
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZnO quantum dot-modified rGO with enhanced electrochemical performance for lithium–sulfur batteries†
Zhixu Jiana,
Shichao Zhang *a,
Xianggang Guana,
Jiajie Lib,
Honglei Lia,
Wenxu Wangc,
Yalan Xinga and
Huaizhe Xub
*a,
Xianggang Guana,
Jiajie Lib,
Honglei Lia,
Wenxu Wangc,
Yalan Xinga and
Huaizhe Xub
aSchool of Materials Science and Engineering, Beihang University, Beijing 100191, PR China. E-mail: csc@buaa.edu.cn; Fax: +86 01082338148; Tel: +86 01082339319
bSchool of Physics, Beihang University, Beijing 100191, PR China
cPulead Technology Industry Co. Ltd, Beijing 102200, PR China
First published on 4th September 2020
Abstract
Lithium–sulfur batteries are considered the most promising next-generation energy storage devices. However, problems like sluggish reaction kinetics and severe shuttle effect need to be solved before the commercialization of Li–S batteries. Here, we successfully prepared ZnO quantum dot-modified reduced graphene oxide (rGO@ZnO QDs), and first introduced it into Li–S cathodes (rGO@ZnO QDs/S). Due to its merits of a catalysis effect and enhancing the reaction kinetics, low surface impedance, and efficient adsorption of polysulfide, rGO@ZnO QDs/S presented excellent rate capacity with clear discharge plateaus even at a high rate of 4C, and superb cycle performance. An initial discharge capacity of 998.8 mA h g−1 was delivered, of which 73.3% was retained after 400 cycles at a high rate of 1C. This work provides a new concept to introduce quantum dots into lithium–sulfur cathodes to realize better electrochemical performance.
1. Introduction
Being widely used in portable electronic devices, electric vehicles and grid energy storage fields, lithium ion batteries based on intercalation compounds are approaching their theoretical energy density ceilings, yet they hardly meet the increasing demands.1–4 Due to the merits of a high theoretical energy density (∼2500 W h kg−1), low cost and environment friendliness, lithium–sulfur (Li–S) batteries are considered the most promising next-generation energy storage devices.4,5 Nevertheless, the commercialization of Li–S batteries is still hindered by some intrinsic challenges. First, the ionic/electronic insulation of sulfur and its discharge products (Li2S and Li2S2) leads to poor reaction kinetics, resulting in low utilization of the active material and large interface resistance. Second, because of the density change, a dramatic volume expansion (∼80%) was generated during the conversion between sulfur and Li2S, which may result in the failure of the construction of cathodes. Third, known as the “shuttle effect”, the soluble intermediate lithium polysulfides (Li2Sx, 4 ≤ x ≤ 8, denoted as LiPSs below) migrate between cathodes and anodes, which would cause the loss of active materials, inducing a low coulombic efficiency, fading of the rapid capacity and an unsatisfying cycle performance.6–8 Therefore, limiting the “shuttle effect” is considered to be the core focus of improving the performance of lithium–sulfur batteries.Over the last decade, massive efforts have been devoted to the design and fabrication of cathodes for Li–S batteries to address these issues. Several strategies have been developed, of which encapsulating sulfur into porous host materials has been proven one of the most effective approaches.9,10 Owing to their advantages of high conductivity, light weight, matured fabrication and modification technologies, carbon materials have become the most indispensable host materials for Li–S batteries.11–13 Mesoporous carbon CMK-3 was first adopted by Nazar et al. as sulfur host material, achieving high capacity and enhanced cycle stability.14 Since then, various carbon materials, including porous carbon,15–17 carbon nanotubes,18–20 and graphene,21–24 have been extensively studied by researchers as cathode carrier materials for Li–S batteries. Thanks to the various elaborately designed structures,25–30 the carbon/sulfur composites achieved better conductivity, less “shuttle effect” by the physical immobilization of LiPSs, and improved electrochemical performance. However, it has been proven that as non-polar materials, carbon materials cannot adsorb polar LiPSs/Li2S species effectively during the charge/discharge process, which may easily cause the detachment of LiPSs, followed by capacity decay and poor rate performance.31 The introduction of polar sites/groups on the surface of carbon materials,9,11,32 such as heteroatom doping,33 polymer material coating,34,35 and metal oxides/metal sulfides loading,32,36–41 has proven to be very effective for the adsorption of LiPSs by the chemical immobilization effect, resulting in the improvement of the electrochemical performance. It is worth noting that by virtue of its high conductivity, high surface area, and facilitating surface modification, graphene has been widely used in Li–S batteries with the polar species loaded as an adsorbent for LiPSs, realizing enhanced cycle performance and better rate capacity.7,42–45 Metal oxide, because of its merits of strong polar surface and stability to the electrolyte, has drawn special attention and been widely adopted in lithium–sulfur cathodes as LiPSs adsorbent.32 MnO2 was adopted into Li–S sulfur cathodes by Nazar et al., which revealed excellent polysulfide entrapment, and achieved excellent cycle stability.41 Tao et al. prepared Al-doped ZnO decorated carbon nanoflakes for use in Li–S batteries, realizing enhanced electrochemical performance.37 Despite those advantages, metal oxides also bring in some detrimental qualities, including extra inactive masses, normally lower conductivity and the additional surface diffusion process of LiPSs, which may hinder the achievement of high performance Li–S batteries.32,37,46 Recently, quantum dots have been regarded as a new approach to settle the drawbacks of Li–S batteries due to their abundant active sites, unique quantum confinement effect and exceptional catalysis effect.47–49 Xu and coworkers adopted black phosphorus (BP) quantum dots (QDs) in Li–S cathodes to chemically immobilize and catalyze the transition of LiPSs.47 They confirmed that BP QDs achieved excellent adsorption ability to LiPSs and unexpected catalytic activity due to their stronger binding energies and the presence of more under-coordinated atomic structures of edge sites.49–51 Pang and partners introduced N-doped carbon quantum dots (NCQD) into a modified separator in Li–S batteries, resulting in enhanced capacity retention and lower self-discharge.48 These quantum dot-based research studies provide new ideas about Li–S batteries.
Herein, we designed and successfully prepared ZnO quantum dot-doped rGO (rGO@ZnO QDs), which was then adopted in Li–S cathodes (rGO@ZnO QDs/S). By diminishing the size of the loaded ZnO to the quantum dot level, the catalysis effect and enhanced reaction kinetics were achieved. In addition, thanks to the reduction of the nonconductive ZnO, lower surface impedance was realized. Furthermore, rGO@ZnO QDs revealed efficient adsorption of polysulfides. All of the merits added up to the enhanced electrochemical performance with excellent rate capacity and good cycle stability. This research also provides a new concept to introduce quantum dots into lithium–sulfur batteries to accomplish better electrochemical performance.
2. Experimental
2.1. Material preparation
RGO@ZnO was synthesized following the same procedure, except without the acid pickling procedure. By increasing the amount of HCl to 100 mL and extending the acid treatment time to 12 hours, rGO was obtained without any other changes to the preparation process.
To prepare the rGO@ZnO QDs/S composite, 30 mg of rGO@ZnO QDs and 70 mg sulfur nanoparticles were dispersed into 30 mL H2O containing 60 μL Triton X-100. After being thoroughly stirred and sonicated, the uniform dispersion was vacuum freeze-dried, and then heated in Ar atmosphere at 155 °C for 12 hours to obtain the final product of rGO@ZnO QDs/S.
