DOI:
10.1039/D0RA05671E
(Paper)
RSC Adv., 2020,
10, 36562-36570
Improving thermal and mechanical properties of biomass-based polymers using structurally ordered polyesters from ricinoleic acid and 4-hydroxycinnamic acids†
Received
29th June 2020
, Accepted 13th September 2020
First published on 12th October 2020
Abstract
Biomass-based copolymers with alternating ricinoleic acid and 4-hydroxycinnamic acid derivatives (p-coumaric acid, ferulic acid, and sinapinic acid) exhibit a repeating structure based on soft and hard segments, derived from ricinoleic and 4-hydroxycinnamic acids, respectively. To achieve this alternating sequence, copolymers were synthesised by the self-condensation of hetero-dimeric monomers derived by the pre-coupling of methyl ricinolate and 4-hydroxycinnamic acid. The glass transition temperature (Tg) was observed to increase as the number of methoxy groups on the main chain increased; the Tg values of poly(coumaric acid-alt-ricinoleic acid), poly(ferulic acid-alt-ricinoleic acid), and poly(sinapinic acid-alt-ricinoleic acid) are −15 °C, −4 °C, and 24 °C respectively, 58 °C, 69 °C, and 97 °C higher than that of poly(ricinoleic acid). The polymers were processed into highly flexible, visually transparent films. Among them, poly(sinapinic acid-alt-ricinoleic acid) bearing two methoxy groups on each cinnamoyl unit, is mechanically the strongest polymer, with an elastic modulus of 126.5 MPa and a tensile strength at break of 15.47 MPa.
1. Introduction
Recently, various biomass-based polymer materials have been reported as alternative and renewable resources for plastics.1–8 While traditional polymers have been manufactured from fossil fuels, biomass-based polymers are carbon-neutral, and thus effectively contribute to the reduction of atmospheric carbon dioxide. Additionally, carbon-neutral polymers can replace petroleum-based polymers and play an important role in a circular economy in the near future. For example, poly(L-lactic acid) (PLLA),9,10 poly(butylene succinate),11,12 and poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) (PHBH)13–15 are typical representatives exhibiting biodegradability, biocompatibility, thermoplasticity, and/or elasticity, and are potential alternative raw materials for various plastic products used in daily life. Those polymers are commonly synthesised from biomass-derived carbohydrates and sugars are environmentally benign resources and promising feedstocks for the production of lactic acid and succinic acid.16 Conversely, PHBHs are directly manufactured by fermentation of plant oils and fats17–19 containing various fatty acids. The fatty acids from plant oil are, therefore, reasonable alternatives to petroleum resources and are utilised to produce polyesters.20–24
Ricinoleic acid (RA) is one of the fatty acids found in the structures of triglycerides in castor oil. It is a structurally characteristic compound, having a hydroxy group at a chiral centre and an unsaturated cis double bond in the alkenyl chain. The triglyceride formed from RA contains more than 90% of the fatty acid in castor oil. Therefore, RA has been widely employed as a starting material for the synthesis of biomass-based polymers25,26 because it is easily extracted after hydrolysis of the triglyceride.27,28 The poly(ricinoleic acid) (PRA) obtained from self-condensation of RA is a viscous liquid at room temperature.29,30 Therefore, PRA has been generally considered suitable for lubricants; it has not been an attractive choice for the scaffold of bioplastics, with the exception of cross-linked PRA30,31 and PRA composites with fillers.32 Numerous copolymers of RA with various comonomers,29,33–36 such as L-lactic acid,37 have been developed with the aim of tuning thermal and mechanical properties. For example, RA copolymers with L-lactic acid are solids with somewhat more desirable physical properties, but there remains room for improvement. In general, the introduction of an aromatic component into the polymer skeleton as a hard segment is expected to improve the thermal and mechanical properties. Gioia et al. have developed a random copolymer from RA with vanillic acid as an aromatic component. However, to the best of our knowledge, a structurally ordered copolyester of RA and aromatic compound has not been reported.38 It is noteworthy that various biorenewable polyesters with alternating aromatic/aliphatic units bearing ether-linkages have been developed by Miller and coworkers.39–41 They indicated that the thermal properties of the biobased polymers are readily tuneable because of an adequate combination of soft and hard segments in the main chain. In particular, the use of ferulic acid and p-coumaric acid as aromatic components contributed to improvements in glass transition temperature (Tg).40
Herein we report that the copolymerisation of RA soft segments with hard segments consisting of 4-hydroxycinnamic acids gives new polyesters that are structurally ordered and fully biomass-based. It has been known that the 4-hydroxycinnamic acid derivatives, i.e. p-coumaric acid, ferulic acid, and sinapinic acid, derived from lignocellulosic biomass resources, are promising aromatic components for preparing fully biomass-based polyesters and other advanced polymers.42 For this reason, our strategy is to use the “hetero-dimeric monomer” consisting of RA and 4-hydroxycinnamic acid to avoid undesired homopolymerisation of either component and to give structurally ordered alternating copolymers. This report describes the synthesis of alternating copolymers using this strategy and provides full details on the thermal properties of the resulting polymers. Furthermore, to evaluate the potential of the product polymers for practical applications such as bioplastics, the mechanical properties based on viscoelastic measurements and tensile testing will also be described.
2. Experimental
2.1. Materials
