
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccelerated trypsin autolysis by affinity polymer templates†
Daniel Smolina,
Niklas Tötsch b,
Jean-Noël Gradb,
Jürgen Lindersa,
Farnusch Kaschanib,
Markus Kaiserb,
Michael Kirsch*c,
Daniel Hoffmann*b and
Thomas Schrader*a
b,
Jean-Noël Gradb,
Jürgen Lindersa,
Farnusch Kaschanib,
Markus Kaiserb,
Michael Kirsch*c,
Daniel Hoffmann*b and
Thomas Schrader*a
aFaculty of Chemistry, University of Duisburg-Essen, 45117 Essen, Germany. E-mail: thomas.schrader@uni-due.de
bFaculty of Biology, University of Duisburg-Essen, 45117 Essen, Germany
cUniversity Clinics, Essen, Germany
First published on 4th August 2020
Abstract
Self-cleavage of proteins is an important natural process that is difficult to control externally. Recently a new mechanism for the accelerated autolysis of trypsin was discovered involving polyanionic template polymers; however it relies on unspecific interactions and is inactive at elevated salt loads. We have now developed affinity copolymers that bind to the surface of proteases by specific recognition of selected amino acid residues. These are highly efficient trypsin inhibitors with low nanomolar IC50 levels and operate at physiological conditions. In this manuscript we show how these affinity copolymers employ the new mechanism of polymer-assisted self-digest (PAS) and act as a template for multiple protease molecules. Their elevated local concentration leads to accelerated autolysis on the accessible surface area and shields complexed areas. The resulting extremely efficient trypsin inhibition was studied by SDS-PAGE, gel filtration, CD, CZE and ESI-MS. We also present a simple theoretical model that simulates most experimental findings and confirms them as a result of multivalency and efficient reversible templating. For the first time, mass spectrometric kinetic analysis of the released peptide fragments gives deeper insight into the underlying mechanism and reveals that polymer-bound trypsin cleaves much more rapidly with low specificity at predominantly uncomplexed surface areas.
Introduction
Protein autolysis – a natural process difficult to control
Most exocrine proteases undergo autolysis – they digest themselves. Prominent examples are trypsin and chymotrypsin, but also alkaline and neutral proteases exhibit a significant autolysis rate especially at elevated temperatures. Under physiological conditions, autolysis may be crucial for their biological function, but it often hampers their use in biotechnology.1External control of enzymatic self-digest in both directions (inhibition or acceleration) is not easily achieved without side-effects: early work by Hatate and Toyomizu reported accelerated autolysis of trypsin after exposure to polymeric oxidized methyl linolenate, but demonstrated that first ε-amino groups formed Schiff bases with carbonyl groups from the polymer producing an enzymatically inactive covalent polymer/protein adduct.2 A somewhat related template effect was recently discovered for CAPN3, a protease in skeletal muscles with unusually rapid autolytic activity. When the autolytic N- and C-terminal fragment form a noncovalent complex, they restore proteolytic function (intermolecular complementation iMOC).3
Neutral proteases are inactivated at higher temperatures because of autolysis, which involves local unfolding of specific solvent-exposed regions. These could be suppressed by surface-located mutations of N-terminal loops in a neutral zinc metalloprotease.4 Recently, a stabilization effect for an alkaline serine protease was observed caused by copper ions (5 mM) as the result of a decrease in both autolysis and thermoinactivation rates.5 Autolysis has been identified as the primary mode of subtilisin activity loss in a heavy-duty liquid detergent (HDLD) formulation; it could in part be repaired by thermodynamic stabilization and/or kinetic inhibition.6 In general, enzymes can be protected from protease digestion by modification with hydrophilic well-hydrated polymers, due to their shielding effects.7 A very interesting contribution along these lines came from Sasai et al., who synthesized highly stabilized polymer–trypsin conjugates with remarkable autolysis resistance. Thus, a vinylmethylether–maleic acid copolymer (VEMAC) was covalently bound to trypsin's lysine amino groups via multi-point attachment. The modified trypsin construct showed strong resistance against autolysis and unaltered conformation (CD spectra), explained by mutual electrostatic repulsion of the negatively charged surfaces of the VEMAC-coated trypsin.8
On the other hand, it may be highly desirable to accelerate autolysis of problematic or even pathogenic proteins. In principle, this may be achieved by increasing their local concentration by assembly on a large template molecule, e.g. an oppositely charged polymer. Such an example has recently been presented for the first time by Lv et al., who were able to accelerate trypsin's self-digest in the presence of polyelectrolytes, most prominently dextrane sulfate.9 In their seminal work the authors demonstrated how direct interaction between trypsin and the negatively charged polymer would greatly diminish the enzymatic activity of the protease, through a rapid self-cleavage into smaller fragments. They called this effect “autolysis-acceleration protease inhibition” (AA-PI) and investigated it with MALDI-TOF, SDS-PAGE, DLS and TEM. From their experiments, the authors deduced that doubly charged monomer units are required, and they also found that, using such polymers, insulin could be protected against trypsin degradation, but only at low ionic strength. Unfortunately, these polymers completely lose their inhibition ability under elevated salt loads, i.e. under physiological conditions, most likely because they primarily rely on multiple electrostatic attraction. At the end of their account, Lv et al.9 conclude that more specific AA-PIs are necessary, and that these may be able to inhibit deleterious pathogenic proteases.