RGO@ZnO/S and rGO/S composites were fabricated following the same path, except replacing the rGO@ZnO QDs with rGO@ZnO/rGO.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 (21 mg and 49 mg, respectively), and added to 250 mL deionized water containing the surfactant, Triton X-100. Then, 10 mg of graphene nanosheets was added to act as a conductive agent (the mass ratio of rGO@ZnO QDs/S to graphene nanosheets was 7:1). After stirring and sonication to full dispersion, the suspension was then vacuum filtered, washed, dried, rolled and peeled. Then, the heat treatment was performed at 155 °C for 12 hours under Ar atmosphere, and finally the rGO@ZnO QDs/S–G self-supporting composite electrode was obtained.
:7 (21 mg and 49 mg, respectively), and added to 250 mL deionized water containing the surfactant, Triton X-100. Then, 10 mg of graphene nanosheets was added to act as a conductive agent (the mass ratio of rGO@ZnO QDs/S to graphene nanosheets was 7:1). After stirring and sonication to full dispersion, the suspension was then vacuum filtered, washed, dried, rolled and peeled. Then, the heat treatment was performed at 155 °C for 12 hours under Ar atmosphere, and finally the rGO@ZnO QDs/S–G self-supporting composite electrode was obtained.2.2. Morphology and structure characterization
Morphology of the samples was measured by field emission scanning electron microscopy (FE-SEM, Hitachi S4800) and transmission electron microscopy (TEM, FEI Tecnai G2 F30). The crystal structure of the samples was analyzed by X-ray diffraction (XRD) carried out by Rigaku D/Max-2400 from 10° to 80° at a scan rate of 6° per min. Raman spectroscopy was collected by a LabRam HR800 with 1.96 eV (632.8 nm) excitation laser. Thermal gravimetric analysis (TGA) was conducted on a thermogravimetric analyzer (STA-449C, NETZSCH) at a ramping rate of 10 °C min−1, with the temperature ranging from 25 °C to 600 °C under an atmosphere of Ar. The Quantachrome Autosorb-1C-VP analyzer was adopted to run nitrogen adsorption isotherms at 77 K to measure the Brunauer–Emmett–Teller (BET) surface areas.2.3. Electrochemical measurements
Standard 2032 coin cells were assembled to test the electrochemical properties. The cathodes were prepared by the following procedure: 70 wt% of active material (rGO@ZnO QDs/S, rGO@ZnO/S, or rGO/S), 20 wt% of carbon black and 10 wt% of polyvinylidene fluoride (PVDF) were mixed by fine grinding, and dispersed in N-methyl-2-pyrrolidinone (NMP) by magnetic stirring overnight to obtain a uniform slurry, which was then casted evenly onto aluminum foil substrates and dried in a vacuum oven at 55 °C for 12 hours, following by being compressed and cut into disks with a diameter of 10 mm. Typically, the areal sulfur loading was ∼1.0 mg cm−2. The cells were assembled in a glovebox filled with Ar (H2O < 0.1 ppm, O2 < 0.1 ppm), with the as-prepared cathodes, lithium foil anodes, Celgard 2300 membrane and 1 M LiTFSI in DOL/DME (1:1, v/v) with additional 0.2 M LiNO3 electrolyte. Cyclic voltammetry (CV, 0.1 mV s−1, 1.7–2.8 V vs. Li/Li+) and the electrochemical impedance spectra test (EIS, 100 kHz to 10 mHz with an amplitude of 5 mV) were conducted on a CHI 660D electrochemical workstation. Galvanostatic discharge/charge tests (voltage range from 1.7 V to 2.8 V, vs. Li/Li+) were carried out by a LAND CT2001A test system at various current densities.
2.4. Visualized tests of polysulfides adsorption
Stoichiometric amounts of sublimed sulfur and Li2S were mixed in solvent DME under magnetic stirring at 60 °C for 12 hours to prepare a uniform solution of Li2S6. All of the procedure was conducted under an atmosphere of Ar. 10 mg of rGO, rGO@ZnO, and rGO@ZnO QDs were separately added into 1 mL Li2S6 solution, kept still for 6 hours, and then photographs were taken. 1 mL Li2S6 solution with no sample added was used as the blank group.3. Results and discussion
As shown in Fig. 1, the rGO@ZnO QDs sample was fabricated according to a modified, reported method.52 First, a piece of polished Zn foil was immersed in a homogeneous GO solution and kept still at room temperature for 4 hours. Since the reduction potential of Zn/Zn2+ is lower than that of rGO/GO, GO would be reduced by Zn spontaneously and form a uniform film on the surface of the Zn foil by self-assembly (Fig. S1†), along with the generation of ZnO particles attaching on the GO flakes as a product of the redox reaction.53,54 Second, the film was detached by sonication, and cleaned with deionized water. Due to the relatively low degree of reduction, the obtained product was denoted as GO@ZnO. After etching by dilute HCl and going through heat treatment of GO@ZnO, the sample of rGO@ZnO QDs was successfully prepared. For comparison, rGO loaded with ZnO particles (denoted as rGO@ZnO) was prepared by the direct annealing of GO@ZnO without an acid etching procedure. rGO was fabricated by the sufficient acid etching of GO@ZnO and subsequent annealing. | ||
| Fig. 1 Schematic illustration of the synthesis of rGO@ZnO QDs. | ||
After comparison of the morphology of ZnO QDs at different etching times (Fig. S2†), the sample with 10 min treatment was selected as the rGO@ZnO QSs. Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) were selected to investigate the morphology and structural features of the rGO@ZnO QDs. As shown in Fig. 2a, ZnO QDs were successfully prepared and uniformly distributed on rGO with a diameter of 5–10 nm. The HRTEM image in Fig. 2b reveals that the lattice fringe spacing of QD is about 0.26 nm, which is consistent with the (002) lattice plane of ZnO. Without the acid treatment, the sample of rGO loaded with ZnO particles was prepared. The scanning electron microscopy (SEM) image in Fig. 2c shows that the surface of rGO was covered with ZnO particles, whose diameters were about 500 nm. The EDS elemental mappings of rGO@ZnO are exhibited in Fig. S3,† of which the images of elemental Zn and O are enriched to be a clear spherical shape consistent with the SEM image, proving the identity of the ZnO particles.
 | ||
| Fig. 2 (a) TEM image and (b) HRTEM image of rGO@ZnO QDs; (c) SEM image of rGO@ZnO; (d) SEM image and (e) EDS elemental mappings of rGO@ZnO QDs/S composite; (f) SEM image of rGO@ZnO/S composite. | ||
In order to get the homogeneous composites of rGO@ZnO QDs/S, rGO@ZnO, and rGO/S, sulfur nanoparticles with a size of approximately 500 nm were first prepared (as shown in Fig. S4†), followed by being uniformly blended with the host materials by dispersion in water and freeze-vacuum-drying. The materials were sealed by heat treatment under Ar atmosphere, and then the target products were finally obtained. With those well-designed procedures, the uniform distribution of sulfur was achieved without agglomeration, which was demonstrated by the SEM images of rGO@ZnO QDs and rGO@ZnO in Fig. 2d and f, respectively. In addition, high-resolution SEM and EDS elemental mappings were adopted to detect the surface element distribution of rGO@ZnO QDs/S (Fig. 2e) and rGO@ZnO/S (Fig. S5†). Both samples presented a homogeneous dispersion of sulfur, while elemental Zn and O showed some difference. For rGO@ZnO QDs/S, the elements O and Zn (especially the latter) were uniformly distributed even though no ZnO particles could be seen on the HRSEM image. This implied the existence of ZnO QDs, which will be further discussed in subsequent XRD measurements. Nevertheless, the elemental mapping of O and Zn in rGO@ZnO formed an obvious spherical morphology, which was consistent with the HRSEM image. From all the morphology investigation, we could draw the conclusions that rGO@ZnO QDs and rGO@ZnO were successfully prepared and uniformly combined with sulfur without agglomeration to obtain rGO@ZnO QDs/S and rGO@ZnO/S. In addition, the TEM, HRTEM, Raman spectrum and EDS elemental mapping results of rGO and rGO/S are presented in Fig. S6.† With sufficient acid etching treatment, no ZnO particles or QDs were left. As shown in Fig. S6b,† the rGO prepared in this paper had a multilayer stack structure, which could be seen vividly from the edge of rGO.
