DOI:
10.1039/D0RA05856D
(Paper)
RSC Adv., 2020,
10, 31909-31935
Retracted Article: The synthesis and biological activity of marine alkaloid derivatives and analogues
Received
5th July 2020
, Accepted 29th July 2020
First published on 28th August 2020
Abstract
The ocean is the origin of life, with a unique ecological environment, which has given birth to a wealth of marine organisms. The ocean is an important source of biological resources and tens of thousands of monomeric compounds have been separated from marine organisms using modern separation technology. Most of these monomeric compounds have some kind of biological activity that has attracted extensive attention from researchers. Marine alkaloids are a kind of compound that can be separated from marine organisms. They have complex and special chemical structures, but at the same time, they can show diversity in biological activities. The biological activities of marine alkaloids mainly manifest in the form of anti-tumor, anti-fungus, anti-viral, anti-malaria, and anti-osteoporosis properties. Many marine alkaloids have good medicinal prospects and can possibly be used as anti-tumor, anti-viral, and anti-fungal clinical drugs or as lead compounds. The limited amounts of marine alkaloids that can be obtained by separation, coupled with the high cytotoxicity and low selectivity of these lead compounds, has restricted the clinical research and industrial development of marine alkaloids. Marine alkaloid derivatives and analogues have been obtained via rational drug design and chemical synthesis, to make up for the shortcomings of marine alkaloids; this has become an urgent subject for research and development. This work systematically reviews the recent developments relating to marine alkaloid derivatives and analogues in the field of medical chemistry over the last 10 years (2010–2019). We divide marine alkaloid derivatives and analogues into five types from the point-of-view of biological activity and elaborated on these activities. We also briefly discuss the optimization process, chemical synthesis, biological activity evaluation, and structure–activity relationship (SAR) of each of these compounds. The abundant SAR data provides reasonable approaches for the design and development of new biologically active marine alkaloid derivatives and analogues.
1. Introduction
Alkaloids are natural compounds (natural products) that contain nitrogen and have significant biological activities.1 They generally have chemical properties similar to bases. Some nitrogenous compounds derived from plants are not alkaline, but are still classified as alkaloids because of their obvious biological activity.2–4 It can be seen from the chemical structure of alkaloids that most of them have a complex nitrogen heterocyclic structure, with only a few organic amine alkaloid compounds not exhibiting such a structure.5–9 The definition of alkaloids by the International Union of Pure and Applied Chemistry (IUPAC) is not strict and exact. For example, some vitamins, amino acids, peptides and other nitrogenous compounds derived from natural products do not belong to the category of alkaloids.10 Alkaloids are widely distributed in nature, especially in plants, and also in animals, microorganisms and marine organisms. There are many known types of alkaloids, with around 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 of them having structural formulas that have not yet been fully determined.11,12 As there are many types of alkaloids, each of which has a different structural formula, their properties are different from each other. However, the levels of alkaloids contained in living organisms are small, and this is similar in human beings. It has long been recognized that some plants or extracts containing alkaloids can cure diseases or be used as poisons.13 Since 1806, when the German pharmacist Serturner isolated morphine from poppies, there have been more than 6000 alkaloids isolated from plants and animals. Through the joint efforts of medical chemists, pharmacologists and other scholars, people have found that a variety of alkaloids have anti-tumor, anti-bacterial, anti-viral and other biological activity.14–16 So far, nearly 100 alkaloid compounds have been previously used or are currently being used in clinical trials. In the treatment of many diseases, many alkaloids make good medicines, but some alkaloids can be used to make pesticides for agriculture.17–19 Because of their wide range of biological activity, alkaloids are of great interest.
000 of them having structural formulas that have not yet been fully determined.11,12 As there are many types of alkaloids, each of which has a different structural formula, their properties are different from each other. However, the levels of alkaloids contained in living organisms are small, and this is similar in human beings. It has long been recognized that some plants or extracts containing alkaloids can cure diseases or be used as poisons.13 Since 1806, when the German pharmacist Serturner isolated morphine from poppies, there have been more than 6000 alkaloids isolated from plants and animals. Through the joint efforts of medical chemists, pharmacologists and other scholars, people have found that a variety of alkaloids have anti-tumor, anti-bacterial, anti-viral and other biological activity.14–16 So far, nearly 100 alkaloid compounds have been previously used or are currently being used in clinical trials. In the treatment of many diseases, many alkaloids make good medicines, but some alkaloids can be used to make pesticides for agriculture.17–19 Because of their wide range of biological activity, alkaloids are of great interest.
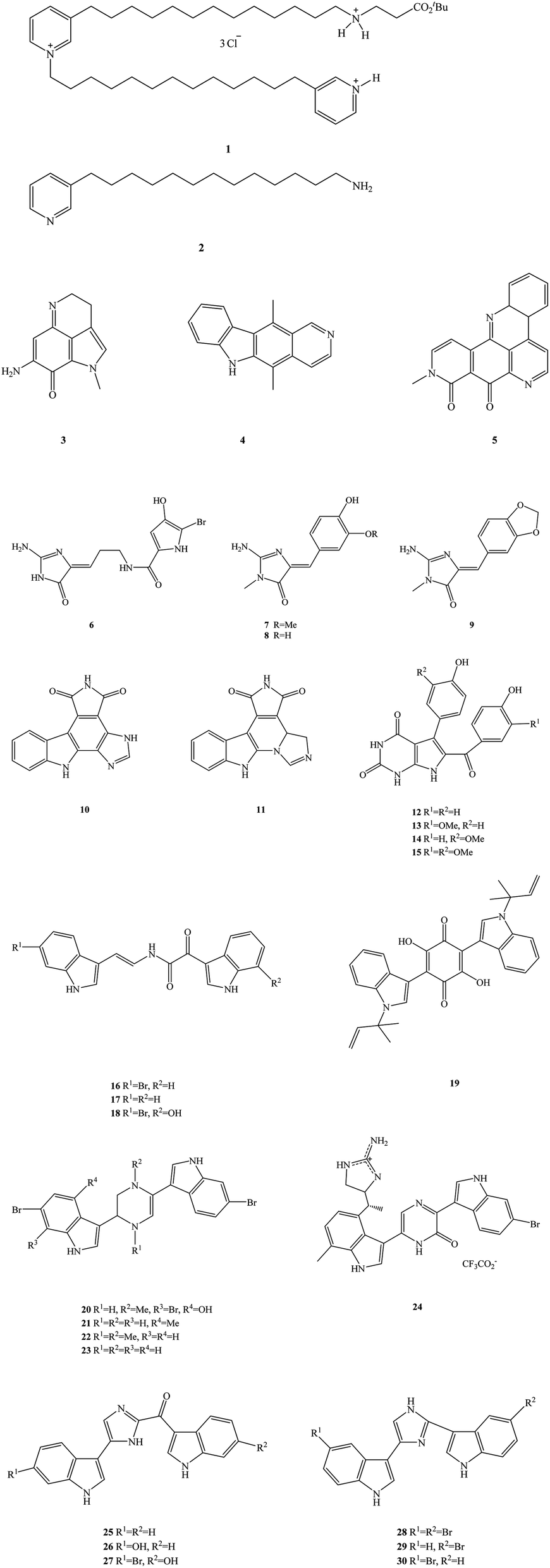
The ocean is the birthplace of life and is rich in biological materials, making it a huge natural products screening resource.20 So far, researchers have found more than 300000 species of marine organisms in the ocean and more than one million new species are thought to be undiscovered.21–24 Marine organisms are secondary metabolites, which have formed and accumulated a large number of special chemical structures, significant biological activity in the process of long-term evolution, metabolism in special environments such as high salt, high pressure, low temperature, hypoxia, and lack of light.25 As such, they show significant anti-viral, anti-inflammatory and anti-tumor properties, among others. Marine alkaloids are a secondary metabolite of marine organisms, mainly derived from marine organisms such as sponges, algae, coelenterates and tunica.26–29 Marine alkaloids are alkaline natural products that have important biological activity, the chemical structures of which include amine nitrogen functional groups and a complex carbon skeleton ring.30 The main biological activities of marine alkaloids are their anti-tumor, anti-viral, anti-malaria, anti-fungal and anti-osteoporosis properties. Many marine alkaloids may be used as anti-tumor, anti-viral and anti-fungal clinical drugs or as the lead compounds for structural modification, as they have good medicinal prospects.31 In recent years, marine drugs research has been paid more and more attention by scholars, especially in the field of marine alkaloid drugs. Many types of marine alkaloids have been found and extracted from marine organisms, but most often the extraction quantity is small and the efficiency is low.32–35 Representative compounds of these marine alkaloids are shown in Fig. 1, including 3-alkyl-pyridinum and pyridine derivatives (e.g. viscosaline, compound 1, and theonelladin C, compound 2, respectively), N-fused heterocycles such as pyrroloquinoline (makaluvamine A, compound 3), carbazoles (ellipticine, compound 4), pyridoacridines (neoamphimedine, compound 5), N-phthalimides (e.g. granulatimide, compound 10, and isogranulatimide, compound 11), pyrrolopyrimidine (rigidin A–D, compounds 12–15), bis-indole derivatives (e.g. coscinamide A–C, compounds 16–18, asterriquinone, compound 19, dragmacidin, compound 20, dragmacidin A–C, compounds 21–23, dragmacidin D, compound 24, topsentin A, B1 and B2, compounds 25–27, nortopsentin A–C, compounds 28–30), and 2-aminoimidazolone derivatives (e.g. dispacamide, compound 6, polyandrocarpamine A and B, compounds 7–8, leucettamine B, compound 9). In addition, these marine alkaloids have the disadvantages of high cytotoxicity, low selectivity index and low activity.36 These problems thus limit the clinical research and industrial development of marine alkaloid drugs.37–39 Therefore, it has become an urgent task in research and development to find marine alkaloid analogues and derivatives that are economical and environmentally friendly. A large number of target compounds can be obtained for activity screening by rational drugs design, structural modification of lead compounds and chemical synthesis.40 In this way, the shortage of natural marine alkaloids may be solved, providing a good platform for the development of marine alkaloid derivatives and analogues.41 This work systematically reviews the recent development of marine alkaloid derivatives and analogues in the field of medical chemistry over the last 10 years (2010–2019). We divide marine alkaloid derivatives and their analogues into five types from the point-of-view of their biological activity, including anti-tumor, anti-malarial, anti-bacterial, anti-viral (HCV), and other types of properties, elaborating on these activities. We also briefly discuss the optimization process, chemical synthesis, biological activity evaluation and structure–activity relationship (SAR) of each type of compound. The SAR is discussed, which provides a reasonable insight into the design and development of novel marine alkaloid derivatives and their analogues.
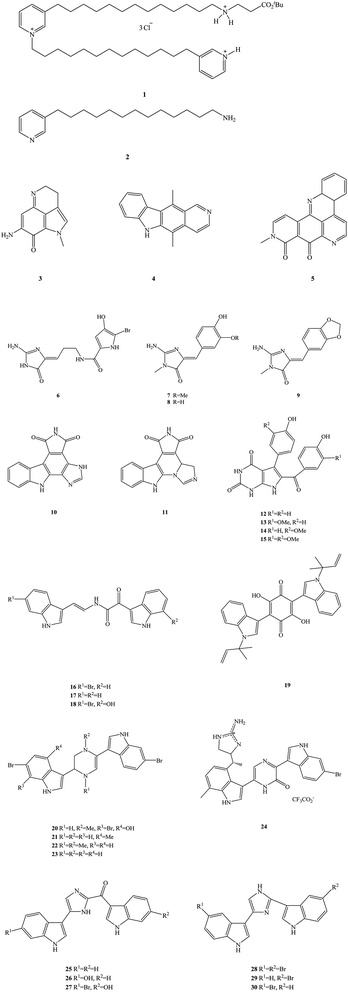 |
| | Fig. 1 The chemical structures of marine alkaloids. | |
2. Synthesis and biological activities
2.1 Anti-tumor activity
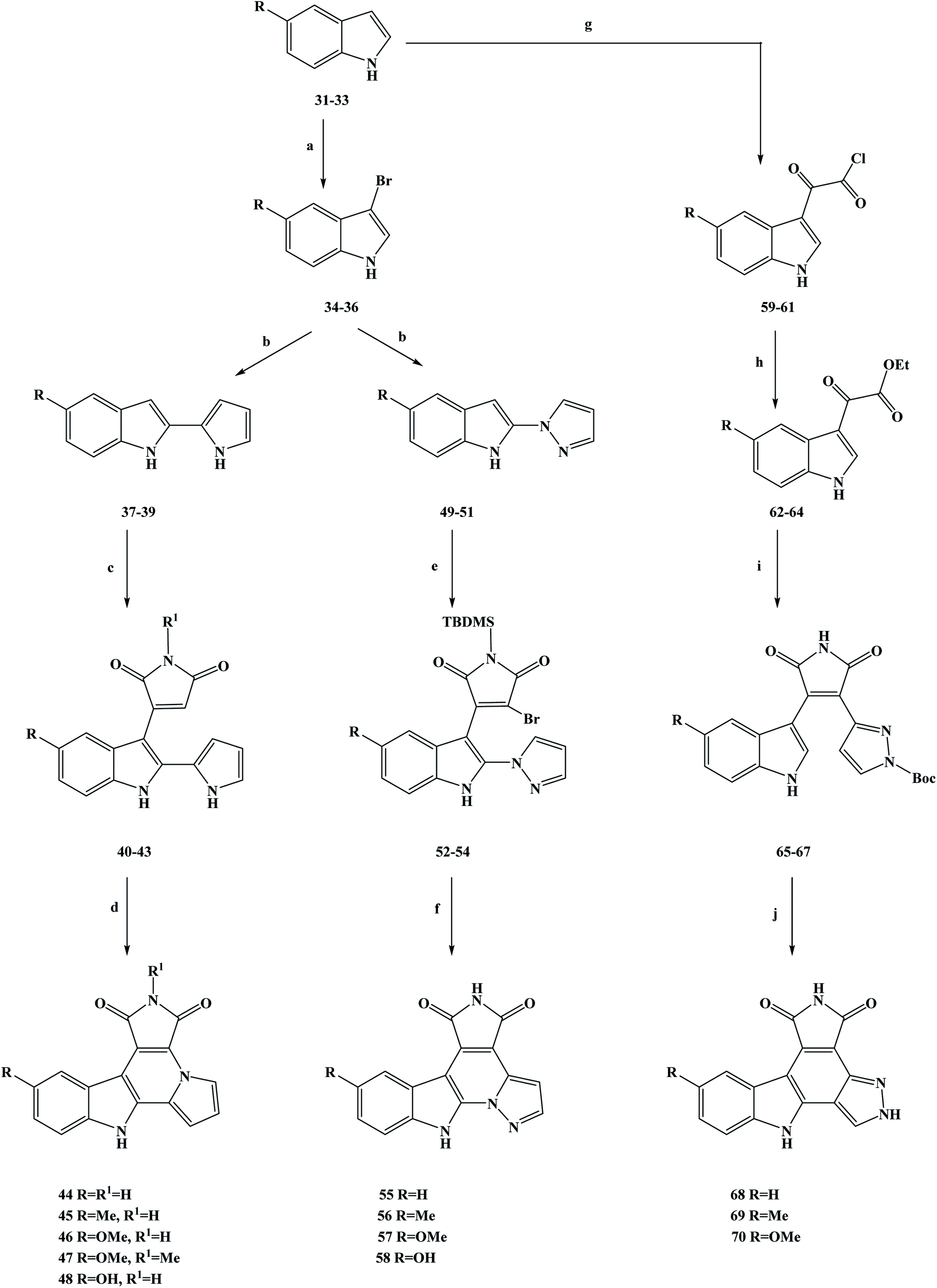
In medicine, cancer refers to a malignant tumor that originates from epithelial tissue. It is the most common kind of disease related to malignant tumors.42 Cancer is characterized by abnormal cell differentiation and proliferation, uncontrolled growth, invasion and metastasis.43–45 Intercellular division is the preparation period for mitosis or meiosis in eukaryotic cells, during which the cell completes the replication of DNA molecules and the synthesis of related proteins. In cell division, the interphase is much longer than the interphase.46 The interphase is usually divided into G1, S and G2 phases. The mitotic interphase is an important preparatory process for the whole process of mitosis. Studies have shown that if the G1 and G2 checkpoints are activated during the intermitotic period, the normal replication of DNA can be disturbed, thereby blocking subsequent cell division.47 In this case, it can provide valuable time to repair normal DNA. A study has shown that in more than 50% of cancer cells, the G1 checkpoint was damaged because of mutations in the p53 anti-oncogene. Relevant research has suggested that if damage to DNA and the G2/M checkpoint inhibitor occurs simultaneously, this may selectively kill cancer cells via premature mitosis. This does not affect normal cell mitosis, with this combination being used in the treatment of cancer as a preliminary and promising method of chemotherapy.48 With this biological background in mind, the marine alkaloids granulatimide (10) and isogranulatimide (11) were isolated and extracted from marine organisms showing G2/M checkpoints inhibiting in the cell cycle.49 The acquisition of these two marine alkaloids has attracted the attention of researchers, and they are a class of inhibitors with great research value. From the perspective of their mechanism of action, these marine alkaloids have been identified as inhibitors that can selectively inhibit checkpoint kinase 1 (Chk1), a key enzyme in the G2/M checkpoint.50,51 Relevant studies have shown that the maleimide groups of these compounds play an important role in affecting their biological activity, and they can establish two hydrogen bonds with Glu85 and Cys87 in the ATP binding pocket of the enzyme.52 On this basis, Deslandes et al.53 designed three series of granulatimide and isogranulatimide analogues for in vitro evaluation of anti-tumor activity, kinase inhibition and G2/M checkpoint abrogation. It can be seen from the chemical structure of the designed target compounds that the newly designed compounds retain the skeleton structure of the lead compounds, and that the main modification was to introduce different substituents into the benzene ring to obtain the active compounds. In the process of synthesizing the target compounds granulatimide and isogranulatimide analogues, they used the process shown in Scheme 1. In the first series of the target compounds 45–48, the substituted indole compounds 31–33 were used as starting materials and brominated with bromine in N,N-dimethylformamide (DMF) to obtain the 3-bromide derivative compounds 34–36. Compounds 34–36 were coupled with pyrrole in methylene dichloride to obtain the intermediate compounds 37–39, which then underwent a condensation reaction with maleimide or methylmaleimide. In this step, the toluene was used as the reaction solvent and SnCl2 as the reaction catalyst, and compounds 40–43 were obtained by Michael addition. Compounds 40–43 were cyclized and prepared by microwave irradiation with Pd as a catalyst to obtain the target compounds 44–47. Meanwhile, the target compound 46 was demethylated in methylene dichloride with tribromo-based boron to obtain the target compound 48. In the second series of target compounds, compounds 34–36 were used as starting materials for the reaction, and the intermediate compounds 49–51 were obtained via a coupling reaction with pyrazole in methylene dichloride. Compounds 49–51 underwent a condensation reaction with N-t-butyldimethylsilyl (TBDMS)-dibromo-maleimide in tetrahydrofuran (THF) to obtain the low yield intermediate compounds 52–54. The intermediate formed in this step does not form a ring, and the TBDMS protective group does not detach during the reaction. Compounds 52–54 were cycloformed in acetonitrile under mild conditions to obtain the target compounds 55–57. Meanwhile, the target compound 57 was demethylated in methylene dichloride with tribromo-based boron to obtain the target compound 58. In the third series of target compounds, compounds 31–33 were first converted into compounds 62–64 in two steps by referring to related literature. Then, compounds 62–64 reacted with N-Boc-pyrazole-3-acetamide in THF to obtain the intermediate compounds 65–67. Finally, compounds 65–67 in acetonitrile, under mild conditions formed a ring reaction, and then the protection with the Boc group (cracking reaction), to obtain the target compounds 68–70. This synthesis route used cheap reagents, the procedures were relatively simple, the functional group protection was easy to achieve, and in the subsequent process deprotection was easy. In addition, microwave irradiation was used in the synthesis method, which reduced the reaction time and increased the yield. However, this synthesis route also has some shortcomings, the yield of some compounds was relatively low (3%), which brings great difficulties for its industrialization in the future. Further optimization of the process conditions is needed to improve the reaction yield and reduce the synthesis cost. In terms of their biological activity, the growth inhibition activity of the synthetic compounds was evaluated in vitro. The test results showed that in cancer cells, these compounds showed a degree of sensitivity and resistance to pro-apoptotic stimuli. Of the three series of target compounds, the second series had the highest average growth inhibition activity on tumor cells, with properties such as those of the target compounds 55 and 58, where the average IC50 growth inhibition concentrations were 11 and 16 nM, respectively. In addition, compounds 40–43 and 52–54 were found to have good anti-tumor activities during the activity screening of the intermediates, with an average IC50 growth inhibition concentration of around 20 nM, among which compound 43 had the best activity (IC50 = 13 nM). Studies and analyses on drug structure–activity relationships (SAR) have shown that isoprenodiamine has weak in vitro anti-tumor activity, while intermediates with open structures, such as the first and second series of target compounds, have good inhibitory activity. The substituents on the benzene ring also showed a certain influence on the activity, among which methoxy substituents had the best anti-tumor activity. At the same time, the anti-tumor activity of the final target compounds was decreased after the open structures of some intermediates were closed. On the basis of in vitro screening, the ability of the compounds to eliminate the activated G2/M checkpoint in HCT116p53 cells was further investigated (Table 1). The test data showed that the target compound 55 had good activity (23.9), while the activity of the other compounds was low. To gain insight into the kinase selectivity of the target compounds 44–47, 55–58 and 68–69, serine/threonine kinases, namely CDK5/p25, CK1, CLK1, DYRK1A and GSK-3 were selected as targets for drug binding to proteins. The results of the selectivity tests showed that these target compounds had weak inhibitory activity and poor selectivity against kinases. Such results have no significant correlation with the anti-tumor activity in vitro, and the ability of the target compounds to inhibit other kinase proteins may be the cause of cytotoxicity. In addition, compounds 55, 40 and 43 were quantitatively examined by electron microscopy. The results showed that compounds 55, 40 and 43 induced cellular inhibition, which reduced cell proliferation by significantly expanding the size of cancer cells, thus reflecting the tissue changes in the actin cytoskeleton. In general, according to the existing data analysis, the target compound 55 and the intermediate compounds 40 and 43 have good anti-tumor activities. These compounds are worthy of further structural modification and other pharmacological studies, to fully understand these drugs and become ideal anti-cancer drugs as soon as possible.
