DOI:
10.1039/D0RA05919F
(Paper)
RSC Adv., 2020,
10, 34815-34824
Synthesis of siRNAs incorporated with cationic peptides R8G7 and R8A7 and the effect of the modifications on siRNA properties†
Received
7th July 2020
, Accepted 14th September 2020
First published on 21st September 2020
Abstract
Small interfering RNA (siRNA) can be used as an innovative next-generation drug. However, there are several challenges in the therapeutic application of siRNAs, including their low cell membrane permeability. In this study, we designed and synthesized siRNAs, incorporating the cationic peptides R8G7 and R8A7 to improve cell membrane permeability of siRNAs. Thermal denaturation studies revealed that R8G7 and R8A7 modifications increased the thermal stability of the siRNA duplexes. Incorporating these peptides at the 3′-ends of the siRNA passenger strands increased the stability of the siRNAs in a buffer containing bovine serum. Further, we found that the peptide–siRNA conjugates did not show sufficient RNA interference (RNAi) activity in the absence of the transfection reagent; however, when the transfection reagent was used, the peptide–siRNA conjugates preserved their RNAi activity.
Introduction
Recently, small interfering RNA (siRNA) drugs, a category of oligonucleotide-based drugs utilizing RNA interference (RNAi), have attracted a lot of attention as next-generation drugs. Generally, siRNAs comprise 19–21 base pairs with two 3′ nucleotide overhangs. After penetrating into cells, siRNA forms an RNA-induced silencing complex (RISC) with a series of proteins including Argonaute 2 (Ago2). In the RISC, one strand (a passenger strand) of the siRNA is cleaved and the remaining strand (a guide strand) forms a duplex with the target mRNA. The target mRNA is then hydrolyzed by the slicer activity of Ago2.1–3
However, there are several challenges in the therapeutic application of siRNAs. siRNAs composed of natural ribonucleotides are easily hydrolyzed by abundant nucleases present inside and outside cells.4 Moreover, siRNA consisting of a polyanionic double-stranded structure can barely penetrate the negatively charged cell membrane.5,6 To overcome these issues, various chemically modified siRNAs have been developed.4,7–9 Attempts to improve the cell membrane permeability of oligonucleotides, by neutralizing their negative charges with positive charges, have been investigated.10–13 For example, it was shown that oligonucleotides with guanidium modifications exhibited remarkable cellular uptake properties.14–18 Moreover, it has been reported that modification of biomolecules by the addition of octaarginine (R8), which is a known cell membrane-penetrating peptide, improved their cell membrane permeability.19,20
Based on this background information, in this study, we designed and synthesized siRNAs with modifications in their basic peptides to improve the low cell membrane permeability of the siRNAs. We designed novel peptides, based on R8, and synthesized siRNAs that were conjugated with these peptides at several positions. Then, we examined the thermal stability, nuclease resistance properties, and RNAi activity of the modified siRNAs.
Result and discussion
Synthesis of the building units
We chose to introduce the modified peptides into RNAs using the Hüisgen cycloaddition reaction. To carry out this reaction, we prepared RNAs and peptides containing an alkynyl group and an azide functional group, respectively.
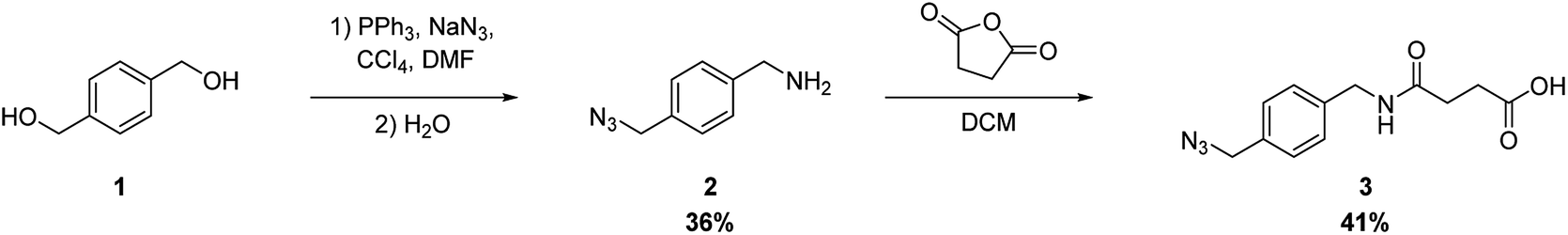
The synthetic route of the azide derivative 3, which is a building unit for introducing the azide functional group into peptides, is shown in Scheme 1. We used 1,4-bis(hydroxymethyl)benzene (1) as the starting material. Azidation of the primary hydroxyl groups of 1 was carried out by treating it with triphenylphosphine, NaN3, and CCl4 in N,N-dimethylformamide. This was followed by selective reduction of one of the 2 azido groups by the Staudinger reaction, which produced 4-(aminomethyl)benzyl azide (2) in 31% yield. Succinylation of 2 by treatment with succinic anhydride in dichloromethane produced 3 in 41% yield.
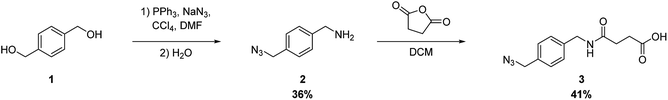 |
| | Scheme 1 | |
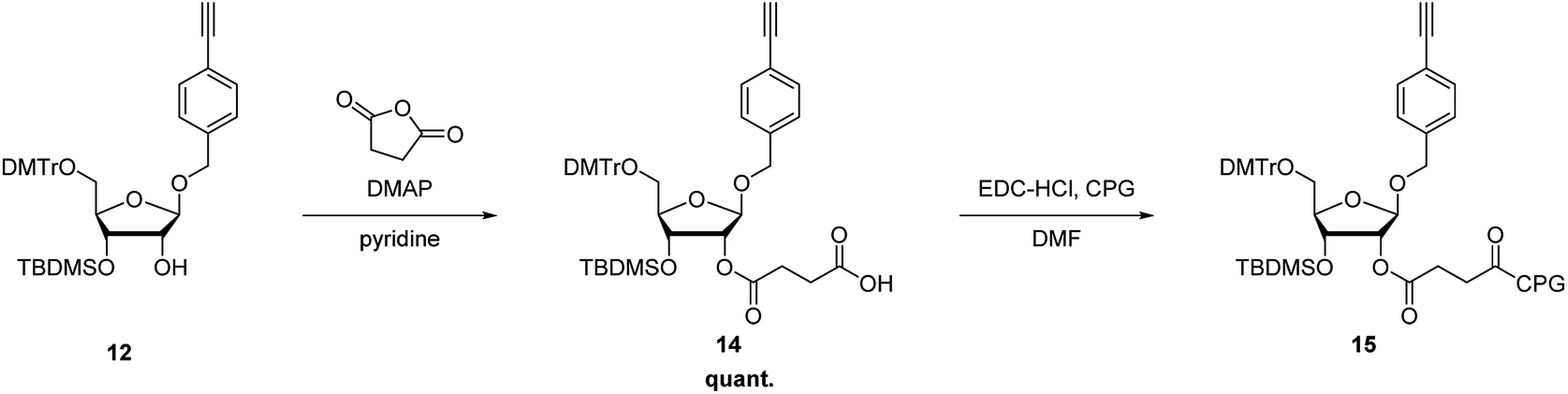
The synthetic route of a phosphoramidite of the nucleoside analog with the alkynyl group is shown in Scheme 2. Here, 4-(hydroxymethyl)iodobenzene (4) was reacted with trimethylsilyl (TMS) acetylene under Sonogashira–Hagihara cross-coupling conditions to quantitatively produce 5. Deprotection of the TMS group of 5 with tetrabutylammonium fluoride in tetrahydrofuran produced the ethynyl derivative 6 in 61% yield. Glycosylation of 1-O-acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose (7) and 6 was performed using TMS trifluoromethanesulfonate as a catalyst, to produce the 1-O-(4-ethynylphenyl)methyl-β-D-ribofuranose derivative 8 in 70% yield. Deprotection of the benzoyl groups of 8 by treatment with CH3ONa in MeOH produced 9 in 64% yield. The 5′-hydroxyl functional group of 9 was protected with a 4,4′-dimethoxytrityl group to produce 10 in 72% yield. Silylation of the hydroxyl group using tert-butyldimethylchlorosilane (TBDMS-Cl) and triethylamine produced the 2′-O-TBDMS derivative, 11, and 3′-O-TBDMS derivative, 12, in 29 and 43% yields, respectively. Then, 11 was phosphorylated by a standard procedure to produce the corresponding phosphoramidite, 13, in 87% yield.