In addition, from the TEM image in Fig. S6a† and the HRTEM image, we could tell that the prepared rGO had good structure integrity. The Raman spectrum of rGO is shown in Fig. S6c.† At ∼2680 cm−1, a flat peak of the 2D band could be observed, which is the second most prominent peak that is always observed in graphite samples.55 The weak intensity of the 2D band confirmed that the rGO prepared in this paper had a relatively low degree of graphitization after heat treatment at a low temperature of 400 °C. Through the same strategy, homogeneous rGO/S was obtained.
X-ray diffraction (XRD) analysis was used to investigate the composition and crystal structure of the host materials. As shown in Fig. 3a, a strong peak at 25.3° (corresponding to the interplanar spacing of 3.52 Å) can be detected for all three samples of rGO, rGO@ZnO QDs, and rGO@ZnO (for rGO@ZnO, the intensity is relatively low because of the high peak intensity of ZnO), which indicates that after reduction by Zn and thermal treatment, GO was partly graphitized. In addition, as can be seen at 31.7°, 34.4°, and 36.2°, the three sharp diffraction peaks of the rGO@ZnO sample are the characteristic peaks of the Wurtzite-structured ZnO (JPCDS card no. 36-1451) for the lattice planes of (100), (002), and (101), respectively.56 Furthermore, the three characterized peaks of ZnO could also be found in the XRD pattern of rGO@ZnO QDs with a faint intensity (due to the low content of the ZnO QDs) and enhanced full width at half maximum (FWHM), which were induced by the size effect of the ZnO QDs.57 As for rGO, because of the thorough acid washing, no signal of ZnO was detected. The Raman spectra of rGO, rGO@ZnO, and rGO@ZnO QDs are shown in Fig. 3b. For all three samples, peaks of the D-band at 1346 cm−1 attributed to the vibration of disordered carbon and the G-band at 1588 cm−1 relating to the vibration of sp2 hybridized carbon–carbon bonds could be clearly observed.58 In addition, the ID/IG ratios of the rGO, rGO@ZnO, and rGO@ZnO QDs are 1.25, 1.26 and 1.24, respectively. The existence of strong G-band and D-band peaks, the same peak position, and the similar ID/IG ratio demonstrate that all three samples are well reduced and partly graphitized, which agree well with the XRD results. Thermal gravimetric analysis was conducted at air atmosphere to measure the ZnO/ZnO QDs content in rGO@ZnO/rGO@ZnOs, and the TGA curves are shown in Fig. 3c. For all three samples, an obvious mass loss at around 500 °C is shown, which corresponds to the burning of rGO in air. Thus, for the rGO@ZnO and rGO@ZnO QDs, the residual materials are ZnO and ZnO QDs, whose contents were calculated to be 71.5% and 6.7%, respectively. Nitrogen adsorption/desorption isotherm was adopted to measure the specific area of rGO@ZnO QDs (Fig. 3d). The continuous N2 uptake below 0.9 (P/P0) and the rapid increase above 0.9 (P/P0) demonstrate the existence of a highly porous structure in the rGO@ZnO QDs,17,25 whose Brunauer–Emmett–Teller (BET) surface area was calculated to be 584.7 m2 g−1. The high specific surface area could achieve a uniform dispersion of sulfur without agglomeration, which makes the rGO@ZnO QDs a very suitable carrier material for lithium–sulfur cathodes. After loading with sulfur, the composites of rGO@ZnO/S and rGO@ZnO QDs/S were prepared and characterized with XRD. From Fig. 3e, we can see that both rGO@ZnO/S and rGO@ZnO QDs/S exhibited the characteristic diffraction peaks of rGO at 25° and the characteristic peaks of sulfur with an Fddd orthorhombic structure (JPCDS card, no. 08-0247). Moreover, in the comparison of the enlarged XRD patterns of rGO@ZnO/S and S in Fig. 3f, the former sample presented three peaks of ZnO corresponding to the lattice planes of (100), (002), and (101), while the latter sample did not. Because of the low content of the ZnO QDs, no signal of ZnO was detected for the rGO@ZnO QDs/S. The results of XRD demonstrate that both rGO@ZnO and rGO@ZnO QDs have been successfully combined with sulfur. It is worth noting that the peak intensity of rGO@ZnO QDs/S is lower than that for rGO@ZnO/S, implying a lower crystalline state, which may be caused by the more uniform distribution of sulfur in rGO@ZnO QDs/S as a result of the relatively higher content of rGO and larger specific surface area of the rGO@ZnO QDs. According to the results of the TGA test in Fig. S7,† the sulfur-loading contents of rGO/S, rGO@ZnO/S, and rGO@ZnO QDs/S are 71.9%, 70.8%, and 71.9%, respectively.
