
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular targets and anticancer activity of quinoline–chalcone hybrids: literature review†
Mamdouh F. A. Mohamed
 *a and
Gamal El-Din A. Abuo-Rahma
*b
*a and
Gamal El-Din A. Abuo-Rahma
*b
aDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Sohag University, 82524 Sohag, Egypt. E-mail: mamdouhfawzy3@yahoo.com; mamdouh.fawzi@pharm.sohag.edu.eg; Tel: (+20)-1018384461
bDepartment of Medicinal Chemistry, Faculty of Pharmacy, Minia University, Minia 61519, Egypt. E-mail: gamal.aborahma@mu.edu.eg; Tel: +201003069431
First published on 21st August 2020
Abstract
α,β-Unsaturated chalcone moieties and quinoline scaffolds play an important role in medicinal chemistry, especially in the identification and development of potential anticancer agents. The multi-target approach or hybridization is considered as a promising strategy in drug design and discovery. Hybridization may improve the affinity and potency while simultaneously decreasing the resistance and/or side effects. The conjugation of quinolines with chalcones has been a promising approach to the identification of potential anticancer agents. Most of these hybrids showed anticancer activities through the inhibition of tubulin polymerization, different kinases, topoisomerases, or by affecting DNA cleavage activity. Accordingly, this class of compounds can be classified based on their molecular modes of action. In this article, the quinolone–chalcone hybrids with potential anticancer activity have been reviewed. This class of compounds might be helpful for the design, discovery and development of new and potential multi-target anticancer agents or drugs.
 Mamdouh F. A. Mohamed | Mamdouh F. A. Mohamed is a Lecturer of Pharmaceutical/Medicinal Chemistry at Sohag University, Egypt. He was born in 1975 in Sohag, Egypt. He was awarded his PhD from Minia University under the supervision of Prof. Gamal El-Din A. Abuo-Rahma. He obtained his Bachelor's and Master's degrees with honors from Al-Azhar University, Assiut, Egypt. He is interested in research on the design and synthesis of small molecules with potential biological activities, especially histone deacetylase inhibitors, the synthesis of compounds with antimicrobial, anti-inflammatory and anticancer activity, quinoline derivatives, and 1,2,4-oxadiazole-containing compounds. He has supervised one Master's thesis. Currently, he is the co-supervisor for 8 Master's theses. He has published more than 10 articles in high-impact international peer-reviewed journals. |
 Gamal El-Din A. Abuo-Rahma | Gamal El-Din A. Abuo-Rahma is a Professor of Pharmaceutical/Medicinal Chemistry at Minia University, Egypt. He was born in 1967 at Assiut, Egypt. He received his PhD through collaborative studies between Minia University and Bonn University under the supervision of Prof. Jochen Lehmann and Prof. Ali El-Emam. He obtained his Bachelor's and Master's degrees with honors from Assiut University, Egypt. He is interested in research on the design and synthesis of small molecules with potential biological activities, especially nitric oxide donor hybrids, quinoline, fluoroquinolone derivatives, and histone deacetylase inhibitors. He has supervised 11 Master's and 7 PhD theses. Currently, he is the main supervisor for 10 PhD and 5 Master's theses. He has published 51 articles in high-impact international peer-reviewed journals. |
1 Introduction
Cancer is an abnormal uncontrollable cell cycle disease characterized by the rapid proliferation of normal cells. Cancer has been ranked as the second leading cause of death all over the world, preceded only by cardiovascular diseases.1,2 The cancer rates increase annually, and it is expected that cancer rates will increase by about 60% over the next 20 years. According to WHO, there are 600 types of cancer, most of which require unique diagnoses and management protocols [file:///C:/Users/compusoft/Downloads/9789240001299-eng.pdf]. In a recent statistic from WHO, about 14 million diagnosed cases and at least 9 million cancer-related deaths occur annually with a ratio of one from every six deaths being due to cancer. The problem of cancer is not only a global health problem but is also a social and economic one. The estimated annual economic cost of cancer was about $1.16 trillion in 2010.3,4 The total cost of cancer in 2020 is estimated to be about $173 billion in the USA [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3107566/]. For the different cancer types, lung, colorectal, skin, stomach, prostate, breast and liver cancers are the most common, and most deaths are caused by colorectal, stomach, liver and breast cancers.5 Although there has been continuous pharmacological progress in the effectiveness of anticancer drugs, there is a continuing urgent need for diverse, novel, targeted molecules to solve the problem of drug resistance and/increase the potency or decrease the side effects of the currently used drugs.6 Consequently, in the literature and clinical trials, large numbers of molecules targeting different sites are under investigation for development as promising anticancer agents.7,8The α,β-unsaturated chalcones are precursors of flavonoid and isoflavonoid-based compounds.9 Chalcones serve as the precursors for the synthesis of various heterocyclic compounds like pyrimidines, pyrazoles,10 2-pyrazolines,11,12 imidazoles,13 and flavonoids.14 The natural or synthetic chalcones exhibit various biological activities, such as anti-inflammatory,15–17 analgesic,18 antioxidant,19–22 antibacterial,22,23 antifungal,24–27 anti-tuberculosis,28 antimalarial,29–32 antiviral,33 parasitic protease inhibitors,34 antioxidant, anti-human immunodeficiency virus (HIV),35 antitumor activities,36–41 HDAC inhibitors,42 and as insulin mimetics in 3T3-L1 adipocytes.43 Moreover, chalcones have been identified as privileged structures in the development process of chalcone-based compounds.44–48 Many chalcone-based compounds have been reported in the last decade as tubulin inhibitors,49–58 in addition to several other possible molecular targets that chalcones directly interact with59 (see ESI†).
On the other hand, quinoline stands out among the most essential N-based biologically active heterocyclic compounds. The presence of nitrogen atoms significantly increases the basic character of quinoline-containing compounds. Moreover, the quinoline nitrogen may engage in hydrogen bonding with the target enzymes. Polarity is an additional significant property that can be used as a method for reducing the lipophilic character, increasing water solubility and thus oral absorption in drug design strategies.60 The quinoline scaffold possesses a variety of biological activities, including anti-HIV,61 antipsychotic,62 antibiotic,63 anti-inflammatory,64–66 PDE4B inhibitors67 and antihypertensive activities.68 Drugs containing the quinoline ring motif, such as mefloquine, chloroquine, quinine, and amodiaquine, are used as effective drugs for the treatment of malaria.69 Moreover, the quinoline nucleus is the milestone structural motif for some important anticancer drugs that are available on the market,8,70,71 Fig. 1.
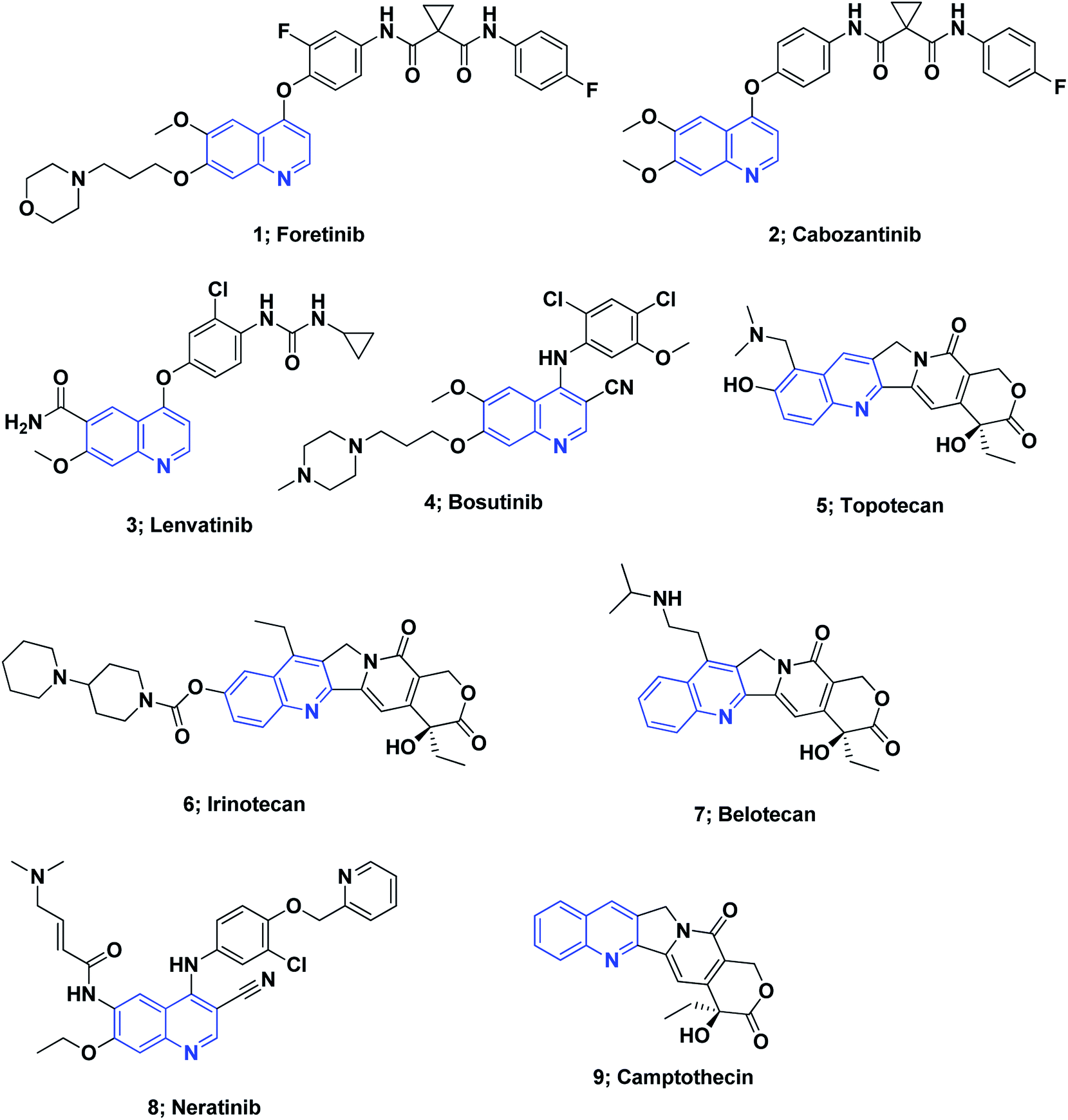 | ||
| Fig. 1 The structures of some approved quinoline-containing anticancer drugs. | ||
Several molecules containing the quinoline framework are in clinical trials as anticancer agents,8,72,73 Fig. 2.
 | ||
| Fig. 2 The structures of some quinoline-containing anticancer agents under clinical trials. | ||
Quinoline derivatives also display excellent anticancer activities via different mechanisms of action, such as the inhibition of tyrosine kinase, alkylating agents, cell cycle arrest, angiogenesis inhibition, apoptosis induction, cell migration disruption, targeting Bcl-2, and inhibition of antimitotic tubulin polymerization74–88 (see ESI†).
Chalcones and quinoline have been researched extensively, with many published review articles.8,89–106 Nonetheless, the precise modes of action for the broad-spectrum biological activities of chalcones and quinoline are still not well known. Hybridization has emerged as one of the most promising approaches in the innovation of new candidates with the potential to enhance the affinity and effectiveness and also to overcome cross-resistance when compared with the parent drugs. In addition to the antineoplastic CX-3543 (quarfloxin), there are numerous quinoline hybrids under clinical trials, such as the antibacterial agents Ro 23-9424, and MCB3837 (ref. 107 and 108) (see ESI†).
The molecular hybridization of quinoline with other pharmacophoric anti-cancer moieties, such as chalcones, is a promising strategy that can lead to new high-potential anticancer agents for drug-sensitive cancers and also for drug-resistant cancers. In the last three decades, substantial efforts have been carried out to innovate novel hybrids of quinoline as anti-cancer candidates with greater safety profiles and better anti-cancer activity. It is obvious from the literature survey that drug molecules binding to more than one target have many advantages in cancer therapy, including synergistic activity, reduced side effects and/or avoiding the problem of drug resistance. There is great interest in multi-target drugs and the combination therapy strategy in cancer drug discovery. Consequently, the aim of this article to review the recent advances of anti-cancer quinolone–chalcone hybrids, discussing their modes of action and structure–activity relationships at the molecular level over the last ten years.
2 Naturally occurring chalcones
Notably, at present, two chalcones are used in clinical practice (see ESI†). Sofalcone is an anti-ulcer agent that increases the amount of mucosal prostaglandins and provides a gastro-protective effect against Helicobacter pylori;109 Metochalcone was approved as a choleretic drug.89 Moreover, hesperidin methylchalcone and hesperidin trimethylchalcone showed promising results in clinical trials in chronic venous lymphatic insufficiency and trunk or branch varicosis, respectively.110Chalcones represent the core of numerous biologically interesting, naturally occurring compounds and have gained considerable research attention.111 There are a large number of natural chalcones such as isoliquiritigenin 16 (anti-cancer, cancer chemopreventive, anti-oxidant and anti-inflammation),112–114 butein 17 (anti-cancer and anti-inflammation),115–117 cardamonin 18 (ATP diphosphohydrolase),118 sappanchalcone 19 (anti-inflammation),119,120 licochalcone A 20 (anti-inflammation and anti-cancer),121,122 and millepachine 21 (anticancer).123 Many other conventional chalcones, dihydrochalcones, chalcone mimics, bichalcones, and fused chalcones have been isolated from natural sources and have potentially promising biological activities,111,124 Fig. 3.