Designed affinity copolymers as specific AA-PIs
We have developed two such specific AA-PIs by supramolecular chemistry. They inhibit their target protease in a highly efficient manner, and do not lose their activity under elevated salt loads. Among the most prominent digestion enzymes they are specific against trypsin and the closely related kallikrein, which they inhibit in a drastically substoichiometric manner. They are part of our project to develop protein-selective copolymers from amino-acid-selective monomer units. In this case, we developed two binding monomers tailored for lysine/arginine and serine, the two most abundant amino acids on the trypsin surface. These recognize their targets differently from simple electrolytes (Fig. 1). A lysine or arginine residue, e.g., is noncovalently bound by the bisphosphonate monomer with a combination of noncovalent interactions, including hydrogen bond reinforced Coulomb attraction, but also π-cation and dispersive interactions as well as the hydrophobic effect (displacement of high energy water). Serine, on the other hand is covalently bound by ester formation with aminomethylphenylboronic acid. Linear copolymerization of one or both co-monomers in the absence of the protein target leads to water-soluble copolymers that are able to complex the protein surface of trypsin by multiple (non)covalent interactions. Since trypsin's active site is not directly flanked with lysines, arginines or serines, its proteolytic activity is not compromised by this complexation event. Up to 10 copies of the serine protease can be accommodated on the linear copolymer (Fig. S11†), leading to a high local concentration that accelerates mutual self-digest. In this paper, we discuss the features of this polymer-assisted self-digest, present a computational model and provide deeper insight into the putative mechanism by proteomics MS analysis.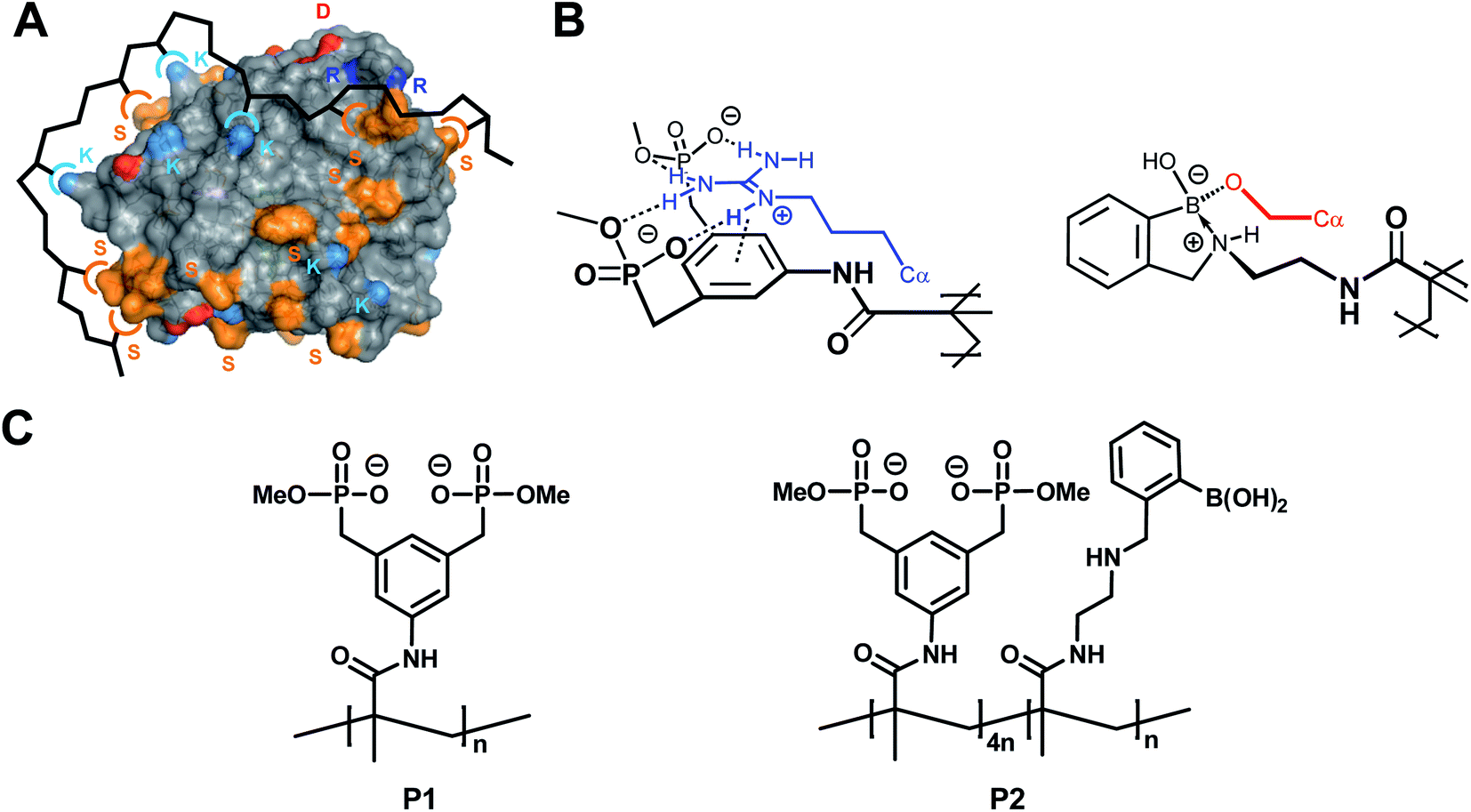 | ||
| Fig. 1 Concept of multivalent protein surface recognition by linear affinity copolymers from amino acid-selective binding monomers. (A) Schematic of the affinity copolymer concept – each color symbol represents an amino acid/binding monomer pair. (B) Molecular recognition pattern for the specific targeting of arginine (left) and serine residues (right) on the protein surface by designed binding sites in affinity copolymers. Note the noncovalent combination of ionic hydrogen bonds and π-cation attraction between bisphosphonate dianion and alkylguanidinium side chain (blue), and reversible covalent formation of a cyclic boronate ester between the aminomethylphenylboronic acid moiety and the primary alcohol (red). (C) Lewis structures of homopolymer P1 and copolymer P2. | ||
The above-mentioned two trypsin-specific polymers represent a larger family of affinity copolymers that are able to engage in multipoint binding10 to hot spots on proteins: they rely on the construction of a series of specific binding monomers for each class of amino acids. Those that are complementary to critical residues on the hot spot, are mixed in defined stoichiometric ratios and subjected to radical copolymerization. The resulting linear copolymers display high affinity and surprising protein specificity by cooperative binding.11 Very recently we developed by this method highly specific polymeric inhibitors for disease-relevant proteases.12 Their specificity comes from the multivalent combination of well-defined binding motifs for critical amino acids on the respective protein surface.13
Results and discussion
Synthesis and properties of P1 and P2
Two powerful polymeric trypsin binders were synthesized from their respective methacrylamide comonomers equipped with a bisphosphonate anion10 and an aminomethylphenylboronic acid moiety.12 Polymerization was started by AIBN and conducted for 24 h in water/DMF. After lyophilization, the crude products were redissolved in ultrapure water and subjected to ultrafiltration to remove monomers and oligomers. Final lyophilization furnished colorless powders with excellent water solubility (yields 55–60%). NMR integration confirmed that the original stoichiometric monomer ratio was retained in P2. GPC produced average molecular weights for both polymers of ∼170 kD with relatively low polydispersities of 1.4–1.5, most likely due to the ultrafiltration cut-off. These molecular weights correspond to a degree of polymerisation DP of 600 for P1 and 730 for P2. The copolymerization parameters were determined earlier for the bisphosphonate monomer and a related polar comonomer with a secondary alcohol. In water, r1 and r2 remained between 0.3 and 2.0 (Fineman–Ross), while in DMF they became 1.0, indicating ideal statistical copolymerization.Tailored affinity polymers P1/P2 accelerate trypsin self-digestion
When we studied the two new trypsin inhibitors with multiple arginine/lysine and serine binding sites (P1: bisphosphonate homopolymer, P2: bisphosphonate/boronate copolymer), we discovered some unusual features. Contrary to most other polymers, including the naturally occurring polypeptides like aprotinin (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), IC50 values dropped to drastically substoichiometric ratios, indicating a 1:10 (P2) or even 1:100 (P1) polymer:protein stoichiometry (see ESI†). In addition, prolonged incubation of enzyme and polymer further lowered IC50 values, consistent with a so-called slow-onset mechanism (see ESI†).14 These initial observations cannot be explained with conventional substrate–polymer competition, but agree very well with the assumed template effect of the affinity polymers for enhanced enzymatic self-cleavage.