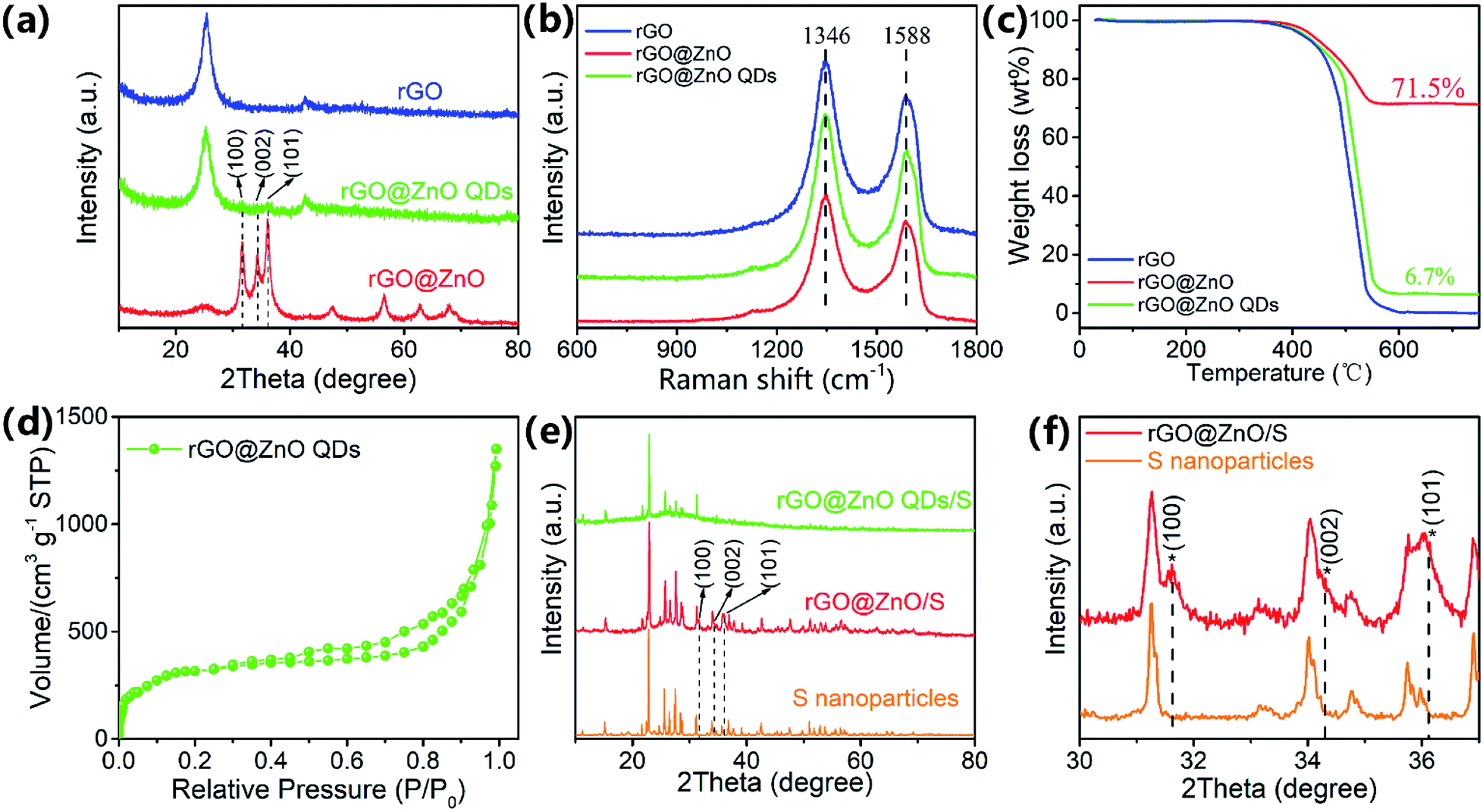 | ||
| Fig. 3 (a) XRD patterns, (b) Raman spectra, and (c) TGA curves of rGO, rGO@ZnO, and rGO@ZnO QDs; (d) N2 adsorption and desorption isotherm of rGO@ZnO QDs; (e) XRD patterns of S nanoparticles, rGO@ZnO/S and rGO@ZnO QDs/S; (f) partially enlarged XRD patterns of S nanoparticles and rGO@ZnO/S. | ||
To study the electrochemical performances, CR2032 coins were assembled with rGO@ZnO QDs/S as the cathodes and lithium foil as the anodes. For comparison, batteries with rGO@ZnO/S and rGO/S as the active material of the cathodes were also prepared, respectively. Cyclic voltammograms (CV) of the rGO@ZnO QDs/S and rGO@ZnO/S were conducted at a scan rate of 0.1 mV s−1, with the results shown in Fig. 4a and b. Both profiles exhibited the typical characteristic peaks of Li–S batteries, which are vivid peaks at around 2.3 V and 2.05 V during the cathodic scans, and partly overlapped peaks at about 2.35 V to 2.37 V during the anodic scans. The two peaks during cathodic scans are considered as reduction peaks, which are thought to correspond to the reduction of molecule S8 to soluble polysulfide Li2Sn (4 ≤ n ≤ 8), and Li2Sn to insoluble Li2S2/Li2S. In addition, the overlapped oxidation peaks are believed to relate to the translation from Li2S2/Li2S to polysulfide, and then S8.6,11 Furthermore, the voltage of the reduction peaks for both samples showed a slightly positive shift after the first cycle, which means a rearrangement of the active materials and lower impedance.26,59 However, the first reduction peak of rGO@ZnO QDs/S is at 2.35 V, which is higher than the reduction peak (2.31 V) of rGO@ZnO/S. This means a lower charge transfer barrier of the conversion of S8 to Li2Sn in rGO@ZnO QDs/S.60 Moreover, with a much sharper second reduction peak, the reaction kinetics of Li2Sn to Li2S2/Li2S is enhanced by rGO@ZnO QDs. Also, the oxidation peaks emerged with a negative shift during the five cycles for rGO@ZnO QDs, while no such phenomenon occurred for rGO@ZnO/S. That indicates the gradual decrease of impedance for Li2Sn to S8 in the rGO@ZnO QDs/S. All of the analysis leads to the conclusion that the presence of ZnO QDs could catalyze the transition between S8 and Li2S2/Li2S, and enhance the reaction kinetics.60,61
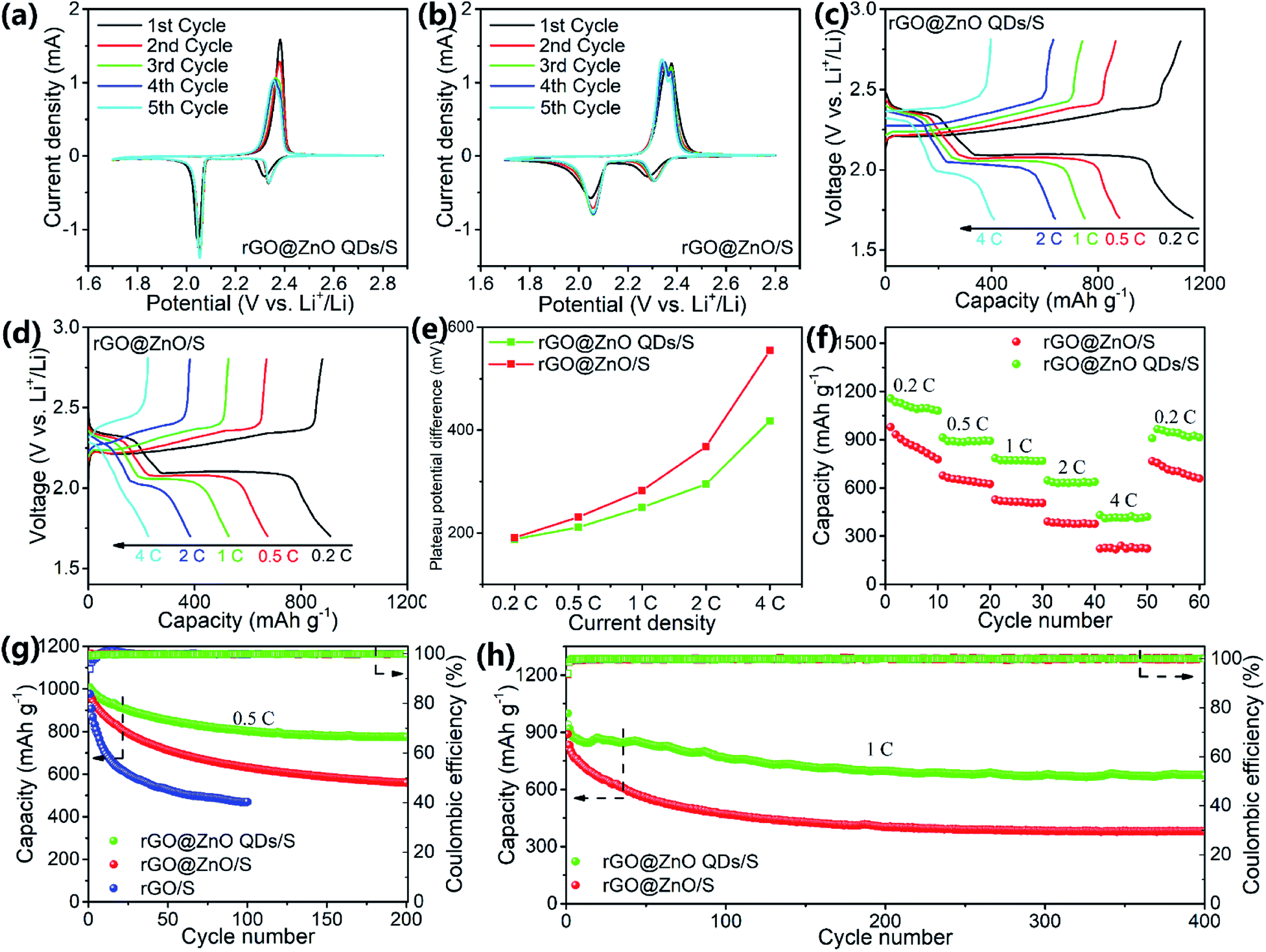 | ||
| Fig. 4 CV curves of (a) rGO@ZnO QDs/S and (b) rGO@ZnO/S; charge/discharge profiles of (c) rGO@ZnO QDs/S and (d) rGO@ZnO/S at various rates of 0.2C, 0.5C, 1C, 2C and 4C; (e) plateau potential difference and (f) rate capacities of rGO@ZnO QDs/S and rGO@ZnO/S at different current densities; cycling performances of rGO@ZnO QDs/S and rGO/S at (g) 0.5C and (h) 1C. | ||
Galvanostatic charge/discharge (GDC) tests were performed at various current rates of 0.2C, 0.5C, 1C, 2C, and 4C, and then switched back to 0.2C with 10 cycles each to examine the rate performance. The GDC curves at different rates for rGO@ZnO QDs/S and rGO@ZnO/S are presented in Fig. 4c and d. The discharge profile of rGO@ZnO QDs/S at 0.2C displayed two plateaus at 2.35 V and 2.1 V, while those of rGO@ZnO/S were at 2.31 V and 2.1 V. This is in agreement with the CV results. It is also worth noting that unlike rGO@ZnO/S, additional discharge/charge phases are shown at the end of the discharge/charge curves of rGO@ZnO QDs/S. This demonstrates that under the impact of the ZnO QDs, more thorough transformations of Li2Sn to Li2S2/Li2S and Li2Sn to S8, respectively, have occurred. Even at a high rate of 4C, the two discharge plateaus could be clearly observed for the rGO@ZnO QDs/S. In contrast, the discharge profile of rGO@ZnO/S at 4C exhibited almost no discharge platform. The overpotentials (η) of the second reduction peak for rGO@ZnO QDs/S and rGO@ZnO/S have been counted, and are presented in Fig. 4e. As we can see, the η of rGO@ZnO QDs/S is lower than that for rGO@ZnO at different rates, which means the polarization for the former sample is much lower than that for the latter one. This indicates the good catalytic effect and higher reaction kinetics of rGO@ZnO QDs.49,62 Due to the excellent catalytic effect and enhanced reaction kinetics, rGO@ZnO QDs/S achieved capacities of 1156.6, 912.9, 784.7, 645.7, and 430.2 mA h g−1 at 0.2C, 0.5C, 1C, 2C, and 4C, respectively, and 965.8 mA h g−1 after the current density was changed back to 