Ferulic acid (99%) and triphenyl phosphine (99%) were purchased from Sigma-Aldrich Japan Co., Ltd. p-Coumaric acid (>98.0%), sinapinic acid (>98.0%), oxalyl chloride (>98.0%), and diisopropyl azodicarboxylate (40% in toluene, ca. 1.9 mol L−1) were purchased from Tokyo Chemical Industry Co., Ltd. Methyl ricinoleate (>97.0%), acetic anhydride (>97.0%), N,N-dimethylaminopyridine (DMAP) (>99.0%), N,N′-diisopropylcarbodiimide (DIC) (>99.0%), pyridine (>99.5%, dehydrated), N,N-dimethylformamide (DMF) (>99.5%, dehydrated), sodium sulphate (Na2SO4) (>99.0%), and tetrahydrofuran (THF) (>99.5%, with stabilizer, for gel permeation chromatography (GPC) grade) were purchased from FUJIFILM Wako Pure Chemical Corporation. p-Toluenesulfonic acid monohydrate (>99.0%) and lithium hydroxide monohydrate (>99.0%) were purchased from KISHIDA CHEMICAL Co., Ltd. Toluene (>99.5%, dehydrated), dichloromethane (CH2Cl2) (>99.5%, dehydrated), and THF (>99.5%, dehydrated stabilizer free) were purchased from Kanto Chemical and purified on a Glass Contour Solvent Purification System (NIKKO HANSEN & Co., Ltd, Japan). Chloroform-d1 (99.8 atom % D with 0.03 vol% tetramethylsilane) was purchased from Kanto Chemical. These chemicals were used without further purification unless otherwise noted. 4-(Dimethylamino)pyridinium p-toluenesulfonate (DPTS) was synthesised according to the literature.43
2.2. Apparatuses
All manipulations of air- and moisture-sensitive compounds were performed in dry argon atmosphere by using a manifold and MBRAUN LABmaster Pro glovebox. The argon in the glovebox was constantly circulated through a copper/molecular sieves catalyst unit. The 1H-, 13C and 2D NMR spectra of the polymers were obtained on a JEOL JNM-ECX400 NMR spectrometer at room temperature. The differential scanning calorimetry (DSC) measurements of the polymers were performed on a DSC7020 (Hitachi High-Tech Science Co., Japan) equipped with a liquid nitrogen cooling accessory. Samples were encapsulated in sealed aluminium pans, passed through a heat/cool/heat cycle at a rate of 5 or 10 °C min−1 in the temperature range −100 °C (or −120 °C) to 100 °C in nitrogen atmosphere. The number average molecular weight (Mn) and the molecular weight distribution (Mw/Mn) of the polymers were determined by GPC using a refractive index (RI) detector. The GPC apparatus consists of DG-2080-54, PU-2085 Plus, AS-2055 Plus, CO-2065 Plus and RI-2031 Plus units (JASCO, Japan). Chromatographic conditions were as follows. The analysis column consisted of three connected columns of TSKgel SuperHZM-M with 6.0 mm I.D. × 15 cm (Tosoh Co., Japan); a polystyrene standard (SM-105 was purchased from Showa Denko K.K.) was used for calibration. 10 standard samples (Mn = 1380 to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 120000, Mw/Mn < 1.08) were used with a THF mobile phase, a flow rate of 0.5 mL min−1 and a column temperature of 40 °C. Thermogravimetric analysis (TGA) was performed on a TG/DTA 220U (Seiko instruments, Japan) using nitrogen as a purge gas. Samples were heated from room temperature to 500 °C at a rate of 10 °C min−1. The polymer films for mechanical tests were prepared by using a vacuum hot press apparatus consisting of IMC-11FD (Imoto machinery, Japan). The films were prepared by hot press apparatus at 95 °C for P(CA-alt-RA) or at 130 °C for P(FA-alt-RA) and P(SA-alt-RA), and then pressed with a force of 1.0 kN. After 60 min, the heaters were turned off, and the samples were cooled to room temperature over 7 h. The test specimens for tensile testing were cut from the resulting polymer films into a dumbbell-shape (width: 2 mm; length: 12 mm; thickness: approximately 1 mm). The gauge length was fixed at 12 mm. Tensile tests were conducted on an apparatus consisting of an EZ-SX (Shimadzu Co., Japan) with a load of 10 kN and a crosshead speed of 20 mm min−1. The tensile strength and tensile modulus were evaluated using three samples. For the elastic strain recovery test, the hysteresis experiments were performed on an Autograph AG-IS (Shimadzu Co., Japan). The specimens were cyclically loaded to the targeted strain (100%) and unloaded to the zero stress point with a rate of 1 mm min−1. The temperature-dependent viscoelastic properties of polymers were characterized with two systems. Measurements in the temperature range from −50 to 50 °C were carried out using an Exstar DMS6100 (Hitachi High-Tech Science Co., Japan) in tensile mode, with a frequency of 1 Hz and a heating rate of 2 °C min−1. Measurements in the temperature range from 20 to 80 °C were performed using a rheometer consisting of an MCR102 instrument (Anton Paar GmbH, Germany). Disposable aluminium parallel plates with 25 mm O.D. were used in the oscillation mode. The strain, frequency, normal force, and heating rate were fixed as 1%, 1 Hz, 0 N and 2 °C min−1, respectively. The cooling rate was 1.92 °C min−1 from 150 °C to 35 °C and 0.75 °C min−1 from 35 °C to 20 °C.
120000, Mw/Mn < 1.08) were used with a THF mobile phase, a flow rate of 0.5 mL min−1 and a column temperature of 40 °C. Thermogravimetric analysis (TGA) was performed on a TG/DTA 220U (Seiko instruments, Japan) using nitrogen as a purge gas. Samples were heated from room temperature to 500 °C at a rate of 10 °C min−1. The polymer films for mechanical tests were prepared by using a vacuum hot press apparatus consisting of IMC-11FD (Imoto machinery, Japan). The films were prepared by hot press apparatus at 95 °C for P(CA-alt-RA) or at 130 °C for P(FA-alt-RA) and P(SA-alt-RA), and then pressed with a force of 1.0 kN. After 60 min, the heaters were turned off, and the samples were cooled to room temperature over 7 h. The test specimens for tensile testing were cut from the resulting polymer films into a dumbbell-shape (width: 2 mm; length: 12 mm; thickness: approximately 1 mm). The gauge length was fixed at 12 mm. Tensile tests were conducted on an apparatus consisting of an EZ-SX (Shimadzu Co., Japan) with a load of 10 kN and a crosshead speed of 20 mm min−1. The tensile strength and tensile modulus were evaluated using three samples. For the elastic strain recovery test, the hysteresis experiments were performed on an Autograph AG-IS (Shimadzu Co., Japan). The specimens were cyclically loaded to the targeted strain (100%) and unloaded to the zero stress point with a rate of 1 mm min−1. The temperature-dependent viscoelastic properties of polymers were characterized with two systems. Measurements in the temperature range from −50 to 50 °C were carried out using an Exstar DMS6100 (Hitachi High-Tech Science Co., Japan) in tensile mode, with a frequency of 1 Hz and a heating rate of 2 °C min−1. Measurements in the temperature range from 20 to 80 °C were performed using a rheometer consisting of an MCR102 instrument (Anton Paar GmbH, Germany). Disposable aluminium parallel plates with 25 mm O.D. were used in the oscillation mode. The strain, frequency, normal force, and heating rate were fixed as 1%, 1 Hz, 0 N and 2 °C min−1, respectively. The cooling rate was 1.92 °C min−1 from 150 °C to 35 °C and 0.75 °C min−1 from 35 °C to 20 °C.