:1), IC50 values dropped to drastically substoichiometric ratios, indicating a 1:10 (P2) or even 1:100 (P1) polymer:protein stoichiometry (see ESI†). In addition, prolonged incubation of enzyme and polymer further lowered IC50 values, consistent with a so-called slow-onset mechanism (see ESI†).14 These initial observations cannot be explained with conventional substrate–polymer competition, but agree very well with the assumed template effect of the affinity polymers for enhanced enzymatic self-cleavage.
First evidence for the postulated polymer accelerated self-digest (PAS) came from gel electrophoresis of the trypsin cleavage products in the absence and presence of affinity polymers. An incubation period of trypsin with P1 or P2 in Tris buffer (37 °C) in the absence of any enzyme substrates was terminated at selected time points by the addition of SDS PAGE loading buffer containing dithiothreitol (DTT), and the integrity of the protein was analyzed by SDS-PAGE (Fig. 2A).15 While native trypsin needed 4 h for complete self-digest (all bands), this stage was already reached after 10–15 min in the presence of 0.1 equiv. of P1 or P2. The largest bands appear to be cleaved faster in the polymer-catalyzed case, leaving a slightly altered molecular weight distribution compared to trypsin alone. The smallest fragments accumulated at the bottom of the gel indicating complete proteolytic destruction into small peptides. When time-dependent CD spectra were recorded at identical protein/polymer ratios (37 °C), the observed effects from gel electrophoresis were strongly supported (Fig. 2B): trypsin self-digest in the absence of polymer needed more than 30 min to dissolve the typical CD band at 212 nm; with P1 or P2, however, this band disappeared already after 5 min (for full CD kinetics see ESI†).
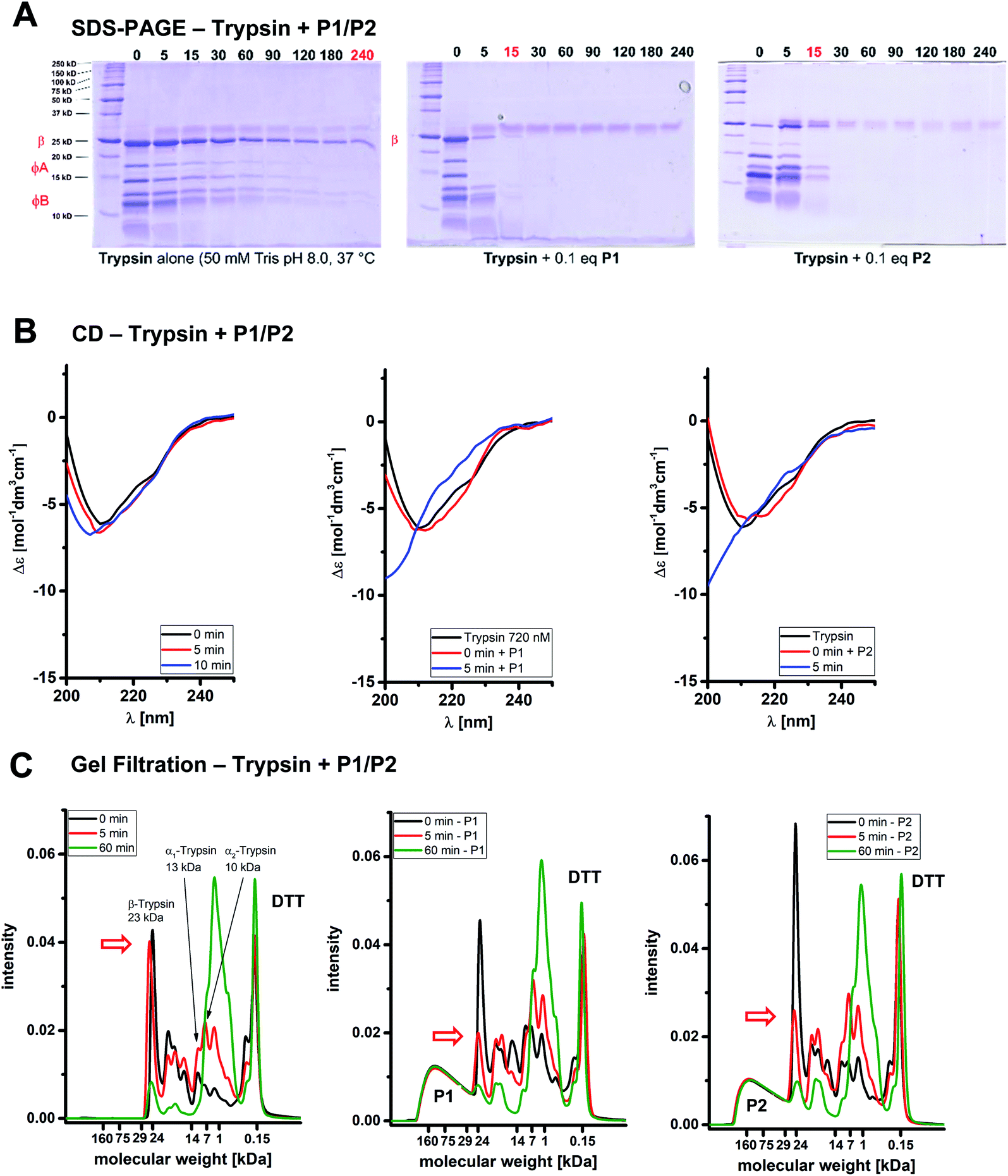 | ||
| Fig. 2 Affinity polymers P1/P2 accelerate trypsin self-digestion. (A) SDS-PAGE. Autolysis of 60 μM trypsin over time (75 mM TRIS buffer, pH 8.0, 37 °C). Left: Trypsin alone; right: trypsin with 6.7 μM P1/P2. (B) CD spectroscopy. Time-dependent CD spectra monitoring the kinetics of polymer-accelerated trypsin autolysis (60 μM trypsin, 75 mM borate buffer). Left: Trypsin alone; right: trypsin with 6.7 μM P1/P2. (C) Gel filtration. Gel filtration chromatograms depicting the molecular weight distribution of trypsin alone and in the presence of P1 during the self-digest process (100 mM borate buffer, denaturated with 6 M urea and 30 mM DTT). Left: Trypsin alone, right: trypsin with P1/P2. P1/P2 concentrations refer to the full polymer molecular weight. | ||
Gel filtration is a powerful method to monitor the molecular weight distribution of proteins;16 we wondered if it could be utilized to demonstrate trypsin self-cleavage into smaller fragments in a time-dependent manner. Our first attempts failed because virtually no changes occurred in the initial chromatogram under the established conditions for catalyzed self-digest (60 μM trypsin, 6.7 μM P1, 100 mM borate buffer, 37 °C). However, in the presence of both DTT (10 mM) which reduces disulfide bonds17 – and a denaturing agent (1 M urea), the desired changes became visible (Fig. 2C): trypsin bands at 18 mL (22 kDa) and 20–23 mL elution volume (5–17 kDa) decreased rapidly only in the presence of P1 or P2 (already after 5 min), and furnished new increasing autolysis bands at 23–28 mL corresponding to molecular weights between 0.5 and 1 kDa. As a control, the affinity polymer band was constant at 11–15 mL (30–100 kDa), and DTT likewise appeared constant at 30 mL (0.3 kDa). Molecular weights were estimated from retention volumes of reference proteins. Intriguingly, in the polymer-catalyzed system, the largest band (β-trypsin)18 was cleaved much more rapidly than the smaller bands. In the growing product bands, molecular weights were gradually shifted towards smaller fragments, illustrating the constant degradation even of medium sized protein fragments on the polymer template.
