
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAddition of 1,3-dicarbonyl compounds to terminal alkynes catalyzed by a cationic cobalt(III) complex†
Mohan Chandra Sau and
Manish Bhattacharjee *
*
Department of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur 721302, India. E-mail: mxb@iitkgp.ac.in
First published on 30th September 2020
Abstract
The Nakamura reaction using a cationic cobalt(III) complex, [Cp*Co(CH3CN)3][SbF6]2 as the catalyst under neutral and aerobic conditions at 110 °C has been described. In solution, the complex is expected to lose a hemilabile acetonitrile ligand to produce a highly electron-deficient cobalt(III) center, and the Lewis acidic nature of the cobalt center has been exploited for the enolization of the dicarbonyl compounds. The reaction of 1,3-dicarbonyl compounds with alkynes affords the corresponding alkenyl derivative. However, the reaction of phenylacetylene and its derivatives with β-ketoesters affords corresponding terphenyl compounds. Details of the mechanisms of the reactions have been proposed based on in situ LCMS measurements.
Introduction
Atom economical carbon–carbon sigma bond formation under neutral and mild conditions is an emerging field in organic synthesis due to the inertness of the C–C bond.1a It was believed that the intermolecular addition of an enolate anion to an unactivated alkene and alkynes is difficult due to the poor polarity of C–C multiple bonds.1b However, Nakamura and coworkers reported the addition of alkynes to β-ketoesters in the presence of In(OTf)3 (0.05 to 5 mol%) at around 140 °C in the absence of any solvent.2 Later on, the same group developed the asymmetric version of the reaction.3 These were followed by several reports using other metal compounds. Lau and coworkers have shown that TpRu[4-CF3C6H4N(PPh2)2](OTf) [Tp = hydro(trispyrazolyl)borate] catalyses the addition 1,3-diketones to terminal alkynes at 120 °C under solvent-free condition.4 It has been shown that RuCl3·3H2O (5 mol%) in the presence of Ag(PF6) (15 mol%) catalyses the addition of 1,3-diketones to alkynes.5 Another report deals with [Ru(CO)3Cl2]2 catalysed addition of diketones to alkynes.6 However, the conversions were poor, and in the cases of cyclic diketones, instead of C–C bond formation, C–O bond formation was observed. Xi et al. reported Nakamura reaction in presence of 0.05 mol% of [Xphos(TA)AuOTf] {TA = 1,2,3-triazole} and 5 mol% of Ga(OTf)3.7 Another report deals with Ir(I) catalysed Nakamura reaction.8 Kuninobu et al. reported that [Re(CO)3Br(THF)] (3 mol%) catalyses the Nakamura reaction at 50 °C.9 It may be noted that to the best of our knowledge to date Nakamura reaction catalysed by an earth abundant first-row transition element has not been reported except a couple of reports, one on Mn(CO)5Br (5 mol%)10 and another on Cp*Co(CO)I2 (5 mol%) + Cu(OTf)2 (5 mol%)11 catalysed addition of dicarbonyl compounds to alkynes and (2 + 2 + 2) cycloaddition. All these methods either suffer from very harsh reaction conditions or require expensive ligands or expensive additives. It may be noted that the reactions catalysed by Cp*Co(CO)I2 and Cu(OTf)2 are carried out at room temperature without using any solvent. However, the catalyst and cocatalyst loading is comparatively high. In the proposed mechanism of the reaction it has been suggested that, Cu(OTf)2 is responsible for the generation of Lewis acidic coordinatively unsaturated species [Cp*Co(OTf)]+.11 Also, some of these metal complexes are air and moister sensitive, so the reactions are carried out in dry solvents under an inert atmosphere.It is anticipated that [Cp*Co(CH3CN)3]2+ can afford a coordinatively unsaturated species, “[Cp*Co]2+” with very high Lewis acidity during the reaction.
