
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUV-protection from chitosan derivatized lignin multilayer thin film†
Thomas J. Kolibabaa,
Daniel L. Stevensa,
Stephen T. Pangburnc,
Olivia Condassamyd,
Martin Camusd,
Etienne Grau*d and
Jaime C. Grunlan *abc
*abc
aDepartment of Chemistry, Texas A&M University, 3255 TAMU, College Station, TX 77843, USA. E-mail: jgrunlan@tamu.edu; Tel: +1-979-845-3027
bDepartment of Materials Science & Engineering, Texas A&M University, 3003 TAMU, College Station, TX 77843, USA
cDepartment of Mechanical Engineering, Texas A&M University, 3123 TAMU, College Station, TX 77843, USA
dLaboratoire de Chimie des Polymères Organiques, Université de Bordeaux, UMR5629, CNRS, Bordeaux INP, ENSCBP, 16 Avenue Pey-Berland, 33607 Cedex Pessac, France. E-mail: etienne.grau@u-bordeaux.fr; Tel: +33-555-684-6189
First published on 4th September 2020
Abstract
Lignin is one of the most abundant renewable materials on the earth. Despite possessing useful antioxidant and UV absorbing properties, its effective utilization in technology has been hampered by its relative insolubility and difficulty to process. In this work, a simple chemical derivatization process is utilized which yields water-soluble lignin possessing anionic carboxylate groups. These carboxylate groups give lignin polyanionic behavior and enable its utilization in the growth of a functional film via layer-by-layer (LbL) assembly with biologically sourced chitosan. The growth mechanism of this film is hypothesized to be a result of both hydrogen bonding and ionic interactions. The film demonstrates excellent UV-absorptive capability. A 100 nm thick chitosan/lignin coating was applied to a poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) film and shown to reduce its degradation sixfold over the course of a 1 hour exposure to harsh UV light. This is the first demonstration of lignin being utilized in a fully biologically derived LbL film. Utilization of lignin in LbL assembly is an important step in the development of renewable nanotechnology.
1. Introduction
In an effort to gain greater independence from fossil fuels, researchers have found wood, and more specifically lignin, to have tremendous potential in both nanotechnology and as a potential chemical feedstock.1–3 Lignin has a high natural abundance in wood, accounting for as much as 25% of some species' total mass.4 Furthermore, as lignin is inedible, its use as a chemical feedstock does not compete with food production. Despite its potential, the functional use of lignin is inhibited by its hydrophobicity and difficulty to process both chemically and physically.5 Lignin is an amorphous, crosslinked network of both carbon–carbon and ether linkages produced from the polymerization of three different lignol monomers.6 Due to the randomness of lignin's polymerization (and variation in the ratios of its constituent monomers among wood species), it has a complex, yet indefinite, three-dimensional structure that is responsible for much of the strength of plant cell walls.7 Despite the difficulty of processing lignin, a great number of uses for functionalized lignin have been developed in recent years. These applications include flame retardants,8–10 cellular imaging aids,11 supercapacitors,12 environmental remediation,13 and UV protection.14,15In recent years, layer-by-layer (LbL) assembly has risen to prominence as a way of depositing functional coatings on almost any substrate.16,17 These coatings impart highly effective functionality from a coating that is often just a few hundred nanometers thick. Many of these functional coatings have incorporated biomaterials and/or renewable materials for many industrially important end-uses.18–22 Despite its relatively high abundance of somewhat acidic protons (and therefore theoretical ease in utilizing it as an anionic ingredient in LbL films), lignin is very challenging to dissolve in an appreciable amount in water without utilizing extremely high pH.23 This restriction allows only strong polycations, such as quaternized amines, to be used. Many polycations are weak (i.e. pH-sensitive) polyamines that will be uncharged at the high pH required to use unmodified lignin in LbL. This is why most of lignin's use in multilayer assemblies are currently very scarce, primarily using poly(diallyldimethylammonium chloride) as the polycation.24–26 One notable exception is the work by Su and coworkers to developed a cationic quaternized lignin that forms films when grown with a polycarboxylic acid.
In the present study, lignin was chemically modified through alkaline oxidation under pressure to yield hydrosoluble lignin. This modified lignin is shown to grow layer-by-layer with biologically-sourced, cationic chitosan (CH) to yield films that have high UV absorbance. The LbL growth is hypothesized to be due to a combination of both hydrogen bonding and ionic interactions between CH and the hydrosoluble lignin. These highly absorbing films were deposited onto spin coated poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) films to demonstrate their potential to protect photovoltaic materials. A 20 bilayer (BL) CH/Lignin film reduces the degradation rate of a PEDOT:PSS film by a factor of 6 over the course of 1 hour of high intensity ultraviolet light exposure.