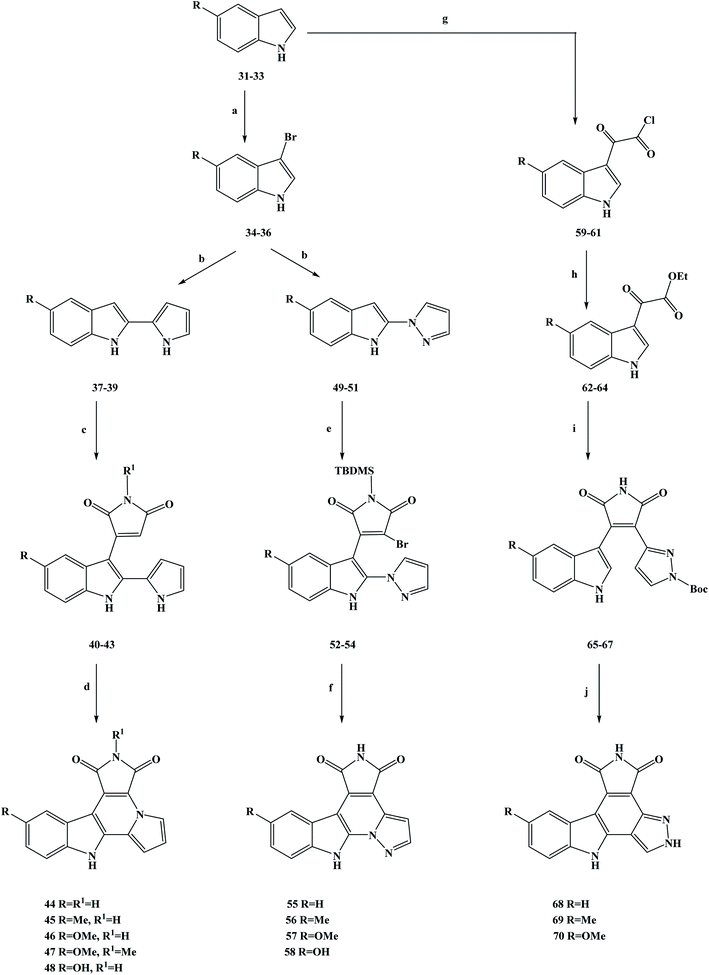 |
| | Scheme 1 Synthesis of the marine alkaloid granulatimide and isogranulatimide analogue compounds 44–48, 55–58, and 68–70. Reagents and conditions: (a) Br2, DMF, 2 h; (b) TFA, CH2Cl2, r.t., 4 h; (c) SnCl2, toluene, reflux, 24 h; (d) Pd, microwave 300 W, 190 °C, 15 min; (e) LiHMDS, THF, −15 °C then r.t., 12 h; (f) hν, CH3CN, r.t., 5 h; (g) (COCl)2, Et2O, 25 °C, 3 h; (h) EtOH, Et3N, 78 °C, 2 h; (i) i: t-BuOK, THF, r.t., 12 h; ii: HCl; (j) hν, CH3CN, r.t., 5 h. | |
Table 1 Evaluation of the G2/M checkpoint abrogation
| Compounds |
Mitotic index |
| 44 |
9.6 |
| 55 |
23.9 |
| 56 |
13.9 |
| 58 |
3.9 |
| 69 |
0 |
| 38 |
5.0 |
| 39 |
8.3 |
| 53 |
5.5 |
| Isogranulatimide |
17.2 |
| Granulatimide |
19.4 |
Natural products with biological activity can be isolated from marine organisms, including effective anti-tumor drugs found in them.54–56 These anti-tumor drugs that are obtained from marine organisms are structurally diverse and complex. Most of these compounds with anti-tumor activity are alkaloids, which are commonly referred to as marine alkaloids.56–59 Among these marine alkaloids, a class of marine alkaloids with a bis-indole structure is well known.60 This kind of bis-indole marine alkaloid is usually isolated from the metabolites of deep-sea sponges, and has been found to have good anti-inflammatory, anti-tumor, anti-viral and anti-bacterial biological activities in screening processes.61 The chemical structure of this type of bis-indole marine alkaloids is characterized by the fact that they are bound by two indoles through their 3-positions to intermediate ligands (including ring and chain structures). Related studies found that nortopsentins A–C (28–30) bis-indole marine alkaloids with imidazole as the five-membered ring as the connector showed IC50 values of 4.5–20.7 μM to P388 cells in vitro during anti-tumor activity testing, while the IC50 of P388 cells was 0.8–2.1 μM for N-methylated derivatives.62–64 The results of in vitro cytotoxicity studies indicated that the methylated derivatives significantly increased the activity of P388.65 In order to obtain a large number of compounds, several complete synthesis methods of nortopsentins and analogues have been reported.66–68 Because of the abundant biological activity of marine alkaloids, they are often regarded as new drugs or lead compounds that have biological activity. Therefore, on this basis, nortopsentin was structurally modified, and dragmacidin (20) analogues were synthesized using six-membered cyclopyridine, pyrimidine, pyrazine and pyrazinone as intermediate ligands. These analogues have been found to have a broad spectrum of anti-tumor activity in human tumor cells. However, nortopsentin analogues have been synthesized using a five-membered-heterocyclic imidazole ring as a ligand.69 In activity testing, it was found that these compounds had significant anti-proliferative activity, and the IC50 value was at the sub-micromolar level. In the process of modifying the structure of the lead compound, one or two indoles were modified in addition to the indoles.70,71 In the activity testing results, it was found that the 3-indolyl-5-phenylpyridine derivative exhibited anti-proliferative activity in the range of 5–15 μM and effectively inhibited CDK1 at the 0.3–0.7 μM. On the basis of previous work, Carbone et al.72 took nortopsentine as the lead compound, modified its structure with pyrrole as the intermediate connector, and modified the substituents on the two indoles to design nortopsentine analogues with a 2,5-bis(3′-indolyl)pyrrole structure. In the process of the synthesis of the target compounds 81–85, they selected the specific synthesis operation shown in Scheme 2. N-methylindoles (compounds 71–75) were used as starting materials, and the compounds were synthesized via a two-step reaction. In the first step reaction, the Vilsmeier–Haack reaction was used to treat compounds 71–75 with phosphorus oxychloride and tetramethylsuccinamide, and the symmetrical intermediate compounds 76–80 were obtained. In the second step, compounds 76–80 were refluxed with ammonium acetate and acetic anhydride in acetic acid, and the corresponding 2,5-bis(3′-indolyl)pyrroles as the target compounds 81–85 were obtained after treatment. The synthesis route required only two steps to obtain the target compounds, is simple to carry out with few reaction steps, the total yields were medium to good, and the reagents used were cheap and easy to obtain, laying a solid foundation for its future industrialization. However, these synthesis methods are all classical chemical reactions, there was no innovation involved. In terms of subsequent process optimization, new synthesis methods can be considered. In the process of screening the biological activity of the synthesized target compounds 81–85, human tumor cell lines were used for monolayer cell survival and proliferation tests were carried out to detect the anti-tumor activity of the target compounds in vitro. Using panels of human tumor cell lines of different source/tissue types, the anti-tumor activity, the selectivity of the tested compounds and evaluation of the eligible target compounds were analyzed for entry into preclinical studies. The in vitro anti-tumor activity of the target compounds 81–85 against 12 human tumor cell lines was screened using a monolayer cell survival and proliferation assay (Table 2). The results of the screening experiments showed that all of the target compounds showed cytotoxicity activity at the maximum test concentration of 100 μM, with average IC50 values ranging from 4.4 to 0.37 μM. The substituents on the indole ring in the target compounds 81–85 are electron-withdrawing and electron-donating groups. The analysis of the drug structure–activity relationship (SAR) shows that the anti-tumor activities of the electron-donating groups on the indole ring in the target compounds were significantly higher than those of the electron-withdrawing groups. At the same time, there was also a SAR observed in which the stronger the ability of electron absorption or electron donation, the better the anti-tumor activity. In addition, the solid space of these substituents also affected the anti-tumor activity of the target compounds. These target compounds showed good in vitro anti-tumor activity, such as the characteristics of the target compounds 81 and 82, so these compounds were selected for further anti-tumor activity study. The target compounds 81 and 82 were further analyzed in monolayer cultures of 42 human tumor cell lines, including 15 different types of solid tumors. The relevant test results showed that the target compounds 81 and 82 had a concentration-dependent inhibitory effect on tumor cell growth and showed obvious cytotoxicity, with average IC50 values of 1.54 and 0.67 μM, respectively. Meanwhile, in in vitro cloning experiments, the target compounds 81 and 82 showed selective anti-tumor activity for human tumor transplantation, and the sensitive tumor models were dispersed among different tumor tissue types. Therefore, combined with these relevant data, the results indicate that the target compounds 81 and 82 are likely to become new anti-tumor drugs, which needs further studies, including evaluation of their in vivo anti-tumor activity and pharmacokinetic properties.
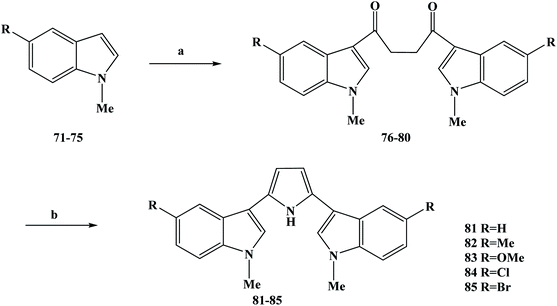 |
| | Scheme 2 Synthesis of 2,5-bis(3′-indolyl)pyrrole analogue compounds 81–85. Reagents and conditions: (a) POCl3, N,N,N′,N′-tetramethylsuccinamide, then 55–60 °C, 8 h or r.t., 20 h; (b) NH4OAc, (CH3CO)2O, CH3COOH, reflux, 4 h. | |
Table 2 In vitro activities of compounds 81–85
| Compounds |
IC50 (μM) |
| 81 |
0.37 |
| 82 |
0.37 |
| 83 |
3.4 |
| 84 |
3.4 |
| 85 |
4.4 |
| Adriamycin |
0.007 |
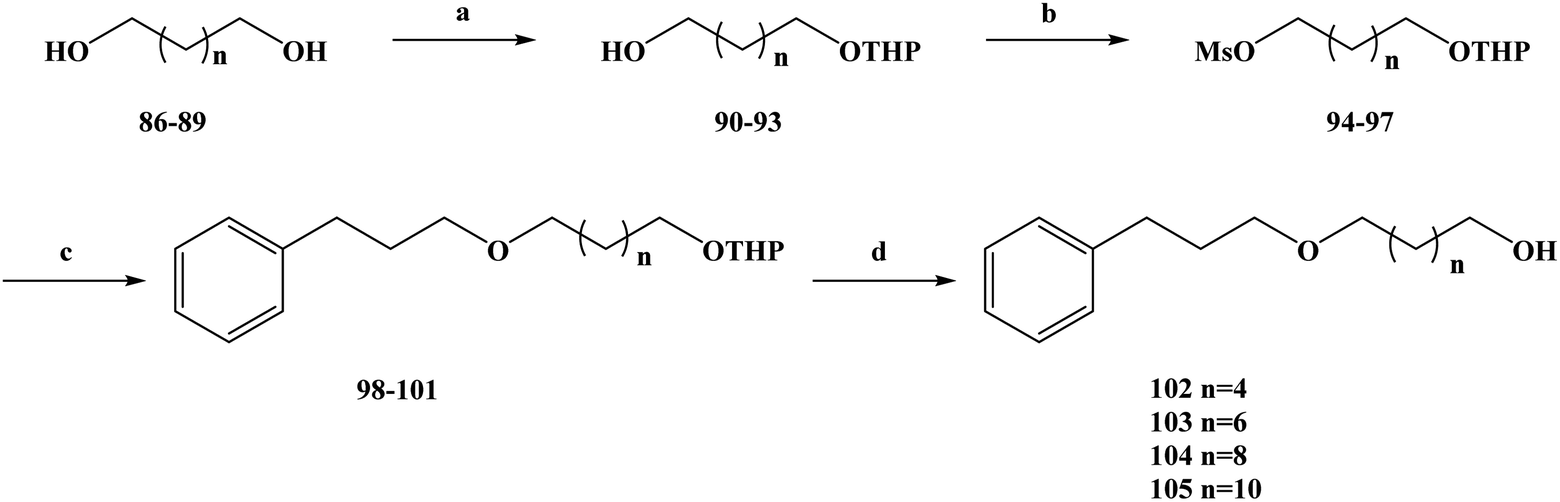
Cancer is one of the biggest threats to health in the world, and the number of deaths from it remains high, especially in developing countries.73 In order to better treat cancer, new drugs with low toxicity and good anti-tumor activity, as well as new treatments, are urgently required.74–76 In the search for new anti-cancer drugs, people have set their sights on natural products. In recent years, many new compounds have been found in the secondary metabolites of marine organisms.77 These marine organisms have been shown to be an important source of new compounds. Many new compounds obtained from marine organisms have been found to have a good inhibitory effect on tumor cell lines during the activity screening process. The secondary metabolites of sponges are abundant in marine organisms, the discovery of which has been widely reported by researchers.78–80 Some compounds were found to have anti-tumor activity during in vitro activity screening, and on this basis, analogues of these metabolites were synthesized.81–83 Many compounds were also found to have anti-tumor activity in activity evaluation. Rich in structural diversity and bioactive peptides were separated from marine sponges, compounds including the cyclopeptide perthamides G–K, glycopeptide theonellamides A–G and 3-alkylpyridine alkaloids (3-APAs) and theonelladines A–D.84–86 Different types of metabolites have been separated from marine sponges, and these compounds and synthetic analogues have been widely investigated in terms of their cytotoxicity activity towards human tumor cell lines. Theonelladine C (2) is an analogue of a 3-alkylpyridine alkaloid, which was found to have anti-protozoa activity and a pro-apoptotic effect on a human colon cancer (RKO-AS-45-1) cell line in activity screening. Therefore, on this basis, Gonçalves et al.87 further modified the structure of theonelladine C to obtain target compounds with anti-tumor activity. During the structural design of the compounds, they introduced oxygen-containing carbon chains into the 3-alkyl chain of the lead compound and changed the length of the chain to alter the anti-tumor activity of the compounds.88,89 They designed marine alkaloid analogues of 3-alkylpyridine with alkyl chains ranging in length from 6 to 12 carbon atoms. At the same time, a hydroxyl group was attached to the end of the alkyl chain to improve the water solubility of the analogues and to provide the necessary preparation for further structural modification in the future. In the synthesis route selection of the new 3-APA analogues compounds 102–105, they used the synthesis shown in Scheme 3. From this synthesis route, it can be seen that the key step in the synthesis of these analogues is the Williamson etherification reaction under mild conditions using phase transfer catalysis (PTC). The first step of this synthesis route was to selectively protect different diols (compounds 86–89) to produce the corresponding tetrahydropyronic acetals (compounds 90–93) in 74–89% yields. Compounds 90–93 were further treated in dichloromethane with methyl sulfonyl chloride to obtain the intermediate compounds 94–97. Next, compounds 94–97 were etherized with 3-(pyrid-3-yl) propan-1-ol using the phase transfer catalyst tetrabutylammonium bromide, and compounds 98–101 with a protective group were obtained. Finally, the protective groups of compounds 98–101 were removed with hydrochloric acid, and the target compounds 102–105 were obtained with in yields of 71–100%. The synthesis route has the advantages of being simple to carry out, low in cost and uses readily available reagents. At the same time, the yields of each step of the synthesis route are relatively high, and the reaction conditions are relatively mild, which is conducive for the use of this process in industrial production in the future. In the course of the anti-tumor biological activity study, the anti-tumor activities of all of the synthesized target compounds 102–105 and intermediates 98–101 were evaluated in vitro for colon cancer (RKO-AS-45-1) and uterine cancer (HeLa) (Table 3). Meanwhile, the target compounds 102–105 and intermediates 98–101 were tested in non-cancerous human lung fibroblasts (WI-26VA4) to evaluate the selectivity index (SI) of the compounds. In vitro, the MTT assay showed that the compounds 100 101, and 104 had good anti-tumor activity. In RKO-AS-45-1 cells, the IC50 values were 5.1, 3.2 and 19.1 μM, respectively. In anti HeLa cells, the IC50 values ranged from 4.0 to 9.4 μM. The drug SAR analysis showed that there was a relationship between the IC50 value and the length of the alkyl chain of the compounds. Upon increasing the length of the carbon alkyl chain, the IC50 values of the two tumor cell lines decreased continuously, among which the compounds with 10 carbon atoms had the best anti-tumor activity (compounds 100 and 104). However, as the carbon chain continued to lengthen, the anti-tumor activity decreased when the carbon chain exceeded 10 carbon atoms. In addition, the selectivity of compound 104 in the in vitro activity evaluation was found to be the highest among the compounds tested, with an SI of 5.18 for RKO-AS-45-1 and 11.65 for HeLa cells. On this basis, compounds 100 and 104 were selected for further related activities. Micronucleus and TUNEL tests showed that compounds 100 and 104 had mutational effects and induced apoptosis. These compounds induced DNA strand breaks and/or aneuploidy under the conditions used. The activation of p53 led to cell cycle arrest, preventing the proliferation of damaged cells and allowing DNA to replicate and undergo mitosis or induce apoptosis to eliminate irreparably damaged cells. At the same time, compound 104 also altered the myogenic RKO-AS-45-1 cells in the cytoskeleton. After treatment with compound 104, the actin polymerization was changed. Drug molecules that can change the dynamics of actin could easily block the mitosis of cells, thus demonstrating the cytotoxicity of compound 104 to block mitosis. Overall, according to the analysis of the existing test results, compounds 100 and 104 could be used as potential anti-tumor drug candidates, or as lead compounds for structural modification, but this needs further research and development.