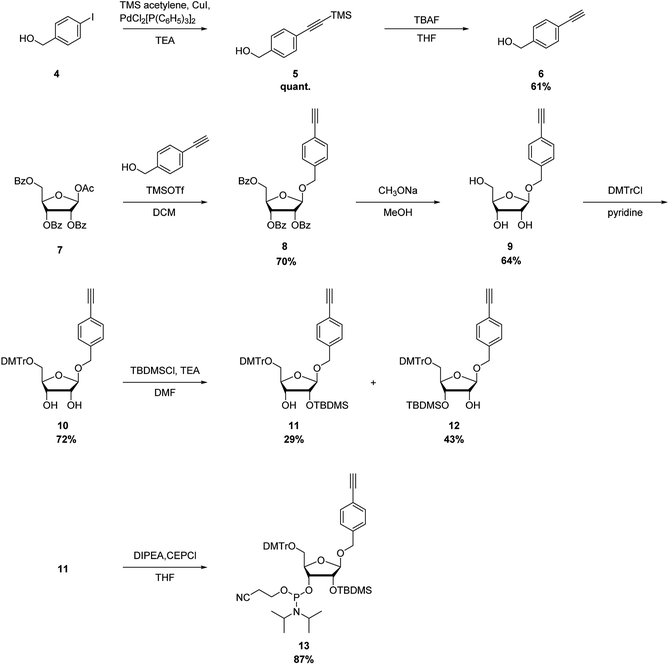 |
| | Scheme 2 | |
To incorporate the nucleoside analog into the 3′-end of RNAs, 12 was further modified to produce the corresponding 3′-succinate, 14, which were then reacted with controlled pore glass (CPG) to give the solid support, 15 (44 μmol g−1) (Scheme 3).
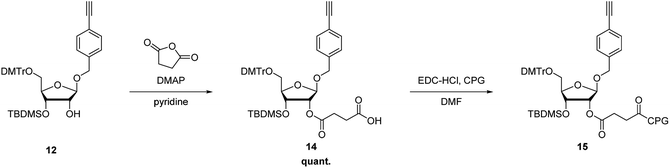 |
| | Scheme 3 | |
Synthesis of peptides
We designed 3 types of peptides (Table 1). R8 comprised octaarginine and building unit 3 at the N-terminal. To control the distance between the guanidino groups, we designed R8G7 and R8A7. R8G7 consisted of a sequence of alternating arginine and glycine, whereas R8A7 comprised a sequence of alternating arginine and β-alanine. The peptides were synthesized by solid-phase synthesis using 9-fluorenylmethyloxycarbonyl (Fmoc) chemistry at a 5.0 μmol scale. A 4-methylbenzhydrylamine resin, which generates an amide functional group at the C-terminal, was used as the solid support for peptide synthesis. Amino acids protected with the Fmoc group were reacted with 1-[bis(dimethylamino)methylene]-1H-benzotriazolium 3-oxide hexafluorophosphate as a coupling reagent, in the presence of 1-hydroxybenzotriazole and N,N′-diisopropylethylamine. Cleavage of the peptide from the solid support and deprotection of the protective groups were performed using a trifluoroacetic acid (TFA) solution containing ethanedithiol, thioanisole, and m-cresol. However, it was found that the azide functional group at the N-terminal of the peptide was reduced and converted to an amino functional group under deprotection conditions. To avoid side reactions, cleavage and deprotection were carried out by treatment with a TFA solution containing only m-cresol. The mass of the peptides was estimated by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). The observed molecular weights from these analyses were found to be consistent with their peptide structures.
Table 1 Sequence of peptides
| Name of peptide |
Abbreviation of peptide |
Sequencea N terminus → C terminus |
Number of amino acids |
Number of charges |
| 3, R, G and A denote azide derivative, arginine, glycine and β-alanine. |
| R8 |
Peptide 1 |
3-RRRRRRRR-NH2 |
8 |
+8 |
| R8G7 |
Peptide 2 |
3-RGRGRGRGRGRGRGR-NH2 |
15 |
+8 |
| R8A7 |
Peptide 3 |
3-RARARARARARARAR-NH2 |
15 |
+8 |
Synthesis of RNA oligomers
RNA oligomers containing the nucleoside analog 9, with the alkynyl group, were synthesized by solid-phase RNA synthesis using a DNA/RNA synthesizer (Table 2). Cleavage from CPG and deprotection of the oligomers were carried out by treatment with a concentrated NH3 solution/40% methylamine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solution at 65 °C for 10 min. Deprotection of 2′-O-TBDMS was performed by treatment with Et3N·3HF in dimethyl sulfoxide (DMSO) at 65 °C for 1.5 h. After cleavage and deprotection, the oligomers were purified by polyacrylamide gel electrophoresis (PAGE) on a 20% denaturing gel. The mass of the purified RNA was estimated by MALDI-TOF MS. The observed molecular weights from these analyses were found to be consistent with their RNA structures.
:1, v/v) solution at 65 °C for 10 min. Deprotection of 2′-O-TBDMS was performed by treatment with Et3N·3HF in dimethyl sulfoxide (DMSO) at 65 °C for 1.5 h. After cleavage and deprotection, the oligomers were purified by polyacrylamide gel electrophoresis (PAGE) on a 20% denaturing gel. The mass of the purified RNA was estimated by MALDI-TOF MS. The observed molecular weights from these analyses were found to be consistent with their RNA structures.
Table 2 Sequence of single stranded (ss) RNAs
| Abbreviation of ssRNA |
Sequencea |
| RE and F denote p-ethynyl analog 8 and fluorescein, respectively. |
| RNA 1 |
5′-GGCCUUUCACUACUCCUACTTRE-3′ |
| RNA 2 |
F-5′-GGCCUUUCACUACUCCUACTTRE-3′ |
| RNA 3 |
5′-REGGCCUUUCACUACUCCUACTT-3′ |
| RNA 4 |
5′-GGCCUUUCACUACUCCUACUU-3′ |
| RNA 5 |
5′-GGCCUUUCACUACUCCUACTTRE-R8G7-3′ |
| RNA 6 |
5′-GGCCUUUCACUACUCCUACTTRE-R8A7-3′ |
| RNA 7 |
5′-R8G7-REGGCCUUUCACUACUCCUACTT-3′ |
| RNA 8 |
5′-R8A7-REGGCCUUUCACUACUCCUACTT-3′ |
| RNA 9 |
5′-GUAGGAGUAGUGAAAGGCCUU-3′ |
| RNA 10 |
F-5′-GUAGGAGUAGUGAAAGGCCUU-3′ |
Synthesis of peptide–RNA conjugates
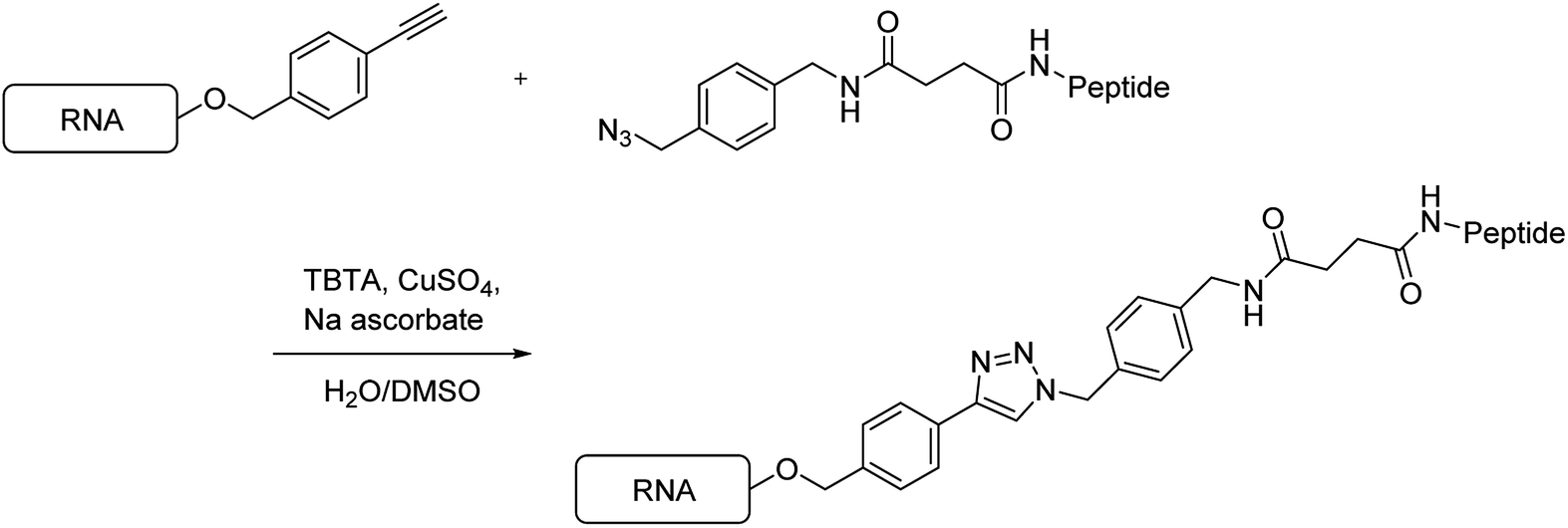
Next, using the Hüisgen cycloaddition reaction (Scheme 4), we tried to connect the RNAs containing the ethynyl group with the peptides having the azide functional group. The RNAs were reacted with the peptides in the presence of tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine, CuSO4, and sodium ascorbate, in a DMSO/H2O solution, at 37 °C for 2 h. Then, the reaction mixtures were analyzed by reversed-phase high-performance liquid chromatography (HPLC).