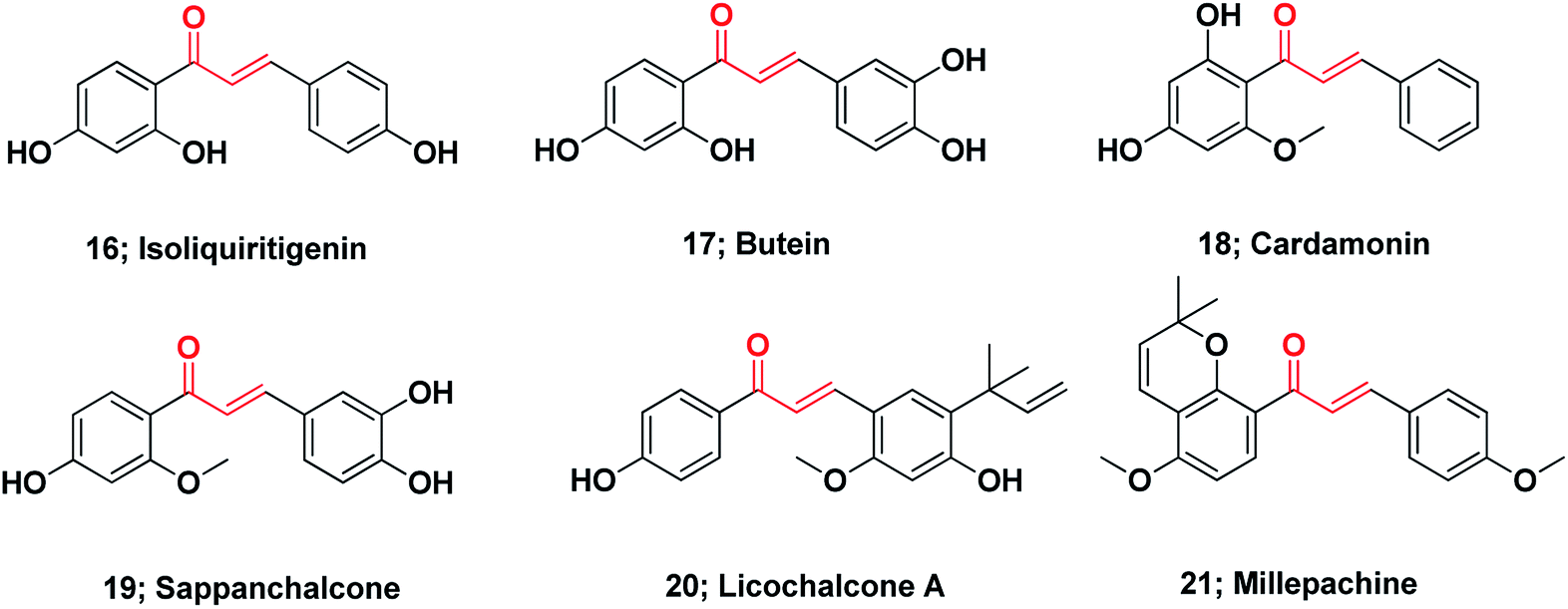 | ||
| Fig. 3 Chemical structures of naturally occurring anticancer chalcones. | ||
3 Synthesis of chalcones
The simple chemistry and the ease of chalcone synthesis allow a diversity of substitutions, which conducted using base- or acid-catalyzed condensation reactions. Despite the ease of chalcone synthesis, currently, diverse new methods and novel procedures have been described due to their various biological activities and also due to the development of different catalysts or reaction conditions. Among all, condensation using the Claisen–Schmidt reaction is the most widely used for chalcone synthesis.125 This reaction is catalyzed by either strong bases or acids in a liquid solvent at 50–100 °C for several hours.95,102,126 Using a base as a catalyst, the chalcone is generated via the dehydration of the produced aldol intermediate in an enolate mechanism; whereas, on using acid as the catalyst, chalcone formation proceeded via an enol mechanism.127 Typically, the conventional Claisen–Schmidt reaction is performed in the liquid phase, but some reactions can be performed in the solid phase or solvent-free phase.128 Additionally, the use of microwaves in liquid and solvent-free Claisen–Schmidt reactions decreases synthesis time and improves chalcone yield (Table 1).129,130| Catalyst | Solvent | Ref. |
|---|---|---|
| NaOH | Ethanol | 131 |
| NaOH | Methanol | 134–136 |
| KOH | Ethanol | |
| Piperazine | Methanol | 138 |
| Ethanol | ||
| Ca(OH)2 | Aprotic solvent | 140 |
| Ba(OH)2 | ||
| Sr(OH)2 | ||
| LiHDMS | THF | 142 |
| NaNO3 | Methanol | 144 and 145 |
| (LiNO3)/natural phosphates | ||
| HCl | Ethanol | 147 |
| AlCl3 | Carbon disulfide | 147 |
| BF3–Et2O | Dioxane | 132 and 133 |
| SOCl2 | Ethanol | 137 |
| p-TsOH | Acetic acid | 139 |
| NaOH/grind | — | 141 |
| KF–Al2O3/grind | — | 143 |
| CaO/MW | — | 146 |
| TBAB | Inorganic alkaline solution | 148 |
| TFAA/H3PO4 | — | 149 |
Because the Claisen–Schmidt condensation has some drawbacks such as the long reaction time, the reaction completion may require several days. The reaction may also give a mixture of products containing the required chalcone, byproducts, and possibly starting materials. Therefore, both the reactants and catalyst have a dramatic effect on the yield with conversion ranging from less than 10% up to nearly 100%.127,150 Thus, for the synthesis of chalcone, more well-recognized reactions such as the photo-Fries rearrangement, cross-coupling, one-pot synthesis from alcohols and Friedel–Crafts acylation were utilized.151–186
4 Chalcone as Michael acceptors
The chemical nature of the structure of chalcones impacts or affects its biological activity.187–190 It is known that Michael acceptors have an electrophile that is involved in many biological processes and regulates important signaling pathways. The α,β-unsaturated carbonyl functional moiety in chalcones is considered a Michael acceptor; it participates in covalent bond formation with thiols or other similar nucleophiles through Michael addition (Fig. 4). For instance, chalcones can modulate Keap1–Nrf2–ARE through covalent bond formation with cysteine. In the design of new drug molecules, it is worth considering the substituents of chalcone aromatic rings, which affect the electron density on the ring and consequently the electrophilicity of the α,β-unsaturated ketone system, and can affect the binding ability and the biological activity of chalcones.190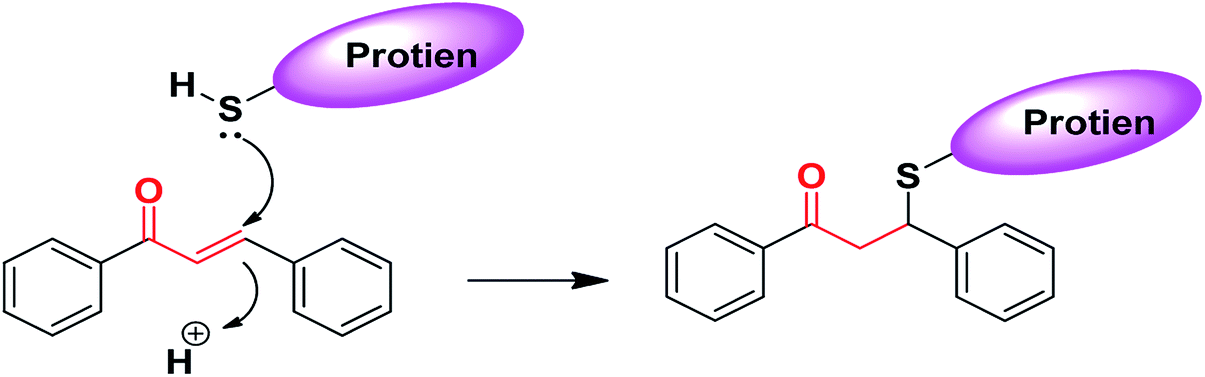 | ||
| Fig. 4 Chalcones as Michael acceptors with cysteine. | ||
5 Quinoline–chalcone hybrids
Tubulin polymerization inhibitors
Microtubules, components of the cytoskeleton, are hollow tubes consisting of α- and β-tubulin. Polymerization of tubulin is necessary to form microtubules that have essential roles in cellular functions, including the maintenance of cell structure, intracellular transport, polarity and mitosis.191 Notably, tubulin also plays a crucial role in non-mitotic cells, which is likely to underlie the overall effectiveness of the tubulin-targeting agents in cancer therapy.192,193It has been documented that hundreds of compounds kill cancer cells by disrupting tubulin polymerization and are known as microtubule-targeting agents (MTAs).194–196 Hence, tubulin polymerization inhibitors are among the most effective targeted anticancer agents. Most tubulin-targeting agents are composed of diverse and sometimes complex structural molecules.197
Recently, Tseng et al. synthesized and investigated the anticancer activity of a series of 3-phenylquinolinyl-chalcones using the XTT assay on three different types of breast cancer cell lines, namely, MCF-7, MDA-MB-231 and SKBR-3, and a non-cancer normal epithelial cell line (H184B5F5/M10).46,198 The results showed that the 3,4,5-trimethoxy analog 22 (Fig. 5) is the most potent compound against the three different types of breast cancer cells growth, showing IC50 values less than 1.05 μM without remarkable cytotoxicity on the normal cell line (IC50 value more than 10 μM). Further studies revealed that compound 22 could induce cell cycle arrest in a time-and concentration-dependent manner. Compound 22, in a dose-dependent manner, caused microtubule disruption with a decrease in the polymer form of tubulins. Also, compound 22 induced apoptosis via the regulation of Bcl-2 and the activation of caspase in MDA-MB-231 cells.
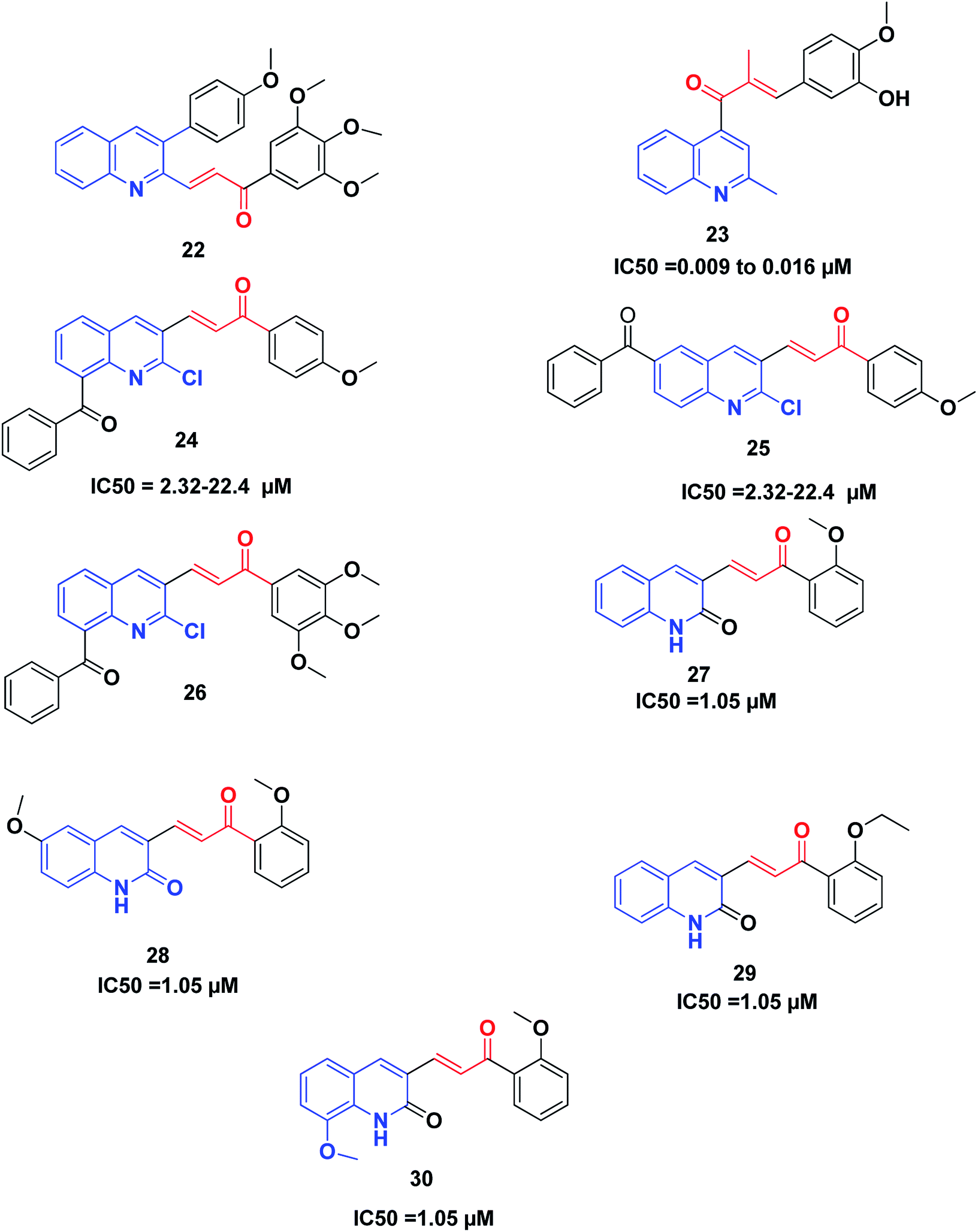 | ||
| Fig. 5 Quinoline–chalcone hybrids as tubulin polymerization inhibitors. | ||
Li et al.199 discovered some novel quinoline–chalcone derivatives and screened their anticancer activities; the results revealed that compound 23 displayed the highest potency (IC50 values 0.009 to 0.016 μM against cancer cell lines panel). Compound 23 also displayed an improved safety profile by intravenous injection, with an LD50 value of 665.62 mg kg−1. The hydrochloride salt 50-HCl was more potent than CA-4 in inhibiting tumor growth in H22 xenograft models without noticeable toxicity. Mechanistically, compound 23 was found to be an inhibitor of tubulin polymerization by binding to the colchicine site of tubulin. Moreover, compound 23 induced cell cycle arrest in the G2/M phase, and induced apoptosis, depolarized mitochondria and induced the generation of reactive oxidative stress in the K562 cell line. Furthermore, compound 23 exhibited potent antimetastasis activity in vitro, as well as antivascular activities in vitro and in vivo. These results revealed that compound 23 is a safe and potent anticancer candidate for cancer therapy, which merits further screening.