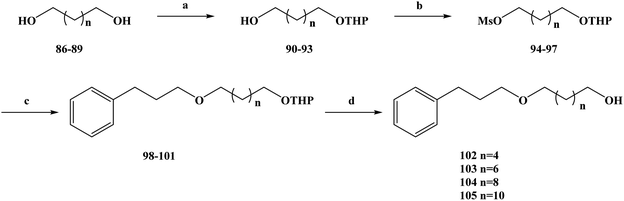 |
| | Scheme 3 Synthesis of the 3-alkylpyridine marine alkaloid analogue compounds 102–105. Reagents and conditions: (a) NaHSO4, DHP, DMSO, hexane, 40 °C, 16 h; (b) MsCl, Et3N, CH2Cl2, r.t., 10 h; (c) 3-(pyrid-3-yl)propan-1-ol, NaOH/H2O, Bu4N+Br−, Et2O, r.t., 72 h; (d) MeOH, HCl, r.t., 12 h. | |
Table 3 In vitro cytotoxicity activities of the compounds
| Compounds |
IC50 (μM) ± SD |
SI |
| PKO AS-45-1 |
HeLa |
WL-26VA4 |
PKO AS-45-1 |
HeLa |
| 98 |
>300 |
>300 |
>300 |
nd |
nd |
| 99 |
23.7 ± 1.7 |
8.1 ± 2.7 |
37.8 ± 5.0 |
1.59 |
4.66 |
| 100 |
5.1 ± 1.1 |
4.0 ± 0.8 |
6.4 ± 0.7 |
1.25 |
1.60 |
| 101 |
3.2 ± 1.7 |
9.4 ± 0.7 |
11.3 ± 1.4 |
3.53 |
1.20 |
| 102 |
>400 |
>400 |
>400 |
nd |
nd |
| 103 |
>300 |
191.8 ± 10.1 |
167.0 ± 10.5 |
nd |
0.87 |
| 104 |
19.1 ± 4.4 |
8.5 ± 2.4 |
99.1 ± 11.2 |
5.18 |
11.65 |
| 105 |
131.9 ± 16.8 |
8.8 ± 1.9 |
34.1 ± 6.5 |
0.25 |
3.87 |
| Etoposide |
1.4 ± 0.6 |
2.7 ± 0.4 |
nd |
— |
— |
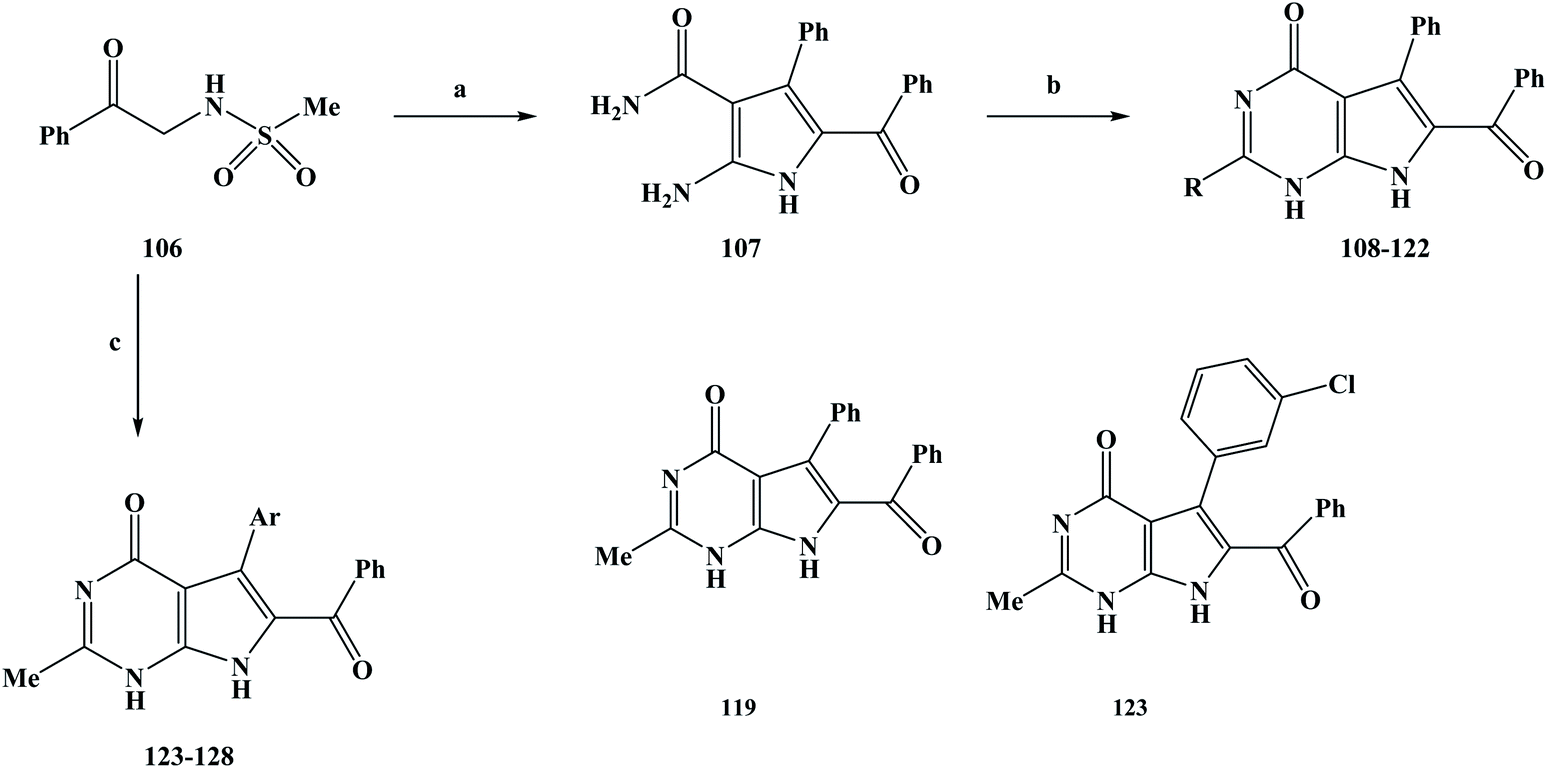
Anti-tumor drugs are a class of drugs that are used to treat cancer, including chemotherapy drugs and biological agents.90 In recent years, the development of molecular oncology and molecular pharmacology has gradually clarified the nature of tumors.91–93 The invention and application of advanced technologies such as large-scale rapid screening, combinatorial chemistry and genetic engineering have accelerated the process of drug development. The research and development of anti-tumor drugs has entered a new era.94 After years of development, many important advances have been made in the development of anti-tumor drugs. However, in the face of the most serious threats to human life and health, which account for more than 90% of malignant tumors, there is still a lack of effective and highly specific drugs.95 On the one hand, the reflects the difficulty in developing anti-tumor drugs. On the other hand, it also means that the development of anti-tumor drugs still needs the application of new ideas, technologies and methods. At present, drug treatment has become one of the most important methods for the clinical treatment of cancer. The sales of anti-tumor drugs has been increasing year on year due to the high incidence and mortality of cancer.96 Marine alkaloids with a pyrrole structure that are obtained from marine organisms have been found to have abundant biological activities during activity screening processes. Among these marine alkaloids, are rigidins A, B, C, and D (12–15). Related studies have shown that these compounds have some potential for biological activity, but few researchers have conducted in-depth studies to explore their biological activity in more detail. Frolova et al.97 completed the total synthesis of rigidins A, B, C and D and carried out a comprehensive study on their biological activities. It was found that L1210 cells in a model of mouse leukemia showed good anti-proliferation activity, but the activity of cultured human cancer cells was very low. On this basis, the structures of 7-deazahypoxanthine, 7-deazaadenine and 7-deazapurine were synthesized by modifying the 7-azathathine skeleton of the rigidins. In the study of the activities of the compounds, the anti-proliferation activity of the tumor cells was greatly improved. These compounds have also been found to damage the microtubule tissue in cancer cells by binding to the colchicine site of β-tubulin. On the basis of previous work, Scott et al.98 further modified the structure of the lead compound and introduced different substituents on the C2 position to change the biological activity of the target compounds. In their structural design, two series of target compounds were designed. During the synthesis of these target compounds, 108–128, they used the synthesis process outlined in Scheme 4. In the first series, the target compounds were obtained via a two-step reaction. N-Methylsulfonamidoacetophenone (compound 106) was used as the starting material in the reaction. The reactant was stirred with benzaldehyde and cyanoacetamide in ethanol, and potassium carbonate was added as a base to obtain the intermediate compound 107. In the second step, a ring formation reaction was carried out. Sodiated ethanol was prepared by reacting Na with ethanol, and compound 107 was reacted with the corresponding ester to obtain the target compounds 108–122. In the second series, the target compounds 123–128 were synthesized in one step. Also, compound 106 was used as the starting material of the reaction, and the corresponding aldehydes, cyanoacetamide and triethyl orthoacetate (MeC(OEt)3) were synthesized in ethanol using a one-pot method. In this step, potassium carbonate was used as a base. A reaction temperature of 90 °C was used for 24 h, followed by 150 °C for 3–6 h, with yields of 40–65% being obtained. The synthesis route has the advantages of being simple to use and low in cost with readily available reagents, and the desired target compounds could be obtained in either one- or two-step reactions. The one-pot method was applied to synthesize the target compounds or intermediates, which greatly improved the reaction efficiency and reduced the synthesis cost. However, this synthesis route also has some shortcomings. The yields of the reactions are generally low, in particular the yield of the target compound 111 was only 18%, which brings great difficulties for future industrialization. Therefore, it was necessary to further optimize the reaction conditions in the subsequent process optimization to increase the yield of the reaction. During the activity study, the HeLa cell line was used as a human cervical adenocarcinoma model and MCF-7 cells were used as a breast adenocarcinoma model to evaluate the anti-tumor proliferation activity of the first series of target compounds in vitro. In vitro screening showed that except for the target compound 108, most of the other target compounds introduced at C2 exhibited anti-tumor cell proliferation activity. The activities of this series of compounds were studied based on drug SAR analysis: the C2-aryl (compounds 111 and 112), C2–OEt (compound 114), and branched C2-alkyl (compound 115) activities were only in the two-digit to single-digit micromolar range; those of the linear C2-alkyl derivative compounds 116–118 showed an inhibitory effect at sub-micromolar concentrations; and the C2-methyl compound (compound 119) could also show nanomolar tumor cell proliferation inhibition activity. When fluorine-containing groups were introduced on C2–Me, the activity gradually decreased with an increase in the space of the C2 position in the order of CH3 (compound 119) >CHC2F (compound 120) >CHF2 (compound 121) >CF3 (compound 123). It can be seen from the activity results of these target compounds that the anti-proliferative activity of this series of compounds is dictated by the spatial properties of the C2 substituents rather than by the electronic properties. In the in vitro screening of the second series of compounds, the anti-proliferative activity of the target compounds was measured against U87 and A549. The results showed that the anti-proliferation activity of this series of compounds was lower than that of the target compound 119 (Table 4), and most of the target compounds exhibited an inhibitory effect at the sub-micromolar concentration, except for the target compound 126. The second series of target compounds is based on target compound 119 with different substituents introduced at the C7. The result of this is that the polarity of the target compound was reduced, which damages the permeability of the cells and thus reduces the anti-tumor proliferation activity of the target compounds. On the basis of previous work, the 7-deazahypoxanthines were proposed to target to the colchicine site on β-tubulin, based on their effective inhibition of [3H] colchicine binding to tubulin. The results of docking studies on the target compounds 117 and 119 showed that linear C2-alkyl substituents on the 7-deazahypoxanthine scaffold could be accommodated in a small channel in the region of Lys352 and Asn258. The co-crystallized colchicine and target compound 119 adopt similar coordination modes in the colchicine pocket. On this basis, target compound 119 with the best activity was selected to study the effect on microtubules in cells. The results showed that 119 achieves the anti-proliferation of tumor cells by acting on microtubule-targeted proteins. According to the existing data, C2-aryl and C2-alkyl-denitroxanthine marine alkaloid analogues have good anti-tumor activities. In particular, 119 could be used as a candidate for anti-tumor drugs and has the potential to be developed into a new anti-tumor drug, which needs further research and development in the future.
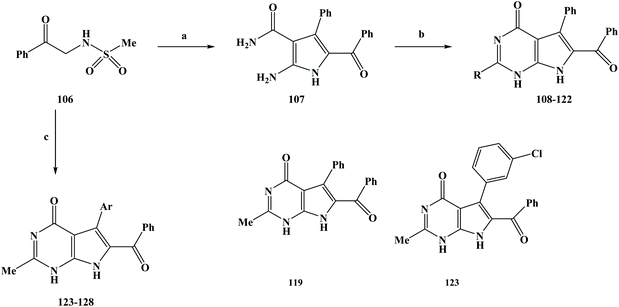 |
| | Scheme 4 Synthesis of the marine alkaloid rigidin analogue compounds 108–128. Reagents and conditions: (a) benzaldehyde, cyanoacetamide, K2CO3, EtOH, reflux, 14 h; (b) the corresponding ester, Na, EtOH, reflux, 10 h; (c) the corresponding aldehydes, cyanoacetamide, MeC(OEt)3, K2CO3, EtOH, 90 °C, 24 h, then 150 °C, 3–6 h. | |
Table 4 Anti-proliferative activities of the synthesized compounds
| Compounds |
IC50 (μM) |
| HeLa |
MCF-7 |
U-87 |
A549 |
| 119 |
0.029 ± 0.001 |
0.035 ± 0.003 |
0.077 ± 0.002 |
0.25 ± 0.01 |
| 123 |
0.27 ± 0.01 |
0.23 ± 0.00 |
0.90 ± 0.16 |
0.60 ± 0.23 |
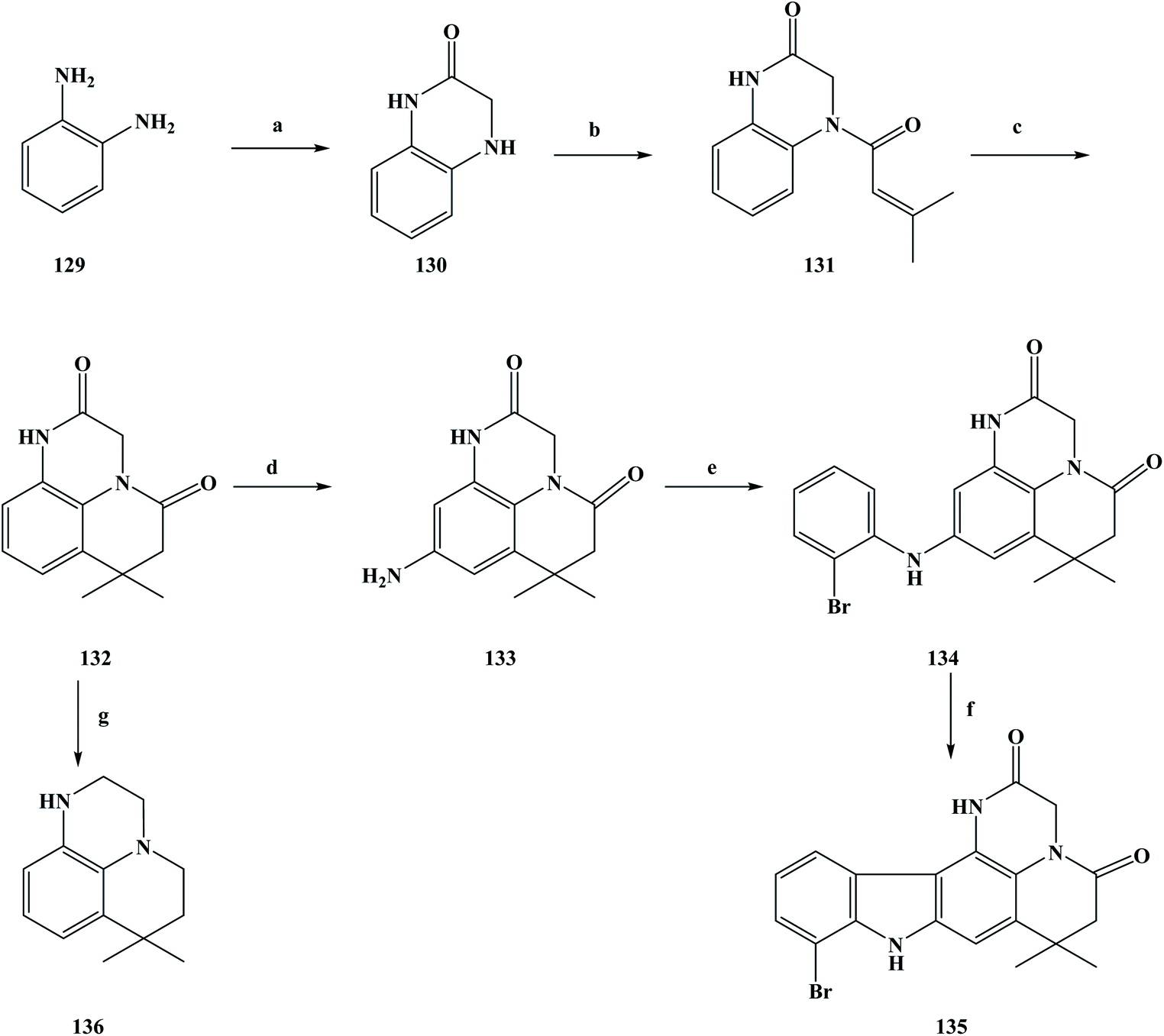
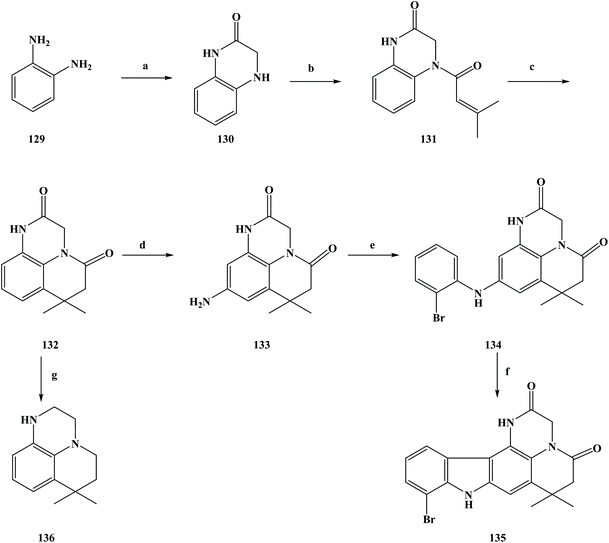
There are many ways in which to discover drugs, but natural products are an important way to obtain new drugs or lead compounds.99 Sources of natural products include compounds with certain biological activity obtained from plants, animals, and microbial metabolites using modern separation techniques. The isolation of drugs with biological activity from naturally occurring secondary metabolites has become an important field in the development of new drugs.100–102 These natural products have a unique chemical structure, and play an important role in drug discovery and innovation. They have irreplaceable significance in the treatment and prevention of human diseases. In natural product extraction and separation, alkaline compounds and alkaloids are widely investigated by researchers. Marine organisms are diverse species with diverse biological activity.103 So far, researchers have found more than 300000 species in the ocean, and it has been estimated that more than 1 million new ocean species have not yet been found. With this quantitative advantage, obtaining new compounds with biological activity from marine organisms has become an important pathway. At present, the research direction of people has turned towards marine organisms, and many marine alkaloids with biological activity have been separated from marine organisms.104 So far, the anti-tumor alkaloids separated from marine sponges include makaluvamine A (3), ellipticine (4) and neoamphimedine (5). Between 2001 and 2010, most of the compounds separated from marine sponges have been alkaloids (marine alkaloids) and macrocyclic compounds. During this period, marine alkaloids with a pyrrolidone structure were separated from marine sponges.105,106 These substances were found to have significant cytotoxicity effects on a variety of tumor cells in vitro in the course of biological activity studies, as well as potential biological activity against topoisomerase II. Based on previous work, Boucle et al.107 designed compounds with a praziquantel structure for anti-tumor activity. As could be seen from its chemical structure, the new compounds were the makaluvamine A analogue compounds 135 and 136. Meanwhile, target compounds with a pentacyclic structure have been investigated as potential anti-topoisomerase II inhibitors. In the synthesis process of the target compounds, the specific operations shown in Scheme 5 were used to synthesize the target compounds. To obtain the target compounds 135 and 136, 1, 2-phenylenediamine (compound 129) was used as the starting material to react with bromoacetyl bromide in THF to produce dihydroquinoxalinone compound 130. The condensation reaction of compound 130 and dimethylacryloyl chloride in pyridine produced compound 131, which was then cyclized in methylene dichloride using anhydrous aluminum chloride as a catalyst to obtain the tricyclic intermediate compound 132 (yield 83%). The intermediate compound 133 containing an amino group was obtained via the two-step nitrification and catalytic hydrogenation of compound 132, in which the nitrification of was carried out with nitric acid in methylene dichloride. Then, the product was reduced via catalytic hydrogenation without separation to obtain compound 133. Compound 133 was reacted with 2-bromoiodobenzene in anhydrous dioxane to obtain compound 134, with a yield of 71%. Finally, compound 134 underwent cyclization, and via the Heck reaction generated pentacyclic compound 135. In the reaction process, anhydrous dioxane was used as the solvent, palladium acetate as the catalyst and potassium carbonate as a base, with a yield of up to 40%. At the same time, compound 132 was reduced by boron/tetrahydrofuran and the target compound 136 was obtained in a yield of 81%. The synthesis route is simple to use, inexpensive and uses readily available raw materials, where moderate to good yields can be obtained at each step. However, this synthesis route also has some shortcomings, as it uses the precious metal Pd as a catalyst, which brings certain difficulties for the future industrialization of this method. At the same time, in the process of the compound 133 synthesis, the yield of this step was only 29%, and low yields increase the synthesis cost, which needs to be improved in the subsequent process optimization. In the process of studying the biological activity of the synthesized compounds, the anti-tumor activity of these compounds and the inhibition efficiency of topoisomerase II were evaluated. First, the compounds were tested against different cancer cell lines to assess their anti-tumor effects (Table 5). The inhibitory efficiency of the synthesized compounds was then evaluated against topoisomerase II in order to determine the mechanism of action of these newly synthesized marine alkaloid analogues. Different human cancer cell lines such as CACO-2, HCT-116, HUH-7, MDA-MB-231, PC-3, and NCI were selected for the in vitro cytotoxicity activity evaluation. The in vitro anti-tumor activity evaluation test results showed that compounds 134–136 have good anti-tumor activity, among which 136 exhibited cytotoxicity to CACO-2, HCT-116, PC-3 and NCI cell lines of 15, 15, 15 and 10 μM, respectively, but had no obvious toxicity to human fibroblasts. However, 134 and 135 also had certain inhibitory effects on human fibroblasts cell, so there was no obvious selectivity for cell inhibition. In the process of screening the anti-topoisomerase II activity of the synthesized compounds, the concentration of the compounds used for electrophoretic analysis was 100 μM. The results of the anti-topoisomerase II activity testing of the compounds showed that 135 better inhibited activity of human DNA topoisomerase II than the positive control doxorubicin at 100 μM. Other synthetic compounds showed no inhibition of topoisomerase II. From the in vitro anti-tumor activity of synthetic compound screening and testing of topoisomerase II activity, it was determined that compounds 135 or 136 could provide a basis for the further design of more analogues. The SAR and drug research can be used to identify compounds that have the best anti-tumor activity and greatest selectivity.