0.2C. In contrast, rGO@ZnO/S showed lower rate capacities. The results of the rate tests have provided strong evidence that by miniaturizing the size of ZnO to the quantum dot level, excellent catalytic effect of the transition between S8 and Li2S2/Li2S and better reaction kinetics could be realized, which is in good agreement with the CV test results. Electrochemical impedance spectrum (EIS) measurements were performed on samples of rGO@ZnO QDs/S, rGO@ZnO/S, and rGO/S before cycling and after 50 cycles at 0.5C to study the interface impedances. As shown in Fig. S8 and Table S1,† due to the good electrical conductivity, rGO/S achieved the lowest resistance of charge transfer (Rct) for 40.4 Ω before the cycling test. With a low doping amount of ZnO QDs, rGO@ZnO QDs/S showed a slightly increased Rct of 44.2 Ω, which still stayed at a low level. For rGO@ZnO, because of the poor conductivity of the ZnO nanoparticles, its Rct increased to 64.8 Ω. This is much higher than the Rct for the former two samples. After 50 cycles at 0.5C, the Rct of the three samples had dropped significantly. This may be triggered by the redistribution of active materials, leading to better contact.59 However, there were still differences between the samples on Rct. Interestingly, instead of rGO/S, the rGO@ZnO QDs/S achieved the lowest Rct of 8.3 Ω, while the Rct of rGO@ZnO/S was still the highest. That should be attributed to the catalytic effect of the ZnO QDs and the enhanced reaction kinetics, which promote the conversion between S8 and Li2S2/Li2S, reducing the accumulation of non-conductive products. The low impedance of the electrode rGO@ZnO QDs/S is a key factor for its excellent rate capacities.
The three samples were discharged and charged at 0.5C to test the cycling performance. As we can see in Fig. 4g, rGO@ZnO QDs/S realized an initial discharge capacity of 1007.2 mA h g−1 and remained at 776.1 mA h g−1 after 200 cycles, by which the capacity retention was calculated to be 77.1%. Nevertheless, rGO@ZnO/S delivered only 968.9 mA h g−1, 57.8% of which was retained after the cycling test, which is much lower than that for rGO@ZnO QDs/S. As for rGO/S, the capacity decayed rapidly from 976.5 to 470.3 mA h g−1 after just 100 cycles. This was caused by the lack of ZnO/ZnO QDs to adsorb the soluble polysulfides, which has been confirmed by the polysulfide adsorption test (Fig. S9†). Thanks to the small size of the ZnO QDs, more surfaces and active sites were exposed. This makes it possible such that even with a low loading content of only 6.7%, an efficient adsorption of polysulfides for rGO@ZnO QDs could be realized.47,63 However, no obvious color change of Li2S6 was observed for rGO, implying a faint ability to restrict the shuttle effect of Li2Sn. To further investigate its cycling stability, rGO@ZnO QDs/S was tested at a high rate of 1C for 400 cylces with rGO@ZnO/S tested for comparison, and the results are shown in Fig. 4h. As we can see, rGO@ZnO QDs/S delivered a discharge capacity of 998.8 mA h g−1 at the first cycle, 918.8 mA h g−1 at the second cycle (the capacity decay was believed to be caused by the form of the equilibrium state of polysulfides in the cathode64), and 673.6 mA h g−1 remained after 400 cycles, realizing a capacity retention ratio of 73.3% (based on the discharge capacity of the second cycle). However, the initial discharge capacity of rGO@ZnO/S was only 890.5 mA h g−1, the second cycle gave a discharge capacity of 831.5 mA h g−1, and the 400th cycle gave 378.7 mA h g−1, with the capacity retention ratio calculated to be 45.5%. This is much lower than rGO@ZnO QDs/S. In addition, both samples achieved a high coulombic efficiency of above 99% during the cycling process. The results of the prolonged cycling tests at 1C have further proven the excellent cycling performance and electrode stability of rGO@ZnO QDs/S. To further examine the cycle performance at high rate, rGO@ZnO QSs/S was cycled at a high rate of 2C for 300 cycles to examine its cycling performance. As shown in Fig. S10,† rGO@ZnO QDs/S realized a capacity of 852.4 mA h g−1. After cycling at 2C for 300 cycles, the discharge capacity was 603.84 mA h g−1. Moreover, no obvious activation process was seen at the beginning of the cycle, which demonstrated the improvement effect of the ZnO QDs on the reaction kinetics.
