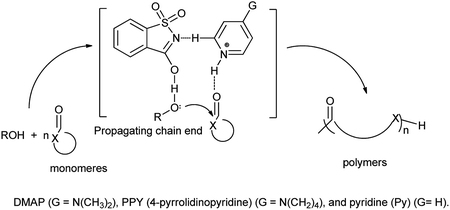 |
| | Scheme 100 Proposed ring-opening polymerization mechanisms of cyclic esters using pyridinium saccharinate as the co-catalyst. | |
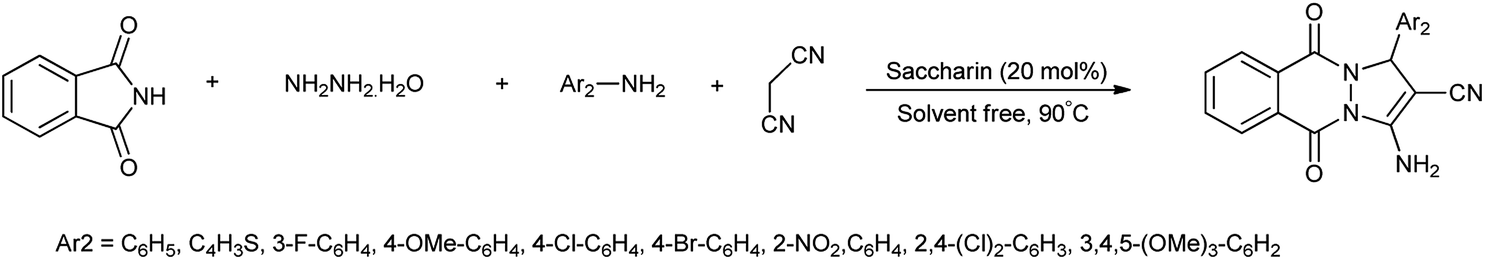
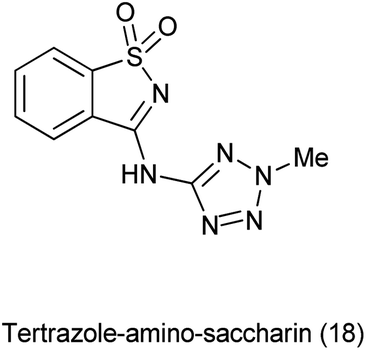
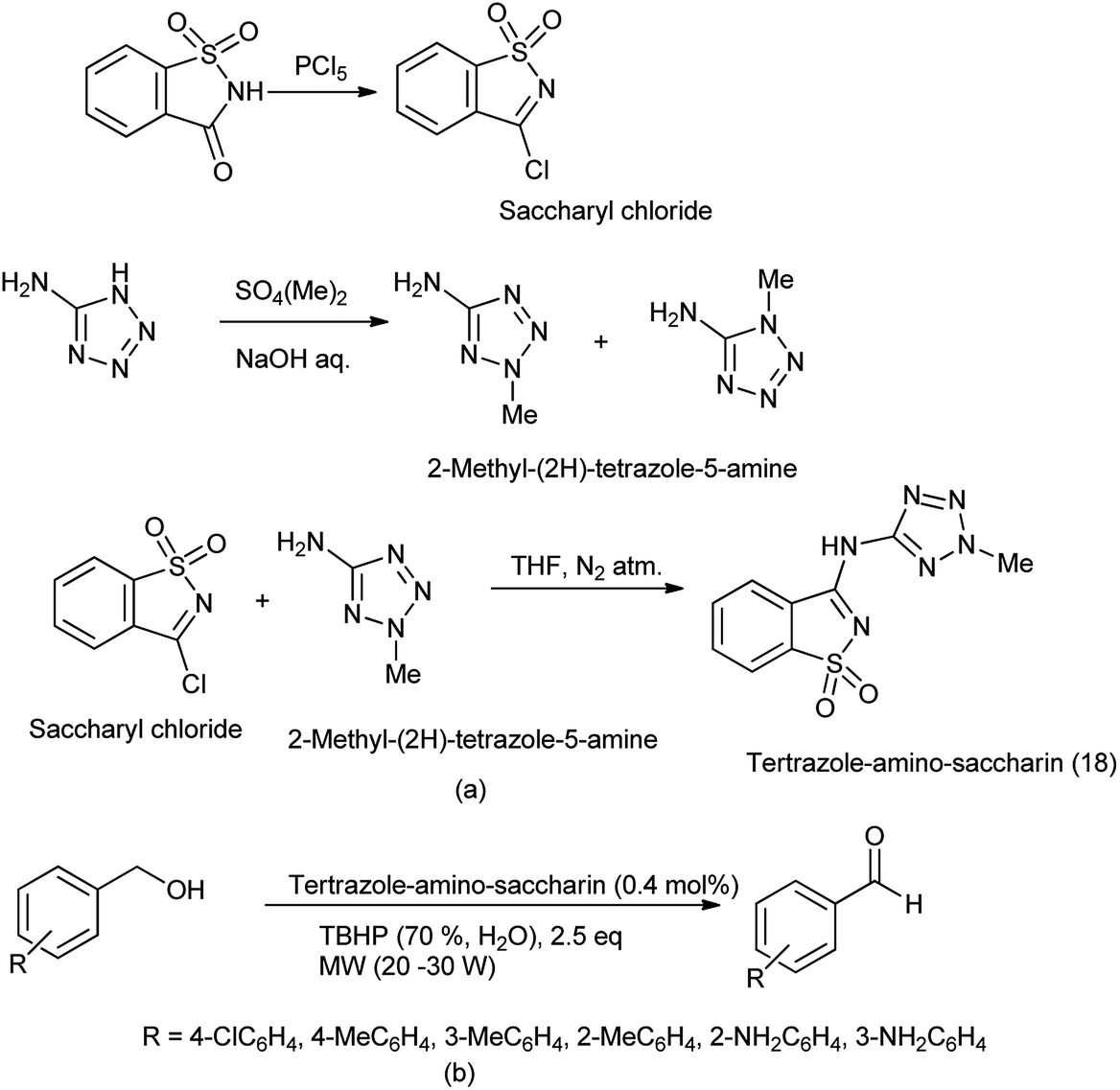
19 Tetrazole-amino-saccharin
Tetrazole-amino-saccharin was synthesized from the reaction of saccharyl chloride (synthesized by saccharin) and 2-methyl-2(H)-tertrazole-5-amine (derived from tetrazole-5-amine) under inert condition as described Scheme 101(a).122
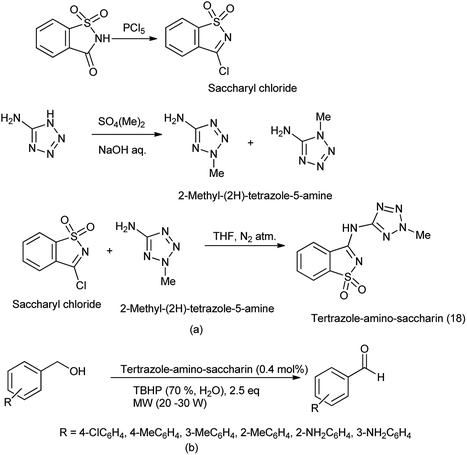 |
| | Scheme 101 (a) Synthesis of tetrazole-amino-saccharin (b) oxidation of benzyl alcohol under microwave condition in presence of tetrazole-amino-saccharin. | |
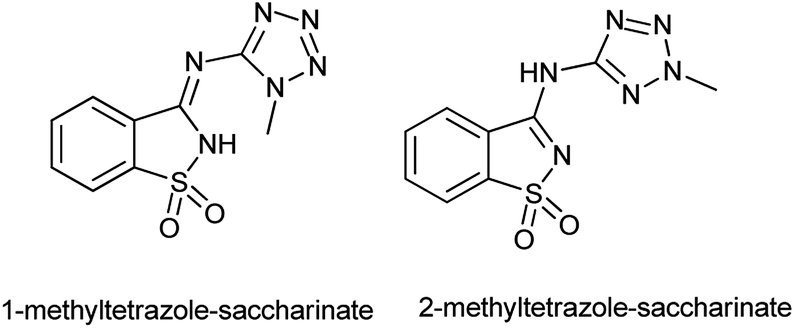
An anaerobic oxidation of benzyl alcohols was reported by Frija et al. in the presence of tetrazole-amino-saccharin as organocatalyst under solvent free conditions and microwave assisted process comprising aqueous tert-butyl hydroperoxide (TBHP) as oxidant (Scheme 101b).123 Ismael et al. reported the structure of nitrogen-linked 1- and 2-methyltetrazole-saccharinates (Fig. 1).124 As both tetrazoles and benzisothiazoles (saccharins) have relevant applications in areas such as agriculture, medicine, and food chemistry, and also act as important building blocks in organic synthesis.125 Currently, some compounds such as 5-chloro-1-phenyl-(1H)-tetrazole and 3-chloro-1,2-benzisothiazole-1,1-dioxides (saccharyl chlorides) are used as derivatizing agents for phenols and alcohols.126