Multisubstituted benzene, especially terphenyl, are a very important class of compounds.12,13 Conventional substitution methods for the synthesis of multisubstituted benzene requires a large number of steps.12 There are many reports on the transition metal-catalysed [2 + 2 + 2] cycloaddition reactions of alkynes.14,15 However, due to the poor regioselectivity of the reaction, it is challenging to synthesize multisubstituted benzene.12 Apart from these reports, a few reports deal with the synthesis of tetrasubstituted benzene from the reaction of β-ketoesters with terminal alkynes catalysed by [ReBr(CO)4(THF)]12,13 in the presence of MS4A and N,N-dimethylacetamide (20 mol%) and [MnBr(CO)5]12 in the presence of MS4A.
Herein we report [Cp*Co(CH3CN)3][SbF6]2 (I) catalysed the addition of terminal alkynes to 1,3-diketones. We also report the synthesis of tetrasubstituted benzene from the reaction of β-ketoesters with terminal alkynes catalysed by I.
Results and discussion
For the reaction of 1,3-diketones with terminal alkynes, the reaction of 2,4-pentane-dione (1a) and phenylacetylene (2a) was chosen as a model reaction for the for optimization. The reactants 1a and 2a were taken in a reaction tube in dichloroethane (DCE), and the reaction solution was heated at different temperatures (room temperature to 110 °C) for 24 hours, but we could not detect any product formation (entry-1, Table 1). To this reaction tube [Cp*Co(CH3CN)3][SbF6]2 (I) (1 mol%) was added and was heated at 110 °C for 12 hours. We could isolate the desired product, 4-hydroxy-3-(1-phenyl vinyl)pent-3-en-2-one (3a) in 48% yield in 12 hours (entry-2, Table 1). When toluene was used as the solvent, 3a was isolated in 59% yield in 12 hours (entry-3, Table 1). All the subsequent reactions were carried out in toluene. We thought that the addition of base might facilitate enolate formation, and thus yield would increase. Keeping this in mind, we carried out the reaction in the presence of K2CO3 (1 equivalent), keeping all other reaction conditions the same. However, the yield of 3a drastically reduced (18%) (Table 1, entry 4). We then varied the concentration of the catalyst. The reaction carried out in the presence of 2 mol% of I at 110 °C afforded 3a in 67% yield in 10 hours (Table 1, entry 5). The reaction carried out in the presence of 3 mol% of I at 90 °C afforded 3a in 68% yield in 7 hours (Table 1, entry 6). When the reaction was carried out at 110 °C using 3 mol% I, the yield of 3a was 76% in 7 hours (Table 1, entry 7). Further decrease in the reaction time to 5 hours did not affect the yield (73%) of the product (Table 1, entry 8). We performed the reaction using 4 mol% of I for 5 hours, but this did not improve the yield of the product (Table 1, entry 9). The reaction was also carried out in the presence of 3 mol% of I at 110 °C for 7 hours. However, the yield was poor (46%) (Table 1, entry 10).| Entry | I (mol%) | Additive (mol%) | Temp (°C) | Time (h) | Solvent | Yieldb (%) |
|---|---|---|---|---|---|---|
| a Reagents and conditions: 1a (0.45 mmol), 2a (0.3 mmol), solvent (4 ml).b Isolated. | ||||||
| 1 | — | — | 90/110 | 12 | DCE | — |
| 2 | 1 | — | 90/110 | 12 | DCE | 48 |
| 3 | 1 | — | 110 | 12 | Toluene | 59 |
| 4 | 1 | K2(CO)3 (1) | 110 | 12 | Toluene | 18 |
| 5 | 2 | — | 110 | 10 | Toluene | 67 |
| 6 | 3 | — | 100 | 7 | Toluene | 68 |
| 7 | 3 | — | 90 | 7 | Toluene | 76 |
| 8 | 3 | — | 110 | 5 | Toluene | 73 |
| 9 | 4 | — | 110 | 5 | Toluene | 75 |
| 10 | 3 | — | 110 | 7 | Neat | 46 |
Having established the reaction condition, we proceeded to investigate the substrate scope of the reaction. First, we carried out the reaction of 2,4-pentane-dione with various alkynes. The products, 3a to 3i, were isolated in 62 to 82% yield (Table 2). The lowest yield (62% in 8 hours) was obtained in the case of 3-[1-(3,4-dichorophenyl)-vinyl]-4-hydroxy-pent-3-en-2-one (3e). Higher conversion to the corresponding product was observed in the cases of 1-ethynyl-4-trifluoromethyl-benzene (2g) (81%, 6 hours) and 1-ethynyl-4-nitro-benzene (2h) (82%, 7 hours). Other products were isolated in approximately 75% yield. When we carried reaction of 2-ethynyl-thiophene (2i) with 2,4-pentanedione (1a) the corresponding product, 4-hydroxy-3-(1-thiophene-2-yl-vinyl)-pent-3-en-2-one (3i) was isolated in 74% yield in 6 hours. However, the reaction of alkynes with the aliphatic side chain failed to give the products. Next, we proceeded to explore the reactions of various acyclic and cyclic diketones with alkynes.