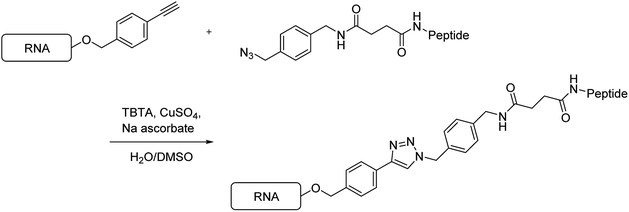 |
| | Scheme 4 | |
When peptide 1 (R8) was reacted with RNA 1, no peak was observed on HPLC. A possible explanation for this was considered that aggregates were formed between the RNA and the peptide by the strong electrostatic interaction between the polyanionic RNA and the polycationic R8. When the solution containing the fluorescein-labeled RNA 2 was mixed with the solution containing peptide 1 (R8), the color of the solution changed from yellow to red, which was attributed to aggregation-induced emission. Thus, we decided to synthesize a new peptide that does not form aggregates with RNA. In 2013, Maeda et al. reported that the affinity of peptides, comprising an alternating sequence of an arginine analog and glycine, to the RNA duplex is less than that of peptides comprising an oligoarginine analog.21 Thus, we designed and synthesized the peptide R8G7, comprising an alternating sequence of arginine and glycine, and the peptide R8A7, comprising an alternating sequence of arginine and β-alanine.
We tried to connect the peptides R8G7 and R8A7 with RNAs by the Hüisgen cycloaddition reaction. When the peptide R8G7 or R8A7 reacted with RNA 1, the peak of RNA 1 disappeared on HPLC after 20 min, and a new peak appeared. The new peak was purified by HPLC, and the molecular weight was measured by MALDI-TOF MS. The observed molecular weights from these analyses were found to be consistent with those of the conjugate RNAs 5 and 6 (Table 2). Similarly, the Hüisgen cycloaddition reaction of RNA 3 with the peptide R8G7 or R8A7 was completed in 20 min. The observed molecular weights from MALDI-TOF MS analyses were found to be consistent with those of the conjugate RNAs 7 and 8. From the above results, it was found that the conjugation reaction of the peptides R8G7 and R8A7 to RNA proceeded rapidly.
Thermal stability of duplexes
Next, we evaluated the effect of the peptide modifications on the thermal stability of the RNA duplexes (siRNAs 1–5). UV melting experiments were performed in a buffer containing 10 mM sodium phosphate (pH 7.0) and 100 mM NaCl (Table 3). The Tm value of the unmodified siRNA (siRNA 1) was 77.6 °C, whereas that of the modified siRNA 2, 3, 4, and 5 was 78.5, 78.1, 79.1, and 78.3 °C, respectively. The ΔTm [Tm(siRNAmod) − Tm(siRNAunmod)] value of siRNA 2, 3, 4, and 5 was +0.9, +0.5, +1.9, and +0.7 °C, respectively. Thus, it was found that incorporating the peptides, R8G7 and R8A7, slightly increased the thermal stability of the siRNA duplexes. Furthermore, the ΔΔTm [Tm(siRNAmod-R8G7) − Tm(siRNAmod-R8A7)] values were −0.4 °C for the 3′-modification and −1.2 °C for the 5′-modification, respectively. These results indicated that the peptide R8G7 stabilized the siRNA duplex more than the peptide R8A7.
Table 3 Sequence of ssRNAs and siRNAs, and Tm values of siRNAsa
| Abbreviation of siRNA |
Abbreviation of ssRNA |
Sequencea |
Tmb (°C) |
ΔTmc (°C) |
ΔΔTmd (°C) |
| RE denote p-ethynyl analog 8. The Tm's were measured in a buffer containing 10 mM sodium phosphate (pH 7.0) and 100 mM NaCl. The concentrations of the duplexes were 3 μM. ΔTm represents [Tm(dsRNAmod) − Tm(dsRNAunmod)]. ΔΔTm represents [Tm(dsRNAmod-R8G7) − Tm(dsRNAmod-R8A7)]. |
| siRNA 1 |
RNA 4 |
5′-GGCCUUUCACUACUCCUACUU-3′ |
77.6 |
— |
— |
| RNA 9 |
3′-UUCCGGAAAGUGAUGAGGAUG-5′ |
| siRNA 2 |
RNA 5 |
5′-GGCCUUUCACUACUCCUACTTRE-R8G7-3′ |
78.5 |
+0.9 |
— |
| RNA 9 |
3′-UUCCGGAAAGUGAUGAGGAUG-5′ |
| siRNA 3 |
RNA 6 |
5′-GGCCUUUCACUACUCCUACTTRE-R8A7-3′ |
78.1 |
+0.5 |
−0.4 |
| RNA 9 |
3′-UUCCGGAAAGUGAUGAGGAUG-5′ |
| siRNA 4 |
RNA 7 |
5′-R8G7-REGGCCUUUCACUACUCCUACTT-3′ |
79.5 |
+1.9 |
— |
| RNA 9 |
3′-UUCCGGAAAGUGAUGAGGAUG-5′ |
| siRNA 5 |
RNA 8 |
5′-R8A7-REGGCCUUUCACUACUCCUACTT-3′ |
78.3 |
+0.7 |
−1.2 |
| RNA 9 |
3′-UUCCGGAAAGUGAUGAGGAUG-5′ |
CD spectra analyses
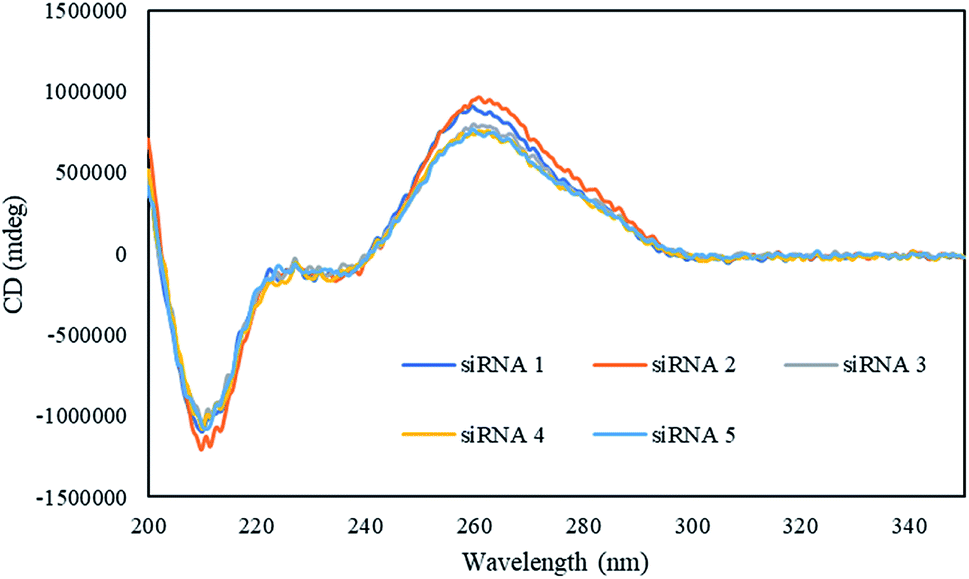
Next, we investigated the effect of the peptides on the global conformations of the RNA duplexes by measuring the circular dichroism (CD) spectra. As shown in Fig. 1, all the duplexes showed positive CD bands, around 260 nm, and negative CD bands, around 210 nm. These results suggested that the global conformation of the siRNA duplexes incorporating the peptides was not significantly different from that of the unmodified siRNA duplex.