Another method for chromatographic separation of proteins from polymers is capillary zone electrophoresis CZE.19 Under the above-described standard conditions, native trypsin reproducibly elutes as a typical mixture of large bands with a characteristic ratio (see ESI†). In the presence of very small amounts of P1 (0.5 mol%) and P2 (0.8 mol%), however, trypsin was fully degraded after less than 20 min, whereas self-digest in their absence remained slow.20 The stoichiometric polymer/protein ratio for these experiments leading to complete trypsin degradation reached 1:185. Parallel enzyme assays confirmed the loss of 95% of the original trypsin activity. These values place P1 and P2 among the most potent trypsin inhibitors known to date.21
Detailed analysis of the inhibition mechanism and the underlying protein polymer recognition event
For a better understanding we investigated the complex formation between trypsin and the polymers more closely.Substoichiometric and slow onset inhibition prevailed in various buffers, as long as the pH was kept slightly acidic or slightly basic (pH 6–9). Preincubation at acidic pH < 3 completely abolished all effects. Under these conditions, self-digest does not occur because trypsin activity is greatly reduced if the pH is below 7 or above 9. Moreover, all bisphosphonate units become protonated and lose their affinity for basic amino acid residues.22 In neutral buffer, however, even at high ionic strength such as 150 mM PBS the inhibition efficiency of P1 and P2 is not lowered, which points to a powerful combination of Coulomb attraction with other (non)covalent interactions, a built-in feature of our amino acid-selective binding monomers. This contrasts with the performance of non-specifically binding polyelectrolytes such as dextrane sulfate, which largely rely on multiple electrostatic attraction, and completely lose their inhibition ability under elevated salt loads.9
Attempted recovery experiments with cationic polymers were frustrated according to both CD spectra as well as enzyme activity assays: irreversible inhibition occurred in various buffers; in other words, the enzyme did not return to its active native state even when the polymer is completely peeled off its surface (see ESI†). Such a reversible denaturation was observed when lysozyme was bound and inhibited by a specific affinity polymer, followed by addition of polyarginine, which turned on enzymatic activity again.23 These experiments rule out a conventional inhibition mechanism that involves reversible coverage of the active site.
In order to exclude unspecific effects from aggregate formation, dynamic light scattering (DLS)24 was performed on trypsinogen (no self-digest) and the isolated polyanionic polymers: monodisperse hydrodynamic radii of 2.4 nm for trypsinogen (lit.: 2.2 nm)25 and 10 nm for both polymers P1/P2 are in good agreement with single protein and polymer molecules in a non-aggregated state. Further confirmation comes from GPC determination of polymer molecular weights at Mn ≈ 100 kDa. Intriguingly, the hydrodynamic radius of P2 decreased from 10 to ≈7 nm when it was complexed with trypsinogen, indicating a more tightly folded state of the polymer (see ESI†).
Very similar results came from PFG-NMR (Pulsed Field Gradient), which produced the hydrodynamic radii of 1 nm for trypsin and 10 nm for the polyanionic polymers, strongly suggesting that polymer and protein did not form aggregates.26 PFG-NMR measurements also were consistent with a drop of polymer radius to about 70% on complex formation with trypsin (see ESI†).27 This effect was independently predicted by a computer model presented below. Fluorescence titrations only led to moderate quenching; for P1 a binding isotherm could be evaluated quantitatively by nonlinear regression and furnished a moderate Kd value of 540 nM at a 9:1 stoichiometry (see ESI†).10 This is very important, because it reveals for the first time that 9 trypsin molecules can be accommodated on one polymer strain simultaneously.28 Since both binding partners carry multiple and opposite charges, electrostatic attraction will most likely be maximized by a substantial folding of the polyanionic polymer around its 9 polycationic trypsin guests leading to a collapsed complex state, as evidenced by DLS and PFG-NMR. Moderate polymer protein affinity enables dissociation of large cleavage products from the complex and secures catalytic turnover, but limits catalytic efficiency. These features are also discussed in the simulation below.