 |
| | Scheme 5 Synthesis of compounds 135–136. Reagents and conditions: (a) bromoacetyl bromide, THF, Et3N, reflux, 2 days; (b) dimethylacryloyl chloride, pyridine, reflux, 2 h; (c) AlCl3, DCM, 24 h; (d) i: HNO3, DCM/DMF, reflux, 24 h; ii: H2, Pd/C, DMF/DMSO, reflux, 18 h; (e) 2-bromoiodobenzene, Pd2dba3, Xantphos, K2CO3, dioxane, Schlenk, 18 h; (f) Pd(OAc)2, Pcy3HBF4, K2CO3, DMA, reflux, 24 h; (g) BH3/THF, THF, reflux, 2 h. | |
Table 5 Cytotoxicities of the synthesized compounds
| Compounds |
IC50 (μM) |
| HUH-7 |
CaCo-2 |
MDA-MB-231 |
HCT-116 |
Pc-3 |
NCI |
Fib.Hum |
| 134 |
20 |
20 |
>25 |
25 |
25 |
>25 |
25 |
| 135 |
20 |
20 |
20 |
10 |
6 |
10 |
6 |
| 136 |
20 |
15 |
20 |
15 |
15 |
10 |
>25 |
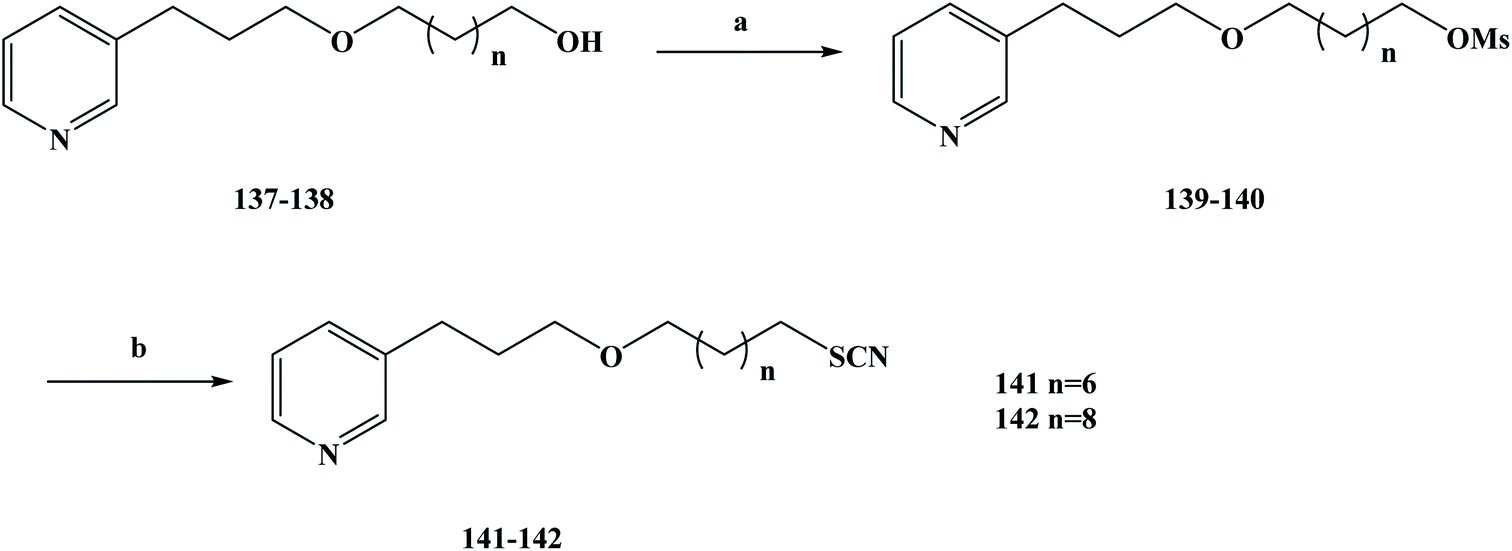
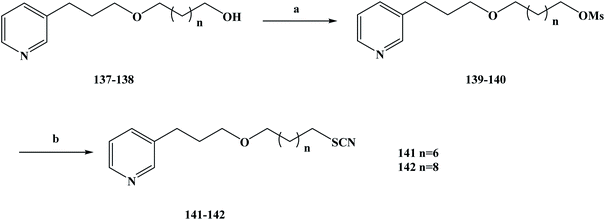
As aforementioned, natural products are compounds rich in biological activity that have the potential to be developed into drugs.108–111 With the research and development of modern drugs, obtaining new drugs or lead compounds from marine organisms has become an important source. Marine sponges have been well studied and many biological activity compounds have been separated from these relatively simple organisms in recent years, including some new structural compounds.112–116 In the process of screening these compounds for biological activity in vitro, the results show that many of them have some specific biological activity and play an important role in the development of new drugs.117 Lithistida is a type of sponge that contains rich compounds that are structurally diverse with biological activity based on 3-alkylpyridine alkaloids (3-APAs), such as viscosaline (1) and theonelladin A–D.118–121 From the analysis of the structures of these obtained marine alkaloids, it was found that 3-APAs have a pyridine structure and an alkyl chain with variable length on the side chain, and the position of this alkyl chain is usually in the 1 or 3 position. The discovery that most marine alkaloids are cytotoxic (i.e. they have anti-tumor activity) has prompted wide study and interest in these.122–126 Previous studies have shown that in the evaluation of their biological activity, synthesized 3-APA analogues exhibit anti-malarial or anti-tumor properties127 and inhibition potential, and some of these compounds may even become ideal new drugs.129–133 Tumors are a major health problem that has drawn worldwide concern.134–136 In previous work, 3-APA analogues were found to promote DNA damage, induce apoptosis, and alter two human tumor cell lines, RKO-AS-45-1 and HeLa actin cytoskeleton, during active screening.137 Naturally occurring molecules containing thiocyanate structures are rare, and these functional groups are mainly present in the formation of cruciferae glucosides during the desaccharification of some natural products with anti-tumor activity. It has also been shown that compounds containing thiocyanate groups also have anti-parasite effects, showing high cytotoxicity in anti-parasite studies. In view of these data, Barbosa et al.138 designed two new 3-APA analogues to study their anti-tumor activity using theoneladin C as a lead compound and introduced thiocyanate groups into its chemical structure. To synthesize the target compounds 141 and 142, they chose the route shown in Scheme 6. They selected the previously reported synthetic compounds 1a–b as the starting materials and synthesized the designed target compounds 141 and 142 in two steps. In the first step, the hydroxyl compounds 137 and 138 were reacted with methyl sulfonyl chloride in methylene dichloromethane to obtain the intermediates 139 and 140, in yields of 52–83% after a simple treatment. The second step was accomplished via SN2 nucleophilic substitution, in which the intermediate compounds 139 and 140 were converted to the corresponding target compounds 141 and 142 containing thiocyanate groups. In this step, 139 and 140 were substituted with potassium thiocyanate in THF and tetrabutylammonium bromide was used as a catalyst. The desired target compounds 141 and 142 were obtained via nucleophilic substitution, in yields of 55–78%. This synthesis route has the advantages of short reaction steps, high yields in each step, and it is easy to control the total yield of the reaction. Also, the reagents used are cheap and easy to obtain, and the reaction conditions are mild, which lays a solid experimental foundation for future industrialization. In the activity screening process of the target compounds 141 and 142, the anti-malarial activity of the target compounds was evaluated (Table 6). In vitro test results showed that target compounds 141 and 142 inhibited the growth of parasites, with IC50 values of 5.5 and 2.3 μM, respectively. These results indicate that the compounds have anti-malarial activity, but their selectivity is not high, which reflects that they have high cytotoxicity to the control cell line and are not conducive to use in proprietary medicine. Because these compounds have good cytotoxicity, they have certain anti-tumor properties. So next, the anti-tumor activity of target compounds 141 and 142 was examined In in vitro activity evaluation, they evaluated the cytotoxicity of the compounds towards human cancer lines of colon cancer (RKO-AS-45-1) and cervical cancer (HeLa) to evaluate the anti-tumor activity of 141 and 142. Meanwhile, the non-tumor human cell line (lung fibroblast, WI-26VA4) was used as a reference to determine the selectivity index of the target compounds, as the selectivity index is one of the most important indexes by which to evaluate the activity of target compounds. The anti-tumor activity of the target compounds 141 and 142 was investigated in vitro. The IC50 values of the target compound 141 against RKO-AS-45-1 and HeLa cells were 0.8 and 12.4 μM, respectively, and the IC50 values of target compound 142 against RKO-AS-45-1 and HeLa cells were 6.36 and 3.8 μM, respectively. Anti-tumor drugs need to be able to be highly selective towards effective (active) compounds, as in the process of killing tumor cells it should be almost impossible to kill normal cells. In determining the selectivity index of target compounds 141 and 142, it was found that 141 was selective for the tumor cell line RKO-AS-45-1 (SI = 3.59). In order to further investigate the cytotoxicity mechanism of 141 and 142, a variety of gene toxicity tests were conducted in vitro, including micronucleus assay, comet assay, ames assay and annexin-V/propidium iodide staining. The results of the relevant experiments showed that the synthetic alkaloids induce the wrong separation of chromosomes and damage DNA during the cell division of human tumor cells, resulting in cell death. These results preliminarily indicate that 141 may be a promising candidate as an anti-tumor chemotherapy drug, which needs to be further studied and developed in the future for its rapid clinical use to treat more cancer patients.
 |
| | Scheme 6 Synthesis of compounds 141 and 142. Reagents and conditions: (a) MsCl, Et3N, CH2Cl2, r.t., 10 h; (b) TBAB, KSCN, THF, reflux, 3 h. | |
Table 6 In vitro inhibitory concentrations (IC50) and selectivity indexes (SI) obtained for the human cancer cell lines RKO-AS45-1 and HeLa cells after exposure to different concentrations of the target compounds 141 and 142
| Compounds |
IC50 (μM) ± SD |
SI |
| PKO-AS45-1 |
HeLa |
WI-26VA4 |
PKO-AS45-1 |
HeLa |
| 141 |
0.80 ± 0.11 |
12.40 ± 2.25 |
2.87 ± 0.85 |
3.59 |
0.23 |
| 142 |
6.36 ± 1.30 |
3.80 ± 0.58 |
4.83 ± 1.14 |
0.76 |
1.27 |
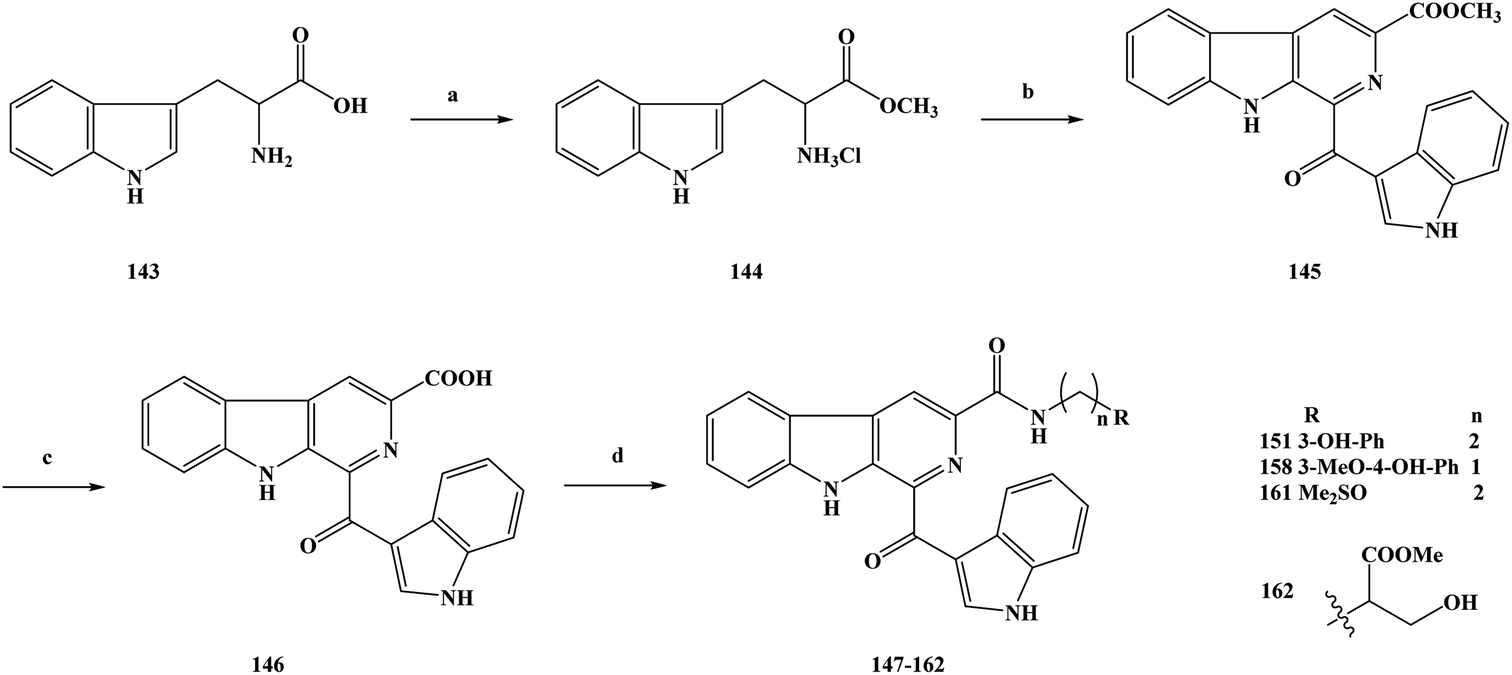
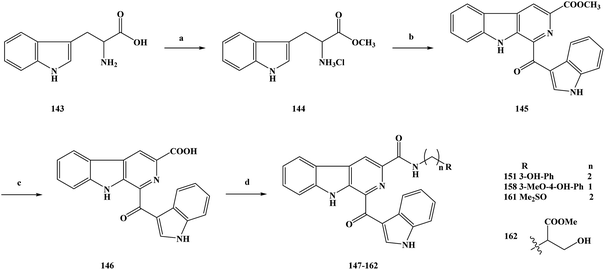
Cancer is one of the major diseases that seriously endanger human health in today's society.139–143 In the treatment of cancer, there are mainly surgical treatment, chemotherapy, radiotherapy and other means, among which chemotherapy is the most important treatment method currently used.144–147 The research and development of novel compounds with a unique chemical structure has become an important research direction in the field of anti-cancer drugs. The discovery of new drugs or lead compounds from natural products is an important route.148 On the basis of natural products, many derivatives or analogues with biological diversity have been synthesized, especially in terms of their anti-tumor activity. Alkaloids are natural products that are widely distributed in plants.149–153 They have a variety of chemical structures and play an important role in medicine. Among these natural alkaloids, β-carboline alkaloids are a type of widely distributed natural product with biological activities, which belong to the indole alkaloids and have a unique structure containing a tricyclic pyrido [3,4-b] indole ring.154–158 Pityriacitrin is a marine alkaloid separated from marine organisms. In terms of chemical structure analysis, it was found that the pityriacitrin tricyclic pyrido [3,4-b] ring structure is connected to the indole ring structure at the C-1 position.159–162 Pityriacitrin has been widely studied because of its unique chemical structure. In recent years, some derivatives and analogues of pityriacitrin have been separated or synthesized by researchers and their biological activities have been studied163 It has been found that these compounds have a wide range of biological activities, especially good anti-tumor activity and cytotoxicity. Natural products are always present in small amounts in nature, so in order to obtain large amounts of these compounds it is impossible to extract them from their sources.164–167 The use of synthetic methods changes this situation, and a large number of desired target compounds can be obtained through synthesis, and then applied to relevant research or used directly as drugs. Xu et al.168 used pityriacitrin as the lead compound to modify its structure to obtain a large number of cytotoxic compounds. From the structure of the designed target compounds, it can be seen that the they retain the chemical structure of pityriacitrin, and different amide groups were introduced at the C-6 position of the tricyclic pyrido [3,4-b] indole ring to obtain marine alkaloid β-carboline analogues. In this work, they combined the pharmacophores of β-carboline with biologically functional amide groups, thus synthesizing target compounds 147–162 with the structure of the β-carboline analogues. For the synthesis of the compounds, they selected the route shown in Scheme 7. Compounds 147–162 were prepared via a four-step reaction using tryptophan (compound 143) as the starting material. In the first step, compound 143 was added to anhydrous methanol and then reacted with sulfoxide chloride to obtain the crude product methyl tryptophan hydrochloride (compound 144), without separation. In the second step, iodine and 1-(1H-indol-3-yl) ethanone were added to dimethylsulfoxide (DMSO) for the reaction, then compound 144 was added to continue the reaction, and 1-(1H-indole-3-carbonyl)-9H-pyrido [3,4-b] indole-3-carboxylate (compound 145) was generated via a simple condensation ring reaction. In the direct nucleophilic substitution compound 145, it was found that the reaction could not be carried out, so the reaction pathway was changed. The third step was to add 145 to a solution of sodium hydroxide. Hydrolysis readily occurred, and the intermediate 146 was obtained. In the final step, 146 was reacted with different substituted amines. DMF was used as the reaction solvent and HOBt/EDCI/Et3N as the reaction catalyst to obtain the target compounds 147–162. This synthesis uses inexpensive and readily available reagents, and the yields of the target compounds were moderate to good, obtained via a simple synthetic route, which lays foundation for its future industrialization. However, there are some problems in terms of the environmental friendliness of this route. Reagents such as chlorinated sulfoxide were used in the experiment, and some polluting gases were generated in the reaction process, which means that the route does not meets the requirements of green chemistry, and the subsequent process conditions therefore need to be optimized. In the course of the biological activity study, the cytotoxic effects of the marine alkaloid β-carboline analogue compounds 147–162 were tested in vitro. The SGC-790, A875, HepG2 and MARC145 cell lines were used as test subjects. In vitro cytotoxicity test results showed that the target compounds exhibited moderate to good inhibitory activity against all four cell lines. The activities of 159 and 160 with aromatic groups directly on their amides were lower than those of the benzylamine (153–158) and phenylethylamine (147–152) substituted compounds. The compound containing a 3-hydroxyl group (151) showed significantly better biological activity than the compound containing a 3-methoxy substituent (148). In order to further study the biological activity of these target compounds and to screen out the more active compounds, the IC50 values of the target compounds were also tested in vitro. The results showed that the target compounds 151, 158, 161 and 162 showed good inhibitory activity (Table 7). The IC50 values of the target compound 161 were 6.82 ± 0.98, 8.43 ± 1.93, 7.69 ± 2.17 and 7.19 ± 1.43 μM, respectively. The SAR results showed that benzylamine substituted compounds usually had better activity than phenylethylamine substituted compounds, and the activities of these two target compounds were obviously better than those of aromatic amine substituted compounds, which demonstrated the importance of the link chain between the aromatic rings and amides. In addition, the biological activities of most of the target compounds were better than those of β-carboline and β-carboline B, which indicated that the presence of amide groups in the compounds was beneficial to the anti-tumor activity of the target compounds. In addition, hydroxyl phenyl substituted compounds (158 and 151) have significantly higher activity than other corresponding compounds, possibly because the polarity of the hydroxyl phenyl group is favorable for binding to the target protein. The target compound 161 contains a special sulfonyl group and has the highest inhibitory activity. The special properties of the hydroxyphenyl and sulfur groups help to improve the biological activities of the target compounds. On the basis of the dose response analysis of 151, 158, 161 and 162 the results showed that these target compounds had a concentration-dependent inhibitory effect on cell lines. In general, these target compounds have good cytotoxic activity and can be used as potential cell agents.
 |
| | Scheme 7 Synthesis of the marine alkaloid oriented β-carboline analogue compounds 147–162. Reagents and conditions: (a) SOCl2, MeOH; (b) 1-(1H-indol-3-yl) ethanone, I2, DMSO; (c) NaOH, MeOH, r.t.; (d) NH2(CH2)nR, HOBt, EDCI, Et3N, DMF, r.t. | |
Table 7 Cytotoxic activities of the target compounds
| Compounds |
IC50 (μM) |
| SGC-7901 |
A875 |
HepG2 |
MARC145 |
| 151 |
17.65 ± 5.84 |
11.91 ± 0.80 |
8.63 ± 3.31 |
13.77 ± 3.75 |
| 158 |
16.18 ± 2.31 |
8.47 ± 2.96 |
11.08 ± 3.33 |
21.77 ± 6.94 |
| 161 |
6.82 ± 0.98 |
8.43 ± 1.93 |
7.69 ± 2.17 |
7.19 ± 1.43 |
| 162 |
14.30 ± 2.57 |
10.46 ± 1.34 |
9.99 ± 1.82 |
16.27 ± 0.07 |
| 5-FU |
53.58 ± 1.99 |
62.12 ± 17.83 |
66.42 ± 12.99 |
115.54 ± 8.30 |
2.2 Anti-malarial activity
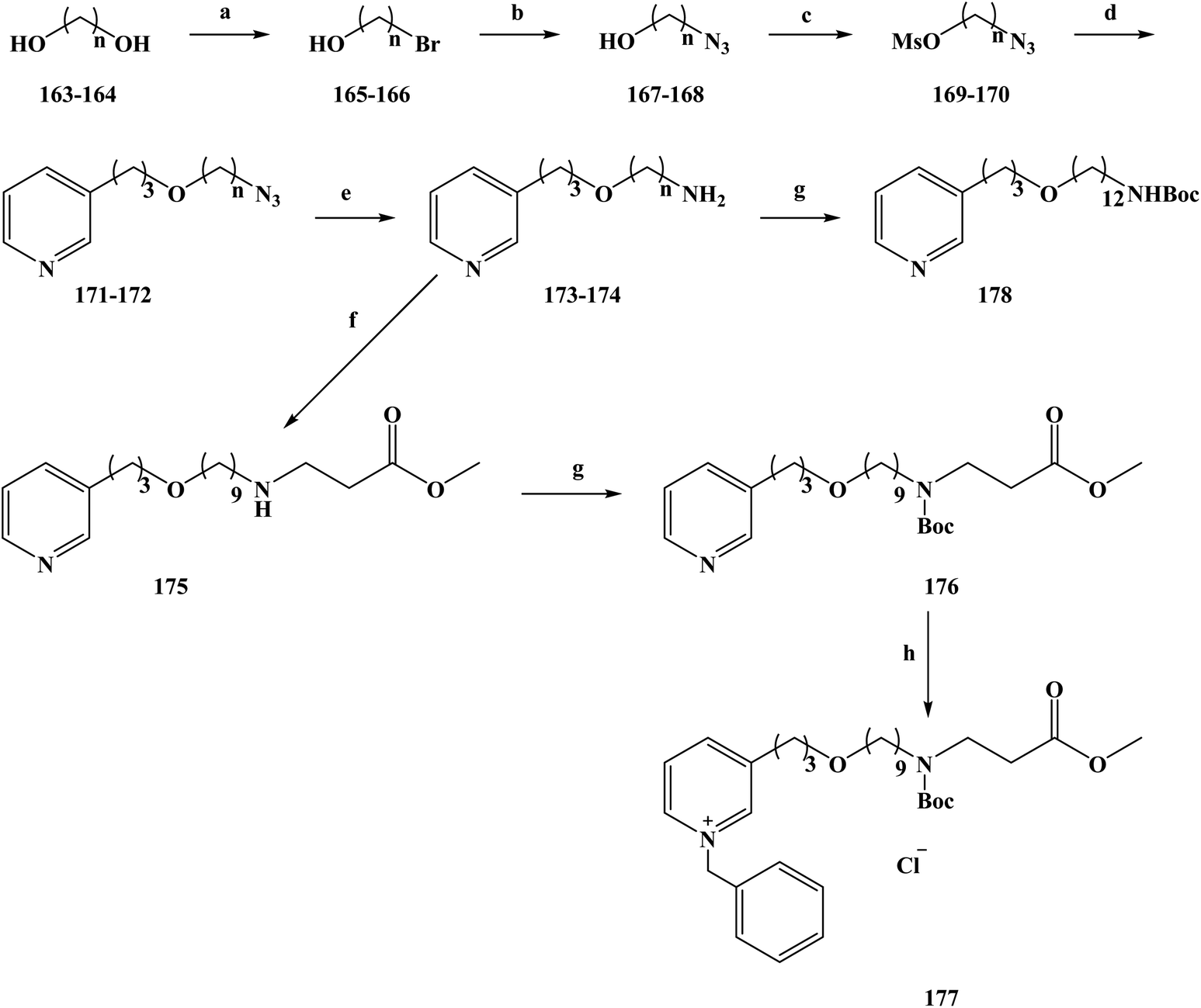
In 2008, the World Health Organization released the world malaria report, which showed that malaria kills about one million people worldwide each year.169–173 Malaria is more serious in countries or regions that have poor living conditions and medical conditions, especially in Africa.174 There is no effective vaccine to control malaria, and control programmes to reduce the spread of the disease still rely on chemotherapy (drug therapy) to prevent malaria and treat malaria-infected people. The existing drugs for the prevention and treatment of malaria mainly include mefloquine and artemisinin analogues, but new drug-resistant strains appear in the clinical, resulting in ineffective drug treatment.175 Therefore, it is urgent to research and develop new and effective anti-malarial drugs for better prevention and treatment of malaria. In the research and development of anti-malarial drugs, medicinal chemists have focused on the research of marine alkaloids and their analogues in recent years, hoping to find effective anti-malarial drugs based on these compounds. In the process of screening the anti-malarial activity of marine alkaloids, makaluvamine A (2) was the first molecule to be studied for anti-malarial activity.176,177 Makaluvamine A is a complex alkaloid with a heterocyclic structure separated from marine resources. However, this marine alkaloid was found to have a very low therapeutic index in an in vitro anti-malarial activity test, which means that it is not an ideal candidate as an anti-malarial drug for clinical treatment. Alkaloids containing piperidine and pyridine structures are widely distributed in marine sponges of different genera, and 3-alkyl pyridine alkaloids have been separated sponges.178 In 2004, Volk and Kcck reported a viscosaline (1) alkaloid separated from a marine sponge, and found that the compound showed high anti-microbial activity towards a variety of bacteria.179,180 In 2007, the Shorey team first reported the total synthesis of viscosaline, and described the key intermediate theonelladin C (2) in the synthesis method. The study found that theonelladin C is also a natural product compounds found in sponges.181 The chemical structures of viscosaline and theonelladin C are relatively simple, with viscosaline showing the highest anti-microbial activity. Hilario et al.182 used viscosaline as a lead compound to modify in order to prepare marine alkaloid viscosaline analogues. These 3-alkylpyridine analogues have a viscosaline pyridine ring in the alkyl chain, and oxygen-containing groups were introduced to alter the biological activity of the analogues. To synthesize compounds 177–178, a relatively simple total synthesis method was chosen. The specific route for the synthesis of the new 3-alkylpyridine compounds is shown in Scheme 8. 1,9-Nonanodiol (compound 163) and 1,12-dodecandiol (compound 164) were used as starting materials in the reaction. In refluxing toluene, the reactants were selectively brominated with hydrobromic acid to obtain the corresponding brominated alcohol compounds 165 (yield 70%) and 166 (yield 80%). Compounds 165 and 166 were converted to their corresponding azides 167 and 168 in dimethyl sulfoxide via SN2 substitution. Then, 167 and 168 were treated with methyl sulfonyl chloride in methylene dichloride to obtain the intermediate compounds 169 and 170. Using tetrabutylammonium bromide as a phase transfer catalyst, compounds 171 and 172 were etherified with 3-pyridine-propanol, in good yields. The amine-containing compounds 173 (72% yield) and 174 (79% yield) were obtained by reducing terminal azides 171 and 172 in THF with lithium aluminum hydride. Compound 173 and methyl acrylate were refluxed in toluene to form 175 (yield 70%). Compounds 174 and 175 were treated with ditert-butyl carbonate in methylene dichloride to obtain 176 (yield 94%) and 178 (yield 75%). Compound 176 was alkylated with benzyl chloride to obtain another target compound, 177. The synthetic enables the total synthesis of the target products, where the raw materials and reagents used are cheap and easy to obtain, and the yield of each step is relatively high, laying a solid foundation for its future industrialization. During the study of anti-malarial biological activity, the anti-malarial effect of the 3-alkylpyridine marine alkaloid analogues in vitro using a [3H]-hypoxanthine doping method (Table 8) was established. On this basis, the cytotoxic effects of seven marine alkaloid analogues (compounds 172–178) were tested using the MTT method to evaluate the safety of the target compounds. The seven tested marine alkaloid analogues were evaluated for their anti-malarial activity at half the maximum inhibitory concentration (IC50) and half the lethal drug concentration (LC50). In vitro anti-malarial results showed that the compounds tested were all active versus chloroquine against the plasmodium falciparum clone W2. Drug SAR studies have shown that the alkyl chain lengths of these tested compounds (compounds 173 and 174) have an impact on their anti-malarial and cytotoxic properties. As the length of the alkyl chain increased from 9 to 12 carbon chains, the anti-malarial activity and selectivity index of the compounds were increasingly improved (compound 174 SI = 10.82). The length of the alkyl chain of compound 175 was also extended, significantly reducing its IC50 value compared with that compound 173, and significantly increasing its selectivity index from 5.48 to 12.36. However, after the amino group of compound 175 was protected with a Boc group, the IC50 value of product 176 was significantly increased, and the selectivity was also significantly reduced (SI = 2.78). These results suggest that the position of the amine plays an important role in the anti-malarial activity of the compounds. At the same time, the target compound 177 of the quaternary ammonium reaction had a significantly reduced IC50 value, and its selectivity was improved, making it the compound with the highest selectivity index (SI > 15.58) among the tested compounds. Preliminary in vitro anti-malarial activity studies showed that 3-alkylpyridine marine alkaloid analogues have ideal anti-malarial activity and cytotoxicity. Among the synthesized compounds, compound 177 had the best anti-malarial activity and the highest selectivity index, thus making it a promising candidate for the development of novel anti-malarial drugs and deserving of further study.