2. Experimental
Materials and substrates
Hydrochloric acid (ACS reagent, 37%), sodium hydroxide (ACS reagent, ≥97.0%), ammonium bisulfate (98%), methanol (ACS reagent, ≥99.5%), chloroform, 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP, 95%), endo-N-hydroxy-5-norbornene-2,3-dicarboxylic acid imide (97%), chromium(III) acetylacetonate, pyridine, N,N-dimethylformamide (DMF), polyethylenimine (PEI, Mn 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and Mw 25000 g mol−1) and poly(diallyldimethylammonium chloride) (PDDA, Mw 400000–500000 g mol−1, 20 wt% in water) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Chitosan (CH, Item FGC-1, 95% deacetylated, 50–60 cP) was purchased from the GTC Bio Corporation (Qingdao, Shandong Province, China). Deuterated chloroform (CDCl3) was purchased from Eurisotop (Saclay, France). Technical lignin was isolated from Pinus pinaster through an ammonium bisulfate cooking process, followed by an alkaline extraction to remove residual lignin from the crude pulp. The lignin used in this study was extracted from the resultant effluent by adjusting its pH to 1 with HCl. A solution of poly(3,4-ethylenedioxythiophene):polystyrene sulfonate (PEDOT:PSS, Clevios PH1000) was purchased from Heraeus Precious Metals (Hanau, Germany). All aqueous solutions were prepared in 18 MΩ deionized (DI) water. Samples for ellipsometry were prepared on single side polished (100) silicon wafers from University Wafer (South Boston, MA, USA). Samples for UV/vis spectroscopy were prepared on quartz slides (Chemglass, Vineland, NJ, USA). Samples for UV degradation studies were deposited on 2.5 × 2.5 × 0.1 cm glass slides (Vistavision™, VWR International, Radnor, PA, USA). All substrates were rinsed sequentially with DI water, methanol, and DI water, and were then dried with a stream of filtered air. Cleaned substrates were then exposed to an air plasma for 5 minutes in a 32 G plasma cleaner (Harrick Plasma Inc. Ithaca, NY, USA).
000 g mol−1 and Mw 25000 g mol−1) and poly(diallyldimethylammonium chloride) (PDDA, Mw 400000–500000 g mol−1, 20 wt% in water) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Chitosan (CH, Item FGC-1, 95% deacetylated, 50–60 cP) was purchased from the GTC Bio Corporation (Qingdao, Shandong Province, China). Deuterated chloroform (CDCl3) was purchased from Eurisotop (Saclay, France). Technical lignin was isolated from Pinus pinaster through an ammonium bisulfate cooking process, followed by an alkaline extraction to remove residual lignin from the crude pulp. The lignin used in this study was extracted from the resultant effluent by adjusting its pH to 1 with HCl. A solution of poly(3,4-ethylenedioxythiophene):polystyrene sulfonate (PEDOT:PSS, Clevios PH1000) was purchased from Heraeus Precious Metals (Hanau, Germany). All aqueous solutions were prepared in 18 MΩ deionized (DI) water. Samples for ellipsometry were prepared on single side polished (100) silicon wafers from University Wafer (South Boston, MA, USA). Samples for UV/vis spectroscopy were prepared on quartz slides (Chemglass, Vineland, NJ, USA). Samples for UV degradation studies were deposited on 2.5 × 2.5 × 0.1 cm glass slides (Vistavision™, VWR International, Radnor, PA, USA). All substrates were rinsed sequentially with DI water, methanol, and DI water, and were then dried with a stream of filtered air. Cleaned substrates were then exposed to an air plasma for 5 minutes in a 32 G plasma cleaner (Harrick Plasma Inc. Ithaca, NY, USA).
Modification of lignin
Lignin modification was performed according to previously published work.22,23 Lignin (2 g) was dissolved in 50 mL of NaOH (0.615 M) and stirred in an autoclave at 120 °C under 10 bar of O2 for 1 hour. After cooling and relieving pressure, the obtained solution was adjusted to pH 2 with 1 M HCl. During this process some material precipitated out of solution, and is referred to as precipitated lignin. The acidified solution was then extracted with chloroform to remove organosoluble lignin. Finally, the lignin solution was dried and washed with methanol to remove residual NaCl to yield the hydrosoluble lignin. This process yielded 58 wt% hydrosoluble lignin (the lignin utilized and referred to throughout the main text of this study) and 42 wt% precipitated lignin, with no appreciable amount of organosoluble material obtained. Lignin molar mass and dispersity were characterized by size-exclusion chromatography in a Tosoh GPC (Tosoh Bioscience GmbH, Griesheim, Germany) with three TSK columns (G3000, G4000, G3000PW) monitored by a 280 nm UV detector. Samples were referenced to a poly(styrene sulfonate) calibration curve. The sample and calibration standards were run in a pH 12 sodium hydroxide solution eluent at 1 mL min−1.The degree of functionality of the lignin was determined with a quantitative 31P NMR technique that phosphorylates hydroxyl groups and references them to a phosphorylated hydroxyl-containing internal standard.27,28 Due to the water sensitivity of this technique, all solvents were dried on 3 Å molecular sieves and all glassware was dried overnight in a 110 °C oven. A dried lignin sample (∼40 mg) was dissolved in 500 μL of anhydrous pyridine/CDCl3 (1.6:1 v/v) mixture, along with an internal standard (endo-N-hydroxy-5-norbornene-2,3-dicarboximide, 2–10 mg) and chromium(III) acetylacetonate (1 mg). To ensure lignin solubility, 500 μL of DMF was added to the mixture. Next, 250 μL of the phosphorylating agent TMDP was added and the mixture was agitated for 1 hour. Finally, 400 μL of this mixture, along with 200 μL of CDCl3 were transferred to an NMR tube for analysis. The 31P NMR study was performed at room temperature in an AVANCE I NMR (Bruker, Billerica, MA, USA) equipped with a 5 mm direct probe (operating at 162.0 MHz). Data acquisition was made with the “reverse gate” zgOig program from the Bruker Topspin database. The spectral window was optimized at 10000 Hz with an acquisition time of 1.6 s and a D1 delay of 5 s. The distribution of hydroxyl groups in the lignin was identified by integrating the following areas: aliphatic hydroxyl (150.0–145.5 ppm), condensed units (144.7–140 ppm), guaiacyl units (140.2–138.6 ppm), p-hydroxyphenyl units (138.4–136.4 ppm), and carboxylic acids (136–134 ppm).