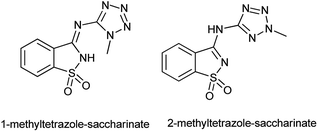 |
| | Fig. 1 Structure of 1- and 2-methyltetrazole-saccharinate. | |
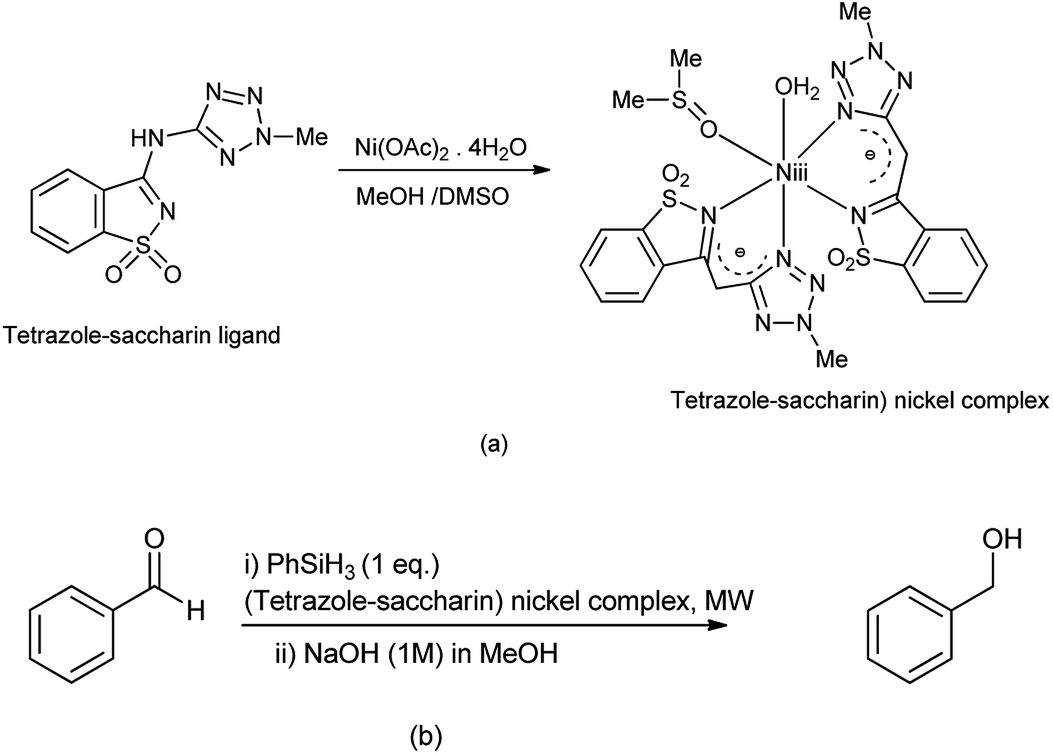
Frija et al. reported the hydrosilative reduction of aldehydes in presence of (tetrazole-saccharin) nickel complex under microwave irradiation (Scheme 102b). The (tetrazole-saccharin) nickel complex can be synthesized under room temperature reacting tetrazole-amino-saccharin with Ni(OAc)2·4H2O salt (Scheme 102a).
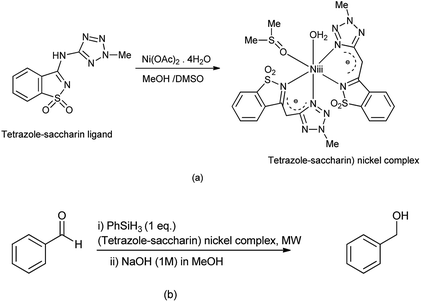 |
| | Scheme 102 (a) Synthesis of (tetrazole-saccharin) nickel complex (b) hydrosilative reductive of aldehydes in presence of nickel complex. | |
This complex consists of two monoanionic bidentate N,N-(tetrazole-saccharin) ligands, one DMSO and water molecule coordinated to Ni(II) ion (Scheme 102a).127 The reaction protocol carried out under environment friendly conditions and is effective.
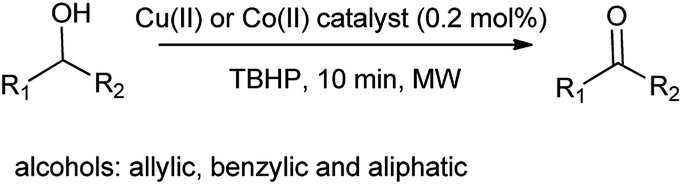
Frija et al. also investigated the oxidation of secondary alcohols such as allylic, benzylic, and aliphatic secondary alcohols to corresponding ketones in the presence of cobalt(II) and copper(II) tetrazole-saccharinate as an effective catalyst (Scheme 103).128 Full conversion was carried out under mild conditions and overcome the major drawback as oxidative (dehydrogenation) activation of alcohol. Moreover allows the wide range of transformation of alcohol.