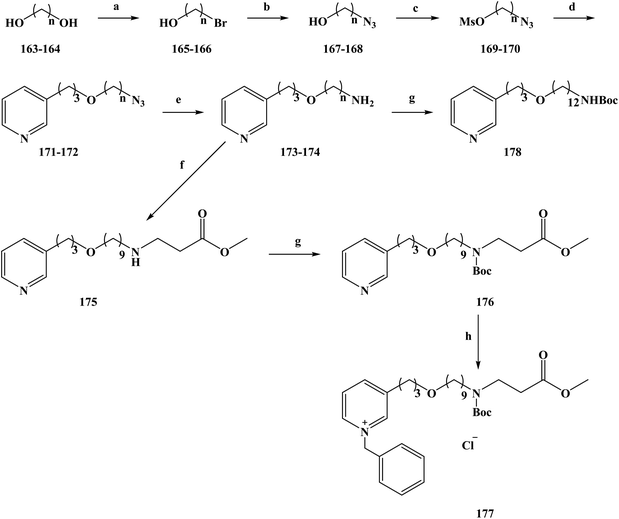 |
| | Scheme 8 Synthesis of the oxygenated 3-alkylpyridine marine alkaloid analogue compounds 177 and 178. Reagents and conditions: (a) HBr (aq), toluene, reflux; (b) NaN3, DMSO, r.t., 12 h; (c) MsCl, Et3N, methylene chloride, stirring, 3 h; (d) NaOH, Bu4N+Br−; (e) LiAlH4, THF; (f) methyl acrylate, toluene, reflux; (g) Boc2O, methylene chloride; (h) BnCl, reflux. | |
Table 8 In vitro anti-plasmodial activities of the oxygenated 3-alkylpyridine marine alkaloid analogues
| Compounds |
IC50 (μM) ± SD |
LC50 (μM) ± SD |
SI |
| 172 |
4.33 ± 0.87 |
45.52 ± 2.71 |
10.51 |
| 173 |
17.96 ± 3.59 |
98.43 ± 3.77 |
5.48 |
| 174 |
6.24 ± 0.31 |
67.54 ± 1.92 |
10.82 |
| 175 |
8.50 ± 0.14 |
105.12 ± 5.73 |
12.36 |
| 176 |
43.04 ± 4.30 |
120.00 ± 8.15 |
2.78 |
| 177 |
<3.38 |
52.67 ± 2.25 |
>15.58 |
| 178 |
<4.76 |
46.78 ± 4.65 |
>9.82 |
| Chloroquine |
0.625 ± 0.003 |
>100 |
>160 |
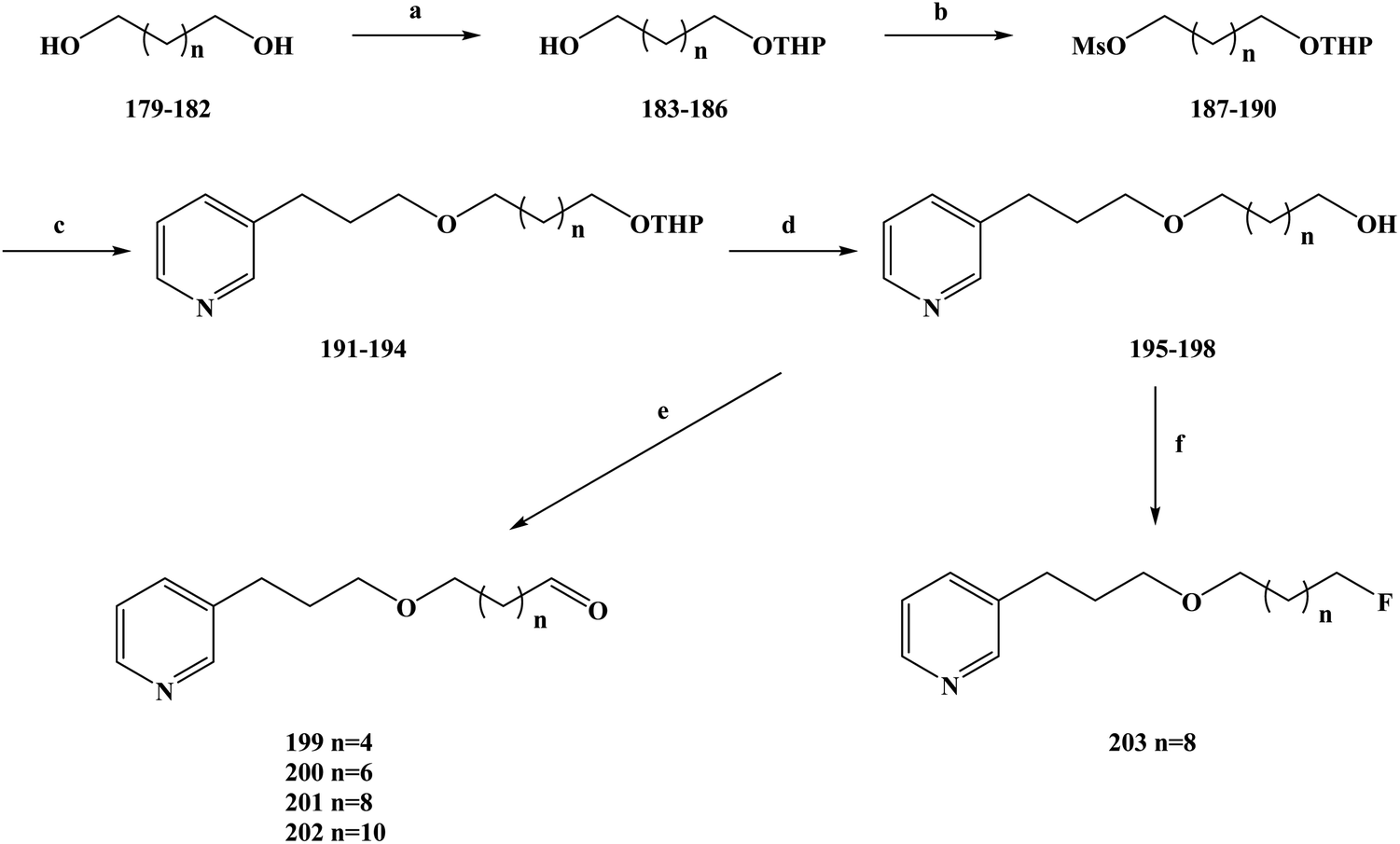
Malaria is a serious infection caused by malaria-carrying parasites and can be transmitted in the blood by female mosquitoes.183 Malaria is a global public disease that has high morbidity and mortality.184 According to the World Health Organization, there are more than 100 species of malaria parasites that cause malaria.185–188 However, only a few species of plasmodium have attracted research attention, including Plasmodium falciparum, Plasmodium malariae, Plasmodium vivax, Plasmodium knowlesi and Plasmodium ovale. Malaria, caused by malignant protozoa, is a severe disease and can cause other syndromes.189 Drug therapy is one of the main strategies for the treatment and control of malaria.190–192 Because of the emergence of artemisinin-resistant parasites in the clinic. It is imperative to find new antimalarial drugs, especially those that are lethal to resistant parasites. In the process of developing new anti-malarial drugs, the target and mechanism of action of the drugs need to be further determined.193–195 Natural products are characterized by their complex structure and biological activity diversity.196 Due to these reasons, research on anti-malarial drugs has shifted to natural products in recent years.197,198 In the process of screening the activity of natural products, 3-alkylpyridine marine alkaloids and their analogues have been found to have anti-malarial activity. On this basis, de Souza Barbosa et al.199 further modified the structure of the lead compound and designed new types of 3-alkylpyridine marine alkaloid analogues. To improve the anti-malarial activity, selectivity and safety of the compounds, they selected different lengths of alkyl chains. The types of functional groups attached to the ends of the chains were used to further investigate the SAR of the designed compounds. At the same time, in order to improve the anti-malarial activity, the selectivity, improve the safety and enhance the affinity of the compounds, the functional groups at the end of the side chain of the compound were fluorinated. During the synthesis route selection of 3-APA analogues, the specific synthesis operations were carried out as shown in Scheme 9. Diols (compounds 179–182) with different length carbon chains were used as the starting materials of the reaction, and their single hydroxyl groups were selectively protected to obtain the corresponding monohydropyronic acetals (compounds 183–186). Then, the hydroxyl groups of compounds 183–186 were further protected to obtain the compounds 187–190. The compounds 187–190 were etherized with 3-(pyrid-3-yl)propan-1-ol using a phase transfer catalyst to obtain compounds 191–194, which were then protected using hydrochloric acid to obtain compounds 195–198. The Swern oxidation was used to convert the alcohols 195–198 to the corresponding aldehydes 199–202, in yields of 60–95%. At the same time, compound 197 and diethylamine-based sulfur trifluoride underwent a fluorine-substituted halogenation reaction in methylene chloride to obtain compound 203 (yield 17%). This synthesis is simple to carry out under mild reaction conditions using cheap and readily available reagents. The yield per step was generally medium to good, but the fluorine substitution produced a low yield of only 16% (compound 203), which would increase the costs of this process if it were to be industrialized. The halogenation reaction will be a key problem in the subsequent process optimization, which needs to be further improved to improve the yield and reduce the reaction costs. Upon in vitro screening of the anti-malarial activity of the abovementioned compounds, they were screened against Plasmodium falciparum (strain W2) and were found to be cytotoxic to human fibroblasts (WI-26VA4). In vitro anti-malarial activity test results showed that the compounds tested had moderate activity against Plasmodium falciparum, with IC50 values ranging from 8.9 to 210.6 μM (Table 9). The results of the SAR showed that there was a certain relationship between the anti-malarial activity and the selectivity index of these compounds. With the extension of the side chain from 6 to 12 carbon atoms, the anti-malarial activity of the side chain increased, and the activity decreased after the chain exceeded 12 carbon atoms. A similar SAR was also observed for the selectivity index, with the length of the carbon chain also affecting the selectivity index of the compounds. At the same time, the compounds containing hydroxyl groups had higher antimalarial activity than those containing other groups. The selectivity index is a key factor in determining whether or not an anti-malarial drug can be used in treatments. Compound 197 had one of the lowest IC50 values (IC50 = 14.7 μM) among the compounds tested, but its selectivity index was the highest (SI = 6.7). Therefore, compound 197 was selected for further structure optimization, and the its hydroxyl group was converted to a fluoride group to obtain compound 203. This substitution was made to improve the anti-malarial activity of compound 197. The in vitro anti-malarial activity of compound 203 was thus significantly increased (5.8 times) compared to that of compound 197. The IC50 value of the chloroquine resistant strain W2 of Plasmodium falciparum was 2.5 μM and the SI value was 11.2. The IC50 value of the chloroquine sensitive strain 3D7 was 2.3 μM and the SI value was 12.2. On this basis, the binding ability of the heme group to compound 203 was further determined. Using an organosilicone method, the results showed that compound 203 more favorably formed stable complexes (hematin/3-APA complex) with heme molecules in a molecular ratio of 2:1 after fluorine was used to replace the hydroxyl groups. In summary, they synthesized a new halogenated 3-APA analogue, compound 203, with good anti-malarial properties and increased activity against Plasmodium falciparum as well as an increased selectivity index. Compound 203 has great development potential as a new anti-malarial drug, and the mechanism of action and pharmacokinetic characteristics of the compound need to be further determined in follow-up work.
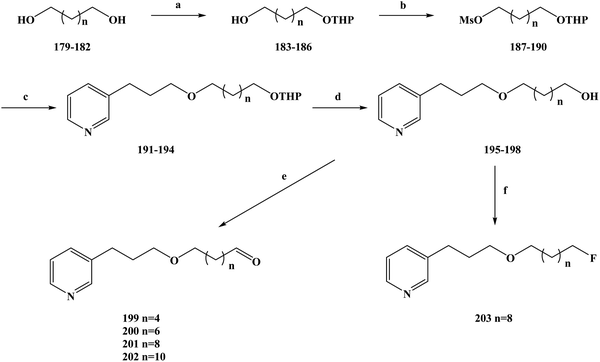 |
| | Scheme 9 Synthesis of the fluorinated 3-alkylpyridine marine alkaloid analogue compounds 199–203. Reagents and conditions: (a) NaHSO4, DHP, DMSO, hexane, 40 °C, 16 h; (b) MsCl, Et3N, CH2Cl2, r.t., 10 h; (c) 3-pyridinepropanol, NaOH/H2O, Bu4N+Br−, Et2O, r.t., 72 h; (d) MeOH, HCl, r.t., 12 h; (e) (COCl)2, Et3N, DMSO, DCM, −60 °C, 25 min; (f) DAST, DCM, 0 °C, 18 h. | |
Table 9 In vitro anti-malarial activities, cytotoxicities and SIs of the 3-APA analogues
| Compounds |
IC50 (μM) ± SD |
SI |
| P. falciparum (W2) |
WI-26VA4 |
| 191 |
53.7 ± 0.7 |
>300 |
>5.6 |
| 192 |
37.7 ± 0.2 |
37.8 ± 5.0 |
1.0 |
| 193 |
14.0 ± 0.6 |
6.4 ± 0.7 |
0.4 |
| 194 |
8.9 ± 0.4 |
11.3 ± 1.4 |
1.2 |
| 195 |
210.6 ± 12.7 |
>400 |
1.9 |
| 196 |
>188 |
167.7 ± 10.5 |
nd |
| 197 |
14.7 ± 0.2 |
99.1 ± 11.2 |
6.7 |
| 198 |
15.1 ± 0.8 |
34.1 ± 6.5 |
2.2 |
| 199 |
180.2 ± 8.3 |
nd |
nd |
| 200 |
52.4 ± 8.9 |
nd |
nd |
| 201 |
20.3 ± 0.2 |
nd |
nd |
| 202 |
30.6 ± 2.7 |
nd |
nd |
| 203 |
2.5 ± 0.1 |
28.2 ± 3.7 |
11.2 |
| Chloroquine |
0.4 ± 0.066 |
>100 |
>250 |
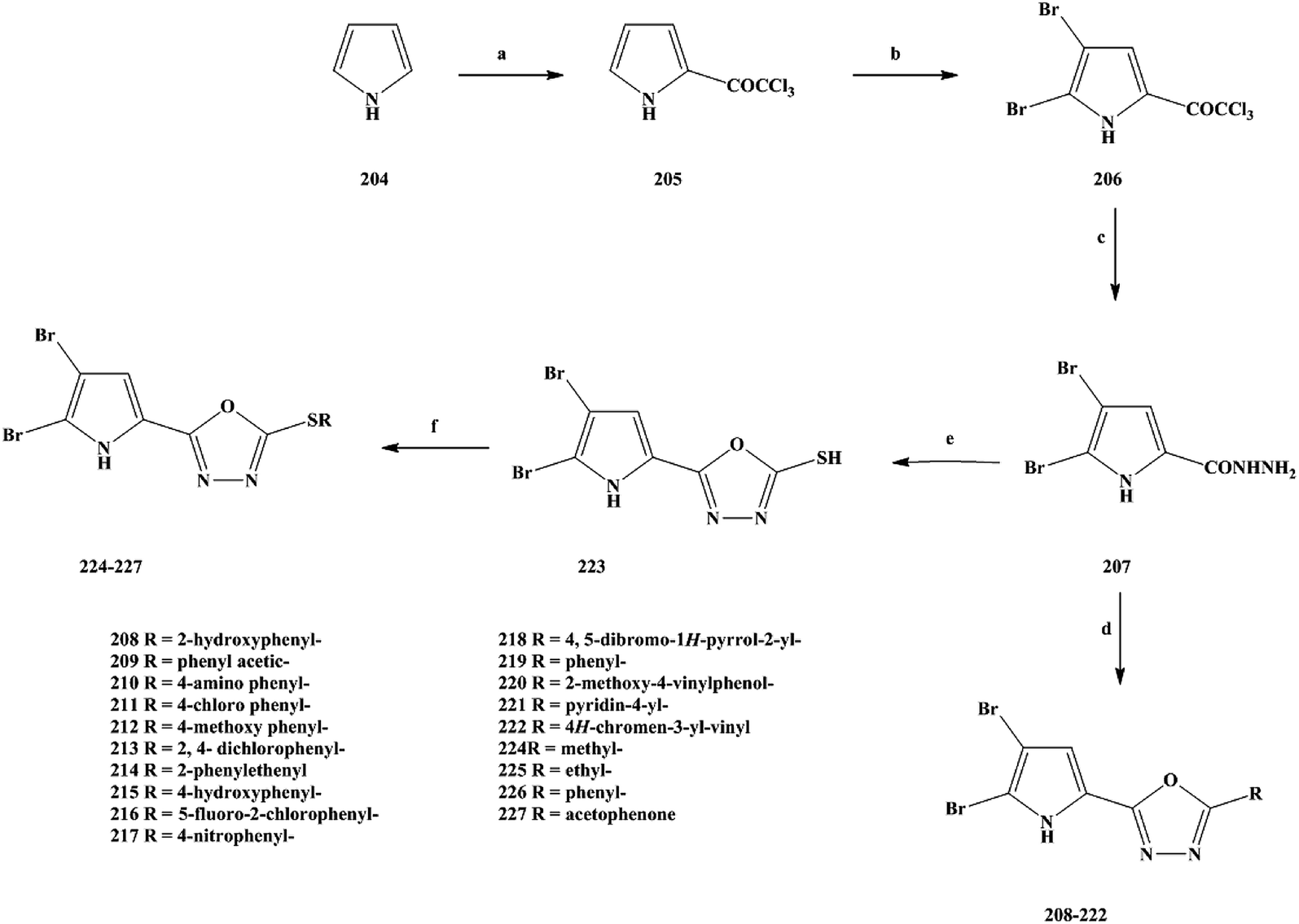
2.3 Anti-bacterial activity
Bacteria are one of the main groups of living organisms and as the most abundant of all living organisms, have a great influence on human activities.200 Bacteria are the cause of many diseases, including tuberculosis, gonorrhea, anthrax poisoning, syphilis, plague, trachoma and other diseases. In plants, bacteria cause leaf spots, fire blight and wilting.201 Anti-microbial drugs generally have bactericidal or antibacterial activity, drugs such as antibiotics, sulfonamides, imidazoles, nitroimidazoles, quinolones and other chemical synthetic drugs. Some products obtained from the culture of microorganisms such as bacteria, actinomycetes and fungi, or the same or similar substances made by chemical semi-synthesis, may also be synthesized chemically.202–204 Anti-microbial agents have inhibitory and killing effects towards pathogens at certain concentrations. 1,3,4-Oxadiazoles are compounds that have special chemical structures205 and biological activities that include anti-inflammatory, hypoglycemic, anti-depressant, anti-tumor cell proliferation, anti-bacterial properties. In molecular docking and pharmacokinetic studies, it was found that the introduction of a 1,3,4-oxadiazole structure into the drug molecule could change its polarity, flexibility and metabolic stability. The chemical structure of 1,3,4-oxadiazole plays an irreplaceable role in drug efficacy.206 Previous work found that heterocycles containing a pyrrole ring system had diverse biological activity, with several pyrrolidone analogues showing obvious anti-microbial activity in the activity evaluation process. Bromopyrrole marine alkaloid, derived from the secondary metabolites of marine sponges, has been widely investigated. These marine alkaloids have a special chemical structure, most of which are composed of 4,5-dibromopyrrole rings.207,208 In the evaluation of the biological activity of these marine alkaloids containing a 4,5-dibromopyrrole ring, it was found that they have diverse biological activities, including blocking serotonin receptor antagonists of α-adrenoceptor receptors, acting as anti-histamines and serotonin receptor antagonists, against myosin aptase, inhibiting kinase activation, and exhibit anti-tumor, anti-microbial, anti-tuberculosis, and anti-fungal properties. On the basis of previous work, Rane et al.209 designed novel bromopyrrole marine alkaloid derivatives. The chemical structures of these designed target compounds showed that two groups could be joined together by splicing to form a hybrid molecule. Specifically, a 4,5-dibromopyrrole ring and 1,3,4-oxadiazole ring were combined via chemical bonds to form a new class of target compounds, 208–222 and 224–227. Different substituents were introduced into the 1,3,4-oxadiazole ring to change its biological activity. To synthesize the target compounds, the synthesis route in Scheme 10 was used. In the first step of the reaction, pyrrole (compound 204) was used as a starting material to conduct an esterification reaction with trichloroacetyl chloride. In the reaction, anhydrous ethyl ether was used as the reaction solvent and potassium carbonate as a base, to obtain a good yield of 2-trichloroacetylpyrrole (compound 205). In the second step, compound 205 was brominated with bromine in chloroform to obtain 4,5-dibromo-2-trichloroacetyl-1H-pyrrole (compound 206) in good yield. The third step was to stir compound 207 with hydrazine hydrate to react at room temperature. In the presence of phosphorus chloride, 2-(4,5-dibromo-1H-pyrrol-2-yl)-5-aryl-1,3,4-oxadiazoles (compounds 208–222) could be prepared via the refluxing of compound 207 with aromatic acids containing different substituents. Meanwhile, 5-(4,5-dibromo-1H-pyrrol-2-yl)-1,3,4-oxadiazole-2-thiol (compound 223) could be obtained by the reaction of compound 207 with carbon disulphide in ethanol under alkaline conditions in the presence of potassium hydroxide. When compound 223 was mixed with different halide substituents in ethanol, in the presence of sodium hydroxide to provide alkaline conditions, an alkylation reaction took place to obtain the target compound S-alkylation derivatives 224–227. The synthetic route can be used to synthesize 20 derivatives, meaning that it has good universality. The reagents used in the synthesis process are cheap, easy to get, the synthesis is simple to carry out and produces compounds in good yield, laying a good foundation for its future industrialization. During the biological activity study, the anti-microbial activity of the target compounds was evaluated in vitro, against Escherichia coli, Staphylococcus aureus, Pseudomonas aeruginosa, and Klebsiella pneumoniae bacterial strains. The minimum concentration (maximum dilution) required to inhibit bacterial growth was used as the minimum inhibitory concentration (MIC) of the target compound to evaluate the activity of the compound. Meanwhile, the anti-fungal activity of candida albicans in DMSO was screened using the agar dilution method. In addition, the synthesized compounds were screened in vitro against Mycobacterium tuberculosis H37Rv to determine the minimum inhibitory concentration. The results of the in vitro anti-microbial activity screening showed that the synthesized compounds have good anti-bacterial, anti-fungal and anti-tuberculous activities. These spliced compounds showed anti-bacterial activity against both Gram-positive and Gram-negative bacteria, showing that they have extensive antibacterial activity. The results of drug SAR showed that the presence of electric-donating groups on the aromatic rings of the compounds, such as amino, chlorine, fluorine and nitro groups, was very important in inhibiting the activity of Escherichia coli. For example, the anti Escherichia coli MIC of compounds 210, 211 and 216–218 was 1.56 μM, and the activity was similar to that of the positive control. For Gram-positive Staphylococcus aureus, compounds 208, 211, 216–218 and 220–222 had an equivalent activity of 1.56 μM at MIC compared with the positive control. However, the compounds 223–227 containing mercaptan groups or S-alkyl/aryl groups were less active against Staphylococcus aureus. All compounds tested had no inhibitory activity against pneumonia bacteria, while compounds 210–219 showed moderate activity against pseudomonas aeruginosa (MIC was 6.25 μM). In the in vitro anti-fungal activity screening, the three compounds with the best anti-fungal activity were 217, 218 and 227, and the MIC was 1.56 μM. In the in vitro evaluation of Mycobacterium tuberculosis activity, it was found that the compounds 213, 218, 221, 222 and 227 showed obvious Mycobacterium tuberculosis activity when the MIC were 1.60, 3.50, 3.50, 2.00 and 1.50 μM, respectively, and the compound 227 showed the highest anti-tuberculosis activity. The SAR analysis showed that by introducing S-aryl substitution and free SH or S-alkyl or S-aryl on the C5 sites of the 1,3,4-oxadiazole ring, the anti-tuberculous activity of the compound increased. At the same time, when heterocyclic rings such as 4,5-dibromopyrrole, pyridine-4-yl and 4H-chromen-3yl-vinyl were introduced, the anti-tuberculosis activities of the compounds were significantly enhanced. For example, the MICs of the compounds 218, 221 and 222 were 3.50, 3.50 and 2.00 μM, respectively. In general, the compounds 221 and 216–218 have good anti-bacterial activity, compounds 217, 218 and 227 have good anti-fungal activity, and compounds 213 and 227 inhibit Mycobacterium tuberculosis at low concentrations. These compounds have been developed into new anti-bacterial, anti-fungal or anti-tuberculosis drugs, which need to be further developed (Table 10).