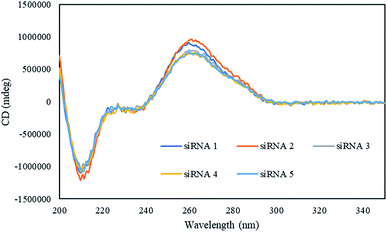 |
| | Fig. 1 CD spectra of the modified and unmodified siRNAs in a buffer containing 10 mM sodium phosphate (pH 7.0) and 100 mM NaCl at 15 °C. The concentrations of duplexes were 4 μM. | |
Nuclease resistance
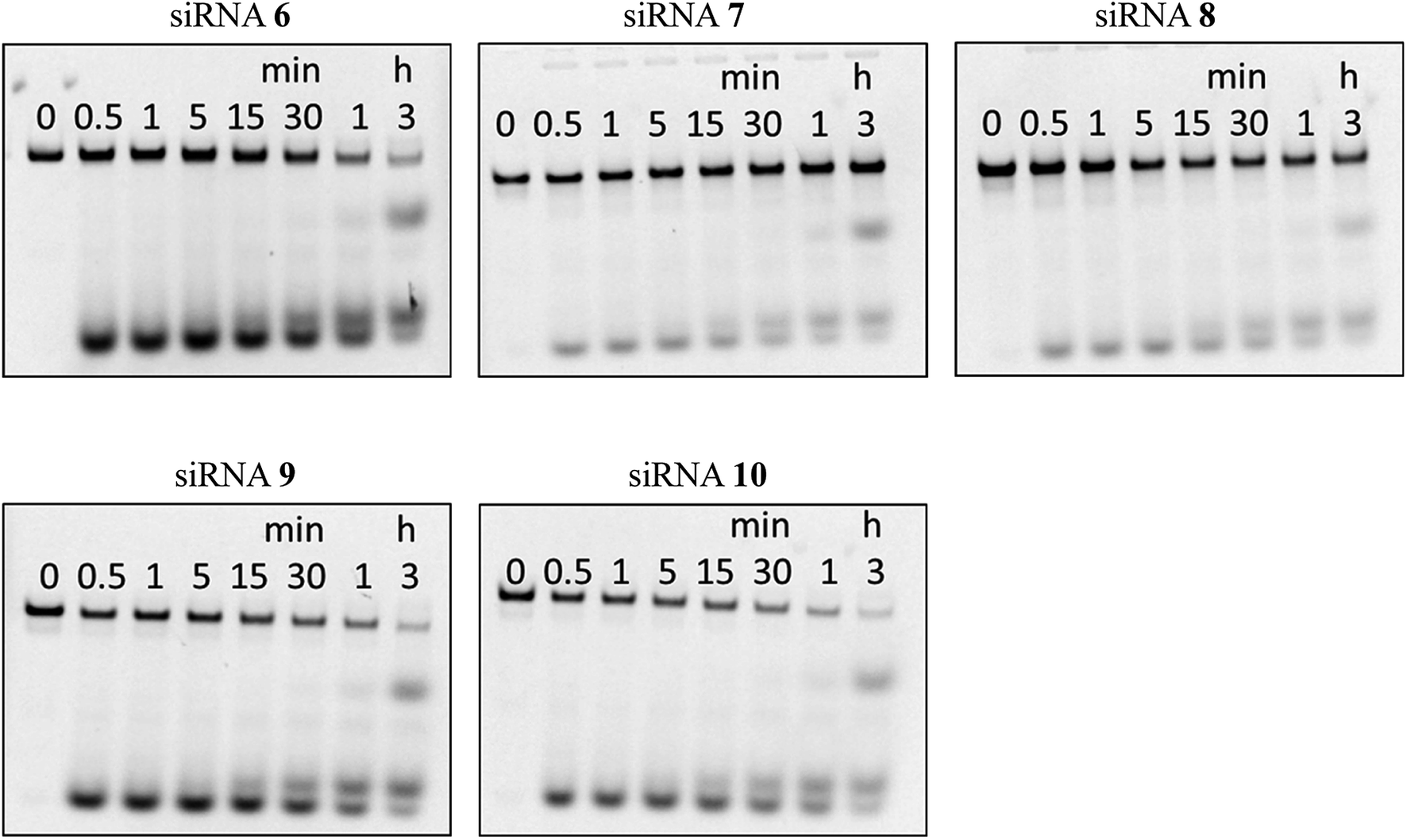
Next, we assessed the stability of the peptide–siRNA conjugates to nucleolytic hydrolysis using nucleases existing in serum. The unmodified siRNA (siRNA 6) and the modified siRNAs incorporating the peptides (siRNAs 7–10) were incubated in a buffer containing 10% bovine serum (BS). Subsequently, the products were analyzed by PAGE. As shown in Fig. 2, the siRNAs were gradually hydrolyzed. After 3 h of incubation, the ratios of the remaining intact siRNAs 6, 7, 8, 9, and 10 were 12, 56, 51, 11, and 10%, respectively. Thus, it was revealed that incorporating the peptides at the 3′-ends of the siRNA passenger strands increased the stability of siRNAs in a buffer containing BS. This could be due to the difference in thermal stability at both ends of the siRNA duplexes. The 5′-GGC-3′/3′-CCG-5′ base pairing is thermally more stable than the 5′-UAC-3′/3′-AUG-5′ base pairing. Therefore, the site of the 5′-UAC-3′/3′-AUG-5′ base pairing was thought to be more nuclease-sensitive than that of the 5′-GGC-3′/3′-CCG-5′ base pairing. Thus, we considered that incorporating the peptides at the nuclease-sensitive sites would improve the nuclease stability of siRNAs more efficiently than that at other sites (Table 4).
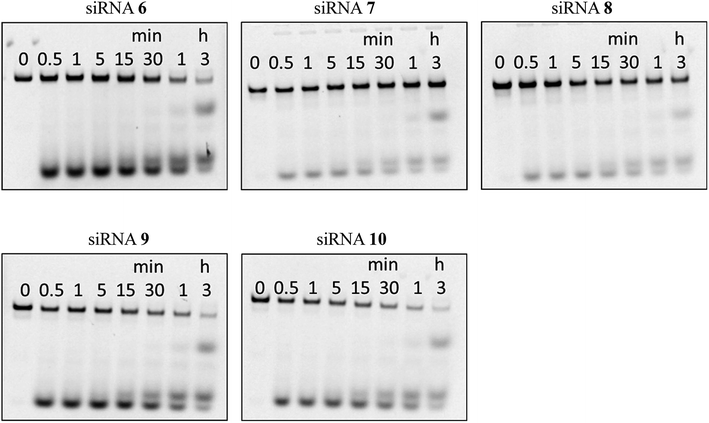 |
| | Fig. 2 Stability of the peptide–siRNA conjugates in a buffer containing bovine serum. | |
Table 4 Sequence of ssRNA and siRNA for nuclease resistance assaya
| RE and F denote p-ethynyl analog 8 and fluorescein. |
| siRNA 6 |
RNA 4 |
5′-GGCCUUUCACUACUCCUACUU-3′ |
| RNA 10 |
3′-UUCCGGAAAGUGAUGAGGAUG-5′-F |
| siRNA 7 |
RNA 5 |
5′-GGCCUUUCACUACUCCUACTTRE-R8G7-3′ |
| RNA 10 |
3′-UUCCGGAAAGUGAUGAGGAUG-5′-F |
| siRNA 8 |
RNA 6 |
5′-GGCCUUUCACUACUCCUACTTRE-R8A7-3′ |
| RNA 10 |
3′-UUCCGGAAAGUGAUGAGGAUG-5′-F |
| siRNA 9 |
RNA 7 |
5′-R8G7-REGGCCUUUCACUACUCCUACTT-3′ |
| RNA 10 |
3′-UUCCGGAAAGUGAUGAGGAUG-5′-F |
| siRNA 10 |
RNA 8 |
5′-R8A7-REGGCCUUUCACUACUCCUACTT-3′ |
| RNA 10 |
3′-UUCCGGAAAGUGAUGAGGAUG-5′-F |
RNAi activity
Finally, we evaluated the RNAi activity of the peptide–siRNA conjugates by a Dual-Luciferase Reporter assay using HeLa cells, in which the target luciferase genes were constitutively expressed. All the siRNAs targeted the Renilla luciferase genes, and firefly luciferase genes were used as controls. All the siRNAs were transfected using RNAiMAX. The expression levels of both types of luciferase genes were analyzed after 24 h of incubation. The ratios of Renilla:firefly luciferase activity with respect to the no siRNA control after 24 h of incubation are shown in Fig. 3. Although the silencing activity of the peptide–siRNA conjugates at 1 nM was slightly weaker than that of the unmodified siRNA, the RNAi activity of the conjugates at 10 nM were almost the same as that of the unmodified siRNA. These data indicate that the peptide modifications at the 3′- or 5′-ends of the siRNA passenger strands were well tolerated by the siRNAs, as indicated by their RNAi activity. On the other hand, in the absence of the transfection reagent, sufficient RNAi activity was not observed.