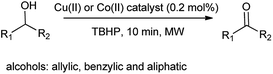 |
| | Scheme 103 Conversion of alcohols to respective ketones. | |
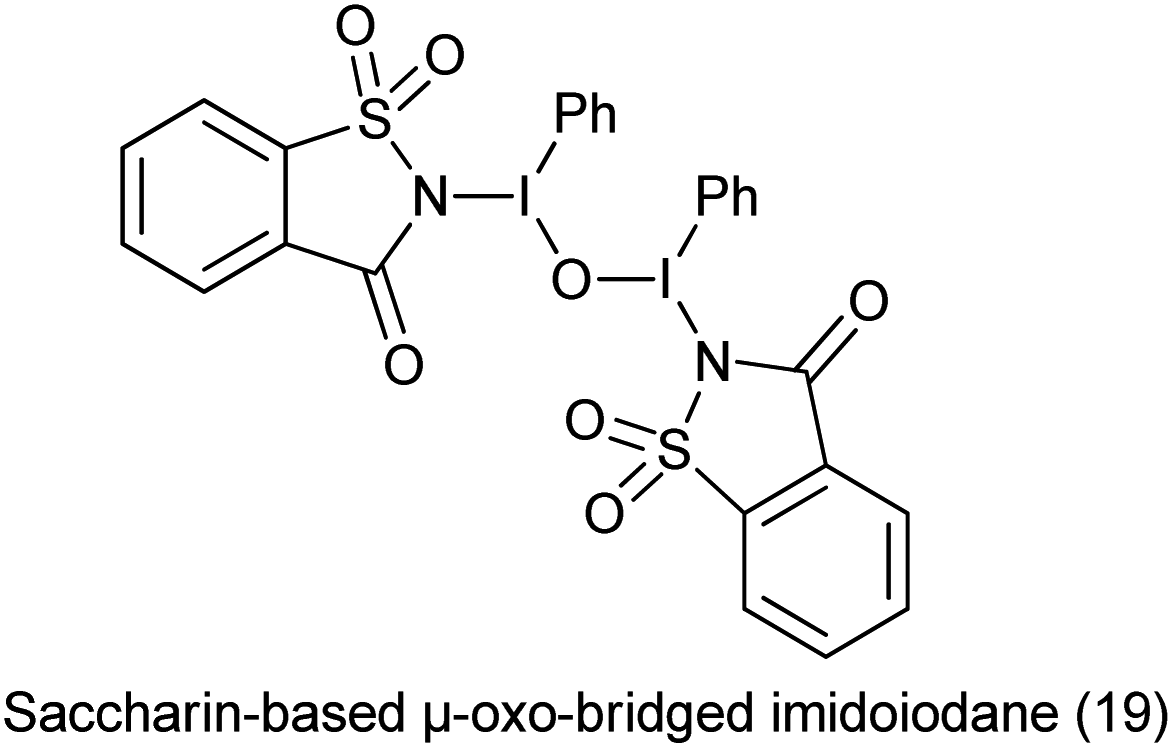
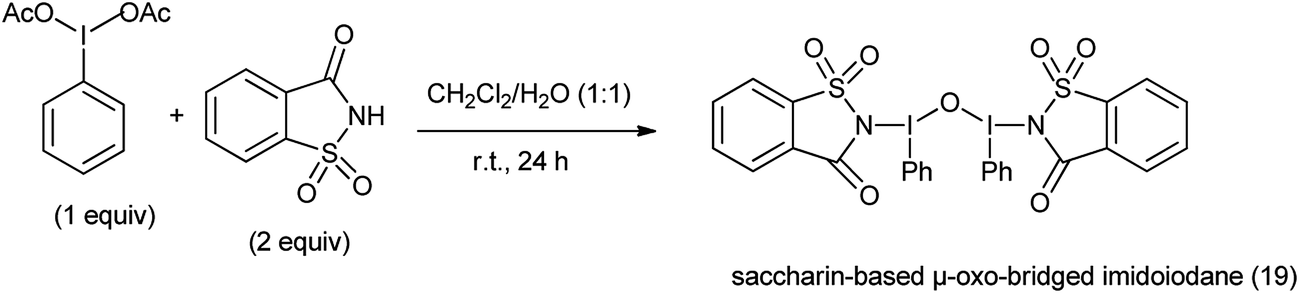
20. Saccharin-based μ-oxo-bridged imidoiodane
Hypervalent iodine reagents have found to be efficient and eco-friendly and efficient oxidizing reagents for various oxidative transformations.129 Saccharin-based μ-oxo-bridged imidoiodane (19) was synthesized by reaction of (diacetoxyiodo)benzene and saccharin (Scheme 104).
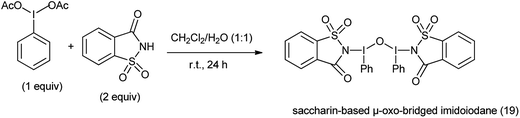 |
| | Scheme 104 Synthesis of saccharin based μ-oxo-bridged imidoiodane. | |
Yoshimura et al. reported Saccharin-based μ-oxo-bridged imidoiodane used for the synthesis of α-aminated carbonyl compounds using silyl enol ethers (Scheme 105).130 The reagent showed high reactivity and selectivity than other reagents.
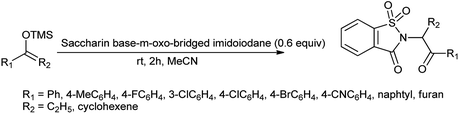 |
| | Scheme 105 Synthesis of α-aminated carbonyl compound using saccharin based μ-oxo-bridged imidoiodane. | |
21. Saccharin as amine source
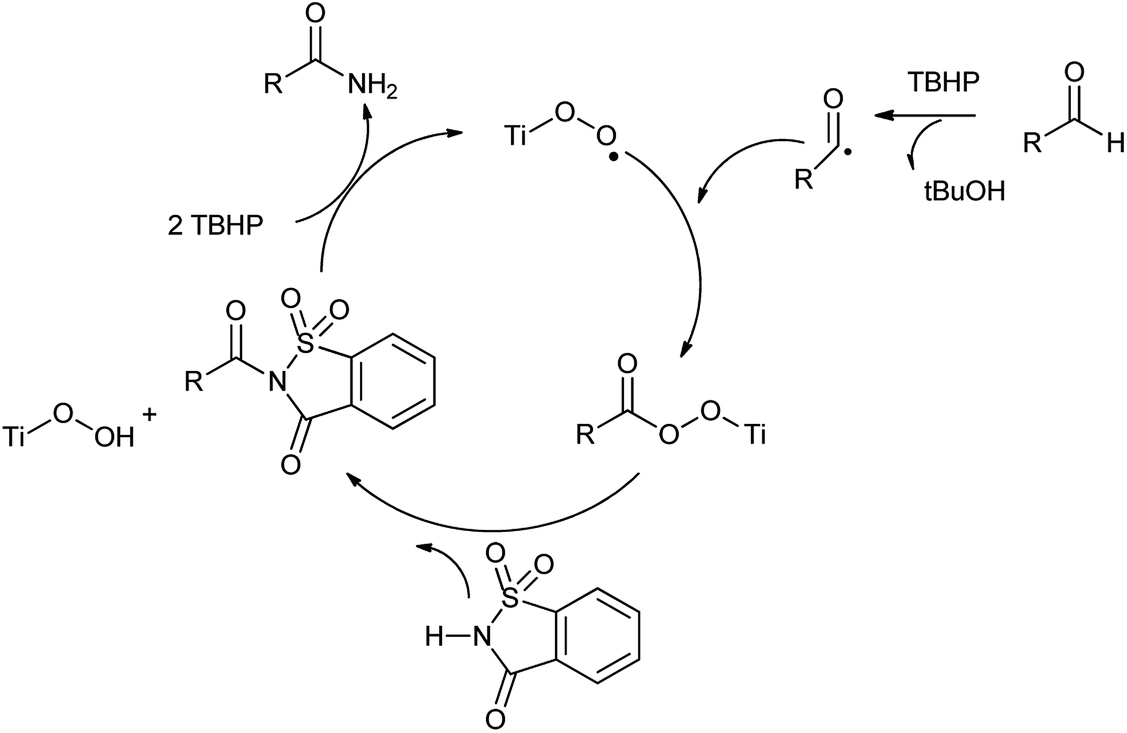
There are many researches recently carried out for the synthesis of organonitrogen chemicals from sustainable sources instead of ammonia. This is greater step towards the greener approach in the organic synthesis. As Pelckmans and coworkers131 reported wide range of amines which were synthesized from biomass-derived substrates by using sustainable heterogeneously catalyzed reaction which could be highly effective in chemical industry. Another study carried by Yan in 2015 showed that waste shrimp, lobster and crab shells could be turn in to nitrogen rich chemicals which would beneficial for environment as well as economy.132 A new heterogeneous catalytic system (saccharin/Ti-super oxide/TBHP) has found to be efficiently catalyzes oxidative amidation of aldehydes to produce various primary amides over Ti-superoxide using TBHP as oxidant by Kamble et al. (Scheme 106).133 Saccharin assesses as amine source and tolerates a wide range of different functional groups.