 |
| | Scheme 10 Synthesis of 2-(4,5-dibromopyrrol-2-yl)-5-substituted-1,3,4-oxadiazole compounds 208–222 and 224–227. Reagents and conditions: (a) CCl3COCl, anhydrous ether, K2CO3; (b) Br2/chloroform; (c) NH2NH2·H2O, r.t., 1 h; (d) R–COOH, POCl3, reflux; (e) CS2, KOH ethanol, reflux, 12 h; (f) alkyl/aryl halide, NaOH, ethanol, stirring, 10 h. | |
Table 10 Anti-microbial test results of the target compounds
| Compounds |
MIC (μM) |
| E. coli |
S. aureus |
P. aeruginosa |
K. pneumoniae |
C. albicans |
Antitubercular activity |
| 211 |
1.56 |
1.56 |
6.25 |
100 |
12.5 |
9.50 |
| 213 |
12.5 |
6.25 |
6.25 |
100 |
12.5 |
1.60 |
| 216 |
1.56 |
1.56 |
6.25 |
100 |
12.5 |
6.50 |
| 217 |
1.56 |
1.56 |
6.25 |
100 |
1.56 |
9.00 |
| 218 |
1.56 |
1.56 |
6.25 |
100 |
1.56 |
3.50 |
| 227 |
3.125 |
12.5 |
12.5 |
100 |
1.56 |
1.50 |
2.4 Anti-viral (HCV) activity
Hepatitis C is a virus hepatitis caused by a hepatitis C virus (HCV) infection, mainly as a result of blood transfusion, acupuncture, drug abuse and other transmissions.210 According to World Health Organization statistics, the global infection rate of HCV is around 3%, and there are an estimated 180 million people infected with HCV, with a 35000 rise in new cases of hepatitis C each year. The mortality rate associated with HCV infection will continue to increase in the next 20 years, which is extremely harmful to the health and life of patients, making it one of the most serious social and public health problems.211 Anti-viral drugs that act directly on the replication cycle of the HCV virus have been developed to a certain extent, offering significant promise for treatment and improvement in patients with HCV infection. At present, the anti-viral drugs that can directly act include non-structural protease 3/4A inhibitors, non-structural 5A complex inhibitors and non-structural 5B polymerase inhibitors. These anti-HCV drugs can act on different viral targets.212,213 At each stage of virus replication, the complementary strategy to directly target viral proteins is to target host cytokines required for the virus life cycle. Targeted host therapy includes inhibition of virus entry, translation, replication, assembly and release. In recent years, it has been found that analogues of marine alkaloid globulin can inhibit the replication of HCV by interacting with cellular chaperone heat shock protein 90 (Hsp90). This compound can bind to its ATP binding site through the N-terminal domain. HSP90 is one of the most common protein types in cytoplasm and plays an important role in eukaryotic cells.214 Almost all viruses need the presence of Hsp90 to successfully spread. Hsp90 inhibitors have been studied in clinical trials for more than 20 years due to their beneficial role in the cell life cycle.215–217 However, the clinical development of Hsp90 inhibitors is also hindered to some extent, mainly because of the untargeted effect of Hsp90 inhibitors and the lack of sufficient specificity and mechanism of action. Previous studies have shown that HCV replication could be inhibited by targeting the host cell protein Hsp90. Preliminary work Lillsunde et al.218 found that compounds with a pyrrolamide and 4,5,6,7-tetrahydrobenzo[1,2-d] thiazole structure could inhibit the replication of HCV type 1b. It was found that compounds containing the unsubstituted 2-amino group had the most effective biological activity in terms of drug SAR. Therefore, they further introduced different substituents on the lead 2-amino compound to change its biological activity. They designed a series of 4,5,6,7-tetrahydrobenzo[1,2-d]thiazole-2-amines with the 2-amino group replaced by a basic group or an N-heterocyclic ring to increase binding to Hsp90, thereby enhancing the inhibition of HCV replication by the target compounds 229–240. In the process of synthesizing the target compounds, they used the route shown in Scheme 11, in which two steps were used to complete the preparation of the desired target compounds, and compound 228 was used as the starting material. The first step in the reaction, the acylation of compound 228 with the corresponding carboxylic acids, 1-ethyl-3-(3-dimethylaminopropyl) carbon diimide (EDC) and 1-hydroxy benzyl triazole (HOBt) as a catalyst, and N,N-dimethylformamide (DMF) as a reaction solvent, the preparation gave the required amide structures needed for the target compounds 229–237. Since some of these compounds contained the Boc protection group in their molecular structure, the second step was performed in order to obtain the compounds with the protective group. In the second reaction, the compounds 230–232 and 236 were placed in acetyl chloride to protect them, and finally the target compounds 237–240 were obtained. Twelve target compounds were prepared according to the synthesis route, showing that it has good universality. The synthetic route is relatively short and relatively easy to use. However, a major disadvantage of this route is that the yield of the reactions was generally low, except for the yields of target compounds 237 and 238, which were high. The yield of the second deprotection reaction was relatively high, while the yield of the first step amide reaction was relatively low, which would bring great difficulties for the future industrialization of this process. Therefore, in the subsequent process optimization, the process should be further improved to increase the yield of the reaction and reduce the synthesis cost. During the biological activity study, a HCV replication cell model was used to assess the anti-viral activity of the marine alkaloid analogue compounds 229–240. The target compounds 229–240 were first tested in a HCV genotype 1b replicator model to preliminarily evaluate the anti-viral activity of the target compounds in vitro. In the initial test results, the inhibition rates of the target compounds 229, 231 and 237–240 on HCV replicons exceeded 90%, and the inhibitory activities of the target compounds 230 and 233–236 were low, ranging from 30% to 87%, while the target compound 232 showed no inhibition. It was found from the SAR that the anti-viral activities of the target compounds containing chain substituents were better than those containing rings. The alkyl chain of the compounds containing the chain substituents was increased from one to two carbon atoms, and it was noted that the anti-viral activity was improved. At the same time, the anti-viral activity of the five-membered heterocyclic substituent was higher than that of the six-membered heterocyclic substituent, and the anti-viral activity of the six-membered heterocyclic azacyclic was higher than that of the benzene ring. Based on in vitro tests, the target compounds 229, 233, 234 and 237–240 were further studied to determine the dose-response (IC50) and cytotoxicity (CC50) of the target compounds to the HCV genotype 1b replication subsystem (Table 11). The results showed that all the selected target compounds inhibited the HCV replicons in a dose-dependent manner and had no toxicity at the replicon-inhibiting concentration. At the same time, the SI results showed that the selectivities of the target compounds 237, 229 and 239 were relatively high, with SI values of 98, 62 and 40, respectively. The selectivities of these target compounds were higher than that of the positive control. To determine whether the target compounds were also effective in inhibiting full-length virus replication, the target compounds were evaluated for their ability to inhibit HCV genotype 2a replicons. The results showed that the target compounds 229, 237 and 239 had anti-viral activity against HCV, with IC50 values ranging from 0.49 to 1.52 μM, while other target compounds had no inhibitory activity. The inhibitory activity of the target compounds 229 and 237 against genotype 2a was even slightly higher than that against genotype 1b. HCV transmission in hepatocytes could be interacted with cellular partners in a variety of ways. Hsp90 could assist in protein maturation, folding, and stabilization and it was used for the replication of HCV. Relevant studies have shown that the target Hsp90 could effectively inhibit the replication of HCV. In order to evaluate the inhibition of Hsp90 as a potential mechanism of action, the binding ability of the target compounds to Hsp90 was evaluated by microscale thermophoresis (MST). The results of the evaluation indicate that Hsp90 is indeed a potential target for the target compounds and may provide important guidance for the further development of anti-viral drugs targeting the progenitor of this cell. The replication and infectivity of different
viruses is the key to the successful development of anti-viral Hsp90 inhibitors. In general, compound 237 has good anti-viral activity and high selectivity, which needs to be further studied.
 |
| | Scheme 11 Synthesis of 4,5,6,7-tetrahydrobenzo [1,2-d] thiazole derivative compounds 229–240. Reagents and conditions: (a) corresponding carboxylic acids, EDC, HOBt, NMM, DMF, r.t., 18 h; (b) acetyl chloride, MeOH, 0 °C, 1 h, then r.t., 18 h. | |
Table 11 Replicon inhibitory activities of the target compounds and control in HCV genotype 1b and 2a replicon models
| Compounds |
HCV replicon genotype |
| 1b IC50 (μM) CC50 (μM) IS |
2a IC50 (μM) |
| 229 |
2.5 ± 0.25 |
154 ± 2.9 |
62 |
1.52 ± 0.15 |
| 233 |
5.5 ± 1.8 |
204 ± 4.9 |
37 |
nd |
| 234 |
21 ± 2.2 |
86 ± 5.2 |
4.1 |
nd |
| 237 |
1.3 ± 0.23 |
128 ± 6.6 |
98 |
0.49 ± 0.06 |
| 238 |
22 ± 2.8 |
67 ± 2.0 |
3.0 |
nd |
| 239 |
1.2 ± 0.41 |
48 ± 2.5 |
40 |
1.32 ± 0.1 |
| 240 |
7.2 ± 0.46 |
72 ± 4.2 |
10 |
nd |
| 17-DMAG |
0.06 ± 0.01 |
1.7 ± 0.25 |
28 |
0.04 ± 0.006 |
| Ribavirin |
64 ± 7.0 |
>300 |
nd |
nd |
2.5 Other types of activity
Natural products from the sea have been paid much attention, especially the compounds with rich biological activity separated from marine sponges.219 In the process of activity screening, the diversity biological activity of these compounds was observed, and these marine natural products were also found to have diverse chemical structures.220 The abundant metabolites produced by marine organisms offer a promising contribution to the discovery of new drugs for the treatment of diseases, or to the development of lead compounds for structural modification. Among these marine natural products, a class of marine alkaloids containing 2-aminoidazolidone structures have been widely studied.221 This chemical structure is an attractive structure with the potential for further structural modification, which can be used as a starting point for the development and research of similar drugs with diverse chemical structures biological activities. These target compounds modified on the basis of the 2-aminoidazolidone structure constitute a huge candidate drug screening library, which can be fully researched and developed.222,223 Dispacamide is a marine alkaloid with anti-histamine activity. Polyandrocarpamine A (7) is selectively cytotoxicity to SF268 cell line (central nervous system) with a GI50 value of 65 μM. These marine alkaloids obtained from marine organisms were found to contain a 2-aminoidazolone structure by chemical analysis. At the same time, leucettamine B (9) is also a marine alkaloid separated from marine sponges, which has been obtained by chemical synthesis224 However, no biological activity related to this substance has been reported so far. These marine alkaloids containing 2-aminoidazolidone structures have been found to have abundant biological activity in activity studies. Based on previous work, Debdab et al.225 designed derivatives of the marine alkaloid leucettamine B. From the chemical structure of the designed target compounds 253–260, it could be seen that these target compounds are derivatives of N-functionalized leucettamine B, in which aryl methyl groups are linked to the 2-amino structure via a short amino-alkyl chain. During the synthesis of the target compounds, they selected the method described in Scheme 12 to synthesis the marine alkaloid Lucettamine B derivatives. The target compounds were synthesized in five steps, the starting material with commercial ethylenediamine (compound 241). The first step was to prepare a single N-Boc protected diamine (compound 242) in methanol with ethylenediamine and di-tert-butyldicarbonate according to a method in the literature, in a yield of 65%. The second step of the reaction is the key step to compounds 242 and (5Z) 5-[(1,3-benzodioxo-5-yl) methylene]-3-methyl-2-ethylsulfanyl-3,5-dihydroimidazol-4-one with a microwave irradiation synthesis used to generate the intermediate compound 243 (yield 69%). The reaction was carried out for 2 h at 105 °C to achieve good conversion. In the third step, the protection group of compound 243 was removed, meaning that the N-Boc group was cracked. Compound 243 was treated with acetyl chloride and methanol in acetonitrile at room temperature, and a high yield of compound 244 (yield 98%) could be obtained by effective deprotection. In the fourth step, compound 244 was reacted with substituted benzaldehyde in methanol, and triethanolamine was used as the catalyst for the reaction. Compounds 245–252 were obtained after reaction at 50 °C for 24 h, reaching yields of 63–81%. In the final step, compounds 245–252 were reacted with NaBH4 in methanol at 50 °C for 24 h to obtain the target compounds 253–260 (yield 72–97%). According to this method, the target compounds 253–260 were prepared in five steps with a total yield of 24% to 31%. This synthesis route is relatively simple to use, the reagents used are cheap and easy to obtain, and the yield is moderate to good, laying a foundation for its future industrialization. At the same time, the new method of microwave irradiation synthesis used in this synthesis route greatly reduced the reaction time and increased the reaction yield. During the biological activity study, the group tested the target compounds 253–260, intermediate compound 244 and 245–252 in vitro for three protein kinases associated with Alzheimer's disease (Table 12). The protein kinases include casein kinase 1α/β (CK1α/β), cyclin-dependent kinase 5 (CDK5)/p25 and glycogen synthase kinase 3α/β (GSK-3α/β). In vitro biological activity test results showed that some marine alkaloid lucettamine B analogues have moderate to strong inhibitory activity of GSK-3α/β, with IC50 values of between 0.86 and 38 μM. The intermediate compound 246 with an IC50 of 0.86 μM, which inhibited GSK-3α/β, was the most active of the compounds tested. These compounds showed poor CK1α/β and CDK5/p25 inhibitory activity compared with GSK-3α/β inhibitory activity; obviously the good selective inhibition of GSK-3α/β exists. The target compounds 253–257 were product obtained from the addition reaction on the basis of the intermediate compounds 245–249, but it was found in the activity test results that with the addition of the imine group, the inhibitory activity on GSK-3α/β was lost. After the comparison of the activity test results of compounds 253–257, a drug SAR was established. The results of SAR showed that the types of substituents and the positions of the substituents on the phenyl groups had certain effects on the biological activity. At the same time, the intermediate compound 244 showed high inhibitory activity to GSK-3α/β, with an IC50 of 1.4 μM, and a good selective inhibitory effect. The amino group in the aminoalkyl chain of compound 244 was replaced, and it retained good inhibitory activity, meaning that it can be further modified as a skeleton structure (the lead compound) for the discovery of novel selective GSK-3α/β inhibitors. In general, according to the available data, compound 246 could be used as a selective GSK-3α/β inhibitor for further in vivo activity evaluation and pharmacokinetic property studies.
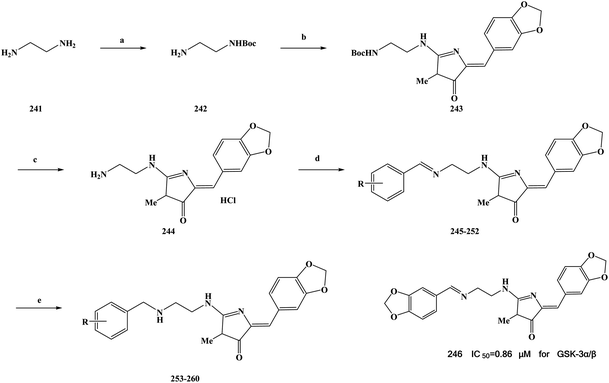 |
| | Scheme 12 Synthesis of the leucettamine B analogue compounds 253–260. Reagents and conditions: (a) (Boc)2O, MeOH, 0 °C, 10 min, then 25 °C, 1 h; (b) 105 °C, 2 h; (c) MeCOCl, MeOH, MeCN, 25 °C, 24 h; (d) Et3N, MeOH, 50 °C, 24 h; (e) NaBH4, MeOH, 50 °C, 24 h. | |
Table 12 The kinases inhibition values
| Compounds |
IC50 (μM) |
| CK1 |
CDK5/p25 |
GSK-3α/β |
| 244 |
>100 |
>100 |
1.4 |
| 245 |
>100 |
>100 |
7.0 |
| 246 |
>100 |
>100 |
0.86 |
| 247 |
>100 |
>100 |
15.0 |
| 248 |
>100 |
>100 |
38.0 |
| 249 |
>100 |
>100 |
7.3 |
| 253 |
>100 |
>100 |
15.0 |
| 254 |
>100 |
>100 |
>10 |
| 255 |
>100 |
>100 |
>10 |
| 256 |
>100 |
>100 |
>10 |
| 257 |
>100 |
>100 |
>10 |
| Leucettamine B |
>100 |
>100 |
>10 |
3. Conclusions and prospects
Many types of compounds with biological activity have been extracted and separated from marine organisms, with marine alkaloids being some of the earliest compounds studied. In vitro or in vivo biological activity screening of marine alkaloids has shown that these compounds have diverse biological activities, including anti-tumor, anti-fungal, anti-viral, anti-malaria, anti-osteoporosis, and other biological activities, with the anti-tumor activity being studied in depth. With the increasing interest in the research of marine drugs, marine alkaloids have a promising future and may be further converted into anti-tumor, anti-viral, and anti-fungal clinical drugs or lead compounds. The amounts of these compounds that can be separated are limited, and it is easy to damage the marine organism environment. In addition, these lead compounds have the disadvantages of high cytotoxicity and low selectivity, which limit their clinical research potential and their industrial development. Therefore, marine alkaloid derivatives or analogues with new structures have been designed by applying modern drug design concepts to the structural modification of lead compounds, and these compounds were obtained via efficient classical or modern methods of chemical synthesis. In the activity screening, these marine alkaloid derivatives or analogues also show biological activity diversity and could make up for the shortcomings of natural marine alkaloids. This has become a focus of research. This work systematically reviewed the recent advances in marine alkaloid derivatives and analogues in the field of medical chemistry over the last 10 years (2010–2019). In the review process, we divided marine alkaloid derivatives and analogues into five types from the perspective of biological activity, including research into anti-tumor, anti-malarial, anti-bacterial, anti-viral (HCV), and other types, and specifically elaborated on these activities. We discussed the optimization process, chemical synthesis, biological activity evaluation and SAR of each type of compound. On this basis, we evaluated the synthesis route of each type of compounds, so as to provide a necessary reference for the future optimization of these processes. The SAR may provide a reasonable approach for the design and development of novel marine alkaloid derivatives or analogues. It is an important way to obtain lead compounds from marine alkaloids and it is very promising to obtain a large number of derivatives and analogues via structural modification. These derivatives and analogues have diverse biological activity, which, combined with the SAR, provides a good research platform for the discovery of new drugs in the future. Researchers should take advantage of the complex and special chemical structures and diverse biological activities of natural marine alkaloids. We should use modern drug design and synthesis methods to obtain more derivatives and analogues for activity screening and clinical research. As a result, the earlier these marine alkaloid derivatives and analogues enter the market, the better they will serve the public to prevent and treat diseases.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The Project was Sponsored by the Postgraduate Research and Innovation Project of Hainan Normal University (Hsyx2018-8) and the Open Foundation Project of Key Laboratory of Tropical Medicinal Resource Chemistry of Ministry of Education (RDZH2019002). The work was also supported by the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry (No. 2015-1098), the Chongqing Key Research Project of Basic Science & Frontier Technology (No. cstc2017jcyjBX0012), the Foundation Project of Chongqing Normal University (No. 14XYY020), the Chongqing General Research Program of Basic Research and Frontier Technology (No. cstc2015jcyjA10054), and the Chongqing Normal University Postgraduate's Research and Innovation Project (No. YKC17004), China.