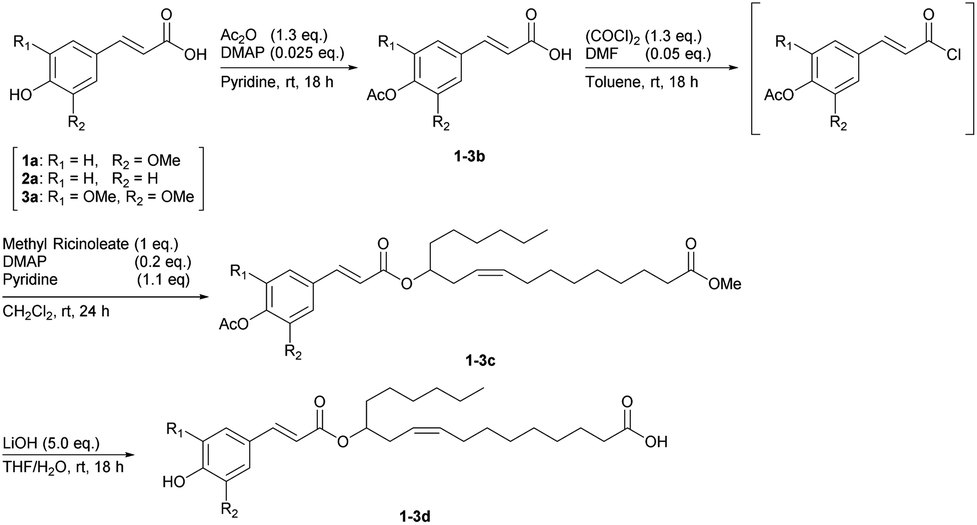
2.3. Synthetic procedures
2.3.1. Typical procedure for the condensation of methyl ricinoleate with 4-hydroxycinnamic acids. [Methyl (R,Z)-12-((E)-3-(4-acetoxy-3-methoxyphenyl)prop-2-enoyloxy)octadec-9-enoate (1c)]. Ferulic acid (1a) (5.8 g, 30 mmol) and DMAP (92 mg, 0.75 mmol) were dissolved in pyridine (20 mL) at 0 °C. Acetic anhydride (3.5 mL, 39 mmol) was added in dropwise fashion to the well-stirred pyridine solution. The mixture was stirred for 18 hours at room temperature, and then acidified with 1.0 M HCl aqueous solution at 0 °C. The solution was then extracted with CH2Cl2. The extract was dried over Na2SO4, filtered, and then solvents were removed under reduced pressure to afford (E)-3-(4-acetoxy-3-methoxyphenyl)prop-2-enoic acid (1b) as a white solid. This crude product was added to toluene (100 mL) at 0 °C. DMF (0.1 mL, 1.5 mmol) and oxalyl chloride (3.3 mL, 39 mmol) were added in dropwise fashion to the toluene solution under an argon atmosphere. This mixture was stirred at room temperature for 18 hours and the volatile components were removed under reduced pressure. Subsequently, CH2Cl2 (180 mL) was added, then pyridine (2.7 mL, 33 mmol) and DMAP (0.73 g, 6.0 mmol) were added to the CH2Cl2 solution at 0 °C. A CH2Cl2 solution (20 mL) of methyl ricinoleate (9.4 g, 30 mmol) was added in dropwise fashion to that solution. The mixture was stirred for 18 hours at room temperature and then acidified with 2.0 M HCl aqueous solution at 0 °C. The solution was extracted with CH2Cl2. The extract was dried over Na2SO4, filtered, and then solvents were removed under reduced pressure. The residue was purified by automated flash column chromatography (Biotage Selekt, Sfär silica HC Duo column, 20 μm silica gel 100 g, hexane:ethyl acetate = 20:1 to 10:1 (v/v)) to afford the desired product 1c (9.7 g, 61%) as colourless oil. 1H NMR (400 MHz; CDCl3; SiMe4): δ ppm 0.88 (t, J = 6.64 Hz, 3H), 1.20–1.40 (m, 17H), 1.54–1.66 (m, 4H), 2.04 (m, 2H), 2.28 (t, J = 7.56 Hz, 2H), 2.32 (s, 3H), 2.34–2.40 (m, 2H), 3.66 (s, 3H), 3.86 (s, 3H), 5.02 (tt, J = 6.18 Hz, 1H), 5.35–5.52 (m, 2H), 6.37 (d, J = 16.0 Hz, 1H), 7.05 (d, J = 8.70 Hz, 1H), 7.10 (s, 1H), 7.11 (d, J = 7.33 Hz, 1H), 7.62 (d, J = 15.6 Hz, 1H). 13C{1H} NMR (100 MHz; CDCl3): δ ppm 14.0, 20.6, 22.6, 24.9, 25.4, 27.3, 29.0–29.2 (4 carbons), 29.5, 31.7, 32.0, 33.7, 34.0, 51.4, 55.9, 74.2, 111.1, 118.8, 121.2, 123.2, 124.1, 132.7, 133.5, 141.3, 143.7, 151.3, 166.5, 168.7, 174.2. Elem. anal. calcd (%) for C31H46O7: C 70.16, H 8.74; found: C 70.36, H 8.84.
2.3.2. Synthesis of methyl (R,Z)-12-((E)-3-(4-acetoxy-phenyl)prop-2-enoyloxy)octadec-9-enoate (2c). 2c was synthesised according to the synthetic procedure for 1c (using p-coumaric acid (2a)), giving a colourless oil (11 g, 72%). 1H NMR (400 MHz; CDCl3; SiMe4): δ ppm 0.88 (t, J = 6.64 Hz, 3H), 1.20–1.40 (m, 17H), 1.54–1.66 (m, 4H), 2.04 (m, 2H), 2.28 (t, J = 7.56 Hz, 2H), 2.31 (s, 3H), 2.34–2.39 (m, 2H), 3.66 (s, 3H), 5.01 (tt, J1 = 6.23, J2 = 6.26 Hz, 1H), 5.35–5.51 (m, 2H), 6.38 (d, J = 16.0 Hz, 1H), 7.12 (d, J = 8.70 Hz, 2H), 7.54 (d, J = 8.70 Hz, 2H), 7.64 (d, J = 16.0 Hz, 1H). 13C{1H} NMR (100 MHz; CDCl3): δ ppm 14.0, 21.1, 22.5, 24.9, 25.4, 27.3, 29.0–29.2 (4 carbons), 29.5, 31.7, 32.0, 33.7, 34.0, 51.4, 74.2, 118.8, 122.1 (2 carbons), 124.2, 129.1 (2 carbons), 132.2, 132.7, 143.2, 152.0, 166.5, 169.1, 174.2. Elem. anal. calcd (%) for C30H44O6: C 71.97, H 8.86; found: C 72.22, H 8.96.
2.3.3. Synthesis of methyl (R,Z)-12-((E)-3-(4-acetoxy-3,5-dimethoxyphenyl)prop-2-enoyloxy)octadec-9-enoate (3c). 3c was synthesised according to the synthetic procedure for 1c (using sinapinic acid (3a)), giving a colourless oil (11 g, 68%). 1H NMR (400 MHz; CDCl3; SiMe4): δ ppm 0.88 (t, J = 6.64 Hz, 3H), 1.20–1.40 (m, 17H), 1.54–1.66 (m, 4H), 2.04 (m, 2H), 2.29 (t, J = 7.56 Hz, 2H), 2.34 (s, 3H), 2.34–2.39 (m, 2H), 3.66 (s, 3H), 3.85 (s, 6H) 5.02 (tt, J = 6.18 Hz, 1H), 5.35–5.52 (m, 2H), 6.37 (d, J = 16.0 Hz, 1H), 6.77 (s, 2H), 7.59 (d, 1H). 13C{1H} NMR (100 MHz; CDCl3): δ ppm 14.0, 20.4, 22.6, 24.9, 25.4, 27.3, 29.0–29.2 (4 carbons), 29.5, 31.7, 32.0, 33.7, 34.0, 51.4, 56.2 (2 carbons), 74.2, 104.6 (2 carbons), 118.9, 124.1, 130.3, 132.7, 132.8 (2 carbons), 144.1, 152.4, 166.4, 168.5, 174.3. Elem. anal. calcd (%) for C32H48O8: C 68.55, H 8.63; found: C 68.81, H 8.75.
2.3.4. Typical procedure for hydrolysis of 1-3c to give hetero-dimeric monomers. [(R,Z)-12-((E)-3-(4-Hydroxy-3-methoxyphenyl)prop-2-enoyloxy)octadec-9-enoic acid (1d)]. An aqueous solution (120 mL) of lithium hydroxide monohydrate (5.3 g, 0.13 mol) was added in dropwise fashion to a THF solution (300 mL) of 1c (13 g, 25 mmol) at 0 °C. The mixture was stirred at room temperature for 18 hours, and then the THF was removed under reduced pressure. The residual aqueous solution was washed with CH2Cl2 to remove starting material, and then acidified with 1.0 M HCl aqueous solution at 0 °C. The aqueous phase was extracted with CH2Cl2. The extract was dried over Na2SO4, filtered, and then solvents were removed under reduced pressure. The residue was purified by automated flash column chromatography (Biotage Selekt, Sfär silica HC Duo column, 20 μm silica gel 100 g, hexane:ethyl acetate = 1:1 (v/v)) to afford the desired product 1d (11 g, 88%) as a colourless oil. 1H NMR (400 MHz; CDCl3; SiMe4): δ ppm 0.87 (t, J = 6.64 Hz, 3H), 1.20–1.40 (m, 17H), 1.54–1.66 (m, 4H), 2.04 (m, 2H), 2.32 (t, J = 7.56 Hz, 2H), 2.35–2.39 (m, 2H), 3.92 (s, 3H), 5.02 (tt, J1 = 6.26 Hz, J2 = 6.43 Hz, 1H), 5.36–5.51 (m, 2H), 6.28 (d, J = 15.8 Hz, 1H), 6.91 (d, J = 7.79 Hz, 1H), 7.02–7.09 (m, 2H), 7.60 (d, J = 15.8 Hz, 1H). 13C{1H} NMR (CDCl3): δ ppm 14.0, 22.5, 24.6, 25.4, 27.3, 29.0–29.1 (4 carbons), 29.5, 31.7, 32.0, 33.7, 34.0, 55.9, 74.0, 109.3, 114.7, 116.0, 123.0, 124.2, 127.0, 132.6, 144.5, 146.8, 147.9, 167.1, 179.8. Elem. anal. calcd (%) for C28H42O6: C 70.86, H 8.92; found: C 70.49, H 9.14.