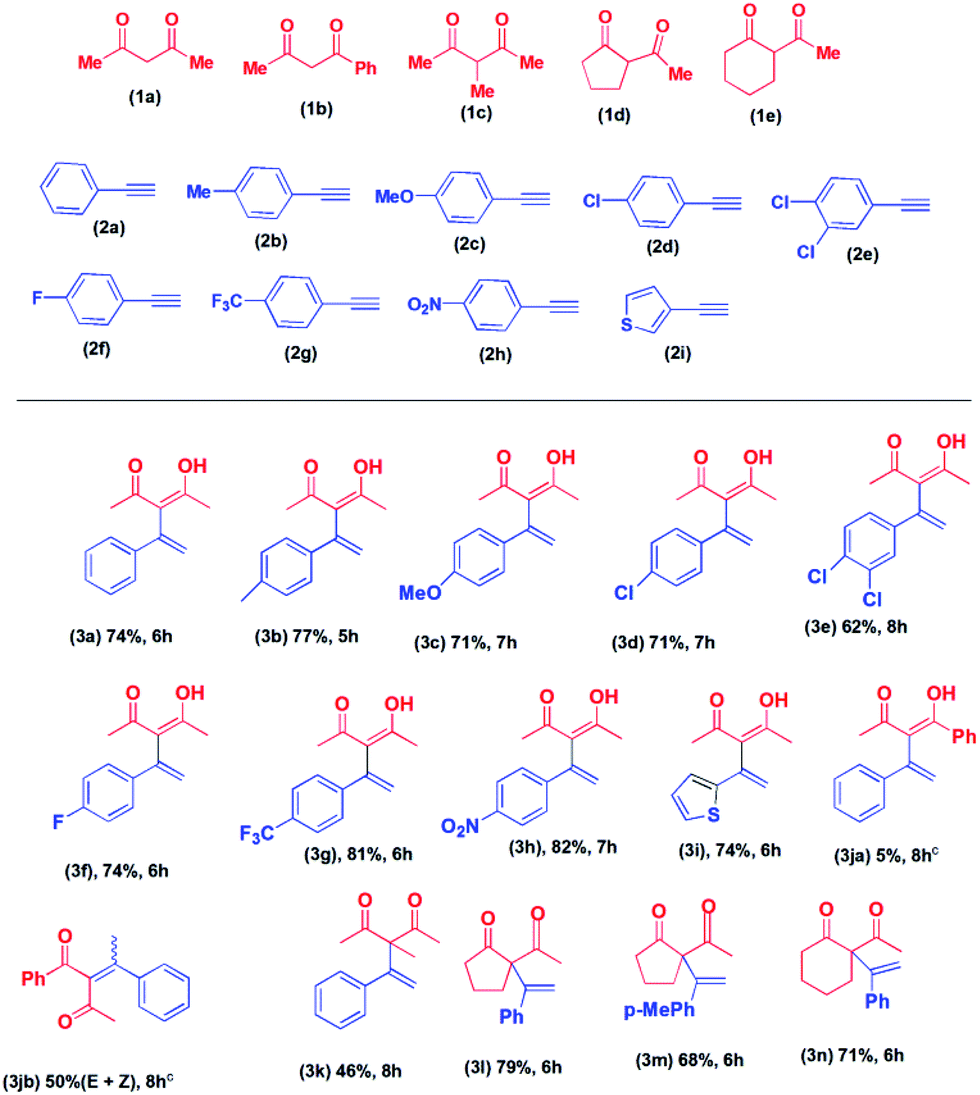
The reaction of 1-phenyl-1,3-pentane-dione with phenylacetylene was carried out under the same reaction condition. To our surprise, we obtained a mixture of E and Z isomers of Markovnikov addition product (3jb) as the major product and anti-Markovnikov addition product (3ja) as the minor product. The reaction of 2-methyl-2,4-pentane-dione with phenylacetylene afforded 3-methyl-3-(1-phenyl vinyl)-pentane-2,4-dione (3k) in 46% yield in 8 hours. The cyclic diketone, 2-acetyl-cyclopentanone (1d) on reaction with phenylacetylene (2a), and 1-ethynyl-4-methyl-benzene (2b) afforded the corresponding products, 3l and 3m in 79% and 68% yield in 6 hours, respectively. The reaction of 2-acetyl-cyclohexanone (1e) with phenylacetylene (2a) afforded 2-acetyl-2-(1-phenyl vinyl)-cyclohexanone (3n) in 71% yield in 6 hours. Our attempted reaction of phenylacetylene with 1,3-diphenyl-propane-1,3-dione failed to afford any product. Similarly, 5,5-dimethyl-cyclohexane-1,3-dione did not afford any product. No product formation was observed when aliphatic alkynes were used.
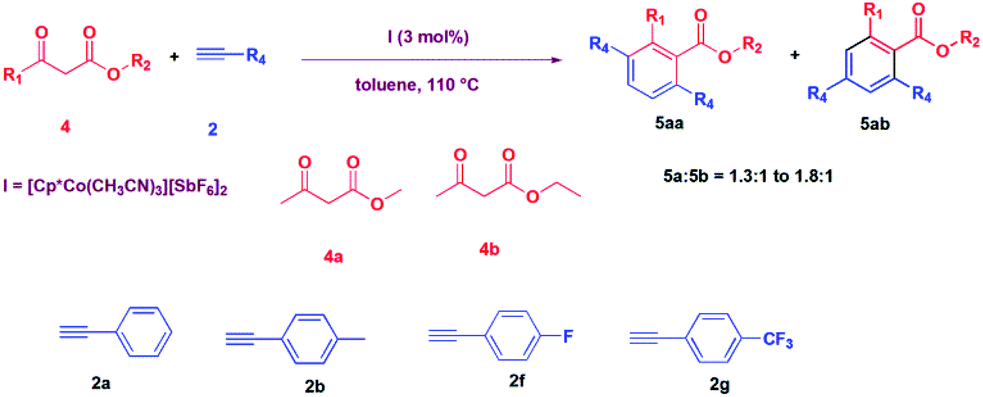
After carrying out the reaction of diketones, we thought to explore the reaction of β-ketoesters with alkynes catalysed by I. Thus, a reaction of 3-oxo-butyric acid methyl ester was carried out with phenylacetylene in the presence of 3 mol% I in toluene at 110 °C. To our surprise, we could not see the formation of the expected product, 3-acetyl-3-phenyl-but-3-enoic acid