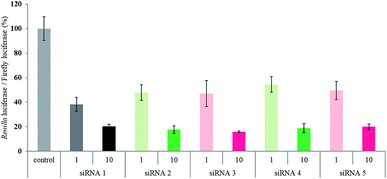 |
| | Fig. 3 RNAi activity of unmodified and modified siRNAs. siRNAs were transfected into HeLa cells at concentrations of 1 and 10 nM. After a 24 h incubation, the activities of Renilla and firefly luciferases in the cells were determined with a Dual-Luciferase Reporter Assay System. The results were confirmed by at least three independent transfection experiments with two cultures each and are expressed as the average from four experiments as mean ± SD. | |
Conclusions
In this study, we demonstrated the synthesis of peptide–siRNA conjugates. We found that introduction of R8 into siRNAs causes aggregation, but R8G7 and R8A7 can be introduced into siRNAs without causing aggregation. Thermal denaturation studies showed that modifications by R8G7 and R8A7 increased the thermal stability of the siRNA duplexes. It was found that incorporating the peptides at the 3′-ends of the siRNA passenger strands increased the stability of siRNAs in a buffer containing BS. Furthermore, the peptide–siRNA conjugates preserved their RNAi activity. However, in the absence of the transfection reagent, the peptide–siRNA conjugates did not show sufficient RNAi activity, probably due to poor cell membrane permeability. Recently, we reported that 4′-C-guanidinomethyl modification of nucleoside moieties increased the cell membrane permeability of RNAs.22 Further, the synthesis of siRNAs, modified with both the 4′-C-guanidinomethyl group and the peptide R8G7 or R8A7, is currently underway in our laboratory.
Experimental section
General remark
Thin layer chromatography (TLC) was performed on silica gel plates precoated with fluorescent indicator with visualization by UV light or by dipping into a solution of 5% (v/v) concentrated H2SO4 in mixture of p-anisaldehyde and methanol and then heating. Silica gel (63–210 mesh) was used for column chromatography. 1H NMR (400, 500 or 600 MHz), 13C NMR (101, 126 or 151 MHz) and 31P NMR (162 MHz) were recorded on 400, 500 or 600 MHz NMR equipment. CDCl3 or DMSO-d6 was used as a solvent for obtaining NMR spectra. Chemical shifts (δ) are given in parts per million (ppm) from CDCl3 (7.26 ppm) or TMS (0.00 ppm) for 1H NMR spectra and from CDCl3 (77.2 ppm) for 13C NMR spectra. The abbreviations s, d, t, q, and m signify singlet, doublet, triplet, quadruplet, and multiplet, respectively. High resolution mass spectra (HRMS) were obtained in positive ion electrospray ionization (ESI-TOF) mode.
4-(Aminomethyl)benzylazide (2)
1,4-Bis(hydroxymethyl)benzene (1) (1.00 g, 7.24 mmol) was added to a solution of triphenylphosphine (2.27 g, 23.9 mmol), NaN3 (1.86 g, 29.0 mmol), CCl4 (2.34 mL, 23.9 mmol) in DMF (20 mL) at 0 °C. After being stirred for 2 h at 0 °C, H2O (5.6 mL) was added to a solution and the mixture was stirred for 26 h at room temperature. The reaction mixture was partitioned between EtOAc and H2O. The organic layer was extracted with saturated NH4Cl aqueous solution. The water layer was adjusted to pH 10 by adding 1 M NaOH aqueous solution. The mixture was partitioned between CHCl3 and saturated NH4Cl aqueous solution. The organic layer was dried over Na2SO4 and concentrated in vacuo. The crude material was purified by column chromatography (15–20% MeOH in CHCl3) to afford 2 as a white solid (0.42 g, 2.61 mmol, 36%). 1H NMR (500 MHz, CDCl3) δ 3.80 (s, 2H), 4.23 (s, 2H), 7.20 (d, 2H, J = 8.0), 7.25 (d, 2H, J = 8.0); 13C NMR (126 MHz, CDCl3) δ 46.2, 54.6, 127.6, 128.6, 133.9, 143.5.
4-({[4-(Azidomethyl)phenyl]methyl}amino)-4-oxobutanoic acid (3)
Compound 2 (0.42 g, 2.61 mmol) was dissolved in CH2Cl2 (3.0 mL). The solution was added to a solution of succinic anhydride (0.31 g, 3.13 mmol) in CH2Cl2 (8.5 mL). The mixture was stirred for 2 h at room temperature and concentrated. The resulting residue was partitioned between EtOAc and 1 M NaOH aqueous solution. The water layer was adjusted to pH 4 by adding 1 M HCl aqueous solution and the mixture was partitioned between EtOAc and H2O. The organic layer was dried over Na2SO4 and concentrated in vacuo. The crude material was purified by column chromatography (25% MeOH in CHCl3) to afford 3 as a white solid (0.28 g, 1.08 mmol, 41%). 1H NMR (500 MHz, DMSO-d6) δ 2.38 (t, 2H, J = 6.9), 2.45 (t, 2H, J = 6.3), 4.26 (d, 2H, J = 5.7), 4.39 (s, 2H), 7.26 (d, 2H, J = 8.1), 7.29 (d, 2H, J = 8.1), 8.36 (t, 1H, J = 5.7); 13C NMR (126 MHz, DMSO-d6) δ 29.67, 30.53, 42.28, 53.89, 128.00, 128.99, 134.45, 140.20, 171.59, 174.41; HRMS (ESI-TOF) m/z calc. for C12H15N4O3 [M + H]+; 263.1144 found 263.1130.
4-(Trimethylsilylethynyl)benzyl alcohol (5)
4-Iodobenzyl alcohol (4) (1.47 g, 6.28 mmol) was dissolved in Et3N (15.0 mL) at room temperature under argon atmosphere. To the solution, PdCl2[P(C6H5)3]2 (0.44 g, 0.63 mmol), CuI (0.12 g, 0.63 mmol) and TMS-acetylene (1.30 mL, 9.93 mmol) were added, and the mixture was stirred at room temperature for 2 h. The mixture was filtered through Celite, and the filtrate was partitioned between EtOAc and H2O. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified by column chromatography (25% ethyl acetate in hexane) to afford 9 as a brown oil (1.28 g, 6.28 mmol, 100%). 1H NMR (600 MHz, CDCl3) δ 0.25 (s, 9H), 1.89 (br s, 1H), 4.66 (s, 2H), 7.28 (d, 2H, J = 7.6), 7.45 (d, 2H, J = 8.2); 13C NMR (151 MHz, CDCl3) δ 0.1, 65.0, 94.3, 105.1, 122.5, 126.7, 132.3, 141.4; HRMS (ESI-TOF) m/z calc. for C12H16OSiNa [M + Na]+; 227.0868 found 227.0867.
4-Ethynylbenzyl alcohol (6)
TBAF (1.0 M in THF) (6.28 mL, 6.28 mmol) was added to a solution of 5 (1.28 g, 6.27 mmol) in THF (31.4 mL) at room temperature under argon atmosphere. After being stirred for 1 h at room temperature, the mixture was concentrated in vacuo, and the resulting residue was purified by column chromatography (25% EtOAc in hexane) to afford 6 as a brown solid (0.51 g, 3.85 mmol, 61%). 1H NMR (400 MHz, CDCl3) δ 2.00 (s, 1H), 3.08 (s, 1H), 4.67 (s, 2H), 7.31 (d, 2H, J = 8.2), 7.48 (d, 2H, J = 8.3); 13C NMR (101 MHz, CDCl3) δ 64.9, 77.3, 83.6, 121.4, 126.8, 132.4, 141.7; HRMS (ESI-TOF) m/z calc. for C9H8ONa [M + Na]+; 155.0528 found 155.0527.