References
- T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Nat. Chem., 2016, 8, 531–541 CrossRef CAS PubMed.
- S. Z. Wang, G. Q. Dong and C. Q. Sheng, Chem. Rev., 2019, 119, 4180–4220 CrossRef CAS PubMed.
- J. Kobayashi, Chem. Pharm. Bull., 2016, 64, 1079–1083 CrossRef CAS PubMed.
- W. B. Wu, Y. Tang, J. L. Yang, E. Idehen and S. M. Sang, J. Agric. Food Chem., 2018, 66, 8005–8014 CrossRef CAS PubMed.
- Y. G. Han, Y. Luo, S. R. Qin, L. Xi, B. Wan and L. F. Du, Pestic. Biochem. Physiol., 2014, 111, 14–18 CrossRef CAS PubMed.
- M. G. Ciulla and K. Kumar, Tetrahedron Lett., 2018, 59, 3223–3233 CrossRef CAS.
- M. J. Martinez, L. M. Olmo and P. B. Benito, Stud. Nat. Prod. Chem., 2015, 30, 393–418 Search PubMed.
- M. M. Gendy, M. Shaaban, K. A. Shaaban, A. M. Bondkly and H. Laatsch, J. Antibiot., 2008, 61, 149–157 CrossRef PubMed.
- U. Battaglia and C. J. Moody, J. Nat. Prod., 2010, 73, 1938–1939 CrossRef CAS PubMed.
- L. H. Stoodley, J. W. Costerton and P. Stoodley, Nat. Rev. Microbiol., 2004, 2, 95–108 CrossRef PubMed.
- E. Tee, T. Karoli, S. Ramu, J. Huang, M. Butler and M. Cooper, J. Nat. Prod., 2010, 73, 1940–1942 CrossRef CAS.
- E. S. Zuniga, A. Korkegian, S. Mullen, E. J. Hembre, P. L. Ornstein, G. Cortez, K. Biswas, N. Kumar, J. Cramer, T. Masquelin, P. A. Hipskind, J. Odingo and T. Parish, Bioorg. Med. Chem., 2017, 25, 3922–3946 CrossRef CAS PubMed.
- J. J. Richards and C. Melander, ChemBioChem, 2010, 10, 2287–2294 CrossRef PubMed.
- W. B. Parker, Virus Res., 2005, 107, 165–171 CrossRef CAS PubMed.
- P. Lv, Y. L. Chen, T. Z. Shi, X. W. Wu, Q. X. Li and R. M. Hua, Pestic. Biochem. Physiol., 2018, 147, 3–10 CrossRef CAS PubMed.
- X. Y. Li, G. F. Hao, Q. M. Wang, Z. Chen, Y. Ding, L. Yu, D. Y. Hu and B. A. Song, Oncotarget, 2017, 8, 82446–82458 CrossRef PubMed.
- T. L. Simmons, E. Andrianasolo, K. M. Phail, P. Flatt and W. H. Gerwick, Mol. Cancer Ther, 2005, 2005(4), 333–342 Search PubMed.
- C. Cuevas and A. Francesch, Nat. Prod. Rep., 2009, 26, 322–337 RSC.
- R. J. Worthington, J. J. Richards and C. Melander, Org. Biomol. Chem., 2012, 10, 7457–7474 RSC.
- B. J. Petek, E. T. Loggers, S. M. Pollack and R. L. Jones, Mar. Drugs, 2015, 13, 974–983 CrossRef CAS PubMed.
- F. Mascilini, G. Amadio, M. G. Stefano, M. Ludovisi, A. D. Legge, C. Conte, R. D. Vincenzo, C. Ricci, V. Masciullo, V. Salutari, G. Scambia and G. Ferrandina, OncoTargets Ther., 2014, 7, 1273–1284 Search PubMed.
- E. Adrian, Curr. Pharm. Des., 2007, 13, 3417–3426 CrossRef PubMed.
- B. Haefner, Drug Discovery Today, 2003, 8, 536–544 CrossRef CAS PubMed.
- K. B. Glaser and A. M. Mayer, Biochem. Pharmacol., 2009, 78, 440–448 CrossRef CAS PubMed.
- R. A. Davis, L. V. Christensen, A. D. Richardson, R. M. Rocha Da and C. M. Ireland, Mar. Drugs, 2003, 1, 27–33 CrossRef CAS.
- T. F. Molinski, D. S. Dalisay, S. L. Lievens and J. P. Saludes, Nat. Rev. Drug Discovery, 2008, 8, 69–85 CrossRef.
- G. Schwartsmann, A. B. Rocha, J. Mattei and R. Lopes, Expert Opin. Invest. Drugs, 2003, 12, 1367–1383 CrossRef CAS PubMed.
- R. Singh, M. Sharma, P. Joshi and D. S. Rawat., Anti-Cancer Agents in Medicinal Chemistry, 2008, 8, 603–617 CrossRef CAS PubMed.
- J. W. Blunt, B. R. Copp, M. H. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2006, 23, 26–78 RSC.
- G. B. Sela, R. Epelbaum and M. Schaffer, Curr. Med. Chem., 2010, 17, 190–197 CrossRef PubMed.
- G. Kuznetsov, K. T. Dyke, M. J. Towle, H. Cheng, J. Liu, J. P. Marsh, S. E. Schiller, M. R. Spyvee, H. Yang, B. M. Seletsky, C. J. Shaffer, V. Marceau, Y. Yao, E. M. Suh, S. Campagna, F. G. Fang, J. J. Kowalczyk and B. A. Littlefield, Mol. Cancer Ther., 2009, 8, 2852–2860 CrossRef CAS PubMed.
- E. A. Guzman, J. D. Johnson, M. K. Carrier, C. I. Meyer, T. P. Pitts, S. P. Gunasekera and A. E. Wright, Anti-Cancer Drugs, 2009, 20, 149–155 CrossRef CAS PubMed.
- S. Deslandes, S. Chassaing and E. Delfourne, Mar. Drugs, 2009, 7, 754–786 CrossRef CAS PubMed.
- R. Dasari and A. Kornienko, Chem. Heterocycl. Compd., 2014, 50, 139–144 CrossRef CAS.
- D. V. La-Barbera, K. Modzelewska, A. I. Glazar, P. D. Gray, M. Kaur, T. Liu, D. Grossman, M. K. Harper, S. K. Kuwada, N. Moghal and C. M. Ireland, Anti-Cancer Drugs, 2009, 20, 425–436 CrossRef CAS.
- R. Campana, L. Casettari, L. Fagioli, M. Cespi, G. Bonacucina and W. Baffone, Int. J. Food Microbiol., 2017, 241, 132–140 CrossRef PubMed.
- F. M. Ranaivoson, B. Gigant, S. Berritt, M. Joullie and M. Knossow, Acta Crystallogr., 2012, 68, 927–934 CrossRef CAS PubMed.
- E. Hamel, Cell Biochem. Biophys., 2003, 38, 1–22 CrossRef CAS.
- J. Kluza, M. A. Gallego, A. Loyens, J. C. Beauvillain, J. M. Sousa-Faro, C. Cuevas, P. Marchetti and C. Bailly, Cancer Res., 2006, 66, 3177–3187 CrossRef CAS PubMed.
- C. Ballot, J. Kluza, S. Lancel, A. Martoriati, S. M. Hassoun, L. Mortier, J. C. Vienne, G. Briand, P. Formstecher, C. Bailly, R. Neviere and P. Marchetti, Apoptosis, 2010, 15, 769–781 CrossRef CAS.
- J. Sun, J. Wu, B. An, N. J. Voogd, W. Cheng and W. Lin, Mar. Drugs, 2018, 16, 1–15 CrossRef PubMed.
- K. Vermeulen, D. R. Bockstaele and Z. N. Berneman, Cell Proliferation, 2003, 36, 131–149 CrossRef CAS.
- A. R. Cuddihy and M. J. Connell, Int. Rev. Cytol., 2003, 222, 99–140 CrossRef.
- C. H. Gowan, Mutat. Res., 2003, 532, 75–84 CrossRef PubMed.
- R. G. Berlinck, R. Britton, E. Piers, L. Lim, M. Roberge, R. Moreira da Rocha and R. Andersen, J. Org. Chem., 1998, 63, 9850–9856 CrossRef CAS.
- X. Jiang, B. Zhao, R. Britton, L. Y. Lim, D. Leong, J. S. Sanghera, B. B. Zhou, E. Piers, R. J. Andersen and M. Roberge, Mol. Cancer Ther., 2004, 3, 1221–1227 CAS.
- R. Boutros, V. Lobjois and B. Ducommun, Nat. Rev. Cancer, 2007, 7, 495–507 CrossRef CAS PubMed.
- S. Deslandes, S. Chassaing and E. Delfourne, Mar. Drugs, 2009, 7, 754–786 CrossRef CAS PubMed.
- B. Hugon, B. Pfeiffer, P. Renard and M. Prudhomme, Tetrahedron Lett., 2003, 44, 3927–3930 CrossRef CAS.
- B. Hugon, F. Anizon, C. Bailly, R. M. Golsteyn, A. Pierré, S. Léonce, J. Hickman, B. Pfeiffer and M. Prudhomme, Bioorg. Med. Chem., 2007, 15, 5965–5980 CrossRef CAS PubMed.
- E. Piers, R. Britton and R. J. Andersen, Improved synthesis of isogranulatimide, a G2 checkpoint inhibitor, J. Org. Chem., 2000, 65, 530–535 CrossRef CAS PubMed.
- M. Contour-Galcéra, A. Sidhu, P. Plas and P. Roubert, ACS Med. Chem. Lett., 2005, 15, 3555–3559 CrossRef PubMed.
- S. Deslandes, D. Lamoral-Theys, C. Frongia, S. Chassaing, C. Bruyère, O. Lozach, L. Meijer, B. Ducommun, R. Kiss and E. Delfourne, Eur. J. Med. Chem., 2012, 54, 626–636 CrossRef CAS PubMed.
- D. J. Faulkner, Nat. Prod. Rep., 2002, 19, 1–48 CAS.
- A. Casapullo, G. Bifulco, I. Bruno and R. Riccio, J. Nat. Prod., 2000, 63, 447–451 CrossRef CAS PubMed.
- B. Bao, Q. Sun, X. Yao, J. Hong, C. Lee, C. J. Sim and J. H. Jung, J. Nat. Prod., 2005, 68, 711–715 CrossRef CAS.
- W. Gul and M. T. Hamann, Life Sci., 2005, 78, 442–453 CrossRef CAS PubMed.
- H. R. Bokesch, L. K. Pannell, T. C. McKee and M. R. Boyd, Tetrahedron Lett., 2000, 41, 6305–6308 CrossRef CAS.
- C. J. Moody and J. R. Roffey, Arkivoc, 2002, 1, 393–401 Search PubMed.
- F. Y. Miyake, K. Yakushijin and D. AHorne, Org. Lett., 2000, 2, 2121–2123 CrossRef CAS PubMed.
- P. M. Fresneda, P. Molina and M. ASanz, Synlett, 2001, 2, 218–221 CrossRef.
- B. Jiang and X. H. Gu, Bioorg. Med. Chem., 2008, 8, 363–371 CrossRef.
- B. Jiang, C. G. Yang, C. Xiong and J. Wang, Bioorg. Med. Chem., 2001, 9, 1149–1154 CrossRef CAS.
- B. Jiang, W. N. Xiong and C. G. Yang, Bioorg. Med. Chem. Lett., 2011, 11, 475–477 CrossRef.
- W. N. Xiong, C. G. Yang and B. Jiang, Bioorg. Med. Chem., 2001, 9, 1773–1780 CrossRef CAS PubMed.
- P. Diana, A. Carbone, P. Barraja, A. Montalbano, A. Martorana, G. Dattolo, O. Gia, L. D. Via and G. Cirrincione, Bioorg. Med. Chem. Lett., 2007, 17, 2342–2346 CrossRef CAS PubMed.
- P. Diana, A. Carbone, P. Barraja, A. Martorana, O. Gia, L. D. Via and G. Cirrincione, Bioorg. Med. Chem. Lett., 2007, 17, 6134–6137 CrossRef CAS PubMed.
- P. Diana, A. Carbone, P. Barraja, H. H. Kelter, G. Fiebig and G. Cirrincione, Bioorg. Med. Chem., 2010, 18, 4524–4529 CrossRef CAS PubMed.
- D. Kumar, N. M. Kumar, K. H. Chang, R. Gupta and K. Shah, Bioorg. Med. Chem. Lett., 2011, 21, 5897–5900 CrossRef CAS PubMed.
- A. Carbone, B. Parrino, G. D. Vita, A. Attanzio, V. Spanò, A. Montalbano, P. Barraja, L. Tesoriere, M. A. Livrea, P. Diana and G. Cirrincione, Mar. Drugs, 2015, 13, 460–492 CrossRef PubMed.
- B. Parrino, A. Carbone, G. D. Vita, C. Ciancimino, A. Attanzio, V. Spanò, A. Montalbano, P. Barraja, L. Tesoriere, M. A. Livrea, P. Diana and G. Cirrincione, Mar. Drugs, 2015, 13, 1901–1924 CrossRef CAS PubMed.
- A. Carbone, B. Parrino, P. Barraja, V. Spanò, G. Cirrincione, P. Diana, A. Maier, G. Kelter and H. H. Fiebig, Mar. Drugs, 2013, 11, 643–654 CrossRef CAS PubMed.
- P. Vineis and C. P. Wild, Lancet, 2014, 383, 549–557 CrossRef.
- W. R. Sawadogo, M. Schumacher, M. H. Teiten, C. Cerella, M. Dicato and M. Diederich, Molecules, 2013, 18, 3641–3673 CrossRef CAS PubMed.
- I. Bhatnagar and S. K. Kim, Mar. Drugs, 2010, 8, 2702–2720 CrossRef CAS PubMed.
- D. Sipkema, M. C. Franssen, R. Osinga, J. Tramper and R. H. Wijffels, Mar. Biotechnol., 2005, 7, 142–162 CrossRef CAS PubMed.
- A. Carbone, M. Pennati, B. Parrino, A. Lopergolo, P. Barraja, A. Montalbano, V. Spanò, S. Sbarra, V. Doldi and M. Cesare, J. Med. Chem., 2013, 56, 7060–7072 CrossRef CAS.
- A. Carbone, M. Pennati, P. Barraja, A. Montalbano, B. Parrino, V. Spanò, A. Lopergolo, S. Sbarra, V. Doldi and N. Zaffaronil, Curr. Med. Chem., 2014, 21, 1654–1666 CrossRef CAS PubMed.
- V. Stonik and S. Fedorov, Mar. Drugs, 2014, 12, 636–671 CrossRef PubMed.
- D. T. Youssef, L. AShaala, G. AMohamed, J. M. Badr, F. H. Bamanie and S. R. Ibrahim, Mar. Drugs, 2014, 12, 1911–1923 CrossRef PubMed.
- J. R. Pereira, F. F. Hilário, A. B. Lima, M. L. Silveira, L. M. Silva, R. BAlves, R. P. Freitas and F. P. Varotti, Prev. Nutr., 2012, 2, 145–148 CrossRef.
- P. AMachado, F. F. Hilário, L. O. Carvalho, M. L. Silveira, R. B. Alves, R. P. Freitas and E. S. Coimbra, Chem. Biol. Drug Des., 2012, 80, 745–751 CrossRef PubMed.
- G. Iannolo, C. Conticello, L. Memeo and R. Maria, Critical Reviews in Oncology/Hematology, 2008, 66, 42–51 CrossRef PubMed.
- D. Rossi and G. Gaidano, Haematologica, 2003, 88, 212–218 CAS.
- I. Decordier, L. Dillen, E. Cundari and M. K. Volders, Mutagenesis, 2002, 17, 337–344 CrossRef CAS PubMed.
- I. Decordier, E. Cundari and M. K. Volders, Mutagenesis, 2005, 20, 173–179 CrossRef CAS PubMed.
- A. M. Gonçalves, A. B. Lima, M. C. Barbosa, L. F. Camargos, J. T. Oliveira, C. S. Barbosa, J. A. Villar, A. C. Costa, I. V. Silva, L. M. Silva, F. P. Varotti, F. V. Santos and G. H. Viana, Mar. Drugs, 2014, 12, 4361–4378 CrossRef PubMed.
- K. H. Khan, M. B. Codesido and L. R. Molife, Critical Reviews in Oncology/Hematology, 2013, 90, 200–219 CrossRef PubMed.
- M. K. Volders, T. Sofuni, M. Aardema, S. Albertini, D. Eastmond, M. Fenech, M. Ishidate, S. Kirchner, E. Lorge and T. Morita, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2003, 540, 153–163 CrossRef PubMed.
- R. A. Hill, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2005, 101, 124–136 RSC.
- Y. Nakao and N. Fusetani, J. Nat. Prod., 2007, 2007(70), 689–710 CrossRef PubMed.
- M. Chittchang, P. Batsomboon, S. Ruchirawat and P. Ploypradith, ChemMedChem, 2009, 4, 457–465 CrossRef CAS PubMed.
- S. Mabuchi, T. Hisamatsu, C. Kawase, M. Hayashi, K. Sawada, K. Mimura, K. Takahashi, T. Takahashi, H. Kurachi and T. Kimura, Clin. Cancer Res., 2011, 17, 4462–4473 CrossRef CAS PubMed.
- M. Sugumaran and W. E. Robinson, Mar. Drugs, 2010, 8, 2906–2935 CrossRef CAS PubMed.
- H. Fan, J. Peng, M. T. Hamann and J. F. Hu, Chem. Rev., 2008, 108, 264–287 CrossRef CAS PubMed.
- M. Tsuda, K. Nozawa, K. Shimbo and J. Kobayashi, J. Nat. Prod., 2003, 66, 292–294 CrossRef CAS PubMed.
- L. V. Frolova, I. V. Magedov, A. E. Romero, M. Karki, I. Otero, K. Hayden, N. M. Evdokimov, L. M. Y. Banuls, S. K. Rastogi, W. R. Smith, S. L. Lu, R. Kiss, C. B. Shuster, E. Hamel, T. Betancourt, S. Rogelj and A. Kornienko, J. Med. Chem., 2013, 56, 6886–6990 CrossRef CAS.
- R. Scott, M. M. R. Karki, M. R. Reisenauer, R. Rodrigues, R. Dasari, W. R. Smith, S. C. Pelly, W. A. Otterlo, C. B. Shuster, S. Rogelj, I. V. Magedov, L. V. Frolova and A. Kornienko, ChemMedChem, 2014, 9, 1428–1435 CrossRef CAS.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS PubMed.
- K. H. Attmann and J. Gertsch, Nat. Prod. Rep., 2007, 24, 327–357 RSC.
- M. F. Mehbub, J. Lei, C. Franco and W. Zhang, Mar. Drugs, 2014, 12, 4539–4577 CrossRef PubMed.
- S. Nag, D. H. Nadkarni, J. J. Qin, S. Voruganti, T. Nguyen, S. Xu, W. Wang, H. Wang, S. E. Velu and R. Zhang, Mol. Cell. Pharmacol., 2012, 4, 69–81 CAS.
- F. M. Deane, E. C. Sullivan, A. R. Maguire, J. Gilbert, J. A. Sakoff, A. M. Cluskey and F. O. Carthy, Org. Biomol. Chem., 2011, 11, 1334–1344 RSC.
- G. Lang, A. Pinkert, J. W. Blunt and M. H. Munro, J. Nat. Prod., 2005, 68, 1796–1798 CrossRef CAS PubMed.
- J. Ponder, B. H. Yoo, A. D. Abraham, Q. Li, A. K. Ashley, C. L. Amerin, Q. Zhou, B. G. Reid, P. Reigan and R. Hromas, Mar. Drugs, 2011, 9, 2397–2408 CrossRef CAS PubMed.
- L. M. West, P. T. Northcote and C. N. Battershill, J. Org. Chem., 2000, 65, 445–449 CrossRef CAS PubMed.
- S. Boucle, C. Melin, M. Clastre and J. Guillard, Mar. Drugs, 2001, 513, 655–665 Search PubMed.
- A. Martins, H. Vieira, H. Gaspar and S. Santos, Mar. Drugs, 2014, 12, 1066–1101 CrossRef PubMed.
- M. Köck, J. Muñoz, C. Cychon, C. Timm and G. Schmidt, Phytochem. Rev., 2013, 12, 391–406 CrossRef.