Layer-by-layer deposition
Solutions of 0.1 wt% PEI and 0.1 wt% PDDA were prepared in DI water and rolled to homogeneity in polyethylene bottles. PDDA was adjusted to pH 4 with 1 M HCl. A solution of 0.1 wt% CH was prepared in pH 1.7 water and stirred overnight, after which it was adjusted to pH 4 with 1 M NaOH. A solution of 0.1 wt% hydrosoluble lignin (henceforth referred to simply as lignin) was prepared in DI water and rolled overnight, bath sonicated for 1 hour in a 5510 ultrasonic cleaning bath (Branson Ultrasonics Corporation, Danbury, CT, USA), and finally rolled one more night to ensure homogeneity. The lignin solution was adjusted to pH 2 with 1 M HCl. All multilayer films were grown via layer-by-layer (LbL) assembly with a homebuilt robotic dipping system.29 After plasma treatment, substrates were first dipped in the PEI solution for 5 minutes, followed by rinsing with DI water and drying with filtered air. The first bilayer (BL) was completed with a 5 minute immersion in the lignin solution, followed by rinsing and drying. After this first bilayer, all immersion times were 1 minute, and the PEI solution was replaced by either CH for CH/lignin films, or by PDDA for PDDA/lignin films. LbL films deposited on PEDOT:PSS films were done without additional plasma treatment of the substrate.PEDOT:PSS thin film preparation
PEDOT:PSS films were prepared via a previously published spin coating method.30 A cleaned, dried and plasma treated glass slide was spin coated (KW-4A Spin-Coater, Chemat Technology, Northridge, CA, USA) with 300 μL of PEDOT:PSS at 500 rpm for 5 seconds then accelerated to 3000 rpm for 15 seconds. The coated films were annealed in a 120 °C oven for 15 minutes. After annealing, 150 μL of methanol was deposited directly onto the substrate in the oven and allowed to further anneal for 10 minutes. The film was then removed from the oven and exposed to another 150 μL of methanol and spun on the spin coater again using the same parameters. The substrate was then annealed in the 120 °C oven for 10 minutes. After coating, the substrate was put in a UV resistant container to avoid exposure to ambient light. The coating was measured by ellipsometry to be ∼80 nm thick.Characterization
Thickness of films on silicon wafers was measured by ellipsometry (Alpha-SE Ellipsometer, J.A. Woollam Co., Inc., Lincoln, NE, USA). UV/vis absorbance of the LbL films on quartz slides was measured using a Hitachi U-4100 UV-vis-NIR spectrometer (Tokyo, Japan). Resistance of films on glass slides was measured on a Keithley 2000 Multimeter (Cleveland, OH, USA). PEDOT:PSS coated glass slides, with and without CH/lignin coatings, were cut in half (ca. 1 × 2 cm2). Contact resistance was reduced by applying silver paint (Electron Microscopy Sciences, Hatfield, PA, USA) on the end of the slide fragments (on both sides), followed by annealing in a Thermofisher TF55030A-1/Blue M Single Segment Tube Furnace (Waltham, MA, USA) under argon at 150 °C for 15 minutes. Films were exposed to ∼250 nm light by being placed immediately under a pair of GERM-3000008 germicidal UV-C light bulbs (1000Bulbs, Garland, TX, USA) attached to a Black-Ray UV Bench Lamp 78 W ballast (UVP, Lipland, CA, USA).3. Results and discussion