Mirzaei et al.200 synthesized and investigated the cytotoxic activity of a new series of quinoline–chalcone hybrids against four human cancer cell lines including A2780, A2780/RCIS, MCF-7, MCF-7/MX and normal HUVEC cells. Compounds 24, 25 and 26 with the benzoyl group displayed notable anticancer activity against resistant cancer cells as well as their parents. Compounds 25 and 26, with IC50 values ranging from 2.32 to 22.4 μM, showed the highest potent anticancer activity. Further studies on the mechanism of action revealed that compounds 25 and 26 were also recognized as tubulin inhibitors, inducing cell cycle arrest at the G2/M phase and also apoptosis. Compound 26 provoked greater arrest in four cancer cell lines at the G2/M phase as compared to compound 25. Finally, the examination of the molecular docking of compound 26 into the colchicine-binding site of tubulin indicated the possibility of this compound interacting in the tubulin active site.
It was established that the quinoline–chalcones 27 and 28 can act as potent anticancer agents. These compounds are capable of binding nicely to the colchicine binding site of tubulin. Moreover, they were found to kill multi-drug resistant cancer cells. Combining these compounds with paclitaxel had a synergistic effect on multi-drug resistant cancer cells. Furthermore, the analogues 29 and 30 disrupted the microtubule dynamics and killed MCF10A breast cancer cells and multi-drug resistant cancer cells,201 Fig. 5.
Quinoline–chalcone hybrids as kinase inhibitors
Protein tyrosine kinases (PTKs) play pivotal roles in signal transduction involving the regulation of many cellular functions such as the cell cycle, cell proliferation, differentiation, cell survival, and angiogenesis. Overexpression or kinase activity mutation is related to the development of different cancer types.202 Therefore, tyrosine kinases are promising targets for the treatment of cancer. Many FDA-approved small molecule inhibitors target different types of tyrosine kinases, such as the vascular endothelial growth factor receptor (VEGFR) and the epidermal growth factor receptor (EGFR), and many others are now at different stages of preclinical and/or clinical development.203,204Rizvi et al. synthesized a series of quinolyl-thienyl chalcones as possible inhibitors of VEGFR-2 kinase, using structure-based virtual screening protocols.205 The in vitro inhibitory activity assay revealed that compound 31 is the most potent, with an IC50 value of 73.41 nM. Moreover, all compounds displayed significant inhibition of the proliferation of human umbilical vein endothelial cells (HUVEC) (compound 31, IC50: 21.78 nM).
The 2-quinolone-benzimidazole hybrid 32 is a potent inhibitor of two important anticancer targets, protein kinase B (α, β and γ) and IkB kinase (α and β), with IC50 of 1.64–44.46 μM.206 SAR revealed that incorporation of the N1-methyl group has a deleterious effect on biological activity. On the other hand, methyl, chloro or bromo substituents retain activity, while halogen substituents on benzimidazole enhance the activity. The hybrid 32 showed the highest activity among this series against protein kinase B (α, β and γ) and IkB kinase (α and β). It also displayed favorable plasma and microsomal stability and acquired potent tumor growth inhibition at dose levels of 10 and 25 mg kg−1 in the xenograft mice model, UACC903 melanoma cells.
George et al.207 synthesized and screened derivatives of quinoline–chalcone for their anti-proliferative activity against cancer cell lines MCF-7, HeLa and DLD1, as well as normal fibroblast WI-38. In addition to good safety towards the normal cell line, the compounds tested showed promising anticancer activity. Compounds 33 and 34 have been screened for their effectiveness as EGFR inhibitors. In comparison to Gefitinib (IC50 = 29.16 nM), they revealed inhibitory activity at the nanomolar level, in particular, compounds 33 with IC50 (37.07 nM).
Novel thienoquinoline carboxamide-chalcone derivatives have been prepared and their anticancer activities were investigated. Thienoquinolines 35 and 36 revealed remarkable broad-spectrum antiproliferative activity. They also showed significant inhibition of EGFR TK with IC50 values ranging between 0.5 and 3.2 μM. Compounds 35 and 36 demonstrated the highest activities among the synthesized series, with better activity than the reference Erlotinib on the melanoma cancer cell line A375. Compound 36 induced pre-G1 apoptosis and cell cycle arrest at the G2/M phase. Docking studies revealed that compound 35 was nicely bound in the active site of EGFR and the thienoquinoline moiety occupied the ATP-binding site, whereas the chalcone moiety was located in the allosteric site; this explains the enhanced activity of these compounds,208 Fig. 6.
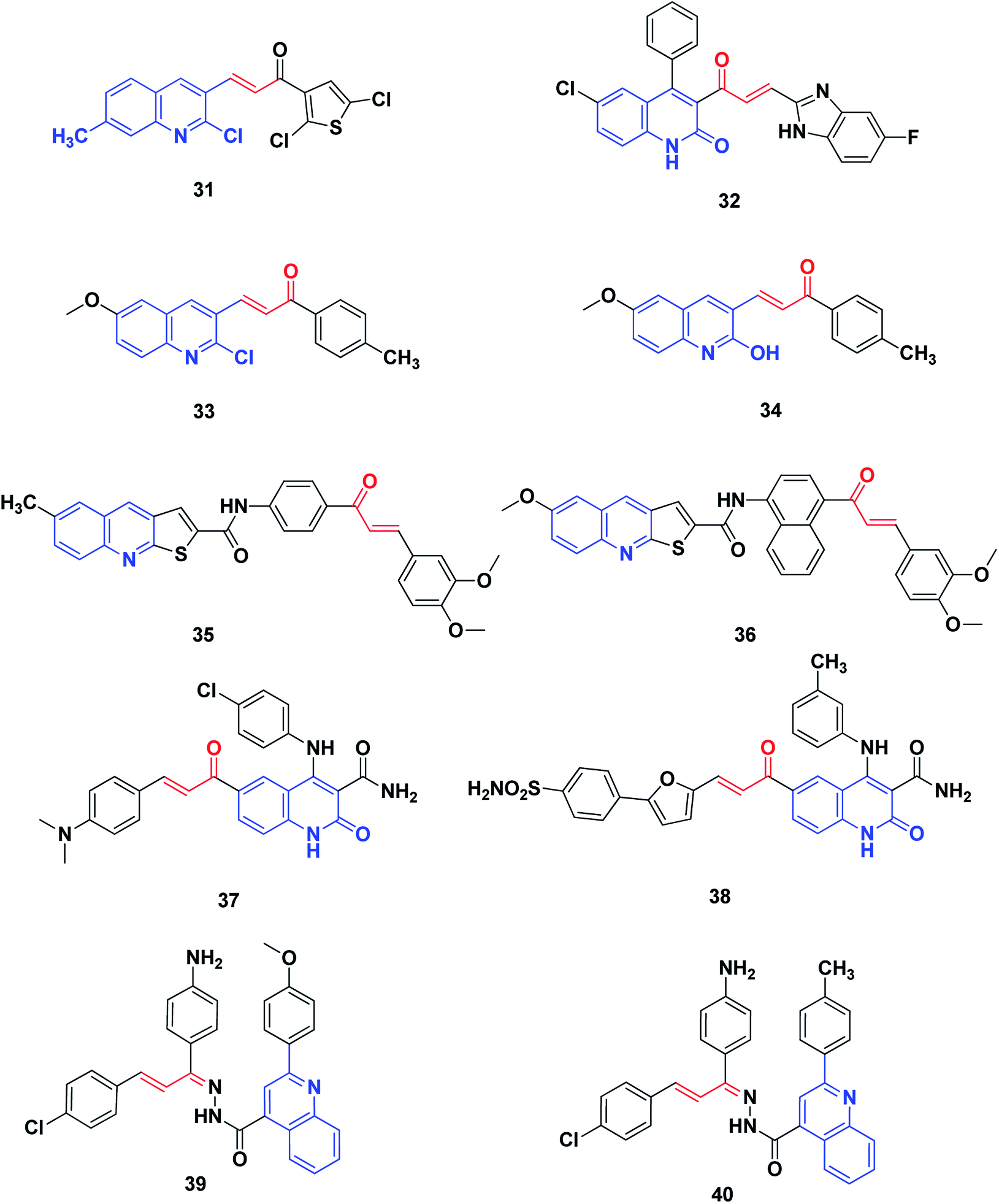 | ||
| Fig. 6 Quinoline–chalcone hybrids as kinase inhibitors. | ||
Ibrahim et al.209 utilized molecular modeling and pharmacophore modeling, docking and binding energy calculations for the design of 4-anilinoquinoline-3-carboxamides. The target-synthesized derivatives were tested as potential anticancer agents with EGFR inhibitory activity. The most promising compound, 37, showed potent anticancer activity (IC50 = 3.46 μM.) and 67% inhibition of EGFR TK.
Aly et al.210 designed compound 38 through molecular modeling techniques as a potential EGFR inhibitor anticancer agent, where some analogues experienced more potent activity. The quinoline-3-carboxamide furan-derivative showed potent EGFR inhibition (IC50 = 2.61 μM) with promising anticancer activity against the MCF-7 cell line (IC50 = 3.35).
Abbas et al.211 synthesized a series of quinoline–chalcone hybrids as potential anti-cancer agents for NSCLC and CML, via inhibiting the PI3K/Akt/mTOR pathway. Cytotoxicity assays revealed that among the synthesized compounds, compounds 39 and 40 were the most active against all cell lines tested, with IC50 values of 1.91 and 5.29 μM against A549 and K-562 cancer cell lines, respectively. Mechanism studies demonstrated that 39 and 40 induced apoptosis and arrested cell cycles at G2/M in both A549 and K562 cells. Moreover, compounds 39 and 40 non-selectively inhibited all PI3K isoforms with IC50 values ranging from 52 to 473 nM, with 39 being the most active against PI3K-γ (IC50 = 52 nM). Analysis of the docking results showed the possible formation of hydrogen bonds with the essential valine residues in the active sites of PI3K-γ isoforms. Furthermore, western blot analysis showed that 39 and 40 inhibited the phosphorylation of PI3K, Akt, mTOR as well as GSK-3β in both A549 and K562 cells, indicating a link between blocking the PI3K/Akt/mTOR pathway and the anticancer activities.
Quinoline–chalcone hybrids affecting DNA cleavage activity
DNA has been recognized as a primary target for anticancer drugs, which can change the DNA conformation and inhibit duplication or transcription.212 DNA is among the most important biological targets for chemotherapeutic agent development.213 There are two broad categories of non-covalent DNA-binding agents, namely, the intercalators and the groove binders. Groove binders fit into the DNA minor groove causing little perturbation of the DNA structure, whereas intercalators bind by inserting a planar aromatic chromophore between adjacent DNA base pairs.213,214Conjugates of α,β-unsaturated ketones, phenyl-butenone, and diaryl-propenones with planar pyrroloquinoline scaffolds were synthesized and evaluated for their anticancer activity toward three lines of human cancer cells. Moreover, their capacity to bind, forming molecular complexes with DNA by linear flow dichroism (LD), and their effects on nuclear enzyme DNA topoisomerase II, were investigated. Potent cytotoxic activity was noticed in the target pyrroloquinolines 41–43, especially against melanoma cell line JR8 (IC50 1.2–3.3 μM). LD experiments confirmed that the pyrroloquinoline scaffold achieved promising results as a carrier for intercalative complexation with DNA. Their ability to intercalate between DNA base pairs appeared to inhibit the relaxation of supercoiled DNA by the topoisomerase II enzyme without remarkable induction of DNA cleavage.215
A series of pyrrolo[2,1-c][1,4]benzodiazepines linked to chalcone hybrids were synthesized by Kamal et al.216,217 Investigation of the potential anticancer activity of hybrid compounds against different lines of human cancer cells, such as leukemia, non-small cell lung, renal, colon, CNS, melanoma, ovarian, breast and prostate cancer, indicated that benzodiazepine-5-one 44 was the most active and demonstrated selective and effective growth inhibition on various cancer cell lines. Further, studies of the thermal denaturation of pyrrolo[2,1-c][1,4]benzodiazepine linked to chalcone hybrids using calf thymus DNA (CT-DNA) exhibited that compound 44 has good DNA binding affinity.