methyl ester, as obtained in the cases of diketones, described above. Instead, we obtained 2-methyl-3,6-diphenyl benzoic acid methyl ester in a 60% yield in 12 hours (Table 3, entry 1). We then explored the substrate scope of the reaction, and the reactions of methyl acetoacetate (4a) and ethyl acetoacetate (4b) with various alkynes were carried out, and the products were isolated in good to moderate yield (52% to 69%) (Table 3).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
:Next, we proceeded to examine the mechanism by in situ LCMS measurements. The in situ LCMS spectra of the reaction mixture of 2,4-pentane-dione with phenylacetylene shows a peak at m/z 293.1, which corresponds to [Cp*Co(acac)]+ ion. Based on this observation, it is suggested that [Cp*Co(CH3CN)3]2+ first loses hemilabile acetonitrile ligands to generate the intermediate [Cp*Co(acac)]+ (A; Scheme 1). Another peak appears at m/z 395.1, which corresponds to the intermediate [Cp*Co(acac)(PhCCH)]+ (B; Scheme 1). The intermediate B is highly electron deficient, and the cobalt enolate attacks the alkyne carbon bearing the substituent and produces species C. It may be noted that we could not distinguish between the species B and C, as they have same m/z value. However, it is suggested that C is produced via the intermediate B. The species C reacts with another alkyne to produce the species D, for which a peak was observed at m/z 497.2. The species D then undergoes protonolysis to produce the product and the intermediate B back.