Technical lignin (i.e. the most common form of extracted lignin) was modified under basic oxidative conditions in an autoclave at 10 bar to yield hydrosoluble lignin.23 Following the functionalization, modified lignin was characterized by size exclusion chromatography and solubility tests. Additionally, the distribution of hydroxyl functionalities (i.e. fraction of aliphatic, phenolic, and carboxylic hydroxyl groups) was determined through derivatization of these groups and analysis with 31P NMR.27,28 The properties of the technical and hydrosoluble lignin are summarized in Table 1. Size exclusion chromatography revealed a considerable decrease in molar mass relative to a poly(styrene sulfonate) calibration curve. After functionalization, the hydrosoluble lignin had considerably greater solubility in water at all pH levels, while technical lignin can only dissolve above pH 12. Furthermore, this modification did not reduce the lignin's solubility in organic solvents, despite the addition of hydrophilic groups.| Property | Technical lignin | Hydrosoluble lignin |
|---|---|---|
| Molar mass (Mw, g mol−1) | 19000 |
4000 |
| Đ | 4.5 | 2.0 |
| Solubility | Water (pH ≥ 12), DMF, DMSO, MeOH | Water (any pH), DMF, DMSO, MeOH |
| Aliphatic –OH groups (mmol g−1) | 1.41 | 0.75 |
| Phenolic –OH groups (mmol g−1) | 1.61 | 0.40 |
| Carboxylic acid groups (mmol g−1) | 0.36 | 2.80 |
It is clear from Table 1 that some of the aliphatic and phenolic hydroxyl groups are oxidized into carboxylates in the hydrosoluble lignin. Scheme 1 shows the reaction conditions, along with hypothetical structures for both technical and hydrosoluble lignin. The improvements in solubility are likely due to the decreased molar mass (likely breaking up some of the crosslinked structure of lignin during oxidation) of the hydrosoluble lignin, which is supported by the Mw values reported in Table 1. The increase in the relative proportion of carboxylic acid groups does not prevent dissolution of the modified lignin in polar organic solvents, indicating that this form of lignin could be used in solvent-based systems if needed. Further improvements in water-solubility are anticipated due to the increased prevalence of ionizable carboxylic acid groups. In an aqueous solution, these acidic groups become negatively charged, causing the hydrosoluble lignin (henceforth simply referred to as ‘lignin’) to behave as a polyanion. This polyanionic behavior is hypothesized to enable the growth of multilayer films containing lignin via layer-by-layer assembly.
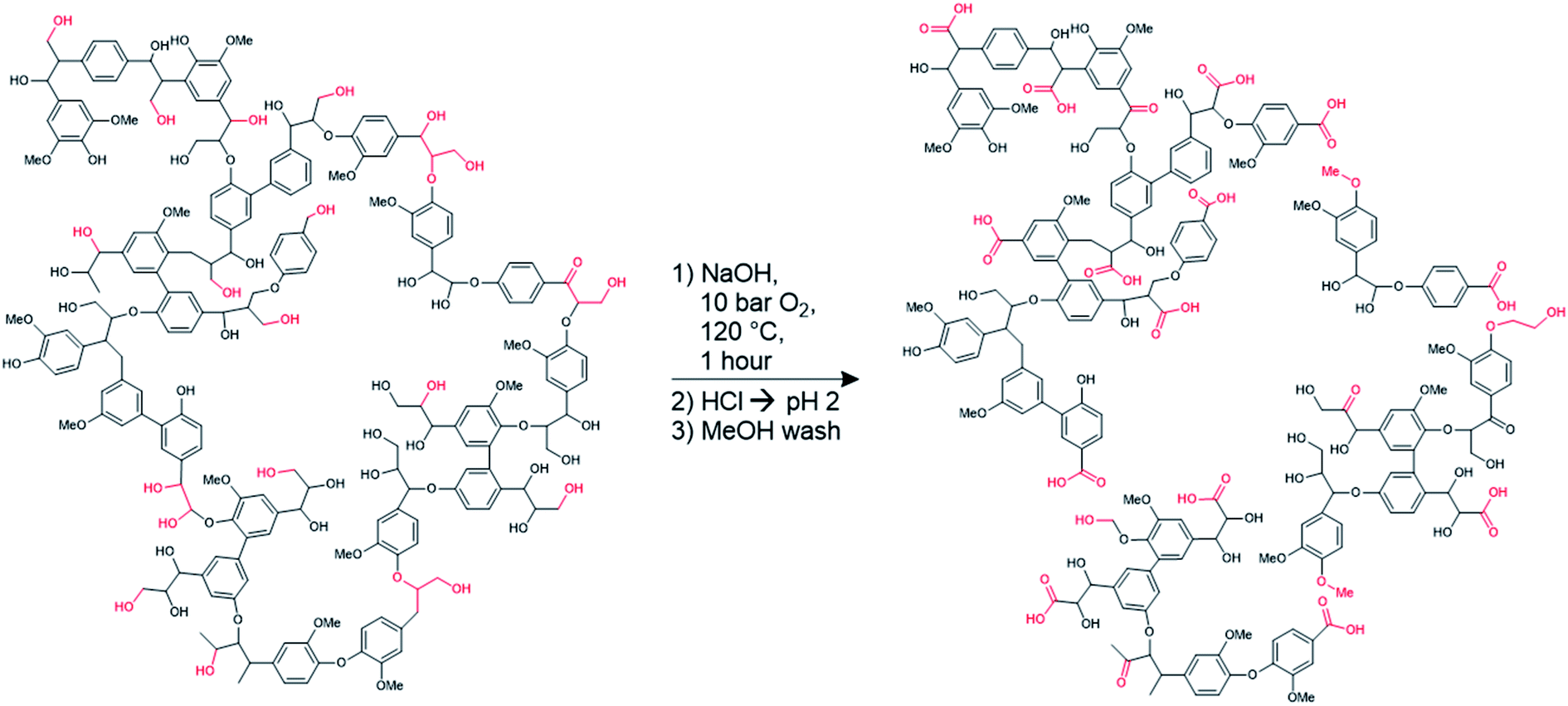 | ||
| Scheme 1 Hypothetical structure of lignin before and after modification to form hydrosoluble lignin.7 Functional groups affected by the transformation are highlighted in red. | ||
Lignin was paired with both chitosan (CH) and poly(diallyldimethylammonium chloride) (PDDA), two polycations used frequently in layer-by-layer assembly,31–34 to study lignin's growth behavior. All solutions were prepared as 0.1 wt% solutions in DI water. Lignin was adjusted to pH 2 (where it would be expected to have a low charge density, as most carboxylate groups will be protonated), while the polycation solutions were both adjusted to pH 4. Fig. 1 shows a schematic of the LbL process and the growth curves of the CH/lignin and PDDA/lignin films. Studying the growth curves reveals that CH/lignin grows thicker than the analogous coating formed by pairing PDDA with lignin. Chitosan is fully charged at pH 4, so this difference in growth rate cannot be attributed