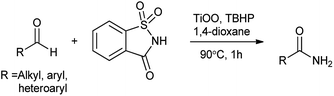 |
| | Scheme 106 Direct oxidative amidation of aldehyde or methyl arenes. | |
This method presented various advantages such as Ti catalyst is recyclable, good functional group compatibility, wide range of substrate scope, no additives and can be easily scaled up, mild reaction conditions and reused for 3 cycles without loss of activity. A possible mechanism and the role of the catalyst in oxidative amidation have shown in Scheme 107.
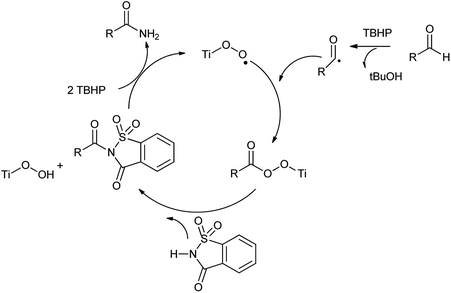 |
| | Scheme 107 Catalytic cycle of oxidative amination of aldehyde. | |
As far as saccharin as amine source is concerned it can also be recycle easily and the reaction are carried under favorable and less hazardous condition moreover it is reported as green catalyst. A procedure of regioselective amination of unsymmetrical 3,5-substituted pyridine N-oxides using saccharin as an ammonium surrogate reported by Farrell et al. (Scheme 108).134 Under mild conditions corresponding saccharin adducts has been achieved. In situ deprotection under acidic conditions allows for a one-pot process to substituted aminopyridines. Herein, saccharin acts as an ammonia surrogate for this application, delivering both high conversions and regioselectivity. This method overcomes challenges linked with direct amination of pyridine N-oxides caused by the competitive reaction of the nucleophile with the activator.
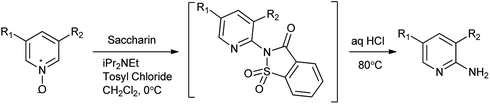 |
| | Scheme 108 Regioselective amination of unsymmetrical 3,5-substituted pyridine N-oxides. | |
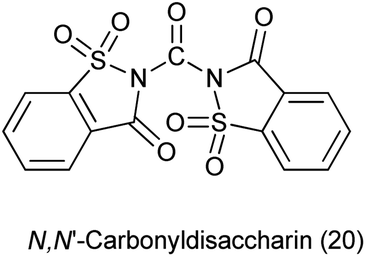
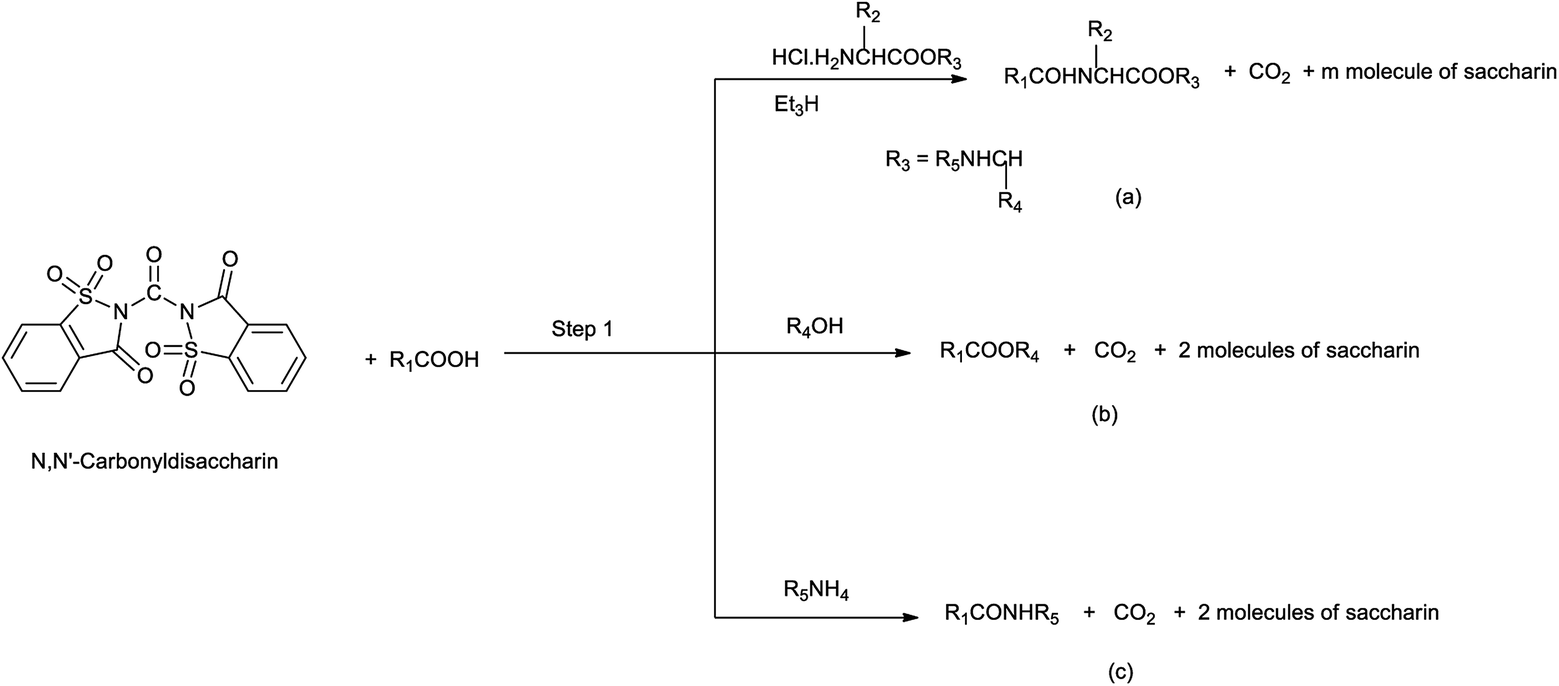
22. N,N′-Carbonyldisaccharin
Yadav et al. in 2003 reported the synthesis of peptides, esters and amides in the presence of new condensing reagent N,N′-carbonyldisaccharin (Scheme 110). This condensing reagent was easily prepared by reacting saccharin and trichloromethyl chloroformate in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio under refluxing toluene as shown in Scheme 109.135
:1 molar ratio under refluxing toluene as shown in Scheme 109.135
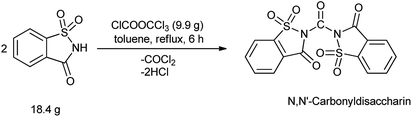 |
| | Scheme 109 Synthesis of N,N′-carbonyldisaccharin. | |
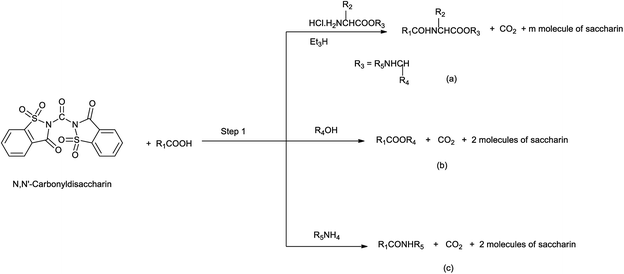 |
| | Scheme 110 Synthesis of (a) peptides, (b) esters, and (c) amides by the use of condensing reagent. | |
This protocol was really helpful as the condensing reagent is easy to handle and sides products as saccharin is easily removed from the product mixture with 1% cold sodium hydrogen carbonate.