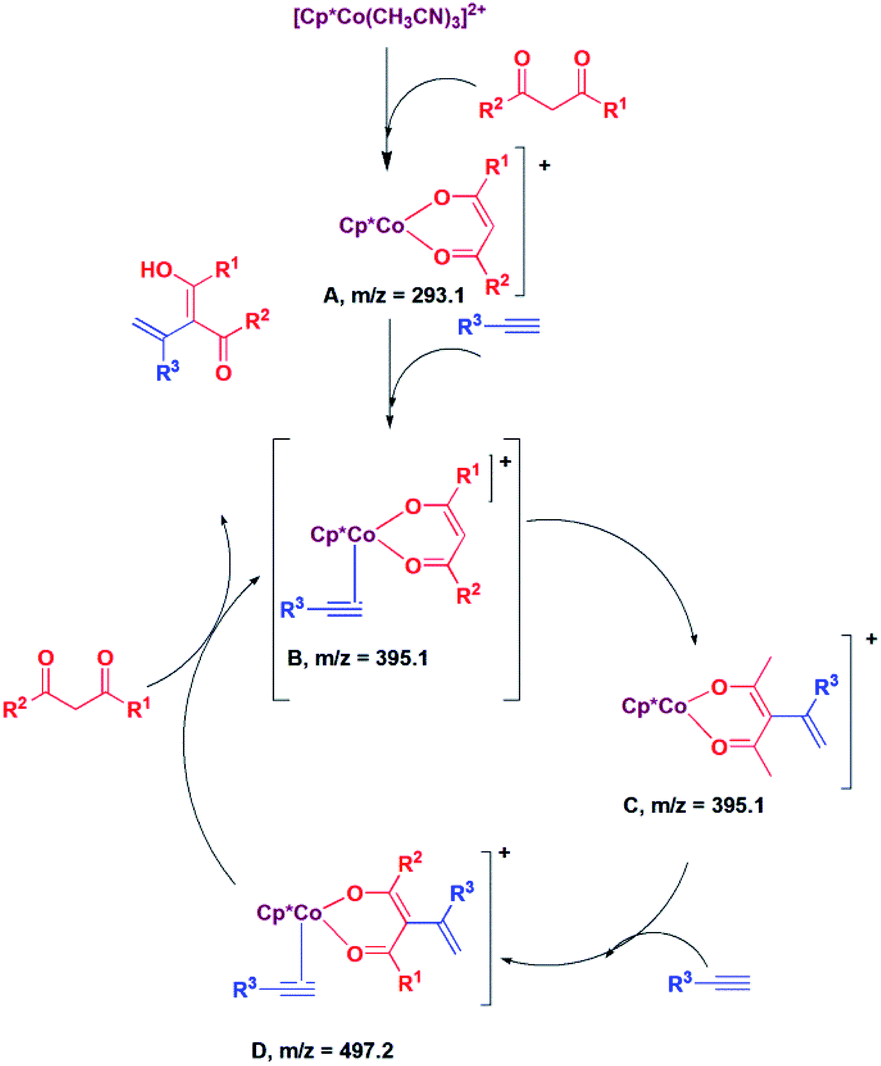 | ||
| Scheme 1 Plausible mechanism of addition 1,3-diketone to alkyne. | ||
In situ LCMS spectra were recorded at different time intervals of the reaction of methyl acetoacetate with phenylacetylene to understand the possible mechanism of the reaction. In the LCMS spectra of the reaction of methyl acetoacetate, a peak was observed at m/z 309.2, which corresponds to the species [Cp*Co(CH3COCHCOOMe)]+ (A, Scheme 2). A peak was observed at m/z 513.4, which may be assigned to [Cp*Co(PhCCH)2(CH3COCHCOOMe)]+ (B; Scheme 2). The species B may undergo addition reaction to afford the species C. Because of this same m/z value, we could not distinguish between B and C. We suggest that C is formed via the intermediate B. It is proposed that the species C undergoes [2 + 2] cycloaddition and subsequent protonolysis to afford the product and the species A.