to differences in the charge densities of the polycations.35 The most likely explanation is that hydrogen bonding plays a role in film growth. Chitosan contains many hydrogen bond accepting and donating sites (i.e. hydroxyl groups), much like lignin, which can pair and cause thicker growth. Other bio-based polyphenolic compounds, such as tannic acid and even sulfonated lignins, have demonstrated LbL growth via hydrogen bonds.36–38 PDDA does not have either hydrogen bond donating or accepting sites, as it bears no functionality besides aliphatic carbons and quaternary ammonium groups. Since the PDDA/lignin coating also grows layer-by-layer, it can be concluded that not all of the film deposition is a result of hydrogen bonding. Ionic bonds are a stronger form of interaction than hydrogen bonds, which suggests that CH/lignin films may be more durable than analogous films composed of other polyphenolics.16,39
 | ||
| Fig. 1 (a) Schematic of the layer-by-layer process to grow CH/lignin films. (b) Growth curves for chitosan/lignin films. (c) UV-visible spectra showing absorbance of 20 and 40 BL films. | ||
The CH/lignin film grows linearly, depositing around 5 nm per bilayer (Fig. 1b). Linear growth in a LbL system typically indicates that there is little interdiffusion of adsorbed species (lignin or chitosan) during the growth process. With a low molar mass, weakly-charged polyelectrolyte like modified lignin, an exponential increase in thickness with respect to deposited bilayers would be expected.40,41 It was reported that diffusion of ionic bonding sites that can occur through local segmental dynamics of a polyelectrolyte, play a greater role in the interdiffusion of exponentially growing polyelectrolyte systems than diffusion of the polymers themselves.42 Despite the relatively low molecular weight of the modified lignin, its segmental motion is limited because of its planar aromatic backbone. This limited segmental motion, which is also true for the stiff cellulosic backbone of chitosan, minimizes interdiffusion of polyelectrolytes during multilayer deposition. It was noted that changing the charge density of lignin can alter film thickness (Fig. S1†), with higher pH solutions of lignin leading to thinner film growth. In all cases the growth curves remain linear, indicating little interdiffusion the of polymers regardless of growth conditions. The thinner growth is due to the increased charge density of lignin as pH rises due to deprotonation of carboxylate groups. A higher charge density requires a smaller amount of lignin to neutralize the surface charge reversal that occurs following the adsorption of chitosan onto the film during the LbL process.16
Deposited CH/lignin films are transparent with a yellow hue. Fig. 1c shows UV-vis absorbance curves for 20 and 40 BL films deposited on quartz slides. As would be expected for a large polyaromatic system, the films display considerable absorbance in the UV range, especially at wavelengths below 350 nm. Beyond ∼350 nm, the films show very low absorbance, so they are highly visibly transparent and there is little scattering of light. The UV-vis spectrum for the 40 BL film is approximately double the 20 BL spectrum's absorbance (Fig. S2†), which suggests a uniform film composition due to the known linear growth. This high UV absorbance, along with lignin's well documented antioxidant properties, suggest that it could have great potential as a protective coating for photo-oxidatively unstable materials, like those used in photovoltaic devices.43–47