The new polymer-accelerated self-digest can be simulated by a simple theoretical model
In line with the above-discussed experimental observations we propose the following mechanistic model which correlates well with that from Lv et al.:9 Polymer Accelerated Self-digest (PAS) is a process in which a polymer in aqueous solution accelerates the self-digest of a protease. Basic ingredients of PAS are a protease that is cleavable by its own kind, and a flexible polymer with affinity to this protease, though not blocking its active site. The polymer has to be long enough to bind several protease molecules. Fig. 3A illustrates the PAS mechanism schematically for the protease trypsin.29 In the solution of polymer and protease (a) a polymer molecule binds several protease molecules (b), often in a multivalent fashion, leading to formation of loops and to polymer collapse. This interaction between polymer and protein concentrates protease molecules in a small volume, causing efficient cannibalistic autolysis. (c) This cannibalistic self-digest is probably incomplete because it requires a small amount of functional protease molecules until the end. (d) The more flexible digest peptides compete with intact, well-structured protease molecules that are still in solution. The result of this competition is likely a release of the digested peptides, which completes the catalytic cycle and allows the polymer to bind further protease molecules. (e) Protease binding, autolysis, and digest release proceed continuously as long as a sufficient number of protease molecules are available in the solution.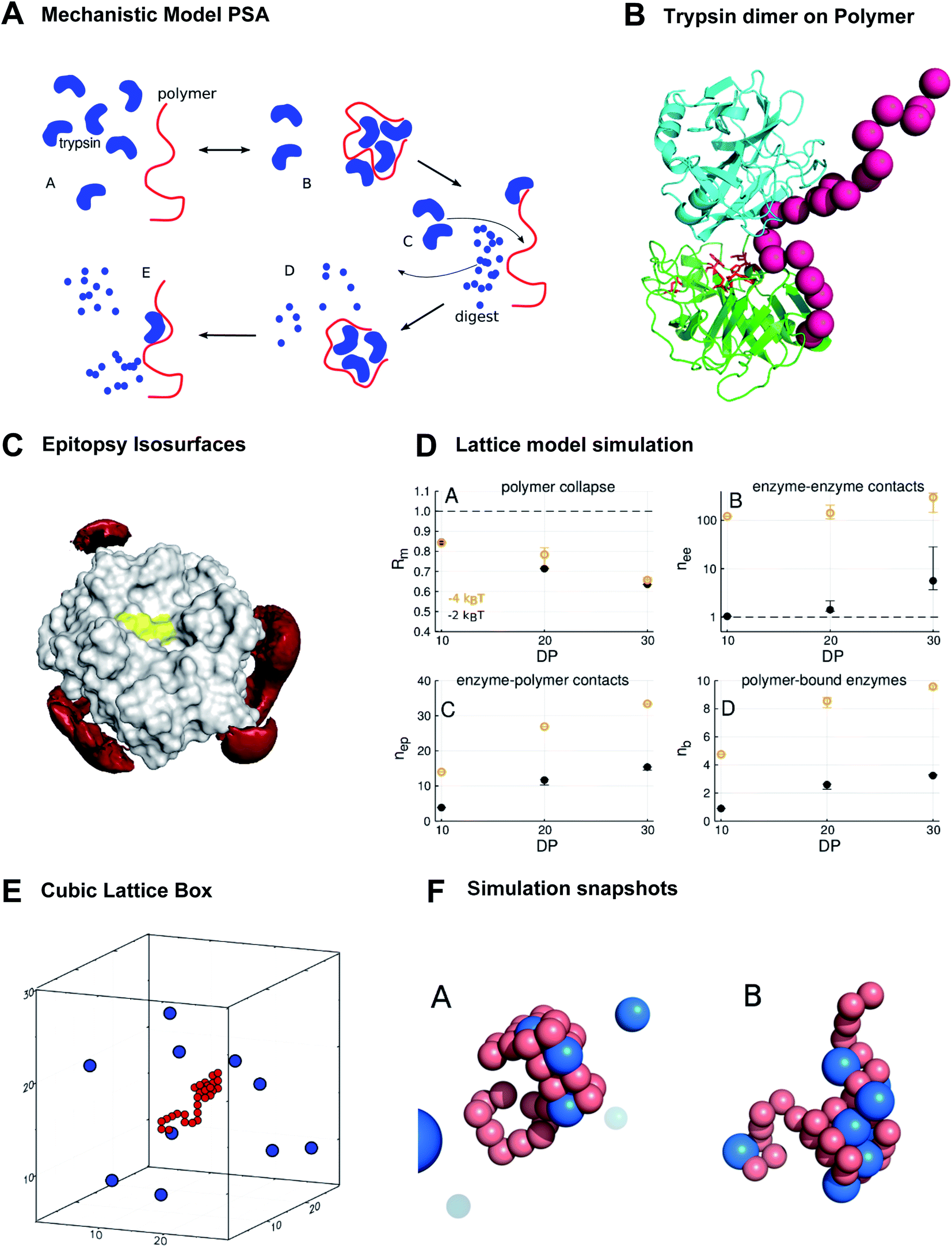 | ||
| Fig. 3 The new polymer-accelerated self-digest can be simulated by a simple computational model. (A) Proposed mechanistic model starting with intact protease and polymer (A), followed by polymer collapse (B), cannibalistic autolysis (C) and released peptide fragments (D and E). (B) Polymer (red) stabilizes a trypsin dimer (blue, green), which favors cannibalistic autolysis (active center in red sticks). (C) Red isosurfaces of increased affinity level (−1kBT) between trypsin and the bisphosphonate monomer, which binds to K/R without shielding the active site (yellow). (D) Lattice model simulation results. DP = degree of polymerization. Polymer contact −2kT/−4kT. (A) Mean monomer–monomer distance Rm; (B) average number nee of enzyme–enzyme contacts; (C) average number nep of enzyme–polymer contacts; (D) average number nb of enzymes bound to polymer (total enzymes = 10). (E) Cubic lattice box with 10 enzymes and polymer of DP.30 (F) Simulation snapshots. (A) 30-mer polymer (red) interacts with trypsin molecules (blue) at contact energies Econt = −2kBT (A) and Econt = −4kBT (B) at the same trypsin concentration. Only in (B) enzymes contact each other, illustrating the importance of strong single interactions. | ||
This mechanism is a beautiful example of the powerful influence of multivalency on biological activity. Only the synergistic binding of many enzyme molecules on the same polymer template leads to the observed high local concentrations and enables the polymer to drastically accelerate trypsin autolysis. A visualization of a stabilized trypsin dimer is given in Fig. 3B (trypsin from PDB 2ptn). These multiple protein–polymer contacts must also be reversible and transient to maintain a catalytic cycle where cleaved peptide fragments dissociate from the polymer template.30 If the PAS mechanism sketched above is correct, we should be able to recapitulate major elements of this mechanism with computational models that share essential features with the real system.