A series of simple quinoline–chalcone hybrids were synthesized by Bindu et al.218 and screened for their nucleolytic activity. Most of the synthesized compounds displayed significant DNA binding and photocleavage activities. However, compounds 45 and 46 showed promising DNA photocleavage against pUC 19 DNA with 85% inhibition at 100 mM concentration and could be potential candidates for future drug discovery and development. The structure–activity relationship analysis of these compounds revealed that the incorporation of an electron-donating group into ring A caused a moderate increase in the DNA binding and photocleavage activities. A series of 4,5-dihydroisoxazoles was prepared from quinoline–chalcones and evaluated for their nucleolytic activities by agarose gel electrophoresis. The results revealed that compound 47 experienced high concentration-dependent DNA cleavage activity. This activity is believed to be enhanced by iminyl and carboxy radicals of 2-chloro-3-(5-aryl-4,5-dihydroisoxazol-3-yl)quinolines,219 Fig. 7.
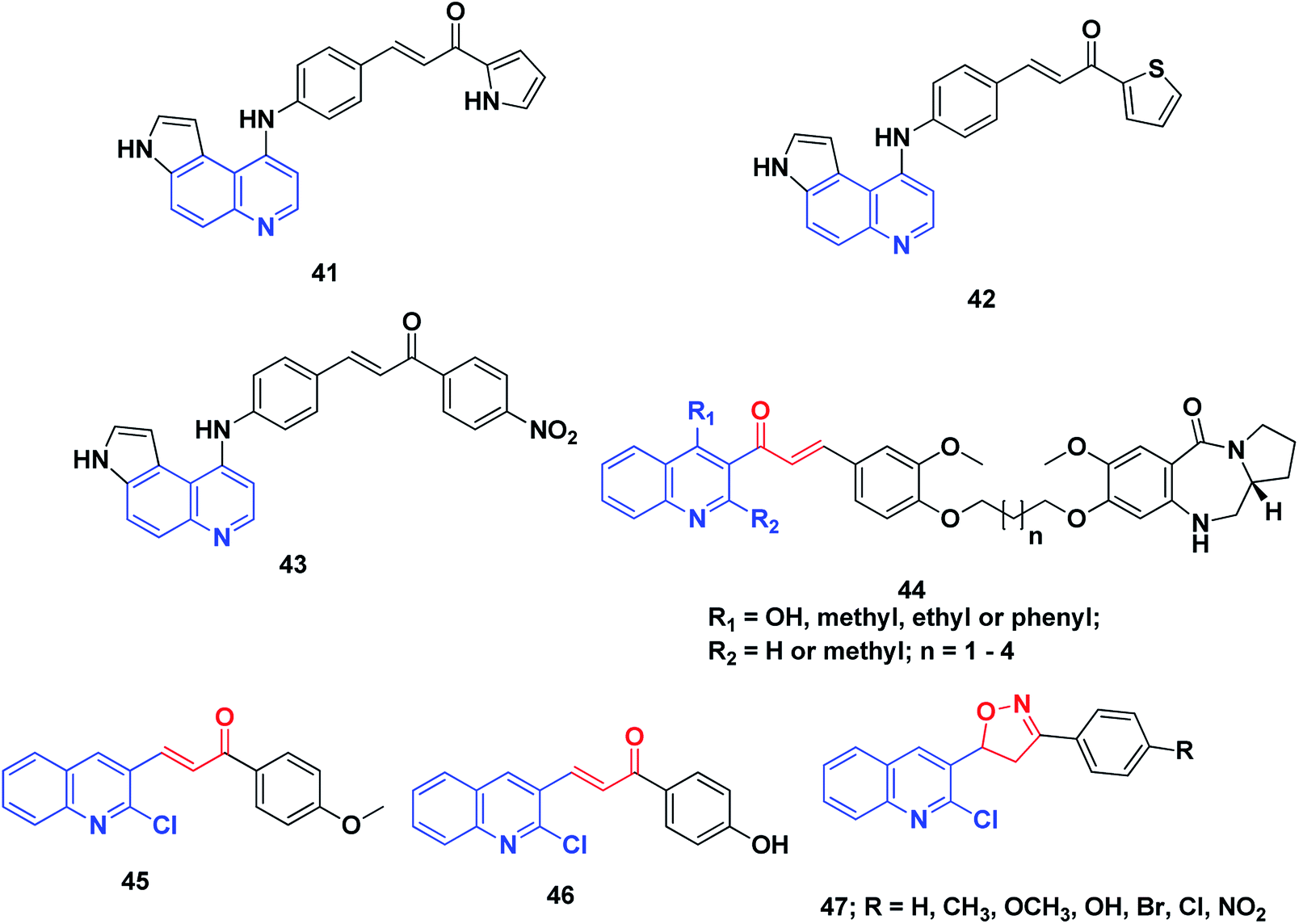 | ||
| Fig. 7 Quinoline–chalcone hybrids affecting DNA cleavage activity. | ||
Quinoline–chalcone hybrids as topoisomerase inhibitors
DNA topoisomerases can be categorized into two classes, namely, type I and type II. They allow DNA strands or double helices to pass through each other, they can solve all of the topological problems of DNA in replication, transcription and other cellular transactions. Over the past three decades, extensive biochemical and structural studies have provided molecular models of how the different sub-families of DNA topoisomerase manipulate DNA. Topoisomerases are considered as important molecular targets of many antimicrobial and anticancer agents. Most drugs targeting topoisomerases, which are currently in clinical use, act through trapping the covalent DNA–enzyme intermediates to convert a normal cellular enzyme to a DNA-damaging agent. Numerous recent reviews of DNA topoisomerases have been published.220–223Abdel-Aziz et al. synthesized a new series of N-4-piperazinyl-ciprofloxacin-chalcone hybrids (Fig. 8) with antitumor and topoisomerase I and II inhibitory activities.224 Compound 48 displayed wide-spectrum anticancer activity without distinct selectivity, whereas compound 49 exhibited significant selectivity against the leukemia subpanel with a selectivity ratio of 6.71 at the GI50 level. Moreover, compounds 50 and 51, compared to etoposide and camptothecin, respectively, exhibited significant topoisomerase I and topoisomerase II inhibitory activities.
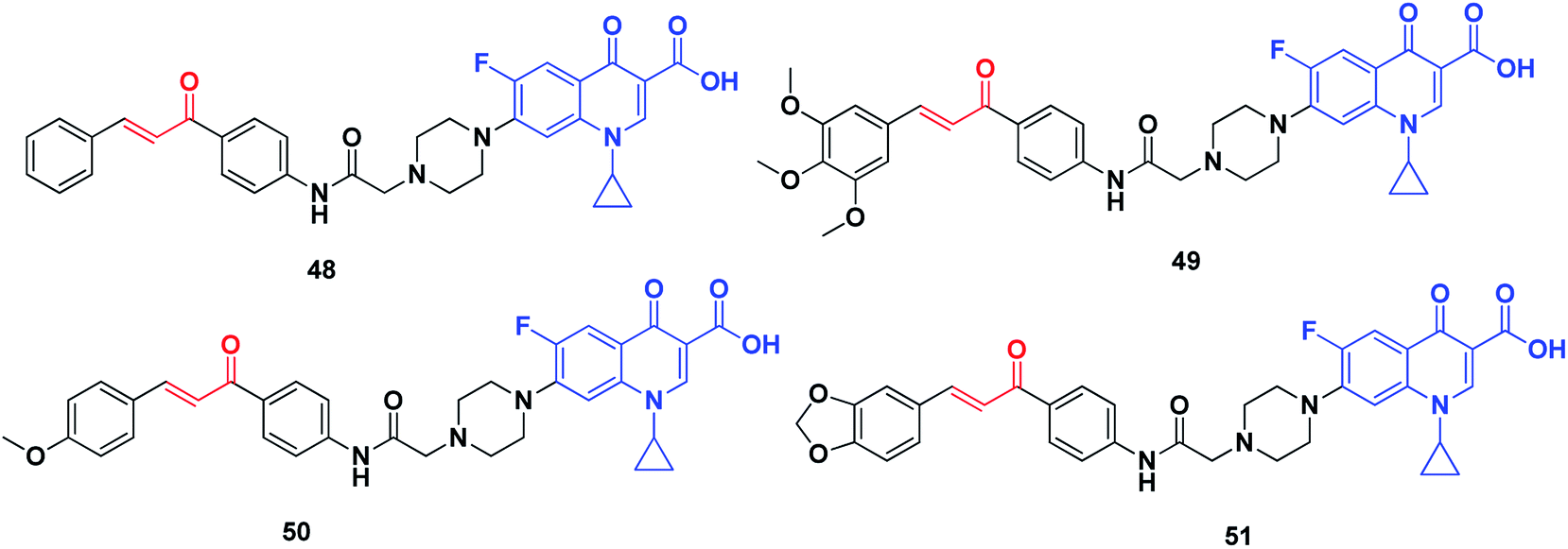 | ||
| Fig. 8 Quinoline–chalcone hybrids as topoisomerase inhibitors. | ||
Quinoline–chalcone hybrids as inducers of cell cycle arrest and apoptosis (Fig. 9)
There are intrinsic and extrinsic pathways for the induction of apoptosis in cancer cells, which are involved in the regulation of the caspase-dependent proteolysis of thousands of cellular proteins, membrane blebbing and endonucleolytic cleavage of chromosomal DNA. Targeting apoptotic pathways in tumor cells is considered a promising anticancer approach because dead tumor cells can contribute to clinical responses but not to tumor relapse. Some of the approved therapeutic drugs, such as venetoclax, directly target the intrinsic apoptosis pathway but most of them indirectly target the apoptosis pathways, such as the proteasome inhibitors, oncogenic signaling pathways inhibitors, and HDAC inhibitors. Novel agents directly targeting the apoptotic pathways are under development and some have promising potential due to high levels of tumor selectivity.225Tseng et al.226 synthesized certain 3-phenylquinolinyl-chalcone derivatives (Fig. 9) and evaluated their anticancer activity against a panel of six cancer cell lines. Among them, two compounds, 52 and 53, were the most potent anticancer agents. Compound 53 was more active against the growth of H1299 (IC50 1.41 μM) and SKBR-3 (IC50 0.70 μM) than the reference topotecan (IC50 values of 6.02 and 8.91 μM, respectively). Compound 53 displayed an IC50 value of less than 0.10 mM against the growth of MDA-MB231, and was non-cytotoxic toward the normal mammary epithelial cells (H184B5F5/M10). Studying the mechanism of action revealed that compound 53 can induce cell cycle arrest at the G2/M phase, followed by the activation of caspase-3, cleavage of PARP and consequently, cell death.226
 | ||
| Fig. 9 Quinoline–chalcone hybrids that induce cell cycle arrest and apoptosis. | ||
Raghavan et al.227 have reported the synthesis of a series of novel curcumin–quinolone hybrids starting with substituted 3-formyl-2-quinolones and vanillin. The in vitro cytotoxicity was determined on four cancer cell lines (A549, MCF7, SKOV3 and H460) using MTT assay. Compound 54, the most potent compound, was analyzed for its mechanism of action using various cell biology experiments. The treatment of SKOV3 cells with compound 54 exhibited distorted cell morphology under phase-contrast imaging and apoptosis induction was confirmed by Annexin V/PE assay. Further experiments on the generation of reactive oxygen species (ROS) and cell cycle analysis revealed that compound 54 induced ROS generation and arrested cell cycle progression in the S and G2/M phases.
The hybridization of quinolines with chloroquine has been recently documented as among the most promising apoptosis-inducing agents used against MCF-7 human breast cancer cells.228,229 All differentiation-inducing quinolines caused growth suppression in MCF-7 and MCF10A cells by the strong suppression of E2F1, which resulted in cell cycle arrest.
Very recently, Jernei et al.230 synthesized some novel hybrids containing chalcone and cinchona fragments linked with triazole linkers and investigated their antitumor/cytotoxic activity against human malignant cell lines PANC-1, COLO-205, A2058 and EBC-1. Among these, compound 55 was recognized as the most potent, exhibiting significant cytotoxicity against all the cancer cell lines used, and characterized by IC50 values in the low-to-submicromolar range. Mechanistically, the most potent compound 55, as well as a compound with markedly low activity, was approached by comparative cell-cycle analyses in PANC-1 cells. These studies revealed that compound 55, with improved antiproliferative activity, exhibited significantly pronounced inhibitory effects in subG1, S and G2/M phases as compared to the less cytotoxic compounds. Additionally, based on the IC50 values, the structure–activity relationships (SAR) could be exploited in rational structure-refinement in the innovation of more potent anticancer candidates with high selectivity, improved activity and well-known mechanisms of action.