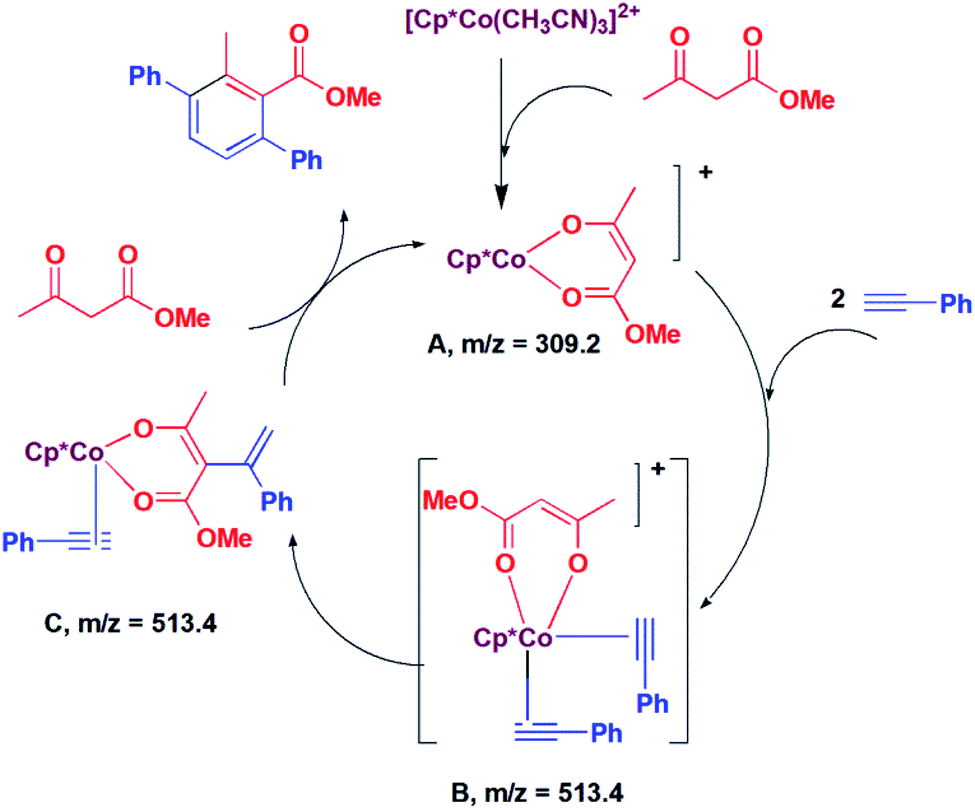 | ||
| Scheme 2 Plausible mechanism of addition β-ketoester to alkyne. | ||
Conclusion
In conclusion, a highly Lewis acidic complex of a first raw transition element, [Cp*Co(CH3CN)3][SbF6]2, has been used as a catalyst for the Nakamura reaction. The same catalyst, when employed for the reaction of β-ketoesters with the terminal alkynes affords aromatic products. The mechanisms of the reactions have been proposed based on the in situ LCMS studies.Experimental section
General methods and materials
All the reactions were performed under aerial atmosphere without further purification of commercially available reagents and solvents. The catalyst [Cp*Co(CH3CN)3][SbF6]2 was synthesized from the literature procedure.16 The 1H NMR (400 MHz) and 13C NMR (100 MHz) were recorded in CDCl3. The LCMS data of the reaction mixture were recorded with a TOF mass spectrometer in ESI+ mode in MeOH and acetonitrile solvent. The HRMS data of newly synthesized compounds were recorded with a TOF mass spectrometer in ESI+ mode in acetonitrile/water mixture. Flash column chromatography was done in silica gel (60–120 mesh) for purification of product from the crude reaction mixture.General procedure for the reaction of alkynes with diketo compounds
The catalyst (0.0070 g, 3 mol%), toluene (2 ml), and the two reactants 1,3-diketone or β-ketoesters (0.3 mmol), terminal alkyne (0.45 mmol) were taken in a screw-cap reaction tube. The reaction tube was placed in an oil-bath having a temperature of 110 °C–115 °C. The reaction solution was stirred for 5–8 hours. The reaction was monitored by TLC. The solvent was evaporated, and the products were purified by silica gel (60–120 mesh) column chromatography using hexane-ethyl acetate (0.2–5%) as eluent.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank SERB for the NMR facility (Grant Number SR/FST/CSII-026/2013).Notes and references
- (a) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS; (b) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333 CrossRef CAS.