23. Conclusions
From this review it is inferred that saccharin and its derivative can provide a vast range of catalytic system for synthesis of various heterocyclic framework, protection of functional groups as well as for functional group conversion. We envision that this review will help to stimulate research in the area of sugar and their derivatives that may release exhilarating perspectives in organic synthesis, material science and pharmaceutical industry. As far the catalytic activity of saccharin and its derivatives is concerned it is found to be more effective with good yield than the standard archetype catalyst. Although there have been noteworthy developments in many aspects of the saccharin derivative as catalyst. However, saccharin and its derivatives offer other opportunities and challenges to the synthetic chemists and biologist for the appropriate use of the same in different way, and we believe that these opportunities will never end because these catalysts will indeed form a huge source for catalytic strategies. From future aspect saccharin and its derivatives can replace other catalyst and favors the green application which is demand of the day as the derivatives can be easily prepared in laboratory and recycled further.
Conflicts of interest
There are no conflict to declare.
Acknowledgements
KK and SS acknowledges Department of Science and Technology (DST) for financial support through Inspire Faculty Award No. IFA-13-CH-123.
References
- A. Kumar, V. D. Tripathi and P. Kumar, Green Chem., 2011, 13, 51–54 RSC.
- B. K. Dhartiben and K. D. Aparnathi, Int. J. Curr. Microbiol. Appl. Sci., 2017, 6(6), 1283–1296 CrossRef CAS.
- T. G. Neltner, N. R. Kulkami, H. M. Alger, M. V. Maffini, E. D. Bongard, N. D. Fortin and E. D. Olson, Compr. Rev. Food Sci. Food Saf., 2011, 10(6), 342–368 CrossRef.
- S. Chattopadhyay, U. Raychaudhuri and R. Chakraborty, J. Food Sci. Technol., 2014, 51(4), 611–621 CrossRef CAS.
-
(a) S. Srivastava, New J. Chem., 2019, 43, 6469–6471 RSC;
(b) A. Kumar, S. Srivastava and G. Gupta, Green Chem., 2012, 14, 3269–3272 RSC;
(c) A. Kumar, S. Srivastava, G. Gupta, V. Chaturvedi, S. Sinha and R. Srivastava, ACS Comb. Sci., 2011, 13, 65–71 CrossRef CAS;
(d) A. Kumar, S. Srivastava and G. Gupta, Tetrahedron Lett., 2010, 51, 517–520 CrossRef CAS;
(e) A. Kumar, G. Gupta and S. Srivastava, J. Comb. Chem., 2010, 12, 458–462 CrossRef CAS;
(f) S. Srivastava, ChemistrySelect, 2020, 5(2), 799–803 CrossRef CAS.
- B. Banerjee, V. Bhardwaj, A. Kaur, G. Kaur and A. Singh, Curr. Org. Chem., 2019, 23, 3191–3205 CrossRef CAS.
-
(a) P. Sanna, A. Carta, M. Loriga, S. Zanetti and L. Sechi, Farmaco, 1999, 54, 161–168 CrossRef CAS;
(b) J. Guillon, P. Grellier, M. Labaied, P. Sonnet, J.-M. Léger, R. Déprez-Poulain, I. Forfar-Bares, P. Dallemagne, N. Lemaître, F. Péhourcq, J. Rochette, C. Sergheraert and C. Jarry, J. Med. Chem., 2004, 47, 1997–2009 CrossRef CAS.
- Y. V. D. Nageswar, K. H. V. Reddy, K. Ramesh and S. N. Murthy, Org. Prep. Proced. Int., 2013, 45, 1–27 CrossRef CAS.
- F. Lassagne, F. Chevallier and F. Mongin, Synth. Commun., 2013, 44, 141–149 CrossRef.
- D. Bandyopadhyay, S. Mukherjee, J. C. Granados, J. D. Short and B. K. Banik, Eur. J. Med. Chem., 2012, 50, 209–215 CrossRef CAS.
- A. Balakrishna, A. Aguiar, P. J. M. Sobral, M. Y. Wani, J. A. E. Silva and A. J. F. N. Sobral, Catal. Rev., 2019, 61, 84–110 CrossRef CAS.
- N. Bhandari and S. L. Gaonkar, Chem. Heterocycl. Compd., 2015, 51, 320–323 CrossRef CAS.
- C. O. Kappe, Eur. J. Med. Chem., 2000, 35(12), 1043–1052 CrossRef CAS.
- F. Mohamadpour, M. T. Maghsoodlou, R. Heydari and M. Lashkari, J. Iran. Chem. Soc., 2016, 13(8), 1549–1560 CrossRef CAS.
- F. Mohamadpour, M. Lashkari and N. Hazeri, Indones. J. Chem., 2018, 18(1), 7–17 CrossRef CAS.
- E. Merino, Chem. Soc. Rev., 2011, 40, 3835–3853 RSC.
-
(a) W. Lewgowd and A. Stanczak, Arch. Pharm. Chem. Life Sci., 2007, 340, 65–80 CrossRef CAS;
(b) M. Szumilak and A. Stanczak, Molecules, 2019, 24(12), 2271 CrossRef CAS.
- N. A. Danilkina, P. S. Vlasov, S. M. Vodianik, A. A. Kruchinin, Y. G. Vlasov and I. A. Balova, Beilstein J. Org. Chem., 2015, 11, 373–384 CrossRef CAS.
- N. G. Khaligh, T. Mihankhah, M. R. Johan and J. J. Ching, Monatsh. Chem., 2018, 149(6), 1083–1087 CrossRef CAS.
- N. G. Khaligh, M. R. Johan and J. J. Ching, Aust. J. Chem., 2018, 71, 186–189 CrossRef CAS.
- N. G. Khaligh, M. R. Johan and J. J. Ching, Green Process. Synth., 2019, 8, 24–29 CAS.
- A. Zare, M. Dianat, N. Pishahang, R. Khanivar, S. Kaman-Torki, F. Fakhraei, Z. Kordrostami and M. Sadeghi-Takallo, Iran. Chem. Commun., 2019, 7, 586–593 Search PubMed.
- F. Shirini, M. A. Zolfigol and M. Abedini, Monatsh. Chem., 2009, 140, 61–64 CrossRef CAS.
- F. Shirini, M. Mamaghani, T. Mostashari-Rad and M. Abedini, Bull. Korean Chem. Soc., 2010, 31, 2399–2401 CrossRef CAS.
- T. W. Greene and P. G. M. Wuts, Greene's Protective Groups in Organic Synthesis, Wiley, New York, 4th edn, 2007 Search PubMed.
-
(a) T. W. Greene and P. G. M. Wuts, Protective groups in organic synthesis, Wiley, New York, 1999 CrossRef;
(b) P. J. Kocienski, R. Endres, R. Noyori and B. M. Trost, Protective groups, Thiemen, Stuttgart, 1994 Search PubMed.
- G. A. Olah, B. G. B. Gupta, S. C. Narang and R. Malhorta, J. Org. Chem., 1979, 44, 1247–1251 CrossRef CAS.