To demonstrate the protective capabilities of CH/lignin nanocoatings, 10 and 20 BL were deposited on PEDOT:PSS thin films that had been methanol treated to improve conductivity.30 CH/lignin was grown on top of an ∼80 nm thick film of PEDOT:PSS. This system was then exposed to UV-C irradiation and the increase in the bulk resistance was used as a means of tracking the degradation of the PEDOT:PSS, as shown in the schematic in Fig. 2a.30,48,49 All films underwent an immediate increase in resistance upon exposure to the UV light, but the unprotected PEDOT:PSS began to rapidly degrade, while the coated samples did not (Fig. 2b). Unprotected PEDOT:PSS resistance increases by more than 1200% with 1 hour of UV exposure, while the 10 and 20 BL films show a resistance increase of only 270% and 200%, respectively. Degradation experiments were stopped after 1 hour because the chosen wavelength leads to rapid degradation of the unprotected PEDOT:PSS films. The achieved R/R0 values in one hour were substantially greater than previously reported degradation experiments of a similar nature.49
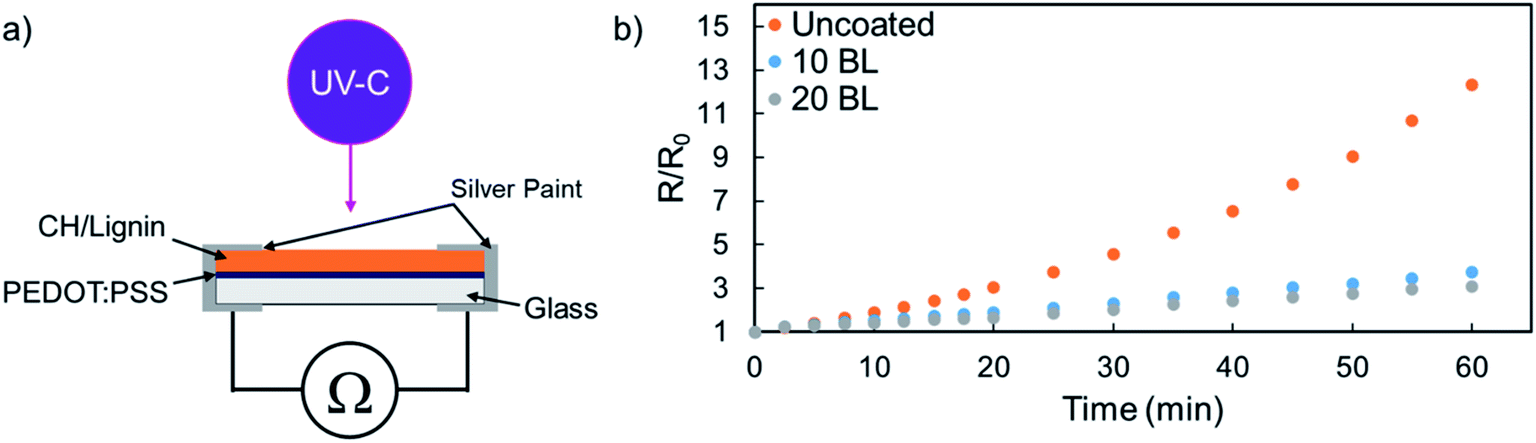 | ||
| Fig. 2 (a) Schematic of the UV degradation experiment, where resistance is measured to determine film degradation rate. (b) Plot of normalized resistance as a function of UV exposure time for unprotected PEDOT:PSS films, as well as films coated with 10 and 20 BL of CH/lignin. | ||
The degradation of PEDOT:PSS is clearly trending exponentially, while the protected films appear to be degrading linearly during this exposure, indicating that protected films would likely compare even more favorably after a longer UV exposure. There are clearly diminishing returns past the initial 10 BL of CH/lignin deposited, as the marginal improvement of the 20 BL film is not commensurate with the increase in coating thickness or added processing steps. The combination of the change in curve shape and the minimal gain from additional coating deposition suggests that the presence of lignin is changing the decomposition mechanism of PEDOT:PSS. It is well known that the photodegradation of PEDOT and other polythiophenes proceeds through oxidation of either the sulfur or side chains which disrupts π-conjugation and ultimately reduces conductivity. Since lignin is a well-known antioxidant,45–47 its mere presence may interrupt this process and protect underlying PEDOT. Further study would be needed to determine the true effect that the coating is having on the underlying PEDOT.
There are several other examples of lignin being used for a UV-protective purpose.15,50,51 None of the techniques used in these studies duplicate the system described here and the present thin films achieve these results at ∼80 (20 BL) and ∼ 190 (40 BL) nm thickness. Most other lignin-based coatings for UV-protection typically have a thicknesses > 1 μm. While the relatively high number of processing steps can appear daunting, the scalability of LbL deposition has been demonstrated numerous times for both roll-to-roll immersion and spray-based processing.52–54 As further applications for lignin-based LbL films are developed, this scalability will be critical to ensure commercial relevance of this technology.