- (a) M. Nakamura, K. Endo and E. Nakamura, J. Am. Chem. Soc., 2003, 125, 13002 CrossRef CAS; (b) K. Endo, T. Hatakeyama, M. Nakamura and E. Nakamura, J. Am. Chem. Soc., 2007, 129, 5264 CrossRef CAS.
- T. Fujimoto, K. Endo, H. Tsuji, M. Nakamura and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 4492 CrossRef CAS.
- H. W. Cheung, C. M. So, K. H. Pun, Z. Zhou and C. P. Lau, Adv. Synth. Catal., 2011, 353, 411 CrossRef CAS.
- M. K. Pennington-Boggio, B. L. Conley and T. J. Williams, J. Organomet. Chem., 2012, 716, 6 CrossRef CAS.
- B. Leotard, T. Fukuyama, Y. Higashibeppu, C. Brancour, H. Okamoto and I. Ryu, Synlett, 2013, 24, 2287 CrossRef CAS.
- Y. Xi, D. Wang, X. Ye, N. G. Akhmedov, J. L. Petersen and X. Shi, Org. Lett., 2014, 16, 306 CrossRef CAS.
- G. Onodera, M. Kato, R. Kawano, Y. Kometani and R. Takeuchi, Org. Lett., 2009, 11, 5038 CrossRef CAS.
- Y. Kuninobu, M. Nishi, S. S. Yudha and K. Takai, Org. Lett., 2008, 10, 3009 CrossRef CAS.
- P. R. Throve, M. M. Guru and B. Maji, J. Org. Chem., 2019, 84, 8185 CrossRef.
- G. K. Verma, M. Rawat and D. S. Rawat, Eur. J. Org. Chem., 2019, 4101 CrossRef CAS.
- Y. Kuninobu, M. Nishi, A. Kawata, H. Takata, Y. Hanatani, S. S. Yudha, A. Iwai and K. Takai, J. Org. Chem., 2010, 75, 334 CrossRef CAS.
- H. Tsuji, K.-i. Yamagata, T. Fujimoto and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 7792 CrossRef CAS.
- Reviews: (a) S. Saito and Y. Yamamato, Chem. Rev., 2000, 100, 2901 CrossRef CAS; (b) D. B. Grotjahn, in Comprehensive Organometallic Chemistry II, Ed. L. S. Hegedus, E. W. Abel, F. G. A. Stone and F. G. Wilkinson, Pergamon, Oxford, UK, 1995, vol. 12, pp. 741–770 Search PubMed; (c) H. Bonnemann and W. Brijoux, in Transition Metals for Organic Synthesis, ed. M. Beller and C. Bolm, WileyVCH, Weinheim, Germany, 2004, vol. 1, pp. 171–197 Search PubMed.
- (a) K. Tanaka, K. Tyoda, A. Wada, K. Shiraska and M. Hirano, Chem. – Eur. J., 2005, 11, 1145 CrossRef CAS; (b) S. Li, H. Qu, H. L. Zhou, K.-i. Kanno, Q. Guo, B. Shen and T. Takahashi, Org. Lett., 2009, 11, 3318 CrossRef CAS.
- D. G. Yu, T. Gensch, F. De Azambuja, S. Vásquez-Céspedes and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17722 CrossRef CAS.
- T. Zhu, P. Zheng, C. Mou, S. Yang, B.-A. Song and Y. R. Chi, Nat. Commun., 2014, 5, 5027 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13CNMR spectra HRMS, and LCMS. See DOI: 10.1039/d0ra05923d |
| This journal is © The Royal Society of Chemistry 2020 |