- K. S. Kochhar, B. S. Bal, R. P. Deshpande, S. N. Rajadhyaksha and H. W. Pinnick, J. Org. Chem., 1983, 48, 1765–1767 CrossRef CAS.
- F. Shirini, M. A. Zolfigol and M. Abedini, Monatsh. Chem., 2009, 140, 1495–1498 CrossRef CAS.
- C. Agami and F. Couty, Tetrahedron, 2002, 58, 2701–2724 CrossRef CAS.
- F. Shirini, M. A. Zolfigol and M. Abedini, J. Iran. Chem. Soc., 2010, 7, 603–607 CrossRef CAS.
-
(a) A. Y. Shen, C. T. Tsai and C. L. Chen, Eur. J. Med. Chem., 1999, 34, 877–882 CrossRef CAS;
(b) M. H. Huang, S.-N. Wa, J. P. Wang, C. H. Lin, S. Lu, L. F. Lian and A. Y. Shen, Drug Dev. Res., 2003, 60, 261 CrossRef CAS.
- A. Zare, H. Kaveh, M. Merajoddin, A. R. Moosavi-Zare, A. Hasaninejad and M. A. Zolfigol, Phosphorus, Sulfur Silicon Relat. Elem., 2013, 188, 573–584 CrossRef CAS.
- A. Zare, M. Mokhlesi, A. Hasaninejad and T. Hekmat-Zadeh, E-J. Chem., 2012, 9(4), 1854–1863 CrossRef CAS.
- J. M. Bachhawat and N. K. Mathur, Tetrahedron Lett., 1971, 12(8), 691–692 CrossRef.
- B. Zajc, Synth. Commun., 1999, 29, 1779–1784 CrossRef CAS.
- E. I. Sănchez and M. J. Fumarola, Synthesis, 1976, 1976(11), 736–737 CrossRef.
- S. P. L. de Souza, J. F. M. da Silva and M. C. S. de Mattos, Synth. Commun., 2003, 33(6), 935–939 CrossRef CAS.
- S. P. L. de Souza, J. F. M. da Silva and M. C. S. de Mattos, J. Braz. Chem. Soc., 2003, 14(5), 832–835 CrossRef CAS.
-
(a) S. C. Bisarya and R. Rao, Synth. Commun., 1993, 23, 779–788 CrossRef CAS;
(b) K. Hisatoshi, A. Katsutomo, O. Tamon and K. Jitsuo, Bull. Chem. Soc. Jpn., 1989, 62, 591–593 CrossRef;
(c) D. Roche, K. Prasad, O. Repic and T. J. Blacklock, Tetrahedron Lett., 2000, 41, 2083–2085 CrossRef CAS;
(d) H. A. Muathen, J. Org. Chem., 1992, 57, 2740–2741 CrossRef CAS;
(e) G. Majetich, R. Hicks and S. Reister, J. Org. Chem., 1997, 62, 4321–4326 CrossRef CAS;
(f) G. Zhang, R. Liu, Q. Xu, L. Ma and X. Liang, Adv. Synth. Catal., 2006, 348, 862–866 CrossRef CAS;
(g) M. Guo, L. Varady, D. Fokas, C. Baldino and L. Yu, Tetrahedron Lett., 2006, 47, 3889–3892 CrossRef CAS.
- H. Alinezhad, S. M. Tavakkoli and F. Salehian, Synth. Commun., 2010, 40, 3226–3232 CrossRef CAS.
- R. Baharfar, H. Alinezhad, S. Azimi and F. Salehian, J. Chil. Chem. Soc., 2011, 56, 863–865 CrossRef CAS.
- M. Abrishamkar, S. N. Azizi and H. Kazemian, Iran. J. Org. Chem., 2010, 2, 338–342 Search PubMed.
- H. Alinezhad, M. Tajbakhsh and S. S. Tehrani, Bull. Korean Chem. Soc., 2011, 32, 1543–1546 CrossRef CAS.
- E. I. Sănchez and M. J. Fumarola, J. Org. Chem., 1982, 47, 1588–1590 CrossRef.
- D. Urankar, I. Rutar, B. Modec and D. Dolenc, Eur. J. Org. Chem., 2005, 2349–2353 CrossRef CAS.
-
(a) B. P. Bandgar, L. B. Kunde and J. L. Thote, Synth. Commun., 1997, 27, 1149–1152 CrossRef CAS;
(b) A. S. Demir, C. Tanyeli and E. Altinel, Tetrahedron Lett., 1997, 38, 7267–7270 CrossRef CAS.
-
(a) C. G. Rao, A. H. Radhakrishna, B. B. Singh and S. P. Bhatnagar, Synthesis, 1983, 1983, 808 CrossRef;
(b) N. B. Barhate, A. S. Gajare, R. D. Wakharkar and A. Sudalai, Tetrahedron Lett., 1997, 38, 653–656 CrossRef CAS.
- A. Khazaei, A. A. Manesh and A. Rostami, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 2483–2486 CrossRef CAS.
- A. Khazaei, A. Rostami and A. A. Manesh, J. Chin. Chem. Soc., 2006, 53, 437–441 CrossRef CAS.
- I. Jain, K. Sharma and V. K. Sharma, Chem. Sci. Rev. Lett., 2016, 5(20), 211–218 CAS.
-
(a) X. Z. Liang, L. Chen, H. Wu, J.-G. Yang and M. Y. He, Sci. China, Ser. B: Chem., 2009, 52, 2166–2170 CrossRef CAS;
(b) E. L. Eliel and S. Morris-Natschke, J. Am. Chem. Soc., 1984, 106, 2937–2942 CrossRef CAS.
- H. Alinezhad and S. Fallahi, Chin. Chem. Lett., 2012, 23, 927–929 CrossRef CAS.
- L. Song, S. Luo and J.-P. Cheng, Org. Chem. Front., 2016, 3, 447–452 RSC.
- L. Song, L. Zhang, S. Luo and J.-P. Cheng, Chem.–Eur.
J., 2014, 20(44), 14231–14234 CrossRef CAS.
- H. Firouzabadi, N. Iranpoor and F. Ebrahimzadeh, J. Iran. Chem. Soc., 2008, 5(3), 400–406 CrossRef CAS.
-
(a) E. B. Merkushev, Synthesis, 1988, 12, 923–937 CrossRef;
(b) S. D. R. Christie, J. Chem. Soc., Perkin Trans. 1, 1999, 737–747 RSC;
(c) A. Kryska and L. Skulski, J. Chem. Res., Synop., 1999, 10, 590–591 RSC;
(d) J. Barluenga, Pure Appl. Chem., 1999, 71, 431–436 CAS.
-
(a) M. Aloui and A. J. Fairbanks, Synlett, 2001, 6, 797–799 CrossRef;
(b) F. Feigl and A. Bondi, Monatsh. Chem., 1929, 53, 508–549 CrossRef;
(c) C. Djerassi and C. T. Lenk, J. Am. Chem. Soc., 1953, 75, 3493–3495 CrossRef CAS.
- D. Dolenc, Synlett, 2000, 4, 544–546 Search PubMed.
- D. Dolenc, Synth. Commun., 2003, 33, 2917–2924 CrossRef CAS.
- A. Rai and L. D. S. Yadav, Tetrahedron Lett., 2010, 51(31), 4045–4049 CrossRef CAS.
- H.-S. Dawn, I. H. Pitman, T. Higuchi and S. Young, J. Pharm. Sci., 1970, 59, 955–959 CrossRef CAS.