Miscellaneous anticancer quinoline–chalcone hybrids
Tavares et al. synthesized novel 6-quinoline–chalcones, 56, 57, quinoline–chalcone salts, 58, and quinoline-N-oxide chalcones, 59–61. All the prepared chalcones were tested against UACC-62 (melanoma), TK-10 (renal), MCF-7 (breast) and leukemic cells, Jurkat and HL60. The results exhibited that compounds 57 and 59 were the most active against MCF-7 and TK-10, while compounds 57, 58, 60 and 61 showed the best activity against leukemic cell lines.231More recently, Kamal et al. synthesized and screened various podophyllotoxin-chalcone conjugates90,232 against A-549 (lung), A375 (melanoma), MCF-7 (breast), HT-29 (colon) and ACHN (renal) cancer cell lines using MTT assay. The results revealed that improvement in the anticancer activity was noticed in quinolone–chalcone-linked podophyllotoxins 62 with IC50 ranging from 2.2 and 15.4 μM in the cell lines screened.
Aly et al. synthesized novel derivatives of quinoline–chalcone hybrids. The target compounds were screened for their anticancer and synergistic anticancer effect with doxorubicin, using a colon cancer cell line (Caco-2). Two compounds, 63 and 64, showed significant anticancer activity with IC50 values of 5.0 and 2.5 μM, respectively.233
Kotra et al.234 synthesized a series of quinoline–chalcone derivatives by reacting quinolinyl and chloroquinolinyl acetophenones with substituted aromatic aldehydes; the compounds were found to exhibit anticancer activity. Among all compounds tested for anticancer activity on RAW cell lines using MTT assay, compound 65 (3-(4-chlorophenyl)-1-(3-methyl-1-phenyl-2-naphthyl)-2-propen-1-one) showed a percentage of inhibition up to 103%.
Kumar et al.235 designed and synthesized a new series of chalcone-fused quinoline derivatives. The results of anticancer activity against three human cancer cell lines, namely, MCF-7 (breast), A-549 (lung), and A375 (melanoma), indicated that all these newly prepared compounds had moderate to good activity on cell lines. In particular, compounds 66a–f, exhibited more potent activity than the positive control doxorubicin.
Ferrer et al.236 synthesized a series of novel [(7-chloroquinolin-4-yl)amino]chalcone derivatives and evaluated their antiproliferative activity against human prostate cancer cell lines (LNCaP). Compounds 67–70 displayed the highest cytotoxic results against human prostate cancer cell proliferation with IC50 values of 7.93 ± 2.05, 7.11 ± 2.06 and 6.95 ± 1.62 μg mL−1, respectively. The effects of these compounds were dose-dependent, showing a lower viable cell number as the drug concentration increased; this effect was also time-dependent, which was evident from 24 hours to 96 hours after drug exposure. Notably, the results showed that the presence of a hydrogen or a halogen on position 3 or 4 as the substituent groups in the aromatic ring improved the antiproliferative activity of these compounds. The antitumor activity of these compounds in human prostate cancer cells confirmed the diverse biological response of these 4-aminoquinolines.
Azad et al.237 synthesized a series of quinolinyl chalcones by the condensation of N-substituted-3-acetyl-4-hydroxyquinolin-2(1H)-ones with different aromatic aldehydes using conventional heating and ultrasound-assisted methods. The synthesized chalcone derivatives were assayed for cytotoxicity and among them, three quinoline chalcone derivatives, 71a, 71b and 71c, were found to possess significantly high cytotoxicity.
Recently, Abonia et al. reported the synthesis and anticancer screening of a new quinoline-2-one-based chalcone series on an NCI panel of 60 different human tumor cell lines. Compounds 72–75 displayed good antiproliferative activity. In particular, compound 72 exhibited remarkable activity and inhibited the proliferation of most of the used human cancer cell lines at sub-micromolar concentrations. Studying the acute toxicity showed that compound 72 was well tolerated intraperitoneally (150 mg per kg per dose) by athymic nude mice (Fig. 10).238
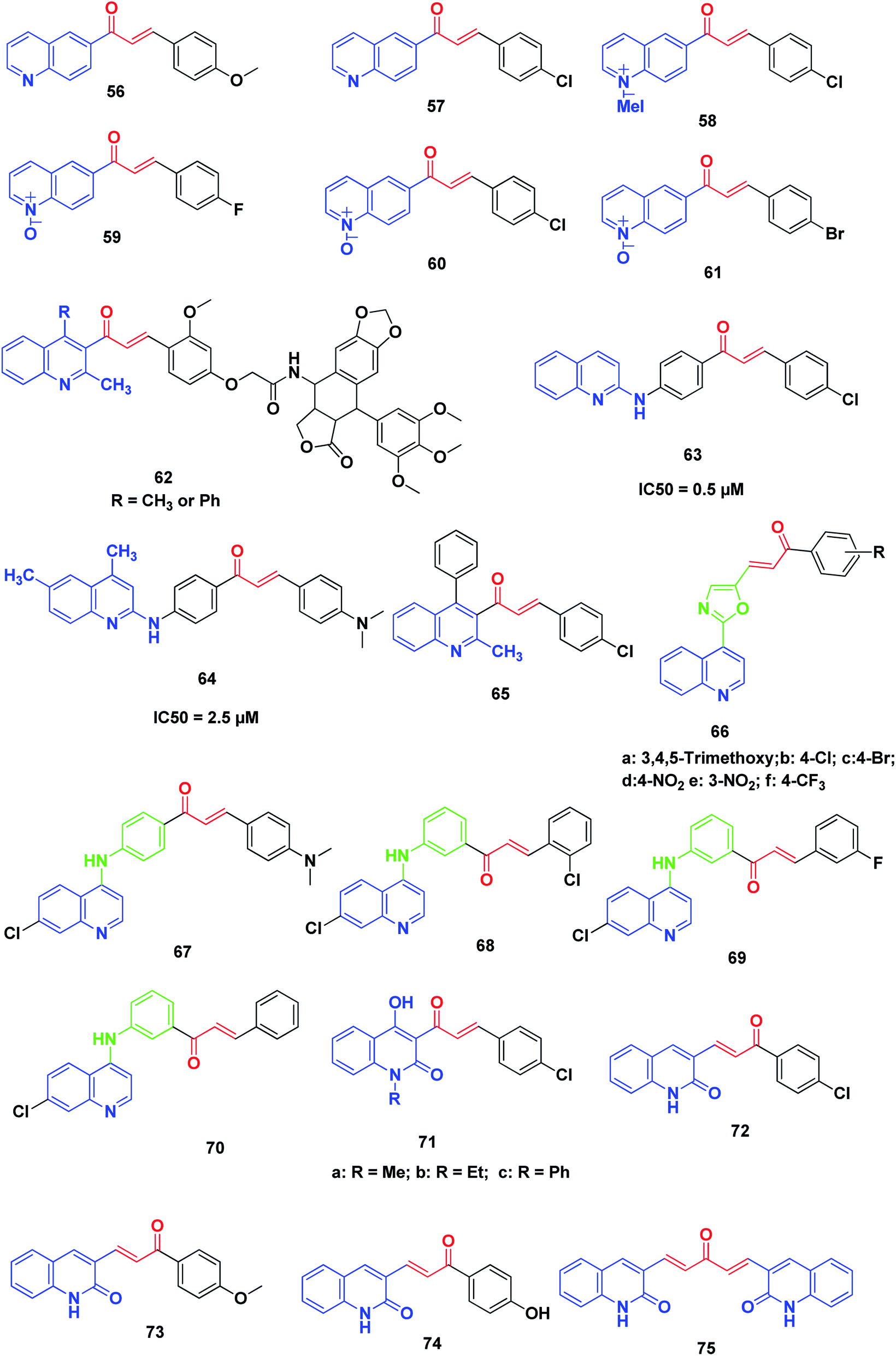 | ||
| Fig. 10 Miscellaneous anticancer quinoline–chalcone hybrids. | ||
6 Conclusion
The simple structures and ease of synthesis of chalcone derivatives, in addition to the characteristic behavior of chalcones in binding with receptors as Michael acceptors, have a great impact on their biological activity. The basic properties and extra-binding ability of the quinoline scaffold play an important role in optimizing the physicochemical properties in the drug design and development strategy. Combining quinoline with chalcone in one molecule revealed the targeted binding to the colchicine binding site of tubulin with synergistic effects and high activity against multi-drug-resistant cancer cells. Such hybrids gave potent EGFR kinase inhibitors with high potency and a good safety profile regarding normal cells. Moreover, hybridization of quinolines with chalcone analogues introduced molecules with potent anticancer activities acting through the apoptosis-inducing effect, intercalation of DNA and/or inhibition of topoisomerase I and II. Quinoline–chalcone hybrids have a prominent place in the medicinal chemistry of anticancer agents. Compounds containing the quinoline scaffold have been broadly and successfully explored, and have resulted in many drug candidates. To date, there have been no quinoline–chalcone candidates in clinical trials but in the near future, continuous efforts will provide further important discoveries in this field.Conflicts of interest
There are no conflicts to declare.References
- H. U. Rashid, Y. Xu, Y. Muhammad, L. Wang and J. Jiang, Eur. J. Med. Chem., 2019, 161, 205–238 CrossRef CAS PubMed.
- J. L. Counihan, E. A. Grossman and D. K. Nomura, Chem. Rev., 2018, 118, 6893–6923 CrossRef CAS PubMed.
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Ca-Cancer J. Clin., 2011, 61, 69–90 CrossRef PubMed.
- W. H. Organization, Cancer Control: Knowledge Into Action: WHO Guide for Effective Programmes. Policy and Advocacy. Module 6, World Health Organization, 2008 Search PubMed.
- T. Tarver, Cancer facts & figures 2012, American cancer society (ACS), Taylor & Francis, Atlanta, GA, 2012 Search PubMed.
- D. S. Shewach and R. D. Kuchta, Chem. Rev., 2009, 109, 2859–2861 CrossRef CAS PubMed.
- X. M. Chu, C. Wang, W. Liu, L. L. Liang, K. K. Gong, C. Y. Zhao and K. L. Sun, Eur. J. Med. Chem., 2019, 161, 101–117 CrossRef CAS PubMed.
- R. Musiol, Expert Opin. Drug Discovery, 2017, 12, 583–597 CrossRef CAS PubMed.
- E. H. Zhang, R. F. Wang, S. Z. Guo and B. Liu, Evid.-Based Complementary Altern. Med., 2013, 2013, 815621 Search PubMed.
- B. A. Bhat, K. L. Dhar, S. C. Puri, A. K. Saxena, M. Shanmugavel and G. N. Qazi, Bioorg. Med. Chem. Lett., 2005, 15, 3177–3180 CrossRef CAS.
- A. Lévai, Arkivok, 2005, 9, 344–352 Search PubMed.
- D. Azarifar and H. Ghasemnejad, Molecules, 2003, 8, 642–648 CrossRef CAS.
- L. Varga, T. Nagy, I. Kövesdi, J. Benet-Buchholz, G. Dormán, L. Ürge and F. Darvas, Tetrahedron, 2003, 59, 655–662 CrossRef CAS.
- Y.-M. Lin, Y. Zhou, M. T. Flavin, L.-M. Zhou, W. Nie and F.-C. Chen, Bioorg. Med. Chem., 2002, 10, 2795–2802 CrossRef CAS PubMed.
- J. Ballesteros, M. Sanz, A. Ubeda, M. Miranda, S. Iborra, M. Paya and M. d. Alcaraz, J. Med. Chem., 1995, 38, 2794–2797 CrossRef CAS.
- S. H. Lee, D. H. Sohn, X. Y. Jin, S. W. Kim, S. C. Choi and G. S. Seo, Biochem. Pharmacol., 2007, 74, 870–880 CrossRef CAS PubMed.
- H.-K. Hsieh, T.-H. Lee, J.-P. Wang, J.-J. Wang and C.-N. Lin, Pharm. Res., 1998, 15, 39–46 CrossRef CAS PubMed.
- G. Viana, M. Bandeira and F. Matos, Phytomedicine, 2003, 10, 189–195 CrossRef CAS PubMed.
- R. J. Anto, K. Sukumaran, G. Kuttan, M. Rao, V. Subbaraju and R. Kuttan, Cancer Lett., 1995, 97, 33–37 CrossRef CAS PubMed.
- I. S. Arty, H. Timmerman, M. Samhoedi and H. van der Goot, Eur. J. Med. Chem., 2000, 35, 449–457 CrossRef CAS PubMed.
- S. Mukherjee, V. Kumar, A. K. Prasad, H. G. Raj, M. E. Bracke, C. E. Olsen, S. C. Jain and V. S. Parmar, Bioorg. Med. Chem., 2001, 9, 337–345 CrossRef CAS PubMed.
- J. Vaya, P. A. Belinky and M. Aviram, Free Radicals Biol. Med., 1997, 23, 302–313 CrossRef CAS PubMed.
- W. Dan and J. Dai, Eur. J. Med. Chem., 2020, 187, 111980 CrossRef CAS PubMed.
- D. Elias, M. Beazely and N. Kandepu, Curr. Med. Chem., 1999, 6, 1125 Search PubMed.
- Z. Nowakowska, Eur. J. Med. Chem., 2007, 42, 125–137 CrossRef CAS.
- M. Go, X. Wu and X. Liu, Curr. Med. Chem., 2005, 12, 483–499 CrossRef CAS PubMed.
- D. Bhakuni and R. Chaturvedi, J. Nat. Prod., 1984, 47, 585–591 CrossRef CAS PubMed.