2.3.5. Synthesis of (R,Z)-12-((E)-3-(4-hydroxyphenyl)prop-2-enoyloxy)octadec-9-enoic acid (2d). 2d was synthesized according to the synthetic procedure for 1d (using 2c), giving a colourless oil (8.5 g, 88%). 1H NMR (400 MHz; CDCl3; SiMe4): δ ppm 0.87 (t, J = 6.64 Hz, 3H), 1.20–1.40 (m, 17H), 1.54–1.66 (m, 4H), 2.04 (m, 2H), 2.31 (t, J = 7.33 Hz, 2H), 2.36 (m, 2H), 5.02 (tt, J = 6.18 Hz, 1H), 5.34–5.51 (m, 2H), 6.28 (d, J = 15.8 Hz, 1H), 6.86 (d, J = 8.24 Hz, 2H), 7.40 (d, J = 8.70 Hz, 2H), 7.61 (d, J = 16.0 Hz, 1H). 13C{1H} NMR (CDCl3): δ ppm 14.0, 22.5, 24.7, 25.4, 27.3, 28.9–29.1 (4 carbons), 29.5, 31.7, 32.0, 33.7, 34.0, 74.4, 115.5, 115.9 (2 carbons), 124.1, 126.9, 129.9 (2 carbons), 132.8, 144.7, 158.2, 167.8, 179.8. Elem. anal. calcd (%) for C27H40O5: C 72.94, H 9.07; found: C 72.55, H 9.18.
2.3.6. Synthesis of (R,Z)-12-((E)-3-(4-hydroxy-3,5-dimethoxyphenyl)prop-2-enoyloxy)octadec-9-enoic acid (3d). 3d was synthesised according to the synthesis procedure of 1d (using 3c), giving a colourless oil (7.4 g, 72%). 1H NMR (400 MHz; CDCl3; SiMe4): δ ppm 0.88 (t, J = 6.87 Hz, 3H), 1.20–1.40 (m, 17H), 1.54–1.66 (m, 4H), 2.02–2.07 (m, 2H), 2.32 (t, J = 7.56 Hz, 2H), 2.35–2.39 (m, 2H), 3.92 (s, 6H) 5.02 (tt, J = 6.18 Hz, 1H), 5.36–5.52 (m, 2H), 6.30 (d, J = 15.6 Hz, 1H), 6.77 (s, 2H), 7.58 (d, J = 15.6 Hz, 2H). 13C{1H} NMR (CDCl3): δ ppm 14.0, 22.5, 24.6, 25.3, 27.3, 29.0–29.1 (4 carbons), 29.5, 31.7, 32.0, 33.7, 34.0, 56.3 (2 carbons), 74.0, 105.0 (2 carbons), 116.3, 124.2, 126.0, 132.6, 137.0, 144.7, 147.2 (2 carbons), 166.9, 179.7. Elem. anal. calcd (%) for C29H44O7: C 69.02, H 8.79; found: C 68.68, H 9.01.
2.3.7. Typical procedure for the self-condensation of hetero-dimeric monomers (Table 1, Run 3). Self-condensation of hetero-dimeric monomers was carried out under argon. The hetero-dimeric monomer 1d (0.23 g, 0.50 mmol) and the DPTS (30 mg, 0.10 mmol) were dissolved in CH2Cl2 (1.0 mL). The reaction mixture was cooled to 0 °C. Then, DIC (0.12 mL, 0.75 mmol) was added dropwise to the well-stirred reaction mixture. The reaction mixture was allowed to stir for 24 hours at room temperature. Diisopropylurea derived from DIC was precipitated in the reaction mixture as the self-condensation proceeded. This urea can be dissolved in methanol; therefore, crude polymer products were dissolved in the CH2Cl2, and reprecipitated in methanol. Solution was removed by decantation. The resulting residue was dried under vacuum at 60 °C to afford a pure polymer product (0.16 g, 73%).
2.3.8. Synthesis of PRA. PRA was synthesised according to the self-condensation procedure from RA, which was prepared by the hydrolysis of methyl ricinoleate, giving a colourless oil (1.3 g, 46%, Mn = 9221, Mw/Mn = 1.9). The polymer product was characterized by comparison to literature spectral data.30
3. Results and discussion
3.1. Synthesis of hetero-dimeric monomers
The condensation of 1a with methyl ricinoleate via the Mitsunobu reaction has previously been reported44 as a strategy for obtaining the hetero-dimeric monomer. We tried to reproduce this known reaction by using the reported method. However, many undesired and structurally unidentified by-products were detected by both thin layer chromatography and 1H-NMR. After extraction followed by chromatography, we eventually confirmed that the desired product was obtained in very low yield.
After this unexpected result, another condensation method using an appropriate protecting group for the phenolic hydroxy group of ferulic acid was examined (Scheme 1). First, the hydroxy group was acetylated, and the crude 1b was used in the next reaction without further purification. Crude 1b was treated with oxalyl chloride to form the corresponding acid chloride in situ and then reacted with methyl ricinoleate to give the heterolytic coupling compound 1c in 61% yield. Finally, both methyl ester and acetyl groups at the termini were simultaneously hydrolysed using excess lithium hydroxide, and the target hetero-dimer product 1d was obtained in 54% total yield from 1a. The syntheses of the other hetero-dimeric monomers 2d (64% yield from 2a) and 3d (49% yield from 3a) were successfully carried out with a procedure similar to that used for 1d.