We used epitopsy31 to computationally screen the surface of trypsin with BP monomers, the main building block of our polymers. The analysis shows several patches on trypsin that have elevated affinity for BP, though the active site of trypsin is not covered (Fig. 3C). This is exactly the pattern expected for a protein that binds multivalently to a BP-based polymer, while the polymer spares the active site region and thus does not inhibit proteolytic activity.
A pattern as the one in Fig. 3C is necessary for PAS but it only accounts for interactions of monomers with a single trypsin molecule. Other observed features of PAS cannot be understood at this level, e.g. the collapse of the polymer in the presence of trypsin molecules, and of course the highly efficient self-digest of trypsin in the presence of the polymer. These aspects have to be addressed with models comprising at least a complete polymer molecule and several trypsin molecules. Since the system of polymer and trypsin molecules in aqueous solution is complex and has a huge configurational space, we opted for a minimalist lattice model that allows for a more thorough sampling of configurational space.32 This lattice model consists of a flexible polymer model surrounded by a number of trypsin molecules, which can bind multivalently to the polymer, with each polymer–trypsin contact contributing a fixed free energy of binding (“contact energy” Econt).
We report here some of the key results from the lattice model simulations (Fig. 3D). These results are in good agreement with experiment and support the PAS model. First, the simulations predict that the interaction with trypsin molecules leads to polymer collapse: the mean monomer–monomer distance Rm, measured as fraction of the mean monomer–monomer distance of the free polymer, drops significantly in the presence of trypsin (Fig. 3D(A)). The drop of Rm becomes more drastic with increasing degree of polymerization (DP), from Rm = 0.84 at DP = 10 to about Rm = 0.65 at DP = 30, which is close to the experimentally observed collapse to 70% of the polymer radius on interaction with trypsin (see ESI†).
Interestingly, it does not need many polymer-bound proteases for a polymer collapse. Even if we have on average only a single protease molecule bound to the polymer molecule with a weak contact energy, as in the case of DP = 10 and Econt = −2kBT (point in lower left corner of Fig. 3D(D)), the value of Rm drops to 0.84 (point in upper left corner of Fig. 3D(A)). This is because protease molecules in the model are typically bound to the polymer multivalently with on average 3 to 5 contacts per protease molecule (compare Fig. 3D(C and D)). Fig. 3F illustrates this with two simulation snapshots. Thus, multivalent protease–polymer binding stabilizes loops and coiling of the polymer and therefore leads to the observed collapse.
The crucial feature of PAS – that is supported by the lattice simulations – is the enrichment of protease–protease contacts in the presence of the polymer (Fig. 3D(B)). The polymer collects proteases from the solution (Fig. 3D(D)) and fixes them by multivalent binding (Fig. 3D(C)) in a small fraction of the available volume. Thus, in comparison to the polymer-free solution, the number nee (with index ee for “enzyme–enzyme”) of protease–protease contacts can increase by orders of magnitude, depending slightly on DP, but strongly on protease–polymer affinity (Fig. 3D(B)).
The increase of nee with DP is essentially due to the higher abundance of binding sites in larger polymers. The increase of nee with protease–polymer affinity (in Fig. 3D(B) shift from black points, −2kBT, to orange points, −4kBT) can be attributed mainly to facilitated protein capture by higher affinity binding sites on the polymer.
However, there is also a more subtle effect that contributes to the upward shift of nee for stronger protease–polymer affinity: for weaker binding energy Econt per protease–polymer contact, protease–polymer complexes with single contacts are thermodynamically unstable. Therefore, stable binding conformations that we observe under these conditions will typically involve per protease several contacts to the polymer. Thus, the polymer will occupy a substantial fraction of the surface of each bound protease, and these regions will then not be available for contacts to other proteases – the polymer partly shields proteases against each other (Fig. 3F(A)). Conversely, for stronger protease–polymer contact energies Econt, even single polymer–protease contacts may be thermodynamically stable, enabling a bound protease to interact with other proteases (Fig. 3F(B)). In our simulations we found that at a contact energy of −2kBT, a protease molecule was on average engaged in 4.3 to 4.7 contacts with the polymer, while at −4kBT the numbers were lower at 2.9 to 3.5, leaving more opportunities for protease–protease contacts.
Protease–polymer aggregation, a key aspect of the above-postulated mechanism, can be observed with the naked eye at elevated concentrations (see ESI†): addition of trypsin to 0.1 equiv. P1 results in instant precipitation of the polymer/protein complex. During the following 15 min, however, the solution becomes gradually less cloudy due to trypsin degradation, finally leading again to a clear solution. Another addition of trypsin repeats the whole cycle until a clear polymer solution is again obtained. For an efficient turnover it is very important, that the trypsin fragments leave the polymer template in order to avoid product inhibition or poisoning of the catalyst.33
A somewhat related principle was recently used to promote B and T cell activation by a polymeric bifunctional multivalent antigen.34 Here, the multivalent presentation of the B cell epitope (DNP) on a ROMP polymer led to efficient oligomerization of the BCR, which in turn induced both signaling and uptake. Similar to our case with PAS, a synthetic polymer acts as a multivalent template to bring about a biologically significant oligomerization event.