4. Conclusion
In this study, an environmentally benign method of lignin functionalization is utilized to facilitate its use as a polyanion in layer-by-layer assembly. This is the first demonstration of lignin in a totally biosourced multilayer film. It was shown that the film grows linearly, despite the low apparent molar mass of the modified lignin, with the rate of growth altered by adjusting the pH of the lignin solution. Furthermore, evidence was presented to suggest that there is both hydrogen and ionic bonding involved in the assembly of lignin with chitosan. The utility of these coatings was demonstrated through the UV-protection of PEDOT:PSS, where protected films degraded at a fraction of the rate of unprotected polymer. Due to the versatile nature of layer-by-layer assembly, there is great promise for the utilization of lignin as a component in a variety of potential renewable, multifunctional films.Funding sources
Prof. Grunlan acknowledges financial support from the Linda & Ralph Schmidt 68 Professorship.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to acknowledge the Texas A&M Materials Characterization Facility for infrastructural support and access to the UV/vis spectrometer. The authors are grateful to the Xyloforest platform for the xylochem reactor (ANR-10-EQPX-16 XYLOFOREST).References
- W. Schutyser, T. Renders, S. Van den Bosch, S.-F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- V. K. Ponnusamy, D. D. Nguyen, J. Dharmaraja, S. Shobana, J. R. Banu, R. G. Saratale, S. W. Chang and G. Kumar, Bioresour. Technol., 2019, 271, 462–472 CrossRef.
- L. A. Berglund and I. Burgert, Adv. Mater., 2018, 30, 1704285 CrossRef.
- Y. Sun and J. Cheng, Bioresour. Technol., 2002, 11 Search PubMed.
- J. D. DeMartini, S. Pattathil, J. S. Miller, H. Li, M. G. Hahn and C. E. Wyman, Energy Environ. Sci., 2013, 6, 898 RSC.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef.
- C. A. S. Hill, Wood Modification: Chemical, Thermal and Other Processes, John Wiley & Sons, Ltd, Chichester, UK, 2006 Search PubMed.
- L. Ferry, G. Dorez, A. Taguet, B. Otazaghine and J. M. Lopez-Cuesta, Polym. Degrad. Stab., 2015, 113, 135–143 CrossRef.
- G. P. Mendis, S. G. Weiss, M. Korey, C. R. Boardman, M. Dietenberger, J. P. Youngblood and J. A. Howarter, Green Mater., 2016, 4, 150–159 CrossRef.
- B. Prieur, M. Meub, M. Wittemann, R. Klein, S. Bellayer, G. Fontaine and S. Bourbigot, Polym. Degrad. Stab., 2016, 127, 32–43 CrossRef.
- A. N. Cauley and J. N. Wilson, Biomater. Sci., 2017, 5, 2114–2121 RSC.
- L. Zhang, T. You, T. Zhou, X. Zhou and F. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 13918–13925 CrossRef.
- Y. Ge and Z. Li, ACS Sustainable Chem. Eng., 2018, 6, 7181–7192 CrossRef.
- Y. Qian, X. Qiu and S. Zhu, Green Chem., 2015, 17, 320–324 RSC.
- M. Farooq, T. Zou, G. Riviere, M. H. Sipponen and M. Österberg, Biomacromolecules, 2019, 20, 693–704 CrossRef CAS.
- G. Decher and J. B. Schlenoff, Multilayer Thin Films: Sequential Assembly of Nanocomposite Materials, John Wiley & Sons, Ltd, 2nd edn, 2012 Search PubMed.
- J. J. Richardson, J. Cui, M. Björnmalm, J. A. Braunger, H. Ejima and F. Caruso, Chem. Rev., 2016, 116, 14828–14867 CrossRef CAS.
- G. Laufer, C. Kirkland, A. A. Cain and J. C. Grunlan, ACS Appl. Mater. Interfaces, 2012, 4, 1643–1649 CrossRef CAS.
- H. Pan, L. Song, L. Ma, Y. Pan, K. M. Liew and Y. Hu, Cellulose, 2014, 21, 2995–3006 CrossRef CAS.
- Y. Pan, J. Zhan, H. Pan, W. Wang, G. Tang, L. Song and Y. Hu, ACS Sustainable Chem. Eng., 2016, 4, 1431–1438 CrossRef.
- D. Shi, M. Ran, L. Zhang, H. Huang, X. Li, M. Chen and M. Akashi, ACS Appl. Mater. Interfaces, 2016, 8, 13688–13697 CrossRef.
- M. Camus, O. Condassamy, F. Ham-Pichavant, C. MIchaud, G. Mignani, S. Mastroianni, E. Grau, H. Cramail and S. Grelier, ChemRxiv, Preprint.
- A. Kalliola, T. Vehmas, T. Liitiä and T. Tamminen, Ind. Crops Prod., 2015, 74, 150–157 CrossRef.
- K. Pillai, F. Navarro Arzate, W. Zhang and S. Renneckar, J. Visualized Exp., 2014, 88, e51257 Search PubMed.