- P. M. Sivakumar, S. K. G. Babu and D. Mukesh, Chem. Pharm. Bull., 2007, 55, 44–49 CrossRef CAS PubMed.
- M. Liu, P. Wilairat, S. L. Croft, A. L.-C. Tan and M.-L. Go, Bioorg. Med. Chem., 2003, 11, 2729–2738 CrossRef CAS.
- V. J. Ram, A. S. Saxena, S. Srivastava and S. Chandra, Bioorg. Med. Chem. Lett., 2000, 10, 2159–2161 CrossRef CAS.
- X. Wu, P. Wilairat and M.-L. Go, Bioorg. Med. Chem. Lett., 2002, 12, 2299–2302 CrossRef CAS.
- T. Narender, K. Tanvir, M. S. Rao, K. Srivastava and S. Puri, Bioorg. Med. Chem. Lett., 2005, 15, 2453–2455 CrossRef CAS.
- J. C. Trivedi, J. B. Bariwal, K. D. Upadhyay, Y. T. Naliapara, S. K. Joshi, C. C. Pannecouque, E. De Clercq and A. K. Shah, Tetrahedron Lett., 2007, 48, 8472–8474 CrossRef CAS.
- R. Li, X. Chen, B. Gong, P. M. Selzer, Z. Li, E. Davidson, G. Kurzban, R. E. Miller, E. O. Nuzum, J. H. McKerrow, R. J. Fletterick, S. A. Gillmor, C. S. Craik, I. D. Kuntz, F. E. Cohen and G. L. Kenyon, Bioorg. Med. Chem., 1996, 4, 1421–1427 CrossRef CAS.
- N. Tiwari and B. Dwivedi, Boll. Chim. Farm., 1989, 128, 332–335 CAS.
- Y. Xia, Z.-Y. Yang, P. Xia, K. F. Bastow, Y. Nakanishi and K.-H. Lee, Bioorg. Med. Chem. Lett., 2000, 10, 699–701 CrossRef CAS PubMed.
- H. A. Abou-Zied, B. G. Youssif, M. F. Mohamed, A. M. Hayallah and M. Abdel-Aziz, Bioorg. Chem., 2019, 89, 102997 CrossRef CAS PubMed.
- S. Ducki, R. Forrest, J. A. Hadfield, A. Kendall, N. J. Lawrence, A. T. McGown and D. Rennison, Bioorg. Med. Chem. Lett., 1998, 8, 1051–1056 CrossRef CAS PubMed.
- J. Loa, P. Chow and K. Zhang, Cancer Chemother. Pharmacol., 2009, 63, 1007–1016 CrossRef CAS PubMed.
- F. Epifano, S. Genovese, L. Menghini and M. Curini, Phytochemistry, 2007, 68, 939–953 CrossRef CAS PubMed.
- R. De Vincenzo, G. Scambia, P. P. Benedetti, F. Ranelletti, G. Bonanno, A. Ercoli, F. Delle Monache, F. Ferrari, M. Piantelli and S. Mancuso, Anti-Cancer Drug Des., 1995, 10, 481 CAS.
- M. F. Mohamed, M. S. A. Shaykoon, M. H. Abdelrahman, B. E. Elsadek, A. S. Aboraia and G. E.-D. A. Abuo-Rahma, Bioorg. Chem., 2017, 72, 32–41 CrossRef CAS PubMed.
- K. J. Jarvill-Taylor, R. A. Anderson and D. J. Graves, J. Am. Coll. Nutr., 2001, 20, 327–336 CrossRef CAS PubMed.
- R. Romagnoli, P. G. Baraldi, T. Sarkar, M. D. Carrion, C. L. Cara, O. Cruz-Lopez, D. Preti, M. A. Tabrizi, M. Tolomeo and S. Grimaudo, J. Med. Chem., 2008, 51, 1464–1468 CrossRef CAS PubMed.
- N. J. Lawrence, D. Rennison, A. T. McGown and J. A. Hadfield, Bioorg. Med. Chem. Lett., 2003, 13, 3759–3763 CrossRef CAS PubMed.
- H. Mirzaei and S. Emami, Eur. J. Med. Chem., 2016, 121, 610–639 CrossRef CAS PubMed.
- W. Li, H. Sun, S. Xu, Z. Zhu and J. Xu, Future Med. Chem., 2017, 9, 1765–1794 CrossRef CAS PubMed.
- S. Ducki, D. Rennison, M. Woo, A. Kendall, J. F. D. Chabert, A. T. McGown and N. J. Lawrence, Bioorg. Med. Chem., 2009, 17, 7698–7710 CrossRef CAS PubMed.
- O. Bueno, G. Tobajas, E. Quesada, J. Estévez-Gallego, S. Noppen, M.-J. Camarasa, J.-F. Díaz, S. Liekens, E.-M. Priego and M.-J. Pérez-Pérez, Eur. J. Med. Chem., 2018, 148, 337–348 CrossRef CAS PubMed.
- C. Dyrager, M. Wickström, M. Fridén-Saxin, A. Friberg, K. Dahlén, E. A. Wallén, J. Gullbo, M. Grøtli and K. Luthman, Bioorg. Med. Chem., 2011, 19, 2659–2665 CrossRef CAS PubMed.
- X. Huang, R. Huang, L. Li, S. Gou and H. Wang, Eur. J. Med. Chem., 2017, 132, 11–25 CrossRef CAS PubMed.
- X. Huang, R. Huang, Z. Wang, L. Li, S. Gou, Z. Liao and H. Wang, Eur. J. Med. Chem., 2018, 146, 435–450 CrossRef CAS PubMed.
- M. T. Konieczny, A. Bulakowska, D. Pirska, W. Konieczny, A. Skladanowski, M. Sabisz, M. Wojciechowski, K. Lemke, A. Pieczykolan and W. Strożek, Eur. J. Med. Chem., 2015, 89, 733–742 CrossRef CAS PubMed.
- L. Li, S. Jiang, X. Li, Y. Liu, J. Su and J. Chen, Eur. J. Med. Chem., 2018, 151, 482–494 CrossRef CAS PubMed.
- N. Shankaraiah, S. Nekkanti, U. R. Brahma, N. P. Kumar, N. Deshpande, D. Prasanna, K. R. Senwar and U. J. Lakshmi, Bioorg. Med. Chem., 2017, 25, 4805–4816 CrossRef CAS PubMed.
- G. Wang, Z. Peng, J. Zhang, J. Qiu, Z. Xie and Z. Gong, Bioorg. Chem., 2018, 78, 332–340 CrossRef CAS PubMed.
- Y.-L. Zhang, B.-Y. Li, R. Yang, L.-Y. Xia, A.-L. Fan, Y.-C. Chu, L.-J. Wang, Z.-C. Wang, A.-Q. Jiang and H.-L. Zhu, Eur. J. Med. Chem., 2019, 163, 896–910 CrossRef CAS.
- M. L. Edwards, D. M. Stemerick and P. S. Sunkara, J. Med. Chem., 1990, 33, 1948–1954 CrossRef CAS.
- B. Zhou and C. Xing, Med. Chem., 2015, 5, 388 Search PubMed.
- L. Aramburu, P. Puebla, E. Caballero, M. González, A. Vicente, M. Medarde and R. Peláez, Curr. Med. Chem., 2016, 23, 1100–1130 CrossRef PubMed.
- C. Bénard, F. Zouhiri, M. Normand-Bayle, M. Danet, D. Desmaële, H. Leh, J.-F. Mouscadet, G. Mbemba, C.-M. Thomas and S. Bonnenfant, Bioorg. Med. Chem. Lett., 2004, 14, 2473–2476 CrossRef PubMed.
- P. Zajdel, A. Partyka, K. Marciniec, A. J. Bojarski, M. Pawlowski and A. Wesolowska, Future Med. Chem., 2014, 6, 57–75 CrossRef CAS PubMed.
- K. Thomas, A. V. Adhikari, S. Telkar, I. H. Chowdhury, R. Mahmood, N. K. Pal, G. Row and E. Sumesh, Eur. J. Med. Chem., 2011, 46, 5283–5292 CrossRef CAS PubMed.
- S. A. El-Feky, Z. K. Abd El-Samii, N. A. Osman, J. Lashine, M. A. Kamel and H. K. Thabet, Bioorg. Chem., 2015, 58, 104–116 CrossRef CAS.
- S. Mukherjee and M. Pal, Drug discovery today, 2013, 18, 389–398 CrossRef CAS PubMed.
- S. Mukherjee and M. Pal, Curr. Med. Chem., 2013, 20, 4386–4410 CrossRef CAS PubMed.
- S. Pal, S. Durgadas, S. B. Nallapati, K. Mukkanti, R. Kapavarapu, C. L. Meda, K. V. Parsa and M. Pal, Bioorg. Med. Chem. Lett., 2011, 21, 6573–6576 CrossRef CAS PubMed.
- N. Muruganantham, R. Sivakumar, N. Anbalagan, V. Gunasekaran and J. T. Leonard, Biol. Pharm. Bull., 2004, 27, 1683–1687 CrossRef CAS PubMed.
- K. Kaur, M. Jain, R. P. Reddy and R. Jain, Eur. J. Med. Chem., 2010, 45, 3245–3264 CrossRef CAS.
- J. Porter, Expert Opin. Ther. Pat., 2010, 20, 159–177 CrossRef CAS.
- F. M. Yakes, J. Chen, J. Tan, K. Yamaguchi, Y. Shi, P. Yu, F. Qian, F. Chu, F. Bentzien and B. Cancilla, Mol. Cancer Ther., 2011, 10, 2298–2308 CrossRef CAS.
- R. Salazar, R. Garcia-Carbonero, S. K. Libutti, A. E. Hendifar, A. Custodio, R. Guimbaud, C. Lombard-Bohas, S. Ricci, H. J. Klümpen and J. Capdevila, Oncologist, 2018, 23, 766 CrossRef CAS PubMed.
- J. Yang, J. Nie, X. Ma, Y. Wei, Y. Peng and X. Wei, Mol. Cancer, 2019, 18, 26 CrossRef PubMed.
- N. Shobeiri, M. Rashedi, F. Mosaffa, A. Zarghi, M. Ghandadi, A. Ghasemi and R. Ghodsi, Eur. J. Med. Chem., 2016, 114, 14–23 CrossRef CAS PubMed.
- W. Li, W. Shuai, H. Sun, F. Xu, Y. Bi, J. Xu, C. Ma, H. Yao, Z. Zhu and S. Xu, Eur. J. Med. Chem., 2019, 163, 428–442 CrossRef CAS PubMed.
- A. Aoyama, R. Katayama, T. Oh-hara, S. Sato, Y. Okuno and N. Fujita, Mol. Cancer Ther., 2014, 13, 2978–2990 CrossRef CAS PubMed.
- C. Pérez-Melero, A. B. Maya, B. del Rey, R. Peláez, E. Caballero and M. Medarde, Bioorg. Med. Chem. Lett., 2004, 14, 3771–3774 CrossRef PubMed.
- D. Carta and M. Grazia Ferlin, Curr. Top. Med. Chem., 2014, 14, 2322–2345 CrossRef CAS PubMed.
- M. S. Abdelbaset, G. E.-D. A. Abuo-Rahma, M. H. Abdelrahman, M. Ramadan, B. G. Youssif, S. N. A. Bukhari, M. F. Mohamed and M. Abdel-Aziz, Bioorg. Chem., 2018, 80, 151–163 CrossRef CAS PubMed.
- L. Zhu, K. Luo, K. Li, Y. Jin and J. Lin, Bioorg. Med. Chem., 2017, 25, 5939–5951 CrossRef CAS PubMed.
- S.-B. Wang, X.-F. Wang, B. Qin, E. Ohkoshi, K.-Y. Hsieh, E. Hamel, M.-T. Cui, D.-Q. Zhu, M. Goto and S. L. Morris-Natschke, Bioorg. Med. Chem., 2015, 23, 5740–5747 CrossRef CAS PubMed.
- I. Khelifi, T. Naret, D. Renko, A. Hamze, G. Bernadat, J. Bignon, C. Lenoir, J. Dubois, J.-D. Brion and O. Provot, Eur. J. Med. Chem., 2017, 127, 1025–1034 CrossRef CAS PubMed.
- V. R Solomon and H. Lee, Curr. Med. Chem., 2011, 18, 1488–1508 CrossRef PubMed.
- D. I. A. Othman, K. B. Selim, M. A. El-Sayed, A. S. Tantawy, Y. Amen, K. Shimizu, T. Okauchi and M. Kitamura, Bioorg. Med. Chem., 2019, 27, 115026 CrossRef PubMed.
- R. Hamdy, S. A. Elseginy, N. I. Ziedan, A. T. Jones and A. D. Westwell, Molecules, 2019, 24, 1274 CrossRef PubMed.
- O. Afzal, S. Kumar, M. R. Haider, M. R. Ali, R. Kumar, M. Jaggi and S. Bawa, Eur. J. Med. Chem., 2015, 97, 871–910 CrossRef CAS PubMed.
- V. Sharma, D. K. Mehta and R. Das, Mini-Rev. Med. Chem., 2017, 17, 1557–1572 CAS.
- M. R. Selim, M. A. Zahran, A. Belal, M. S. Abusaif, S. A. Shedid, A. Mehany, G. A. Elhagali and Y. A. Ammar, Anti-Cancer Agents Med. Chem., 2019, 19, 439–452 CrossRef CAS PubMed.