Mass spectrometry: trypsin bound to polymers P1/P2 cleaves with low specificity predominantly at uncomplexed surface areas
For deeper insight into the templated cleavage process triggered by the addition of the polymer P1 we performed a mass spectrometric analysis of the released peptide fragments. In a bottom-up approach we initiated trypsin self-digest under optimized conditions (borate buffer pH 8.0) in the presence and absence of P1 and at varying reaction times. The reactions were terminated (5% formic acid) and subsequently, an excess of acetone was added which is known to precipitate undigested proteins and partially digested polypeptides while smaller oligopeptides generated by autolysis at the different time points remain in solution (Fig. S19†).35 After removing insoluble matter by centrifugation acetone was evaporated and the remaining peptides were analyzed by LC-MS/MS. After stringent filtering 49 specific trypsin degradation peptides were identified, whose intensities were normalized (Fig. S20 and File S20†). Their majority (31 peptides) showed a marked increase in abundance upon treatment with P1, a small fraction (6 peptides) showed partially decreased abundance, and 12 peptides showed no marked effect.Interestingly, the surface-exposed cleavage sites from peptides with increased or unchanged abundance upon P1 treatment are predominantly in regions with low affinity towards BP according to the epitopsy calculation and therefore expected to remain accessible if P1 is present (Fig. 4A–C). The agreement between MS and epitopsy is encouraging although there are major approximations (see sections on MS and epitopsy in the ESI and Fig. S21†). P1 apparently increases local concentration without blocking the active site or covering too many peptide bonds susceptible to proteolysis which is a requirement for our suggested mechanism.
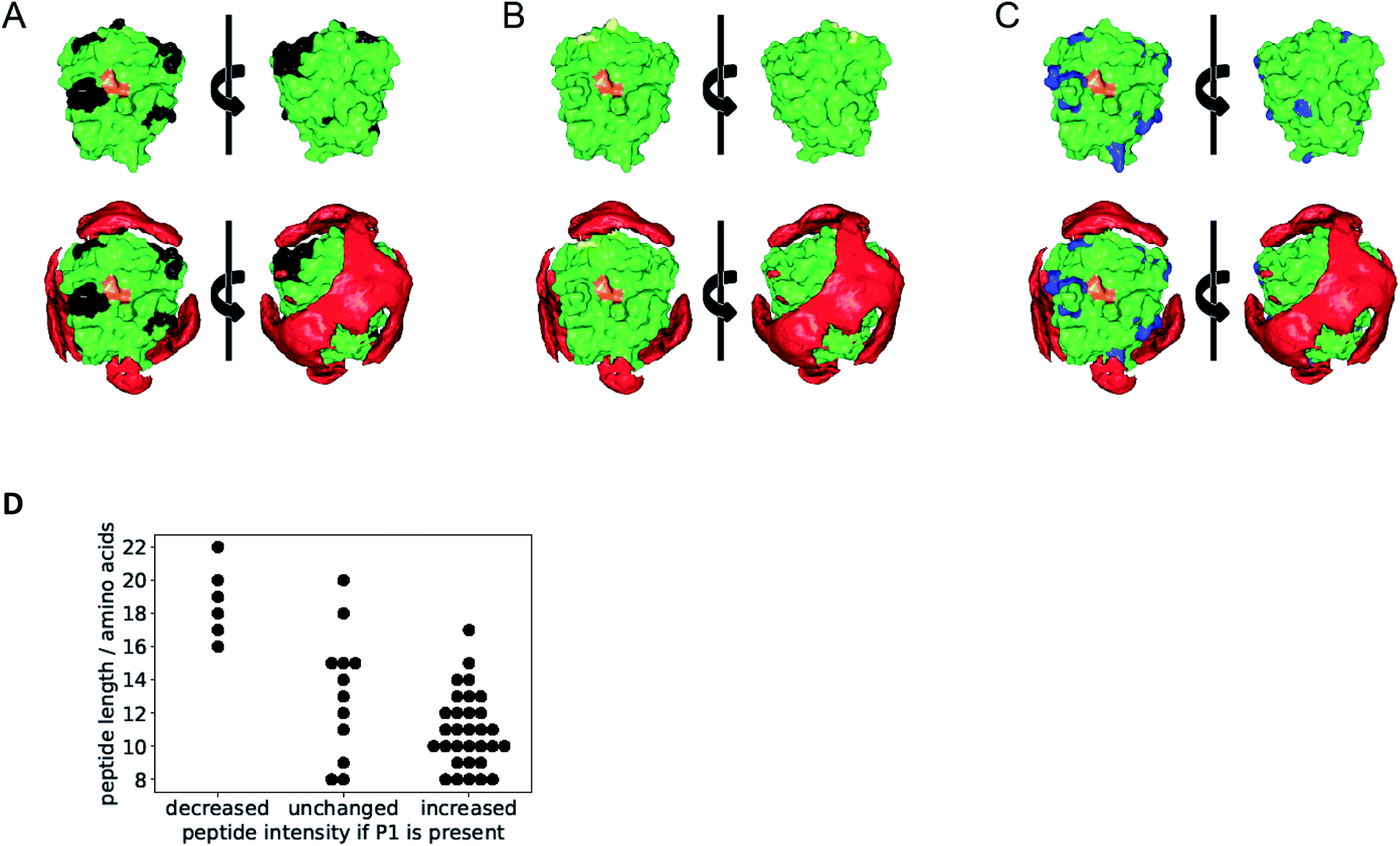 | ||
| Fig. 4 Overlay of epitopsy calculation with cleavage sites identified by MS. The active site of the enzyme is depicted in orange and represents the front of the enzyme. (A) Black cleavage sites found in peptides produced with increasing concentration upon treatment with P1. (B) Yellow cleavage sites from peptides with decreased concentration upon treatment with P1. (C) Blue peptides with no significant abundance change upon P1 treatment. (D) Accelerated autolysis leads to shorter peptide fragments. Each point corresponds to one specific digest peptide. Presence of P1 decreases abundance of longer peptides and increases abundance of shorter peptides. | ||
Digested peptides decreasing in abundance if P1 is present were substantially longer than other peptides (Fig. 4D). Thus our results suggest a promotion of trypsin autolysis by P1, either by a more thorough digest leading to smaller products, or a faster autolysis, or a combination of both.
Surprisingly many peptides were not fully tryptic (ESI File S20†); most identified peptides were semi-tryptic. We can conclude that in the presence of P1 cleavage specificity was low. The reason for unspecific trypsin autolysis in the presence of P1/P2 is unclear at present.