1-O-(4-Ethynylphenyl)methyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose (8)
1-O-Acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose (7) (1.40 g, 2.72 mmol) was dissolved in CH2Cl2 (10 mL). To the solution, TMSOTf (0.7 mL, 4.08 mmol) was slowly added at −35 °C. Then, a solution of 6 (0.30 g, 2.27 mmol) in CH2Cl2 (4 mL) was added slowly to the solution. After being stirred at −35 °C for 3.5 h, saturated NaHCO3 aqueous solution was added to the reaction mixture. The mixture was partitioned between CHCl3 and H2O. The organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude material was purified by column chromatography (20% EtOAc in hexane) to afford 8 as a yellow oil (1.10 g, 1.91 mmol, 70%). 1H NMR (600 MHz, CDCl3) δ 3.07 (s, 1H), 4.51–4.55 (m, 1H), 4.58 (d, 1H, J = 12.4), 4.73–4.77 (m, 2H), 4.80 (d, 1H, J = 12.4), 5.32 (s, 1H), 5.77 (d, 1H, J = 4.8), 5.94 (m, 1H), 7.24–7.26 (m, 2H), 7.29–7.33 (m, 4H), 7.40–7.43 (m, 4H), 7.49–7.51 (m, 2H), 7.56–7.58 (m, 1H), 7.89 (d, 2H, J = 7.6), 8.00–8.02 (m, 4H); 13C NMR (151 MHz, CDCl3) δ 64.6, 69.3, 72.3, 75.8, 79.4, 83.6, 104.8, 121.6, 127.8, 128.5, 128.5, 128.6, 129.0, 129.2, 129.6, 129.8, 129.8, 129.8, 132.2, 133.2, 133.4, 133.5, 137.6, 165.3, 165.4, 166.3; HRMS (ESI-TOF) m/z calc. for C35H28O8Na [M + Na]+; 599.1682 found 599.1681.
1-O-(4-Ethynylphenyl)methyl-β-D-ribofuranose (9)
NaOCH3 (6 drops) was added to a solution of 8 (1.10 g, 1.90 mmol) in MeOH (15.0 mL) at room temperature. After being stirred for 22 h at room temperature, the mixture was concentrated in vacuo, and the resulting residue was purified by column chromatography (10% MeOH in CHCl3) to afford 9 as a white solid (0.32 g, 1.21 mmol, 64%). 1H NMR (600 MHz, DMSO-d6) δ 2.54 (s, 1H), 3.41–3.43 (m, 1H), 3.63–3.58 (m, 1H), 3.82–3.84 (m, 2H), 3.94–3.98 (m, 1H), 4.19 (s, 1H), 4.47 (d, 1H, J = 12.4), 4.69 (t, 1H, J = 5.9), 4.74 (d, 1H, J = 12.4), 4.85–4.86 (m, 1H), 5.07–5.08 (d, 1H, J = 4.1), 7.37 (d, 2H, J = 7.6), 7.47 (d, 2H, J = 8.2); 13C NMR (151 MHz, DMSO-d6) δ 63.6, 68.0, 71.5, 75.0, 81.2, 83.9, 84.3, 106.8, 121.2, 128.3, 132.1, 139.7; HRMS (ESI-TOF) m/z calc. for C14H16O5Na [M + Na]+; 287.0986 found 287.0995.
1-O-(4-Ethynylphenyl)methyl-5-O-[bis(4-methoxyphenyl)phenylmethyl]-β-D-ribofuranose (10)
DMTr-Cl (0.54 g, 1.59 mmol) was added to a solution of 9 (0.32 g, 1.21 mmol) in pyridine (5.0 mL) at room temperature. After being stirred at room temperature for 2 h, the mixture was partitioned between EtOAc and saturated NaHCO3 aqueous solution. The organic layer was washed with H2O, brine, dried over Na2SO4, and concentrated in vacuo. The crude material was purified by column chromatography (50% EtOAc in hexane) to afford 10 as a yellow solid (0.50 g, 0.88 mmol, 72%). 1H NMR (600 MHz, CDCl3) δ 2.62 (br s, 1H), 2.88 (br s, 1H), 3.07 (s, 1H), 3.31 (d, 2H, J = 5.5), 3.76 (s, 6H), 4.12–4.14 (m, 2H), 4.34 (t, 1H, J = 5.5), 4.44 (d, 1H, J = 11.7), 4.71 (d, 1H, J = 12.4), 5.05 (s, 1H), 6.79 (dd, 4H, J = 4.1 and 8.3), 7.16 (d, 2H, J = 8.3), 7.21 (t, 1H, J = 7.2), 7.27 (t, 2H, J = 7.6), 7.35 (d, 4H, J = 8.9), 7.42 (d, 2H, J = 8.2), 7.47 (d, 2H, J = 7.6); 13C NMR (151 MHz, CDCl3) δ 55.3, 64.9, 69.0, 72.8, 75.5, 82.5, 83.6, 86.3, 106.6, 113.3, 121.4, 126.9, 127.8, 128.0, 128.3, 130.2, 132.3, 136.1, 138.5, 144.8, 158.6; HRMS (ESI-TOF) m/z calc. for C35H34O7Na [M + Na]+; 589.2229 found 589.2229.
2-O-(tert-Buthyldimethylsilyl)-1-O-(4-ethynylphenyl)methyl-5-O-[bis(4-methoxyphenyl)phenylmethyl]-β-D-ribofuranose (11)
TEA (0.40 mL, 2.87 mmol) and TBDMS-Cl (2.0 g, 1.31 mmol) were added to a solution of 10 (0.50 g, 0.876 mmol) in DMF (10 mL) at room temperature. After being stirred at room temperature for 24 h, the mixture was partitioned between EtOAc and H2O. The organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude material was purified by column chromatography (15–20% EtOAc in hexane) to afford 11 as a colorless oil (0.17 g, 0.254 mmol, 29%). 1H NMR (500 MHz, CDCl3) δ 0.11 (s, 6H), 0.91 (s, 9H), 2.47 (d, 1H, J = 8.0), 3.06 (s, 1H), 3.16–3.18 (m, 1H), 3.35–3.37 (m, 1H), 3.76 (s, 6H), 4.11–4.21 (m, 3H), 4.49 (d, 1H, J = 12.1), 4.79 (d, 1H, J = 12.1), 4.97 (d, 1H, J = 1.75), 6.79 (dd, 4H, J = 2.3 and 9.2), 7.17–7.21 (m, 3H), 7.27 (t, 2H, J = 7.4), 7.38 (d, 4H, J = 9.2), 7.44 (d, 2H, J = 8.6), 7.50 (d, 2H, J = 7.5); 13C NMR (126 MHz, CDCl3) δ −4.5, 18.2, 25.8, 55.2, 64.7, 69.1, 72.3, 76.6, 76.8, 83.8, 86.0, 106.7, 113.1, 121.4, 126.7, 127.6, 127.8, 128.3, 130.1, 132.2, 136.2, 136.2, 138.5, 145.0, 158.4; HRMS (ESI-TOF) m/z calc. for C41H48O7SiNa [M + Na]+; 703.3067 found 703.3080.
3-O-(tert-Butyldimethylsilyl)-1-O-(4-ethynylphenyl)methyl-5-O-[bis(4-methoxyphenyl)phenylmethyl]-β-D-ribofuranose (12)
TEA (0.40 mL, 2.87 mmol) and TBDMS-Cl (2.0 g, 1.31 mmol) were added to a solution of 10 (0.50 g, 0.876 mmol) in DMF (10 mL) at room temperature. After being stirred at room temperature for 24 h, the mixture was partitioned between EtOAc and H2O. The organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude material was purified by column chromatography (15–20% EtOAc in hexane) to afford 12 as a colorless oil (0.26 g, 0.38 mmol, 43%). 1H NMR (500 MHz, CDCl3) δ −0.13 (s, 3H), −0.004 (s, 3H), 0.81 (s, 9H), 2.76 (d, 1H, J = 2.3), 3.06 (s, 1H), 3.07–3.09 (m, 1H), 3.37–3.40 (m, 1H), 3.76 (s, 6H), 3.94–3.98 (m, 1H), 4.12–4.15 (m, 1H), 4.40 (t, 1H, J = 5.7), 4.53 (d, 1H, J = 12.0), 4.80 (d, 1H, J = 12.1), 5.11 (s, 1H), 6.79 (dd, 4H, J = 5.8 and 8.6), 7.18–7.20 (m, 3H), 7.27 (t, 2H, J = 7.4), 7.38 (d, 4H, J = 9.2), 7.42 (d, 2H, J = 8.1), 7.50 (d, 2H, J = 7.5); 13C NMR (126 MHz, CDCl3) δ −4.8, 17.9, 25.7, 55.2, 63.8, 69.2, 72.4, 75.5, 76.8, 83.0, 83.2, 85.9, 106.6, 113.1, 126.8, 127.8, 127.9, 128.3, 130.1, 132.2, 136.2, 136.2, 138.2, 145.5, 158.5; HRMS (ESI-TOF) m/z calc. for C41H48O7SiNa [M + Na]+; 703.3067 found 703.3071.