-
(a) P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772–3789 CrossRef CAS;
(b) M. J. Earle and K. R. Seddon, Pure Appl. Chem., 2000, 72, 1391–1398 CAS;
(c) C. M. Gordon, Appl. Catal., A, 2001, 222, 101–117 CrossRef CAS.
- A. Kumar, G. Gupta and S. Srivastava, Green Chem., 2011, 13, 2459–2463 RSC.
- P. Nockemann, B. Thijs, K. Driesen, C. R. Janssen, K. V. Hecke, L. V. Meervelt, S. Kossmann, B. Kirchner and K. Binnemans, J. Phys. Chem. B, 2007, 111, 5254–5263 CrossRef CAS.
- A. Kumar, P. Kumar, V. D. Tripathi and S. Srivastava, RSC Adv., 2012, 2, 11641–11644 RSC.
- H. Sharma and S. Srivastava, RSC Adv., 2018, 8, 38974–38979 RSC.
- H. Sharma and S. Srivastava, New J. Chem., 2019, 43, 12054–12058 RSC.
- L. Ming, G. Wei-Si, W. Li-Rong, L. Ya-Feng and Y. Hua-Zheng, J. Mol. Catal. A: Chem., 2006, 258, 133–138 CrossRef.
- A. Kumar, S. Srivastava, G. Gupta, P. Kumar and J. Sarkar, RSC Adv., 2013, 3, 3548–3552 RSC.
-
(a) H. Ni, D. J. Aaserud, W. J. Simonsick Jr and M. D. Soucek, Polymer, 2000, 41, 57–71 CrossRef CAS;
(b) M. Modesti and A. Lorenzetti, Eur. Polym. J., 2000, 37, 949–954 CrossRef.
-
(a) J. Tang, T. Mohan and J. G. Verkade, J. Org. Chem., 1994, 59, 4931–4938 CrossRef CAS;
(b) A. P. Murray and M. J. Miller, J. Org. Chem., 2003, 68, 191–194 CrossRef CAS.
- F. M. Moghaddam, G. R. Koozehgiri and M. G. Dekamin, Monatsh. Chem., 2004, 135, 849–851 CrossRef CAS.
-
(a) D. V. Vorobyeva, N. M. Karimova, I. L. Odinets, G. V. Roschenthaler and S. N. Osipov, Org. Biomol. Chem., 2011, 9, 7335–7342 RSC;
(b) S. Batra and A. K. Roy, Synthesis, 2004, 15, 2550–2554 CrossRef;
(c) S. Zhu, S. Shi and S. W. Gerritz, Tetrahedron Lett., 2011, 52, 4001–4004 CrossRef CAS.
- H. Kiyani and F. Ghorbani, Heterocycl. Lett., 2013, 3, 359–369 CAS.
- L. Moradi and M. A. Sadegh, Acta Chim. Slov., 2017, 64, 506–512 CrossRef CAS.
-
(a) P. H. Gehrtz, V. Hirschbeck and I. A. Fleischer, Chem. Commun., 2015, 51, 12574–12577 RSC;
(b) I. Chiyomaru, E. Yoshinaga and H. Ito, Jp. Pat., 48008500, 1973.
-
(a) L. Ashfield and C. F. J. Bernard, Org. Process Res. Dev., 2007, 11, 39–43 CrossRef CAS;
(b) A. Brennfhrer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef.
-
(a) P. Hermange, A. T. Lindhardt, R. H. Taaning, K. Bjerglund, D. Lupp and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 6061 CrossRef CAS;
(b) S. D. Friis, R. H. Taaning, A. T. Lindhardt and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 18114–18117 CrossRef CAS;
(c) T. Morimoto, K. Yamasaki, A. Hirano, K. Tsutsumi, N. Kagawa, K. Kakiuchi, Y. Harada, Y. Fukumoto, N. Chatani and T. Nishioka, Org. Lett., 2009, 11, 1777–1780 CrossRef CAS.
- T. Cochet, V. Bellosta, A. Greiner, D. Roche and J. Cossy, Synlett, 2011, 13, 1920–1922 Search PubMed.
- H.-P. Fang, C.-C. Fu, C.-K. Tai, K.-H. Chang, R.-H. Yang, M.-J. Wu, H.-C. Chen, C.-J. Li, S.-Q. Huang, W.-H. Lien, C.-H. Chen, C.-H. Hsieh, B.-C. Wang, S.-F. Cheung and P.-S. Pan, RSC Adv., 2016, 6, 30362–30371 RSC.
- T. Ueda, H. Konishi and K. Manabe, Angew. Chem., Int. Ed., 2013, 52, 8611–8615 CrossRef CAS.
- T. Ueda, H. Konishi and K. Manabe, Org. Lett., 2013, 15, 5370–5373 CrossRef CAS.
- P. Gautam, P. Kathe and B. M. Bhanage, Green Chem., 2017, 19, 823–830 RSC.
- V. K. Yadav, V. P. Srivastava and L. D. S. Yadav, Synlett, 2016, 27, 2826–2830 CrossRef CAS.
- V. Hirschbeck, P. H. Gehrtz and I. Fleischer, J. Am. Chem. Soc., 2016, 138, 16794–16799 CrossRef CAS.
- X. Fan, M. Shi and Y. Wei, Adv. Synth. Catal., 2019, 361, 5677–5683 CrossRef CAS.
- C. F. Perez, E. L. Calandri, M. R. Mazzieri, B. Arguello, A. R. Suarez and M. J. Fumarola, Org. Prep. Proced. Int., 1984, 16(1), 37–47 CrossRef CAS.
- N. S. Ramegowda, M. N. Modi, A. K. Koul, J. M. Bora, C. K. Narang and N. K. Mathur, Tetrahedron, 1973, 29, 3985–3986 CrossRef CAS.
- F. Micheel and M. Lorenz, Tetrahedron Lett., 1963, 4, 2119–2122 CrossRef.
- H. Wu, Y. Li, M. Cui, J. Jian and Z. Zeng, Adv. Synth. Catal., 2016, 358(23), 3876–3880 CrossRef CAS.
- C. Liu, G. Meng, Y. Liu, R. Liu, R. Lalancette, R. Szostak and M. Szostak, Org. Lett., 2016, 18(17), 4194–4197 CrossRef CAS.
- H. Wu, T. Liu, M. Cui, Y. Li, J. Jian, H. Wang and Z. Zeng, Org. Biomol. Chem., 2017, 15(3), 536–540 RSC.
- C. Liu, G. Meng and M. Szostak, J. Org. Chem., 2016, 81, 12023–12030 CrossRef CAS.
- S. S. Malunavar, S. M. Sutar, P. K. Prabhala, R. G. Kalkhambkar and K. K. Laali, Tetrahedron Lett., 2020, 61, 151987–151991 CrossRef CAS.
- A. P. Koskin, I. V. Mishakov and A. A. Vedyagin, Resour. Technol., 2016, 2, 118–125 Search PubMed.
- R. Calvo, K. Zhang, A. Passera and D. Katayev, Nat. Commun., 2019, 10, 3410 CrossRef.
-
(a) S. Munavalli, D. K. Rohrbaugh, F. R. Longo and H. D. Durst, Phosphorus, Sulfur Silicon Relat. Elem., 2002, 177, 1073–1083 CrossRef CAS;
(b) S. Munavalli, D. K. Rohrbaugh, D. I. Rossman, W. G. Wagner and H. D. Durst, Phosphorus, Sulfur Silicon Relat. Elem., 2002, 177, 1021–1031 CrossRef CAS.