Conclusion and outlook
In summary, we have demonstrated how linear affinity copolymers that specifically recognize the protein surface lead to accelerated autolysis. Presumably, they act as a template for multiple protease molecules. Local protease concentration on the accessible surface area is substantially higher than in the bulk solution and complexed areas are shielded from digestion. The extremely efficient trypsin inhibition has been monitored by SDS-PAGE, gel filtration, CD, CZE and ESI-MS. Such a controlled and specific enzyme self-destruction may be helpful to counteract trypsin upregulation in ischemia and reperfusion injuries of the small intestines as well as in acute pancreatitis. It may also be directed against other problematic proteases involved in diseases (e.g., retroviral protease or β-secretase). We have recently discovered that affinity copolymers tailored for kallikrein also exhibit drastically substoichiometric protein inhibition and will likewise examine their mechanism of action supported with calculations. Moreover, controlling protease activity is highly desirable to protect therapeutic peptides. Another potential application involves the accelerated digest of other selected proteins in the presence of a small amount of trypsin and an appropriate affinity polymer. Employing living radical copolymerization, we envisage the construction of block copolymers for simultaneous trypsin and protein substrate docking and accelerated substrate digest. Experiments in this direction are underway in our laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft DFG by funding the Collaborative Research Centre CRC 1093 Supramolecular Chemistry on Proteins.References
- D. C. Whitcomb, M. Gorry and R. Preston, et al., Nat. Genet., 1996, 14, 141–145 CrossRef CAS PubMed
.
- H. Hatate and M. Toyomizu, Bull. Jpn. Soc. Sci. Fish., 1985, 51, 627–633 CrossRef CAS
- Y. Ono, M. Shindo, N. Doi, F. Kitamura, C. C. Gregorio and H. Sorimachi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E5527–E5536 CrossRef CAS PubMed
- S. A. Shirdel, K. Khajeh, S. M. Asghari and H. R. Karbalaei-Heidari, Eng. Life Sci., 2014, 14, 229–234 CrossRef CAS
- N. C. Ozturk, D. Kazan, A. A. Denizci and A. Erarslan, Eng. Life Sci., 2012, 12, 662–671 CrossRef CAS
- M. R. Stoner, D. A. Dale, P. J. Gualfetti, T. Becker, M. C. Manning, J. F. Carpenter and T. W. Randolph, Enzyme Microb. Technol., 2004, 34, 114–125 CrossRef CAS
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS PubMed
- Y. Sasai, H. Kanno, N. Doi, Y. Yamauchi, M. Kuzuya and S.-I. Kondo, Catalysts, 2017, 7, 4–13 CrossRef
- Y. Lv, J. Zhang, H. Wu, S. Zhao, Y. Song, S. Wang, B. Wang, G. Lv and X. Ma, Chem. Commun., 2015, 51, 5959–5962 RSC
- C. Renner, J. Piehler and T. Schrader, J. Am. Chem. Soc., 2006, 128, 620–628 CrossRef CAS PubMed
- S. Koch, C. Renner, X. Xie and T. Schrader, Angew. Chem., 2006, 38, 6500–6503 CrossRef
- P. Gilles, K. Wenck, I. Stratmann, M. Kirsch, D. A. Smolin, T. Schaller, H. de Groot, A. Kraft and T. Schrader, Biomacromolecules, 2017, 18, 1772–1784 CrossRef CAS PubMed
- T. Schrader, Chem.–Eur. J., 1997, 3, 1537–1541 CrossRef CAS
-
(a) A. Baici, Slow-Onset Enzyme Inhibition, Kinetics of Enzyme-Modifier Interactions, Springer, Wien, 2015, pp. 367–444 CrossRef
- J. S. Hanspal, G. R. Bushell and P. Ghosh, Anal. Biochem., 1983, 132, 288–293 CrossRef CAS PubMed
- P. Hong, S. Koza and E. S. P. Bouvier, J. Liq. Chromatogr. Relat. Technol., 2012, 35, 2923–2950 CrossRef CAS PubMed
- U. T. Ruegg and J. Rudinger, Methods Enzymol., 1977, 47, 111–116 CAS
- M. Vestling, C. M. Murphy and C. Fenselau, Anal. Chem., 1990, 62, 2391–2394 CrossRef CAS PubMed
- Capillary Electrophoresis of Proteins and Peptides, Methods and Protocols, Methods in Mol. Biology, ed. N. T. Tran and M. Taverna, Humana Press, Springer, New York, 2016 Search PubMed
- This somewhat contrasts the 20% rest activity found in enzyme assays; however, CZE does not measure activity but molecule charge and size..
- It has to be acknowledged that the polymers do not act as classical inhibitors, but rather as catalytic accelerators of tryptic self-digest by means of a template effect..
- The pKa value of the bisphosphonic acid was determined at 2.5..
- K. Wenck, S. Koch, C. Renner, W. Sun and T. Schrader, J. Am. Chem. Soc., 2007, 129, 16015–16019 CrossRef CAS PubMed
-
(a) J. Mogridge, Methods Mol. Biol., 2004, 261, 113–118 CAS
- G. Caracciolo, et al., Eur. Biophys. J., 2001, 30, 163–170 CrossRef CAS PubMed
- J. Linders, C. Mayer, T. Sekine and H. Hoffmann, J. Phys. Chem. B, 2012, 116, 11459–11465 CrossRef CAS PubMed
- M. Tirrell, ACS Cent. Sci., 2018, 4, 532–533 CrossRef CAS PubMed
- C. Y. Huang, Methods Enzymol., 1982, 87, 509–525 CAS
- J. Walter, W. Steigemann, T. P. Singh, H. Bartunik, W. Bode and R. Huber, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 1462–1472 CrossRef
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed
- J.-N. Grad, A. Gigante, C. Wilms, J. N. Dybowski, L. Ohl, C. Ottmann, C. Schmuck and D. Hoffmann, J. Chem. Inf. Model., 2018, 58, 315–327 CrossRef CAS PubMed
-
(a) D. P. Landau and K. Binder, A Guide to Monte Carlo Simulations in Statistical Physics, Cambridge University Press, Cambridge, 2009 CrossRef
- This reversible turbidity increase exactly parallels the behaviour of dextrane sulfate in ref. 9..
- N. R. Bennett, D. B. Zwick, A. H. Courtney and L. L. Kiessling, ACS Chem. Biol., 2015, 10, 1817–1824 CrossRef CAS PubMed
- K. Bravo-Rodriguez, B. Hagemeier, L. Drescher, M. Lorenz, J. Rey, M. Meltzer, F. Kaschani, M. Kaiser and M. Ehrmann, Rapid Commun. Mass Spectrom., 2018, 32, 1659–1667 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, enzyme assays, fluorescence and ITC titrations, CD spectroscopy, DLS, gel filtration, CZE, SDS-PAGE, PFG-NMR, ESI-MS fragment analysis, computation. See DOI: 10.1039/d0ra05827k |
| This journal is © The Royal Society of Chemistry 2020 |