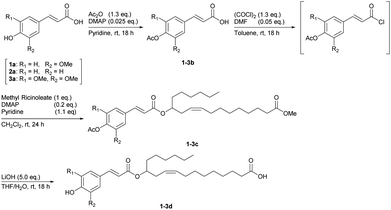 |
| | Scheme 1 Synthesis of hetero-dimeric monomers from 4-hydroxycinnamic acids (1-3a) and methyl ricinoleate via acid chloride intermediates. | |
3.2. Self-condensation of hetero-dimeric monomers
To obtain the corresponding biomass-based polyester with alternating structure, the self-condensation of 1d was attempted with the use of DIC and various catalysts in CH2Cl2, according to the procedure used for the self-polymerisation of hydroxy acids.43 As a result, polymerisations using various reaction conditions afforded several P(FA-alt-RA) samples, as expected. The Mn and Mw/Mn of the products were determined by GPC using polystyrene standards. The results are summarised in Table 1. Initially, we examined the reaction using DMAP as a catalyst (Run 1). The Mn was determined to be 17700, a moderate molecular weight. Mn was decreased to 9500 when pyridine was used instead of DMAP (Run 2). The 1H-NMR spectrum of the product showed a small doublet corresponding to the methyl groups of the isopropyl substituent in N-acyl urea (Fig. S12 in ESI†). The N-acyl urea is often generated as a by-product in the presence of DMAP or pyridine catalysts when using DIC.43 It was thus considered that a rearrangement reaction involving the terminal ester group and diisopropylurea was the likely source of the undesired urea unit on the end of the polymer chain and that this inhibited further chain growth. Eventually, we found that the use of DPTS as catalyst resulted in the highest observed molecular weight (Mn = 24300) (Run 3). Similar molecular weight improvements resulting from the use of DPTS have been reported for PLLA synthesis using DIC in CH2Cl2.45 It should be noted that CH2Cl2 has been a reliable solvent for this type of condensation reaction. In addition, catalyst, urea, and solvent are easily removed by washing with methanol and vacuum drying after polymerisation. However, CH2Cl2 is classified as a hazardous solvent. Therefore, the self-condensation was attempted in other solvents, using a combination of DIC and DPTS as standard condensation reagents. In THF, a polymer with a relatively narrow molecular weight distribution (Mw/Mn = 1.8) was produced in the highest yield (80%, Run 4), but Mn was decreased by ∼30%, relative to that seen in CH2Cl2, to 16800. With toluene, Mn was 20200, and Mw/Mn was increased to 2.2 (Run 5). In DMF, while the Mn and Mw/Mn values were almost identical with those seen for the reaction in CH2Cl2, the yield of polymer was slightly decreased to 69% (Run 6). These results indicated that, at least for the present, the use of DPTS catalyst in CH2Cl2 results in the optimum average molecular weight. Using these conditions, we carried out the self-condensation of hetero-dimeric monomer 1d on a 10-fold larger scale to obtain a quantity of polymer sample necessary for the examination of thermal and mechanical properties. The larger scale self-condensation of 1d proceeded smoothly to afford moderately high molecular weight polymer, although the Mn was decreased by approximately 20% relative to that seen with the small scale (Run 7). The relatively large scale self-condensations of 2d and 3d were also carried out in a manner similar to that used for 1d and gave the corresponding polymers (P(CA-alt-RA) and P(SA-alt-RA)) (Table 1, Runs 8 and 9). These three polymers are monomodally distributed (Fig. S13 in ESI†). We also confirmed that there are no significant differences in the degrees of polymerisation between P(CA-alt-RA), P(FA-alt-RA), and P(SA-alt-RA) (Table S1 in ESI†). Based on our present results, we conclude that differences in the molecular weights of the polymers have little effect on the thermal, viscoelastic, and mechanical properties described below.
Table 1 Self-condensation of 1-3da
|
| Run |
Monomer |
Catalyst |
Solvent |
Yield (%) |
Mnb (×103) |
Mw/Mnb |
| 1d, 0.5 mmol; catalyst, 0.1 mmol; DIC, 0.75 mmol; solvent, 1 mL. Determined by GPC in THF at 40 °C. Monomer, 10 mmol; DPTS, 2 mmol; DIC, 15 mmol; CH2Cl2, 20 mL. |
| 1 |
1d |
DMAP |
CH2Cl2 |
77 |
17.7 |
1.9 |
| 2 |
1d |
Pyridine |
CH2Cl2 |
57 |
9.5 |
1.3 |
| 3 |
1d |
DPTS |
CH2Cl2 |
73 |
24.3 |
2.4 |
| 4 |
1d |
DPTS |
THF |
80 |
16.8 |
1.8 |
| 5 |
1d |
DPTS |
Toluene |
73 |
20.2 |
2.2 |
| 6 |
1d |
DPTS |
DMF |
69 |
23.9 |
2.4 |
| 7c |
1d |
DPTS |
CH2Cl2 |
70 |
19.3 |
2.0 |
| 8c |
2d |
DPTS |
CH2Cl2 |
75 |
12.7 |
2.2 |
| 9c |
3d |
DPTS |
CH2Cl2 |
65 |
22.3 |
2.2 |
3.3. Thermal properties of polymers
The thermal properties of P(CA-alt-RA), P(FA-alt-RA) and P(SA-alt-RA) were analysed by DSC, as summarised in Table 2. The three polymers exhibited Tg of −15 °C, −4 °C and 24 °C, respectively. These values are much higher than is that of PRA (Tg = −73 °C). In addition, the Tg depended strongly on the number of methoxy groups on the cinnamoyl unit. A small increase (4 °C) has been reported for the minimal effect of a methoxy group on the Tg of polyethylene ferulate.40 On the contrary, clear and gradual increases in Tg were observed in the present study; there was an 11 °C increase on going from P(CA-alt-RA) to P(FA-alt-RA) and a 28 °C increase on going from P(FA-alt-RA) to P(SA-alt-RA). Therefore, we concluded that the effect of methoxy groups on Tg could not be ignored and that the Tg could be tuned by the number of the methoxy groups on the cinnamoyl unit of the backbone, at least for the present polymers. The introduction of methoxy groups onto the main chain has often been observed to lead to lower Tg values. For example, the observed decrease in Tg with increasing numbers of methoxy groups in polyalkylenehydroxybenzoates39 and aromatic/aliphatic polyesters46 was accounted for by increases in the free volumes associated with the polymer backbones. Clearly, our present results show the opposite trend to those previously reported. However, the Tg values of some poly(ether-amide)s composed of 4-hydroxycinnamic acid units have been reported to increase as the number of methoxy groups in their main chains increased. In this case, the increase in Tg was explained in terms of limited polymer-chain rotational flexibility due to steric effect.47 At present, the increase in Tg through the introduction of methoxy groups in our study is believed to be due to the latter reason. Note that the crystallisation temperature (Tc) and the melting temperature (Tm) were observed only for P(CA-alt-RA) (Fig. 1). The lack of methoxy groups in the main chain is clearly the main structural difference between P(CA-alt-RA) and the other two polymers. Therefore, we conclude that the introduction of methoxy groups significantly reduces the crystallinities of the corresponding polymers due to steric hindrance in a manner similar to that of a main-chain aromatic polyester.46 Moreover, during heating, the Tc was observed at 15 °C, and the Tm was observed at 58 °C, but neither of them was observed during cooling (Fig. 2). This monotropic behaviour could reasonably be attributed to the very slow crystallisation from the molten state, similar to the thermal behaviour seen for PLLA.48 We will consider this issue again in the discussion of the rheological studies to be described later.