2-O-(tert-Butyldimethylsilyl)-3-O-[(2-cyanoethoxy)(diisopropylamino)phosphanyl-1-O-(4-ethynylphenyl)methyl]-5-O-[bis(4-methoxyphenyl)phenylmethyl]-β-D-ribofuranose (13)
Chloro(2-cyanoethoxy)(N,N-diisopropylamino)phosphine (0.51 mL, 2.28 mmol) and N,N-diisopropylethylamine (1.0 mL, 5.70 mmol) were added to a solution of 11 (0.78 g, 1.14 mmol) in THF (8 mL) under argon atmosphere and the mixture was stirred for 1.5 h at room temperature. The mixture was partitioned between saturated NaHCO3 solution and CHCl3. The organic layer was washed with brine, dried over Na2SO4 and concentrated in vacuo. The crude material was purified by column chromatography (25% EtOAc in hexane) to afford 13 as a colorless oil (0.87 g, 0.99 mmol, 87%). 1H NMR (600 MHz, CDCl3) δ 0.065 (s, 1H), 0.079 (d, 3H, J = 4.2), 0.10 (d, 2H, J = 3.0), 0.11 (s, 0.5H), 0.89 (d, 9H, J = 6.6), 0.91 (s, 2H), 0.96 (d, 2H, J = 6.6), 1.09 (d, 3H, J = 7.2), 1.12 (t, 6H, J = 7.8), 2.31 (t, 1H, J = 6.9), 2.57 (q, 0.5H, J = 7.0), 3.06 (d, 1H, J = 3.6), 3.10–3.08 (m, 0.5H), 3.39 (dd, 0.5H, J = 10.2 and 1.8), 3.53–3.47 (m, 2H), 3.74 (s, 1.5H), 3.75 (d, 5.5H, J = 2.4), 4.16 (d, 0.5H, J = 3.6), 4.21–4.19 (m, 0.5H), 4.33–4.24 (m, 1.5H), 4.49 (d, 0.5H, J = 3.6), 4.52 (d, 0.5H, J = 13.2), 4.82 (dd, 0.5H, J = 12.6, 4.2), 4.95 (s, 0.5H), 4.97 (d, 0.5H, J = 2.4), 6.78–6.74 (m, 4.5H), 7.25–7.16 (m, 6H), 7.39–7.35 (m, 4.5H), 7.42 (d, 2H, J = 7.2), 7.51–7.48 (m, 2H); 13C NMR (151 MHz, CDCl3) δ −4.53, −4.42, 2.01, 18.28, 20.12, 20.40, 24.67, 24.78, 25.92, 43.06, 43.13, 43.24, 43.32, 55.28, 55.31, 58.23, 58.34, 58.46, 64.08, 64.77, 68.92, 69.23, 72.39, 72.56, 72.62, 73.46, 75.76, 76.04, 76.74, 76.95, 77.37, 81.46, 81.57, 83.71, 83.91, 85.94, 86.02, 86.11, 106.77, 107.01, 113.10, 113.14, 113.19, 117.50, 117.66, 121.32, 121.45, 126.79, 127.65, 127.67, 127.74, 127.83, 127.86, 127.90, 128.40, 128.45, 128.52, 130.26, 132.22, 136.27, 136.39, 136.53, 138.87, 145.06, 145.13, 158.49; 31P NMR (202 MHz, CDCl3) δ 149.2, 149.9; HRMS (ESI-TOF) m/z calc. for C50H65N2O8PSiK [M + K]+; 919.3885 found 919.3879.
Synthesis of controlled pore glass support (15)
N,N-Dimethyl-4-aminopyridine (93 mg, 0.76 mmol) and succinic anhydride (0.15 g, 1.52 mmol) were added to a solution of compound 12 (0.26 g, 0.38 mmol) in pyridine (3.0 mL). After being stirred for 24 h at room temperature, the mixture was partitioned between EtOAc and H2O. The organic layer was washed with saturated NaHCO3 aqueous solution and brine, dried over Na2SO4, and concentrated in vacuo. The resulting residue was dissolved in DMF (4.1 mL). Aminopropyl controlled pore glass (0.66 g, CPG) and 1-(3-(dimethylamino)propyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl, 78 mg, 0.41 mmol) were added to the solution of 14, and the mixture was kept at room temperature for 3 days. After the resin was washed with pyridine, 15 mL of capping solution (0.1 M DMAP in pyridine/Ac2O 9:1) was added to the resin and the mixture was kept at room temperature for 1 day. The resin was washed with pyridine, EtOH, and CH3CN and dried under vacuum to give solid support 15. The amount loaded nucleoside 14 to the solid support was 44 μmol g−1 from calculation of released dimethoxytrityl cation by a solution of 70% HClO4:EtOH (3:2, v/v).
Solid-phase peptide synthesis
The peptides were synthesized by solid-phase synthesis with the 9-fluorenylmethyloxycarbonyl (Fmoc) chemistry in a 5.0 μmol scale. A 4-methylbenzhydrylamine resin, which generates an amide function at the C terminal, was used as the solid support for the peptide synthesis. Amino acids protected with the Fmoc group were reacted by using 1-[bis(dimethylamino)methylene]-1H-benzotriazolium 3-oxide hexafluorophosphate (HBTU) as a coupling reagent in the presence of 1-hydroxybenzotriazole (HOBt) and N,N′-diisopropylethylamine (DIPEA). The Fmoc group was deprotected by 25% piperidine in DMF and each coupling reaction was performed for 30 min. Cleavage of the peptide from the resin and deprotection of the side chain were performed by treating with a TFA solution containing m-cresol (95/5, v/v) for 1 h. Diethyl ether (Et2O) was added to the solution. The resulting precipitate was washed several times with Et2O to give the peptide. The peptides were successfully identified by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF/MS). Peptide 1 m/z = 1511.97 (calcd for C60H112N37O10 [M + H]+, 1511.86); peptide 2 m/z = 1910.03 (calcd for C74H133N44O17 [M + H]+, 1910.24); peptide 3 m/z = 2007.92 (calcd for C81H147N44O17 [M + H]+, 2008.42).
Solid-phase oligonucleotide synthesis
The synthesis was carried out with a DNA/RNA synthesizer by the phosphoramidite method. After the synthesis, the oligomers were cleaved from CPG beads and deprotected by treatment with concentrated NH3 solution/40% methylamine (1:1, v/v) for 10 min at 65 °C. 2′-O-TBDMS groups were removed by Et3N·3HF (125 μL) in DMSO (100 μL) at 65 °C for 1.5 h. The reaction was quenched with 0.1 M triethylammonium acetate (TEAA) buffer (pH 7.0), and the mixture was desalted using a Sep-Pak C18 cartridge. The oligonucleotides were purified by 20% PAGE containing 7 M urea to give highly purified oligonucleotides. The oligonucleotides were successfully identified by MALDI-TOF/MS. RNA 1 m/z = 6829.07 (calcd for C210H264N65O153P20 [M − H]−, 6828.14); RNA 2 m/z = 6827.91 (calcd for C210H264N65O153P20 [M − H]−, 6828.14).
Peptide–RNA conjugates
The oligonucleotide (1.0 nmol) dissolved in sterile H2O was mixed with tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) (0.6 μL, 30 nmol, 50 mM), CuSO4 (0.6 μL, 15 nmol, 25 mM), and Na ascorbate (0.4 μL, 20.0 nmol, 50 mM). After adding DMSO to the solution, the mixture was incubated at 37 °C for 30 min. Then, peptide 1, 2 or 3 (1.0 μL, 5.0 nmol, 5 mM) dissolved in H2O was added, and the reaction solution was incubated at 37 °C for 2 h. The reaction solution was diluted with 1 M TEAA buffer, and the peptide–RNA conjugate was purified by the reversed-phase HPLC (1 M TEAA–MeCN). The peptide–RNA conjugates were successfully identified by MALDI-TOF/MS. RNA 5 m/z = 8737.66 (calcd for C284H396N109O170P20 [M − H]−, 8737.38); RNA 6 m/z = 8837.57 (calcd for C291H410N109O170P20 [M − H]−, 8835.56); RNA 7 m/z = 8739.32 (calcd for C284H396N109O170P20 [M − H]−, 8737.38); RNA 8 m/z = 8835.99 (calcd for C291H410N109O170P20 [M − H]−, 8835.56).