The self-supporting composite membrane rGO@ZnO QDs/S–G with a high sulfur loading of 3.8 mg cm−2 was prepared. From Fig. 5a, we could see that the thickness of rGO@ZnO QDs/S–G was about 65 μm. After heat treatment, sulfur was evenly distributed on the surface of rGO@ZnO QDs, forming a 3D interconnected structure of the cathode (Fig. 5b–d). The XRD pattern of rGO@ZnO QDs/S–G in Fig. S12† shows both characteristic peaks of sulfur, rGO and graphene. Due to the low content of ZnO QDs, no peak of ZnO was detected. The results vividly indicated that rGO@ZnO QDs/S–G was successfully loaded with sulfur. Furthermore, the S loading was calculated to be 3.8 mg cm−2. The rGO@ZnO QDs/S–G was assembled into a 2032 coin cell and cycled at 0.2C. As shown in Fig. 5d, at the first cycle, rGO@ZnO QDs/S–G realized a discharge capacity of 742.6 mA h g−1. Subsequently, after about 20 cycles of activation, a specific capacity of 724.4 mA h g−1 was achieved. After another 100 cycles, the discharge capacity was 572.8 mA h g−1, with a capacity retention rate of 79.1%. The cycle test at 0.2C shows that the rGO@ZnO QDs/S–G self-supporting composite electrode has good cycle stability.
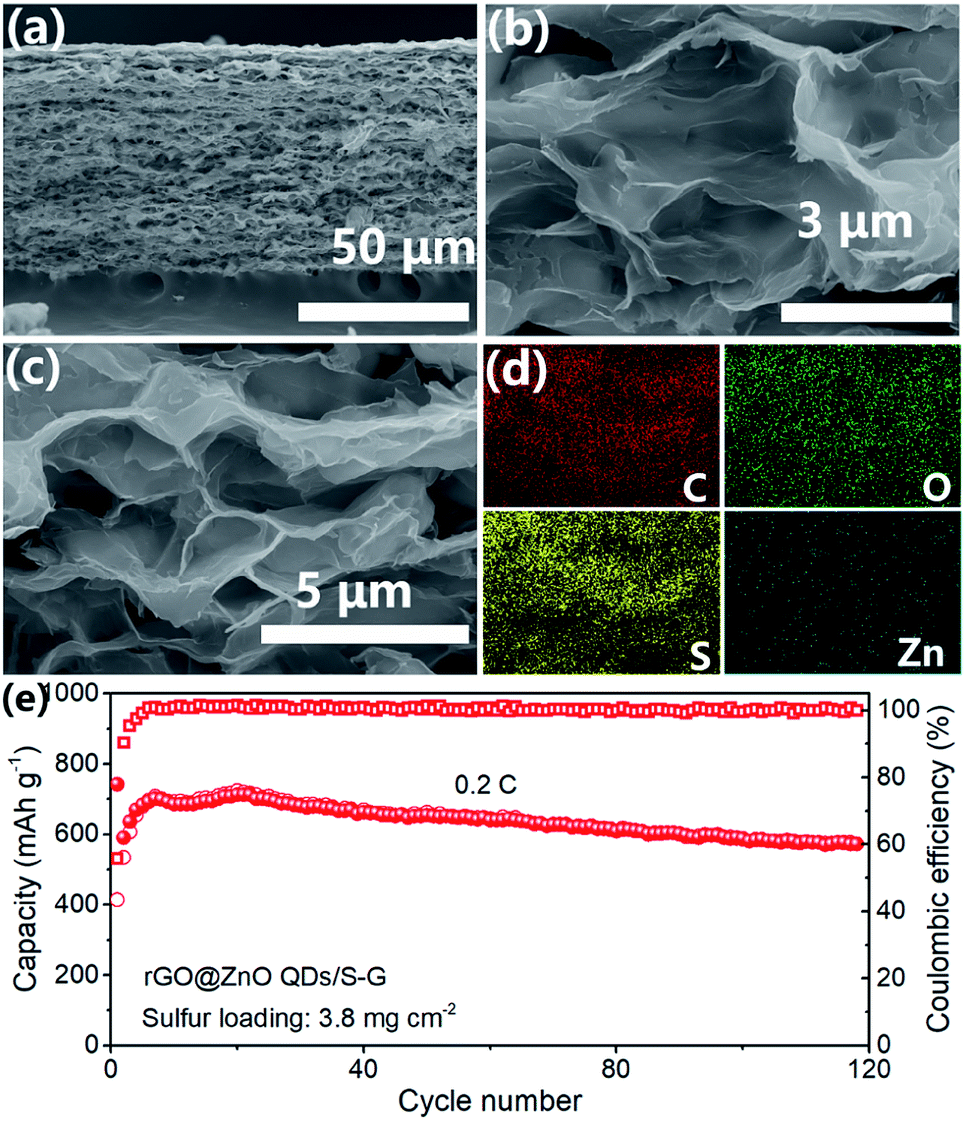 | ||
| Fig. 5 (a–c) SEM images and (d) EDS mapping results of rGO@ZnO QDs/S–G. (e) Cycling performance of rGO@ZnO QDs/S–G self-supporting composite electrode at 0.2C. | ||
Based on all the above analysis, we can see that rGO@ZnO QDs/S showed superb electrochemical performance, which could be attributed to three key factors, as shown in Fig. 6. First, ZnO QDs could catalyze the transition between S8 and Li2S2/Li2S, resulting in the enhanced reaction kinetics. The catalytic effect of ZnO QDs may be derived from the abundant edge sites of the ZnO quantum dots, which are believed to possess higher electrochemical catalytic activity because of their undercoordinated atomic structures.47,50,51 As reported by Zhang et al., reducing the particle size to the quantum dot level could significantly improve the exposed edge density, which would improve the catalytic effect.47 A similar opinion was also expressed by Zhou et al.49 Second, due to the reduced size of ZnO to quantum dots, rGO@ZnO QDs could realize lower surface impedance than rGO@ZnO with sub-micro sized ZnO when being used in lithium–sulfur cathodes. Third, with more exposed surfaces and active sites, ZnO QDs were able to adsorb polysulfides efficiently, suppressing the shuttle effect and improving the cycling performance.