- J. R. Pereira, F. F. Hilário, A. B. Lima, M. L. Silveira, L. M. Silva, R. B. Alves, R. P. De Freitas, F. P. Varotti and G. H. Viana, Biomedicine & Preventive Nutrition, 2012, 2, 145–148 Search PubMed.
- R. R. Soares, J. M. Silva, B. C. Carlos, C. C. Fonseca, L. S. Souza, F. V. Lopes, R. M. Paula Dias, P. O. Moreira, C. Abramo, G. H. Viana, F. P. Varotti, A. D. Silva and K. K. Scopel, Bioorg. Med. Chem. Lett., 2015, 25, 2308–2313 CrossRef CAS PubMed.
- I. Leto, M. Coronnello, C. Righeschi, M. C. Bergonzi, E. Mini and A. R. Bilia, ChemMedChem, 2016, 11, 1745–1751 CrossRef CAS PubMed.
- M. Burow, A. Bergner, J. Gershenzon and U. Wittstock, Plant Mol. Biol., 2007, 63, 49–61 CrossRef CAS PubMed.
- R. M. Viana, A. P. Butera, E. S. Santos, C. A. Tischer, R. B. Alves, R. P. Freitas, L. Guimaraes, F. Varotti, G. H. Viana and C. S. Nascimento, J. Chem. Inf. Model., 2016, 56, 571–579 CrossRef PubMed.
- J. Beard, Nature, 2013, 92, 60–61 CrossRef.
- M. Lu, L. Sun, J. Zhou and J. Yang, Tumor Biol., 2014, 35, 5307–5314 CrossRef CAS PubMed.
- M. Fenech, Nat. Protoc., 2007, 2, 1084–1104 CrossRef CAS PubMed.
- J. T. Oliveira, M. C. Barbosa, L. F. Camargos, I. V. Silva, F. P. Varotti, L. M. Silva, L. M. Moreira, J. P. Lyon, V. J. Santos and F. V. Santos, Cytotechnology., 2017, 69, 699–710 CrossRef PubMed.
- E. Elhalem, B. N. Bailey, R. Docampo, S. H. Szajnman and J. B. Rodriguez, J. Med. Chem., 2002, 86, 3984–3999 CrossRef PubMed.
- G. G. Liñares, S. Gismondi, N. O. Codesido, S. N.Moreno, R. Docampo and J. B. Rodriguez, Bioorg. Med. Chem. Lett., 2007, 17, 5068–5071 CrossRef PubMed.
- H. Noedl, C. Wongsrichanalai, R. S. Miller, K. S. Myint, S. Looareesuwan, Y. Sukthana, V. Wongchotigul, H. Kollaritsch, G. Wiedermann and W. H. Wernsdorfer, Exp. Parasitol., 2002, 102, 157–163 CrossRef CAS PubMed.
- R. H. Lee, B. Kim, I. Choi, H. Kim, S. Choi, K. Suh, Y. C. Bae and J. S. Jung, Cell. Physiol. Biochem., 2004, 14, 311–324 CrossRef CAS PubMed.
- F. V. Santos, M. Andreo, A. L. Nasser, L. M. Moreira, W. Vilegas, I. M. Cólus and E. A. Varanda, Arch. Biol. Sci., 2013, 65, 191–195 CrossRef.
- D. M. Cottrell, J. Capers, M. M. Salem, K. DeLuca-Fradley, S. L. Croft and K. A. Werbovetz, Bioorg. Med. Chem., 2004, 12, 2815–2824 CrossRef CAS PubMed.
- C. G. Havens, N. Bryant, L. Asher, L. Lamoreaux, S. Perfetto, J. J. Brendle and K. A. Werbovetz, Mol. Biochem. Parasitol., 2000, 110, 223–236 CrossRef CAS.
- C. C. Gomes, L. M. Moreira, V. J. Santos, A. S. Ramos, J. P. Lyon, C. P. Soares and F. V Santos, Genet. Mol. Biol., 2011, 34, 116–121 CrossRef CAS PubMed.
- P. L. Olive and J. P. Banáth, J. Nat. Prod., 2006, 1, 23–29 CAS.
- A. R. Collins, Mol. Biotechnol., 2004, 26, 249–261 CrossRef CAS PubMed.
- V. S. Gontijo, P. F. Espuri, R. B. Alves, L. F. Camargos, F. V. Dos Santos, W. A. Souza Judice, M. J. Marques and R. P. Freitas, Eur. J. Med. Chem., 2015, 101, 24–33 CrossRef CAS PubMed.
- L. Recio, C. Hobbs, W. Caspary and K. L. Witt, J. Toxicol. Sci., 2010, 35, 149–162 CrossRef CAS PubMed.
- C. Aiub, A. Giannerini, F. Ferreira, J. Mazzei, L. Stankevicins, G. L. Hajdu, P. Guimarães, E. Hajdu and I. Felzenszwalb, Mutat. Res., 2006, 611, 34–41 CAS.
- V. Alcolea, D. Plano, I. Encío, J. A. Palop, A. K. Sharma and C. Sanmartín, Eur. J. Med. Chem., 2016, 123, 407–418 CrossRef CAS.
- R. Abbotts, N. Thompson and S. Madhusudan, Cancer Manage. Res., 2014, 6, 77–92 Search PubMed.
- M. K. Volders, S. Bonassi, S. Knasmueller, N. Holland, C. Bolognesi and M. F. Fenech, Mutat. Res., 2014, 759, 49–58 Search PubMed.
- P. Villani, M. Spanò, F. Pacchierotti, M. Weimer and E. Cordelli, Reprod. Toxicol., 2010, 30, 44–49 CrossRef CAS PubMed.
- L. Stankevicins, C. Aiub, L. C. Santa Maria, G. L. Hajdu and I. Felzenszwalb, Toxicol. In Vitro, 2008, 22, 1869–1877 CrossRef CAS PubMed.
- K. Utani, Y. Kohno, A. Okamoto and N. Shimizu, PLoS One, 2010, 5, 10089 CrossRef PubMed.
- M. C. Barbosa, C. S. Barbosa, J. T. Oliveira, N. C. Moreira, N. R. Martins, G. K. Gomes, C. A. Caldeira, M. L. Costa, D. S. Martins, L. Guimaraes, C. S. Nascimento, F. P. Varotti, G. H. Viana and F. V. Santos, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2018, 825, 31–39 CrossRef CAS PubMed.
- R. Tandon, I. Singh, V. Luxami, N. Tandon and K. Paul, Chem. Rec., 2018, 18, 1–33 CrossRef.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 7–30 CrossRef PubMed.
- Y. Sumii, N. Kotoku, A. Fukuda, T. Kawachi, M. Arai and M. Kobayashi, Mar. Drugs, 2015, 13, 7419–7432 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, Future Med. Chem., 2009, 1, 1415–1427 CrossRef CAS PubMed.
- D. D. Luo, K. A. Carter, D. Miranda and J. F. Lovell, Adv. Sci., 2017, 4, 1600106 CrossRef PubMed.
- K. V. Kudryavtsev, C. C. Yu, P. M. Ivantcova, V. I. Polshakov, A. V. Churakov, S. Brase, N. S. Zefirov and J. H. Guh, Chem.–Asian J., 2015, 10, 383–389 CrossRef CAS PubMed.
- B. C. Gerwick and T. C. Sparks, Natural products for pest control: an analysis of their role, value and future, Pest Manage. Sci., 2014, 70, 1169–1185 CrossRef CAS PubMed.
- O. F. Hüter, Phytochem. Rev., 2011, 10, 185–194 CrossRef.
- T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Nat. Chem., 2016, 8, 531–541 CrossRef CAS PubMed.
- K. Kaur, V. Kumar, A. K. Sharma and G. K. Gupta, Eur. J. Med. Chem., 2014, 77, 121–133 CrossRef CAS PubMed.
- I. Boulogne, P. Petit, H. Ozier-Lafontaine, L. Desfontaines and G. Loranger-Merciris, Environ. Chem. Lett., 2012, 10, 325–347 CrossRef CAS.
- T. C. Sparks, D. R. Hahn and N. V. Garizi, Pest Manage. Sci., 2017, 73, 700–715 CrossRef CAS PubMed.
- S. Miresmailli and M. B. Isman, Trends Plant Sci., 2014, 19, 29–35 CrossRef CAS PubMed.
- J. H. Schrittwieser and V. Resch, RSC Adv., 2013, 3, 17602–17632 RSC.
- B. Parrino, A. Attanzio, V. Spano, S. Cascioferro, A. Montalbano, P. Barraja, L. Tesoriere, P. Diana, G. Cirrincione and A. Carbone, Eur. J. Med. Chem., 2017, 138, 371–383 CrossRef CAS PubMed.
- M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2015, 32, 1389–1471 RSC.
- M. Salmoun, C. Devijver, D. Daloze, J. C. Braekman and R. W. Soest, J. Nat. Prod., 2002, 65, 1173–1176 CrossRef CAS PubMed.
- V. Spano, A. Attanzio, S. Cascioferro, A. Carbone, A. Montalbano, P. Barraja, L. Tesoriere, G. Cirrincione, P. Diana and B. Parrino, Mar. Drugs, 2016, 14, 226 CrossRef PubMed.
- S. Cascioferro, A. Attanzio, V. D. Sarno, S. Musella, L. Tesoriere, G. Cirrincione, P. Diana and B. Parrino, Mar. Drugs, 2019, 17, 35 CrossRef PubMed.
- N. Mexia, G. Gaitanis, A. Velegraki, A. Soshilov, M. S. Denison and P. Magiatis, Arch. Biochem. Biophys., 2015, 571, 16–20 CrossRef CAS PubMed.
- C. Tan, Y. Zhang, N. Kong, Y. Di and X. Hao, Helv. Chim. Acta, 2015, 98, 72–77 CrossRef CAS.
- X. Shang, X. Guo, B. Li, H. Pan, J. Zhang, Y. Zhang and X. Miao, J. Ethnopharmacol., 2016, 192, 350–361 CrossRef CAS PubMed.
- H. M. Viart, A. Bachmann, W. Kayitare and R. Sarpong, J. Am. Chem. Soc., 2017, 139, 1325–1329 CrossRef CAS PubMed.
- P. Zhang, X. Sun, B. Xu, K. Bijian, S. Wan, G. Li, M. A. Jamali and T. Jiang, Eur. J. Med. Chem., 2011, 46, 6089–6097 CrossRef CAS PubMed.
- S. Manda, S. Sharma, A. Wani, P. Joshi, V. Kumar, S. K. Guru, S. S. Bharate, S. Bhushan, R. A. Vishwakarma, A. Kumar and S. B. Bharate, Eur. J. Med. Chem., 2016, 107, 1–11 CrossRef CAS PubMed.
- T. Suzuki, M. N. Khan, H. Sawada, E. Imai, Y. Itoh, K. Yamatsuta, N. Tokuda, J. Takeuchi, T. Seko, H. Nakagawa and N. Miyata, J. Med. Chem., 2012, 55, 5760–5773 CrossRef CAS PubMed.
- N. Pradidphol, N. Kongkathip, P. Sittikul, N. Boonyalai and B. Kongkathip, Eur. J. Med. Chem., 2012, 49, 253–270 CrossRef CAS PubMed.
- S. Ke, L. Shi, Z. Zhang and Z. Yang, Sci. Rep., 2017, 7, 44439 CrossRef PubMed.
- T. Xu, L. Shi, Y. Zhang, K. Wang, Z. Yang and S. Ke, Eur. J. Med. Chem., 2019, 168, 293–300 CrossRef CAS PubMed.
- C. P. Sanchez, A. Dave, W. D. Stein and M. Lanzer, Int. J. Parasitol., 2010, 40, 1109–1118 CrossRef CAS PubMed.
- M. Dhanawat, N. Das, R. C. Nagarwal and S. K. Shrivastava, Mini-Rev. Med. Chem., 2009, 9, 1447–1469 CrossRef CAS PubMed.
- N. V. Dharia, A. Chatterjee and E. A. Winzeler, Mini-Rev. Med. Chem., 2010, 11, 131–138 CAS.
- K. Sepcic, J. Toxicol., Toxin Rev., 2000, 19, 139–160 CrossRef CAS.
- J. Rodriguez, Stud. Nat. Prod. Chem., 2000, 24, 573–681 CAS.
- C. Timm and M. Kock, Synthesis, 2006, 113, 2580–2584 Search PubMed.
- D. L. Gardiner, T. S. Skinner-Adams, C. L. Brown, K. T. Andrews, C. M. Stack, J. S. Carthy, J. P. Dalton and K. R. Trenhome, Expert
Rev. Anti-Infect. Ther.c, 2009, 7, 1087–1098 CrossRef CAS PubMed.
- E. Fattorusso and O. Taglialatela-Scafati, Mar. Drugs, 2009, 7, 130–152 CrossRef CAS PubMed.
- R. C. Larock and Y. Wang, Tetrahedron Lett., 2002, 43, 21–23 CrossRef CAS.
- Y. Wang, X. Y. Dong and R. C. Larock, J. Org. Chem., 2003, 68, 3090–3098 CrossRef CAS PubMed.
- C. A. Volk and M. Kock, Org. Biomol. Chem., 2004, 2, 1827–1830 RSC.
- B. J. Shorey, V. Lee and J. E. Baldwin, Tetrahedron, 2007, 63, 5587–5592 CrossRef CAS.
- J. B. Lekana-Douki, J. B. Bongui, S. L. Liabagui, S. E. Edou, R. Zatra and U. Bisvigou, J. Ethnopharmacol., 2011, 133, 1103–1108 CrossRef PubMed.
- F. F. Hilario, R. C. Paula, M. L. Silveira, G. H. Viana, R. B. Alves, J. R. Pereira, L. M. Silva, R. P. Freitas and F. P. Varotti, Chem. Biol. Drug Des., 2011, 78, 477–482 CrossRef CAS PubMed.
- I. NNkumama, W. P. Meara and F. H. Osier, Trends Parasitol., 2017, 33, 128–140 CrossRef PubMed.
- J. N. Burrows, S. Duparc, W. E. Gutteridge, R. H. Huijsduijnen, W. Kaszubska, F. Macintyre, S. Mazzuri, J. J. Möhrle and T. N. CWells, Malar. J., 2017, 16, 26 CrossRef PubMed.
- T. S. Skinner-Adams, S. D. Sumanadasa, G. M. Fisher, R. A. Davis, D. L. Doolan and K. T. Andrews, Drug Discovery Today, 2016, 21, 725–739 CrossRef CAS PubMed.
- K. C. Nicolaou, Angew. Chem., Int. Ed., 2014, 53, 9128–9140 CrossRef CAS PubMed.
- A. Rossati, O. Bargiacchi, V. Kroumova, M. Zaramella, A. Caputo and P. L. Garavelli, Infez. Med., 2016, 24, 93–104 Search PubMed.
- N. Tiberti, S. L. Latham, S. Bush, A. Cohen, R. O. Opoka, C. C. John, A. Juillard, G. E. Grau and V. Combes, Sci. Rep., 2016, 6, 37871 CrossRef CAS PubMed.
- V. C. Corey, A. K. Lukens, E. S. Istvan, M. C. Lee, V. Franco, P. Magistrado, O. Coburn-Flynn, T. Sakata-Kato, O. Fuchs and E. A. Winzeler, Nature Communications, 2016, 7, 11901 CrossRef CAS PubMed.
- R. M. Ribeiro-Viana, A. P. Butera, E. S. Santos, C. A. Tischer, R. B. Alves, R. P. Freitas, L. Guimaraes, F. P. Varotti, G. H. Viana and C. S. Nascimento, J. Chem. Inf. Model., 2016, 56, 571–579 CrossRef CAS PubMed.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- A. R. Collins, Mol. Biotechnol., 2004, 26, 249–261 CrossRef CAS PubMed.
- M. Fenech, Nat. Protoc., 2007, 2, 1084–1104 CrossRef CAS PubMed.
- C. C. Gomes, L. M. Moreira, V. J. Santos, A. S. Ramos, J. P. Lyon, C. P. Soares and F. V. Santos, Genet. Mol. Biol., 2011, 34, 116–121 CrossRef CAS PubMed.
- F. V. Santos, M. Andreo, A. L. Nasser, L. M. Moreira, W. Vilegas, I. M. Cylus and E. A. Varanda, Arch. Biol. Sci., 2013, 65, 191–195 CrossRef.
- K. Katsuno, J. N. Burrows, K. Duncan, R. H. Huijsduijnen, T. Kaneko, K. Kita, C. E. Mowbray, D. Schmatz, P. Warner and B. T. Slingsby, Nat. Rev. Drug Discovery, 2015, 14, 751–758 CrossRef CAS.
- A. Carbone, B. Parrino, M. G. Cusimano, V. Spanò, A. Montalbano, P. Barraja, D. Schillaci, G. Cirrincione, P. Diana and S. Cascioferro, Mar. Drugs, 2018, 16, 274 CrossRef PubMed.
- R. P. Araldi, T. C. Melo, T. B. Mendes, P. L. SáJunior, B. H. Nozima, E. TIto, R. F. Carvalho, E. B. Souza and R. C. Stocco, Biomed. Pharmacother., 2015, 72, 74–82 CrossRef CAS PubMed.
- C. S. Barbosa, D. S. Guimaraes, A. M. Goncalves, M. C. Barbosá, M. L. Costa, C. S. Nascimento, L. Guimaraes, R. M. Ribeiro-Viana, F. V. Santos, C. F. Brito, F. P. Varotti and G. H. Viana, ACS Omega, 2017, 2, 8264–8272 CrossRef CAS PubMed.
- P. J. Durai, V. Padma, P. Vijaya, P. N. Sylaja and J. M. Murthy, Stroke and thrombolysis in developing countries, Int. J. Stroke, 2007, 2, 17–26 CrossRef PubMed.
- J. Wu, J. Ling, X. Wang, T. Li, J. Liu, Y. Lai, H. Ji, S. Peng, J. Tian and Y. Zhang, J. Med. Chem., 2012, 55, 7173 CrossRef CAS PubMed.
- G. Bartosz, Biochem. Pharmacol., 2009, 77, 1303–1315 CrossRef CAS.
- P. Talalay, A. T. Dinkova-Kostova and W. D. Holtzclaw, Adv. Enzyme Regul., 2003, 43, 121–134 CrossRef CAS.
- R. Sane, S. Agarwal and W. F. Elmquist, Drug Metab. Dispos., 2012, 40, 1612 CrossRef CAS.
- P. Paumard, J. Vaillier, B. Coulary, J. Schaeffer, V. Soubannier, D. M. Mueller, D. Brethes, R. J. Di and J. Velours, EMBO J., 2002, 21, 221–230 CrossRef CAS.
- E. R. Stadtman, Role of oxidant species in aging, Curr. Med. Chem., 2004, 11, 1105–1112 CrossRef CAS.
- H. Chen, H. Yoshioka, G. S. Kim, J. E. Jung, N. Okami, H. Sakata, C. M. Maier, P. Narasimhan, C. E. Goeders and P. H. Chan, Antioxid. Redox Signaling, 2011, 14, 1505–1517 CrossRef CAS PubMed.
- H. Liu, C. J. Li, J. Z. Yang, N. Na, Y. K. Si, L. Li, N. H. Chen and D. M. Zhang, J. Nat. Prod., 2012, 75, 677–682 CrossRef CAS PubMed.
- R. A. Rane, S. D. Gutte and N. U. Sahu, Bioorg. Med. Chem. Lett., 2012, 22, 6429–6432 CrossRef CAS PubMed.
- J. M. Pawlotsky, Gastroenterology, 2014, 146, 1176–1192 CrossRef CAS PubMed.
- M. B. Zeisel, E. Crouchet, T. F. Baumert and C. Schuster, Viruses., 2015, 7, 5659–5685 CrossRef CAS PubMed.
- K. E. Lillsunde, T. Tomasic, D. Kikelj and P. Tammela, Chem. Biol. Drug Des., 2017, 90, 1147–1154 CrossRef CAS PubMed.
- J. I. MacPherson, B. Sidders, S. Wieland, J. Zhong, P. Targett-Adams, V. Lohmann, P. Backes, O. Delpuech-Adams, F. Chisari, M. Lewis, T. Parkinson and D. L. Robertson, PLoS One, 2011, 6, 25584 CrossRef PubMed.
- P. T. Dolan, C. Zhang, S. Khadka, V. Arumugaswami, A. D. Vangeloff, N. S. Heaton, S. Sahasrabudhe, G. Randall, R. Sun and D. J. LaCount, Mol. BioSyst., 2019, 3, 3199–3209 Search PubMed.
- M. Taipale, D. F. Jarosz and S. Lindquist, Nat. Rev. Mol. Cell Biol., 2010, 11, 515–528 CrossRef CAS PubMed.
- R. Geller, S. Taguwa and J. Frydman, Biochim. Biophys. Acta, 2012, 1823, 698–706 CrossRef CAS PubMed.
- M. A. Germain, L. Chatel-Chaix, B. Gagn, P. Bonneil, P. Thibault, F. Pradezynski, B. Chassey, L. Meyniel-Schicklin, V. Lotteau, M. Baril and D. Lamarre, Mol. Cell. Proteomics., 2014, 13, 184–203 CrossRef CAS PubMed.
- K. E. Lillsunde, T. Tomašič, P. Schult, V. Lohmann, D. Kikelj and P. Tammela, ChemMedChem., 2019, 14, 334–342 CrossRef CAS PubMed.
- J. R. Chérouvrier, F. Carreaux and J. P. Bazureau, Tet. Lett., 2002, 43, 3581–3584 CrossRef.
- M. Rahaus, N. Desloges and M. H. Wolff, Cellular Signalling, 2007, 19, 312–320 CrossRef CAS PubMed.
- B. W. Doble, S. Patel, G. A. Wood, L. K. Kockeritz and J. R. Woodgett, Deve. Cell., 2007, 12, 957–971 CrossRef CAS PubMed.
- Q. Wang, Y. Zhou and B. M. Evers, Neoplasia, 2006, 8, 781–787 CrossRef CAS.
- S. Leclerc, M. Garnier, R. Hoessel, D. Marko, J. A. Bibb, G. L. Snyder, P. Greengard, J. Biernat, E.-M. Mandelkow, G. Eisenbrand and L. Meijer, J. Biol. Chem., 2001, 276, 251–260 CrossRef CAS.
- A. Primot, B. Baratte, M. Gompel, A. Borgne, S. Liabeuf, J. L. Romette, F. Costantini and L. Meijer, Protein Expression Purif., 2000, 20, 394–404 CrossRef CAS.
- M. Debdab, S. Renault, O. Lozach, L. Meijer, L. Paquin, F. Carreaux and J. P. Bazureau, Eur. J. Med. Chem., 2010, 45, 805–810 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a