Thermal denaturation study
The solution containing 3.0 μM duplex in a buffer of 10 mM sodium phosphate (pH 7.0) containing 100 mM NaCl was heated at 100 °C and then cooled gradually to room temperature and used for the thermal denaturation study. Thermally induced transitions were monitored at 260 nm with a UV/vis spectrometer fitted with temperature controller in quartz cuvettes with a path length of 1.0 cm. The sample temperature was increased by 0.5 °C min−1.
CD spectroscopy
All CD spectra were recorded at 25 °C. The following instrument settings were used: resolution, 0.1 nm; response, 1.0 s; speed, 50 nm min−1; accumulation, 10.
Nuclease resistance of siRNA
Fluorescein labeled siRNAs (600 pmol) were dissolved in 20 μL of buffer of 10 mM sodium phosphate (pH 7.0) containing 100 mM NaCl, and the solutions were heated at 100 °C and then cooled gradually to room temperature and used for the serum stability test. 100 μL of Opti-MEM and 12 μL of bovine serum were added, and the mixture was incubated at 37 °C for the required time. Aliquots of 6.7 μL were diluted with a stop solution (65 mM EDTA, 12% glycerol, 8.0 μL). Samples were subjected to electrophoresis in 15% polyacrylamide-TBE under nondenaturing conditions and quantified by a Luminescent Image Analyzer LAS-4000 (Fujifilm).
Dual-luciferase assay
HeLa cells were transfected with the psiCHECK-2 (Promega) reporter and the pcDNA3.1 containing a hygromycin resistance gene (Thermo Fisher Scientific). Cells were cultured in the presence of 0.5 mg mL−1 hygromycin for 1 week. Stable HeLa-psiCHECK-2 cells expressing both Renilla and firefly luciferases were grown in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% bovine serum (BS) and 0.25 mg mL−1 hygromycin at 37 °C. HeLa-psiCHECK-2 cells (8.0 × 104/mL) were cultured on a 96-well plate (100 μL per well) for 24 h and transfected with siRNA targeting the Renilla luciferase gene using lipofectamine RNAiMax in Opti-MEM reduced serum medium. Transfection without siRNA was used as a control. After 1 h, DMEM (50 μL) containing 10% BS was added to each well and cells were further incubated for another 24 h. The activities of Renilla and firefly luciferases in the cells were determined with the Dual-Luciferase Reporter Assay System (Promega) according to a manufacture's protocol. The activity of Renilla luciferase was normalized by the firefly luciferase activity. The results were confirmed by at least three independent transfection experiments with two cultures each and are expressed as the average from four experiments as mean ± SD.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Japan Agency for Medical Research and Development (AMED) through its Funding Program for Basic Science and Platform Technology Program for Innovative Biological Medicine, development of siRNA conjugates with tissue-specific delivery functions (18am0301022h0004).
References
- A. Fire, S. Q. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans, Nature, 1998, 391, 806–811 CrossRef CAS.
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Duplexes of 21±nucleotide RNAs mediate RNA interference in cultured mammalian cells, Nature, 2001, 411, 494–498 CrossRef CAS.
- S. M. Elbashir, W. Lendeckel and T. Tuschl, RNA interference is mediated by 21- and 22-nucleotide RNAs, Genes Dev., 2001, 15, 188–200 CrossRef CAS.
- A. Khvorova and J. K. Watts, The chemical evolution of oligonucleotide therapies of clinical utility, Nat. Biotechnol., 2017, 35, 238–248 CrossRef CAS.
- S. Benizri, A. Gissot, N. Sasaki, B. Vialet, M. W. Grinstaff and P. Barthélémy, Bioconjugated oligonucleotides: recent developments and therapeutic applications, Bioconjugate Chem., 2019, 30, 366–383 CrossRef CAS.
- S. F. Dowdy, Overcoming cellular barriers for RNA therapeutics, Nat. Biotechnol., 2017, 35, 222–229 CrossRef CAS.
- M. Manoharan, RNA interference and chemically modified small interfering RNAs, Curr. Opin. Chem. Biol., 2004, 8, 570–579 CrossRef CAS.
- Y. L. Chiu and T. M. Rana, siRNA function in RNAi: a chemical modification analysis, RNA, 2003, 9, 1034–1048 CrossRef CAS.
- G. F. Deleavey and M. J. Damha, Designing chemically modified oligonucleotides for targeted gene silencing, Chem. Biol., 2012, 19, 937–954 CrossRef CAS.
- K. T. Gagnon, J. K. Watts, H. M. Pendergraff, C. Montaillier, D. Thai, P. Potier and D. R. Corey, Antisense and antigene inhibition of gene expression by cell-permeable oligonucleotide–oligospermine conjugates, J. Am. Chem. Soc., 2011, 133, 8404–8407 CrossRef CAS.
- C. Paris, V. Moreau, G. Deglane, L. Karim, B. Couturier, M.-E. Bonnet, V. Kedinger, M. Messmer, A.-L. Bolcato-Bellemin, J.-P. Behr, P. Erbacher and N. Lenne-Samuel, Conjugating phosphospermines to siRNAs for improved stability in serum, intracellular delivery and RNAi-mediated gene silencing, Mol. Pharmaceutics, 2012, 9, 3464–3475 CrossRef CAS.
- M. Nothisen, J. Bagilet, J.-P. Behr, J.-S. Remy and M. Kotera, Structure tuning of cationic oligospermine–siRNA conjugates for carrier-free gene silencing, Mol. Pharmaceutics, 2016, 13(8), 2718–2728 CrossRef CAS.
- S. Milton, D. Honcharenko, C. S. J. Rocha, P. M. D. Moreno, C. I. Edvard Smith and R. Strömberg, Nuclease resistant oligonucleotides with cell penetrating properties, Chem. Commun., 2015, 51, 4044–4047 RSC.
- P. Zhou, M. Wang, L. Du, G. W. Fisher, A. Waggoner and D. H. Ly, Novel binding and efficient cellular uptake of guanidine-based peptide nucleic acids (GPNA), J. Am. Chem. Soc., 2003, 125, 6878–6879 CrossRef CAS.
- T. Ohmichi, M. Kuwahara, N. Sasaki, M. Hasegawa, T. Nishikata, H. Sawai and N. Sugimoto, Nucleic acid with guanidinium modification exhibits efficient cellular uptake, Angew. Chem., Int. Ed., 2005, 44, 6682–6685 CrossRef CAS.
- G. Deglane, S. Abes, T. Michel, P. Prévot, E. Vives, F. Debart, I. Barvik, B. Lebleu and J. J. Vasseur, Impact of the guanidinium group on hybridization and cellular uptake of cationic oligonucleotides, ChemBioChem, 2006, 7, 684–692 CrossRef CAS.
- A. R. Shrestha, Y. Kotobuki, Y. Hari and S. Obika, Guanidine bridged nucleic acid (GuNA): an effect of a cationic bridged
nucleic acid on DNA binding affinity, Chem. Commun., 2014, 50, 575–577 RSC.
- K. Skakuj, K. E. Bujold and C. E. Mirkin, Mercury-free automated synthesis of guanidinium backbone oligonucleotides, J. Am. Chem. Soc., 2019, 141, 20171–20176 CrossRef CAS.
- S. Futaki, T. Suzuki, W. Ohashi, T. Yagami, S. Tanaka, K. Ueda and Y. Sugiura, Arginine-rich peptides an abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery, J. Biol. Chem., 2001, 276, 5836–5840 CrossRef CAS.
- I. Nakase, M. Niwa, T. Takeuchi, K. Sonomura, N. Kawabata, Y. Koike, M. Takehashi, S. Tanaka, K. Ueda, J. C. Simpson, A. T. Jones, Y. Sugiura and S. Futaki, Cellular uptake of arginine-rich peptides: roles for macropinocytosis and actin rearrangement, Mol. Ther., 2004, 10, 1011–1022 CrossRef CAS.
- Y. Maeda, R. Iwata and T. Wada, Synthesis and properties of cationic oligopeptides with different side chain lengths that bind to RNA duplexes, Bioorg. Med. Chem., 2013, 21, 1717–1723 CrossRef CAS.
- A. Uematsu, R. Kajino, Y. Maeda and Y. Ueno, Synthesis and characterization of 4′-C-guanidinomethyl-2′-O-methyl-modified RNA oligomers, Nucleosides Nucleotides Nucleic Acids, 2020, 39, 280–291 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: UV melting profiles. 1H, 13C and 19F NMR spectra of compounds 3, 5, 6, 8–12 and 31P NMR spectrum of compound 13. See DOI: 10.1039/d0ra05919f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc
*abc