- N. K Sahu, S. S Balbhadra, J. Choudhary and D. V Kohli, Curr. Med. Chem., 2012, 19, 209–225 CrossRef PubMed.
- P. Singh, A. Anand and V. Kumar, Eur. J. Med. Chem., 2014, 85, 758–777 CrossRef CAS PubMed.
- V. Sharma, V. Kumar and P. Kumar, Anti-Cancer Agents Med. Chem., 2013, 13, 422–432 CAS.
- A. Boumendjel, X. Ronot and J. Boutonnat, Curr. Drug Targets, 2009, 10, 363–371 CrossRef PubMed.
- A. J Leon-Gonzalez, N. Acero, D. Muñoz-Mingarro, I. Navarro and C. Martín-Cordero, Curr. Med. Chem., 2015, 22, 3407–3425 CrossRef PubMed.
- D. K. Mahapatra, V. Asati and S. K. Bharti, Eur. J. Med. Chem., 2015, 92, 839–865 CrossRef CAS PubMed.
- D. K. Mahapatra, S. K. Bharti and V. Asati, Eur. J. Med. Chem., 2015, 101, 496–524 CrossRef CAS.
- D. K. Mahapatra, S. K. Bharti and V. Asati, Eur. J. Med. Chem., 2015, 98, 69–114 CrossRef CAS PubMed.
- A. Kamal, M. Kashi Reddy and A. Viswanath, Expert Opin. Drug Discovery, 2013, 8, 289–304 CrossRef CAS PubMed.
- M. J. Matos, S. Vazquez-Rodriguez, E. Uriarte and L. Santana, Expert Opin. Ther. Pat., 2015, 25, 351–366 CrossRef CAS PubMed.
- M. Das and K. Manna, J. Toxicol., 2016, 2016, 1–14 Search PubMed.
- D. K. Mahapatra and S. K. Bharti, Life Sci., 2016, 148, 154–172 CrossRef CAS PubMed.
- S. Nasir Abbas Bukhari, S. G. Franzblau, I. Jantan and M. Jasamai, Med. Chem., 2013, 9, 897–903 CrossRef PubMed.
- S. Nasir Abbas Bukhari, M. Jasamai and I. Jantan, Mini-Rev. Med. Chem., 2012, 12, 1394–1403 Search PubMed.
- C. Kontogiorgis, M. Mantzanidou and D. Hadjipavlou-Litina, Mini-Rev. Med. Chem., 2008, 8, 1224 CrossRef CAS PubMed.
- D. Kumar, M. Kumar, A. Kumar and S. Kumar Singh, Mini-Rev. Med. Chem., 2013, 13, 2116–2133 CrossRef CAS.
- H. H. Mohammed, G. E.-D. A. Abuo-Rahma, S. H. Abbas and E.-S. Abdelhafez, Curr. Med. Chem., 2019, 26, 3132–3149 CrossRef CAS PubMed.
- L. M. Nainwal, S. Tasneem, W. Akhtar, G. Verma, M. F. Khan, S. Parvez, M. Shaquiquzzaman, M. Akhter and M. M. Alam, Eur. J. Med. Chem., 2019, 164, 121–170 CrossRef CAS PubMed.
- J. H. Xu, Y. L. Fan and J. Zhou, J. Heterocycl. Chem., 2018, 55, 1854–1862 CrossRef CAS.
- Q.-C. Ren, C. Gao, Z. Xu, L.-S. Feng, M.-L. Liu, X. Wu and F. Zhao, Curr. Top. Med. Chem., 2018, 18, 101–113 CrossRef CAS PubMed.
- K. Higuchi, T. Watanabe, T. Tanigawa, K. Tominaga, Y. Fujiwara and T. Arakawa, J. Gastroenterol. Hepatol., 2010, 25, S155–S160 CrossRef CAS.
- R. Beltramino, A. Penenory and A. M. Buceta, Angiology, 2000, 51, 535–544 CrossRef CAS PubMed.
- C. Zhuang, W. Zhang, C. Sheng, W. Zhang, C. Xing and Z. Miao, Chem. Rev., 2017, 117, 7762–7810 CrossRef CAS PubMed.
- M. Funakoshi-Tago, K. Okamoto, R. Izumi, K. Tago, K. Yanagisawa, Y. Narukawa, F. Kiuchi, T. Kasahara and H. Tamura, Int. Immunopharmacol., 2015, 25, 189–198 CrossRef CAS.
- S. K. Jung, M.-H. Lee, J. E. Kim, P. Singh, S.-Y. Lee, C.-H. Jeong, T.-G. Lim, H. Chen, Y.-I. Chi and J. K. Kundu, J. Biol. Chem., 2014, 289, 35839–35848 CrossRef CAS.
- Z. Wang, N. Wang, S. Han, D. Wang, S. Mo, L. Yu, H. Huang, K. Tsui, J. Shen and J. Chen, PLoS One, 2013, 8, e68566 CrossRef CAS.
- E.-B. Yang, K. Zhang, L. Y. Cheng and P. Mack, Biochem. Biophys. Res. Commun., 1998, 245, 435–438 CrossRef CAS PubMed.
- E. B. Yang, Y. J. Guo, K. Zhang, Y. Z. Chen and P. Mack, BBA, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2001, 1550, 144–152 CrossRef CAS.
- J. C. Milne and J. M. Denu, Curr. Opin. Chem. Biol., 2008, 12, 11–17 CrossRef CAS.
- C. C. de Castro, P. S. Costa, G. T. Laktin, P. H. de Carvalho, R. B. Geraldo, J. de Moraes, P. L. Pinto, M. R. Couri, P. d. F. Pinto and A. A. Da Silva Filho, Phytomedicine, 2015, 22, 921–928 CrossRef CAS PubMed.
- M. Washiyama, Y. Sasaki, T. Hosokawa and S. Nagumo, Biol. Pharm. Bull., 2009, 32, 941–944 CrossRef CAS PubMed.
- L.-c. Fu, X.-a. Huang, Z.-y. Lai, Y.-j. Hu, H.-j. Liu and X.-l. Cai, Molecules, 2008, 13, 1923–1930 CrossRef CAS PubMed.
- Y. Cui, M. Ao, J. Hu and L. Yu, Z. Naturforsch., C: J. Biosci., 2008, 63, 361–365 CAS.
- M. Funakoshi-Tago, S. Tanabe, K. Tago, H. Itoh, T. Mashino, Y. Sonoda and T. Kasahara, Mol. Pharmacol., 2009, 76, 745–753 CrossRef CAS PubMed.
- W. Wu, H. Ye, L. Wan, X. Han, G. Wang, J. Hu, M. Tang, X. Duan, Y. Fan and S. He, Carcinogenesis, 2013, 34, 1636–1643 CrossRef CAS PubMed.
- M. N. Gomes, E. N. Muratov, M. Pereira, J. C. Peixoto, L. P. Rosseto, P. V. Cravo, C. H. Andrade and B. J. Neves, Molecules, 2017, 22, 1210 CrossRef PubMed.
- D. G. Powers, D. S. Casebier, D. Fokas, W. J. Ryan, J. R. Troth and D. L. Coffen, Tetrahedron, 1998, 54, 4085–4096 CrossRef CAS.
- S. R. Waldvogel, Synthesis, 2010, 50(05), 892 CrossRef.
- A. T. Nielsen and W. J. Houlihan, Org. React., 2004, 16, 1–438 Search PubMed.
- F. Fringuelli, F. Pizzo, C. Vittoriani and L. Vaccaro, Chem. Commun., 2004, 2756–2757 RSC.
- Y. Srivastava, Rasayan J. Chem., 2008, 1, 884–886 CAS.
- D. Kakati and J. C. Sarma, Chem. Cent. J., 2011, 5, 8 CrossRef CAS PubMed.
- Q.-B. Song, X.-N. Li, T.-H. Shen, S.-D. Yang, G.-R. Qiang, X.-L. Wu and Y.-X. Ma, Synth. Commun., 2003, 33, 3935–3941 CrossRef CAS.
- T. Narender and K. P. Reddy, Tetrahedron Lett., 2007, 48, 3177–3180 CrossRef CAS.
- D. S. Breslow and C. R. Hauser, J. Am. Chem. Soc., 1940, 62, 2385–2388 CrossRef CAS.
- Y.-S. Kil, S.-K. Choi, Y.-S. Lee, M. Jafari and E.-K. Seo, J. Nat. Prod., 2015, 78, 2481–2487 CrossRef CAS PubMed.
- Y. Ren, C. Yuan, Y. Qian, H.-B. Chai, X. Chen, M. Goetz and A. D. Kinghorn, J. Nat. Prod., 2014, 77, 550–556 CrossRef CAS PubMed.
- D. N. Dhar, The chemistry of chalcones and related compounds, John Wiley & Sons, 1981 Search PubMed.
- O. Petrov, Y. Ivanova and M. Gerova, Catal. Commun., 2008, 9, 315–316 CrossRef CAS.
- D. Armesto, W. M. Horspool, N. Martin, A. Ramos and C. Seoane, J. Org. Chem., 1989, 54, 3069–3072 CrossRef CAS.
- C. Tan, R. Zheng, S. Luo and Q. Xin, Chem. Ind., 2012, 41, 23–25 CAS.
- D. D. Patel and J. M. Lee, Chem. Rec., 2012, 12, 329–355 CrossRef CAS PubMed.
- N. M. Rateb and H. F. Zohdi, Synth. Commun., 2009, 39, 2789–2794 CrossRef CAS.
- J. B. Daskiewicz, G. Comte, D. Barron, A. Di Pietro and F. Thomasson, Tetrahedron Lett., 1999, 40, 7095–7098 CrossRef CAS.
- X. Liu and D. Shi, Appl. Chem. Ind., 2009, 38, 1210–1213 CAS.
- S. d. Sebti, A. Solhy, A. Smahi, A. Kossir and H. Oumimoun, Catal. Commun., 2002, 3, 335–339 CrossRef CAS.
- S. d. Sebti, A. Solhy, R. Tahir, S. d. Boulaajaj, J. A. Mayoral, J. M. Fraile, A. Kossir and H. Oumimoun, Tetrahedron Lett., 2001, 42, 7953–7955 CrossRef CAS.
- P. Kulkarni, Curr. Microwave Chem., 2015, 2, 144–149 CrossRef CAS.
- N. Calloway and L. D. Green, J. Am. Chem. Soc., 1937, 59, 809–811 CrossRef CAS.
- J. Yang, G. Ji, H. Liu and Y. Lian, Chemical Abstracts, 2013 Search PubMed; J. Yang, G. Ji, H. Liu and Y. Lian, CN Patent 103408494A, Chem. Abstr., 2013, 160, 33884 Search PubMed.
- K. Kankanala, L. Venkata Reddy, V. Ranga Reddy, K. Mukkanti and S. Pal, Lett. Org. Chem., 2011, 8, 53–59 CrossRef CAS.
- T. Mukaiyama, Organic reactions, 1982 Search PubMed.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- S. Eddarir, N. Cotelle, Y. Bakkour and C. Rolando, Tetrahedron Lett., 2003, 44, 5359–5363 CrossRef CAS.
- N. A. Bumagin and D. N. Korolev, Tetrahedron Lett., 1999, 40, 3057–3060 CrossRef CAS.
- M. Haddach and J. R. McCarthy, Tetrahedron Lett., 1999, 40, 3109–3112 CrossRef CAS.
- K. R. Buszek and N. Brown, Org. Lett., 2007, 9, 707–710 CrossRef CAS PubMed.
- R. F. Heck and J. Nolley Jr, J. Org. Chem., 1972, 37, 2320–2322 CrossRef CAS.
- T. Mizoroki, K. Mori and A. Ozaki, Bull. Chem. Soc. Jpn., 1971, 44, 581 CrossRef CAS.
- G. Zou, J. Guo, Z. Wang, W. Huang and J. Tang, Dalton Trans., 2007, 3055–3064 RSC.
- A. Bianco, C. Cavarischia and M. Guiso, Eur. J. Org. Chem., 2004, 2004, 2894–2898 CrossRef.
- X.-F. Wu, H. Neumann, A. Spannenberg, T. Schulz, H. Jiao and M. Beller, J. Am. Chem. Soc., 2010, 132, 14596–14602 CrossRef CAS PubMed.
- X. F. Wu, H. Neumann and M. Beller, Chem.–Asian J., 2012, 7, 282–285 CrossRef CAS PubMed.
- X. F. Wu, H. Neumann and M. Beller, Angew. Chem., 2010, 122, 5412–5416 CrossRef.
- P. Hermange, T. M. Gøgsig, A. T. Lindhardt, R. H. Taaning and T. Skrydstrup, Org. Lett., 2011, 13, 2444–2447 CrossRef CAS PubMed.
- A. Bianco, C. Cavarischia, A. Farina, M. Guiso and C. Marra, Tetrahedron Lett., 2003, 44, 9107–9109 CrossRef CAS.
- H. J. Bestmann and B. Arnason, Chem. Ber., 1962, 95, 1513–1527 CrossRef CAS.
- F. Ramirez and S. Dershowitz, J. Org. Chem., 1957, 22, 41–45 CrossRef CAS.
- C. Xu, G. Chen and X. Huang, Org. Prep. Proced. Int., 1995, 27, 559–561 CrossRef CAS.