Table 2 Thermal properties of alternating copolyesters
| Run |
Polymer sample |
Tga (°C) |
Tma (°C) |
Tca (°C) |
Td5b (°C) |
Td50b (°C) |
Tdmax1b (°C) |
Tdmax2b (°C) |
| Glass transition (Tg), melting (Tm) and crystallisation (Tc) temperatures were determined by DSC. Temperatures of 5% weight loss (Td5), 50% weight loss (Td50), first maximum rate of weight loss (Tdmax1) and second maximum rate of weight loss (Tdmax2) were determined by TGA. PRA was synthesised by the self-condensation of RA, using RA, 10 mmol; DPTS, 2 mmol; DIC, 15 mmol; CH2Cl2, 20 mL, rt, 24 h. Not observed. |
| 1 |
PRAc |
−73 |
—d |
—d |
328 |
352 |
353 |
—d |
| 2 |
P(CA-alt-RA) |
−15 |
58 |
15 |
319 |
357 |
341 |
448 |
| 3 |
P(FA-alt-RA) |
−4 |
—d |
—d |
319 |
355 |
345 |
439 |
| 4 |
P(SA-alt-RA) |
24 |
—d |
—d |
312 |
356 |
346 |
429 |
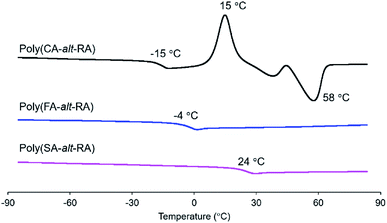 |
| | Fig. 1 DSC profile of polymers in the 2nd heating from −90 °C to 90 °C. | |
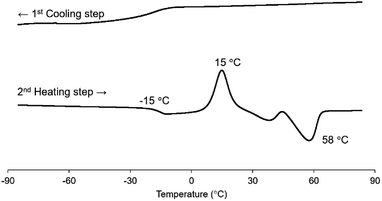 |
| | Fig. 2 Detailed thermal profile of P(CA-alt-RA) in 1st cooling step from 100 °C to −100 °C, and then in 2nd heating increase to 100 °C (DSC scan). | |
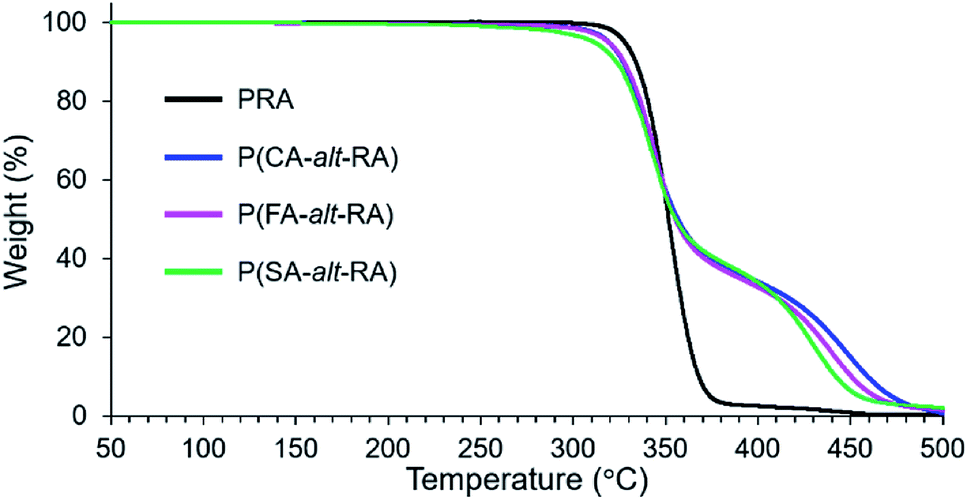
The thermal stability of the polymers as reflected by their heat resistance was also investigated with TGA under a nitrogen atmosphere. The results are summarised in Table 2 and illustrated in Fig. 3. Interestingly, all of these polymers showed a two-step decomposition process, in contrast with the single-step decomposition of PRA. The temperatures corresponding to 5% weight loss (Td5) and 50% weight loss (Td50), and the first maximum rate of weight loss (Tdmax1) were almost identical for P(CA-alt-RA), P(FA-alt-RA), and P(SA-alt-RA): Td5 = 312–319 °C, Td50 = 355–357 °C, and Tdmax1 = 341–346 °C (Table 2 and Fig. 3). The thermal stabilities are close to that of PRA, showing no significant change for the initial stage of the thermal decomposition. P(CA-alt-RA), P(FA-alt-RA) and P(SA-alt-RA) exhibited second weight loss steps (Tdmax2) with maximum rates at 448 °C, 439 °C, and 429 °C, respectively, corresponding to the degradation temperature of the 4-hydroxycinnamic acid unit.49 Thus, the introduction of the various 4-hydroxycinnamic acid units into the backbone obviously increased the decomposition temperature of PRA, although the intrinsic thermal stability (e.g. Td5) of PRA was maintained. Iwata et al. also reported a similar two-step decomposition process by TGA for a structurally related polymer.50 These authors developed an alternating poly(ester-amide) derived from ferulic acid and glycine and attributed the first decomposition step to carbonization and the second step to thermal cracking of the aromatic units. However, their proposed decomposition mechanism does not fully explain our decomposition process because even the monomer showed clear two-step decomposition behaviour by TGA in their study. This observation is not fully consistent with the present study where at least two of the hetero-dimeric monomer precursors (compounds 1c and 2c) showed mainly one-step decomposition processes, with a small shoulder observed in the high-temperature region (above 350 °C) as the second step in each case (Fig. S19 in ESI†). In the case of 3c, a clear two-step decomposition process was observed; however, the weight-loss pattern was not identical to that of the corresponding polymer. In other words, the two-step decomposition process observed in the present study is partly the result of the nature of the heterolytic structure of the monomer, as observed in the former study. Moreover, the observed trend is clearly due to the continuous linkage of monomers during polymerization. Hence, the origin of the two-step decomposition process remains unclear.
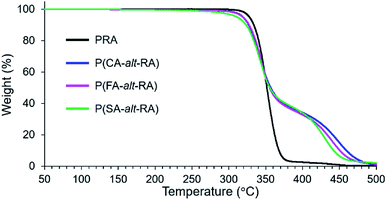 |
| | Fig. 3 TGA curves of PRA and resulting polymers. | |
3.4. Mechanical properties of polymers
The temperature-dependent viscoelastic properties of the polymers were also characterised by dynamic mechanical analysis (DMA) in a certain temperature range. We used two measurement modes, depending on temperature; the measurements in the temperature range from −50 to 50 °C were carried out in the tensile mode. The measurements in the temperature range 20–80 °C were carried out in the shear mode by using a rheometer. Polymer film samples for measurements were prepared using a heating press under vacuum at 95 °C for P(CA-alt-RA) or at 130 °C for P(FA-alt-RA) and P(SA-alt-RA).
The films from all three polymers were visually transparent and colourless (Fig. 4). The effect of temperature on the storage modulus (E′ and G′), loss modulus (E′′ and G′′) and loss factor (tanδ) are illustrated in Fig. 5 and 6. The Tg values were observed from the peak of tanδ, and occurred at −10.8 °C, 7.7 °C, and 31.3 °C, respectively (Fig. 5). The higher temperatures seen with the increasing number of methoxy groups were consistent with the trend in the DSC results. A rubbery plateau region was observed after the peak in E′′ for all polymers. The dramatic decrease in the G′ of P(CA-alt-RA) above 55.4 °C (Fig. 6a) meant that the polymer became a viscous liquid above that temperature. The melting temperature corresponded well with the Tm of P(CA-alt-RA) observed by DSC analysis. The intersection of G′ and G′′ was observed at 30.1 °C during cooling from high temperatures, a point lower than that seen during the heating process (Fig. S20 in ESI†). These results suggest that the crystallisation from a molten state was relatively slow, as described in the discussion of DSC results. In fact, the Tc was not observed in DSC studies with a cooling rate of 10 °C min−1 and the film sample of P(CA-alt-RA) did not show crystallisation behaviour at 15 °C in DMA experiments (Fig. 5a). The film sample used in the DMA measurement was slowly cooled to room temperature from 95 °C for 7 h during the pressing process. It is thought that the film sample had already crystallised during that process. Actually, the Tc of the film sample was not observed by DSC during heating from −100 °C (Fig. S16 in ESI†). P(FA-alt-RA) and P(SA-alt-RA) behave like viscous liquids above 70.6 °C, a temperature higher than that seen for P(CA-alt-RA).