- C. Xu, B. Ma and Q. Shen, Angew. Chem., Int. Ed., 2014, 53, 9316–9320 CrossRef CAS.
- C. Xu and Q. Shen, Org. Lett., 2015, 17(18), 4561–4563 CrossRef CAS.
- X. Liu, R. An, X. Zhang, J. Luo and X. Zhao, Angew. Chem., Int. Ed., 2016, 55, 5846–5850 CrossRef CAS.
- C. Mc Carthy, M. Tacke and X. Zhu, Eur. J. Org. Chem., 2019, 2729–2734 CrossRef.
-
(a) S. T. Purrington, B. S. Kagen and T. B. Patrick, Chem. Rev., 1986, 86, 997–1018 CrossRef CAS;
(b) S. Rozen and R. Filler, Tetrahedron, 1985, 41, 1111–1153 CrossRef CAS;
(c) Z.-Q. Xu, D. D. DesMarteau and Y. Gotoh, J. Chem. Soc., Chem. Commun., 1991, 179–181 RSC;
(d) S. T. Purrington and W. A. Jones, J. Org. Chem., 1983, 48, 761–762 CrossRef CAS;
(e) W. E. Barnette, J. Am. Chem. Soc., 1984, 106, 452–454 CrossRef CAS;
(f) T. Umemoto, K. Kawada and K. Tomita, Tetrahedron Lett., 1986, 27, 4465–4468 CrossRef CAS;
(g) E. Differding and R. W. Lang, Tetrahedron Lett., 1988, 29, 6087 CrossRef CAS.
- E. Differding and R. W. Lang, Helv. Chim. Acta, 1989, 72, 1248–1252 CrossRef CAS.
- K. C. Nicolaou, N. A. Petasis and D. A. Claremon, Tetrahedron, 1985, 41, 4835–4841 CrossRef CAS.
- M. Tingoli, M. Tiecco, L. Testaferri and A. Temperini, Synth. Commun., 1998, 28, 1769–1778 CrossRef CAS.
- M. Tiecco, L. Testaferri, M. Tingoli, D. Chianelli and D. Bartoli, Tetrahedron Lett., 1989, 30, 1417–1420 CrossRef CAS.
- M. Tingoli, R. Diana and B. Panunzi, Tetrahedron Lett., 2006, 47, 7529–7531 CrossRef CAS.
- M. Pawliczek, L. K. B. Garve and D.-B. Werz, Org. Lett., 2015, 17, 1716–1719 CrossRef CAS.
- T. Castanheiro, J. Suffert, M. Donnard and M. Gulea, Chem. Soc. Rev., 2016, 45, 494–505 RSC.
- D. Wu, J. Qiu, P. G. Karmaker, H. Yin and F.-X. Chen, J. Org. Chem., 2018, 83, 1576–1583 CrossRef CAS.
-
(a) P. Zhou, B.-C. Chen and F. A. Davis, Tetrahedron, 2004, 60, 8003–8030 CrossRef CAS;
(b) J.-P. Begue, D. Bonnet-Delpon, B. Crousse and J. Legros, Chem. Soc. Rev., 2005, 34, 562–572 RSC.
- K. Y. Lee, C. G. Lee and J. N. Kim, Tetrahedron Lett., 2003, 44, 1231–1234 CrossRef CAS.
- R. Patel, V. P. Srivastava and L. D. S. Yadav, Adv. Synth. Catal., 2010, 352, 1610–1614 CrossRef CAS.
-
(a) K. H. Jensen and M. S. Sigman, Org. Biomol. Chem., 2008, 6, 4083–4088 RSC;
(b) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981–3019 CrossRef CAS.
- C. Martínez, E. G. Pérez, Á. Iglesias, E. C. Escudero-Adán and K. Muñiz, Org. Lett., 2016, 18(12), 2998–3001 CrossRef.
- A. Iglesias, E. G. Prez and K. Muiz, Angew. Chem., Int. Ed., 2010, 49, 8109–8111 CrossRef CAS.
- N. Lu, W.-H. Chang, W.-H. Tu and C.-K. Li, Chem. Commun., 2011, 47, 7227–7229 RSC.
- N. Lu, W.-H. Chang, R.-J. Wei, Y.-C. Fang, T.-W. Han, G.-Q. Wang, J.-Y. Chang, Y.-S. Wen and L.-K. Liu, Tetrahedron, 2016, 72, 3468–3476 CrossRef CAS.
- J. Lal, R. Green and S. Ellis, J. Polym. Sci., 1957, 25, 75–84 CrossRef.
- F. Wei, H. Zhu, Z. Li, H. Wang, Y. Zhu, L. Zhang, Z. Yao, Z. Luo, C. Zhang and K. Guo, Adv. Synth. Catal., 2019, 361(6), 1335–1347 CrossRef CAS.
- L. M. T. Frija, E. C. B. A. Alegria, M. Sutradhar, M. L. S. Cristiano, A. Ismael, M. N. Kopylovich and A. J. L. Pombeiro, J. Mol. Catal. A: Chem., 2016, 425, 283–290 CrossRef CAS.
- L. M. T. Frija, M. L. Kuznetsov, B. G. M. Rocha, L. Cabral, M. L. S. Cristiano, M. N. Kopylovich and A. J. L. Pombeiro, Mol. Catal., 2017, 442, 57–65 CrossRef CAS.
- A. Ismael, J. A. Paixão, R. Fausto and M. L. S. Cristiano, J. Mol. Struct., 2012, 1023, 128–142 CrossRef CAS.
- Y. Tamura, F. Watanabe, T. Nakatani, K. Yasui, M. Fuji, T. Komurasaki, H. Tsuzuki, R. Maekawa, T. Yoshioka, K. Kawada, K. Sugita and M. Ohtani, J. Med. Chem., 1998, 41, 640–649 CrossRef CAS.
- S. C. S. Bugalho, L. Lapinski, M. L. S. Cristiano, L. M. T. Frija and R. Fausto, Vib. Spectrosc., 2002, 30, 213–225 CrossRef CAS.
- L. M. T. Frija, B. G. M. Rocha, M. L. Kuznetsov, L. I. L. Cabral, M. L. S. Cristiano and A. J. L. Pombeiro, Pure Appl. Chem., 2020, 92, 151–166 CAS.
- L. M. T. Frija, E. C. B. A. Alegria, M. Sutradhar, M. L. S. Cristiano, A. Ismael, M. N. Kopylovich and A. J. L. Pombeiro, J. Mol. Catal. A: Chem., 2016, 425, 283–290 CrossRef CAS.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116(5), 3328–3435 CrossRef CAS.
- A. Yoshimura, S. R. Koski, J. M. Fuchs, A. Saito, V. N. Nemykin and V. V. Zhdankin, Chem.–Eur. J., 2015, 21, 5328–5331 CrossRef CAS.
- M. Pelckmans, T. Renders, S. Van de Vyver and B. F. Sels, Green Chem., 2017, 19, 5303–5307 RSC.
- N. Yan and X. Chin, Nature, 2015, 524, 155–157 CrossRef CAS.
- R. B. Kamble, K. D. Mane, B. R. Rupanawar, P. Korekar, A. Sudalai and G. Suryavanshi, RSC Adv., 2020, 10, 724–728 RSC.
- R. P. Farrell, M. W. Silva Elipe, M. D. Bartberger, J. S. Tedrow and F. Vounatsos, Org. Lett., 2013, 15, 168–171 CrossRef CAS.
- L. D. S. Yadav, S. Dubey and A. Singh, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2003, 42, 2601–2603 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Suman Srivastava
and
Suman Srivastava