- A. Kumar, S. Sharma, V. D. Tripathi and S. Srivastava, Tetrahedron, 2010, 66, 9445–9449 CrossRef CAS.
- M. Nielsen, C. B. Jacobsen, N. Holub, M. W. Paixao and K. A. Jørgensen, Angew. Chem., Int. Ed., 2010, 49, 2668–2679 CrossRef CAS.
- D. A. Alonso, M. Fuensanta, C. Nájera and M. Varea, J. Org. Chem., 2005, 70, 6404–6416 CrossRef CAS.
- N. Zhang, D. Yang, W. Wei, L. Yuan, F. Nie, L. Tian and H. Wang, J. Org. Chem., 2015, 80, 3258–3263 CrossRef CAS PubMed.
- Y. Unoh, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2013, 78, 5096–5102 CrossRef CAS PubMed.
- M. Al-Masum, E. Ng and M. C. Wai, Tetrahedron Lett., 2011, 52, 1008–1010 CrossRef CAS.
- J. Zhou, G. Wu, M. Zhang, X. Jie and W. Su, Chem.–Eur. J., 2012, 18, 8032–8036 CrossRef CAS PubMed.
- J. D. Weaver, A. Recio III, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846–1913 CrossRef CAS.
- R. Shotter, K. Johnston and J. Jones, Tetrahedron, 1978, 34, 741–746 CrossRef CAS.
- M. Sisa, S. L. Bonnet, D. Ferreira and J. H. Van der Westhuizen, Molecules, 2010, 15, 5196–5245 CrossRef CAS.
- H. Obara and H. Takahashi, Bull. Chem. Soc. Jpn., 1967, 40, 1012 CrossRef CAS.
- H. Obara, H. Takahashi and H. Hirano, Bull. Chem. Soc. Jpn., 1969, 42, 560–561 CrossRef CAS.
- J.-i. Onodera and H. Obara, Bull. Chem. Soc. Jpn., 1974, 47, 240–241 CrossRef CAS.
- V. K. Bhatia and J. Kagan, A photochemical synthesis of 2′,6′-dihydroxy-4′-methoxy- and 2′,4′- dihydroxy-6′-methoxychalcones, Chemistry & Industry, Belgrave Square, London SW1X 8PS, 1970, vol. 14, pp. 1203–1204 Search PubMed.
- V. Ramakrishnan and J. Kagan, J. Org. Chem., 1970, 35, 2901–2904 CrossRef CAS.
- Y. Li and D. Chen, Chin. J. Chem., 2011, 29, 2086–2090 CrossRef CAS.
- M. S. Kwon, N. Kim, S. H. Seo, I. S. Park, R. K. Cheedrala and J. Park, Angew. Chem., Int. Ed., 2005, 44, 6913–6915 CrossRef CAS.
- S. Kim, S. W. Bae, J. S. Lee and J. Park, Tetrahedron, 2009, 65, 1461–1466 CrossRef CAS.
- A. T. Dinkova-Kostova, M. A. Massiah, R. E. Bozak, R. J. Hicks and P. Talalay, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3404–3409 CrossRef CAS.
- A. Dinkova-Kostova, J. Cheah, A. Samouilov, J. Zweier, R. Bozak, R. Hicks and P. Talalay, Med. Chem., 2007, 3, 261–268 CrossRef CAS.
- F.-F. Gan, K. K. Kaminska, H. Yang, C.-Y. Liew, P.-C. Leow, C.-L. So, L. N. Tu, A. Roy, C.-W. Yap and T.-S. Kang, Antioxid. Redox Signaling, 2013, 19, 1149–1165 CrossRef CAS PubMed.
- A. T. Dinkova-Kostova, C. Abeygunawardana and P. Talalay, J. Med. Chem., 1998, 41, 5287–5296 CrossRef CAS PubMed.
- B. Srinivasan, T. E. Johnson, R. Lad and C. Xing, J. Med. Chem., 2009, 52, 7228–7235 CrossRef CAS PubMed.
- L. Munoz, Nat. Rev. Drug Discovery, 2017, 16, 424 CrossRef CAS PubMed.
- E. Komlodi-Pasztor, D. Sackett, J. Wilkerson and T. Fojo, Nat. Rev. Clin. Oncol., 2011, 8, 244–250 CrossRef CAS PubMed.
- A. L. Parker, M. Kavallaris and J. A. McCarroll, Front. Oncol., 2014, 4, 153 Search PubMed.
- M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS PubMed.
- Z.-l. Liu, W. Tian, Y. Wang, S. Kuang, X.-m. Luo and Q. Yu, Acta Pharmacol. Sin., 2012, 33, 261–270 CrossRef CAS PubMed.
- K. Chan, C. G. Koh and H. Li, Cell Death Dis., 2012, 3, e411 CrossRef PubMed.
- C. Dumontet and M. A. Jordan, Nat. Rev. Drug Discovery, 2010, 9, 790–803 CrossRef CAS PubMed.
- C.-H. Tseng, C.-C. Tzeng, C.-Y. Hsu, C.-M. Cheng, C.-N. Yang and Y.-L. Chen, Eur. J. Med. Chem., 2015, 97, 306–319 CrossRef CAS PubMed.
- W. Li, F. Xu, W. Shuai, H. Sun, H. Yao, C. Ma, S. Xu, H. Yao, Z. Zhu and D.-H. Yang, J. Med. Chem., 2018, 62, 993–1013 CrossRef PubMed.
- S. Mirzaei, F. Hadizadeh, F. Eisvand, F. Mosaffa and R. Ghodsi, J. Mol. Struct., 2020, 1202, 127310 CrossRef CAS.
- H. Lee, P. Trivedi, C. Karthikeyan and I. K. Lindamulage, Quinolone chalcone compounds and uses thereof, US Pat., Application 15/777,468, 2018.
- Y. Teng, K. Lu, Q. Zhang, L. Zhao, Y. Huang, A. M. Ingarra, H. Galons, T. Li, S. Cui and P. Yu, Eur. J. Med. Chem., 2019, 183, 111641 CrossRef CAS PubMed.
- Y. Yarden and G. Pines, Nat. Rev. Cancer, 2012, 12, 553–563 CrossRef CAS PubMed.
- M. Shibuya, J. Biochem., 2013, 153, 13–19 CrossRef CAS PubMed.
- S. U. F. Rizvi, H. L. Siddiqui, M. Nisar, N. Khan and I. Khan, Bioorg. Med. Chem. Lett., 2012, 22, 942–944 CrossRef CAS PubMed.
- E. Barile, S. K. De, Y. Feng, V. Chen, L. Yang, Z. e. Ronai and M. Pellecchia, Chem. Biol. Drug Des., 2013, 82, 520–533 CrossRef CAS PubMed.
- R. F. George, E. M. Samir, M. N. Abdelhamed, H. A. Abdel-Aziz and S. E. Abbas, Bioorg. Chem., 2019, 83, 186–197 CrossRef CAS PubMed.
- M. S. Abdelbaset, M. Abdel-Aziz, M. Ramadan, M. H. Abdelrahman, S. N. A. Bukhari, T. F. Ali and G. E.-D. A. Abuo-Rahma, Bioorg. Med. Chem., 2019, 27, 1076–1086 CrossRef CAS PubMed.
- D. A. Ibrahim, D. A. Abou El Ella, A. M. El-Motwally and R. M. Aly, Eur. J. Med. Chem., 2015, 102, 115–131 CrossRef CAS PubMed.
- R. M. Aly, R. A. Serya, A. M. El-Motwally, A. Esmat, S. Abbas and D. A. Abou El Ella, Bioorg. Chem., 2017, 75, 368–392 CrossRef CAS PubMed.
- S. H. Abbas, A. A. Abd El-Hafeez, M. E. Shoman, M. M. Montano and H. A. Hassan, Bioorg. Chem., 2019, 82, 360–377 CrossRef CAS PubMed.
- L. Li, W. Cao, W. Zheng, C. Fan and T. Chen, Dalton Trans., 2012, 41, 12766–12772 RSC.
- A. Rescifina, C. Zagni, M. G. Varrica, V. Pistarà and A. Corsaro, Eur. J. Med. Chem., 2014, 74, 95–115 CrossRef CAS PubMed.
- J. Chaires, Curr. Opin. Struct. Biol., 1998, 314, 8 Search PubMed.
- L. Dalla Via, O. Gia, G. Chiarelotto and M. G. Ferlin, Eur. J. Med. Chem., 2009, 44, 2854–2861 CrossRef CAS PubMed.
- A. Kamal, B. R. Prasad and A. M. Reddy, Chalcone linked pyrrolo [2,1-C][1, 4] benzodiazepine hybrids as potential anticancer agents and process for the preparation thereof, US Pat., 8,461,150, issued June 11, 2013.
- C. Karthikeyan, N. S. H. Narayana Moorthy, S. Ramasamy, U. Vanam, E. Manivannan, D. Karunagaran and P. Trivedi, Recent Pat. Anti-Cancer Drug Discovery, 2015, 10, 97–115 CrossRef CAS.
- P. Bindu, K. Mahadevan, T. R. Naik and B. Harish, MedChemComm, 2014, 5, 1708–1717 RSC.
- P. Bindu, K. Mahadevan and T. R. Naik, Bioorg. Med. Chem. Lett., 2012, 22, 6095–6098 CrossRef CAS PubMed.
- J. C. Wang, Nat. Rev. Mol. Cell Biol., 2002, 3, 430–440 CrossRef CAS PubMed.
- R. J. Lewis, F. T. Tsai and D. B. Wigley, Bioessays, 1996, 18, 661–671 CrossRef CAS PubMed.
- A. Maxwell, DNA gyrase as a drug target, Portland Press Ltd., 1999, pp. 48–53 Search PubMed.
- T.-K. Li and L. F. Liu, Annu. Rev. Pharmacol. Toxicol., 2001, 41, 53–77 CrossRef CAS PubMed.
- M. Abdel-Aziz, S.-E. Park, G. E.-D. A. Abuo-Rahma, M. A. Sayed and Y. Kwon, Eur. J. Med. Chem., 2013, 69, 427–438 CrossRef CAS PubMed.
- B. A. Carneiro and W. S. El-Deiry, Nat. Rev. Clin. Oncol., 2020, 1–23 Search PubMed.
- C.-H. Tseng, Y.-L. Chen, C.-Y. Hsu, T.-C. Chen, C.-M. Cheng, H.-C. Tso, Y.-J. Lu and C.-C. Tzeng, Eur. J. Med. Chem., 2013, 59, 274–282 CrossRef CAS PubMed.
- S. Raghavan, P. Manogaran, K. K. G. Narasimha, B. K. Kuppusami, P. Mariyappan, A. Gopalakrishnan and G. Venkatraman, Bioorg. Med. Chem. Lett., 2015, 25, 3601–3605 CrossRef CAS PubMed.
- A. R. Martirosyan, R. Rahim-Bata, A. B. Freeman, C. D. Clarke, R. L. Howard and J. S. Strobl, Biochem. Pharmacol., 2004, 68, 1729–1738 CrossRef CAS.
- S. Jain, V. Chandra, P. K. Jain, K. Pathak, D. Pathak and A. Vaidya, Arabian J. Chem., 2019, 12, 4920–4946 CrossRef CAS.
- T. Jernei, C. Duró, A. Dembo, E. Lajkó, A. Takács, L. Kőhidai, G. Schlosser and A. Csámpai, Molecules, 2019, 24, 4077 CrossRef PubMed.
- L. de Carvalho Tavares, S. Johann, T. M. de Almeida Alves, J. C. Guerra, E. M. de Souza-Fagundes, P. S. Cisalpino, A. J. Bortoluzzi, G. F. Caramori, R. de Mattos Piccoli and H. T. Braibante, Eur. J. Med. Chem., 2011, 46, 4448–4456 CrossRef.
- A. Kamal, A. Mallareddy, P. Suresh, V. L. Nayak, R. V. Shetti, N. S. Rao, J. R. Tamboli, T. B. Shaik, M. Vishnuvardhan and S. Ramakrishna, Eur. J. Med. Chem., 2012, 47, 530–545 CrossRef CAS.
- M. R. Aly, E.-S. I. Ibrahim, F. A. El Shahed, H. A. Soliman, Z. S. Ibrahim and S. A. El-Shazly, Russ. J. Bioorg. Chem., 2012, 38, 428–434 CrossRef CAS.
- V. Kotra, S. Ganapaty and S. R. Adapa, Indian J. Chem., 2010, 49B, 1109–1116 CAS.
- L. Kumar, A. Jha and G. Sridhar, Chem. Data Collect., 2019, 22, 100236 CrossRef.
- R. Ferrer, G. Lobo, N. Gamboa, J. Rodrigues, C. Abramjuk, K. Jung, M. Lein and J. E. Charris, Sci. Pharm., 2009, 77, 725–742 CrossRef CAS.
- M. Azad, M. A. Munawar and M. Athar, J. Appl. Sci., 2007, 7, 1620–1625 CrossRef CAS.
- R. Abonia, D. Insuasty, J. Castillo, B. Insuasty, J. Quiroga, M. Nogueras and J. Cobo, Eur. J. Med. Chem., 2012, 57, 29–40 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05594h |
| This journal is © The Royal Society of Chemistry 2020 |