 |
| | Fig. 4 Photographic images of dumbbell-shaped polymer films prepared for mechanical testing (width: 2 mm; length: 12 mm; thickness: approximately 1 mm). Polymer films are overlaid on coloured text: “AIST” (National Institute of Advanced Industrial Science and Technology). | |
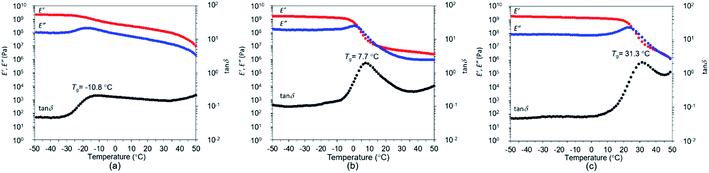 |
| | Fig. 5 Temperature dependence (range: −50–50 °C) of storage modulus (E′), loss modulus (E′′) and tanδ. (a) P(CA-alt-RA), (b) P(FA-alt-RA), (c) P(SA-alt-RA). | |
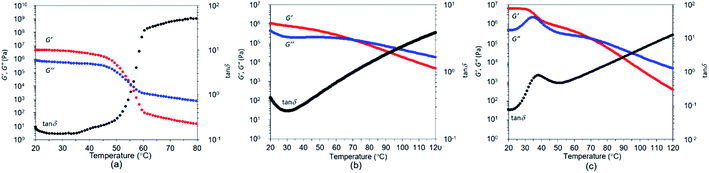 |
| | Fig. 6 Temperature dependence (range: 20–80 °C) of storage modulus (G′), loss modulus (G′′) and tanδ. (a) P(CA-alt-RA), (b) P(FA-alt-RA), (c) P(SA-alt-RA). | |
In considering practical uses for a biomass-based polymer, mechanical properties are among the most critical issues. The tensile strength test with dumbbell-shaped films was thus performed at room temperature, with samples prepared by a heating press under vacuum at 95 °C for P(CA-alt-RA) or at 130 °C for P(FA-alt-RA) and P(SA-alt-RA). The results are summarised in Table 3. The number of methoxy group on the cinnamoyl unit affected the elastic modules, tensile strengths and elongations at break for the polymers. P(CA-alt-RA) exhibited an elastic modulus of 61.9 MPa and a tensile strength of 2.33 MPa, indicating moderate mechanical toughness. The elongation degree, however, was relatively low (37%) among the three polymers. Conversely, P(FA-alt-RA) showed an elastic modulus (26.8 MPa) and tensile strength (0.38 MPa) lower than those of P(CA-alt-RA). P(FA-alt-RA) did not break, even after stretching by more than 800%, and this very high flexibility is quite characteristic for P(FA-alt-RA). The P(SA-alt-RA) sample exhibited the highest values for elastic modulus (126 MPa) and tensile strength (15.5 MPa). In addition, no yield point was observed, and the degree of elongation reached 585%. It was noteworthy that these mechanical properties were superior to those of the corresponding copolymer of RA with L-lactic acid (PLLA content: 45 wt%; elastic modulus: 42 MPa; tensile strength at break: 2.4 MPa; elongation at break: 17%).37 This means that P(SA-alt-RA) is reasonably tough and flexible, as compared to conventional PRA-based polymers. Furthermore, the recovery of strain for P(SA-alt-RA) at zero stress point was observed to range up to 13% from target strain (100%) in the cycle test with a rate of 1 mm min−1 (Fig. S24 in ESI†). The P(SA-alt-RA) sample slowly reverted to its original length after stretching. Thus far, the elongation behaviour of various PHAs have been reported,8 although their recovery of strain has not been examined in detail. Note that the present results indicate that P(SA-alt-RA) shows promise as a new biomass-based polymer, and also an elastic and shape-recoverable material. Moreover, as a preliminary experiment, the reversible bending behaviour of the P(SA-alt-RA) films and others were also examined (Fig. 7).
Table 3 Mechanical properties of P(CA-alt-RA), P(FA-alt-RA) and P(SA-alt-RA)
| Polymer |
Elastic modulus (MPa) |
Tensile strength at break (MPa) |
Elongation at break (%) |
| P(CA-alt-RA) |
61.9 ± 9.6 |
2.33 ± 0.15 |
37 ± 2 |
| P(FA-alt-RA) |
26.8 ± 4.3 |
0.38 ± 0.07 |
>800 |
| P(SA-alt-RA) |
126.5 ± 46.9 |
15.47 ± 0.43 |
585 ± 23 |
 |
| | Fig. 7 Reversible bending behaviour of the polymer films. | |
4. Conclusions
Carbon-neutral and abundant biomass-derived chemicals such as castor oil-derived RA and lignin-derived ferulic acid, p-coumaric acid, and sinapinic acid have been transformed into novel biomass-based polyesters via self-condensation of the corresponding hetero-dimeric monomers. The copolymers consist of two repeating characteristic units: one is a RA unit with a long cis alkenylene chain functioning as a soft segment, and the other is a hydroxycinnamic acid unit functioning as a hard segment. We found that the introduction of alternating soft and hard sequences in the polymer chain led to improvements in both thermal and mechanical properties, as expected. In studies of thermal properties, the resulting polymers showed a trend in Tg that depended on the structure of hydroxycinnamic acid units; this was comparable to the behaviour of PRA seen by DSC analysis. Among the substrates studied here, P(SA-alt-RA), bearing two methoxy groups in the monomer unit, exhibited the highest Tg value and this was 80 °C higher than that of the original PRA. It is suggested that Tg could be tuned by varying the number of methoxy groups on the cinnamoyl unit of the backbone. In addition, the temperature for full decomposition of the polymers seen in TGA studies was also increased by approximately 100 °C relative to that of PRA. However, the Td5, Td50 and Tdmax1 values were almost identical to those of PRA. All three polymers could be moulded as flexible films; this behaviour differed from that of PRA, which is a viscous liquid at ambient temperatures. Studies of their mechanical properties showed that P(FA-alt-RA) did not break, even when stretched by a factor exceeding 800%. Among the materials considered here, the P(SA-alt-RA) sample exhibited the highest values for both elastic modulus (126.5 MPa) and tensile strength (15.47 MPa). Moreover, P(SA-alt-RA) reverted to its original length after being stretched, and its recovery of strain was confirmed to be up to 13% in the cycle test. These results suggested that P(SA-alt-RA) may be regarded as an elastic and shape-recoverable polymer. The present study revealed new possibilities for biomass-based polymers, with improved thermal and mechanical properties seen for structures with alternating RA and 4-hydroxycinnamic acid substrates. The origins of the thermal stability and unique elasticity are under investigation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This paper is based on results obtained with the support of the RIKEN–AIST Joint Research Fund (Full research). The authors also appreciate the support provided by the fundamental research fund of AIST. The authors are grateful to Ms Sayo Osanai for her technical assistance with the syntheses. The authors are also grateful to Prof. Kazuo Koike (Faculty of Pharmaceutical Sciences, Toho University), Tsuyoshi Kawakami and Shiori Takeuchi (Medicinal Plant Garden, Faculty of Pharmaceutical Sciences, Toho University), and Mr Masaru Akatsuka (Media Net Center, Toho University) for providing useful information and images of castor seeds and plants.
Notes and references
- P. A. Wilbon, F. Chu and C. Tang, Macromol. Rapid Commun., 2013, 34, 8–37 CrossRef CAS.
- S. Muñoz-Guerra, C. Lavilla, C. Japu and A. Martínez de Ilarduya, Green Chem., 2014, 16, 1716–1739 RSC.