- L. Su, S. Zhang, G. Jiang, J. Pang, D. Wang, J. Shi and G. Fang, J. Appl. Polym. Sci., 2017, 134(9), 44416 Search PubMed.
- P. K. Mishra and R. Wimmer, Ultrason. Sonochem., 2017, 35, 45–50 CrossRef CAS.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- H. Sadeghifar, C. Cui and D. S. Argyropoulos, Ind. Eng. Chem. Res., 2012, 51, 16713–16720 CrossRef CAS.
- W.-S. Jang and J. C. Grunlan, Rev. Sci. Instrum., 2005, 76, 103904 CrossRef.
- D. Alemu, H.-Y. Wei, K.-C. Ho and C.-W. Chu, Energy Environ. Sci., 2012, 5, 9662 RSC.
- S. Li, X. Lin, Y. Liu, R. Li, X. Ren and T.-S. Huang, Cellulose, 2019, 26, 4213–4223 CrossRef.
- Y. Zhao, C. Gao and B. Van der Bruggen, Nanoscale, 2019, 11, 2264–2274 RSC.
- D. L. Stevens, G. A. Gamage, Z. Ren and J. C. Grunlan, RSC Adv., 2020, 10, 11800–11807 RSC.
- J. D. Delgado and J. B. Schlenoff, Macromolecules, 2019, 52, 7812–7820 CrossRef CAS.
- R. Pelton, Langmuir, 2014, 30, 15373–15382 CrossRef CAS.
- I. Erel-Unal and S. A. Sukhishvili, Macromolecules, 2008, 41, 3962–3970 CrossRef CAS.
- T. Shutava, M. Prouty, D. Kommireddy and Y. Lvov, Macromolecules, 2005, 38, 2850–2858 CrossRef CAS.
- Y. Deng, T. Wang, Y. Guo, X. Qiu and Y. Qian, ACS Sustainable Chem. Eng., 2015, 3, 1215–1220 CrossRef CAS.
- Y. Fu, S. Bai, S. Cui, D. Qiu, Z. Wang and X. Zhang, Macromolecules, 2002, 35, 9451–9458 CrossRef CAS.
- D. L. Elbert, C. B. Herbert and J. A. Hubbell, Langmuir, 1999, 15, 5355–5362 CrossRef CAS.
- S. S. Shiratori and M. F. Rubner, Macromolecules, 2000, 33, 4213–4219 CrossRef CAS.
- H. M. Fares and J. B. Schlenoff, J. Am. Chem. Soc., 2017, 139, 14656–14667 CrossRef.
- V. K. Thakur, M. K. Thakur, P. Raghavan and M. R. Kessler, ACS Sustainable Chem. Eng., 2014, 2, 1072–1092 CrossRef.
- P. Figueiredo, K. Lintinen, J. T. Hirvonen, M. A. Kostiainen and H. A. Santos, Prog. Mater. Sci., 2018, 93, 233–269 CrossRef.
- C. G. Boeriu, D. Bravo, R. J. A. Gosselink and J. E. G. van Dam, Ind. Crops Prod., 2004, 20, 205–218 CrossRef.
- V. Aguié-Béghin, L. Foulon, P. Soto, D. Crônier, E. Corti, F. Legée, L. Cézard, B. Chabbert, M.-N. Maillard, W. J. J. Huijgen and S. Baumberger, J. Agric. Food Chem., 2015, 63, 10022–10031 CrossRef.
- K. Crouvisier-Urion, P. R. Bodart, P. Winckler, J. Raya, R. D. Gougeon, P. Cayot, S. Domenek, F. Debeaufort and T. Karbowiak, ACS Sustainable Chem. Eng., 2016, 4, 6371–6381 CrossRef CAS.
- T. J. Dawidczyk, M. D. Walton, W.-S. Jang and J. C. Grunlan, Langmuir, 2008, 24, 8314–8318 CrossRef.
- T. Guin, J. H. Cho, F. Xiang, C. J. Ellison and J. C. Grunlan, ACS Macro Lett., 2015, 4, 335–338 CrossRef.
- A. Hambardzumyan, L. Foulon, B. Chabbert and V. Aguié-Béghin, Biomacromolecules, 2012, 13, 4081–4088 CrossRef.
- M. Parit, P. Saha, V. A. Davis and Z. Jiang, ACS Omega, 2018, 3, 10679–10691 CrossRef.
- A. J. Mateos, A. A. Cain and J. C. Grunlan, Ind. Eng. Chem. Res., 2014, 53, 6409–6416 CrossRef.
- F. Xiang, T. M. Givens and J. C. Grunlan, Ind. Eng. Chem. Res., 2015, 54, 5254–5260 CrossRef CAS.
- P. C. Suarez-Martinez, J. Robinson, H. An, R. C. Nahas, D. Cinoman and J. L. Lutkenhaus, Macromol. Mater. Eng., 2017, 302, 1600552 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05829g |
| This journal is © The Royal Society of Chemistry 2020 |