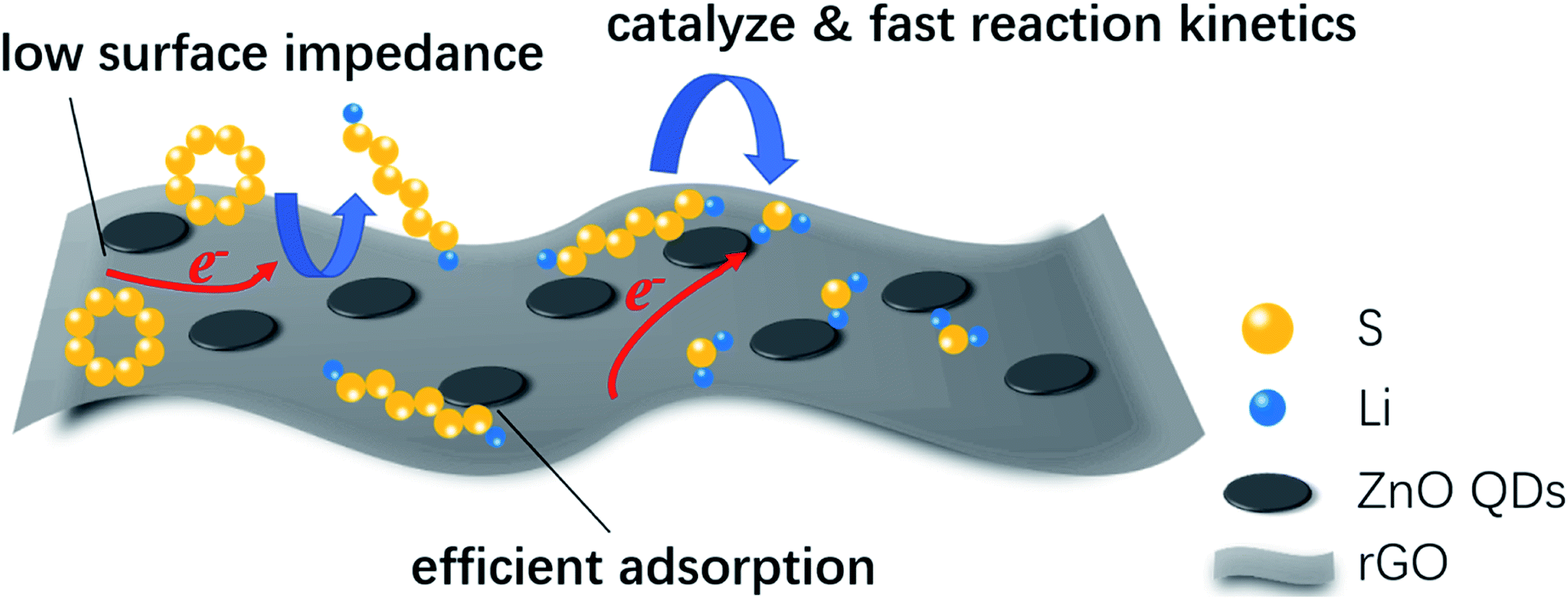 | ||
| Fig. 6 Schematic illustration of the advantages of rGO@ZnO QDs for use in lithium–sulfur cathodes. | ||
4. Conclusion
In conclusion, rGO loaded with ZnO QDs was successfully prepared and first introduced into lithium–sulfur cathodes. Benefitting from the unique characteristics of ZnO QDs, the conversion between S8 and Li2S2/Li2S was catalyzed, and the reaction kinetics was improved significantly. In addition, the reduced loading of the non-conductive, inactive ZnO decreased the surface impedance, resulting in lower polarization. The ZnO QDs could also adsorb soluble polysulfides efficiently, restraining the shuttle effect. Owing to the advantages of the rGO@ZnO QDs, excellent rate capacity and cyclic performance were realized. Our research provides new concepts for introducing quantum dots into the cathodes of lithium–sulfur batteries, which has been proven to have broad research prospects. We believe that based on our research ideas, a variety of quantum dots can be obtained and introduced into lithium–sulfur cathodes to achieve better catalytic effects and outstanding electrochemical performances.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2019YFA0705700), National Natural Science Foundation of China (51774017 and 5190401), Key Program of Equipment Pre-Research Foundation of China (6140721020103) and the Scientific Research Foundation of SGCC (52170217000L).Notes and references
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS.
- F. X. Wu and G. Yushin, Energy Environ. Sci., 2017, 10, 435–459 RSC.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- A. Manthiram, Y. Z. Fu and Y. S. Su, Acc. Chem. Res., 2013, 46, 1125–1134 CrossRef CAS.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef.
- S.-H. Chung, C.-H. Chang and A. Manthiram, Adv. Funct. Mater., 2018, 28, 1801188 CrossRef.
- Z. W. Seh, Y. M. Sun, Q. F. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969–A1976 CrossRef CAS.
- Y. Wang, X. Huang, S. Zhang and Y. Hou, Small Methods, 2018, 1700345, DOI:10.1002/smtd.201700345.
- H. W. Chen, C. H. Wang, Y. F. Dai, S. Q. Qiu, J. L. Yang, W. Lu and L. W. Chen, Nano Lett., 2015, 15, 5443–5448 CrossRef CAS.
- Z.-L. Xu, J.-K. Kim and K. Kang, Nano Today, 2018, 19, 84–107 CrossRef CAS.
- Y. Yang, G. Y. Zheng and Y. Cui, Chem. Soc. Rev., 2013, 42, 3018–3032 RSC.
- J. Zhang, H. Huang, J. Bae, S.-H. Chung, W. Zhang, A. Manthiram and G. Yu, Small Methods, 2017, 1700279, DOI:10.1002/smtd.201700279.
- X. L. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS.
- S. R. Chen, Y. P. Zhai, G. L. Xu, Y. X. Jiang, D. Y. Zhao, J. T. Li, L. Huang and S. G. Sun, Electrochim. Acta, 2011, 56, 9549–9555 CrossRef.
- X. L. Li, Y. L. Cao, W. Qi, L. V. Saraf, J. Xiao, Z. M. Nie, J. Mietek, J. G. Zhang, B. Schwenzer and J. Liu, J. Mater. Chem., 2011, 21, 16603–16610 RSC.
- J. W. F. To, Z. Chen, H. Yao, J. He, K. Kim, H.-H. Chou, L. Pan, J. Wilcox, Y. Cui and Z. Bao, ACS Cent. Sci., 2015, 1, 68–76 CrossRef.
- Y. S. Su, Y. Z. Fu and A. Manthiram, Phys. Chem. Chem. Phys., 2012, 14, 14495–14499 RSC.
- J. J. Chen, X. Jia, Q. J. She, C. Wang, Q. A. Zhang, M. S. Zheng and Q. F. Dong, Electrochim. Acta, 2010, 55, 8062–8066 CrossRef.
- L. X. Yuan, H. P. Yuan, X. P. Qiu, L. Q. Chen and W. T. Zhu, J. Power Sources, 2009, 189, 1141–1146 CrossRef CAS.
- C. Wang, X. S. Wang, Y. J. Wang, J. T. Chen, H. H. Zhou and Y. H. Huang, Nano Energy, 2015, 11, 678–686 CrossRef.
- B. Papandrea, X. Xu, Y. X. Xu, C. Y. Chen, Z. Y. Lin, G. M. Wang, Y. Z. Luo, M. Liu, Y. Huang, L. Q. Mai and X. F. Duan, Nano Res., 2016, 9, 240–248 CrossRef.
- G. M. Zhou, L. Li, D. W. Wang, X. Y. Shan, S. F. Pei, F. Li and H. M. Cheng, Adv. Mater., 2015, 27, 641–647 CrossRef.
- G. M. Zhou, L. Li, C. Q. Ma, S. G. Wang, Y. Shi, N. Koratkar, W. C. Ren, F. Li and H. M. Cheng, Nano Energy, 2015, 11, 356–365 CrossRef.
- F. Pei, L. Lin, D. Ou, Z. Zheng, S. Mo, X. Fang and N. Zheng, Nat. Commun., 2017, 8, 482 CrossRef.
- Z. Jian, H. Li, R. Cao, H. Zhou, H. Xu, G. Zhao, Y. Xing and S. Zhang, Electrochim. Acta, 2019, 319, 359–365 CrossRef CAS.
- J. Zhang, C. P. Yang, Y. X. Yin, L. J. Wan and Y. G. Guo, Adv. Mater., 2016, 28, 9539–9544 CrossRef CAS.
- J. Zhang, H. Ye, Y. X. Yin and Y. G. Guo, J. Energy Chem., 2014, 23, 308–314 CrossRef CAS.