- B. Lochab, S. Shukla and I. K. Varma, RSC Adv., 2014, 4, 21712–21752 RSC.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- J. A. Galbis, M. de G. García-Martín, M. Violante de Paz and E. Galbis, Chem. Rev., 2016, 116, 1600–1636 CrossRef CAS.
- X. Zhou, P. Wang, Y. Zhang, X. Zhang and Y. Jiang, ACS Sustainable Chem. Eng., 2016, 4, 5585–5593 CrossRef CAS.
- H. Ye, K. Zhang, D. Kai, Z. Li and X. J. Loh, Chem. Soc. Rev., 2018, 47, 4545–4580 RSC.
- A. Larranaga and E. Lizundia, Eur. Polym. J., 2019, 121, 109296 CrossRef CAS.
- A. Södergårg and M. Stolt, Prog. Polym. Sci., 2002, 27, 1123–1163 Search PubMed.
- S. Farah, D. G. Anderson and R. Langer, Adv. Drug Delivery Rev., 2016, 107, 367–392 CrossRef CAS.
- V. Tserki, P. Matzinos, E. Pavlidou, D. Vachliotis and C. Panayiotou, Polym. Degrad. Stab., 2006, 91, 367–376 CrossRef CAS.
- M. Gigli, M. Fabbri, N. Lotti, R. Gamberini, B. Rimini and A. Munari, Eur. Polym. J., 2016, 75, 431–460 CrossRef CAS.
- J. J. Fischer, Y. Aoyagi, M. Enoki, Y. Doi and T. Iwata, Polym. Degrad. Stab., 2004, 83, 453–460 CrossRef CAS.
- G.-Q. Chen, Chem. Soc. Rev., 2009, 38, 2434–2446 RSC.
- Y. Tabata and H. Abe, Macromolecules, 2014, 47, 7354–7361 CrossRef CAS.
- J. J. Bozell and G. R. Peterson, Green Chem., 2010, 12, 539–554 RSC.
- S. Sato, T. Fujiki and K. Matsumoto, J. Biosci. Bioeng., 2013, 116, 677–681 CrossRef CAS.
- S. L. Riedel, J. Lu, U. Stahl and C. J. Brigham, Appl. Microbiol. Biotechnol., 2014, 98, 1469–1483 CrossRef CAS.
- G. Kutralam-Muniasamy and F. Peréz-Guevara, World J. Microbiol. Biotechnol., 2018, 34, 79–91 CrossRef.
- U. Biermann, W. Friedt, S. Lang, W. Lühs, G. Machmüller, J. O. Metzger, M. R. G. Klaas, H. J. Schäfer and M. P. Schneider, Angew. Chem., Int. Ed., 2000, 39, 2206–2224 CrossRef CAS.
- M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788–1802 RSC.
- L. Maisonneuve, T. Lebarbé, E. Grau and H. Cramail, Polym. Chem., 2013, 4, 5472–5517 RSC.
- F. Stempfle, B. S. Ritter, R. Mülhaupt and S. Mecking, Green Chem., 2014, 16, 2008–2014 RSC.
- S. L. Kristufek, K. T. Wacker, Y.-Y. T. Tsao, L. Su and K. L. Wooley, Nat. Prod. Rep., 2017, 34, 433–459 RSC.
- K. R. Kunduru, A. Basu, M. H. Zada and A. J. Domb, Biomacromolecules, 2015, 16, 2572–2587 CrossRef CAS.
- B.-X. Zhang, J. Azuma, S. Takeno, N. Suzuki, Y. Nakazawa and H. Uyama, Polym. J., 2016, 48, 821–827 CrossRef CAS.
- D. S. Ogunniyi, Bioresour. Technol., 2006, 97, 1086–1091 CrossRef CAS.
- M. B. Mensah, J. A. M. Awudza and P. O'Brien, R. Soc. Open Sci., 2018, 5, 180824 CrossRef CAS.
- R. Slivniak and A. J. Domb, Biomacromolecules, 2005, 6, 1679–1688 CrossRef CAS.
- H. Ebata, K. Toshima and S. Matsumura, Macromol. Biosci., 2007, 7, 798–803 CrossRef CAS.
- A. Kazariya and S. Matsumura, Polymers, 2012, 4, 486–500 CrossRef.
- H. Ebata, M. Yasuda, K. Toshima and S. Matsumura, J. Oleo Sci., 2008, 57, 315–320 CrossRef CAS.
- D. Teomim, A. Nyska and A. J. Domb, J. Biomed. Mater. Res., 1999, 45, 258–267 CrossRef CAS.
- M. Haim-Zada, A. Basu, T. Hagigit, R. Schlinger, M. Grishko, A. Kraminsky, E. Hanuka and A. J. Domb, Biomacromolecules, 2016, 17, 2253–2259 CrossRef CAS.
- P. Rajalakshmi, J. M. Marie and A. J. M. Xavier, Polym. Degrad. Stab., 2019, 170, 109016 CrossRef.
- W.-X. Wu, J. Li, X.-L. Yang, N. Wang and X.-Q. Yu, Eur. Polym. J., 2019, 121, 109315 CrossRef CAS.
- T. Lebarbé, E. Ibarboure, B. Gadenne, C. Alfos and H. Cramail, Polym. Chem., 2013, 4, 3357–3369 RSC.
- C. Gioia, M. B. Banella, G. Totaro, M. Vannini, P. Marchese, M. Colonna, L. Sisti and A. Celli, J. Renewable Mater., 2018, 6, 126–135 CrossRef CAS.
- L. Mialon, R. Vanderhenst, A. G. Pemba and S. A. Miller, Macromol. Rapid Commun., 2011, 32, 1386–1392 CrossRef CAS.
- H. T. H. Nguyen, M. H. Reis, P. Qi and S. A. Miller, Green Chem., 2015, 17, 4512–4517 RSC.
- G. N. Short, H. T. H. Nguyen, P. I. Scheurle and S. A. Miller, Polym. Chem., 2018, 9, 4113–4119 RSC.
- A. C. Fonseca, M. S. Lima, A. F. Sousa, A. J. Silvestre, J. F. J. Coelho and A. C. Serra, Polym. Chem., 2019, 10, 1696–1723 RSC.
- J. S. Moore and S. I. Stupp, Macromolecules, 1990, 23, 65–70 CrossRef CAS.
- K. K. Reddy, T. Ravinder and S. Kanjilal, Food Chem., 2012, 134, 2201–2207 CrossRef CAS.
- S. Shyamroy, B. Garnaik and S. Sivaram, Polym. Bull., 2015, 72, 405–415 CrossRef CAS.
- J.-I. Jin and C.-S. Kang, Polymer, 1993, 34, 2407–2412 CrossRef CAS.
- B. M. Upton and A. M. Kasko, Biomacromolecules, 2019, 20, 758–766 CrossRef CAS.
- Z. Kulinski and E. Piorkowska, Polymer, 2005, 46, 10290–10300 CrossRef CAS.
- H. T. H. Nguyen, E. R. Suda, E. M. Bradic, J. A. Hvozdovich and S. A. Miller, Polyesters from Bio-Aromatics, ACS Symposium Series Green Polymer Chemistry III: Biobased Materials and Biocatalysis, ed. H. N. Cheng, R. Gross and P. Smith, 2015, ch. 24, vol. 1192, pp. 401–409 Search PubMed.
- T. Goto, D. Ishii, Y. Enomoto-Rogers, A. Takemura and T. Iwata, Polymer, 2017, 112, 385–392 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and polymer characterization data. See DOI: 10.1039/d0ra05671e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Koji Nemoto*ab,
Masaru Yoshida
a,
Koji Nemoto*ab,
Masaru Yoshida