- Y. You, W. C. Zeng, Y. X. Yin, J. Zhang, C. P. Yang, Y. W. Zhu and Y. G. Guo, J Mater Chem A, 2015, 3, 4799–4802 RSC.
- Y. Chen, S. T. Lu, X. H. Wu and J. Liu, J. Phys. Chem. C, 2015, 119, 10288–10294 CrossRef.
- G. Y. Zheng, Q. F. Zhang, J. J. Cha, Y. Yang, W. Y. Li, Z. W. Seh and Y. Cui, Nano Lett., 2013, 13, 1265–1270 CrossRef.
- X. Liu, J. Q. Huang, Q. Zhang and L. Mai, Adv. Mater., 2017, 29, 1601759 CrossRef.
- C. Tang, Q. Zhang, M. Q. Zhao, J. Q. Huang, X. B. Cheng, G. L. Tian, H. J. Peng and F. Wei, Adv. Mater., 2014, 26, 6100–6105 CrossRef.
- F. Yin, J. Ren, Y. Zhang, T. Tan and Z. Chen, Nanoscale Res. Lett., 2018, 13, 307 CrossRef.
- W. D. Zhou, Y. C. Yu, H. Chen, F. J. DiSalvo and H. D. Abruna, J. Am. Chem. Soc., 2013, 135, 16736–16743 CrossRef CAS.
- T. Ma, M. Liu, T. Huang and A. Yu, J. Power Sources, 2018, 398, 75–82 CrossRef CAS.
- Y. Kong, J. Luo, C. Jin, H. Yuan, O. Sheng, L. Zhang, C. Fang, W. Zhang, H. Huang, Y. Xia, C. Liang, J. Zhang, Y. Gan and X. Tao, Nano Res., 2017, 11, 477–489 CrossRef.
- X. J. Gao, X. F. Yang, M. S. Li, Q. Sun, J. N. Liang, J. Luo, J. W. Wang, W. H. Li, J. W. Liang, Y. L. Liu, S. Z. Wang, Y. F. Hu, Q. F. Xiao, R. Y. Li, T. K. Sham and X. L. Sun, Adv. Funct. Mater., 2019, 29, 1806724 CrossRef.
- H. B. Yao, G. Y. Zheng, P. C. Hsu, D. S. Kong, J. J. Cha, W. Y. Li, Z. W. Seh, M. T. McDowell, K. Yan, Z. Liang, V. K. Narasimhan and Y. Cui, Nat. Commun., 2014, 5, 3943 CrossRef CAS.
- X. Liang and L. F. Nazar, ACS Nano, 2016, 10, 4192–4198 CrossRef CAS.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 5682 CrossRef.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2015, 14, 271–279 CrossRef CAS.
- G. Zhao, X. Li, M. Huang, Z. Zhen, Y. Zhong, Q. Chen, X. Zhao, Y. He, R. Hu, T. Yang, R. Zhang, C. Li, J. Kong, J. B. Xu, R. S. Ruoff and H. Zhu, Chem. Soc. Rev., 2017, 46, 4417–4449 RSC.
- J. Cao, C. Chen, Q. Zhao, N. Zhang, Q. Q. Lu, X. Y. Wang, Z. Q. Niu and J. Chen, Adv. Mater., 2016, 28, 9629–9636 CrossRef CAS.
- Z. Cheng, Z. Xiao, H. Pan, S. Wang and R. Wang, Adv. Energy Mater., 2018, 8, 1702337 CrossRef.
- X. Y. Tao, J. G. Wang, C. Liu, H. T. Wang, H. B. Yao, G. Y. Zheng, Z. W. Seh, Q. X. Cai, W. Y. Li, G. M. Zhou, C. X. Zu and Y. Cui, Nat. Commun., 2016, 7, 11203 CrossRef CAS.
- Z. L. Xu, S. Lin, N. Onofrio, L. Zhou, F. Shi, W. Lu, K. Kang, Q. Zhang and S. P. Lau, Nat. Commun., 2018, 9, 4164 CrossRef.
- Y. Pang, J. Wei, Y. Wang and Y. Xia, Adv. Energy Mater., 2018, 8, 1702288 CrossRef.
- G. Zhou, S. Zhao, T. Wang, S. Z. Yang, B. Johannessen, H. Chen, C. Liu, Y. Ye, Y. Wu, Y. Peng, C. Liu, S. P. Jiang, Q. Zhang and Y. Cui, Nano Lett., 2020, 20, 1252–1261 CrossRef CAS.
- H. T. Wang, Q. F. Zhang, H. B. Yao, Z. Liang, H. W. Lee, P. C. Hsu, G. Y. Zheng and Y. Cui, Nano Lett., 2014, 14, 7138–7144 CrossRef CAS.
- G. Babu, N. Masurkar, H. Al Salem and L. M. Arava, J. Am. Chem. Soc., 2017, 139, 171–178 CrossRef CAS.
- Z. B. Jiao, X. G. Guan, M. Wang, Q. M. Wang, B. H. Xu, Y. P. Bi and X. S. Zhao, Chem. Eng. J., 2019, 356, 781–790 CrossRef.
- X. Cao, D. Qi, S. Yin, J. Bu, F. Li, C. F. Goh, S. Zhang and X. Chen, Adv. Mater., 2013, 25, 2957–2962 CrossRef CAS.
- U. N. Maiti, J. Lim, K. E. Lee, W. J. Lee and S. O. Kim, Adv. Mater., 2014, 26, 615–619 CrossRef CAS.
- K. Krishnamoorthy, M. Veerapandian, R. Mohan and S. J. Kim, Appl. Phys. Mater. Sci. Process, 2012, 106, 501–506 CrossRef CAS.
- W. Kang, X. Jimeng and W. Xitao, Appl. Surf. Sci., 2016, 360, 270–275 CrossRef CAS.
- K.-F. Lin, H.-M. Cheng, H.-C. Hsu, L.-J. Lin and W.-F. Hsieh, Chem. Phys. Lett., 2005, 409, 208–211 CrossRef CAS.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Nat. Commun., 2010, 1, 73 CrossRef.
- Y. S. Su and A. Manthiram, Chem. Commun., 2012, 48, 8817–8819 RSC.
- X. Huang, Z. Wang, R. Knibbe, B. Luo, S. A. Ahad, D. Sun and L. Wang, Energy Technol., 2019, 7, 1801001 Search PubMed.
- D. H. Liu, C. Zhang, G. M. Zhou, W. Lv, G. W. Ling, L. J. Zhi and Q. H. Yang, Adv. Sci., 2018, 5, 1700270 CrossRef.
- Z. Sun, J. Zhang, L. Yin, G. Hu, R. Fang, H. M. Cheng and F. Li, Nat. Commun., 2017, 8, 14627 CrossRef.
- G. M. Zhou, H. Z. Tian, Y. Jin, X. Y. Tao, B. F. Liu, R. F. Zhang, Z. W. Seh, D. Zhuo, Y. Y. Liu, J. Sun, J. Zhao, C. X. Zu, D. S. Wu, Q. F. Zhang and Y. Cui, Proc. Natl. Acad. Sci. U.S.A., 2017, 114, 840–845 CrossRef CAS.
- M. N. Liu, F. M. Ye, W. F. Li, H. F. Li and Y. G. Zhang, Nano Res., 2016, 9, 94–116 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04986g |
| This journal is © The Royal Society of Chemistry 2020 |